
Beyond Pathogenic RUNX1 Germline Variants: The Spectrum of Somatic Alterations in RUNX1-Familial Platelet Disorder with Predisposition to Hematologic Malignancies


Abstract
:Simple Summary
Abstract
1. Introduction
2. Methods
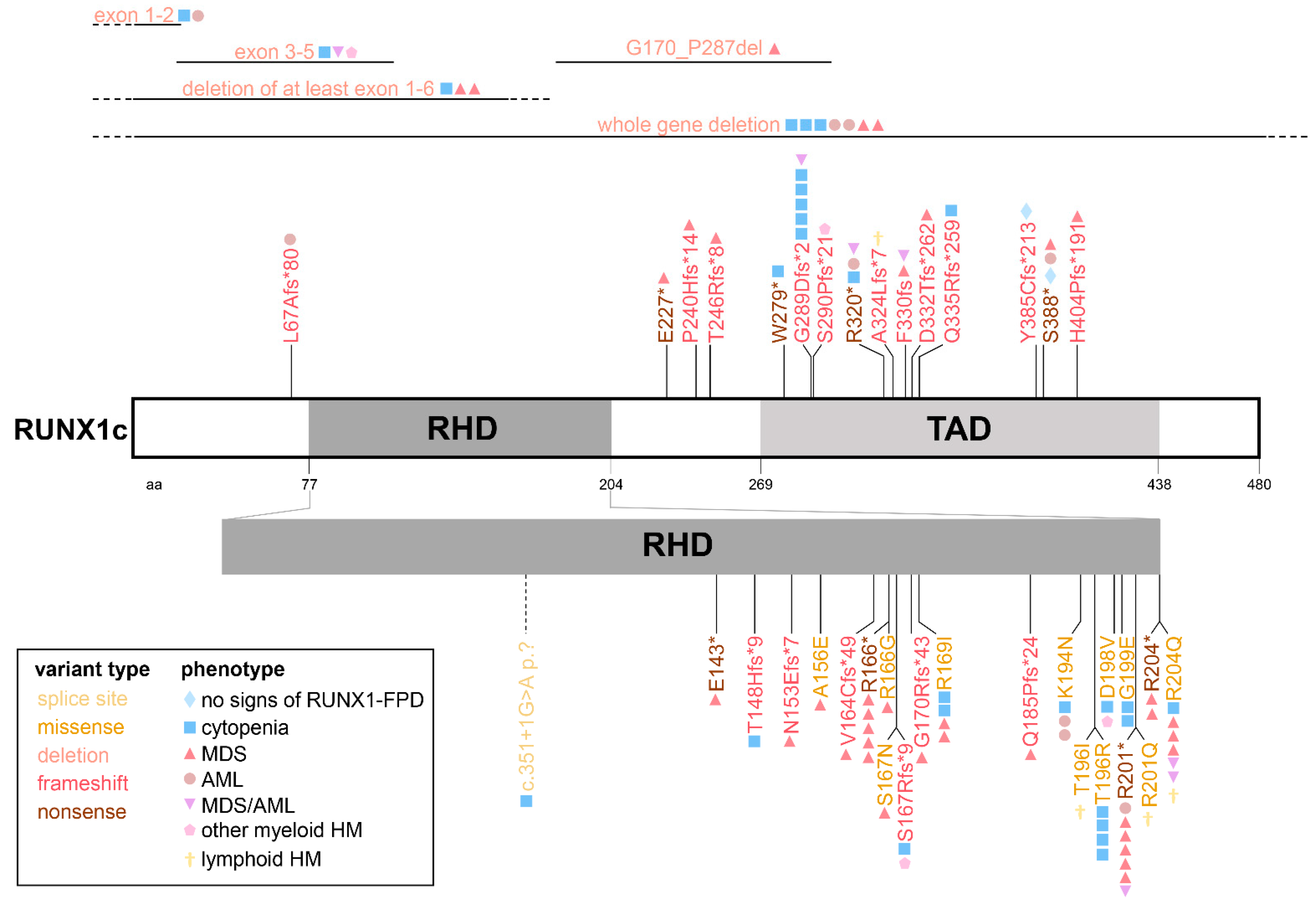
3. Disease-Causing RUNX1 Germline Variants and Associated Phenotypes in RUNX1-FPD
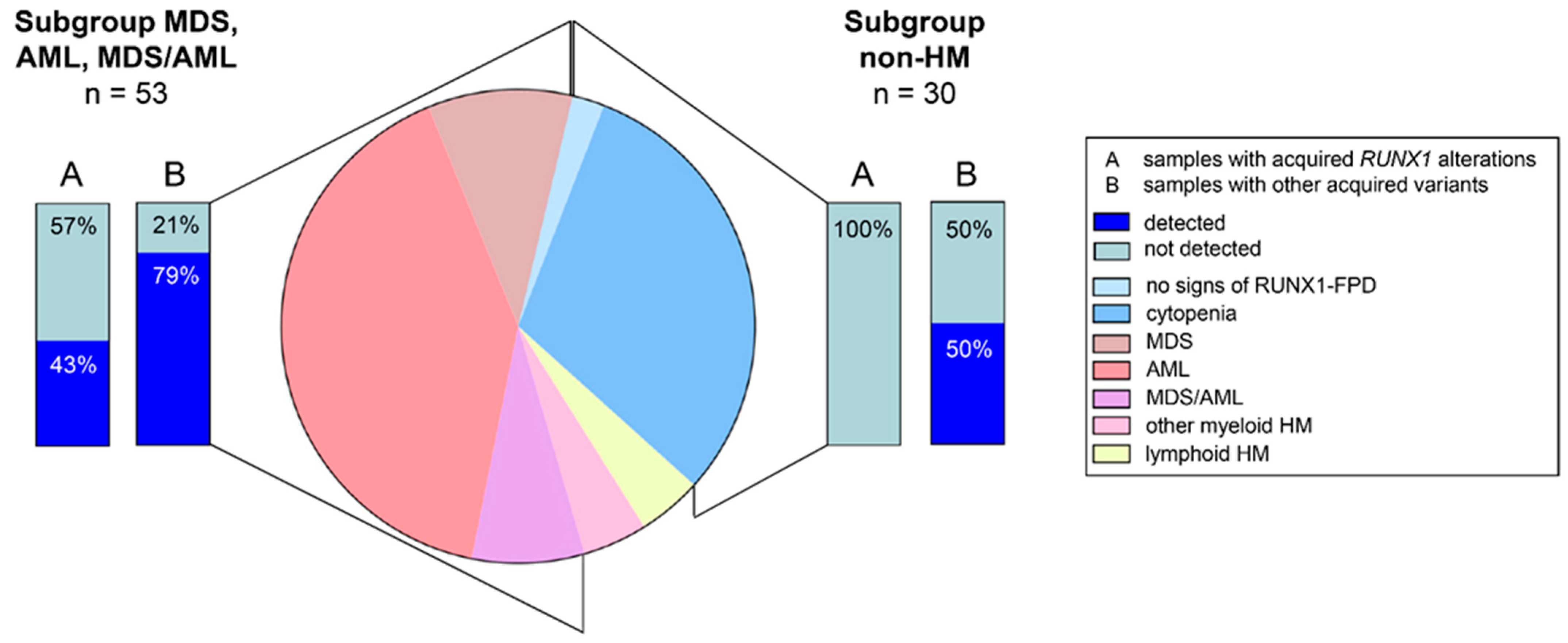
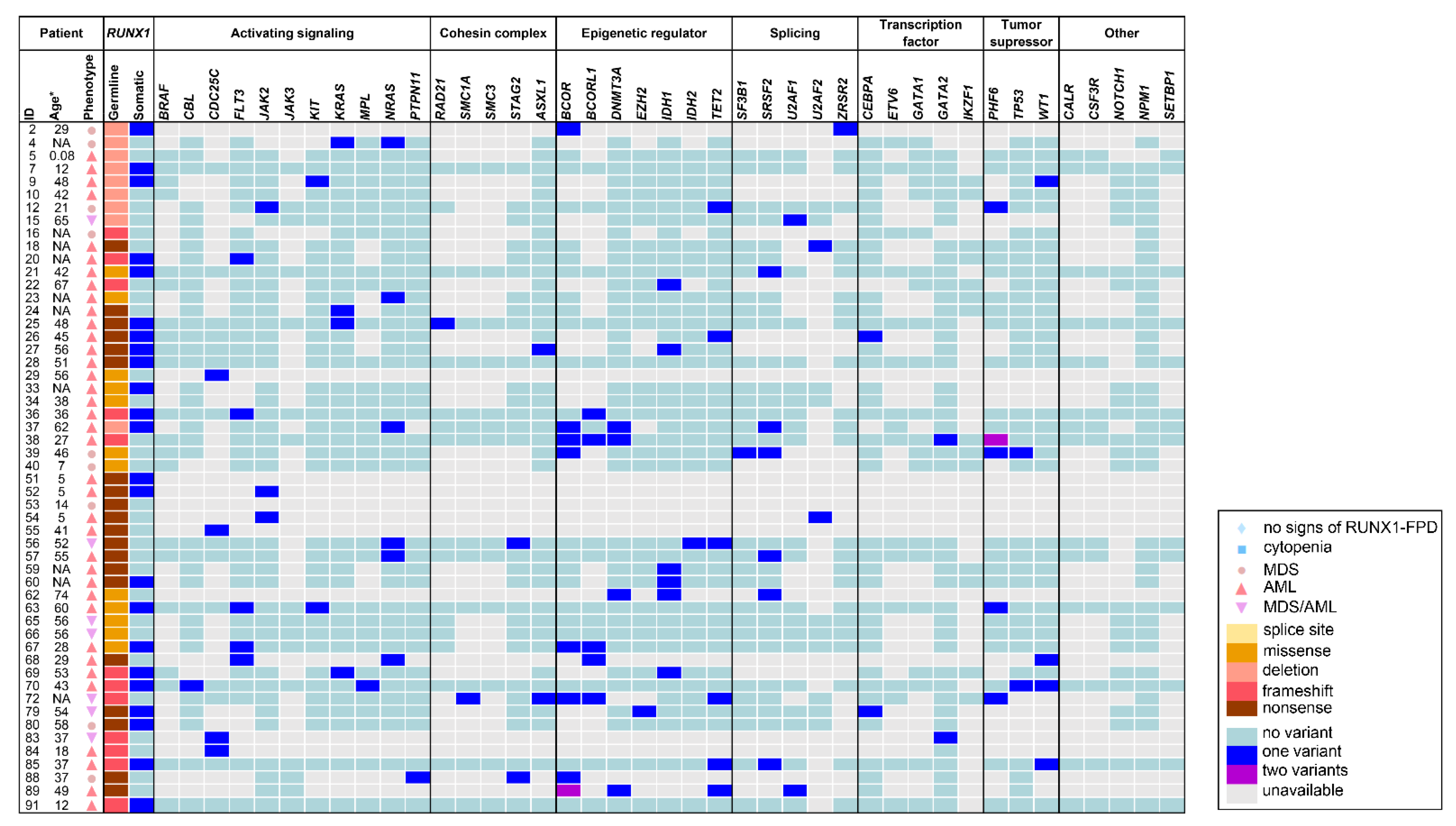
4. Spectrum of Somatic Variants and Affected Genes in RUNX1-FPD
5. Prospective Surveillance Strategies for RUNX1-FPD Patients
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Appendix A
References
- Sood, R.; Kamikubo, Y.; Liu, P. Role of RUNX1 in hematological malignancies. Blood 2017, 129, 2070–2082. [Google Scholar] [CrossRef] [Green Version]
- Miyoshi, H.; Ohira, M.; Shimizu, K.; Mitani, K.; Hirai, H.; Imai, T.; Yokoyama, K.; Soeda, E.; Ohki, M. Alternative splicing and genomic structure of the AML1 gene involved in acute myeloid leukemia. Nucleic Acids Res. 1995, 23, 2762–2769. [Google Scholar] [CrossRef] [Green Version]
- Kagoshima, H.; Shigesada, K.; Satake, M.; Ito, Y.; Miyoshi, H.; Ohki, M.; Pepling, M.; Gergen, P. The Runt domain identifies a new family of heteromeric transcriptional regulators. Trends Genet. 1993, 9, 338–341. [Google Scholar] [CrossRef]
- Wang, S.; Wang, Q.; Crute, B.E.; Melnikova, I.N.; Keller, S.R.; Speck, N.A. Cloning and characterization of subunits of the T-cell receptor and murine leukemia virus enhancer core-binding factor. Mol. Cell Biol. 1993, 13, 3324–3339. [Google Scholar] [CrossRef]
- Ogawa, E.; Inuzuka, M.; Maruyama, M.; Satake, M.; Naito-Fujimoto, M.; Ito, Y.; Shigesada, K. Molecular cloning and characterization of PEBP2 beta, the heterodimeric partner of a novel Drosophila runt-related DNA binding protein PEBP2 alpha. Virology 1993, 194, 314–331. [Google Scholar] [CrossRef]
- Bravo, J.; Li, Z.; Speck, N.A.; Warren, A.J. The leukemia-associated AML1 (Runx1)—CBF beta complex functions as a DNA-induced molecular clamp. Nat. Struct. Biol. 2001, 8, 371–378. [Google Scholar] [CrossRef]
- Huang, G.; Shigesada, K.; Ito, K.; Wee, H.J.; Yokomizo, T.; Ito, Y. Dimerization with PEBP2beta protects RUNX1/AML1 from ubiquitin-proteasome-mediated degradation. Embo J. 2001, 20, 723–733. [Google Scholar] [CrossRef] [Green Version]
- Kanno, T.; Kanno, Y.; Chen, L.F.; Ogawa, E.; Kim, W.Y.; Ito, Y. Intrinsic transcriptional activation-inhibition domains of the polyomavirus enhancer binding protein 2/core binding factor alpha subunit revealed in the presence of the beta subunit. Mol. Cell Biol. 1998, 18, 2444–2454. [Google Scholar] [CrossRef] [Green Version]
- Lutterbach, B.; Westendorf, J.J.; Linggi, B.; Isaac, S.; Seto, E.; Hiebert, S.W. A mechanism of repression by acute myeloid leukemia-1, the target of multiple chromosomal translocations in acute leukemia. J. Biol. Chem. 2000, 275, 651–656. [Google Scholar] [CrossRef] [Green Version]
- Durst, K.L.; Hiebert, S.W. Role of RUNX family members in transcriptional repression and gene silencing. Oncogene 2004, 23, 4220–4224. [Google Scholar] [CrossRef] [Green Version]
- Hong, D.; Fritz, A.J.; Gordon, J.A.; Tye, C.E.; Boyd, J.R.; Tracy, K.M.; Frietze, S.E.; Carr, F.E.; Nickerson, J.A.; Van Wijnen, A.J.; et al. RUNX1-dependent mechanisms in biological control and dysregulation in cancer. J. Cell Physiol. 2019, 234, 8597–8609. [Google Scholar] [CrossRef]
