
Myeloid Derived Suppressor Cells Migrate in Response to Flow and Lymphatic Endothelial Cell Interaction in the Breast Tumor Microenvironment

 ,
,
Abstract
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Cell Culture
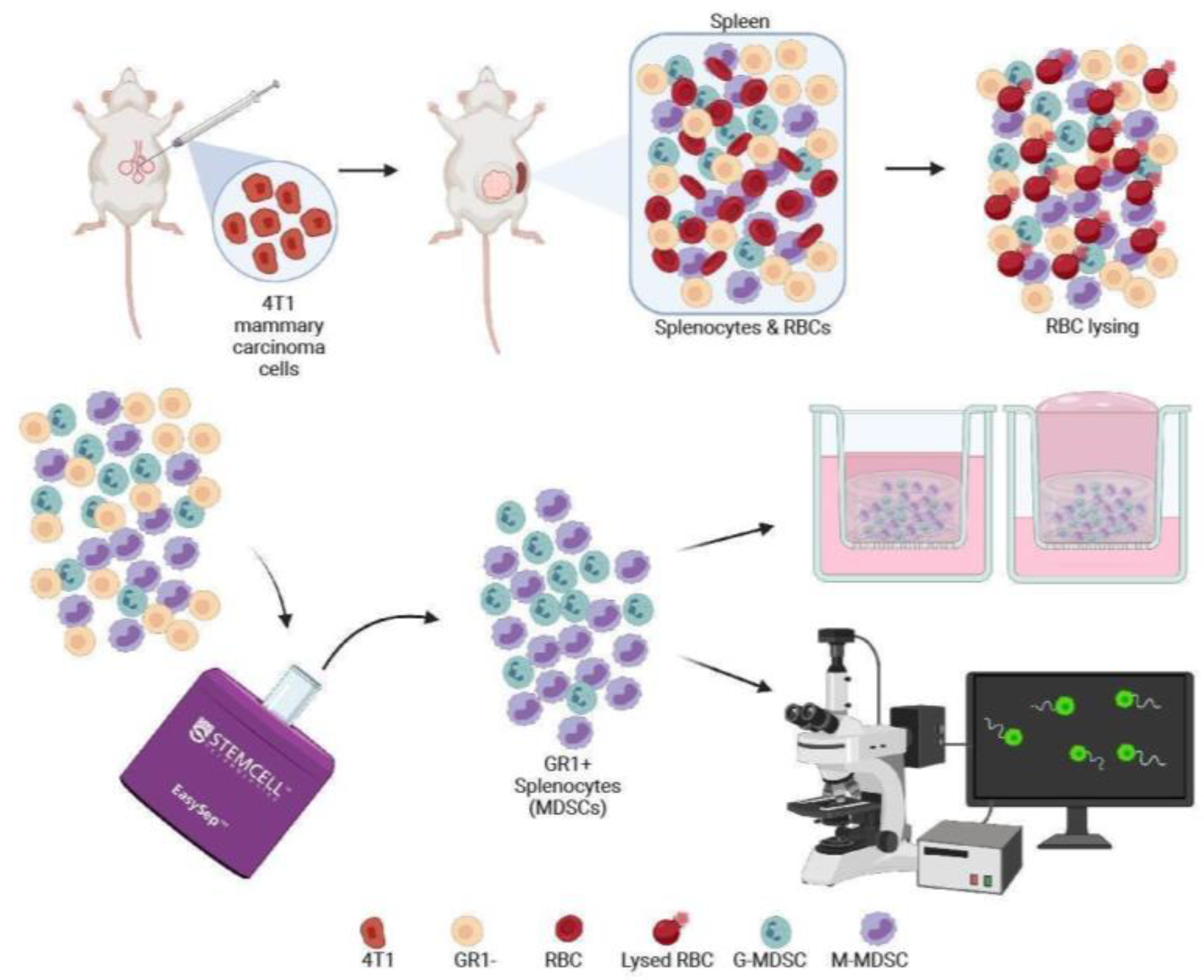
2.3. Splenocyte Isolation and Sorting
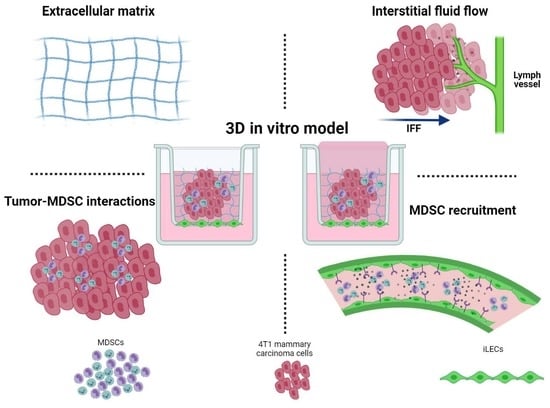
2.4. Three-Dimensional Cell Culture Model
2.5. Time-Lapse Microscopy
2.6. Cell Tracking
2.7. Invasion Assay
2.8. Invasion Analysis
2.9. Statistical Analysis
3. Results
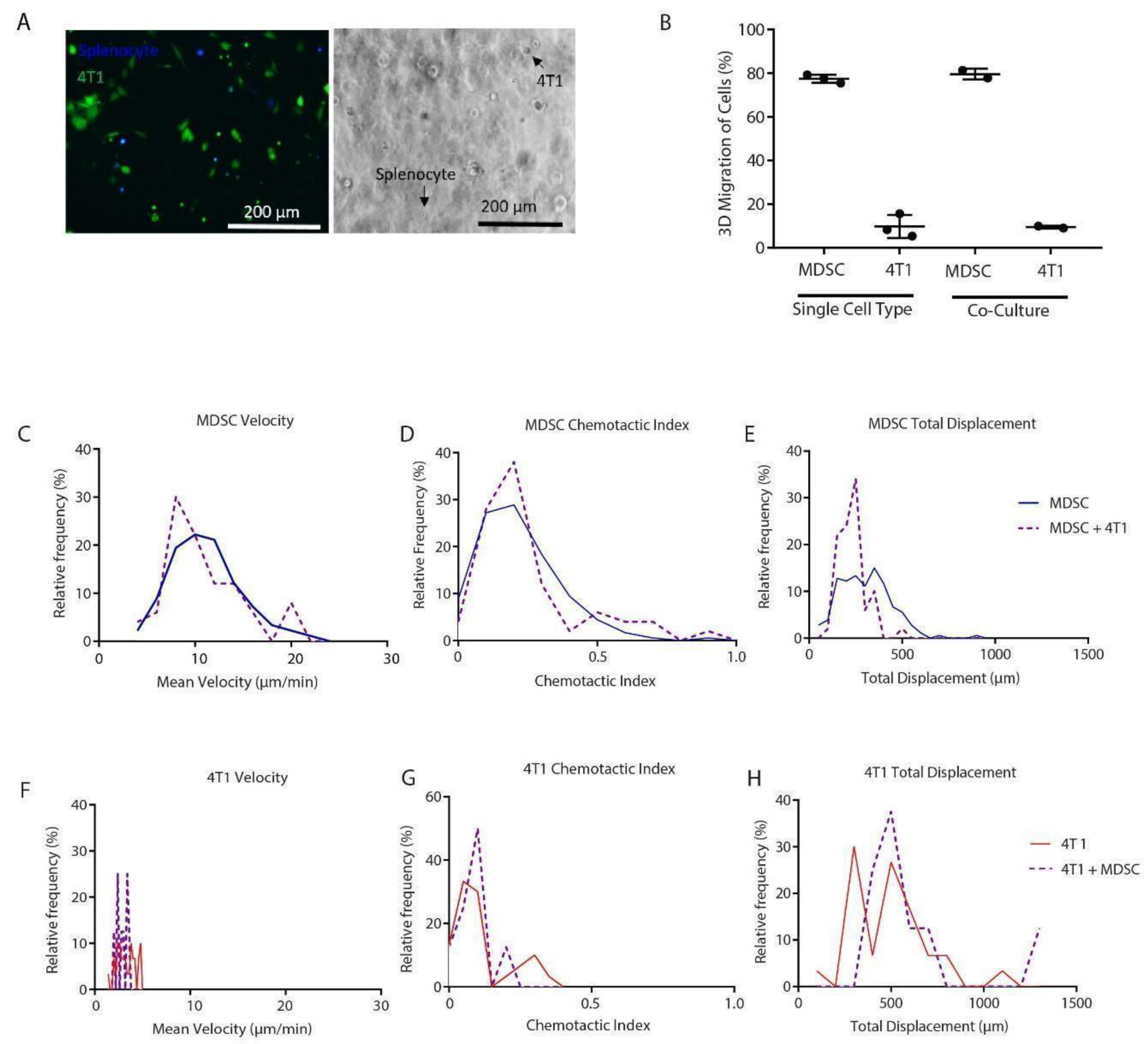
3.1. Development of a 3D Culture Model for MDSCs
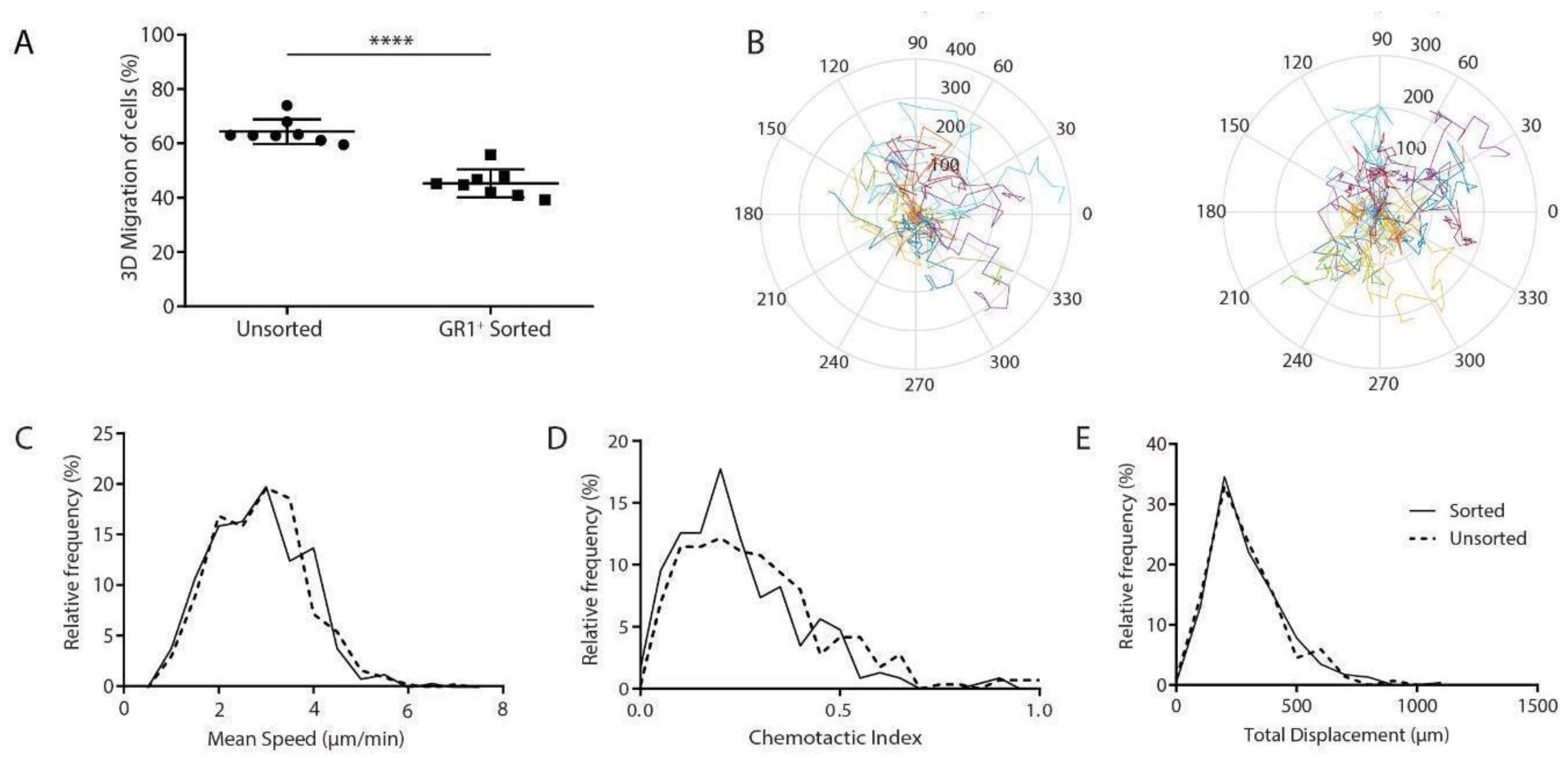
3.2. GR1+ Cells Sorted from Splenocytes have Reduced Overall Migration
3.3. Migration Characteristics of MDSCs Are Sensitive to Harvest
3.4. MDSCs Migrate Differently in the Presence of Tumor Cells
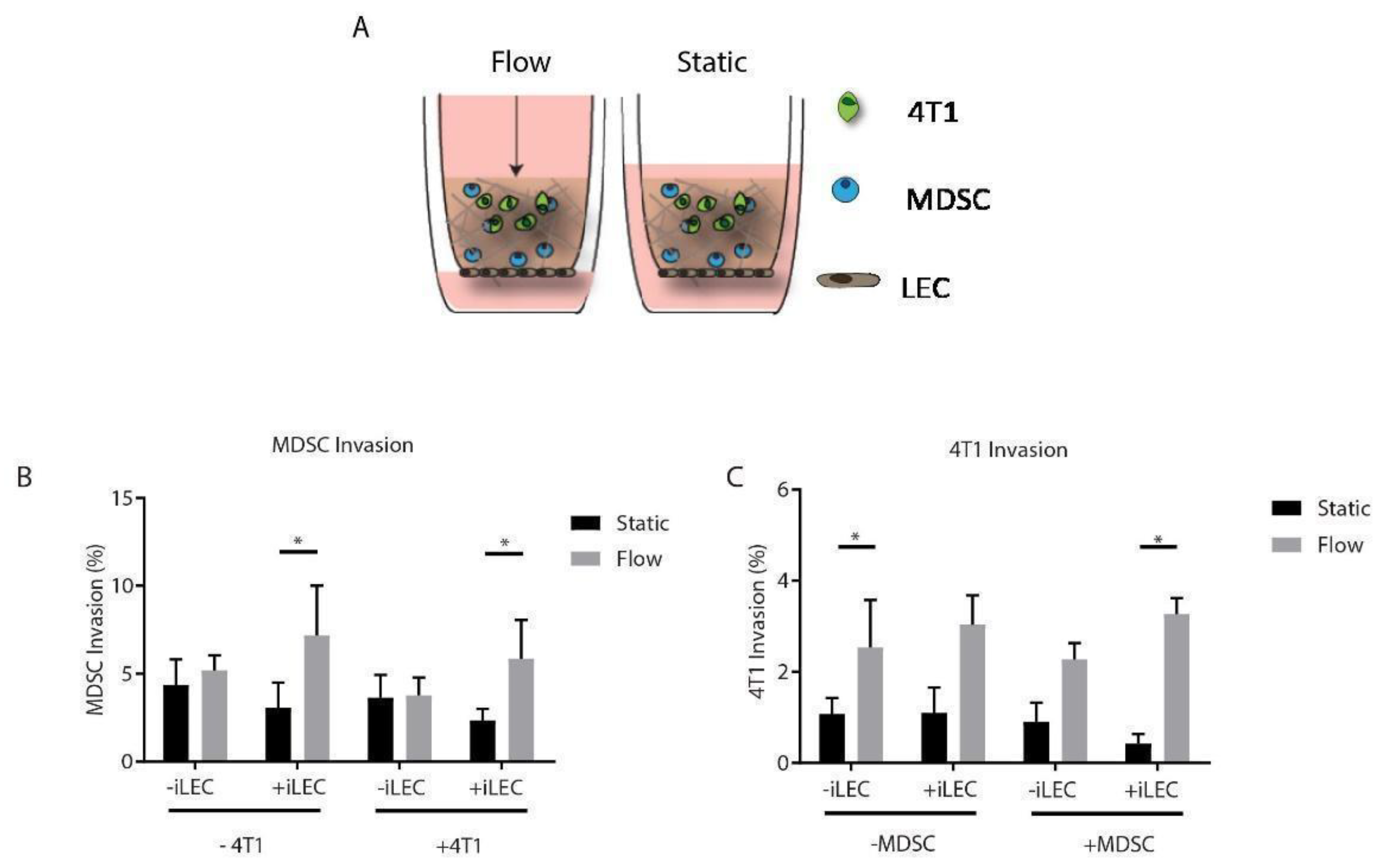
3.5. MDSCs Respond to Both Interstitial Fluid Flow and Lymphatic Endothelial Cells by Increasing Their Migration
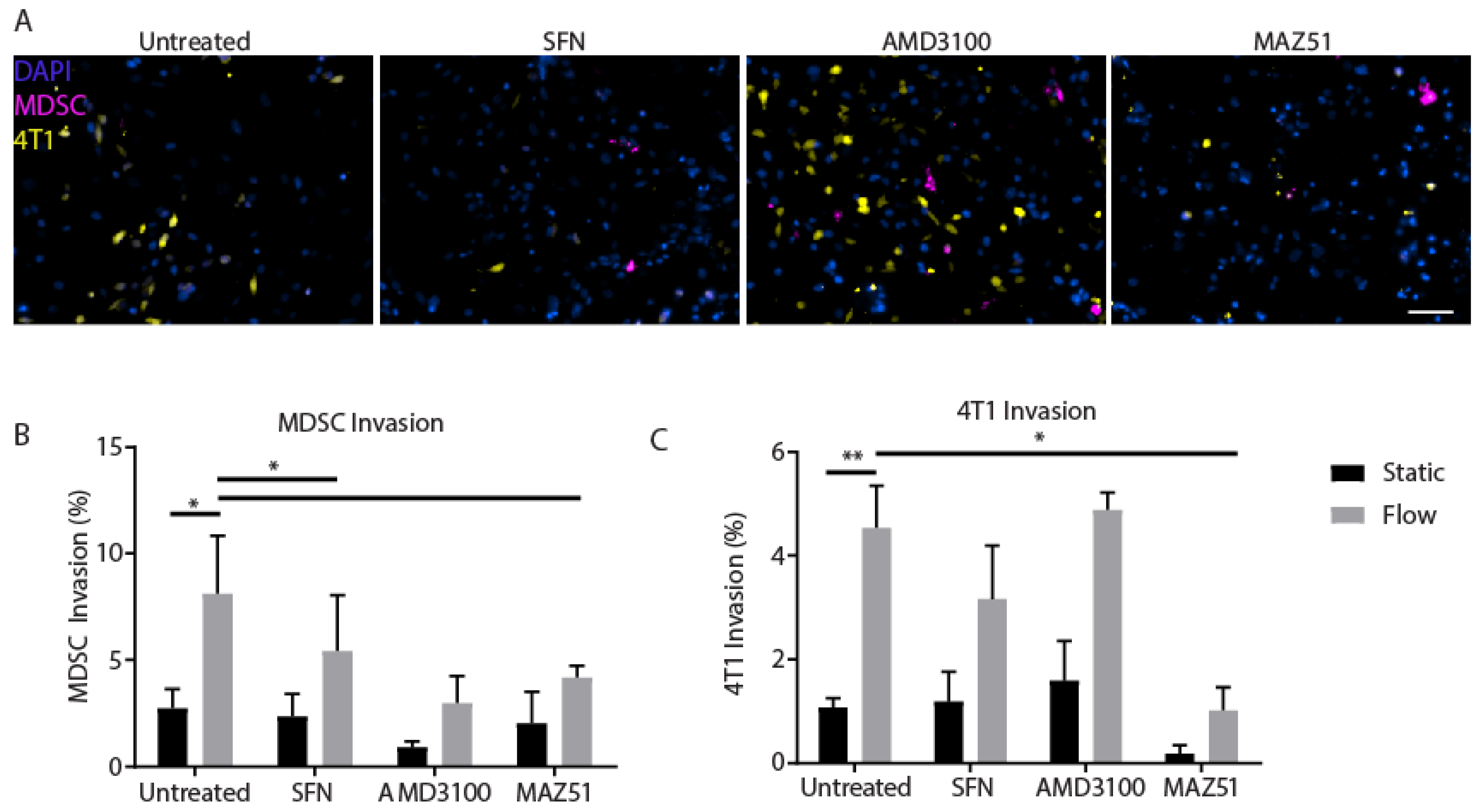
3.6. Inhibition of VEGFR3 Blocks Migration of MDSCs and 4T1s
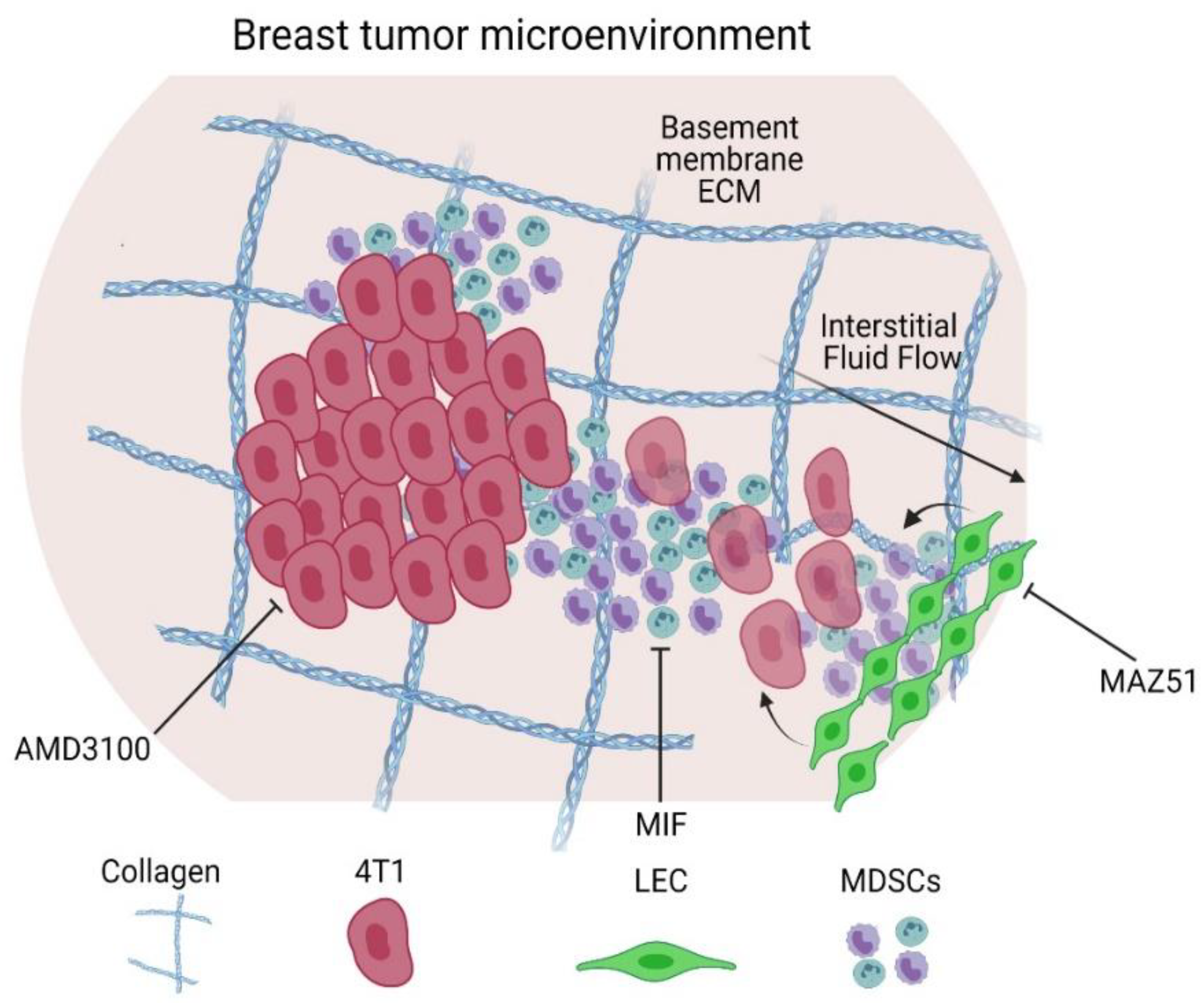
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Veglia, F.; Sanseviero, E.; Gabrilovich, D.I. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat. Rev. Immunol. 2021, 21, 485–498. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef] [Green Version]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived-suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162. [Google Scholar] [CrossRef] [Green Version]
- Bergenfelz, C.; Roxå, A.; Mehmeti, M.; Leandersson, K.; Larsson, A.-M. Clinical relevance of systemic monocytic-MDSCs in patients with metastatic breast cancer. Cancer Immunol. Immunother. 2020, 69, 435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, K.-T.; Hsieh, C.-C.; Huang, T.-Y.; Chen, P.-C.; Shih, H.-N.; Lee, M.S.; Chang, P.-J. Staphylococcus aureus biofilm elicits the expansion, activation and polarization of myeloid-derived suppressor cells in vivo and in vitro. PLoS ONE 2017, 12, e0183271. [Google Scholar] [CrossRef] [Green Version]
