
Comprehensive Analysis of Genomic Alterations in Hepatoid Adenocarcinoma of the Stomach and Identification of Clinically Actionable Alterations

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients and Samples
2.2. DNA Extraction and Library Construction
2.3. Single-Nucleotide Variants (SNVs) and Copy Number Variants (CNVs) Calling
2.4. Statistical Analyses
3. Results
3.1. Clinicopathological Characteristics of HAS
3.2. HAS Was Distinct in the Somatic Mutation Landscape
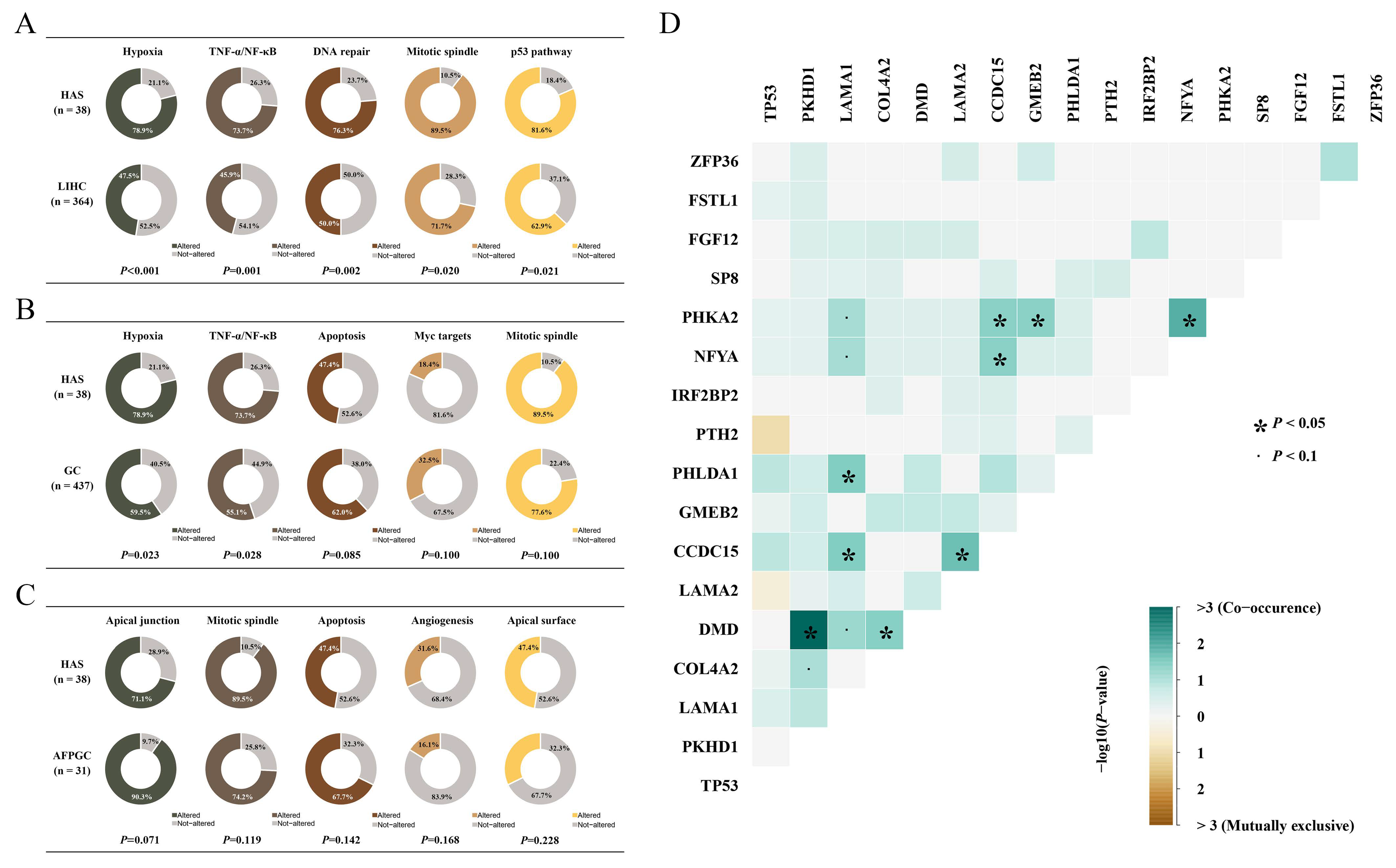
3.3. Mutated Pathway Analyses in HAS
3.4. Mutagenesis Mechanism Heterogeneities among HAS, LIHC, GC, and AFPGC
3.5. Copy Number Variants Correlated with Outcomes of HAS
3.6. Overview of Clinically Actionable Alterations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, Y.; Sun, L.; Li, Z.; Gao, J.; Ge, S.; Zhang, C.; Yuan, J.; Wang, X.; Li, J.; Lu, Z.; et al. Hepatoid adenocarcinoma of the stomach: A unique subgroup with distinct clinicopathological and molecular features. Gastric Cancer 2019, 22, 1183–1192. [Google Scholar] [CrossRef] [Green Version]
- The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014, 513, 202–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Y.; Zhao, Z.; Li, P.; Wang, Y.; Guo, C.; Wang, X.; Tang, W.; Liu, Q.; Lu, N.; Xue, L.; et al. Hepatoid adenocarcinoma of the stomach is a special and easily misdiagnosed or missed diagnosed subtype of gastric cancer with poor prognosis but curative for patients of pN0/1: The experience of a single center. Int. J. Clin. Exp. Med. 2015, 8, 6762–6772. [Google Scholar] [PubMed]
- Zeng, X.Y.; Yin, Y.P.; Xiao, H.; Zhang, P.; He, J.; Liu, W.Z.; Gao, J.B.; Shuai, X.M.; Wang, G.B.; Wu, X.L.; et al. Clinicopathological Characteristics and Prognosis of Hepatoid Adenocarcinoma of the Stomach: Evaluation of a Pooled Case Series. Curr. Med. Sci. 2018, 38, 1054–1061. [Google Scholar] [CrossRef]
- Shoenfeld, Y. Experimental and induced animal models of systemic lupus erythematosus and Sjogren’s syndrome. Curr. Opin. Rheumatol. 1989, 1, 360–368. [Google Scholar] [CrossRef] [PubMed]
- Fujii, H.; Ichikawa, K.; Takagaki, T.; Nakanishi, Y.; Ikegami, M.; Hirose, S.; Shimoda, T. Genetic evolution of alpha fetoprotein producing gastric cancer. J. Clin. Pathol. 2003, 56, 942–949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akiyama, S.; Tamura, G.; Endoh, Y.; Fukushima, N.; Ichihara, Y.; Aizawa, K.; Kawata, S.; Motoyama, T. Histogenesis of hepatoid adenocarcinoma of the stomach: Molecular evidence of identical origin with coexistent tubular adenocarcinoma. Int. J. Cancer 2003, 106, 510–515. [Google Scholar] [CrossRef] [PubMed]