- Okuda, T.; van Deursen, J.; Hiebert, S.W.; Grosveld, G.; Downing, J.R. AML1, the target of multiple chromosomal translocations in human leukemia, is essential for normal fetal liver hematopoiesis. Cell 1996, 84, 321–330. [Google Scholar] [CrossRef] [Green Version]
- Yzaguirre, A.D.; de Bruijn, M.F.; Speck, N.A. The Role of Runx1 in Embryonic Blood Cell Formation. Adv. Exp. Med. Biol. 2017, 962, 47–64. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Stacy, T.; Binder, M.; Marin-Padilla, M.; Sharpe, A.H.; Speck, N.A. Disruption of the Cbfa2 gene causes necrosis and hemorrhaging in the central nervous system and blocks definitive hematopoiesis. Proc. Natl. Acad. Sci. USA 1996, 93, 3444–3449. [Google Scholar] [CrossRef] [Green Version]
- Bellissimo, D.C.; Speck, N.A. RUNX1 Mutations in Inherited and Sporadic Leukemia. Front. Cell Dev. Biol. 2017, 5, 111. [Google Scholar] [CrossRef]
- Metzeler, K.H.; Herold, T.; Rothenberg-Thurley, M.; Amler, S.; Sauerland, M.C.; Görlich, D.; Schneider, S.; Konstandin, N.P.; Dufour, A.; Bräundl, K.; et al. Spectrum and prognostic relevance of driver gene mutations in acute myeloid leukemia. Blood 2016, 128, 686–698. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, Y.; Harada, Y.; Huang, G.; Harada, H. Myeloid neoplasms with germ line RUNX1 mutation. Int. J. Hematol. 2017, 106, 183–188. [Google Scholar] [CrossRef]
- Hayashi, Y.; Harada, Y.; Harada, H. Myeloid neoplasms and clonal hematopoiesis from the RUNX1 perspective. Leukemia 2022, 36, 1203–1214. [Google Scholar] [CrossRef]
- Huret, J.L.; Ahmad, M.; Arsaban, M.; Bernheim, A.; Cigna, J.; Desangles, F.; Guignard, J.C.; Jacquemot-Perbal, M.C.; Labarussias, M.; Leberre, V.; et al. Atlas of genetics and cytogenetics in oncology and haematology in 2013. Nucleic Acids Res. 2013, 41, D920–D924. [Google Scholar] [CrossRef]
- Stengel, A.; Kern, W.; Meggendorfer, M.; Nadarajah, N.; Perglerovà, K.; Haferlach, T.; Haferlach, C. Number of RUNX1 mutations, wild-type allele loss and additional mutations impact on prognosis in adult RUNX1-mutated AML. Leukemia 2018, 32, 295–302. [Google Scholar] [CrossRef]
- Yamato, G.; Shiba, N.; Yoshida, K.; Hara, Y.; Shiraishi, Y.; Ohki, K.; Okubo, J.; Park, M.J.; Sotomatsu, M.; Arakawa, H.; et al. RUNX1 mutations in pediatric acute myeloid leukemia are associated with distinct genetic features and an inferior prognosis. Blood 2018, 131, 2266–2270. [Google Scholar] [CrossRef]
- Mendler, J.H.; Maharry, K.; Radmacher, M.D.; Mrózek, K.; Becker, H.; Metzeler, K.H.; Schwind, S.; Whitman, S.P.; Khalife, J.; Kohlschmidt, J.; et al. RUNX1 mutations are associated with poor outcome in younger and older patients with cytogenetically normal acute myeloid leukemia and with distinct gene and MicroRNA expression signatures. J. Clin. Oncol. 2012, 30, 3109–3118. [Google Scholar] [CrossRef] [Green Version]
- Grossmann, V.; Kern, W.; Harbich, S.; Alpermann, T.; Jeromin, S.; Schnittger, S.; Haferlach, C.; Haferlach, T.; Kohlmann, A. Prognostic relevance of RUNX1 mutations in T-cell acute lymphoblastic leukemia. Haematologica 2011, 96, 1874–1877. [Google Scholar] [CrossRef] [Green Version]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Song, W.J.; Sullivan, M.G.; Legare, R.D.; Hutchings, S.; Tan, X.; Kufrin, D.; Ratajczak, J.; Resende, I.C.; Haworth, C.; Hock, R.; et al. Haploinsufficiency of CBFA2 causes familial thrombocytopenia with propensity to develop acute myelogenous leukaemia. Nat. Genet. 1999, 23, 166–175. [Google Scholar] [CrossRef]