- Ribechini, E.; Eckert, I.; Beilhack, A.; Plessis, N.D.; Walzl, G.; Schleicher, U.; Ritter, U.; Lutz, M.B. Heat-killed Mycobacterium tuberculosis prime-boost vaccination induces myeloid-derived suppressor cells with spleen dendritic cell–killing capability. JCI Insight 2020, 4, e128664. [Google Scholar] [CrossRef] [Green Version]
- Cassetta, L.; Baekkevold, E.S.; Brandau, S.; Bujko, A.; Cassatella, M.A.; Dorhoi, A.; Krieg, C.; Lin, A.; Loré, K.; Marini, O.; et al. Deciphering myeloid-derived suppressor cells: Isolation and markers in humans, mice and non-human primates. Cancer Immunol. Immunother. 2019, 68, 687–697. [Google Scholar] [CrossRef] [Green Version]
- Budhwar, S.; Verma, P.; Verma, R.; Rai, S.; Singh, K. The Yin and Yang of Myeloid Derived Suppressor Cells. Front. Immunol. 2018, 9, 2776. [Google Scholar] [CrossRef]
- Bronte, V.; Brandau, S.; Chen, S.-H.; Colombo, M.P.; Frey, A.B.; Greten, T.F.; Mandruzzato, S.; Murray, P.J.; Ochoa, A.; Ostrand-Rosenberg, S.; et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat. Commun. 2016, 7, 12150. [Google Scholar] [CrossRef] [Green Version]
- Lv, M.; Wang, K.; Huang, X. Myeloid-derived suppressor cells in hematological malignancies: Friends or foes. J. Hematol. Oncol. 2019, 12, 105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruger, A.M.; Dorhoi, A.; Esendagli, G.; Barczyk-Kahlert, K.; van der Bruggen, P.; Lipoldova, M.; Perecko, T.; Santibanez, J.; Saraiva, M.; Van Ginderachter, J.A.; et al. How to measure the immunosuppressive activity of MDSC: Assays, problems and potential solutions. Cancer Immunol. Immunother. 2018, 68, 631–644. [Google Scholar] [CrossRef] [PubMed]
- Law, A.M.K.; Valdes-Mora, F.; Gallego-Ortega, D. Myeloid-Derived Suppressor Cells as a Therapeutic Target for Cancer. Cells 2020, 9, 561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alshetaiwi, H.; Pervolarakis, N.; McIntyre, L.L.; Ma, D.; Nguyen, Q.; Rath, J.A.; Nee, K.; Hernandez, G.; Evans, K.; Torosian, L.; et al. Defining the emergence of myeloid-derived suppressor cells in breast cancer using single-cell transcriptomics. Sci. Immunol. 2020, 5, eaay6017. [Google Scholar] [CrossRef]
