
OATD-02 Validates the Benefits of Pharmacological Inhibition of Arginase 1 and 2 in Cancer

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemical Compounds
2.2. Recombinant Arginase Inhibition Assays
2.3. Cell-Based Arginase Inhibition Assays
2.4. Cell Culture
2.5. ARG1 and ARG2 Detection
2.6. Murine Tumour Models
2.7. Flow Cytometry
2.8. Bioanalytics
2.9. Statistical Analysis
3. Results
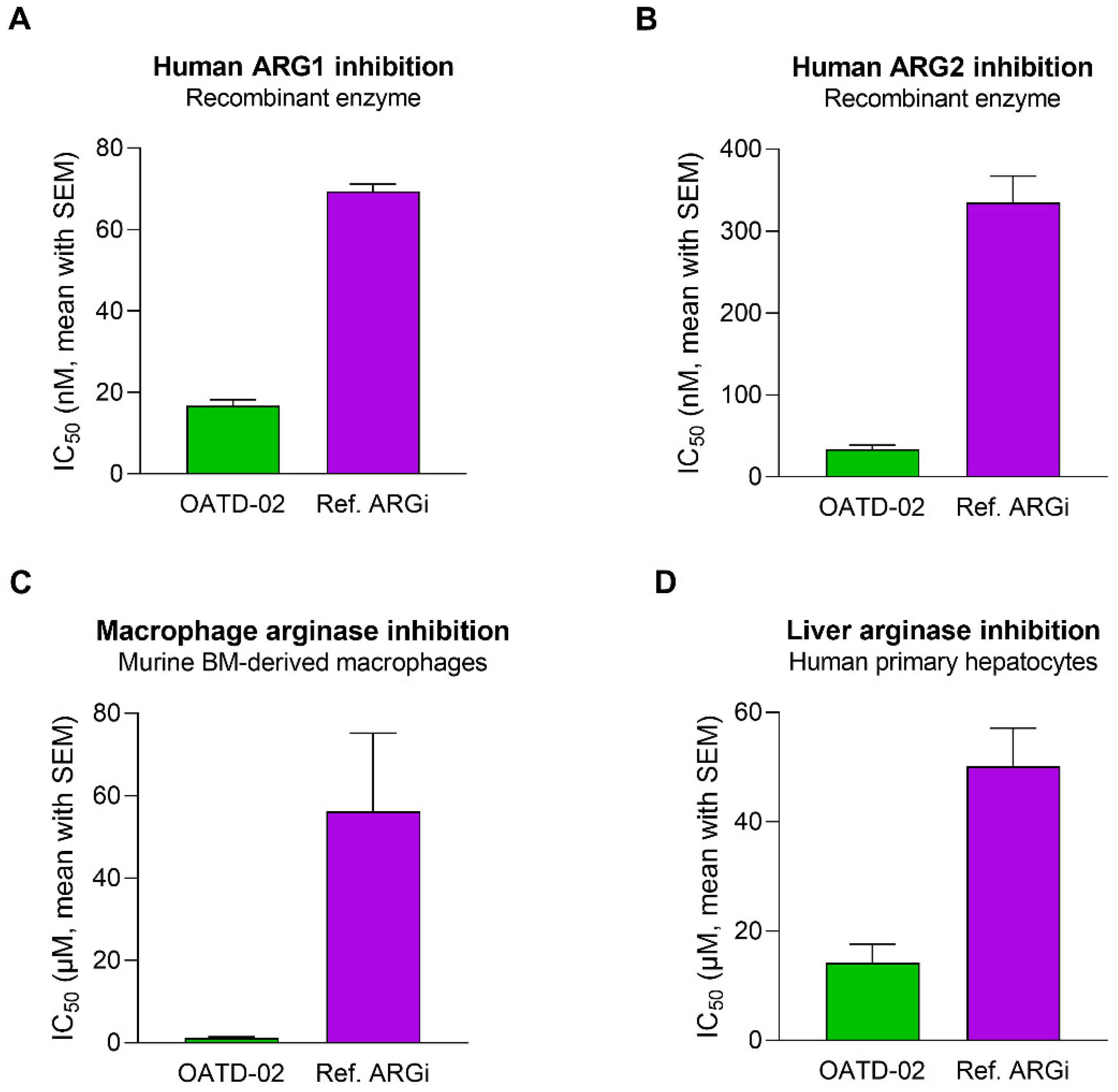
3.1. OATD-02 Is a Highly Potent Dual ARG1/ARG2 Small-Molecule Inhibitor
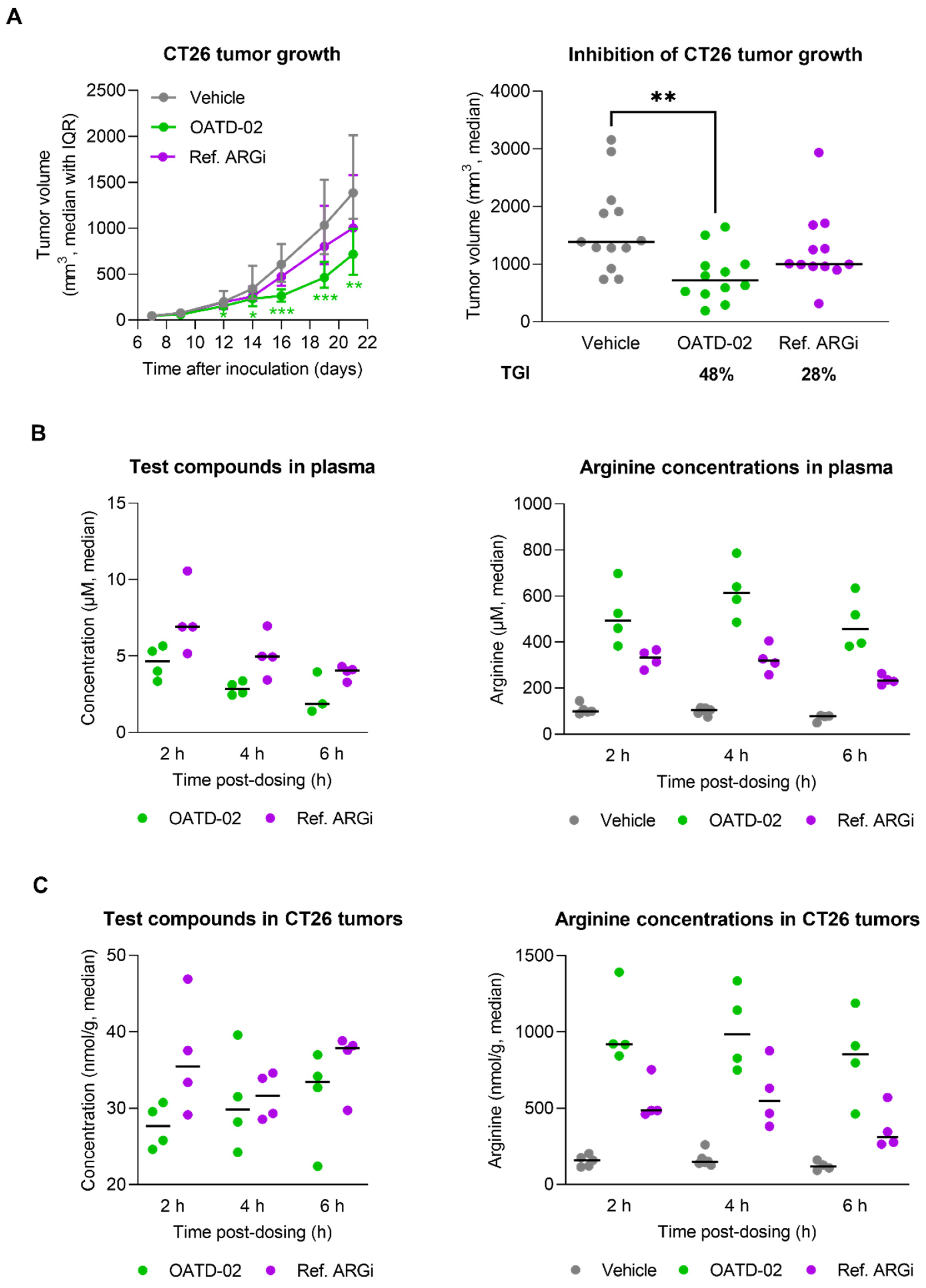
3.2. OATD-02 Showed Superior Activity in Combination with ICI and IDO Inhibitor
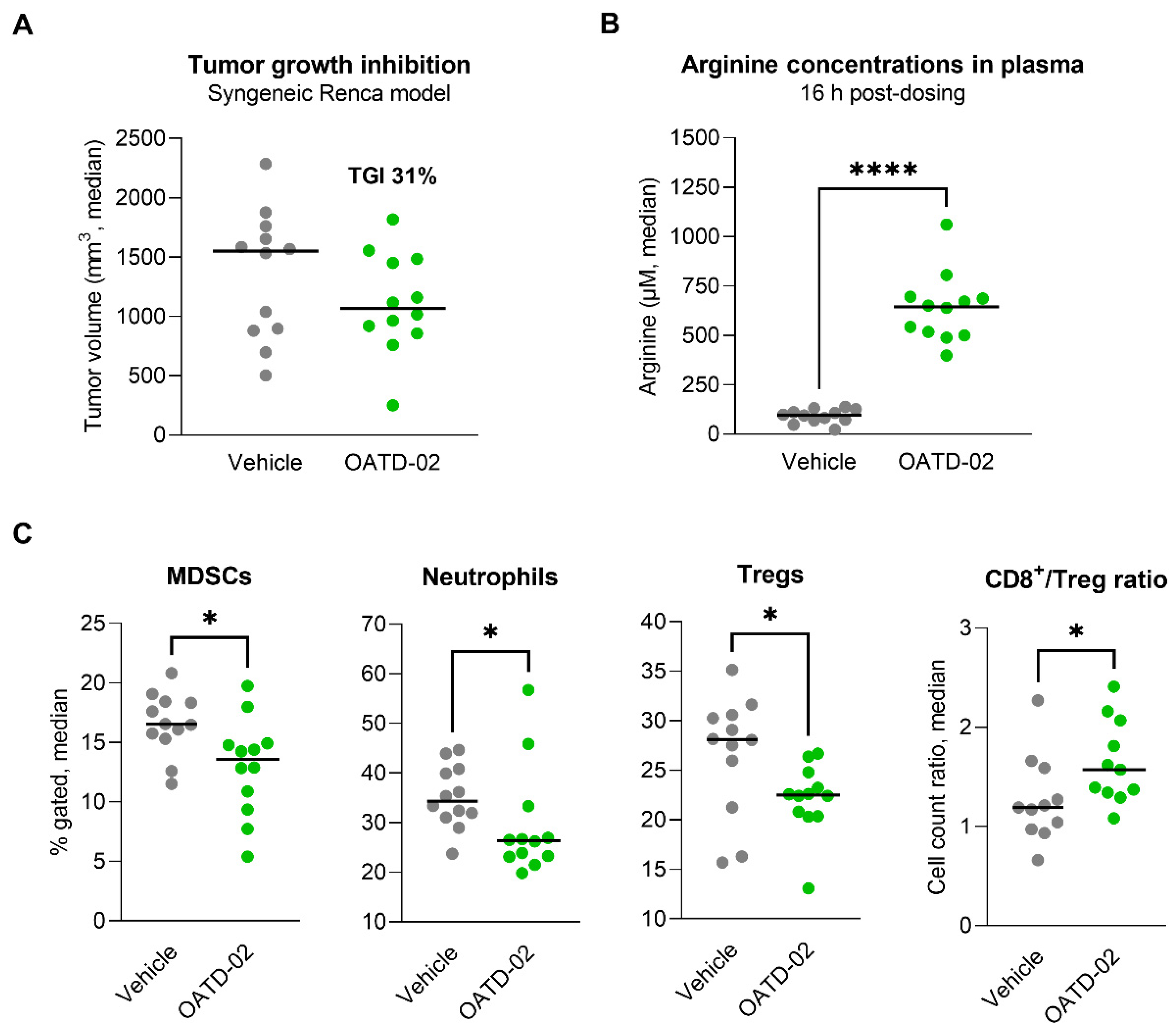
3.3. OATD-02 Altered the TME of ARG2-Positive Renca Tumours
3.4. OATD-02 Showed Direct Antitumour Effect on Human ARG2-Positive K562 Cells
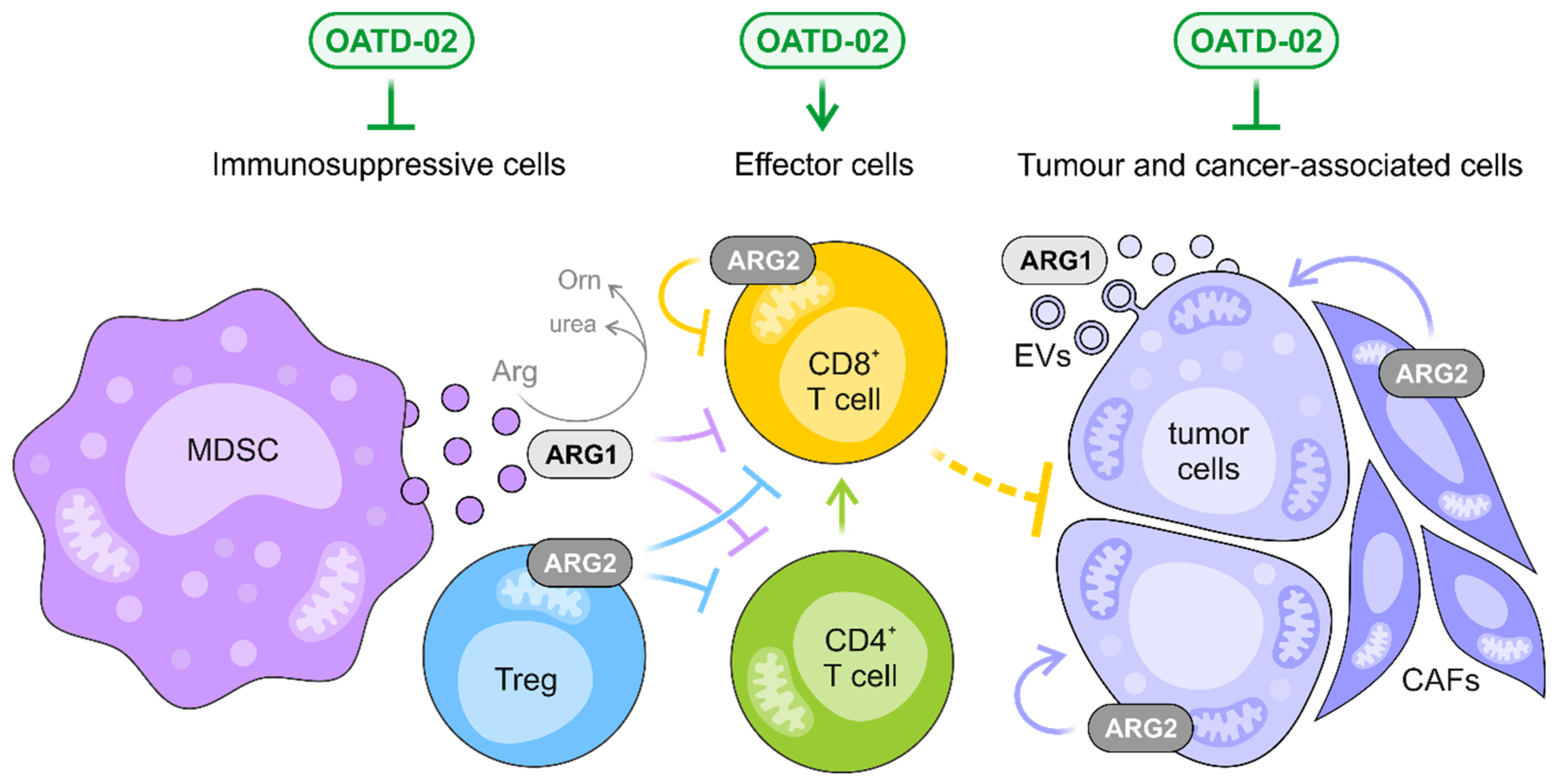
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Farkona, S.; Diamandis, E.P.; Blasutig, I.M. Cancer immunotherapy: The beginning of the end of cancer? BMC Med. 2016, 14, 73. [Google Scholar] [CrossRef]
- Labani-Motlagh, A.; Ashja-Mahdavi, M.; Loskog, A. The Tumor Microenvironment: A Milieu Hindering and Obstructing Antitumor Immune Responses. Front. Immunol. 2020, 11, 940. [Google Scholar] [CrossRef]
- Heintzman, D.R.; Fisher, E.L.; Rathmell, J.C. Microenvironmental influences on T cell immunity in cancer and inflammation. Cell. Mol. Immunol. 2022, 19, 316–326. [Google Scholar] [CrossRef]
- Sosnowska, A.; Chlebowska-Tuz, J.; Matryba, P.; Pilch, Z.; Greig, A.; Wolny, A.; Grzywa, T.M.; Rydzynska, Z.; Sokolowska, O.; Rygiel, T.P.; et al. Inhibition of arginase modulates T-cell response in the tumor microenvironment of lung carcinoma. Oncoimmunology 2021, 10, 1956143. [Google Scholar] [CrossRef]
- Pilanc, P.; Wojnicki, K.; Roura, A.J.; Cyranowski, S.; Ellert-Miklaszewska, A.; Ochocka, N.; Gielniewski, B.; Grzybowski, M.M.; Błaszczyk, R.; Stańczak, P.S.; et al. A Novel Oral Arginase 1/2 Inhibitor Enhances the Antitumor Effect of PD-1 Inhibition in Murine Experimental Gliomas by Altering the Immunosuppressive Environment. Front. Oncol. 2021, 11, 703465. [Google Scholar] [CrossRef]
- Kelly, B.; Pearce, E.L. Amino Assets: How Amino Acids Support Immunity. Cell Metab. 2020, 32, 154–175. [Google Scholar] [CrossRef]
- Lieu, E.L.; Nguyen, T.; Rhyne, S.; Kim, J. Amino acids in cancer. Exp. Mol. Med. 2020, 52, 15–30. [Google Scholar] [CrossRef]
- Sullivan, M.R.; Danai, L.V.; Lewis, C.A.; Chan, S.H.; Gui, D.Y.; Kunchok, T.; Dennstedt, E.A.; Vander Heiden, M.G.; Muir, A. Quantification of microenvironmental metabolites in murine cancers reveals determinants of tumor nutrient availability. Elife 2019, 8, e44235. [Google Scholar] [CrossRef]