- Deuitch, N.; Broadbridge, E.; Cunningham, L.; Liu, P. RUNX1 Familial Platelet Disorder with Associated Myeloid Malignancies. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Mirzaa, G.M., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Latger-Cannard, V.; Philippe, C.; Bouquet, A.; Baccini, V.; Alessi, M.C.; Ankri, A.; Bauters, A.; Bayart, S.; Cornillet-Lefebvre, P.; Daliphard, S.; et al. Haematological spectrum and genotype-phenotype correlations in nine unrelated families with RUNX1 mutations from the French network on inherited platelet disorders. Orphanet J. Rare Dis. 2016, 11, 49. [Google Scholar] [CrossRef] [Green Version]
- Godley, L.A. Inherited predisposition to acute myeloid leukemia. Semin. Hematol. 2014, 51, 306–321. [Google Scholar] [CrossRef] [Green Version]
- Churpek, J.E.; Lorenz, R.; Nedumgottil, S.; Onel, K.; Olopade, O.I.; Sorrell, A.; Owen, C.J.; Bertuch, A.A.; Godley, L.A. Proposal for the clinical detection and management of patients and their family members with familial myelodysplastic syndrome/acute leukemia predisposition syndromes. Leuk. Lymphoma 2013, 54, 28–35. [Google Scholar] [CrossRef]
- West, A.H.; Godley, L.A.; Churpek, J.E. Familial myelodysplastic syndrome/acute leukemia syndromes: A review and utility for translational investigations. Ann. N. Y. Acad. Sci. 2014, 1310, 111–118. [Google Scholar] [CrossRef]
- Simon, L.; Spinella, J.F.; Yao, C.Y.; Lavallée, V.P.; Boivin, I.; Boucher, G.; Audemard, E.; Bordeleau, M.E.; Lemieux, S.; Hébert, J.; et al. High frequency of germline RUNX1 mutations in patients with RUNX1-mutated AML. Blood 2020, 135, 1882–1886. [Google Scholar] [CrossRef]
- Feurstein, S.; Zhang, L.; DiNardo, C.D. Accurate germline RUNX1 variant interpretation and its clinical significance. Blood Adv. 2020, 4, 6199–6203. [Google Scholar] [CrossRef]
- Bąk, A.; Skonieczka, K.; Jaśkowiec, A.; Junkiert-Czarnecka, A.; Heise, M.; Pilarska-Deltow, M.; Potoczek, S.; Czyżewska, M.; Haus, O. Searching for germline mutations in the RUNX1 gene among Polish patients with acute myeloid leukemia. Leuk. Lymphoma 2021, 62, 1749–1755. [Google Scholar] [CrossRef]
- Ernst, M.P.T.; Kavelaars, F.G.; Löwenberg, B.; Valk, P.J.M.; Raaijmakers, M. RUNX1 germline variants in RUNX1-mutant AML: How frequent? Blood 2021, 137, 1428–1431. [Google Scholar] [CrossRef]
- Homan, C.C.; King-Smith, S.L.; Lawrence, D.M.; Arts, P.; Feng, J.; Andrews, J.; Armstrong, M.; Ha, T.; Dobbins, J.; Drazer, M.W.; et al. The RUNX1 database (RUNX1db): Establishment of an expert curated RUNX1 registry and genomics database as a public resource for familial platelet disorder with myeloid malignancy. Haematologica 2021, 106, 3004–3007. [Google Scholar] [CrossRef]
- Brown, A.L.; Arts, P.; Carmichael, C.L.; Babic, M.; Dobbins, J.; Chong, C.E.; Schreiber, A.W.; Feng, J.; Phillips, K.; Wang, P.P.S.; et al. RUNX1-mutated families show phenotype heterogeneity and a somatic mutation profile unique to germline predisposed AML. Blood Adv. 2020, 4, 1131–1144. [Google Scholar] [CrossRef] [Green Version]
- Brown, A.L.; Hahn, C.N.; Scott, H.S. Secondary leukemia in patients with germline transcription factor mutations (RUNX1, GATA2, CEBPA). Blood 2020, 136, 24–35. [Google Scholar] [CrossRef]
- Luo, X.; Feurstein, S.; Mohan, S.; Porter, C.C.; Jackson, S.A.; Keel, S.; Chicka, M.; Brown, A.L.; Kesserwan, C.; Agarwal, A.; et al. ClinGen Myeloid Malignancy Variant Curation Expert Panel recommendations for germline RUNX1 variants. Blood Adv. 2019, 3, 2962–2979. [Google Scholar] [CrossRef] [Green Version]
- Decker, M.; Lammens, T.; Ferster, A.; Erlacher, M.; Yoshimi, A.; Niemeyer, C.M.; Ernst, M.P.T.; Raaijmakers, M.; Duployez, N.; Flaum, A.; et al. Functional classification of RUNX1 variants in familial platelet disorder with associated myeloid malignancies. Leukemia 2021, 35, 3304–3308. [Google Scholar] [CrossRef]