- Sangaletti, S.; Chiodoni, C.; Tripodo, C.; Colombo, M.P. Common extracellular matrix regulation of myeloid cell activity in the bone marrow and tumor microenvironments. Cancer Immunol. Immunother. 2017, 66, 1059–1067. [Google Scholar] [CrossRef] [Green Version]
- Petrova, V.; Annicchiarico-Petruzzelli, M.; Melino, G.; Amelio, I. The hypoxic tumour microenvironment. Oncogenesis 2018, 7, 10. [Google Scholar] [CrossRef]
- Liu, T.; Han, C.; Wang, S.; Fang, P.; Ma, Z.; Xu, L.; Yin, R. Cancer-associated fibroblasts: An emerging target of anti-cancer immunotherapy. J. Hematol. Oncol. 2019, 12, 86. [Google Scholar] [CrossRef]
- Swartz, M.A.; Lund, A.W. Lymphatic and interstitial flow in the tumour microenvironment: Linking mechanobiology with immunity. Nat. Rev. Cancer. 2012, 12, 210–219. [Google Scholar] [CrossRef]
- Kataru, R.P.; Ly, C.L.; Shin, J.; Park, H.J.; Baik, J.E.; Rehal, S.; Ortega, S.; Lyden, D.; Mehrara, B.J. Tumor Lymphatic Function Regulates Tumor Inflammatory and Immunosuppressive Microenvironments. Cancer Immunol. Res. 2019, 7, 1345–1358. [Google Scholar] [CrossRef]
- Xu, Y.; Fang, F.; Jiao, H.; Zheng, X.; Huang, L.; Yi, X.; Zhao, W. Activated hepatic stellate cells regulate MDSC migration through the SDF-1/CXCR4 axis in an orthotopic mouse model of hepatocellular carcinoma. Cancer Immunol. Immunother. 2019, 68, 1959–1969. [Google Scholar] [CrossRef]
- Jiang, K.; Li, J.; Zhang, J.; Wang, L.; Zhang, Q.; Ge, J.; Guo, Y.; Wang, B.; Huang, Y.; Yang, T.; et al. SDF-1/CXCR4 axis facilitates myeloid-derived suppressor cells accumulation in osteosarcoma microenvironment and blunts the response to anti-PD-1 therapy. Int. Immunopharmacol. 2019, 75, 105818. [Google Scholar] [CrossRef] [PubMed]
- Groth, C.; Arpinati, L.; Shaul, M.E.; Winkler, N.; Diester, K.; Gengenbacher, N.; Weber, R.; Arkhypov, I.; Lasser, S.; Petrova, V.; et al. Blocking Migration of Polymorphonuclear Myeloid-Derived Suppressor Cells Inhibits Mouse Melanoma Progression. Cancers 2021, 13, 726. [Google Scholar] [CrossRef] [PubMed]