- Tsuruta, S.; Ohishi, Y.; Fujiwara, M.; Ihara, E.; Ogawa, Y.; Oki, E.; Nakamura, M.; Oda, Y. Gastric hepatoid adenocarcinomas are a genetically heterogenous group; most tumors show chromosomal instability, but MSI tumors do exist. Hum. Pathol. 2019, 88, 27–38. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, A.; Pu, Y.; Li, Z.; Xue, R.; Zhang, C.; Xiang, X.; E, J.-Y.; Bu, Z.; Bai, F.; et al. Genomic and transcriptomic profiling of hepatoid adenocarcinoma of the stomach. Oncogene 2021, 40, 5705–5717. [Google Scholar] [CrossRef]
- Lu, J.; Ding, Y.; Chen, Y.; Jiang, J.; Chen, Y.; Huang, Y.; Wu, M.; Li, C.; Kong, M.; Zhao, W.; et al. Whole-exome sequencing of alpha-fetoprotein producing gastric carcinoma reveals genomic profile and therapeutic targets. Nat. Commun. 2021, 12, 3946. [Google Scholar] [CrossRef]
- Liberzon, A.; Birger, C.; Thorvaldsdottir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [Green Version]
- Alexandrov, L.; Kim, J.; Haradhvala, N.; Huang, M.; Tian Ng, A.; Wu, Y.; Boot, A.; Covington, K.; Gordenin, D.; Bergstrom, E.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef] [Green Version]
- Huet-Duvillier, G.; Gomes, V.; Tetaert, D.; Mathon, P.; Boersma, A.; Degand, P. Trypanosoma brucei brucei: Variability in the association of some variant surface glycoproteins. Exp. Parasitol. 1988, 67, 31–38. [Google Scholar] [CrossRef]
- McGlynn, K.A.; Petrick, J.L.; El-Serag, H.B. Epidemiology of Hepatocellular Carcinoma. Hepatology 2021, 73 (Suppl. S1), 4–13. [Google Scholar] [CrossRef]
- Kang, J.U. Chromosome 8q as the most frequent target for amplification in early gastric carcinoma. Oncol. Lett. 2014, 7, 1139–1143. [Google Scholar] [CrossRef] [Green Version]
- Shi, Z.Z.; Shang, L.; Jiang, Y.Y.; Hao, J.J.; Zhang, Y.; Zhang, T.T.; Lin, D.C.; Liu, S.G.; Wang, B.S.; Gong, T.; et al. Consistent and differential genetic aberrations between esophageal dysplasia and squamous cell carcinoma detected by array comparative genomic hybridization. Clin. Cancer Res. 2013, 19, 5867–5878. [Google Scholar] [CrossRef] [Green Version]
- Saleh, M.N.; Patel, M.R.; Bauer, T.M.; Goel, S.; Falchook, G.S.; Shapiro, G.I.; Chung, K.Y.; Infante, J.R.; Conry, R.M.; Rabinowits, G.; et al. Phase 1 Trial of ALRN-6924, a Dual Inhibitor of MDMX and MDM2, in Patients with Solid Tumors and Lymphomas Bearing Wild-Type TP53. Clin. Cancer Res. 2021, 27, 5236–5247. [Google Scholar] [CrossRef]
