
Evidence for Recombinant GRP78, CALR, PDIA3 and GPI as Mediators of Genetic Instability in Human CD34+ Cells

 , ,
, , 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Preparation of Femoral Heads
2.2. Isolation of Human CD34+ Cells
2.3. Culture of CD34+ Cells
2.4. Analysis of Genetic Instability in CD34+ Cells
2.5. Statistical Analysis
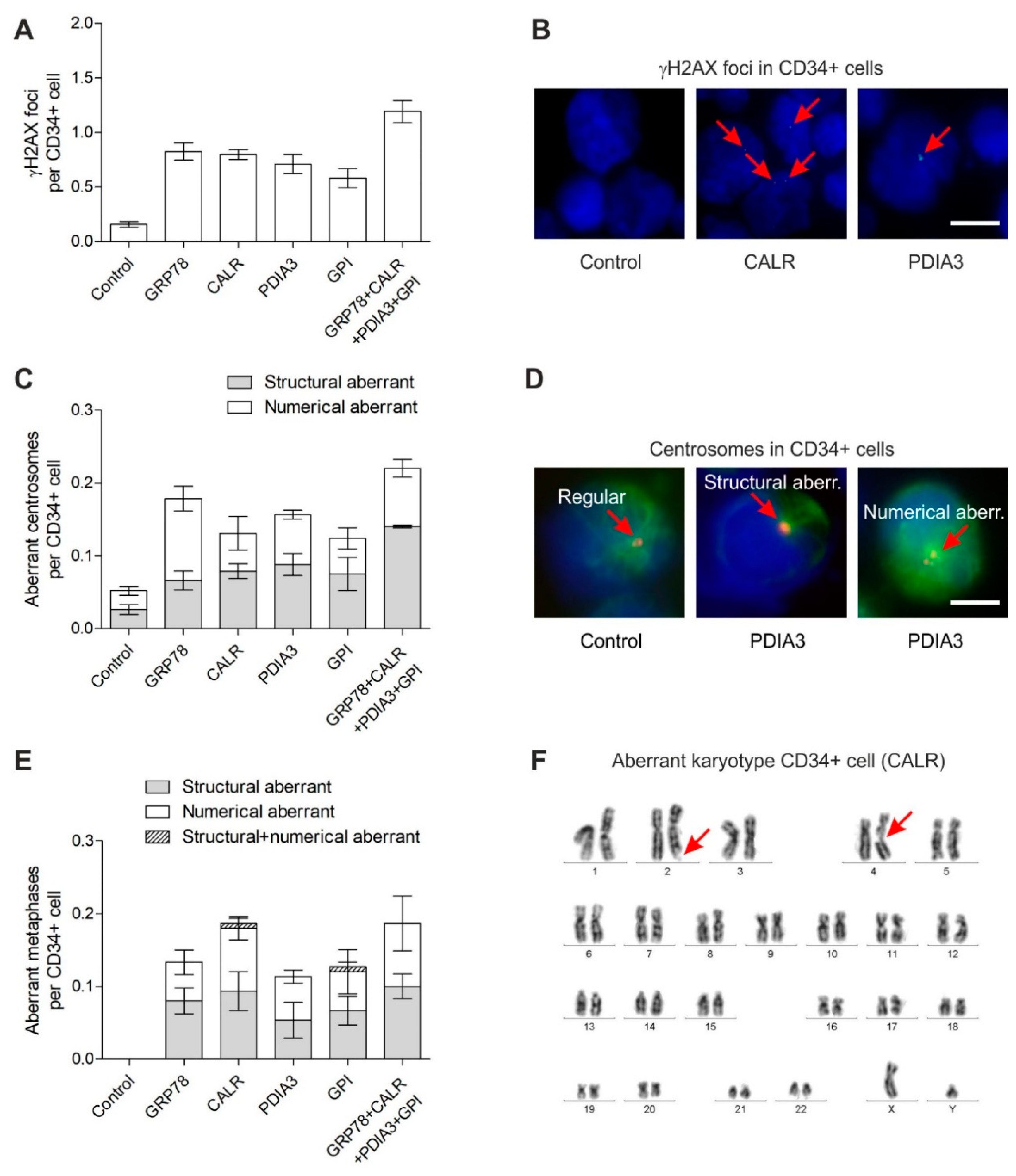
3. Results
3.1. DNA Damage in CD34+ Cells
3.2. Centrosome Aberrations in CD34+ Cells
3.3. Chromosomal Instability in CD34+ Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hu, L.; Yin, X.; Zhang, Y.; Pang, A.; Xie, X.; Yang, S.; Zhu, C.; Li, Y.; Zhang, B.; Huang, Y.; et al. Radiation-induced bystander effects impair transplanted human hematopoietic stem cells via oxidative DNA damage. Blood 2021, 137, 3339–3350. [Google Scholar] [CrossRef] [PubMed]
- Mothersill, C.; Rusin, A.; Seymour, C. Relevance of Non-Targeted Effects for Radiotherapy and Diagnostic Radiology; A Historical and Conceptual Analysis of Key Players. Cancers 2019, 11, 1236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikitaki, Z.; Mavragani, I.V.; Laskaratou, D.A.; Gika, V.; Moskvin, V.P.; Theofilatos, K.; Vougas, K.; Stewart, R.D.; Georgakilas, A.G. Systemic mechanisms and effects of ionizing radiation: A new ‘old’ paradigm of how the bystanders and distant can become the players. Semin. Cancer Biol. 2016, 37–38, 77–95. [Google Scholar] [CrossRef] [PubMed]
- Heeran, A.B.; Berrigan, H.P.; O’Sullivan, J. The Radiation-Induced Bystander Effect (RIBE) and its Connections with the Hallmarks of Cancer. Radiat. Res. 2019, 192, 668–679. [Google Scholar] [CrossRef] [PubMed]
- Lorimore, S.A.; McIlrath, J.M.; Coates, P.J.; Wright, E.G. Chromosomal instability in unirradiated hemopoietic cells resulting from a delayed in vivo bystander effect of gamma radiation. Cancer Res. 2005, 65, 5668–5673. [Google Scholar] [CrossRef] [Green Version]
- Lorimore, S.A.; Chrystal, J.A.; Robinson, J.I.; Coates, P.J.; Wright, E.G. Chromosomal instability in unirradiated hemaopoietic cells induced by macrophages exposed in vivo to ionizing radiation. Cancer Res. 2008, 68, 8122–8126. [Google Scholar] [CrossRef] [Green Version]
- Kohl, V.; Drews, O.; Costina, V.; Bierbaum, M.; Jawhar, A.; Roehl, H.; Weiss, C.; Brendel, S.; Kleiner, H.; Flach, J.; et al. Proteins Marking the Sequence of Genotoxic Signaling from Irradiated Mesenchymal Stromal Cells to CD34+ Cells. Int. J. Mol. Sci. 2021, 22, 5844. [Google Scholar] [CrossRef]