- Alfaro, C.; Teijeira, A.; Oñate, C.; Pérez, G.P.; Sanmamed, M.F.; Andueza, M.P.; Alignani, D.; Labiano, S.; Azpilikueta, A.; Rodriguez-Paulete, A.; et al. Biology of Human Tumors Tumor-Produced Interleukin-8 Attracts Human Myeloid-Derived Suppressor Cells and Elicits Extrusion of Neutrophil Extracellular Traps (NETs). Clin. Cancer Res. 2016, 22, 3924–3936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.-Y.; Lai, Y.-S.; Chu, P.-Y.; Chan, S.-H.; Wang, L.-H.; Hung, W.-C. Cancer-Derived VEGF-C Increases Chemokine Production in Lymphatic Endothelial Cells to Promote CXCR2-Dependent Cancer Invasion and MDSC Recruitment. Cancers 2019, 11, 1120. [Google Scholar] [CrossRef] [Green Version]
- Lahmar, Q.; Schouppe, E.; Morias, Y.; Van Overmeire, E.; De Baetselier, P.; Movahedi, K.; Laoui, D.; Sarukhan, A.; Van Ginderachter, J.A. Monocytic myeloid-derived suppressor cells home to tumor-draining lymph nodes via CCR2 and locally modulate the immune response. Cell. Immunol. 2021, 362, 104296. [Google Scholar] [CrossRef] [PubMed]
- Munson, J.M.; Shieh, A.C. Interstitial fluid flow in cancer: Implications for disease progression and treatment. Cancer Manag. Res. 2014, 6, 317. [Google Scholar] [CrossRef] [Green Version]
- Vigl, B.; Aebischer, D.; Nitschké, M.; Iolyeva, M.; Röthlin, T.; Antsiferova, O.; Halin, C. Tissue inflammation modulates gene expression of lymphatic endothelial cells and dendritic cell migration in a stimulus-dependent manner. Blood 2011, 118, 205–215. [Google Scholar] [CrossRef]
- Youn, J.-I.; Nagaraj, S.; Collazo, M.; Gabrilovich, D.I. Subsets of Myeloid-Derived Suppressor Cells in Tumor Bearing Mice. J. Immunol. 2008, 181, 5791. [Google Scholar] [CrossRef]
- Jordan, K.R.; Kapoor, P.; Spongberg, E.; Tobin, R.P.; Gao, D.; Borges, V.F.; McCarter, M.D. Immunosuppressive myeloid-derived suppressor cells are increased in splenocytes from cancer patients. Cancer Immunol. Immunother. 2017, 66, 503. [Google Scholar] [CrossRef] [Green Version]
