
Novel Treatments of Uveal Melanoma Identified with a Synthetic Lethal CRISPR/Cas9 Screen

 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Cell Viability Assay
2.3. Treatments of Samples for Western Blot
2.4. Western Blot
2.5. Flow Cytometry
2.6. CRISPR/Cas9 Synthetic Lethality Screen
2.7. Generation of CRISPR/Cas9 Knockout Cell Lines
2.8. Generation of Metastatic UM-Patient-Derived Zebrafish Xenograft Model
3. Results
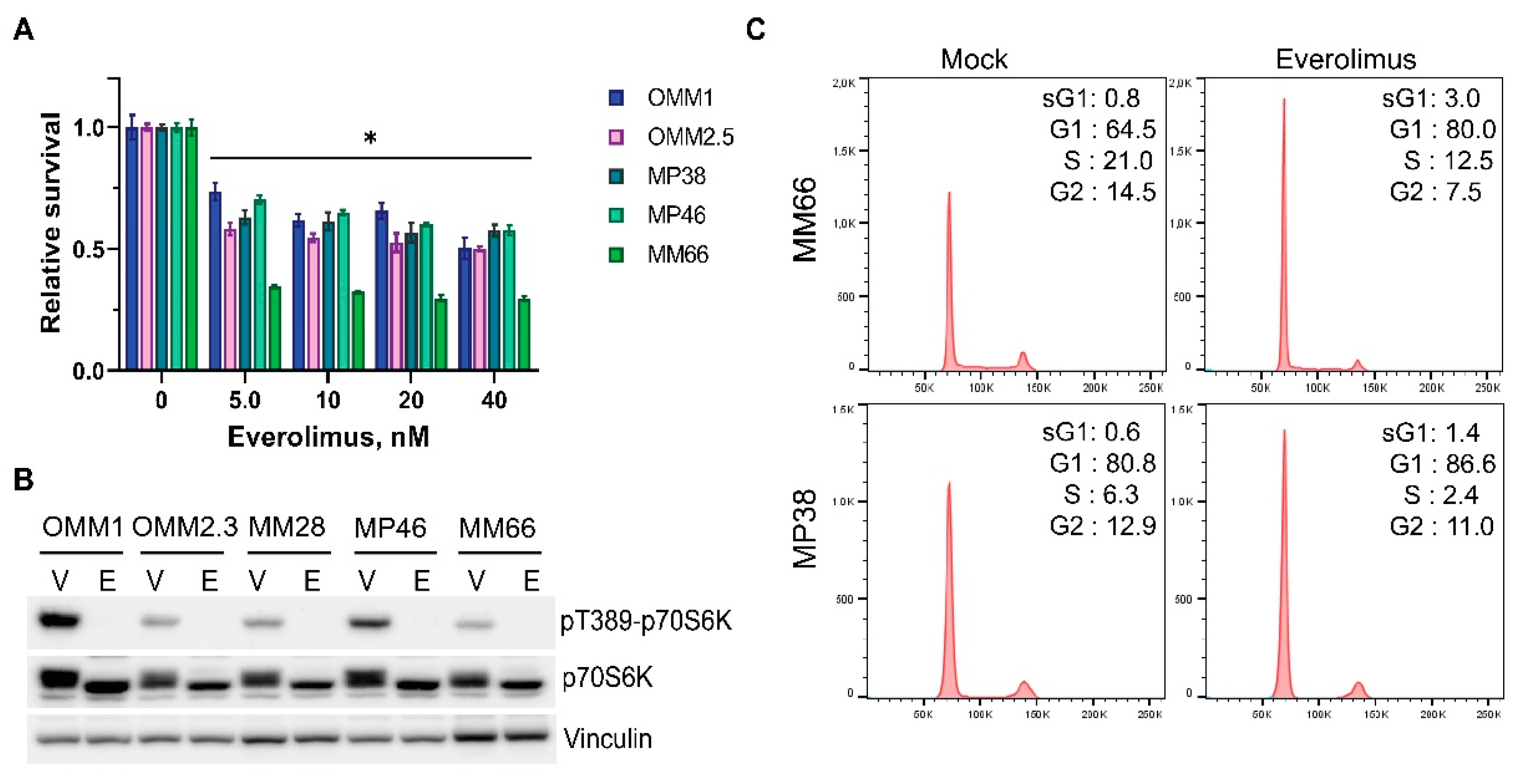
3.1. Everolimus Has Cytostatic Effect on UM Cell Lines

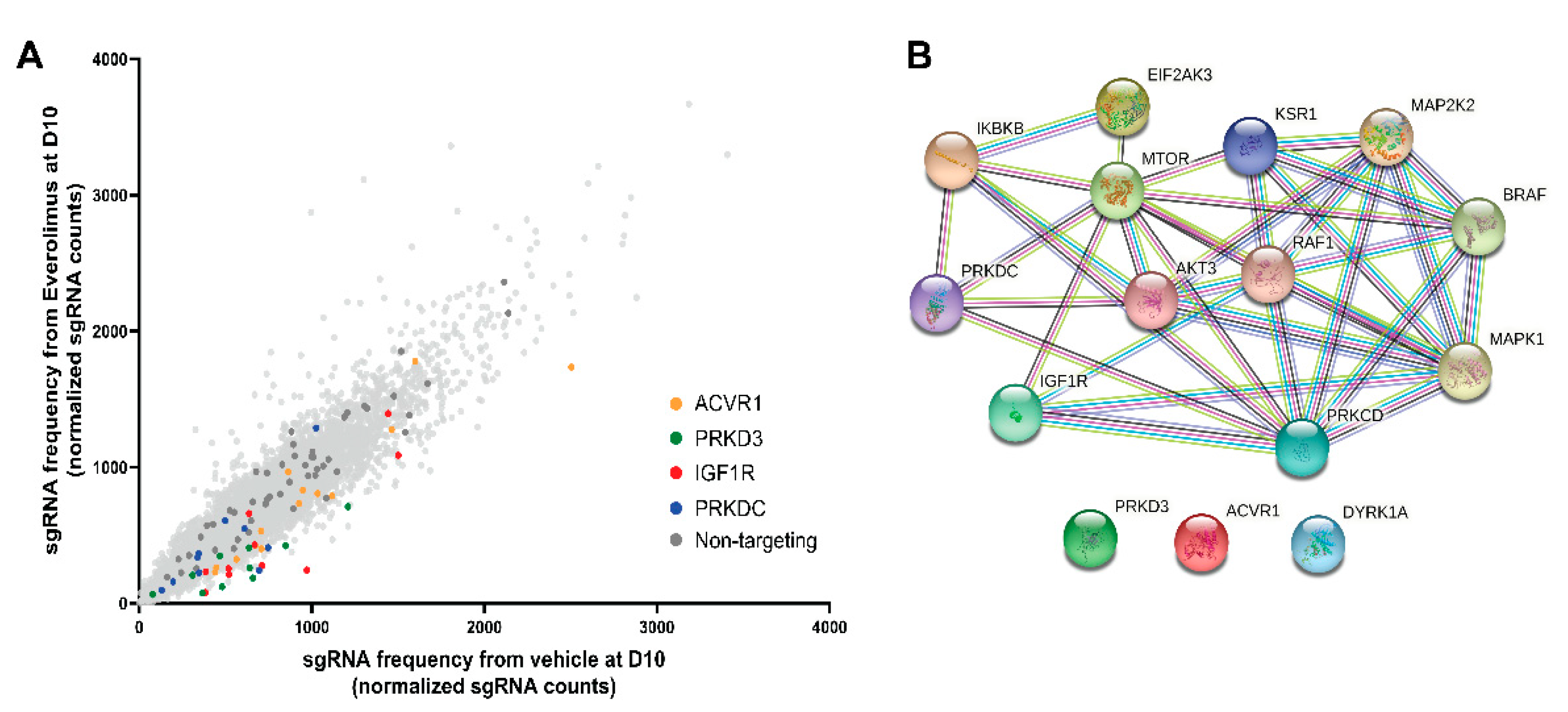
3.2. Synthetic Lethal CRISPR-Cas9 Screen
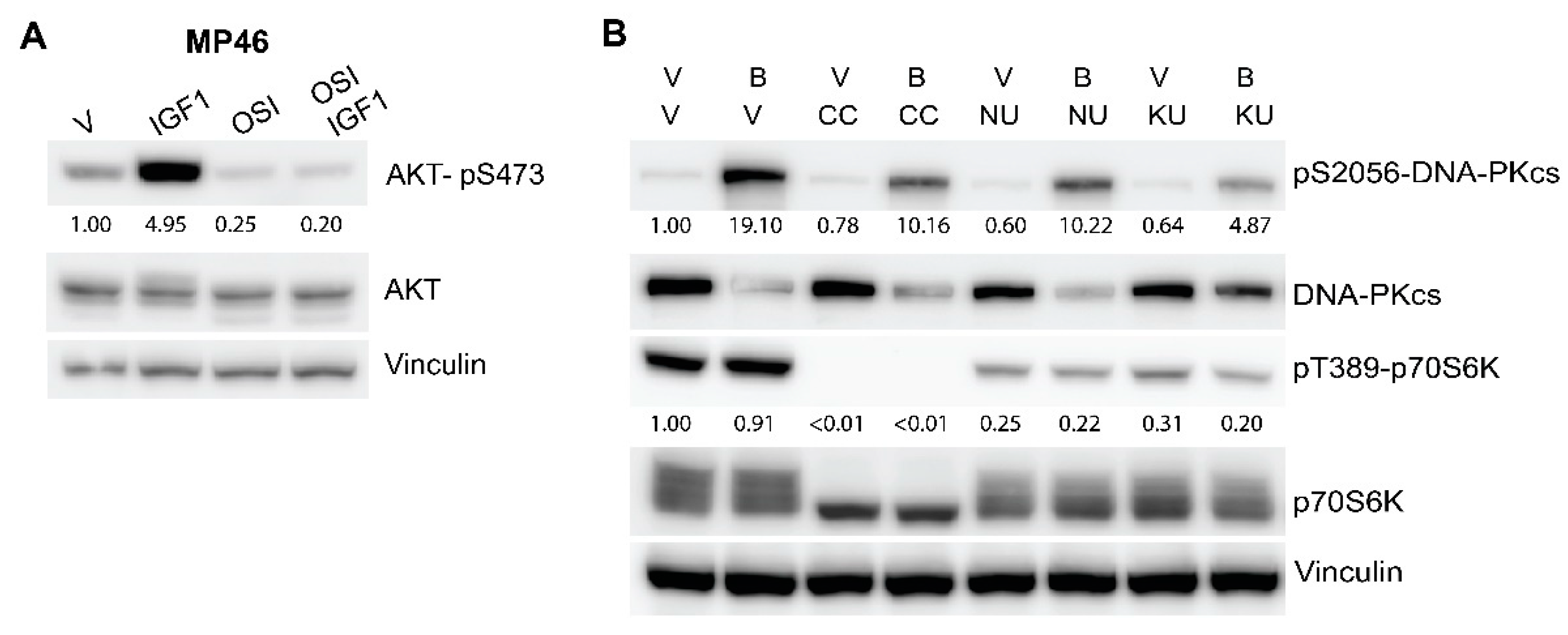
3.3. Inhibitors of DNA-PKcs or IGF1R Synergistically Enhance Cytostatic Effect of Everolimus
3.4. The Combinations of Everolimus with DNA-PKcs or IGF1R Inhibitors Induce Cell Cycle Arrest in UM Cell Lines
3.5. mTOR Active Site Inhibitor KU0063794 Is, Similar to Everolimus, Synergistic in Combination with DNA-PKcs or IGF1R Inhibitors
3.6. Genetic Verification of the Hits
3.7. CC-115 Efficiently Inhibits Growth of UM Cells in an In Vivo Model
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Virgili, G.; Gatta, G.; Ciccolallo, L.; Capocaccia, R.; Biggeri, A.; Crocetti, E.; Lutz, J.M.; Paci, E.; Eurocare Working Group. Incidence of uveal melanoma in Europe. Ophthalmology 2007, 114, 2309–2315. [Google Scholar] [CrossRef] [PubMed]
- Kujala, E.; Makitie, T.; Kivela, T. Very long-term prognosis of patients with malignant uveal melanoma. Investig. Ophthalmol. Vis. Sci. 2003, 44, 4651–4659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koutsandrea, C.; Moschos, M.M.; Dimissianos, M.; Georgopoulos, G.; Ladas, I.; Apostolopoulos, M. Metastasis rates and sites after treatment for choroidal melanoma by proton beam irradiation or by enucleation. Clin. Ophthalmol. 2008, 2, 989–995. [Google Scholar]
- Rietschel, P.; Panageas, K.; Hanlon, C.; Patel, A.; Chapman, P.B. Variates of survival for stage IV uveal melanoma. J. Clin. Oncol. 2005, 23, 8076–8080. [Google Scholar] [CrossRef] [PubMed]
- Diener-West, M.; Reynolds, S.M.; Agugliaro, D.J.; Caldwell, R.; Cumming, K.; Earle, J.D.; Green, D.L.; Hawkins, B.S.; Hayman, J.; Jaiyesimi, I.; et al. Screening for metastasis from choroidal melanoma: The Collaborative Ocular Melanoma Study Group report 23. J. Clin. Oncol. 2004, 22, 2438–2444. [Google Scholar] [CrossRef] [PubMed]