- Kohl, V.; Fabarius, A.; Drews, O.; Bierbaum, M.; Jawhar, A.; Darwich, A.; Weiss, C.; Flach, J.; Brendel, S.; Kleiner, H.; et al. Genotoxic Bystander Signals from Irradiated Human Mesenchymal Stromal Cells Mainly Localize in the 10-100 kDa Fraction of Conditioned Medium. Cells 2021, 10, 827. [Google Scholar] [CrossRef]
- Chen, S.; Zhao, Y.; Han, W.; Zhao, G.; Zhu, L.; Wang, J.; Bao, L.; Jiang, E.; Xu, A.; Hei, T.K.; et al. Mitochondria-dependent signalling pathway are involved in the early process of radiation-induced bystander effects. Br. J. Cancer 2008, 98, 1839–1844. [Google Scholar] [CrossRef] [Green Version]
- Lyng, F.M.; Howe, O.L.; McClean, B. Reactive oxygen species-induced release of signalling factors in irradiated cells triggers membrane signalling and calcium influx in bystander cells. Int. J. Radiat. Biol. 2011, 87, 683–695. [Google Scholar] [CrossRef]
- Tartier, L.; Gilchrist, S.; Burdak-Rothkamm, S.; Folkard, M.; Prise, K.M. Cytoplasmic irradiation induces mitochondrial-dependent 53BP1 protein relocalization in irradiated and bystander cells. Cancer Res. 2007, 67, 5872–5879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, C.; Stewart, V.; Folkard, M.; Michael, B.D.; Prise, K.M. Nitric oxide-mediated signaling in the bystander response of individually targeted glioma cells. Cancer Res. 2003, 63, 8437–8442. [Google Scholar] [PubMed]
- Jella, K.K.; Moriarty, R.; McClean, B.; Byrne, H.J.; Lyng, F.M. Reactive oxygen species and nitric oxide signaling in bystander cells. PLoS ONE 2018, 13, e0195371. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; He, M.; Shen, B.; Yuan, D.; Shao, C. Alpha particle-induced bystander effect is mediated by ROS via a p53-dependent SCO2 pathway in hepatoma cells. Int. J. Radiat. Biol. 2013, 89, 1028–1034. [Google Scholar] [CrossRef]
- Desai, S.; Kumar, A.; Laskar, S.; Pandey, B.N. Cytokine profile of conditioned medium from human tumor cell lines after acute and fractionated doses of gamma radiation and its effect on survival of bystander tumor cells. Cytokine 2013, 61, 54–62. [Google Scholar] [CrossRef]
- Shao, C.; Folkard, M.; Prise, K.M. Role of TGF-beta1 and nitric oxide in the bystander response of irradiated glioma cells. Oncogene 2008, 27, 434–440. [Google Scholar] [CrossRef] [Green Version]
- Gow, M.D.; Seymour, C.B.; Ryan, L.A.; Mothersill, C.E. Induction of bystander response in human glioma cells using high-energy electrons: A role for TGF-beta1. Radiat. Res. 2010, 173, 769–778. [Google Scholar] [CrossRef]
- Peng, Y.; Zhang, M.; Zheng, L.; Liang, Q.; Li, H.; Chen, J.T.; Guo, H.; Yoshina, S.; Chen, Y.Z.; Zhao, X.; et al. Cysteine protease cathepsin B mediates radiation-induced bystander effects. Nature 2017, 547, 458–462. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Ivanov, V.N.; Lien, Y.C.; Davidson, M.; Hei, T.K. Mitochondrial function and nuclear factor-kappaB-mediated signaling in radiation-induced bystander effects. Cancer Res. 2008, 68, 2233–2240. [Google Scholar] [CrossRef] [Green Version]