- Morris, S.M. Recent advances in arginine metabolism: Roles and regulation of the arginases. Br. J. Pharmacol. 2009, 157, 922–930. [Google Scholar] [CrossRef]
- Groth, C.; Hu, X.; Weber, R.; Fleming, V.; Altevogt, P.; Utikal, J.; Umansky, V. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumour progression. Br. J. Cancer 2019, 120, 16–25. [Google Scholar] [CrossRef]
- Serafini, P.; Mgebroff, S.; Noonan, K.; Borrello, I. Myeloid-derived suppressor cells promote cross-tolerance in B-cell lymphoma by expanding regulatory T cells. Cancer Res. 2008, 68, 5439–5449. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.C.; Mattila, J.T. “Of mice and men”: Arginine metabolism in macrophages. Front. Immunol. 2014, 5, 479. [Google Scholar] [CrossRef] [PubMed]
- Mussai, F.; De Santo, C.; Abu-Dayyeh, I.; Booth, S.; Quek, L.; McEwen-Smith, R.M.; Qureshi, A.; Dazzi, F.; Vyas, P.; Cerundolo, V. Acute myeloid leukemia creates an arginase-dependent immunosuppressive microenvironment. Blood 2013, 122, 749–758. [Google Scholar] [CrossRef] [PubMed]
- Mussai, F.; Egan, S.; Hunter, S.; Webber, H.; Fisher, J.; Wheat, R.; McConville, C.; Sbirkov, Y.; Wheeler, K.; Bendle, G.; et al. Neuroblastoma Arginase Activity Creates an Immunosuppressive Microenvironment That Impairs Autologous and Engineered Immunity. Cancer Res. 2015, 75, 3043–3053. [Google Scholar] [CrossRef]
- Czystowska-Kuzmicz, M.; Sosnowska, A.; Nowis, D.; Ramji, K.; Szajnik, M.; Chlebowska-Tuz, J.; Wolinska, E.; Gaj, P.; Grazul, M.; Pilch, Z.; et al. Small extracellular vesicles containing arginase-1 suppress T-cell responses and promote tumor growth in ovarian carcinoma. Nat. Commun. 2019, 10, 3000. [Google Scholar] [CrossRef]
- Dunand-Sauthier, I.; Irla, M.; Carnesecchi, S.; Seguín-Estévez, Q.; Vejnar, C.E.; Zdobnov, E.M.; Santiago-Raber, M.L.; Reith, W. Repression of arginase-2 expression in dendritic cells by microRNA-155 is critical for promoting T cell proliferation. J. Immunol. 2014, 193, 1690–1700. [Google Scholar] [CrossRef]
- Ino, Y.; Yamazaki-Itoh, R.; Oguro, S.; Shimada, K.; Kosuge, T.; Zavada, J.; Kanai, Y.; Hiraoka, N. Arginase II expressed in cancer-associated fibroblasts indicates tissue hypoxia and predicts poor outcome in patients with pancreatic cancer. PLoS ONE 2013, 8, e55146. [Google Scholar] [CrossRef]
- Zaytouni, T.; Tsai, P.Y.; Hitchcock, D.S.; DuBois, C.D.; Freinkman, E.; Lin, L.; Morales-Oyarvide, V.; Lenehan, P.J.; Wolpin, B.M.; Mino-Kenudson, M.; et al. Critical role for arginase 2 in obesity-associated pancreatic cancer. Nat. Commun. 2017, 8, 242. [Google Scholar] [CrossRef]