- Decker, M.; Agarwal, A.; Benneche, A.; Churpek, J.E.; Duployez, N.; DuVall, A.; Ernst, M.P.T.; Förster, A.; Høberg Vetti, H.; Nash, M.; et al. Validation and clinical application of transactivation assays for RUNX1 variant classification. Blood Adv. 2022, 6, 3195–3200. [Google Scholar] [CrossRef]
- Ripperger, T.; Steinemann, D.; Göhring, G.; Finke, J.; Niemeyer, C.M.; Strahm, B.; Schlegelberger, B. A novel pedigree with heterozygous germline RUNX1 mutation causing familial MDS-related AML: Can these families serve as a multistep model for leukemic transformation? Leukemia 2009, 23, 1364–1366. [Google Scholar] [CrossRef] [Green Version]
- Osato, M.; Yanagida, M.; Shigesada, K.; Ito, Y. Point mutations of the RUNx1/AML1 gene in sporadic and familial myeloid leukemias. Int. J. Hematol. 2001, 74, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Gaidzik, V.I.; Teleanu, V.; Papaemmanuil, E.; Weber, D.; Paschka, P.; Hahn, J.; Wallrabenstein, T.; Kolbinger, B.; Köhne, C.H.; Horst, H.A.; et al. RUNX1 mutations in acute myeloid leukemia are associated with distinct clinico-pathologic and genetic features. Leukemia 2016, 30, 2160–2168. [Google Scholar] [CrossRef] [PubMed]
- Yoshimi, A.; Toya, T.; Kawazu, M.; Ueno, T.; Tsukamoto, A.; Iizuka, H.; Nakagawa, M.; Nannya, Y.; Arai, S.; Harada, H.; et al. Recurrent CDC25C mutations drive malignant transformation in FPD/AML. Nat. Commun. 2014, 5, 4770. [Google Scholar] [CrossRef] [PubMed]
- Preudhomme, C.; Renneville, A.; Bourdon, V.; Philippe, N.; Roche-Lestienne, C.; Boissel, N.; Dhedin, N.; André, J.M.; Cornillet-Lefebvre, P.; Baruchel, A.; et al. High frequency of RUNX1 biallelic alteration in acute myeloid leukemia secondary to familial platelet disorder. Blood 2009, 113, 5583–5587. [Google Scholar] [CrossRef] [Green Version]
- Meisel, M.; Hinterleitner, R.; Pacis, A.; Chen, L.; Earley, Z.M.; Mayassi, T.; Pierre, J.F.; Ernest, J.D.; Galipeau, H.J.; Thuille, N.; et al. Microbial signals drive pre-leukaemic myeloproliferation in a Tet2-deficient host. Nature 2018, 557, 580–584. [Google Scholar] [CrossRef]
- Avagyan, S.; Brown, A.L. To T or not to B: Germline RUNX1 mutation preferences in pediatric ALL predisposition. J. Clin. Invest. 2021, 131. [Google Scholar] [CrossRef]
- Antony-Debré, I.; Duployez, N.; Bucci, M.; Geffroy, S.; Micol, J.B.; Renneville, A.; Boissel, N.; Dhédin, N.; Réa, D.; Nelken, B.; et al. Somatic mutations associated with leukemic progression of familial platelet disorder with predisposition to acute myeloid leukemia. Leukemia 2016, 30, 999–1002. [Google Scholar] [CrossRef]
- Six, K.A.; Gerdemann, U.; Brown, A.L.; Place, A.E.; Cantor, A.B.; Kutny, M.A.; Avagyan, S. B-cell acute lymphoblastic leukemia in patients with germline RUNX1 mutations. Blood Adv. 2021, 5, 3199–3202. [Google Scholar] [CrossRef]
- DiFilippo, E.C.; Coltro, G.; Carr, R.M.; Mangaonkar, A.A.; Binder, M.; Khan, S.P.; Rodriguez, V.; Gangat, N.; Wolanskyj, A.; Pruthi, R.K.; et al. Spectrum of abnormalities and clonal transformation in germline RUNX1 familial platelet disorder and a genomic comparative analysis with somatic RUNX1 mutations in MDS/MPN overlap neoplasms. Leukemia 2020, 34, 2519–2524. [Google Scholar] [CrossRef]
- Harada, H.; Harada, Y.; Niimi, H.; Kyo, T.; Kimura, A.; Inaba, T. High incidence of somatic mutations in the AML1/RUNX1 gene in myelodysplastic syndrome and low blast percentage myeloid leukemia with myelodysplasia. Blood 2004, 103, 2316–2324. [Google Scholar] [CrossRef]