- Simpson, K.D.; Templeton, D.J.; Cross, J.V. Macrophage Migration Inhibitory Factor promotes tumor growth and metastasis by inducing Myeloid Derived Suppressor Cells in the tumor microenvironment. J. Immunol. 2012, 189, 5533. [Google Scholar] [CrossRef] [Green Version]
- Marigo, I.; Bosio, E.; Solito, S.; Mesa, C.; Fernandez, A.; Dolcetti, L.; Ugel, S.; Sonda, N.; Bicciato, S.; Falisi, E.; et al. Tumor-Induced Tolerance and Immune Suppression Depend on the C/EBPβ Transcription Factor. Immunity 2010, 32, 790–802. [Google Scholar] [CrossRef] [PubMed]
- Munson, J.M.; Bellamkonda, R.V.; Swartz, M.A. Interstitial flow in a 3d microenvironment increases glioma invasion by a cxcr4-dependent mechanism. Cancer Res. 2013, 73, 1536–1546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haessler, U.; Teo, J.C.M.; Foretay, D.; Renaud, P.; Swartz, M.A. Migration dynamics of breast cancer cells in a tunable 3D interstitial flow chamber. Integr. Biol. 2012, 4, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Harris, A.R.; Perez, M.J.; Munson, J.M. Docetaxel facilitates lymphatic-tumor crosstalk to promote lymphangiogenesis and cancer progression. BMC Cancer 2018, 18, 718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, V.; Patel, S.; Tcyganov, E.; Gabrilovich, D.I. The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol. 2016, 37, 208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damuzzo, V.; Pinton, L.; Desantis, G.; Solito, S.; Marigo, I.; Bronte, V.; Mandruzzato, S. Complexity and Challenges in Defining Myeloid-Derived Suppressor Cells. Cytom. B Clin. Cytom. 2015, 88, 77. [Google Scholar] [CrossRef] [PubMed]
- Shields, J.D.; Fleury, M.E.; Yong, C.; Tomei, A.A.; Randolph, G.J.; Swartz, M.A. Autologous Chemotaxis as a Mechanism of Tumor Cell Homing to Lymphatics via Interstitial Flow and Autocrine CCR7 Signaling. Cancer Cell 2007, 11, 526–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, S.; Sarrou, E.; Podgrabinska, S.; Cassella, M.; Mungamuri, S.K.; Feirt, N.; Gordon, R.; Nagi, C.S.; Wang, Y.; Entenberg, D.; et al. Tumor cell entry into the lymph node is controlled by CCL1 chemokine expressed by lymph node lymphatic sinuses. J. Exp. Med. 2013, 210, 1509–1528. [Google Scholar] [CrossRef] [Green Version]