- Lyng, F.M.; Maguire, P.; McClean, B.; Seymour, C.; Mothersill, C. The involvement of calcium and MAP kinase signaling pathways in the production of radiation-induced bystander effects. Radiat. Res. 2006, 165, 400–409. [Google Scholar] [CrossRef]
- Xu, S.; Wang, J.; Ding, N.; Hu, W.; Zhang, X.; Wang, B.; Hua, J.; Wei, W.; Zhu, Q. Exosome-mediated microRNA transfer plays a role in radiation-induced bystander effect. RNA Biol. 2015, 12, 1355–1363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ariyoshi, K.; Miura, T.; Kasai, K.; Fujishima, Y.; Nakata, A.; Yoshida, M. Radiation-Induced Bystander Effect is Mediated by Mitochondrial DNA in Exosome-Like Vesicles. Sci. Rep. 2019, 9, 9103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirolikar, S.; Prasannan, P.; Raghuram, G.V.; Pancholi, N.; Saha, T.; Tidke, P.; Chaudhari, P.; Shaikh, A.; Rane, B.; Pandey, R.; et al. Prevention of radiation-induced bystander effects by agents that inactivate cell-free chromatin released from irradiated dying cells. Cell Death Dis. 2018, 9, 1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, C.; Furusawa, Y.; Aoki, M.; Ando, K. Role of gap junctional intercellular communication in radiation-induced bystander effects in human fibroblasts. Radiat. Res. 2003, 160, 318–323. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [Google Scholar] [CrossRef]
- Meusser, B.; Hirsch, C.; Jarosch, E.; Sommer, T. ERAD: The long road to destruction. Nat. Cell Biol. 2005, 7, 766–772. [Google Scholar] [CrossRef]
- Ni, M.; Zhang, Y.; Lee, A.S. Beyond the endoplasmic reticulum: Atypical GRP78 in cell viability, signalling and therapeutic targeting. Biochem. J. 2011, 434, 181–188. [Google Scholar] [CrossRef]
- Kern, J.; Untergasser, G.; Zenzmaier, C.; Sarg, B.; Gastl, G.; Gunsilius, E.; Steurer, M. GRP-78 secreted by tumor cells blocks the antiangiogenic activity of bortezomib. Blood 2009, 114, 3960–3967. [Google Scholar] [CrossRef] [Green Version]
- Kelber, J.A.; Panopoulos, A.D.; Shani, G.; Booker, E.C.; Belmonte, J.C.; Vale, W.W.; Gray, P.C. Blockade of Cripto binding to cell surface GRP78 inhibits oncogenic Cripto signaling via MAPK/PI3K and Smad2/3 pathways. Oncogene 2009, 28, 2324–2336. [Google Scholar] [CrossRef] [Green Version]