- Michaud, J.; Wu, F.; Osato, M.; Cottles, G.M.; Yanagida, M.; Asou, N.; Shigesada, K.; Ito, Y.; Benson, K.F.; Raskind, W.H.; et al. In vitro analyses of known and novel RUNX1/AML1 mutations in dominant familial platelet disorder with predisposition to acute myelogenous leukemia: Implications for mechanisms of pathogenesis. Blood 2002, 99, 1364–1372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matheny, C.J.; Speck, M.E.; Cushing, P.R.; Zhou, Y.; Corpora, T.; Regan, M.; Newman, M.; Roudaia, L.; Speck, C.L.; Gu, T.L.; et al. Disease mutations in RUNX1 and RUNX2 create nonfunctional, dominant-negative, or hypomorphic alleles. Embo J. 2007, 26, 1163–1175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fenwarth, L.; Caulier, A.; Lachaier, E.; Goursaud, L.; Marceau-Renaut, A.; Fournier, E.; Lebon, D.; Boyer, T.; Berthon, C.; Marolleau, J.P.; et al. Hereditary Predisposition to Acute Myeloid Leukemia in Older Adults. Hemasphere 2021, 5, e552. [Google Scholar] [CrossRef] [PubMed]
- Feurstein, S.; Drazer, M.; Godley, L.A. Germline predisposition to hematopoietic malignancies. Hum. Mol. Genet. 2021, 30, R225–R235. [Google Scholar] [CrossRef]
- DeRoin, L.; Cavalcante de Andrade Silva, M.; Petras, K.; Arndt, K.; Phillips, N.; Wanjari, P.; Subramanian, H.P.; Montes, D.; McElherne, J.; Theissen, M.; et al. Feasibility and limitations of cultured skin fibroblasts for germline genetic testing in hematologic disorders. Hum. Mutat. 2022, 43, 950–962. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Gerstung, M.; Malcovati, L.; Tauro, S.; Gundem, G.; Van Loo, P.; Yoon, C.J.; Ellis, P.; Wedge, D.C.; Pellagatti, A.; et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 2013, 122, 3616–3627; quiz 3699. [Google Scholar] [CrossRef]
- Haferlach, T.; Nagata, Y.; Grossmann, V.; Okuno, Y.; Bacher, U.; Nagae, G.; Schnittger, S.; Sanada, M.; Kon, A.; Alpermann, T.; et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia 2014, 28, 241–247. [Google Scholar] [CrossRef] [Green Version]
- Churpek, J.E.; Pyrtel, K.; Kanchi, K.L.; Shao, J.; Koboldt, D.; Miller, C.A.; Shen, D.; Fulton, R.; O’Laughlin, M.; Fronick, C.; et al. Genomic analysis of germ line and somatic variants in familial myelodysplasia/acute myeloid leukemia. Blood 2015, 126, 2484–2490. [Google Scholar] [CrossRef] [Green Version]
- Haslam, K.; Langabeer, S.E.; Hayat, A.; Conneally, E.; Vandenberghe, E. Targeted next-generation sequencing of familial platelet disorder with predisposition to acute myeloid leukaemia. Br. J. Haematol. 2016, 175, 161–163. [Google Scholar] [CrossRef] [Green Version]
- Tawana, K.; Wang, J.; Király, P.A.; Kállay, K.; Benyó, G.; Zombori, M.; Csomor, J.; Al Seraihi, A.; Rio-Machin, A.; Matolcsy, A.; et al. Recurrent somatic JAK-STAT pathway variants within a RUNX1-mutated pedigree. Eur. J. Hum. Genet. 2017, 25, 1020–1024. [Google Scholar] [CrossRef] [Green Version]
- Delhommeau, F.; Dupont, S.; Della Valle, V.; James, C.; Trannoy, S.; Massé, A.; Kosmider, O.; Le Couedic, J.P.; Robert, F.; Alberdi, A.; et al. Mutation in TET2 in myeloid cancers. N. Engl. J. Med. 2009, 360, 2289–2301. [Google Scholar] [CrossRef] [PubMed]