- Alexander, H.R., Jr.; Libutti, S.K.; Pingpank, J.F.; Steinberg, S.M.; Bartlett, D.L.; Helsabeck, C.; Beresneva, T. Hyperthermic isolated hepatic perfusion using melphalan for patients with ocular melanoma metastatic to liver. Clin. Cancer Res. 2003, 9, 6343–6349. [Google Scholar] [CrossRef]
- Noter, S.L.; Rothbarth, J.; Pijl, M.E.; Keunen, J.E.; Hartgrink, H.H.; Tijl, F.G.; Kuppen, P.J.; van de Velde, C.J.; Tollenaar, R.A. Isolated hepatic perfusion with high-dose melphalan for the treatment of uveal melanoma metastases confined to the liver. Melanoma Res. 2004, 14, 67–72. [Google Scholar] [CrossRef]
- Meijer, T.S.; Burgmans, M.C.; de Leede, E.M.; de Geus-Oei, L.F.; Boekestijn, B.; Handgraaf, H.J.M.; Hilling, D.E.; Lutjeboer, J.; Vuijk, J.; Martini, C.H.; et al. Percutaneous Hepatic Perfusion with Melphalan in Patients with Unresectable Ocular Melanoma Metastases Confined to the Liver: A Prospective Phase II Study. Ann. Surg. Oncol. 2021, 28, 1130–1141. [Google Scholar] [CrossRef]
- de Leede, E.M.; Burgmans, M.C.; Martini, C.H.; Tijl, F.G.J.; van Erkel, A.R.; Vuyk, J.; Kapiteijn, E.; Verhoef, C.; van de Velde, C.J.H.; Vahrmeijer, A.L. Percutaneous Hepatic Perfusion (PHP) with Melphalan as a Treatment for Unresectable Metastases Confined to the Liver. J. Vis. Exp. 2016, 113, e53795. [Google Scholar] [CrossRef]
- Robertson, A.G.; Shih, J.L.; Yau, C.; Gibb, E.A.; Oba, J.; Mungall, K.L.; Hess, J.M.; Uzunangelov, V.; Walter, V.; Danilova, L.; et al. Integrative Analysis Identifies Four Molecular and Clinical Subsets in Uveal Melanoma. Cancer Cell 2018, 33, 204–220. [Google Scholar] [CrossRef] [Green Version]
- Van Raamsdonk, C.D.; Griewank, K.G.; Crosby, M.B.; Garrido, M.C.; Vemula, S.; Wiesner, T.; Obenauf, A.C.; Wackernagel, W.; Green, G.; Bouvier, N.; et al. Mutations in GNA11 in uveal melanoma. N. Engl. J. Med. 2010, 363, 2191–2199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansson, P.; Aoude, L.G.; Wadt, K.; Glasson, W.J.; Warrier, S.K.; Hewitt, A.W.; Kiilgaard, J.F.; Heegaard, S.; Isaacs, T.; Franchina, M.; et al. Deep sequencing of uveal melanoma identifies a recurrent mutation in PLCB4. Oncotarget 2016, 7, 4624–4631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, A.R.; Ceraudo, E.; Sher, J.J.; Guan, Y.X.; Shoushtari, A.N.; Chang, M.T.; Zhang, J.Q.; Walczak, E.G.; Kazmi, M.A.; Taylor, B.S.; et al. Recurrent activating mutations of G-protein-coupled receptor CYSLTR2 in uveal melanoma. Nat. Genet. 2016, 48, 675–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kilic, E.; van Gils, W.; Lodder, E.; Beverloo, H.B.; van Til, M.E.; Mooy, C.M.; Paridaens, D.; de Klein, A.; Luyten, G.P. Clinical and cytogenetic analyses in uveal melanoma. Investig. Ophthalmol. Vis. Sci. 2006, 47, 3703–3707. [Google Scholar] [CrossRef] [PubMed]