- Lowe, M.M.; Boothby, I.; Clancy, S.; Ahn, R.S.; Liao, W.; Nguyen, D.N.; Schumann, K.; Marson, A.; Mahuron, K.M.; Kingsbury, G.A.; et al. Regulatory T cells use arginase 2 to enhance their metabolic fitness in tissues. JCI Insight 2019, 4, e129756. [Google Scholar] [CrossRef]
- Martí i Líndez, A.A.; Dunand-Sauthier, I.; Conti, M.; Gobet, F.; Núñez, N.; Hannich, J.T.; Riezman, H.; Geiger, R.; Piersigilli, A.; Hahn, K.; et al. Mitochondrial arginase-2 is a cell-autonomous regulator of CD8+ T cell function and antitumor efficacy. JCI Insight 2019, 4, e132975. [Google Scholar] [CrossRef]
- Sade-Feldman, M.; Yizhak, K.; Bjorgaard, S.L.; Ray, J.P.; de Boer, C.G.; Jenkins, R.W.; Lieb, D.J.; Chen, J.H.; Frederick, D.T.; Barzily-Rokni, M.; et al. Defining T Cell States Associated with Response to Checkpoint Immunotherapy in Melanoma. Cell 2018, 175, 998–1013.e1020, Erratum in Cell 2019, 176, 404. [Google Scholar] [CrossRef]
- Borek, B.; Gajda, T.; Golebiowski, A.; Blaszczyk, R. Boronic acid-based arginase inhibitors in cancer immunotherapy. Bioorg. Med. Chem. 2020, 28, 115658. [Google Scholar] [CrossRef]
- Muller, J.; Cardey, B.; Zedet, A.; Desingle, C.; Grzybowski, M.; Pomper, P.; Foley, S.; Harakat, D.; Ramseyer, C.; Girard, C.; et al. Synthesis, evaluation and molecular modelling of piceatannol analogues as arginase inhibitors. RSC Med. Chem. 2020, 11, 559–568. [Google Scholar] [CrossRef]
- Steggerda, S.M.; Bennett, M.K.; Chen, J.; Emberley, E.; Huang, T.; Janes, J.R.; Li, W.; MacKinnon, A.L.; Makkouk, A.; Marguier, G.; et al. Inhibition of arginase by CB-1158 blocks myeloid cell-mediated immune suppression in the tumor microenvironment. J. Immunother. Cancer 2017, 5, 101. [Google Scholar] [CrossRef]
- Baggio, R.; Elbaum, D.; Kanyo, Z.F.; Carroll, P.J.; Cavalli, R.C.; Ash, D.E.; Christianson, D.W. Inhibition of Mn2+2-Arginase by Borate Leads to the Design of a Transition State Analogue Inhibitor, 2(S)-Amino-6-boronohexanoic Acid. J. Am. Chem. Soc. 1997, 119, 8107–8108. [Google Scholar] [CrossRef]
- Van Zandt, M.; Whitehouse, D.; Golebiowski, A.; Ji, M.; Zhang, M.; Beckett, R.; Jagdmann, G.; Ryder, T.; Sheeler, R.; Andreoli, M.; et al. Discovery of (R)-2-Amino-6-borono-2-(2-(piperidin-1-yl)ethyl)hexanoic Acid and Congeners As Highly Potent Inhibitors of Human Arginases I and II for Treatment of Myocardial Reperfusion Injury. J. Med. Chem. 2013, 56, 2568–2580. [Google Scholar] [CrossRef]
- Golebiowski, A.; Whitehouse, D.; Beckett, R.; Van Zandt, M.; Ji, M.; Ryder, T.; Jagdmann, E.; Andreoli, M.; Lee, Y.; Sheeler, R.; et al. Synthesis of quaternary alpha-amino acid-based arginase inhibitors via the Ugi reaction. Bioorganic Med. Chem. Lett. 2013, 23, 4837–4841. [Google Scholar] [CrossRef]