- Konopleva, M.; Martinelli, G.; Daver, N.; Papayannidis, C.; Wei, A.; Higgins, B.; Ott, M.; Mascarenhas, J.; Andreeff, M. MDM2 inhibition: An important step forward in cancer therapy. Leukemia 2020, 34, 2858–2874. [Google Scholar] [CrossRef]
- Takahashi, S.; Fujiwara, Y.; Nakano, K.; Shimizu, T.; Tomomatsu, J.; Koyama, T.; Ogura, M.; Tachibana, M.; Kakurai, Y.; Yamashita, T.; et al. Safety and pharmacokinetics of milademetan, a MDM2 inhibitor, in Japanese patients with solid tumors: A phase I study. Cancer Sci. 2021, 112, 2361–2370. [Google Scholar] [CrossRef]
- Chao, J.; Fuchs, C.S.; Shitara, K.; Tabernero, J.; Muro, K.; Van Cutsem, E.; Bang, Y.J.; De Vita, F.; Landers, G.; Yen, C.J.; et al. Assessment of Pembrolizumab Therapy for the Treatment of Microsatellite Instability-High Gastric or Gastroesophageal Junction Cancer Among Patients in the KEYNOTE-059, KEYNOTE-061, and KEYNOTE-062 Clinical Trials. JAMA Oncol. 2021, 7, 895–902. [Google Scholar] [CrossRef]
- Valero, C.; Lee, M.; Hoen, D.; Zehir, A.; Berger, M.F.; Seshan, V.E.; Chan, T.A.; Morris, L.G.T. Response Rates to Anti-PD-1 Immunotherapy in Microsatellite-Stable Solid Tumors With 10 or More Mutations per Megabase. JAMA Oncol. 2021, 7, 739–743. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.K.; Yu, T.; de la Monte, S.M.; Wands, J.R.; Derdak, Z.; Kim, M. Restoration of Wnt/β-catenin signaling attenuates alcoholic liver disease progression in a rat model. J. Hepatol. 2015, 63, 191–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaughan, C.A.; Singh, S.; Subler, M.A.; Windle, J.J.; Inoue, K.; Fry, E.A.; Pillappa, R.; Grossman, S.R.; Windle, B.; Yeudall, W.A.; et al. The oncogenicity of tumor-derived mutant p53 is enhanced by the recruitment of PLK3. Nat. Commun. 2021, 12, 704. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Shen, J.; Jiang, Y. Circ_0027599/PHDLA1 suppresses gastric cancer progression by sponging miR-101-3p.1. Cell Biosci. 2018, 8, 58. [Google Scholar] [CrossRef]
- Fearon, A.E.; Carter, E.P.; Clayton, N.S.; Wilkes, E.H.; Baker, A.M.; Kapitonova, E.; Bakhouche, B.A.; Tanner, Y.; Wang, J.; Gadaleta, E.; et al. PHLDA1 Mediates Drug Resistance in Receptor Tyrosine Kinase-Driven Cancer. Cell Rep. 2018, 22, 2469–2481. [Google Scholar] [CrossRef] [Green Version]