- Staquicini, D.I.; D’Angelo, S.; Ferrara, F.; Karjalainen, K.; Sharma, G.; Smith, T.L.; Tarleton, C.A.; Jaalouk, D.E.; Kuniyasu, A.; Baze, W.B.; et al. Therapeutic targeting of membrane-associated GRP78 in leukemia and lymphoma: Preclinical efficacy in vitro and formal toxicity study of BMTP-78 in rodents and primates. Pharm. J. 2018, 18, 436–443. [Google Scholar] [CrossRef] [Green Version]
- Hebbar, N.; Epperly, R.; Vaidya, A.; Thanekar, U.; Moore, S.E.; Umeda, M.; Ma, J.; Patil, S.L.; Langfitt, D.; Huang, S.; et al. CAR T cells redirected to cell surface GRP78 display robust anti-acute myeloid leukemia activity and do not target hematopoietic progenitor cells. Nat. Commun. 2022, 13, 587. [Google Scholar] [CrossRef] [PubMed]
- Fucikova, J.; Spisek, R.; Kroemer, G.; Galluzzi, L. Calreticulin and cancer. Cell Res. 2020, 31, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Obeid, M.; Tesniere, A.; Ghiringhelli, F.; Fimia, G.M.; Apetoh, L.; Perfettini, J.L.; Castedo, M.; Mignot, G.; Panaretakis, T.; Casares, N.; et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med. 2007, 13, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Prins, D.; Gonzalez Arias, C.; Klampfl, T.; Grinfeld, J.; Green, A.R. Mutant Calreticulin in the Myeloproliferative Neoplasms. Hemasphere 2020, 4, e333. [Google Scholar] [CrossRef]
- Bourdi, M.; Demady, D.; Martin, J.L.; Jabbour, S.K.; Martin, B.M.; George, J.W.; Pohl, L.R. cDNA cloning and baculovirus expression of the human liver endoplasmic reticulum P58: Characterization as a protein disulfide isomerase isoform, but not as a protease or a carnitine acyltransferase. Arch. Biochem. Biophys. 1995, 323, 397–403. [Google Scholar] [CrossRef]
- Oliver, J.D.; van der Wal, F.J.; Bulleid, N.J.; High, S. Interaction of the thiol-dependent reductase ERp57 with nascent glycoproteins. Science 1997, 275, 86–88. [Google Scholar] [CrossRef]
- Chichiarelli, S.; Altieri, F.; Paglia, G.; Rubini, E.; Minacori, M.; Eufemi, M. ERp57/PDIA3: New insight. Cell Mol. Biol. Lett. 2022, 27, 12. [Google Scholar] [CrossRef]
- Coe, H.; Jung, J.; Groenendyk, J.; Prins, D.; Michalak, M. ERp57 modulates STAT3 signaling from the lumen of the endoplasmic reticulum. J. Biol. Chem. 2010, 285, 6725–6738. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.; Lee, D.H. Emerging roles of protein disulfide isomerase in cancer. BMB Rep. 2017, 50, 401–410. [Google Scholar] [CrossRef] [Green Version]
- Ye, Q.; Fu, P.; Dou, J.; Wang, N. Downregulation of PDIA3 inhibits proliferation and invasion of human acute myeloid leukemia cells. Onco Targets Ther. 2018, 11, 2925–2935. [Google Scholar] [CrossRef] [Green Version]