- Cabioglu, N.; Yazici, M.S.; Arun, B.; Broglio, K.R.; Hortobagyi, G.N.; Price, J.E.; Sahin, A. CCR7 and CXCR4 as novel biomarkers predicting axillary lymph node metastasis in T1 breast cancer. Clin. Cancer Res. 2005, 11, 5686–5693. [Google Scholar] [CrossRef] [Green Version]
- Simpson, K.D.; Cross, J.V. MIF: Metastasis/MDSC-inducing factor? Oncoimmunology 2013, 2, e23337. [Google Scholar] [CrossRef] [Green Version]
- Balogh, K.N.; Templeton, D.J.; Cross, J.V. Macrophage Migration Inhibitory Factor protects cancer cells from immunogenic cell death and impairs anti-tumor immune responses. PLoS ONE 2018, 13, e0197702. [Google Scholar] [CrossRef]
- Alban, T.J.; Bayik, D.; Otvos, B.; Rabljenovic, A.; Leng, L.; Jia-Shiun, L.; Roversi, G.; Lauko, A.; Momin, A.A.; Mohammadi, A.M.; et al. Glioblastoma Myeloid-Derived Suppressor Cell Subsets Express Differential Macrophage Migration Inhibitory Factor Receptor Profiles That Can Be Targeted to Reduce Immune Suppression. Front. Immunol. 2020, 11, 1191. [Google Scholar] [CrossRef] [PubMed]
- Kingsmore, K.M.; Logsdon, D.K.; Floyd, D.H.; Peirce, S.M.; Purow, B.W.; Munson, J.M. Interstitial flow differentially increases patient-derived glioblastoma stem cell invasion: Via CXCR4, CXCL12, and CD44-mediated mechanisms. Integr. Biol. 2016, 8, 1246–1260. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.D.; Bouchard, M.J.; Shieh, A.C. Interstitial Fluid Flow Increases Hepatocellular Carcinoma Cell Invasion through CXCR4/CXCL12 and MEK/ERK Signaling. PLoS ONE 2015, 10, e0142337. [Google Scholar] [CrossRef] [PubMed]
- Polacheck, W.J.; Charest, J.L.; Kamm, R.D. Interstitial flow influences direction of tumor cell migration through competing mechanisms. Proc. Natl. Acad. Sci. USA 2011, 108, 11115–11120. [Google Scholar] [CrossRef] [Green Version]
- Baeyens, N.; Nicoli, S.; Coon, B.G.; Ross, T.D.; Van Den Dries, K.; Han, J.; Lauridsen, H.M.; Mejean, C.O.; Eichmann, A.; Thomas, J.L.; et al. Vascular remodeling is governed by a vegfr3-dependent fluid shear stress set point. Elife 2015, 4, e04645. [Google Scholar] [CrossRef]
