
Adenosine Targeting as a New Strategy to Decrease Glioblastoma Aggressiveness

 , , , , and
, , , , and
Abstract
:Simple Summary
Abstract
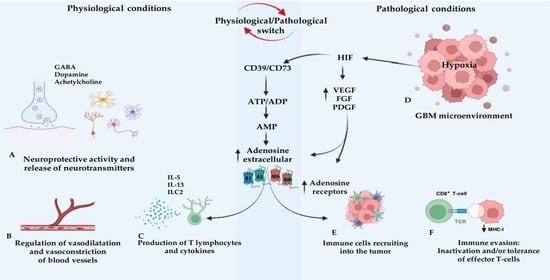
1. Introduction
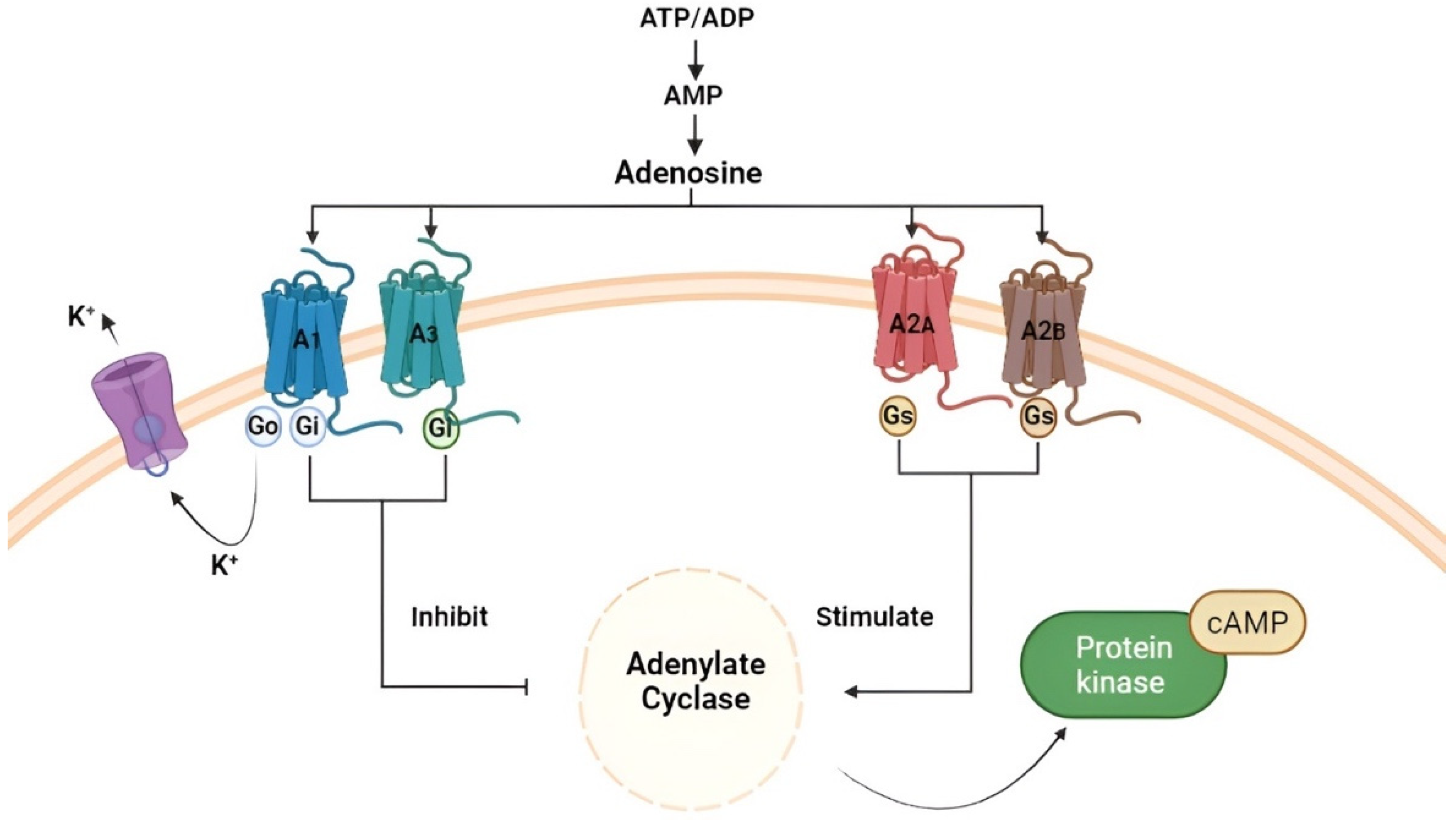
2. Adenosine and Adenosine Receptors (ARs)
3. The Role of Adenosine in Glioblastoma Multiforme
4. Adenosine Receptor Antagonists
4.1. Xanthine Derivates
4.2. Polyheterocyclic Nitrogen System
4.3. Enhancement of Immunotherapy Induced by Adenosine Receptor Antagonists
5. Future Perspectives
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bondy, M.L.; Scheurer, M.E.; Malmer, B.; Barnholtz-Sloan, J.S.; Davis, F.G.; Il’yasova, D.; Kruchko, C.; McCarthy, B.J.; Rajaraman, P.; Schwartzbaum, J.A.; et al. Brain tumor epidemiology: Consensus from the Brain Tumor Epidemiology Consortium. Cancer 2008, 113, 1953–1968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daneman, R.; Zhou, L.; Kebede, A.A.; Barres, B.A. Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature 2010, 468, 562–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, K.L.; Chan, S.H.; Chan, J.Y. Neuroinflammation and oxidative stress in rostral ventrolateral medulla contribute to neurogenic hypertension induced by systemic inflammation. J. Neuroinflamm. 2012, 9, 212. [Google Scholar] [CrossRef] [Green Version]
- McFaline-Figueroa, J.R.; Lee, E.Q. Brain Tumors. Am. J. Med. 2018, 131, 874–882. [Google Scholar] [CrossRef] [PubMed]
- Ansell, P.; Johnston, T.; Simpson, J.; Crouch, S.; Roman, E.; Picton, S. Brain tumor signs and symptoms: Analysis of primary health care records from the UKCCS. Pediatrics 2010, 125, 112–119. [Google Scholar] [CrossRef] [Green Version]
- Minniti, G.; Traish, D.; Ashley, S.; Gonsalves, A.; Brada, M. Risk of second brain tumor after conservative surgery and radiotherapy for pituitary adenoma: Update after an additional 10 years. J. Clin. Endocrinol. Metab. 2005, 90, 800–804. [Google Scholar] [CrossRef] [Green Version]
- Wirsching, H.G.; Galanis, E.; Weller, M. Glioblastoma. Handb. Clin. Neurol. 2016, 134, 381–397. [Google Scholar] [CrossRef]
- Kaplan, K.; Kaya, Y.; Kuncan, M.; Ertunc, H.M. Brain tumor classification using modified local binary patterns (LBP) feature extraction methods. Med. Hypotheses 2020, 139, 109696. [Google Scholar] [CrossRef]
- Ardizzone, A.; Scuderi, S.A.; Giuffrida, D.; Colarossi, C.; Puglisi, C.; Campolo, M.; Cuzzocrea, S.; Esposito, E.; Paterniti, I. Role of Fibroblast Growth Factors Receptors (FGFRs) in Brain Tumors, Focus on Astrocytoma and Glioblastoma. Cancers 2020, 12, 3825. [Google Scholar] [CrossRef]
- Jiang, Y.; Uhrbom, L. On the origin of glioma. Upsala J. Med. Sci. 2012, 117, 113–121. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, T.; Nobusawa, S.; Kleihues, P.; Ohgaki, H. IDH1 mutations are early events in the development of astrocytomas and oligodendrogliomas. Am. J. Pathol. 2009, 174, 1149–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Ali, F.; Hendon, A.J.; Liepman, M.K.; Wisniewski, J.L.; Krinock, M.J.; Beckman, K. Oligodendroglioma metastatic to bone marrow. AJNR Am. J. Neuroradiol. 2005, 26, 2410–2414. [Google Scholar] [PubMed]
- Neumann, J.E.; Spohn, M.; Obrecht, D.; Mynarek, M.; Thomas, C.; Hasselblatt, M.; Dorostkar, M.M.; Wefers, A.K.; Frank, S.; Monoranu, C.M.; et al. Molecular characterization of histopathological ependymoma variants. Acta Neuropathol. 2020, 139, 305–318. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Yoo, H.; Shin, S.H.; Gwak, H.S.; Lee, S.H. Extraneural Metastases of Glioblastoma without Simultaneous Central Nervous System Recurrence. Brain Tumor Res. Treat. 2014, 2, 124–127. [Google Scholar] [CrossRef] [Green Version]
- Scuderi, S.A.; Casili, G.; Ardizzone, A.; Forte, S.; Colarossi, L.; Sava, S.; Paterniti, I.; Esposito, E.; Cuzzocrea, S.; Campolo, M. KYP-2047, an Inhibitor of Prolyl-Oligopeptidase, Reduces GlioBlastoma Proliferation through Angiogenesis and Apoptosis Modulation. Cancers 2021, 13, 3444. [Google Scholar] [CrossRef]
- Ohgaki, H.; Kleihues, P. Genetic pathways to primary and secondary glioblastoma. Am. J. Pathol. 2007, 170, 1445–1453. [Google Scholar] [CrossRef] [Green Version]
- Louis, D.N. Molecular pathology of malignant gliomas. Annu. Rev. Pathol. 2006, 1, 97–117. [Google Scholar] [CrossRef] [Green Version]
- Colwell, N.; Larion, M.; Giles, A.J.; Seldomridge, A.N.; Sizdahkhani, S.; Gilbert, M.R.; Park, D.M. Hypoxia in the glioblastoma microenvironment: Shaping the phenotype of cancer stem-like cells. Neuro Oncol. 2017, 19, 887–896. [Google Scholar] [CrossRef]
- Jovcevska, I. Genetic secrets of long-term glioblastoma survivors. Bosn. J. Basic Med. Sci. 2019, 19, 116–124. [Google Scholar] [CrossRef]
- Kim, H.J.; Park, J.W.; Lee, J.H. Genetic Architectures and Cell-of-Origin in Glioblastoma. Front. Oncol. 2020, 10, 615400. [Google Scholar] [CrossRef]