- Funasaka, T.; Haga, A.; Raz, A.; Nagase, H. Tumor autocrine motility factor is an angiogenic factor that stimulates endothelial cell motility. Biochem. Biophys. Res. Commun. 2001, 285, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Tsutsumi, S.; Gupta, S.K.; Hogan, V.; Collard, J.G.; Raz, A. Activation of small GTPase Rho is required for autocrine motility factor signaling. Cancer Res. 2002, 62, 4484–4490. [Google Scholar] [PubMed]
- Araki, K.; Shimura, T.; Yajima, T.; Tsutsumi, S.; Suzuki, H.; Okada, K.; Kobayashi, T.; Raz, A.; Kuwano, H. Phosphoglucose isomerase/autocrine motility factor promotes melanoma cell migration through ERK activation dependent on autocrine production of interleukin-8. J. Biol. Chem. 2009, 284, 32305–32311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsutsumi, S.; Hogan, V.; Nabi, I.R.; Raz, A. Overexpression of the autocrine motility factor/phosphoglucose isomerase induces transformation and survival of NIH-3T3 fibroblasts. Cancer Res. 2003, 63, 242–249. [Google Scholar]
- Fu, M.; Li, L.; Albrecht, T.; Johnson, J.D.; Kojic, L.D.; Nabi, I.R. Autocrine motility factor/phosphoglucose isomerase regulates ER stress and cell death through control of ER calcium release. Cell Death Differ. 2011, 18, 1057–1070. [Google Scholar] [CrossRef] [Green Version]
- Heim, S.; Mitelman, F. Cancer Cytogenetics, 3rd ed.; Wiley-Blackwell: Hoboken, NJ, USA, 2009; Volume 3, pp. 9–16. [Google Scholar]
- McGowan-Jordan, J.; Hastings, R.J.; Moore, S. An International System for Human Cytogenetic Nomenclature; Karger: Basel, Switzerland, 2020. [Google Scholar]
- La, X.; Zhang, L.; Li, H.; Li, Z.; Song, G.; Yang, P.; Yang, Y. Ajuba receptor mediates the internalization of tumor-secreted GRP78 into macrophages through different endocytosis pathways. Oncotarget 2018, 9, 15464–15479. [Google Scholar] [CrossRef] [Green Version]
- Roukos, V.; Voss, T.C.; Schmidt, C.K.; Lee, S.; Wangsa, D.; Misteli, T. Spatial dynamics of chromosome translocations in living cells. Science 2013, 341, 660–664. [Google Scholar] [CrossRef]
- Popp, H.D.; Naumann, N.; Brendel, S.; Henzler, T.; Weiss, C.; Hofmann, W.K.; Fabarius, A. Increase of DNA damage and alteration of the DNA damage response in myelodysplastic syndromes and acute myeloid leukemias. Leuk Res. 2017, 57, 112–118. [Google Scholar] [CrossRef]
- Ruppenthal, S.; Kleiner, H.; Nolte, F.; Fabarius, A.; Hofmann, W.K.; Nowak, D.; Seifarth, W. Increased separase activity and occurrence of centrosome aberrations concur with transformation of MDS. PLoS ONE 2018, 13, e0191734. [Google Scholar] [CrossRef] [Green Version]