- Blaszczyk, R.; Brzezinska, J.; Dymek, B.; Stanczak, P.S.; Mazurkiewicz, M.; Olczak, J.; Nowicka, J.; Dzwonek, K.; Zagozdzon, A.; Golab, J.; et al. Discovery and Pharmacokinetics of Sulfamides and Guanidines as Potent Human Arginase 1 Inhibitors. ACS Med. Chem. Lett. 2020, 11, 433–438. [Google Scholar] [CrossRef]
- Jung, D.; Biggs, H.; Erikson, J.; Ledyard, P.U. New Colorimetric reaction for end-point, continuous-flow, and kinetic measurement of urea. Clin. Chem. 1975, 21, 1136–1140. [Google Scholar] [CrossRef]
- Prendergast, G.C.; Malachowski, W.P.; DuHadaway, J.B.; Muller, A.J. Discovery of IDO1 Inhibitors: From Bench to Bedside. Cancer Res. 2017, 77, 6795–6811. [Google Scholar] [CrossRef]
- Prendergast, G.C.; Malachowski, W.J.; Mondal, A.; Scherle, P.; Muller, A.J. Indoleamine 2,3-Dioxygenase and Its Therapeutic Inhibition in Cancer. Int. Rev. Cell Mol. Biol. 2018, 336, 175–203. [Google Scholar] [CrossRef]
- Long, G.V.; Dummer, R.; Hamid, O.; Gajewski, T.F.; Caglevic, C.; Dalle, S.; Arance, A.; Carlino, M.S.; Grob, J.J.; Kim, T.M.; et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): A phase 3, randomised, double-blind study. Lancet Oncol. 2019, 20, 1083–1097. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Gettinger, S.; Chow, L.Q.M.; Gordon, M.; Awad, M.M.; Cha, E.; Gong, X.; Zhou, G.; Walker, C.; Leopold, L.; et al. Phase 1 study of epacadostat in combination with atezolizumab for patients with previously treated advanced nonsmall cell lung cancer. Int. J. Cancer 2020, 147, 1963–1969. [Google Scholar] [CrossRef]
- Naing, A.; Powderly, J.D.; Nemunaitis, J.J.; Luke, J.J.; Mansfield, A.S.; Messersmith, W.A.; Sahebjam, S.; LoRusso, P.M.; Garrido-Laguna, I.; Leopold, L.; et al. Exploring the safety, effect on the tumor microenvironment, and efficacy of itacitinib in combination with epacadostat or parsaclisib in advanced solid tumors: A phase I study. J. Immunother. Cancer 2022, 10, e004223. [Google Scholar] [CrossRef]
- Sun, J.Y.; Zhang, D.; Wu, S.; Xu, M.; Zhou, X.; Lu, X.J.; Ji, J. Resistance to PD-1/PD-L1 blockade cancer immunotherapy: Mechanisms, predictive factors, and future perspectives. Biomark Res. 2020, 8, 35. [Google Scholar] [CrossRef]
- Chang, C.H.; Pearce, E.L. Emerging concepts of T cell metabolism as a target of immunotherapy. Nat. Immunol. 2016, 17, 364–368. [Google Scholar] [CrossRef]
- Renner, K.; Singer, K.; Koehl, G.E.; Geissler, E.K.; Peter, K.; Siska, P.J.; Kreutz, M. Metabolic Hallmarks of Tumor and Immune Cells in the Tumor Microenvironment. Front. Immunol. 2017, 8, 248. [Google Scholar] [CrossRef]