- Dirkse, A.; Golebiewska, A.; Buder, T.; Nazarov, P.V.; Muller, A.; Poovathingal, S.; Brons, N.H.C.; Leite, S.; Sauvageot, N.; Sarkisjan, D.; et al. Stem cell-associated heterogeneity in Glioblastoma results from intrinsic tumor plasticity shaped by the microenvironment. Nat. Commun. 2019, 10, 1787. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.M.; Simon, M.C. The tumor microenvironment. Curr. Biol. 2020, 30, R921–R925. [Google Scholar] [CrossRef] [PubMed]
- Heiland, D.H.; Ravi, V.M.; Behringer, S.P.; Frenking, J.H.; Wurm, J.; Joseph, K.; Garrelfs, N.W.C.; Strahle, J.; Heynckes, S.; Grauvogel, J.; et al. Tumor-associated reactive astrocytes aid the evolution of immunosuppressive environment in glioblastoma. Nat. Commun. 2019, 10, 2541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zong, H.; Verhaak, R.G.; Canoll, P. The cellular origin for malignant glioma and prospects for clinical advancements. Expert Rev. Mol. Diagn. 2012, 12, 383–394. [Google Scholar] [CrossRef] [Green Version]
- Ohta, A.; Sitkovsky, M. Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature 2001, 414, 916–920. [Google Scholar] [CrossRef] [Green Version]
- Koritzinsky, M.; Seigneuric, R.; Magagnin, M.G.; van den Beucken, T.; Lambin, P.; Wouters, B.G. The hypoxic proteome is influenced by gene-specific changes in mRNA translation. Radiother. Oncol. 2005, 76, 177–186. [Google Scholar] [CrossRef]
- Liebelt, B.D.; Shingu, T.; Zhou, X.; Ren, J.; Shin, S.A.; Hu, J. Glioma Stem Cells: Signaling, Microenvironment, and Therapy. Stem. Cells Int. 2016, 2016, 7849890. [Google Scholar] [CrossRef] [Green Version]
- Kietzmann, T.; Mennerich, D.; Dimova, E.Y. Hypoxia-Inducible Factors (HIFs) and Phosphorylation: Impact on Stability, Localization, and Transactivity. Front. Cell Dev. Biol. 2016, 4, 11. [Google Scholar] [CrossRef]
- Das, S.; Marsden, P.A. Angiogenesis in glioblastoma. N. Engl. J. Med. 2013, 369, 1561–1563. [Google Scholar] [CrossRef] [Green Version]
- Gabler, L.; Jaunecker, C.N.; Katz, S.; van Schoonhoven, S.; Englinger, B.; Pirker, C.; Mohr, T.; Vician, P.; Stojanovic, M.; Woitzuck, V.; et al. Fibroblast growth factor receptor 4 promotes glioblastoma progression: A central role of integrin-mediated cell invasiveness. Acta Neuropathol. Commun. 2022, 10, 65. [Google Scholar] [CrossRef]
- Westermark, B. Platelet-derived growth factor in glioblastoma-driver or biomarker? Upsala J. Med. Sci. 2014, 119, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Pearson, J.R.D.; Cuzzubbo, S.; McArthur, S.; Durrant, L.G.; Adhikaree, J.; Tinsley, C.J.; Pockley, A.G.; McArdle, S.E.B. Immune Escape in Glioblastoma Multiforme and the Adaptation of Immunotherapies for Treatment. Front. Immunol. 2020, 11, 582106. [Google Scholar] [CrossRef] [PubMed]
- Chouaib, S.; El Hage, F.; Benlalam, H.; Mami-Chouaib, F. Immunotherapy of cancer: Promise and reality. Med. Sci. 2006, 22, 755–759. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, K.; Smyth, M.J. Myeloid immunosuppression and immune checkpoints in the tumor microenvironment. Cell. Mol. Immunol. 2020, 17, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Goswami, S.; Walle, T.; Cornish, A.E.; Basu, S.; Anandhan, S.; Fernandez, I.; Vence, L.; Blando, J.; Zhao, H.; Yadav, S.S.; et al. Immune profiling of human tumors identifies CD73 as a combinatorial target in glioblastoma. Nat. Med. 2020, 26, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Fishman, P.; Bar-Yehuda, S.; Synowitz, M.; Powell, J.D.; Klotz, K.N.; Gessi, S.; Borea, P.A. Adenosine receptors and cancer. Handb. Exp. Pharmacol. 2009, 193, 399–441. [Google Scholar] [CrossRef] [Green Version]
- Borea, P.A.; Gessi, S.; Merighi, S.; Vincenzi, F.; Varani, K. Pharmacology of adenosine receptors: The state of the art. Physiol. Rev. 2018, 98, 1591–1625. [Google Scholar] [CrossRef]
- O’Hayre, M.; Vazquez-Prado, J.; Kufareva, I.; Stawiski, E.W.; Handel, T.M.; Seshagiri, S.; Gutkind, J.S. The emerging mutational landscape of G proteins and G-protein-coupled receptors in cancer. Nat. Rev. Cancer 2013, 13, 412–424. [Google Scholar] [CrossRef]
- Geraldo, L.H.M.; Garcia, C.; da Fonseca, A.C.C.; Dubois, L.G.F.; de Sampaio, E.S.T.C.L.; Matias, D.; de Camargo Magalhaes, E.S.; do Amaral, R.F.; da Rosa, B.G.; Grimaldi, I.; et al. Glioblastoma Therapy in the Age of Molecular Medicine. Trends Cancer 2019, 5, 46–65. [Google Scholar] [CrossRef]
- Stupp, R.; Dietrich, P.Y.; Ostermann Kraljevic, S.; Pica, A.; Maillard, I.; Maeder, P.; Meuli, R.; Janzer, R.; Pizzolato, G.; Miralbell, R.; et al. Promising survival for patients with newly diagnosed glioblastoma multiforme treated with concomitant radiation plus temozolomide followed by adjuvant temozolomide. J. Clin. Oncol. 2002, 20, 1375–1382. [Google Scholar] [CrossRef]
- Ghiaseddin, A.; Peters, K.B. Use of bevacizumab in recurrent glioblastoma. CNS Oncol. 2015, 4, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, G.; Caccese, M.; Padovan, M.; Cerretti, G.; Pintacuda, G.; Manara, R.; Di Sarra, F.; Zagonel, V. Regorafenib in Recurrent Glioblastoma Patients: A Large and Monocentric Real-Life Study. Cancers 2021, 13, 4731. [Google Scholar] [CrossRef] [PubMed]