- Law, E.K.; Levin-Klein, R.; Jarvis, M.C.; Kim, H.; Argyris, P.P.; Carpenter, M.A.; Starrett, G.J.; Temiz, N.A.; Larson, L.K.; Durfee, C.; et al. APOBEC3A catalyzes mutation and drives carcinogenesis in vivo. J. Exp. Med. 2020, 217, e20200261. [Google Scholar] [CrossRef]
- Yang, J.D.; Hainaut, P.; Gores, G.J.; Amadou, A.; Plymoth, A.; Roberts, L.R. A global view of hepatocellular carcinoma: Trends, risk, prevention and management. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 589–604. [Google Scholar] [CrossRef]
- Kim, H.; Nguyen, N.P.; Turner, K.; Wu, S.; Gujar, A.D.; Luebeck, J.; Liu, J.; Deshpande, V.; Rajkumar, U.; Namburi, S.; et al. Extrachromosomal DNA is associated with oncogene amplification and poor outcome across multiple cancers. Nat. Genet. 2020, 52, 891–897. [Google Scholar] [CrossRef]
- Killcoyne, S.; Gregson, E.; Wedge, D.C.; Woodcock, D.J.; Eldridge, M.D.; de la Rue, R.; Miremadi, A.; Abbas, S.; Blasko, A.; Kosmidou, C.; et al. Genomic copy number predicts esophageal cancer years before transformation. Nat. Med. 2020, 26, 1726–1732. [Google Scholar] [CrossRef]
- Jogo, T.; Nakamura, Y.; Shitara, K.; Bando, H.; Yasui, H.; Esaki, T.; Terazawa, T.; Satoh, T.; Shinozaki, E.; Nishina, T.; et al. Circulating Tumor DNA Analysis Detects FGFR2 Amplification and Concurrent Genomic Alterations Associated with FGFR Inhibitor Efficacy in Advanced Gastric Cancer. Clin. Cancer Res. 2021, 27, 5619–5627. [Google Scholar] [CrossRef]
- Arakawa, N.; Sugai, T.; Habano, W.; Eizuka, M.; Sugimoto, R.; Akasaka, R.; Toya, Y.; Yamamoto, E.; Koeda, K.; Sasaki, A.; et al. Genome-wide analysis of DNA copy number alterations in early and advanced gastric cancers. Mol. Carcinog. 2017, 56, 527–537. [Google Scholar] [CrossRef]
- Zhou, H.T.; Shi, Z.Z.; Zhou, Z.X.; Jiang, Y.Y.; Hao, J.J.; Zhang, T.T.; Shi, F.; Xu, X.; Wang, M.R.; Zhang, Y. Genomic changes in rectal adenocarcinoma associated with liver metastasis. Cancer Biomark. 2013, 13, 281–288. [Google Scholar] [CrossRef]
- Gallo, D.; Young, J.T.F.; Fourtounis, J.; Martino, G.; Alvarez-Quilon, A.; Bernier, C.; Duffy, N.M.; Papp, R.; Roulston, A.; Stocco, R.; et al. CCNE1 amplification is synthetic lethal with PKMYT1 kinase inhibition. Nature 2022, 604, 749–756. [Google Scholar] [CrossRef]
- Geier, C.B.; Piller, A.; Eibl, M.M.; Ciznar, P.; Ilencikova, D.; Wolf, H.M. Terminal 14q32.33 deletion as a novel cause of agammaglobulinemia. Clin. Immunol. 2017, 183, 41–45. [Google Scholar] [CrossRef]
- Gruber, S.B.; Entius, M.M.; Petersen, G.M.; Laken, S.J.; Longo, P.A.; Boyer, R.; Levin, A.M.; Mujumdar, U.J.; Trent, J.M.; Kinzler, K.W.; et al. Pathogenesis of adenocarcinoma in Peutz-Jeghers syndrome. Cancer Res. 1998, 58, 5267–5270. [Google Scholar]