- Ala, M. The footprint of kynurenine pathway in every cancer: A new target for chemotherapy. Eur. J. Pharmacol. 2021, 896, 173921. [Google Scholar] [CrossRef]
- Liu, V.C.; Wong, L.Y.; Jang, T.; Shah, A.H.; Park, I.; Yang, X.; Zhang, Q.; Lonning, S.; Teicher, B.A.; Lee, C. Tumor evasion of the immune system by converting CD4+CD25- T cells into CD4+CD25+ T regulatory cells: Role of tumor-derived TGF-beta. J. Immunol. 2007, 178, 2883–2892. [Google Scholar] [CrossRef]
- Lechner, M.G.; Karimi, S.S.; Barry-Holson, K.; Angell, T.E.; Murphy, K.A.; Church, C.H.; Ohlfest, J.R.; Hu, P.; Epstein, A.L. Immunogenicity of murine solid tumor models as a defining feature of in vivo behavior and response to immunotherapy. J. Immunother. 2013, 36, 477–489. [Google Scholar] [CrossRef]
- Yu, J.W.; Bhattacharya, S.; Yanamandra, N.; Kilian, D.; Shi, H.; Yadavilli, S.; Katlinskaya, Y.; Kaczynski, H.; Conner, M.; Benson, W.; et al. Tumor-immune profiling of murine syngeneic tumor models as a framework to guide mechanistic studies and predict therapy response in distinct tumor microenvironments. PLoS ONE 2018, 13, e0206223. [Google Scholar] [CrossRef] [PubMed]
- Tate, D.J.; Vonderhaar, D.J.; Caldas, Y.A.; Metoyer, T.; Patterson, J.R.; Aviles, D.H.; Zea, A.H. Effect of arginase II on L-arginine depletion and cell growth in murine cell lines of renal cell carcinoma. J. Hematol. Oncol. 2008, 1, 14. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Meng, Y.; Wu, X.; Sun, Y. Polyamines and related signaling pathways in cancer. Cancer Cell Int. 2020, 20, 539. [Google Scholar] [CrossRef] [PubMed]
- Cederbaum, S.D.; Yu, H.; Grody, W.W.; Kern, R.M.; Yoo, P.; Iyer, R.K. Arginases I and II: Do their functions overlap? Mol. Genet. Metab. 2004, 81 (Suppl. S1), S38–S44. [Google Scholar] [CrossRef]
- Chen, C.L.; Hsu, S.C.; Ann, D.K.; Yen, Y.; Kung, H.J. Arginine Signaling and Cancer Metabolism. Cancers 2021, 13, 3541. [Google Scholar] [CrossRef]
- Giatromanolaki, A.; Harris, A.L.; Koukourakis, M.I. The prognostic and therapeutic implications of distinct patterns of argininosuccinate synthase 1 (ASS1) and arginase-2 (ARG2) expression by cancer cells and tumor stroma in non-small-cell lung cancer. Cancer Metab. 2021, 9, 28. [Google Scholar] [CrossRef]
- Clemente, G.S.; van Waarde, A.; Antunes, I.F.; Dömling, A.; Elsinga, P.H. Arginase as a Potential Biomarker of Disease Progression: A Molecular Imaging Perspective. Int. J. Mol. Sci. 2020, 21, 5291. [Google Scholar] [CrossRef]
- Wu, C.W.; Chi, C.W.; Lin, E.C.; Lui, W.Y.; P’eng, F.K.; Wang, S.R. Serum arginase level in patients with gastric cancer. J. Clin. Gastroenterol. 1994, 18, 84–85. [Google Scholar] [CrossRef]