- Green, A.L.; Mulcahy Levy, J.M.; Vibhakar, R.; Hemenway, M.; Madden, J.; Foreman, N.; Dorris, K. Tumor treating fields in pediatric high-grade glioma. Childs Nerv. Syst. 2017, 33, 1043–1045. [Google Scholar] [CrossRef] [PubMed]
- Mehta, M.; Wen, P.; Nishikawa, R.; Reardon, D.; Peters, K. Critical review of the addition of tumor treating fields (TTFields) to the existing standard of care for newly diagnosed glioblastoma patients. Crit. Rev. Oncol. Hematol. 2017, 111, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Ram, Z.; Culver, K.W.; Oshiro, E.M.; Viola, J.J.; DeVroom, H.L.; Otto, E.; Long, Z.; Chiang, Y.; McGarrity, G.J.; Muul, L.M.; et al. Therapy of malignant brain tumors by intratumoral implantation of retroviral vector-producing cells. Nat. Med. 1997, 3, 1354–1361. [Google Scholar] [CrossRef]
- Klatzmann, D.; Valery, C.A.; Bensimon, G.; Marro, B.; Boyer, O.; Mokhtari, K.; Diquet, B.; Salzmann, J.L.; Philippon, J. A phase I/II study of herpes simplex virus type 1 thymidine kinase "suicide" gene therapy for recurrent glioblastoma. Study Group on Gene Therapy for Glioblastoma. Hum. Gene Ther. 1998, 9, 2595–2604. [Google Scholar] [CrossRef]
- Raffel, C.; Culver, K.; Kohn, D.; Nelson, M.; Siegel, S.; Gillis, F.; Link, C.J.; Villablanca, J.G.; Anderson, W.F. Gene therapy for the treatment of recurrent pediatric malignant astrocytomas with in vivo tumor transduction with the herpes simplex thymidine kinase gene/ganciclovir system. Hum. Gene Ther. 1994, 5, 863–890. [Google Scholar] [CrossRef]
- Zhou, W.; Qing, H.; Tong, Y.; Song, W. BACE1 gene expression and protein degradation. Ann. N. Y. Acad. Sci. 2004, 1035, 49–67. [Google Scholar] [CrossRef]
- Cloughesy, T.F.; Mochizuki, A.Y.; Orpilla, J.R.; Hugo, W.; Lee, A.H.; Davidson, T.B.; Wang, A.C.; Ellingson, B.M.; Rytlewski, J.A.; Sanders, C.M.; et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat. Med. 2019, 25, 477–486. [Google Scholar] [CrossRef]
- Bagley, S.J.; Desai, A.S.; Linette, G.P.; June, C.H.; O’Rourke, D.M. CAR T-cell therapy for glioblastoma: Recent clinical advances and future challenges. Neuro Oncol. 2018, 20, 1429–1438. [Google Scholar] [CrossRef] [Green Version]
- Decking, U.K.; Schlieper, G.; Kroll, K.; Schrader, J. Hypoxia-induced inhibition of adenosine kinase potentiates cardiac adenosine release. Circ. Res. 1997, 81, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Fenton, R.A.; Wheeler, H.B.; Powell, C.C.; Peyton, B.D.; Cutler, B.S.; Dobson, J.G., Jr. Adenosine A2a receptors increase arterial endothelial cell nitric oxide. J. Surg. Res. 1998, 80, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Bouma, M.G.; Stad, R.K.; van den Wildenberg, F.A.; Buurman, W.A. Differential regulatory effects of adenosine on cytokine release by activated human monocytes. J. Immunol. 1994, 153, 4159–4168. [Google Scholar] [PubMed]
- Van der Graaf, P.H.; Van Schaick, E.A.; Visser, S.A.; De Greef, H.J.; Ijzerman, A.P.; Danhof, M. Mechanism-based pharmacokinetic-pharmacodynamic modeling of antilipolytic effects of adenosine A(1) receptor agonists in rats: Prediction of tissue-dependent efficacy in vivo. J. Pharmacol. Exp. Ther. 1999, 290, 702–709. [Google Scholar] [PubMed]
- Williams-Karnesky, R.L.; Stenzel-Poore, M.P. Adenosine and stroke: Maximizing the therapeutic potential of adenosine as a prophylactic and acute neuroprotectant. Curr. Neuropharmacol. 2009, 7, 217–227. [Google Scholar] [CrossRef] [Green Version]
- Franco, R.; Rivas-Santisteban, R.; Navarro, G.; Reyes-Resina, I. Adenosine Receptor Antagonists to Combat Cancer and to Boost Anti-Cancer Chemotherapy and Immunotherapy. Cells 2021, 10, 2831. [Google Scholar] [CrossRef]
- Coney, A.M.; Marshall, J.M. Role of adenosine and its receptors in the vasodilatation induced in the cerebral cortex of the rat by systemic hypoxia. J. Physiol. 1998, 509 Pt 2, 507–518. [Google Scholar] [CrossRef]
- Manjunath, S.; Sakhare, P.M. Adenosine and adenosine receptors: Newer therapeutic perspective. Indian J. Pharmacol. 2009, 41, 97–105. [Google Scholar] [CrossRef]
- Marcelino, H.; Carvalho, T.M.A.; Tomas, J.; Teles, F.I.; Honorio, A.C.; Rosa, C.B.; Costa, A.R.; Costa, B.M.; Santos, C.R.A.; Sebastiao, A.M.; et al. Adenosine Inhibits Cell Proliferation Differently in Human Astrocytes and in Glioblastoma Cell Lines. Neuroscience 2021, 467, 122–133. [Google Scholar] [CrossRef]
- Deussen, A.; Stappert, M.; Schafer, S.; Kelm, M. Quantification of extracellular and intracellular adenosine production: Understanding the transmembranous concentration gradient. Circulation 1999, 99, 2041–2047. [Google Scholar] [CrossRef]