- Vaahtomeri, K.; Karaman, S.; Mäkinen, T.; Alitalo, K. Lymphangiogenesis guidance by paracrine and pericellular factors. Genes Dev. 2017, 31, 1615–1634. [Google Scholar] [CrossRef]
- Bunt, S.K.; Sinha, P.; Clements, V.K.; Leips, J.; Ostrand-Rosenberg, S. Inflammation Induces Myeloid-Derived Suppressor Cells that Facilitate Tumor Progression. J. Immunol. 2006, 176, 284–290. [Google Scholar] [CrossRef] [Green Version]
- Ostrand-Rosenberg, S.; Ostrand-Rosenberg, S. Myeloid-derived suppressor cells: More mechanisms for inhibiting antitumor immunity. Cancer Immunol. Immunother. 2010, 59, 1593–1600. [Google Scholar] [CrossRef] [Green Version]
- Liao, S.; von der Weid, P.-Y.; Inflammation Research Network, Snyder Institute for Chronic Diseases. Lymphatic System: An Active Pathway for Immune Protection. Semin. Cell Dev. Biol. 2015, 38, 83. [Google Scholar] [CrossRef] [Green Version]
- Karin, N. The Development and Homing of Myeloid-Derived Suppressor Cells: From a Two-Stage Model to a Multistep Narrative. Front. Immunol. 2020, 11, 2447. [Google Scholar] [CrossRef]
- Wiig, H.; Swartz, M.A. Interstitial fluid and lymph formation and transport: Physiological regulation and roles in inflammation and cancer. Physiol. Rev. 2012, 92, 1005–1060. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Wang, M.; Yin, T.; Zhao, Y.; Wei, X. Myeloid-Derived Suppressor Cells Promote Metastasis in Breast Cancer After the Stress of Operative Removal of the Primary Cancer. Front. Oncol. 2019, 9, 855. [Google Scholar] [CrossRef]
- Dawod, B.; Liu, J.; Gebremeskel, S.; Yan, C.; Sappong, A.; Johnston, B.; Hoskin, D.W.; Marshall, J.S.; Wang, J. Myeloid-derived suppressor cell depletion therapy targets IL-17A-expressing mammary carcinomas. Sci. Rep. 2020, 10, 13343. [Google Scholar] [CrossRef]
- Miteva, D.O.; Rutkowski, J.M.; Dixon, J.B.; Kilarski, W.; Shields, J.D.; Swartz, M.A. Transmural Flow Modulates Cell and Fluid Transport Functions of Lymphatic Endothelium. Circ. Res. 2010, 106, 920–931. [Google Scholar] [CrossRef]