- Khalili, A.; Yadegari, A.H.; Delavari, S.; Yazdani, R.; Abolhassani, H. Disseminated Intravascular Coagulation Associated with Large Deletion of Immunoglobulin Heavy Chain. Iran. J. Allergy Asthma Immunol. 2021, 20, 778–783. [Google Scholar] [CrossRef]
- Serra, G.; Felice, S.; Antona, V.; Di Pace, M.R.; Giuffre, M.; Piro, E.; Corsello, G. Cardio-facio-cutaneous syndrome and gastrointestinal defects: Report on a newborn with 19p13.3 deletion including the MAP 2 K2 gene. Ital. J. Pediatr. 2022, 48, 65. [Google Scholar] [CrossRef]
- Farshidfar, F.; Rhrissorrakrai, K.; Levovitz, C.; Peng, C.; Knight, J.; Bacchiocchi, A.; Su, J.; Yin, M.; Sznol, M.; Ariyan, S.; et al. Integrative molecular and clinical profiling of acral melanoma links focal amplification of 22q11.21 to metastasis. Nat. Commun. 2022, 13, 898. [Google Scholar] [CrossRef]
- Prasad, R.B.; Hosking, F.J.; Vijayakrishnan, J.; Papaemmanuil, E.; Koehler, R.; Greaves, M.; Sheridan, E.; Gast, A.; Kinsey, S.E.; Lightfoot, T.; et al. Verification of the susceptibility loci on 7p12.2, 10q21.2, and 14q11.2 in precursor B-cell acute lymphoblastic leukemia of childhood. Blood 2010, 115, 1765–1767. [Google Scholar] [CrossRef] [Green Version]
- Chakravarty, D.; Gao, J.; Phillips, S.M.; Kundra, R.; Zhang, H.; Wang, J.; Rudolph, J.E.; Yaeger, R.; Soumerai, T.; Nissan, M.H.; et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precis. Oncol. 2017. [Google Scholar] [CrossRef]
- Bayrak, M.; Olmez, O.F.; Kurt, E.; Cubukcu, E.; Evrensel, T.; Kanat, O.; Manavoglu, O. Prognostic significance of c-erbB2 overexpression in patients with metastatic gastric cancer. Clin. Transl. Oncol. 2013, 15, 307–312. [Google Scholar] [CrossRef]
- Bang, Y.J.; Van Cutsem, E.; Feyereislova, A.; Chung, H.C.; Shen, L.; Sawaki, A.; Lordick, F.; Ohtsu, A.; Omuro, Y.; Satoh, T.; et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): A phase 3, open-label, randomised controlled trial. Lancet 2010, 376, 687–697. [Google Scholar] [CrossRef]
- Meric-Bernstam, F.; Johnson, A.M.; Dumbrava, E.E.I.; Raghav, K.; Balaji, K.; Bhatt, M.; Murthy, R.K.; Rodon, J.; Piha-Paul, S.A. Advances in HER2-Targeted Therapy: Novel Agents and Opportunities Beyond Breast and Gastric Cancer. Clin. Cancer Res. 2019, 25, 2033–2041. [Google Scholar] [CrossRef] [Green Version]