- Collins, J.F.; Ghishan, F.K. Molecular cloning, functional expression, tissue distribution, and in situ hybridization of the renal sodium phosphate (Na+/P(i)) transporter in the control and hypophosphatemic mouse. FASEB J. 1994, 8, 862–868. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.F.; Eltzschig, H.K.; Fredholm, B.B. Adenosine receptors as drug targets—What are the challenges? Nat. Rev. Drug Discov. 2013, 12, 265–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmer, T.M.; Benovic, J.L.; Stiles, G.L. Molecular basis for subtype-specific desensitization of inhibitory adenosine receptors. Analysis of a chimeric A1-A3 adenosine receptor. J. Biol. Chem. 1996, 271, 15272–15278. [Google Scholar] [CrossRef] [Green Version]
- Sidders, B.; Zhang, P.; Goodwin, K.; O’Connor, G.; Russell, D.L.; Borodovsky, A.; Armenia, J.; McEwen, R.; Linghu, B.; Bendell, J.C.; et al. Adenosine Signaling Is Prognostic for Cancer Outcome and Has Predictive Utility for Immunotherapeutic Response. Clin. Cancer Res. 2020, 26, 2176–2187. [Google Scholar] [CrossRef]
- Luongo, L.; Guida, F.; Imperatore, R.; Napolitano, F.; Gatta, L.; Cristino, L.; Giordano, C.; Siniscalco, D.; Di Marzo, V.; Bellini, G.; et al. The A1 adenosine receptor as a new player in microglia physiology. Glia 2014, 62, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Synowitz, M.; Glass, R.; Farber, K.; Markovic, D.; Kronenberg, G.; Herrmann, K.; Schnermann, J.; Nolte, C.; van Rooijen, N.; Kiwit, J.; et al. A1 adenosine receptors in microglia control glioblastoma-host interaction. Cancer Res. 2006, 66, 8550–8557. [Google Scholar] [CrossRef] [Green Version]
- Marti Navia, A.; Dal Ben, D.; Lambertucci, C.; Spinaci, A.; Volpini, R.; Marques-Morgado, I.; Coelho, J.E.; Lopes, L.V.; Marucci, G.; Buccioni, M. Adenosine Receptors as Neuroinflammation Modulators: Role of A1 Agonists and A2A Antagonists. Cells 2020, 9, 1739. [Google Scholar] [CrossRef]
- Muroi, Y.; Ishii, T.; Teramoto, K.; Hori, M.; Nishimura, M. Calcineurin contributes to the enhancing effect of adenosine on nerve growth factor-induced neurite outgrowth via the decreased duration of p38 mitogen-activated protein kinase phosphorylation. J. Pharmacol. Sci. 2004, 95, 124–131. [Google Scholar] [CrossRef] [Green Version]
- Barkan, K.; Lagarias, P.; Stampelou, M.; Stamatis, D.; Hoare, S.; Safitri, D.; Klotz, K.N.; Vrontaki, E.; Kolocouris, A.; Ladds, G. Pharmacological characterisation of novel adenosine A3 receptor antagonists. Sci. Rep. 2020, 10, 20781. [Google Scholar] [CrossRef]
- Rocha, R.; Torres, A.; Ojeda, K.; Uribe, D.; Rocha, D.; Erices, J.; Niechi, I.; Ehrenfeld, P.; San Martin, R.; Quezada, C. The Adenosine A(3) Receptor Regulates Differentiation of Glioblastoma Stem-Like Cells to Endothelial Cells under Hypoxia. Int. J. Mol. Sci. 2018, 19, 1228. [Google Scholar] [CrossRef] [Green Version]
- Weaver, D.R. A2a adenosine receptor gene expression in developing rat brain. Brain Res. Mol. Brain Res. 1993, 20, 313–327. [Google Scholar] [CrossRef]
- Singh, B.L.; Chen, L.; Cai, H.; Shi, H.; Wang, Y.; Yu, C.; Chen, X.; Han, X.; Cai, X. Activation of adenosine A2a receptor accelerates and A2a receptor antagonist reduces intermittent hypoxia induced PC12 cell injury via PKC-KATP pathway. Brain Res. Bull. 2019, 150, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Luongo, L.; Guida, F.; Maione, S.; Jacobson, K.A.; Salvemini, D. Adenosine Metabotropic Receptors in Chronic Pain Management. Front. Pharmacol. 2021, 12, 651038. [Google Scholar] [CrossRef] [PubMed]
- Cunha, R.A.; Almeida, T.; Ribeiro, J.A. Modification by arachidonic acid of extracellular adenosine metabolism and neuromodulatory action in the rat hippocampus. J. Biol. Chem. 2000, 275, 37572–37581. [Google Scholar] [CrossRef] [Green Version]
- Rebola, N.; Rodrigues, R.J.; Oliveira, C.R.; Cunha, R.A. Different roles of adenosine A1, A2A and A3 receptors in controlling kainate-induced toxicity in cortical cultured neurons. Neurochem. Int. 2005, 47, 317–325. [Google Scholar] [CrossRef]
- Li, X.X.; Nomura, T.; Aihara, H.; Nishizaki, T. Adenosine enhances glial glutamate efflux via A2a adenosine receptors. Life Sci. 2001, 68, 1343–1350. [Google Scholar] [CrossRef]
- Melani, A.; Cipriani, S.; Vannucchi, M.G.; Nosi, D.; Donati, C.; Bruni, P.; Giovannini, M.G.; Pedata, F. Selective adenosine A2a receptor antagonism reduces JNK activation in oligodendrocytes after cerebral ischaemia. Brain 2009, 132, 1480–1495. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Apasov, S.; Koshiba, M.; Sitkovsky, M. Role of A2a extracellular adenosine receptor-mediated signaling in adenosine-mediated inhibition of T-cell activation and expansion. Blood 1997, 90, 1600–1610. [Google Scholar] [CrossRef]
- Young, A.; Ngiow, S.F.; Gao, Y.; Patch, A.M.; Barkauskas, D.S.; Messaoudene, M.; Lin, G.; Coudert, J.D.; Stannard, K.A.; Zitvogel, L.; et al. A2AR Adenosine Signaling Suppresses Natural Killer Cell Maturation in the Tumor Microenvironment. Cancer Res. 2018, 78, 1003–1016. [Google Scholar] [CrossRef] [Green Version]