- Chrzanowska, A.; Graboń, W.; Mielczarek-Puta, M.; Barańczyk-Kuźma, A. Significance of arginase determination in body fluids of patients with hepatocellular carcinoma and liver cirrhosis before and after surgical treatment. Clin. Biochem. 2014, 47, 1056–1059. [Google Scholar] [CrossRef]
- Chrzanowska, A.; Krawczyk, M.; Barańczyk-Kuźma, A. Changes in arginase isoenzymes pattern in human hepatocellular carcinoma. Biochem. Biophys. Res. Commun. 2008, 377, 337–340. [Google Scholar] [CrossRef]
- Niu, F.; Yu, Y.; Li, Z.; Ren, Y.; Ye, Q.; Liu, P.; Ji, C.; Qian, L.; Xiong, Y. Arginase: An emerging and promising therapeutic target for cancer treatment. Biomed. Pharmacother. 2022, 149, 112840. [Google Scholar] [CrossRef] [PubMed]
- Zea, A.H.; Rodriguez, P.C.; Culotta, K.S.; Hernandez, C.P.; DeSalvo, J.; Ochoa, J.B.; Park, H.J.; Zabaleta, J.; Ochoa, A.C. L-Arginine modulates CD3zeta expression and T cell function in activated human T lymphocytes. Cell. Immunol. 2004, 232, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Nan, H.; Shi, N.; Hao, W.; Dong, J.; Chen, H. Chlorogenic acid inhibits proliferation in human hepatoma cells by suppressing noncanonical NF-κB signaling pathway and triggering mitochondrial apoptosis. Mol. Biol. Rep. 2021, 48, 2351–2364. [Google Scholar] [CrossRef] [PubMed]
- Ng, K.P.; Manjeri, A.; Lee, L.M.; Chan, Z.E.; Tan, C.Y.; Tan, Q.D.; Majeed, A.; Lee, K.L.; Chuah, C.; Suda, T.; et al. The arginase inhibitor Nω-hydroxy-nor-arginine (nor-NOHA) induces apoptosis in leukemic cells specifically under hypoxic conditions but CRISPR/Cas9 excludes arginase 2 (ARG2) as the functional target. PLoS ONE 2018, 13, e0205254. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Pervin, S.; Karimi, A.; Cederbaum, S.; Chaudhuri, G. Arginase activity in human breast cancer cell lines: N(omega)-hydroxy-L-arginine selectively inhibits cell proliferation and induces apoptosis in MDA-MB-468 cells. Cancer Res. 2000, 60, 3305–3312. [Google Scholar]
- Singh, R.; Avliyakulov, N.K.; Braga, M.; Haykinson, M.J.; Martinez, L.; Singh, V.; Parveen, M.; Chaudhuri, G.; Pervin, S. Proteomic identification of mitochondrial targets of arginase in human breast cancer. PLoS ONE 2013, 8, e79242. [Google Scholar] [CrossRef]
- Yu, Y.; Ladeiras, D.; Xiong, Y.; Boligan, K.F.; Liang, X.; von Gunten, S.; Hunger, R.E.; Ming, X.F.; Yang, Z. Arginase-II promotes melanoma migration and adhesion through enhancing hydrogen peroxide production and STAT3 signaling. J. Cell. Physiol. 2020, 235, 9997–10011. [Google Scholar] [CrossRef]
- Patil, M.D.; Bhaumik, J.; Babykutty, S.; Banerjee, U.C.; Fukumura, D. Arginine dependence of tumor cells: Targeting a chink in cancer’s armor. Oncogene 2016, 35, 4957–4972. [Google Scholar] [CrossRef]