- Döhner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weissmann, S.; Alpermann, T.; Grossmann, V.; Kowarsch, A.; Nadarajah, N.; Eder, C.; Dicker, F.; Fasan, A.; Haferlach, C.; Haferlach, T.; et al. Landscape of TET2 mutations in acute myeloid leukemia. Leukemia 2012, 26, 934–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ley, T.J.; Ding, L.; Walter, M.J.; McLellan, M.D.; Lamprecht, T.; Larson, D.E.; Kandoth, C.; Payton, J.E.; Baty, J.; Welch, J.; et al. DNMT3A mutations in acute myeloid leukemia. N. Engl. J. Med. 2010, 363, 2424–2433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walter, M.J.; Ding, L.; Shen, D.; Shao, J.; Grillot, M.; McLellan, M.; Fulton, R.; Schmidt, H.; Kalicki-Veizer, J.; O’Laughlin, M.; et al. Recurrent DNMT3A mutations in patients with myelodysplastic syndromes. Leukemia 2011, 25, 1153–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grossmann, V.; Haferlach, C.; Weissmann, S.; Roller, A.; Schindela, S.; Poetzinger, F.; Stadler, K.; Bellos, F.; Kern, W.; Haferlach, T.; et al. The molecular profile of adult T-cell acute lymphoblastic leukemia: Mutations in RUNX1 and DNMT3A are associated with poor prognosis in T-ALL. Genes Chromosomes Cancer 2013, 52, 410–422. [Google Scholar] [CrossRef] [PubMed]
- Itzykson, R.; Fenaux, P.; Bowen, D.; Cross, N.C.P.; Cortes, J.; De Witte, T.; Germing, U.; Onida, F.; Padron, E.; Platzbecker, U.; et al. Diagnosis and Treatment of Chronic Myelomonocytic Leukemias in Adults: Recommendations From the European Hematology Association and the European LeukemiaNet. Hemasphere 2018, 2, e150. [Google Scholar] [CrossRef]
- Tefferi, A. Primary myelofibrosis: 2017 update on diagnosis, risk-stratification, and management. Am. J. Hematol. 2016, 91, 1262–1271. [Google Scholar] [CrossRef]
- Vannucchi, A.M.; Lasho, T.L.; Guglielmelli, P.; Biamonte, F.; Pardanani, A.; Pereira, A.; Finke, C.; Score, J.; Gangat, N.; Mannarelli, C.; et al. Mutations and prognosis in primary myelofibrosis. Leukemia 2013, 27, 1861–1869. [Google Scholar] [CrossRef]
- Tefferi, A.; Vannucchi, A.M. Genetic Risk Assessment in Myeloproliferative Neoplasms. Mayo Clin. Proc. 2017, 92, 1283–1290. [Google Scholar] [CrossRef]
- Bolouri, H.; Farrar, J.E.; Triche, T., Jr.; Ries, R.E.; Lim, E.L.; Alonzo, T.A.; Ma, Y.; Moore, R.; Mungall, A.J.; Marra, M.A.; et al. The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specific mutational interactions. Nat. Med. 2018, 24, 103–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastor, V.; Hirabayashi, S.; Karow, A.; Wehrle, J.; Kozyra, E.J.; Nienhold, R.; Ruzaike, G.; Lebrecht, D.; Yoshimi, A.; Niewisch, M.; et al. Mutational landscape in children with myelodysplastic syndromes is distinct from adults: Specific somatic drivers and novel germline variants. Leukemia 2017, 31, 759–762. [Google Scholar] [CrossRef] [PubMed]
- Kanagal-Shamanna, R.; Loghavi, S.; DiNardo, C.D.; Medeiros, L.J.; Garcia-Manero, G.; Jabbour, E.; Routbort, M.J.; Luthra, R.; Bueso-Ramos, C.E.; Khoury, J.D. Bone marrow pathologic abnormalities in familial platelet disorder with propensity for myeloid malignancy and germline RUNX1 mutation. Haematologica 2017, 102, 1661–1670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lachowiez, C.; Bannon, S.; Loghavi, S.; Wang, F.; Kanagal-Shamanna, R.; Mehta, R.; Daver, N.; Borthakur, G.; Pemmaraju, N.; Ravandi, F.; et al. Clonal evolution and treatment outcomes in hematopoietic neoplasms arising in patients with germline RUNX1 mutations. Am. J. Hematol. 2020, 95, E313–E315. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef] [Green Version]
- Marnell, C.S.; Bick, A.; Natarajan, P. Clonal hematopoiesis of indeterminate potential (CHIP): Linking somatic mutations, hematopoiesis, chronic inflammation and cardiovascular disease. J. Mol. Cell Cardiol. 2021, 161, 98–105. [Google Scholar] [CrossRef]