- Ott, M.; Tomaszowski, K.H.; Marisetty, A.; Kong, L.Y.; Wei, J.; Duna, M.; Blumberg, K.; Ji, X.; Jacobs, C.; Fuller, G.N.; et al. Profiling of patients with glioma reveals the dominant immunosuppressive axis is refractory to immune function restoration. JCI Insight 2020, 5. [Google Scholar] [CrossRef]
- Block, E.T.; Cronstein, B.N. Interferon-gamma inhibits adenosine A2A receptor function in hepatic stellate cells by STAT1-mediated repression of adenylyl cyclase. Int. J. Interferon Cytok. Mediat. Res. 2010, 2010, 113–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, H.B.; Ward, A.; Hamidzadeh, K.; Ravid, K.; Mosser, D.M. IFN-gamma Prevents Adenosine Receptor (A2bR) Upregulation To Sustain the Macrophage Activation Response. J. Immunol. 2015, 195, 3828–3837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arslan, G.; Kontny, E.; Fredholm, B.B. Down-regulation of adenosine A2A receptors upon NGF-induced differentiation of PC12 cells. Neuropharmacology 1997, 36, 1319–1326. [Google Scholar] [CrossRef]
- Orr, A.G.; Orr, A.L.; Li, X.J.; Gross, R.E.; Traynelis, S.F. Adenosine A(2A) receptor mediates microglial process retraction. Nat. Neurosci. 2009, 12, 872–878. [Google Scholar] [CrossRef]
- Simoes, A.P.; Duarte, J.A.; Agasse, F.; Canas, P.M.; Tome, A.R.; Agostinho, P.; Cunha, R.A. Blockade of adenosine A2A receptors prevents interleukin-1beta-induced exacerbation of neuronal toxicity through a p38 mitogen-activated protein kinase pathway. J. Neuroinflammation 2012, 9, 204. [Google Scholar] [CrossRef] [Green Version]
- Real, J.I.; Simoes, A.P.; Cunha, R.A.; Ferreira, S.G.; Rial, D. Adenosine A2A receptors modulate the dopamine D2 receptor-mediated inhibition of synaptic transmission in the mouse prefrontal cortex. Eur. J. Neurosci. 2018, 47, 1127–1134. [Google Scholar] [CrossRef]
- Ray, C.J.; Marshall, J.M. The cellular mechanisms by which adenosine evokes release of nitric oxide from rat aortic endothelium. J. Physiol. 2006, 570, 85–96. [Google Scholar] [CrossRef]
- Peng, W.; Wu, Z.; Song, K.; Zhang, S.; Li, Y.; Xu, M. Regulation of sleep homeostasis mediator adenosine by basal forebrain glutamatergic neurons. Science 2020, 369, eabb0556. [Google Scholar] [CrossRef]
- Antonioli, L.; Yegutkin, G.G.; Pacher, P.; Blandizzi, C.; Hasko, G. Anti-CD73 in cancer immunotherapy: Awakening new opportunities. Trends Cancer 2016, 2, 95–109. [Google Scholar] [CrossRef] [Green Version]
- Li, X.Y.; Moesta, A.K.; Xiao, C.; Nakamura, K.; Casey, M.; Zhang, H.; Madore, J.; Lepletier, A.; Aguilera, A.R.; Sundarrajan, A.; et al. Targeting CD39 in Cancer Reveals an Extracellular ATP- and Inflammasome-Driven Tumor Immunity. Cancer Discov. 2019, 9, 1754–1773. [Google Scholar] [CrossRef] [Green Version]
- Maj, T.; Wang, W.; Crespo, J.; Zhang, H.; Wang, W.; Wei, S.; Zhao, L.; Vatan, L.; Shao, I.; Szeliga, W.; et al. Oxidative stress controls regulatory T cell apoptosis and suppressor activity and PD-L1-blockade resistance in tumor. Nat. Immunol. 2017, 18, 1332–1341. [Google Scholar] [CrossRef] [PubMed]
- Arab, S.; Hadjati, J. Adenosine Blockage in Tumor Microenvironment and Improvement of Cancer Immunotherapy. Immune. Netw. 2019, 19, e23. [Google Scholar] [CrossRef] [PubMed]
- MacKenzie, W.M.; Hoskin, D.W.; Blay, J. Adenosine suppresses alpha(4)beta(7) integrin-mediated adhesion of T lymphocytes to colon adenocarcinoma cells. Exp. Cell Res. 2002, 276, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Seydyousefi, M.; Moghanlou, A.E.; Metz, G.A.S.; Gursoy, R.; Faghfoori, M.H.; Mirghani, S.J.; Faghfoori, Z. Exogenous adenosine facilitates neuroprotection and functional recovery following cerebral ischemia in rats. Brain Res. Bull. 2019, 153, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Elsaadi, S.; Misund, K.; Abdollahi, P.; Vandsemb, E.N.; Moen, S.H.; Kusnierczyk, A.; Slupphaug, G.; Standal, T.; Waage, A.; et al. Conversion of ATP to adenosine by CD39 and CD73 in multiple myeloma can be successfully targeted together with adenosine receptor A2A blockade. J. Immunother. Cancer 2020, 8, e000610. [Google Scholar] [CrossRef]
- Volmer, J.B.; Thompson, L.F.; Blackburn, M.R. Ecto-5′-nucleotidase (CD73)-mediated adenosine production is tissue protective in a model of bleomycin-induced lung injury. J. Immunol. 2006, 176, 4449–4458. [Google Scholar] [CrossRef] [Green Version]
- Antonioli, L.; Fornai, M.; Pellegrini, C.; D’Antongiovanni, V.; Turiello, R.; Morello, S.; Hasko, G.; Blandizzi, C. Adenosine Signaling in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2021, 1270, 145–167. [Google Scholar] [CrossRef]
- Koussemou, M.; Lorenz, K.; Klotz, K.N. The A2B adenosine receptor in MDA-MB-231 breast cancer cells diminishes ERK1/2 phosphorylation by activation of MAPK-phosphatase-1. PLoS ONE 2018, 13, e0202914. [Google Scholar] [CrossRef]
- Ohta, A.; Gorelik, E.; Prasad, S.J.; Ronchese, F.; Lukashev, D.; Wong, M.K.; Huang, X.; Caldwell, S.; Liu, K.; Smith, P.; et al. A2A adenosine receptor protects tumors from antitumor T cells. Proc. Natl. Acad. Sci. USA 2006, 103, 13132–13137. [Google Scholar] [CrossRef] [Green Version]