- Hou, H.; Sun, D.; Zhang, X. The role of MDM2 amplification and overexpression in therapeutic resistance of malignant tumors. Cancer Cell Int. 2019, 19, 216. [Google Scholar] [CrossRef] [Green Version]
- Duffy, M.J.; Synnott, N.C.; O′Grady, S.; Crown, J. Targeting p53 for the treatment of cancer. Semin. Cancer Biol. 2022, 79, 58–67. [Google Scholar] [CrossRef]
- Levine, A.J. p53: 800 million years of evolution and 40 years of discovery. Nat. Rev. Cancer 2020, 20, 471–480. [Google Scholar] [CrossRef]
- Stein, E.M.; DeAngelo, D.J.; Chromik, J.; Chatterjee, M.; Bauer, S.; Lin, C.C.; Suarez, C.; de Vos, F.; Steeghs, N.; Cassier, P.A.; et al. Results from a First-in-Human Phase I Study of Siremadlin (HDM201) in Patients with Advanced Wild-Type TP53 Solid Tumors and Acute Leukemia. Clin. Cancer Res. 2022, 28, 870–881. [Google Scholar] [CrossRef]
- Poveda, A.; Floquet, A.; Ledermann, J.A.; Asher, R.; Penson, R.T.; Oza, A.M.; Korach, J.; Huzarski, T.; Pignata, S.; Friedlander, M.; et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): A final analysis of a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2021, 22, 620–631. [Google Scholar] [CrossRef]
- Chida, K.; Kawazoe, A.; Kawazu, M.; Suzuki, T.; Nakamura, Y.; Nakatsura, T.; Kuwata, T.; Ueno, T.; Kuboki, Y.; Kotani, D.; et al. A Low Tumor Mutational Burden and PTEN Mutations Are Predictors of a Negative Response to PD-1 Blockade in MSI-H/dMMR Gastrointestinal Tumors. Clin. Cancer Res. 2021, 27, 3714–3724. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, R.; Li, H.; Ge, W.; Zhu, X.; Zhu, L.; Wan, X.; Wang, G.; Pan, H.; Lu, J.; Han, W. Comprehensive Analysis of Genomic Alterations in Hepatoid Adenocarcinoma of the Stomach and Identification of Clinically Actionable Alterations. Cancers 2022, 14, 3849. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14163849
Zhao R, Li H, Ge W, Zhu X, Zhu L, Wan X, Wang G, Pan H, Lu J, Han W. Comprehensive Analysis of Genomic Alterations in Hepatoid Adenocarcinoma of the Stomach and Identification of Clinically Actionable Alterations. Cancers. 2022; 14(16):3849. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14163849
Chicago/Turabian StyleZhao, Rongjie, Hongshen Li, Weiting Ge, Xiuming Zhu, Liang Zhu, Xiangbo Wan, Guanglan Wang, Hongming Pan, Jie Lu, and Weidong Han. 2022. "Comprehensive Analysis of Genomic Alterations in Hepatoid Adenocarcinoma of the Stomach and Identification of Clinically Actionable Alterations" Cancers 14, no. 16: 3849. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14163849