- Cook, E.K.; Luo, M.; Rauh, M.J. Clonal hematopoiesis and inflammation: Partners in leukemogenesis and comorbidity. Exp. Hematol. 2020, 83, 85–94. [Google Scholar] [CrossRef] [Green Version]
- Falini, B.; Sportoletti, P.; Brunetti, L.; Martelli, M.P. Perspectives for therapeutic targeting of gene mutations in acute myeloid leukaemia with normal cytogenetics. Br. J. Haematol. 2015, 170, 305–322. [Google Scholar] [CrossRef]
- Sakurai, M.; Kasahara, H.; Yoshida, K.; Yoshimi, A.; Kunimoto, H.; Watanabe, N.; Shiraishi, Y.; Chiba, K.; Tanaka, H.; Harada, Y.; et al. Genetic basis of myeloid transformation in familial platelet disorder/acute myeloid leukemia patients with haploinsufficient RUNX1 allele. Blood Cancer J. 2016, 6, e392. [Google Scholar] [CrossRef]
- Béri-Dexheimer, M.; Latger-Cannard, V.; Philippe, C.; Bonnet, C.; Chambon, P.; Roth, V.; Grégoire, M.J.; Bordigoni, P.; Lecompte, T.; Leheup, B.; et al. Clinical phenotype of germline RUNX1 haploinsufficiency: From point mutations to large genomic deletions. Eur. J. Hum. Genet. 2008, 16, 1014–1018. [Google Scholar] [CrossRef] [PubMed]
- Bluteau, D.; Gilles, L.; Hilpert, M.; Antony-Debré, I.; James, C.; Debili, N.; Camara-Clayette, V.; Wagner-Ballon, O.; Cordette-Lagarde, V.; Robert, T.; et al. Down-regulation of the RUNX1-target gene NR4A3 contributes to hematopoiesis deregulation in familial platelet disorder/acute myelogenous leukemia. Blood 2011, 118, 6310–6320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duarte, B.K.L.; Yamaguti-Hayakawa, G.G.; Medina, S.S.; Siqueira, L.H.; Snetsinger, B.; Costa, F.F.; Rauh, M.J.; Ozelo, M.C. Longitudinal sequencing of RUNX1 familial platelet disorder: New insights into genetic mechanisms of transformation to myeloid malignancies. Br. J. Haematol. 2019, 186, 724–734. [Google Scholar] [CrossRef]
- Bagla, S.; Regling, K.A.; Wakeling, E.N.; Gadgeel, M.; Buck, S.; Zaidi, A.U.; Flore, L.A.; Chicka, M.; Schiffer, C.A.; Chitlur, M.B.; et al. Distinctive phenotypes in two children with novel germline RUNX1 mutations-one with myeloid malignancy and increased fetal hemoglobin. Pediatr. Hematol. Oncol. 2021, 38, 65–79. [Google Scholar] [CrossRef] [PubMed]
- Ng, I.K.; Lee, J.; Ng, C.; Kosmo, B.; Chiu, L.; Seah, E.; Mok, M.M.H.; Tan, K.; Osato, M.; Chng, W.J.; et al. Preleukemic and second-hit mutational events in an acute myeloid leukemia patient with a novel germline RUNX1 mutation. Biomark. Res. 2018, 6, 16. [Google Scholar] [CrossRef] [Green Version]
- Manchev, V.T.; Bouzid, H.; Antony-Debré, I.; Leite, B.; Meurice, G.; Droin, N.; Prebet, T.; Costello, R.T.; Vainchenker, W.; Plo, I.; et al. Acquired TET2 mutation in one patient with familial platelet disorder with predisposition to AML led to the development of pre-leukaemic clone resulting in T2-ALL and AML-M0. J. Cell Mol. Med. 2017, 21, 1237–1242. [Google Scholar] [CrossRef] [PubMed]
- Rajpal, S.; Jain, A.; Jamwal, M.; Jain, N.; Sachdeva, M.U.S.; Malhotra, P.; Varma, N.; Das, R. A novel germline RUNX1 mutation with co-occurrence of somatic alterations in a case of myeloid neoplasm with familial thrombocytopenia: First report from India. Leuk. Lymphoma 2019, 60, 2568–2571. [Google Scholar] [CrossRef] [PubMed]
- Staňo Kozubík, K.; Radová, L.; Pešová, M.; Réblová, K.; Trizuljak, J.; Plevová, K.; Fiamoli, V.; Gumulec, J.; Urbánková, H.; Szotkowski, T.; et al. C-terminal RUNX1 mutation in familial platelet disorder with predisposition to myeloid malignancies. Int. J. Hematol. 2018, 108, 652–657. [Google Scholar] [CrossRef]