- Overwijk, W.W.; Restifo, N.P. Creating therapeutic cancer vaccines: Notes from the battlefield. Trends Immunol. 2001, 22, 5–7. [Google Scholar] [CrossRef]
- Ablamunits, V. The importance of APC. J. Autoimmune Dis. 2005, 2, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez, H.; Hagerling, C.; Werb, Z. Roles of the immune system in cancer: From tumor initiation to metastatic progression. Genes Dev. 2018, 32, 1267–1284. [Google Scholar] [CrossRef] [Green Version]
- Leone, R.D.; Lo, Y.C.; Powell, J.D. A2aR antagonists: Next generation checkpoint blockade for cancer immunotherapy. Comput. Struct. Biotechnol. J. 2015, 13, 265–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mapara, M.Y.; Sykes, M. Tolerance and cancer: Mechanisms of tumor evasion and strategies for breaking tolerance. J. Clin. Oncol. 2004, 22, 1136–1151. [Google Scholar] [CrossRef] [PubMed]
- Sitkovsky, M.; Lukashev, D.; Deaglio, S.; Dwyer, K.; Robson, S.C.; Ohta, A. Adenosine A2A receptor antagonists: Blockade of adenosinergic effects and T regulatory cells. Br. J. Pharmacol. 2008, 153 (Suppl. S1), S457–S464. [Google Scholar] [CrossRef] [Green Version]
- Gabrilovich, D.I.; Chen, H.L.; Girgis, K.R.; Cunningham, H.T.; Meny, G.M.; Nadaf, S.; Kavanaugh, D.; Carbone, D.P. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat. Med. 1996, 2, 1096–1103. [Google Scholar] [CrossRef]
- Antonioli, L.; Lucarini, E.; Lambertucci, C.; Fornai, M.; Pellegrini, C.; Benvenuti, L.; Di Cesare Mannelli, L.; Spinaci, A.; Marucci, G.; Blandizzi, C.; et al. The Anti-Inflammatory and Pain-Relieving Effects of AR170, an Adenosine A3 Receptor Agonist, in a Rat Model of Colitis. Cells 2020, 9, 1509. [Google Scholar] [CrossRef]
- Litchfield, K.; Reading, J.L.; Puttick, C.; Thakkar, K.; Abbosh, C.; Bentham, R.; Watkins, T.B.K.; Rosenthal, R.; Biswas, D.; Rowan, A.; et al. Meta-analysis of tumor- and T cell-intrinsic mechanisms of sensitization to checkpoint inhibition. Cell 2021, 184, 596–614.e514. [Google Scholar] [CrossRef]
- Boison, D. Adenosine kinase: Exploitation for therapeutic gain. Pharmacol. Rev. 2013, 65, 906–943. [Google Scholar] [CrossRef] [Green Version]
- Reisser, D.; Martin, F. CD4+ T cells recovered from a mixed immune lymphocyte-tumor cell culture induce thymidine incorporation by naive rat lymphocytes in response to tumor cells. Int. J. Cancer 1994, 57, 254–258. [Google Scholar] [CrossRef]
- Mediavilla-Varela, M.; Luddy, K.; Noyes, D.; Khalil, F.K.; Neuger, A.M.; Soliman, H.; Antonia, S.J. Antagonism of adenosine A2A receptor expressed by lung adenocarcinoma tumor cells and cancer associated fibroblasts inhibits their growth. Cancer Biol. Ther. 2013, 14, 860–868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres, A.; Erices, J.I.; Sanchez, F.; Ehrenfeld, P.; Turchi, L.; Virolle, T.; Uribe, D.; Niechi, I.; Spichiger, C.; Rocha, J.D.; et al. Extracellular adenosine promotes cell migration/invasion of Glioblastoma Stem-like Cells through A3 Adenosine Receptor activation under hypoxia. Cancer Lett. 2019, 446, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Waickman, A.T.; Alme, A.; Senaldi, L.; Zarek, P.E.; Horton, M.; Powell, J.D. Enhancement of tumor immunotherapy by deletion of the A2A adenosine receptor. Cancer Immunol. Immunother. 2012, 61, 917–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Huang, P. Adenosine A2B Receptor: From Cell Biology to Human Diseases. Front. Chem. 2016, 4, 37. [Google Scholar] [CrossRef] [PubMed]
- Vigano, S.; Alatzoglou, D.; Irving, M.; Menetrier-Caux, C.; Caux, C.; Romero, P.; Coukos, G. Targeting Adenosine in Cancer Immunotherapy to Enhance T-Cell Function. Front. Immunol. 2019, 10, 925. [Google Scholar] [CrossRef] [Green Version]
- Vijayan, D.; Young, A.; Teng, M.W.L.; Smyth, M.J. Targeting immunosuppressive adenosine in cancer. Nat. Rev. Cancer 2017, 17, 709–724. [Google Scholar] [CrossRef]
- Hatfield, S.M.; Kjaergaard, J.; Lukashev, D.; Belikoff, B.; Schreiber, T.H.; Sethumadhavan, S.; Abbott, R.; Philbrook, P.; Thayer, M.; Shujia, D.; et al. Systemic oxygenation weakens the hypoxia and hypoxia inducible factor 1alpha-dependent and extracellular adenosine-mediated tumor protection. J. Mol. Med. 2014, 92, 1283–1292. [Google Scholar] [CrossRef]
- Yan, A.; Joachims, M.L.; Thompson, L.F.; Miller, A.D.; Canoll, P.D.; Bynoe, M.S. CD73 Promotes Glioblastoma Pathogenesis and Enhances Its Chemoresistance via A2B Adenosine Receptor Signaling. J. Neurosci. 2019, 39, 4387–4402. [Google Scholar] [CrossRef] [Green Version]
- Daniele, S.; Zappelli, E.; Natali, L.; Martini, C.; Trincavelli, M.L. Modulation of A1 and A2B adenosine receptor activity: A new strategy to sensitise glioblastoma stem cells to chemotherapy. Cell Death Dis. 2014, 5, e1539. [Google Scholar] [CrossRef] [Green Version]