- Matos, A.; Carvalho, M.; Bicho, M.; Ribeiro, R. Arginine and Arginases Modulate Metabolism, Tumor Microenvironment and Prostate Cancer Progression. Nutrients 2021, 13, 4503. [Google Scholar] [CrossRef]
- Cheng, C.T.; Qi, Y.; Wang, Y.C.; Chi, K.K.; Chung, Y.; Ouyang, C.; Chen, Y.R.; Oh, M.E.; Sheng, X.; Tang, Y.; et al. Arginine starvation kills tumor cells through aspartate exhaustion and mitochondrial dysfunction. Commun. Biol. 2018, 1, 178. [Google Scholar] [CrossRef]
- Delage, B.; Fennell, D.A.; Nicholson, L.; McNeish, I.; Lemoine, N.R.; Crook, T.; Szlosarek, P.W. Arginine deprivation and argininosuccinate synthetase expression in the treatment of cancer. Int. J. Cancer 2010, 126, 2762–2772. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Zhang, H.; Li, D.; Childers, M.; Pu, Q.; Palte, R.L.; Gathiaka, S.; Lyons, T.W.; Palani, A.; Fan, P.W.; et al. Structure-Based Discovery of Proline-Derived Arginase Inhibitors with Improved Oral Bioavailability for Immuno-Oncology. ACS Med. Chem. Lett. 2021, 12, 1380–1388. [Google Scholar] [CrossRef] [PubMed]
- Bron, L.; Jandus, C.; Andrejevic-Blant, S.; Speiser, D.E.; Monnier, P.; Romero, P.; Rivals, J.P. Prognostic value of arginase-II expression and regulatory T-cell infiltration in head and neck squamous cell carcinoma. Int. J. Cancer 2013, 132, E85–E93. [Google Scholar] [CrossRef] [PubMed]
- Roci, I.; Watrous, J.D.; Lagerborg, K.A.; Lafranchi, L.; Lindqvist, A.; Jain, M.; Nilsson, R. Mapping Metabolic Events in the Cancer Cell Cycle Reveals Arginine Catabolism in the Committed SG. Cell Rep. 2019, 26, 1691–1700.e1695. [Google Scholar] [CrossRef]
- Weis-Banke, S.E.; Hübbe, M.L.; Holmström, M.O.; Jørgensen, M.A.; Bendtsen, S.K.; Martinenaite, E.; Carretta, M.; Svane, I.M.; Ødum, N.; Pedersen, A.W.; et al. The metabolic enzyme arginase-2 is a potential target for novel immune modulatory vaccines. Oncoimmunology 2020, 9, 1771142. [Google Scholar] [CrossRef]
- Grzywa, T.M.; Sosnowska, A.; Matryba, P.; Rydzynska, Z.; Jasinski, M.; Nowis, D.; Golab, J. Myeloid Cell-Derived Arginase in Cancer Immune Response. Front. Immunol. 2020, 11, 938. [Google Scholar] [CrossRef]
- Rodriguez, P.C.; Zea, A.H.; Culotta, K.S.; Zabaleta, J.; Ochoa, J.B.; Ochoa, A.C. Regulation of T cell receptor CD3zeta chain expression by L-arginine. J. Biol. Chem. 2002, 277, 21123–21129. [Google Scholar] [CrossRef]
- Rodriguez, P.C.; Zea, A.H.; DeSalvo, J.; Culotta, K.S.; Zabaleta, J.; Quiceno, D.G.; Ochoa, J.B.; Ochoa, A.C. L-arginine consumption by macrophages modulates the expression of CD3 zeta chain in T lymphocytes. J. Immunol. 2003, 171, 1232–1239. [Google Scholar] [CrossRef]