- Neben, K.; Giesecke, C.; Schweizer, S.; Ho, A.D.; Kramer, A. Centrosome aberrations in acute myeloid leukemia are correlated with cytogenetic risk profile. Blood 2003, 101, 289–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stolzel, F.; Mohr, B.; Kramer, M.; Oelschlagel, U.; Bochtler, T.; Berdel, W.E.; Kaufmann, M.; Baldus, C.D.; Schafer-Eckart, K.; Stuhlmann, R.; et al. Karyotype complexity and prognosis in acute myeloid leukemia. Blood Cancer J. 2016, 6, e386. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Goto, H.; Nishimura, Y.; Kasahara, K.; Mizoguchi, A.; Inagaki, M. Tetraploidy in cancer and its possible link to aging. Cancer Sci. 2018, 109, 2632–2640. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Wang, S.A.; DiNardo, C.; Li, S.; Hu, S.; Xu, J.; Zhou, W.; Goswami, M.; Medeiros, L.J.; Tang, G. Tetraploidy/near-tetraploidy acute myeloid leukemia. Leuk Res. 2017, 53, 20–27. [Google Scholar] [CrossRef]
- Wey, S.; Luo, B.; Tseng, C.C.; Ni, M.; Zhou, H.; Fu, Y.; Bhojwani, D.; Carroll, W.L.; Lee, A.S. Inducible knockout of GRP78/BiP in the hematopoietic system suppresses Pten-null leukemogenesis and AKT oncogenic signaling. Blood 2012, 119, 817–825. [Google Scholar] [CrossRef] [Green Version]
- Henry, M.K.; Lynch, J.T.; Eapen, A.K.; Quelle, F.W. DNA damage-induced cell-cycle arrest of hematopoietic cells is overridden by activation of the PI-3 kinase/Akt signaling pathway. Blood 2001, 98, 834–841. [Google Scholar] [CrossRef] [Green Version]
- Plo, I.; Laulier, C.; Gauthier, L.; Lebrun, F.; Calvo, F.; Lopez, B.S. AKT1 inhibits homologous recombination by inducing cytoplasmic retention of BRCA1 and RAD51. Cancer Res. 2008, 68, 9404–9412. [Google Scholar] [CrossRef] [Green Version]
- Tonic, I.; Yu, W.N.; Park, Y.; Chen, C.C.; Hay, N. Akt activation emulates Chk1 inhibition and Bcl2 overexpression and abrogates G2 cell cycle checkpoint by inhibiting BRCA1 foci. J. Biol. Chem. 2010, 285, 23790–23798. [Google Scholar] [CrossRef] [Green Version]
- Nam, H.J.; Chae, S.; Jang, S.H.; Cho, H.; Lee, J.H. The PI3K-Akt mediates oncogenic Met-induced centrosome amplification and chromosome instability. Carcinogenesis 2010, 31, 1531–1540. [Google Scholar] [CrossRef] [Green Version]
- Berenjeno, I.M.; Pineiro, R.; Castillo, S.D.; Pearce, W.; McGranahan, N.; Dewhurst, S.M.; Meniel, V.; Birkbak, N.J.; Lau, E.; Sansregret, L.; et al. Oncogenic PIK3CA induces centrosome amplification and tolerance to genome doubling. Nat. Commun. 2017, 8, 1773. [Google Scholar] [CrossRef] [Green Version]