- Wink, M.R.; Lenz, G.; Braganhol, E.; Tamajusuku, A.S.; Schwartsmann, G.; Sarkis, J.J.; Battastini, A.M. Altered extracellular ATP, ADP and AMP catabolism in glioma cell lines. Cancer Lett. 2003, 198, 211–218. [Google Scholar] [CrossRef]
- Cascalheira, J.F.; Goncalves, M.; Barroso, M.; Castro, R.; Palmeira, M.; Serpa, A.; Dias-Cabral, A.C.; Domingues, F.C.; Almeida, S. Association of the transcobalamin II gene 776C → G polymorphism with Alzheimer’s type dementia: Dependence on the 5, 10-methylenetetrahydrofolate reductase 1298A → C polymorphism genotype. Ann. Clin. Biochem. 2015, 52, 448–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semmler, A.; Simon, M.; Moskau, S.; Linnebank, M. The methionine synthase polymorphism c.2756A>G alters susceptibility to glioblastoma multiforme. Cancer Epidemiol. Biomark. Prev. 2006, 15, 2314–2316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmeliet, P.; Dor, Y.; Herbert, J.M.; Fukumura, D.; Brusselmans, K.; Dewerchin, M.; Neeman, M.; Bono, F.; Abramovitch, R.; Maxwell, P.; et al. Role of HIF-1alpha in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Nature 1998, 394, 485–490. [Google Scholar] [CrossRef]
- Kust, B.M.; Biber, K.; van Calker, D.; Gebicke-Haerter, P.J. Regulation of K+ channel mRNA expression by stimulation of adenosine A2a-receptors in cultured rat microglia. Glia 1999, 25, 120–130. [Google Scholar] [CrossRef]
- Heese, K.; Fiebich, B.L.; Bauer, J.; Otten, U. Nerve growth factor (NGF) expression in rat microglia is induced by adenosine A2a-receptors. Neurosci. Lett. 1997, 231, 83–86. [Google Scholar] [CrossRef]
- Fiebich, B.L.; Biber, K.; Lieb, K.; van Calker, D.; Berger, M.; Bauer, J.; Gebicke-Haerter, P.J. Cyclooxygenase-2 expression in rat microglia is induced by adenosine A2a-receptors. Glia 1996, 18, 152–160. [Google Scholar] [CrossRef]
- Ma, S.R.; Deng, W.W.; Liu, J.F.; Mao, L.; Yu, G.T.; Bu, L.L.; Kulkarni, A.B.; Zhang, W.F.; Sun, Z.J. Blockade of adenosine A2A receptor enhances CD8(+) T cells response and decreases regulatory T cells in head and neck squamous cell carcinoma. Mol. Cancer 2017, 16, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.Z.; Wang, X.; Bai, Y.F.; Liao, H.Z.; Qiu, S.C.; Yang, Y.Q.; Yan, X.H.; Chen, J.; Guo, H.B.; Zhang, S.Z. The HIF-2alpha dependent induction of PAP and adenosine synthesis regulates glioblastoma stem cell function through the A2B adenosine receptor. Int. J. Biochem. Cell Biol. 2014, 49, 8–16. [Google Scholar] [CrossRef]
- Wrensch, M.; Jenkins, R.B.; Chang, J.S.; Yeh, R.F.; Xiao, Y.; Decker, P.A.; Ballman, K.V.; Berger, M.; Buckner, J.C.; Chang, S.; et al. Variants in the CDKN2B and RTEL1 regions are associated with high-grade glioma susceptibility. Nat. Genet. 2009, 41, 905–908. [Google Scholar] [CrossRef]
- Jacobson, K.A.; Nikodijevic, O.; Padgett, W.L.; Gallo-Rodriguez, C.; Maillard, M.; Daly, J.W. 8-(3-Chlorostyryl)caffeine (CSC) is a selective A2-adenosine antagonist in vitro and in vivo. FEBS Lett. 1993, 323, 141–144. [Google Scholar] [CrossRef] [Green Version]
- Borodovsky, A.; Barbon, C.M.; Wang, Y.; Ye, M.; Prickett, L.; Chandra, D.; Shaw, J.; Deng, N.; Sachsenmeier, K.; Clarke, J.D.; et al. Small molecule AZD4635 inhibitor of A2AR signaling rescues immune cell function including CD103(+) dendritic cells enhancing anti-tumor immunity. J. Immunother. Cancer 2020, 8, e000417. [Google Scholar] [CrossRef] [PubMed]
- Harmse, R.; van der Walt, M.M.; Petzer, J.P.; Terre’Blanche, G. Discovery of 1,3-diethyl-7-methyl-8-(phenoxymethyl)-xanthine derivatives as novel adenosine A1 and A2A receptor antagonists. Bioorg. Med. Chem. Lett. 2016, 26, 5951–5955. [Google Scholar] [CrossRef] [PubMed]
- Schwabe, U.; Ukena, D.; Lohse, M.J. Xanthine derivatives as antagonists at A1 and A2 adenosine receptors. Naunyn Schmiedebergs Arch. Pharmacol. 1985, 330, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.F.; Xu, K.; Petzer, J.P.; Staal, R.; Xu, Y.H.; Beilstein, M.; Sonsalla, P.K.; Castagnoli, K.; Castagnoli, N., Jr.; Schwarzschild, M.A. Neuroprotection by caffeine and A(2A) adenosine receptor inactivation in a model of Parkinson’s disease. J. Neurosci. 2001, 21, RC143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szopa, A.; Bogatko, K.; Serefko, A.; Wyska, E.; Wosko, S.; Swiader, K.; Doboszewska, U.; Wlaz, A.; Wrobel, A.; Wlaz, P.; et al. Agomelatine and tianeptine antidepressant activity in mice behavioral despair tests is enhanced by DMPX, a selective adenosine A2A receptor antagonist, but not DPCPX, a selective adenosine A1 receptor antagonist. Pharmacol. Rep. 2019, 71, 676–681. [Google Scholar] [CrossRef] [PubMed]
- Masjedi, A.; Ahmadi, A.; Ghani, S.; Malakotikhah, F.; Nabi Afjadi, M.; Irandoust, M.; Karoon Kiani, F.; Heydarzadeh Asl, S.; Atyabi, F.; Hassannia, H.; et al. Silencing adenosine A2a receptor enhances dendritic cell-based cancer immunotherapy. Nanomedicine 2020, 29, 102240. [Google Scholar] [CrossRef] [PubMed]