- Sisley, K.; Rennie, I.G.; Parsons, M.A.; Jacques, R.; Hammond, D.W.; Bell, S.M.; Potter, A.M.; Rees, R.C. Abnormalities of chromosomes 3 and 8 in posterior uveal melanoma correlate with prognosis. Genes Chromosomes Cancer 1997, 19, 22–28. [Google Scholar] [CrossRef]
- Kalirai, H.; Dodson, A.; Faqir, S.; Damato, B.E.; Coupland, S.E. Lack of BAP1 protein expression in uveal melanoma is associated with increased metastatic risk and has utility in routine prognostic testing. Br. J. Cancer 2014, 111, 1373–1380. [Google Scholar] [CrossRef] [Green Version]
- Shoushtari, A.N.; Carvajal, R.D. GNAQ and GNA11 mutations in uveal melanoma. Melanoma Res. 2014, 24, 525–534. [Google Scholar] [CrossRef]
- Chen, X.; Wu, Q.X.; Depeille, P.; Chen, P.R.; Thornton, S.; Kalirai, H.; Coupland, S.E.; Roose, J.P.; Bastian, B.C. RasGRP3 Mediates MAPK Pathway Activation in GNAQ Mutant Uveal Melanoma. Cancer Cell 2017, 31, 685–696. [Google Scholar] [CrossRef] [Green Version]
- Xie, P.; Browning, D.D.; Hay, N.; Mackman, N.; Ye, R.D. Activation of NF-kappa B by bradykinin through a Galpha(q)- and Gbeta gamma-dependent pathway that involves phosphoinositide 3-kinase and Akt. J. Biol. Chem. 2000, 275, 24907–24914. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Li, J.; Zhu, M.; Fletcher, J.A.; Hodi, F.S. Protein kinase C inhibitor AEB071 targets ocular melanoma harboring GNAQ mutations via effects on the PKC/Erk1/2 and PKC/NF-kappaB pathways. Mol. Cancer Ther. 2012, 11, 1905–1914. [Google Scholar] [CrossRef] [Green Version]
- Yu, F.X.; Luo, J.; Mo, J.S.; Liu, G.B.; Kim, Y.C.; Meng, Z.P.; Zhao, L.; Peyman, G.; Ouyang, H.; Jiang, W.; et al. Mutant Gq/11 Promote Uveal Melanoma Tumorigenesis by Activating YAP. Cancer Cell 2014, 25, 822–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falchook, G.S.; Lewis, K.D.; Infante, J.R.; Gordon, M.S.; Vogelzang, N.J.; DeMarini, D.J.; Sun, P.; Moy, C.; Szabo, S.A.; Roadcap, L.T.; et al. Activity of the oral MEK inhibitor trametinib in patients with advanced melanoma: A phase 1 dose-escalation trial. Lancet Oncol. 2012, 13, 782–789. [Google Scholar] [CrossRef] [Green Version]