- Jackson, D.G. Leucocyte Trafficking via the Lymphatic Vasculature—Mechanisms and Consequences. Front. Immunol. 2019, 10, 471. [Google Scholar] [CrossRef]
- Shieh, A.C.; Rozansky, H.A.; Hinz, B.; Swartz, M.A. Tumor cell invasion is promoted by interstitial flow-induced matrix priming by stromal fibroblasts. Cancer Res. 2011, 71, 790–800. [Google Scholar] [CrossRef] [Green Version]
- Ng, C.P.; Swartz, M.A. Fibroblast alignment under interstitial fluid flow using a novel 3-D tissue culture model. Am. J. Physiol.-Heart Circ. Physiol. 2003, 284, 1771–1777. [Google Scholar] [CrossRef]
- Ng, C.P.; Hinz, B.; Swartz, M.A. Interstitial fluid flow induces myofibroblast differentiation and collagen alignment in vitro. J. Cell Sci. 2005, 118, 4731–4739. [Google Scholar] [CrossRef] [Green Version]
- Xiang, H.; Ramil, C.P.; Hai, J.; Zhang, C.; Wang, H.; Watkins, A.A.; Afshar, R.; Georgiev, P.; Sze, M.A.; Song, X.S.; et al. Cancer-associated fibroblasts promote immunosuppression by inducing ROS-generating monocytic MDSCs in lung squamous cell carcinoma. Cancer Immunol. Res. 2020, 8, 436–450. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roberts, L.M.; Perez, M.J.; Balogh, K.N.; Mingledorff, G.; Cross, J.V.; Munson, J.M. Myeloid Derived Suppressor Cells Migrate in Response to Flow and Lymphatic Endothelial Cell Interaction in the Breast Tumor Microenvironment. Cancers 2022, 14, 3008. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14123008
Roberts LM, Perez MJ, Balogh KN, Mingledorff G, Cross JV, Munson JM. Myeloid Derived Suppressor Cells Migrate in Response to Flow and Lymphatic Endothelial Cell Interaction in the Breast Tumor Microenvironment. Cancers. 2022; 14(12):3008. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14123008
Chicago/Turabian StyleRoberts, LaDeidra Monét, Matthew J. Perez, Kristen N. Balogh, Garnett Mingledorff, Janet V. Cross, and Jennifer M. Munson. 2022. "Myeloid Derived Suppressor Cells Migrate in Response to Flow and Lymphatic Endothelial Cell Interaction in the Breast Tumor Microenvironment" Cancers 14, no. 12: 3008. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14123008