- Pollack, A.E.; Fink, J.S. Adenosine antagonists potentiate D2 dopamine-dependent activation of Fos in the striatopallidal pathway. Neuroscience 1995, 68, 721–728. [Google Scholar] [CrossRef]
- Fong, L.; Hotson, A.; Powderly, J.D.; Sznol, M.; Heist, R.S.; Choueiri, T.K.; George, S.; Hughes, B.G.M.; Hellmann, M.D.; Shepard, D.R.; et al. Adenosine 2A Receptor Blockade as an Immunotherapy for Treatment-Refractory Renal Cell Cancer. Cancer Discov. 2020, 10, 40–53. [Google Scholar] [CrossRef] [Green Version]
- Chiappori, A.A.; Creelan, B.; Tanvetyanon, T.; Gray, J.E.; Haura, E.B.; Thapa, R.; Barlow, M.L.; Chen, Z.; Chen, D.T.; Beg, A.A.; et al. Phase I Study of Taminadenant (PBF509/NIR178), an Adenosine 2A Receptor Antagonist, with or without Spartalizumab (PDR001), in Patients with Advanced Non-Small Cell Lung Cancer. Clin. Cancer Res. 2022, 28, 2313–2320. [Google Scholar] [CrossRef]
- Yu, J.; Zhong, Y.; Shen, X.; Cheng, Y.; Qi, J.; Wang, J. In vitro effect of adenosine A2A receptor antagonist SCH 442416 on the expression of glutamine synthetase and glutamate aspartate transporter in rat retinal Muller cells at elevated hydrostatic pressure. Oncol. Rep. 2012, 27, 748–752. [Google Scholar] [CrossRef]
- Poucher, S.M.; Keddie, J.R.; Brooks, R.; Shaw, G.R.; McKillop, D. Pharmacodynamics of ZM 241385, a potent A2a adenosine receptor antagonist, after enteric administration in rat, cat and dog. J. Pharm. Pharmacol. 1996, 48, 601–606. [Google Scholar] [CrossRef] [PubMed]
- Poucher, S.M.; Keddie, J.R.; Singh, P.; Stoggall, S.M.; Caulkett, P.W.; Jones, G.; Coll, M.G. The in vitro pharmacology of ZM 241385, a potent, non-xanthine A2a selective adenosine receptor antagonist. Br. J. Pharmacol. 1995, 115, 1096–1102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Todde, S.; Moresco, R.M.; Simonelli, P.; Baraldi, P.G.; Cacciari, B.; Spalluto, G.; Varani, K.; Monopoli, A.; Matarrese, M.; Carpinelli, A.; et al. Design, radiosynthesis, and biodistribution of a new potent and selective ligand for in vivo imaging of the adenosine A(2A) receptor system using positron emission tomography. J. Med. Chem. 2000, 43, 4359–4362. [Google Scholar] [CrossRef]
- Gillespie, R.J.; Cliffe, I.A.; Dawson, C.E.; Dourish, C.T.; Gaur, S.; Jordan, A.M.; Knight, A.R.; Lerpiniere, J.; Misra, A.; Pratt, R.M.; et al. Antagonists of the human adenosine A2A receptor. Part 3: Design and synthesis of pyrazolo[3,4-d]pyrimidines, pyrrolo[2,3-d]pyrimidines and 6-arylpurines. Bioorg. Med. Chem. Lett. 2008, 18, 2924–2929. [Google Scholar] [CrossRef]
- Willingham, S.B.; Hotson, A.N.; Miller, R.A. Targeting the A2AR in cancer; early lessons from the clinic. Curr. Opin. Pharmacol. 2020, 53, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Gnad, T.; Navarro, G.; Lahesmaa, M.; Reverte-Salisa, L.; Copperi, F.; Cordomi, A.; Naumann, J.; Hochhauser, A.; Haufs-Brusberg, S.; Wenzel, D.; et al. Adenosine/A2B Receptor Signaling Ameliorates the Effects of Aging and Counteracts Obesity. Cell Metab. 2020, 32, 56–70.e57. [Google Scholar] [CrossRef] [PubMed]
- Hinz, S.; Navarro, G.; Borroto-Escuela, D.; Seibt, B.F.; Ammon, Y.C.; de Filippo, E.; Danish, A.; Lacher, S.K.; Cervinkova, B.; Rafehi, M.; et al. Adenosine A2A receptor ligand recognition and signaling is blocked by A2B receptors. Oncotarget 2018, 9, 13593–13611. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Summary of Adenosine Receptor Antagonists | |||||
|---|---|---|---|---|---|
| Adenosine Receptor Antagonists | A1 | A2A | A2B | A3 | References |
| Xanthine derivates | |||||
| Caffeine | + | + | + | + | [132] |
| Theophylline | + | + | + | + | [132] |
| DMPX | + | +++ | ++ | + | [135] |
| Istradefylline | + | +++ | + | + | [130] |
| Taminadenant | + | ++ | + | ++ | [139] |
| Non-Xanthine derivates | |||||
| Ciforadenant | +++ | +++ | ++ | + | [138] |
| Imaradenant | ++ | +++ | + | + | [131] |
| SCH442416 | + | +++ | ++ | + | [140] |
| ZM241385 | + | +++ | ++ | ++ | [141] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bova, V.; Filippone, A.; Casili, G.; Lanza, M.; Campolo, M.; Capra, A.P.; Repici, A.; Crupi, L.; Motta, G.; Colarossi, C.; et al. Adenosine Targeting as a New Strategy to Decrease Glioblastoma Aggressiveness. Cancers 2022, 14, 4032. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14164032
Bova V, Filippone A, Casili G, Lanza M, Campolo M, Capra AP, Repici A, Crupi L, Motta G, Colarossi C, et al. Adenosine Targeting as a New Strategy to Decrease Glioblastoma Aggressiveness. Cancers. 2022; 14(16):4032. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14164032
Chicago/Turabian StyleBova, Valentina, Alessia Filippone, Giovanna Casili, Marika Lanza, Michela Campolo, Anna Paola Capra, Alberto Repici, Lelio Crupi, Gianmarco Motta, Cristina Colarossi, and et al. 2022. "Adenosine Targeting as a New Strategy to Decrease Glioblastoma Aggressiveness" Cancers 14, no. 16: 4032. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14164032