- Shoushtari, A.N.; Kudchadkar, R.R.; Panageas, K.; Murthy, R.K.; Jung, M.; Shah, R.; O’Donnell, B.; Khawaja, T.T.; Shames, Y.; Prempeh-Keteku, N.A.; et al. A randomized phase 2 study of trametinib with or without GSK2141795 in patients with advanced uveal melanoma. J. Clin. Oncol. 2016, 34, 9511. [Google Scholar] [CrossRef]
- Carvajal, R.D.; Piperno-Neumann, S.; Kapiteijn, E.; Chapman, P.B.; Frank, S.; Joshua, A.M.; Piulats, J.M.; Wolter, P.; Cocquyt, V.; Chmielowski, B.; et al. Selumetinib in Combination With Dacarbazine in Patients With Metastatic Uveal Melanoma: A Phase III, Multicenter, Randomized Trial (SUMIT). J. Clin. Oncol. 2018, 36, 1232–1239. [Google Scholar] [CrossRef] [Green Version]
- Piperno-Neumann, S.; Larkin, J.; Carvajal, R.D.; Luke, J.J.; Schwartz, G.K.; Hodi, F.S.; Sablin, M.P.; Shoushtari, A.N.; Szpakowski, S.; Chowdhury, N.R.; et al. Genomic Profiling of Metastatic Uveal Melanoma and Clinical Results of a Phase I Study of the Protein Kinase C Inhibitor AEB071. Mol. Cancer Ther. 2020, 19, 1031–1039. [Google Scholar] [CrossRef]
- Kapiteijn, E.; Carlino, M.; Boni, V.; Loirat, D.; Speetjens, F.; Park, J.; Calvo, E.; Carvajal, R.; Nyakas, M.; Gonzalez-Maffe, J.; et al. A Phase I trial of LXS196, a novel PKC inhibitor for metastatic uveal melanoma. Cancer Res. 2019, 79, CT068. [Google Scholar] [CrossRef]
- Populoa, H.; Soares, P.; Rocha, A.S.; Silva, P.; Lopes, J.M. Evaluation of the mTOR pathway in ocular (uvea and conjunctiva) melanoma. Melanoma Res. 2010, 20, 107–117. [Google Scholar] [CrossRef]
- Amirouchene-Angelozzi, N.; Nemati, F.; Gentien, D.; Nicolas, A.; Dumont, A.; Carita, G.; Camonis, J.; Desjardins, L.; Cassoux, N.; Piperno-Neumann, S.; et al. Establishment of novel cell lines recapitulating the genetic landscape of uveal melanoma and preclinical validation of mTOR as a therapeutic target. Mol. Oncol. 2014, 8, 1508–1520. [Google Scholar] [CrossRef]
- Babchia, N.; Calipel, A.; Mouriaux, F.; Faussat, A.M.; Mascarelli, F. The PI3K/Akt and mTOR/P70S6K Signaling Pathways in Human Uveal Melanoma Cells: Interaction with B-Raf/ERK. Investig. Ophthalmol. Vis. Sci. 2010, 51, 421–429. [Google Scholar] [CrossRef] [Green Version]
- Populo, H.; Tavares, S.; Faustino, A.; Nunes, J.B.; Lopes, J.M.; Soares, P. GNAQ and BRAF mutations show differential activation of the mTOR pathway in human transformed cells. PeerJ 2013, 1, e104. [Google Scholar] [CrossRef]