- Onishi, K.; Higuchi, M.; Asakura, T.; Masuyama, N.; Gotoh, Y. The PI3K-Akt pathway promotes microtubule stabilization in migrating fibroblasts. Genes Cells 2007, 12, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Muller, H.; Schmidt, D.; Steinbrink, S.; Mirgorodskaya, E.; Lehmann, V.; Habermann, K.; Dreher, F.; Gustavsson, N.; Kessler, T.; Lehrach, H.; et al. Proteomic and functional analysis of the mitotic Drosophila centrosome. EMBO J. 2010, 29, 3344–3357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salati, S.; Prudente, Z.; Genovese, E.; Pennucci, V.; Rontauroli, S.; Bartalucci, N.; Mannarelli, C.; Ruberti, S.; Zini, R.; Rossi, C.; et al. Calreticulin Affects Hematopoietic Stem/Progenitor Cell Fate by Impacting Erythroid and Megakaryocytic Differentiation. Stem. Cells Dev. 2018, 27, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Kamhi-Nesher, S.; Shenkman, M.; Tolchinsky, S.; Fromm, S.V.; Ehrlich, R.; Lederkremer, G.Z. A novel quality control compartment derived from the endoplasmic reticulum. Mol. Biol. Cell 2001, 12, 1711–1723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helassa, N.; Nugues, C.; Rajamanoharan, D.; Burgoyne, R.D.; Haynes, L.P. A centrosome-localized calcium signal is essential for mammalian cell mitosis. FASEB J. 2019, 33, 14602–14610. [Google Scholar] [CrossRef] [Green Version]
- Krynetskaia, N.F.; Phadke, M.S.; Jadhav, S.H.; Krynetskiy, E.Y. Chromatin-associated proteins HMGB1/2 and PDIA3 trigger cellular response to chemotherapy-induced DNA damage. Mol. Cancer Ther. 2009, 8, 864–872. [Google Scholar] [CrossRef] [Green Version]
- Cooper, J.; Giancotti, F.G. Integrin Signaling in Cancer: Mechanotransduction, Stemness, Epithelial Plasticity, and Therapeutic Resistance. Cancer Cell 2019, 35, 347–367. [Google Scholar] [CrossRef]
- Nicolay, N.H.; Lopez Perez, R.; Saffrich, R.; Huber, P.E. Radio-resistant mesenchymal stem cells: Mechanisms of resistance and potential implications for the clinic. Oncotarget 2015, 6, 19366–19380. [Google Scholar] [CrossRef] [Green Version]
- Sugrue, T.; Lowndes, N.F.; Ceredig, R. Mesenchymal stromal cells: Radio-resistant members of the bone marrow. Immunol. Cell Biol. 2013, 91, 5–11. [Google Scholar] [CrossRef]


{kind=link}
| Pt | Age/ Sex | Cytogenetics (ISCN) CD34+ Cells | |||||
|---|---|---|---|---|---|---|---|
| Control | +GRP78 | +CALR | +PDIA3 | +GPI | +GRP78 + CALR +PDIA3 + GPI | ||
| #1 | 78/♀ | 46,XX[25] | 46,XX[23] 46,XX,chtb(2q)[1] 92,XXXX[1] | 46,XX[20] 46,XX,chtb(Xq)[1] 46,XX,+min[1] 47,XX,+14[1] 92,XXXX[2] | 46,XX[21] 46,XX,chtb(5q)[1] 46,XX,chtb(7p), +min[1] 46,XX,chtb(10q)[1] 47,XX,+21[1] | 46,XX[21] 46,XX,chtb(3q)[1] 46,XX,chtb(4q)[1] 46,XX,chtb(10q)[1] 47,XX,+15[1] | 46,XX[20] 46,XX,chtb(4q)[1] 92,XXXX[4] |
| #2 | 71/♂ | 46,XY[25] | 46,XY[22] 46,XY,chtb(5q)[1] 46,XY,chtb(18q)[1] 54,XXY,+6,+7,+9, +12,+16,+17,+18[1] | 46,XY[19] 46,XY,chtb(2q), chtb(4q)[1] 47,XY,+2, chtb(2q)[1] 47,XY,+1[1] 46,XY,+min[1] 69,XXY[2] | 46,XY[23] 92,XXYY[2] | 46,XY[21] 46,XY,chtb(10q)[1] 45,XY,-5[1] 46,XY,chtb(7q), chtb(18q)[1] 45,XY,der(5)t(5;18) (q35;q23)[1] | 46,XY[20] 46,XY,chtb(2q)[2] 46,XY,chtb(16q)[1] 47,XY,+8[1] 46,XY,chtb(1p)[1] |