- Lamas, B.; Vergnaud-Gauduchon, J.; Goncalves-Mendes, N.; Perche, O.; Rossary, A.; Vasson, M.P.; Farges, M.C. Altered functions of natural killer cells in response to L-Arginine availability. Cell. Immunol. 2012, 280, 182–190. [Google Scholar] [CrossRef]
- Yan, X.; Takahara, M.; Xie, L.; Gondo, C.; Setsu, N.; Oda, Y.; Takeuchi, S.; Tu, Y.; Moroi, Y.; Furue, M. Arginine metabolism in soft tissue sarcoma. J. Dermatol. Sci. 2011, 61, 211–215. [Google Scholar] [CrossRef]
- Dowling, J.K.; Afzal, R.; Gearing, L.J.; Cervantes-Silva, M.P.; Annett, S.; Davis, G.M.; De Santi, C.; Assmann, N.; Dettmer, K.; Gough, D.J.; et al. Mitochondrial arginase-2 is essential for IL-10 metabolic reprogramming of inflammatory macrophages. Nat. Commun. 2021, 12, 1460. [Google Scholar] [CrossRef] [PubMed]
- Preston, C.C.; Maurer, M.J.; Oberg, A.L.; Visscher, D.W.; Kalli, K.R.; Hartmann, L.C.; Goode, E.L.; Knutson, K.L. The ratios of CD8+ T cells to CD4+CD25+ FOXP3+ and FOXP3- T cells correlate with poor clinical outcome in human serous ovarian cancer. PLoS ONE 2013, 8, e80063. [Google Scholar] [CrossRef] [PubMed]
- Grzywa, T.M.; Sosnowska, A.; Rydzynska, Z.; Lazniewski, M.; Plewczynski, D.; Klicka, K.; Malecka-Gieldowska, M.; Rodziewicz-Lurzynska, A.; Ciepiela, O.; Justyniarska, M.; et al. Potent but transient immunosuppression of T-cells is a general feature of CD71. Commun. Biol. 2021, 4, 1384. [Google Scholar] [CrossRef] [PubMed]
- Mumenthaler, S.M.; Yu, H.; Tze, S.; Cederbaum, S.D.; Pegg, A.E.; Seligson, D.B.; Grody, W.W. Expression of arginase II in prostate cancer. Int. J. Oncol. 2008, 32, 357–365. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grzybowski, M.M.; Stańczak, P.S.; Pomper, P.; Błaszczyk, R.; Borek, B.; Gzik, A.; Nowicka, J.; Jędrzejczak, K.; Brzezińska, J.; Rejczak, T.; et al. OATD-02 Validates the Benefits of Pharmacological Inhibition of Arginase 1 and 2 in Cancer. Cancers 2022, 14, 3967. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14163967
Grzybowski MM, Stańczak PS, Pomper P, Błaszczyk R, Borek B, Gzik A, Nowicka J, Jędrzejczak K, Brzezińska J, Rejczak T, et al. OATD-02 Validates the Benefits of Pharmacological Inhibition of Arginase 1 and 2 in Cancer. Cancers. 2022; 14(16):3967. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14163967
Chicago/Turabian StyleGrzybowski, Marcin Mikołaj, Paulina Seweryna Stańczak, Paulina Pomper, Roman Błaszczyk, Bartłomiej Borek, Anna Gzik, Julita Nowicka, Karol Jędrzejczak, Joanna Brzezińska, Tomasz Rejczak, and et al. 2022. "OATD-02 Validates the Benefits of Pharmacological Inhibition of Arginase 1 and 2 in Cancer" Cancers 14, no. 16: 3967. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14163967