- Cheng, H.Y.; Chua, V.; Liao, C.; Purwin, T.J.; Terai, M.; Kageyama, K.; Davies, M.A.; Sato, T.; Aplin, A.E. Co-targeting HGF/cMET Signaling with MEK Inhibitors in Metastatic Uveal Melanoma. Mol. Cancer Ther. 2017, 16, 516–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- All-Ericsson, C.; Girnita, L.; Muller-Brunotte, A.; Brodin, B.; Seregard, S.; Ostman, A.; Larsson, O. c-Kit-dependent growth of uveal melanoma cells: A potential therapeutic target? Investig. Ophthalmol. Vis. Sci. 2004, 45, 2075–2082. [Google Scholar] [CrossRef] [PubMed]
- Missotten, G.S.O.; Notting, I.C.; Schlingemann, R.O.; Zijlmans, H.J.; Lau, C.; Eilers, P.H.C.; Keunen, J.E.E.; Jager, M.J. Vascular endothelial growth factor A in eyes with uveal melanoma. Arch. Ophthalmol. 2006, 124, 1428–1434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdel-Rahman, M.H.; Yang, Y.; Zhou, X.P.; Craig, E.L.; Davidorf, F.H.; Eng, C. High frequency of submicroscopic hemizygous deletion is a major mechanism of loss of expression of PTEN in uveal melanoma. J. Clin. Oncol. 2006, 24, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Amirouchene-Angelozzi, N.; Frisch-Dit-Leitz, E.; Carita, G.; Dahmani, A.; Raymondie, C.; Liot, G.; Gentien, D.; Nemati, F.; Decaudin, D.; Roman-Roman, S.; et al. The mTOR inhibitor Everolimus synergizes with the PI3K inhibitor GDC0941 to enhance anti-tumor efficacy in uveal melanoma. Oncotarget 2016, 7, 23633–23646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carita, G.; Frisch-Dit-Leitz, E.; Dahmani, A.; Raymondie, C.; Cassoux, N.; Piperno-Neumann, S.; Nemati, F.; Laurent, C.; De Koning, L.; Halilovic, E.; et al. Dual inhibition of protein kinase C and p53-MDM2 or PKC and mTORC1 are novel efficient therapeutic approaches for uveal melanoma. Oncotarget 2016, 7, 33542–33556. [Google Scholar] [CrossRef] [Green Version]
- Jerusalem, G.; de Boer, R.H.; Hurvitz, S.; Yardley, D.A.; Kovalenko, E.; Ejlertsen, B.; Blau, S.; Ozguroglu, M.; Landherr, L.; Ewertz, M.; et al. Everolimus Plus Exemestane vs Everolimus or Capecitabine Monotherapy for Estrogen Receptor-Positive, HER2-Negative Advanced Breast Cancer The BOLERO-6 Randomized Clinical Trial. JAMA Oncol. 2018, 4, 1367–1374. [Google Scholar] [CrossRef]
- Buti, S.; Leonetti, A.; Dallatomasina, A.; Bersanelli, M. Everolimus in the management of metastatic renal cell carcinoma: An evidence-based review of its place in therapy. Core Evid. 2016, 11, 23–36. [Google Scholar] [CrossRef] [Green Version]