- Shiba, N.; Hasegawa, D.; Park, M.J.; Murata, C.; Sato-Otsubo, A.; Ogawa, C.; Manabe, A.; Arakawa, H.; Ogawa, S.; Hayashi, Y. CBL mutation in chronic myelomonocytic leukemia secondary to familial platelet disorder with propensity to develop acute myeloid leukemia (FPD/AML). Blood 2012, 119, 2612–2614. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| All Cases (n = 91): Number (Range or %) | No Signs of RUNX1-FPD (n = 2): Number (Range or %) | Cytopenia (n = 28): Number (Range or %) | MDS (n = 9): Number (Range or %) | AML (n = 37): Number (Range or %) | MDS/AML (n = 7): Number (Range or %) | Other Myeloid HM a (n = 4): Number (Range or %) | Lymphoid HM b (n = 4): Number (Range or %) | |
|---|---|---|---|---|---|---|---|---|
| Characteristics | ||||||||
| median age at diagnosis (years) c | 42 (0.08–74) | 35.5 (18–53) | 52.5 (3–71) | 29 (7–58) | 42 (0.08–74) | 55 (37–65) | 37.5 (10–63) | 29 (16–42) |
| age at diagnosis, NA | 28 (31%) | 0 (0%) | 18 (64%) | 2 (22%) | 7 (19%) | 1 (14%) | 0 (0%) | 0 (0%) |
| Germline RUNX1 variant type | ||||||||
| missense | 27 (30%) | 0 (0%) | 11 (39%) | 2 (22%) | 8 (22%) | 2 (29%) | 1 (25%) | 3 (75%) |
| nonsense | 23 (25%) | 1 (50%) | 2 (7%) | 3 (33%) | 15 (41%) | 2 (29%) | 0 (0%) | 0 (0%) |
| frameshift | 24 (26%) | 1 (50%) | 8 (29%) | 1 (11%) | 9 (24%) | 2 (29%) | 2 (50%) | 1 (25%) |
| deletion d | 16 (18%) | 0 (0%) | 6 (21%) | 3 (33%) | 5 (14%) | 1 (14%) | 1 (25%) | 0 (0%) |
| splice site | 1 (1%) | 0 (0%) | 1 (4%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) |
| Karyotype | ||||||||
| normal | 25 (27%) | 0 (0%) | 9 (32%) | 2 (22%) | 9 (24%) | 2 (29%) | 1 (25%) | 1 (25%) |
| abnormal | 31 (34%) | 0 (0%) | 0 (0%) | 6 (67%) | 19 (51%) | 2 (25%) | 1 (25%) | 3 (75%) |
| NA | 35 (38%) | 2 (100%) | 19 (68%) | 1 (11%) | 9 (24%) | 3 (43%) | 2 (50%) | 0 (0%) |
| Somatic RUNX1 alteration | ||||||||
| detected | 23 (25%) | 0 (0%) | 0 (0%) | 2 (22%) | 20 (54%) | 1 (14%) | 0 (0%) | 0 (0%) |
| not detected | 66 (73%) | 2 (100%) | 28 (100%) | 7 (78%) | 17 (46%) | 6 (86%) | 4 (100%) | 4 (100%) |
| NA | 2 (2%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) |
| Additional somatic variants | ||||||||
| median number of analyzed genes | 28 (1–51) | 27 (21–33) | 33 (1–43) | 23 (2–38) | 27 (1–51) | 38 (17–48) | 16.5 (3–33) | 35 (1–43) |
| median number of somatic variants | 2 (0–32) | 3 (0–6) | 0.5 (0–6) | 2 (0–20) | 2 (0–12) | 2 (0–10) | 2.5 (1–3) | 2 (1–32) |
| no variants detected | 25 (27%) | 1 (50%) | 14 (50%) | 3 (33%) | 6 (16%) | 1 (29%) | 0 (0%) | 0 (0%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Förster, A.; Decker, M.; Schlegelberger, B.; Ripperger, T. Beyond Pathogenic RUNX1 Germline Variants: The Spectrum of Somatic Alterations in RUNX1-Familial Platelet Disorder with Predisposition to Hematologic Malignancies. Cancers 2022, 14, 3431. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14143431
Förster A, Decker M, Schlegelberger B, Ripperger T. Beyond Pathogenic RUNX1 Germline Variants: The Spectrum of Somatic Alterations in RUNX1-Familial Platelet Disorder with Predisposition to Hematologic Malignancies. Cancers. 2022; 14(14):3431. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14143431
Chicago/Turabian StyleFörster, Alisa, Melanie Decker, Brigitte Schlegelberger, and Tim Ripperger. 2022. "Beyond Pathogenic RUNX1 Germline Variants: The Spectrum of Somatic Alterations in RUNX1-Familial Platelet Disorder with Predisposition to Hematologic Malignancies" Cancers 14, no. 14: 3431. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14143431