| #3 | 44/♂ | 46,XY[25] | 46,XY[21] 46,XY,chtb(2q)[1] 46,XY,chtb(4q)[1] 46,XY,chtb(15q)[1] 46,XY,chtb(5q)[1] | 46,XY[22] 45,XY,-5[1] 46,XY,chtb(14q)[1] 46,XY,chtb(15q)[1] | 46,XY[21] 45,XY,chtb(20p)[1] 46,XY,chtb(5q)[1] 45,XY,-5[1] 92,XXYY[1] | 46,XY[23] 45,XY,-14,-17,+f[1] 46,XY,chtb(8q)[1] | 46,XY[22] 45,XY,-5[1] 46,XY,chtb(10q)[1] 46,XY,chtb(3q)[1] |
| #4 | 78/♀ | 46,XX[25] | 46,XX[21] 46,XX,chtb(2q)[1] 47,XX,+8[1] 47,XX,+20[1] 48,XX,+8,+20[1] | 46,XX[20] 46,XX,chtb(1p)[1] 46,XX,chtb(15q)[1] 46,XX,chtb(7p)[1] 50,XX,+1,+6,+11, +17[1] 92,XXXX[1] | 46,XX[21] 46,XX, del(7)(p11)[1] 46,XX,chtb(1q)[1] 46,XX,chtb(7q)[1] 47,XX,+18[1] | 46,XX[23] 45,XX,chtb(4q)[1] 46,XX,chtb(16q)[1] | 46,XX[16] 92,XXXX[5] 47,XX,+16[1] 46,XX,chtb(4q)[1] 46,XX,chtb(10q)[1] 46,XX,chtb(9q)[1] |
| #5 | 53/♂ | 46,XY[25] | 46,XY[22] 47,XY,+12[1] 46,XY,chtb(7q)[1] 46,XY,chsb(5q)[1] | 46,XY[24] 47,XY,+19[1] | 46,XY[23] 92,XXYY[1] 184,XXXXYYYY[1] | 46,XY[24] 47,XY,+9[1] | 46,XY[23] 46,XY,chtb(7q)[1] 46,XY,chtb(12q)[1] |
| #6 | 73/♀ | 46,XX[25] | 46,XX[21] 46,XX,chtb(14q)[1] 46,XX,chtb(Xq)[1] 47,XX,+9[1] 47,XX,+14[1] | 46,XX[17] 47,XX,+9[1] 46,XX,chsb(3p)[1] 46,XX,chtb(5p)[2] 46,XX,chtb(4q)[2] 92,XXXX[2] | 46,XX[24] 92,XXXX[1] | 46,XX[19] 46,XX,chtb(8q)[1] 48,XX,+3,+16[1] 92,XXXX[3] 184,XXXXXXXX[1] | 46,XX[21] 46,XX,chtb(8q)[1] 46,XX,chtb(13q)[1] 47,XX,+f[1] 92,XXXX[1] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fabarius, A.; Samra, V.; Drews, O.; Mörz, H.; Bierbaum, M.; Darwich, A.; Weiss, C.; Brendel, S.; Kleiner, H.; Seifarth, W.; et al. Evidence for Recombinant GRP78, CALR, PDIA3 and GPI as Mediators of Genetic Instability in Human CD34+ Cells. Cancers 2022, 14, 2883. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14122883
Fabarius A, Samra V, Drews O, Mörz H, Bierbaum M, Darwich A, Weiss C, Brendel S, Kleiner H, Seifarth W, et al. Evidence for Recombinant GRP78, CALR, PDIA3 and GPI as Mediators of Genetic Instability in Human CD34+ Cells. Cancers. 2022; 14(12):2883. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14122883
Chicago/Turabian StyleFabarius, Alice, Vanessa Samra, Oliver Drews, Handan Mörz, Miriam Bierbaum, Ali Darwich, Christel Weiss, Susanne Brendel, Helga Kleiner, Wolfgang Seifarth, and et al. 2022. "Evidence for Recombinant GRP78, CALR, PDIA3 and GPI as Mediators of Genetic Instability in Human CD34+ Cells" Cancers 14, no. 12: 2883. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14122883