- Lee, L.; Ito, T.; Jensen, R.T. Everolimus in the treatment of neuroendocrine tumors: Efficacy, side-effects, resistance, and factors affecting its place in the treatment sequence. Expert Opin. Pharmacother. 2018, 19, 909–928. [Google Scholar] [CrossRef]
- Shoushtari, A.N.; Ong, L.T.; Schoder, H.; Singh-Kandah, S.; Abbate, K.T.; Postow, M.A.; Callahan, M.K.; Wolchok, J.; Chapman, P.B.; Panageas, K.S.; et al. A phase 2 trial of everolimus and pasireotide long-acting release in patients with metastatic uveal melanoma. Melanoma Res. 2016, 26, 272–277. [Google Scholar] [CrossRef] [Green Version]
- Luyten, G.P.M.; Naus, N.C.; Mooy, C.M.; Hagemeijer, A.; KanMitchell, J.; VanDrunen, E.; Vuzevski, V.; DeJong, P.T.V.M.; Luider, T.M. Establishment and characterization of primary and metastatic uveal melanoma cell lines. Int. J. Cancer 1996, 66, 380–387. [Google Scholar] [CrossRef]
- Bliss, C.I. Calculation of Microbial Assays. Bacteriol. Rev. 1956, 20, 243–258. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Jin, H.J.; Gao, D.M.; Wang, L.Q.; Evers, B.; Xue, Z.; Jin, G.Z.; Lieftink, C.; Beijersbergen, R.L.; Qin, W.X.; et al. A CRISPR screen identifies CDK7 as a therapeutic target in hepatocellular carcinoma. Cell Res. 2018, 28, 690–692. [Google Scholar] [CrossRef]
- Li, W.; Xu, H.; Xiao, T.F.; Cong, L.; Love, M.I.; Zhang, F.; Irizarry, R.A.; Liu, J.S.; Brown, M.; Liu, X.S. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 2014, 15, 554. [Google Scholar] [CrossRef] [PubMed]
- Groenewoud, A.; Yin, J.; Gelmi, M.C.; Alsafadi, S.; Nemati, F.; Decaudin, D.; Roman-Roman, S.; Kalirai, H.; Coupland, S.E.; Jochemsen, A.G.; et al. Patient-Derived Zebrafish Xenograft Models Reveal Ferroptosis as a Fatal and Druggable Weakness in Metastatic Uveal Melanoma. 2021. Available online: https://www.biorxiv.org/content/10.1101/2021.10.26.465874v1.full (accessed on 28 November 2021).
- Lawson, N.D.; Weinstein, B.M. In vivo imaging of embryonic vascular development using transgenic zebrafish. Dev. Biol. 2002, 248, 307–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groenewoud, A.; Yin, J.; Snaar-Jagalska, B.E. Ortho- and Ectopic Zebrafish Xeno-Engraftment of Ocular Melanoma to Recapitulate Primary Tumor and Experimental Metastasis Development. J. Vis. Exp. 2021, 175, e62356. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Groenewoud, A. JoVE Three Channel (c3) Analysis (MaxEntropy) Macro for FIJI. 2020. Available online: https://zenodo.org/record/4290225#.YoToEKhBxaQ (accessed on 10 December 2021).
- Griewank, K.G.; Yu, X.; Khalili, J.; Sozen, M.M.; Stempke-Hale, K.; Bernatchez, C.; Wardell, S.; Bastian, B.C.; Woodman, S.E. Genetic and molecular characterization of uveal melanoma cell lines. Pigment. Cell Melanoma Res. 2012, 25, 182–187. [Google Scholar] [CrossRef] [Green Version]
- Munster, P.; Mita, M.; Mahipal, A.; Nemunaitis, J.; Massard, C.; Mikkelsen, T.; Cruz, C.; Paz-Ares, L.; Hidalgo, M.; Rathkopf, D.; et al. First-In-Human Phase I Study Of A Dual mTOR Kinase And DNA-PK Inhibitor (CC-115) In Advanced Malignancy. Cancer Manag. Res. 2019, 11, 10463–10476. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Martinez, J.M.; Moran, J.; Clarke, R.G.; Gray, A.; Cosulich, S.C.; Chresta, C.M.; Alessi, D.R. Ku-0063794 is a specific inhibitor of the mammalian target of rapamycin (mTOR). Biochem. J. 2009, 421, 29–42. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, N.G.; Castro, M.; Rodrigues, A.S.; Gil, O.M.; Toscano-Rico, J.M.; Rueff, J. DNA-PK inhibitor wortmannin enhances DNA damage induced by bleomycin in V79 Chinese hamster cells. Teratog. Carcinog. Mutagenes. 2002, 22, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Thomas, H.D.; Batey, M.A.; Cowell, I.G.; Richardson, C.J.; Griffin, R.J.; Calvert, A.H.; Newell, D.R.; Smith, G.C.M.; Curtin, N.J. Preclinical evaluation of a potent novel DNA-dependent protein kinase inhibitor NU7441. Cancer Res. 2006, 66, 5354–5362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambrosini, G.; Musi, E.; Ho, A.L.; de Stanchina, E.; Schwartz, G.K. Inhibition of Mutant GNAQ Signaling in Uveal Melanoma Induces AMPK-Dependent Autophagic Cell Death. Mol. Cancer Ther. 2013, 12, 768–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campone, M.; Bachelot, T.; Treilleux, I.; Pistilli, B.; Salleron, J.; Seegers, V.; Arnedos, M.; Loussouarn, D.; Wang, Q.; Vanlemmens, L.; et al. A phase II randomised study of preoperative trastuzumab alone or combined with everolimus in patients with early HER2-positive breast cancer and predictive biomarkers (RADHER trial). Eur. J. Cancer 2021, 158, 169–180. [Google Scholar] [CrossRef]
- Watson, A.L.; Anderson, L.K.; Greeley, A.D.; Keng, V.W.; Rahrmann, E.P.; Halfond, A.L.; Powell, N.M.; Collins, M.H.; Rizvi, T.; Moertel, C.L.; et al. Co-targeting the MAPK and PI3K/AKT/mTOR pathways in two genetically engineered mouse models of schwann cell tumors reduces tumor grade and multiplicity. Oncotarget 2014, 5, 1502–1514. [Google Scholar] [CrossRef] [Green Version]
- Carracedo, A.; Ma, L.; Teruya-Feldstein, J.; Rojo, F.; Salmena, L.; Alimonti, A.; Egia, A.; Sasaki, A.T.; Thomas, G.; Kozma, S.C.; et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J. Clin. Investig. 2008, 118, 3065–3074. [Google Scholar] [CrossRef]
- Steeb, T.; Wessely, A.; Ruzicka, T.; Heppt, M.V.; Berking, C. How to MEK the best of uveal melanoma: A systematic review on the efficacy and safety of MEK inhibitors in metastatic or unresectable uveal melanoma. Eur. J. Cancer 2018, 103, 41–51. [Google Scholar] [CrossRef]
- Yoshida, M.; Selvan, S.; McCue, P.A.; DeAngelis, T.; Baserga, R.; Fujii, A.; Rui, H.; Mastrangelo, M.J.; Sato, T. Expression of insulin-like growth factor-1 receptor in metastatic uveal melanoma and implications for potential autocrine and paracrine tumor cell growth. Pigment. Cell Melanoma Res. 2014, 27, 297–308. [Google Scholar] [CrossRef]
- Mattei, J.; Ballhausen, A.; Bassett, R.; Shephard, M.; Chattopadhyay, C.; Hudgens, C.; Tetzlaff, M.; Woodman, S.; Sato, T.; Patel, S.P. A phase II study of the insulin-like growth factor type I receptor inhibitor IMC-A12 in patients with metastatic uveal melanoma. Melanoma Res. 2020, 30, 574–579. [Google Scholar] [CrossRef]
- Quek, R.H.; Morgan, J.A.; Shapiro, G.; Butrynski, J.E.; Wang, Q.; Huftalen, T.; Jederlinic, N.; Wagner, A.J.; Demetri, G.D.; George, S. Combination mTOR plus IGF-IR inhibition: Phase I trial of everolimus and CP-751871 in patients with advanced sarcomas and other solid tumors. J. Clin. Oncol. 2010, 28, 10002. [Google Scholar] [CrossRef]
- Bendell, J.C.; Jones, S.F.; Hart, L.; Spigel, D.R.; Lane, C.M.; Earwood, C.; Infante, J.R.; Barton, J.; Burris, H.A. A phase Ib study of linsitinib (OSI-906), a dual inhibitor of IGF-1R and IR tyrosine kinase, in combination with everolimus as treatment for patients with refractory metastatic colorectal cancer. Investig. New Drugs 2015, 33, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.J.; Wu, H.Y.; Hao, J.L.; Wu, Y.L.; Yang, B. Inhibition of DNA-PKcs activity re-sensitizes uveal melanoma cells to radio- and chemotherapy. Biochem. Biophys. Res. Commun. 2020, 522, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Doherty, R.E.; Bryant, H.E.; Valluru, M.K.; Rennie, I.G.; Sisley, K. Increased Non-Homologous End Joining Makes DNA-PK a Promising Target for Therapeutic Intervention in Uveal Melanoma. Cancers 2019, 11, 1278. [Google Scholar] [CrossRef] [Green Version]
- Dogrusoz, M.; Trasel, A.R.; Cao, J.F.; Colak, S.; van Pelt, S.I.; Kroes, W.G.M.; Teunisse, A.E.A.S.; Alsafadi, S.; van Duinen, S.G.; Luyten, G.P.M.; et al. Differential Expression of DNA Repair Genes in Prognostically-Favorable versus Unfavorable Uveal Melanoma. Cancers 2019, 11, 1104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honegger, A.; Humbel, R.E. Insulin-Like Growth Factor-I and Factor-Ii in Fetal and Adult Bovine Serum-Purification, Primary Structures, and Immunological Cross-Reactivities. J. Biol. Chem. 1986, 261, 569–575. [Google Scholar] [CrossRef]
- Jiang, W.X.; Crowe, J.L.; Liu, X.Y.; Nakajima, S.; Wang, Y.Y.; Li, C.; Lee, B.J.; Dubois, R.L.; Liu, C.; Yu, X.C.; et al. Differential Phosphorylation of DNA-PKcs Regulates the Interplay between End-Processing and End-Ligation during Nonhomologous End-Joining. Mol. Cell 2015, 58, 172–185. [Google Scholar] [CrossRef] [Green Version]
- Tsuji, T.; Sapinoso, L.M.; Tran, T.; Gaffney, B.; Wong, L.; Sankar, S.; Raymon, H.K.; Mortensen, D.S.; Xu, S.C. CC-115, a dual inhibitor of mTOR kinase and DNA-PK, blocks DNA damage repair pathways and selectively inhibits ATM-deficient cell growth in vitro. Oncotarget 2017, 8, 74688–74702. [Google Scholar] [CrossRef] [Green Version]
- Rathkopf, D.E.; Autio, K.A.; Antonarakis, E.S.; Cheng, H.H.; Arauz, G.; Slack, A.; Hullings, M.; Scher, H.I.; Feng, F.Y.; Knudsen, K.E. c15-160: Enzalutamide (ENZA) plus CC-115 in men with metastatic castration-resistant prostate cancer (mCRPC): A phase 1b Prostate Cancer Clinical Trials Consortium study. J. Clin. Oncol. 2018, 36, 5045. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Origin | GNAQ Mutation | GNA11 Mutation | BAP1 Expression | Reference |
|---|---|---|---|---|---|
| OMM2.3 | Liver metastases | c.626 A > C | - | Yes | [41,50] |
| OMM2.5 | Liver metastases | c.626 A > C | - | Yes | [28,41,50] |
| OMM1 | Subcutaneous metastasis | - | c.626 A > T | Yes | [28,41,50] |
| MM66 | PDX established from liver metastasis | - | c.626 A > T | Yes | [28] |
| MM28 | PDX established from liver metastasis | - | c.626 A > T | No | [28] |
| MP46 | PDX established fromPrimary tumor | c.626 A > T | - | No | [28] |
| MP38 | Primary tumor | c.626 A > T | - | No | [28] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Glinkina, K.; Groenewoud, A.; Teunisse, A.F.A.S.; Snaar-Jagalska, B.E.; Jochemsen, A.G. Novel Treatments of Uveal Melanoma Identified with a Synthetic Lethal CRISPR/Cas9 Screen. Cancers 2022, 14, 3186. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14133186
Glinkina K, Groenewoud A, Teunisse AFAS, Snaar-Jagalska BE, Jochemsen AG. Novel Treatments of Uveal Melanoma Identified with a Synthetic Lethal CRISPR/Cas9 Screen. Cancers. 2022; 14(13):3186. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14133186
Chicago/Turabian StyleGlinkina, Kseniya, Arwin Groenewoud, Amina F. A. S. Teunisse, B. Ewa Snaar-Jagalska, and Aart G. Jochemsen. 2022. "Novel Treatments of Uveal Melanoma Identified with a Synthetic Lethal CRISPR/Cas9 Screen" Cancers 14, no. 13: 3186. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14133186