
Temsirolimus Enhances Anti-Cancer Immunity by Inducing Autophagy-Mediated Degradation of the Secretion of Small Extracellular Vesicle PD-L1


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Cell Lines and Cell Culture
2.3. Cell Viability Assay
2.4. Isolation of sEVs
2.5. Transmission Electron Microscopy Analysis
2.6. Nanoparticle Tracking Analysis (NTA)
2.7. Western Blotting
2.8. Immunofluorescence Staining
2.9. CD8+ T Cell-Mediated Cancer Cell Killing Assay
2.10. Enzyme-Linked Immunosorbent Assay (ELISA)
2.11. Flow Cytometric Analysis for Immune Phenotyping
2.12. Animal Studies
2.13. Statistical Analysis
3. Results
3.1. TEM Inhibits sEV PD-L1 Secretion through the Additive Effect of Suppressing sEV Secretion and Cellular PD-L1 Expression
3.2. TEM Suppresses sEV PD-L1 Secretion through the Regulation of Rab Protein Levels and Activation of Autophagy
3.3. TEM Improves CD8+ T Cell-Mediated Anti-Cancer Effects
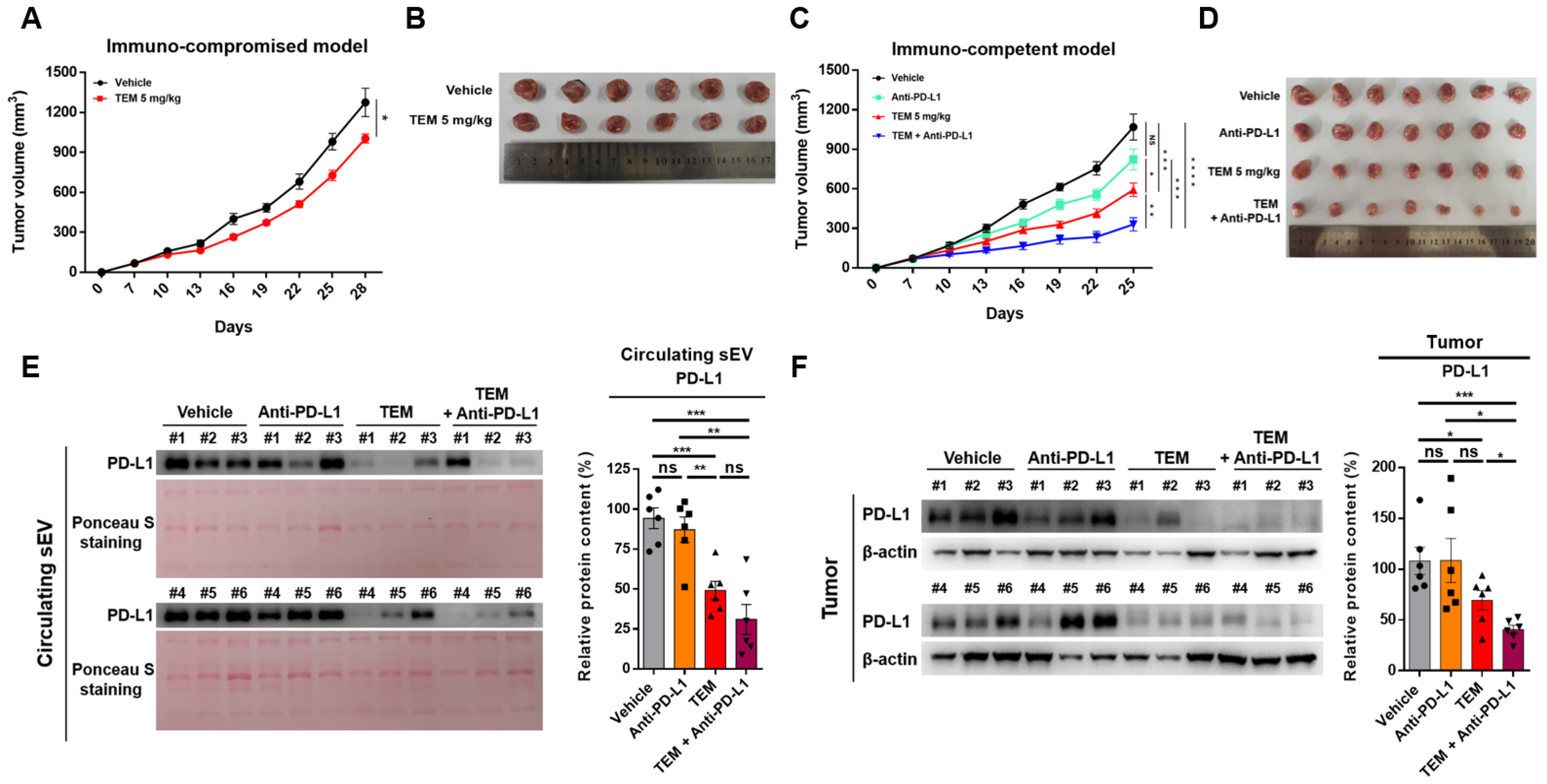
3.4. TEM Enhances the Efficacy of Anti-PD-L1 Therapy
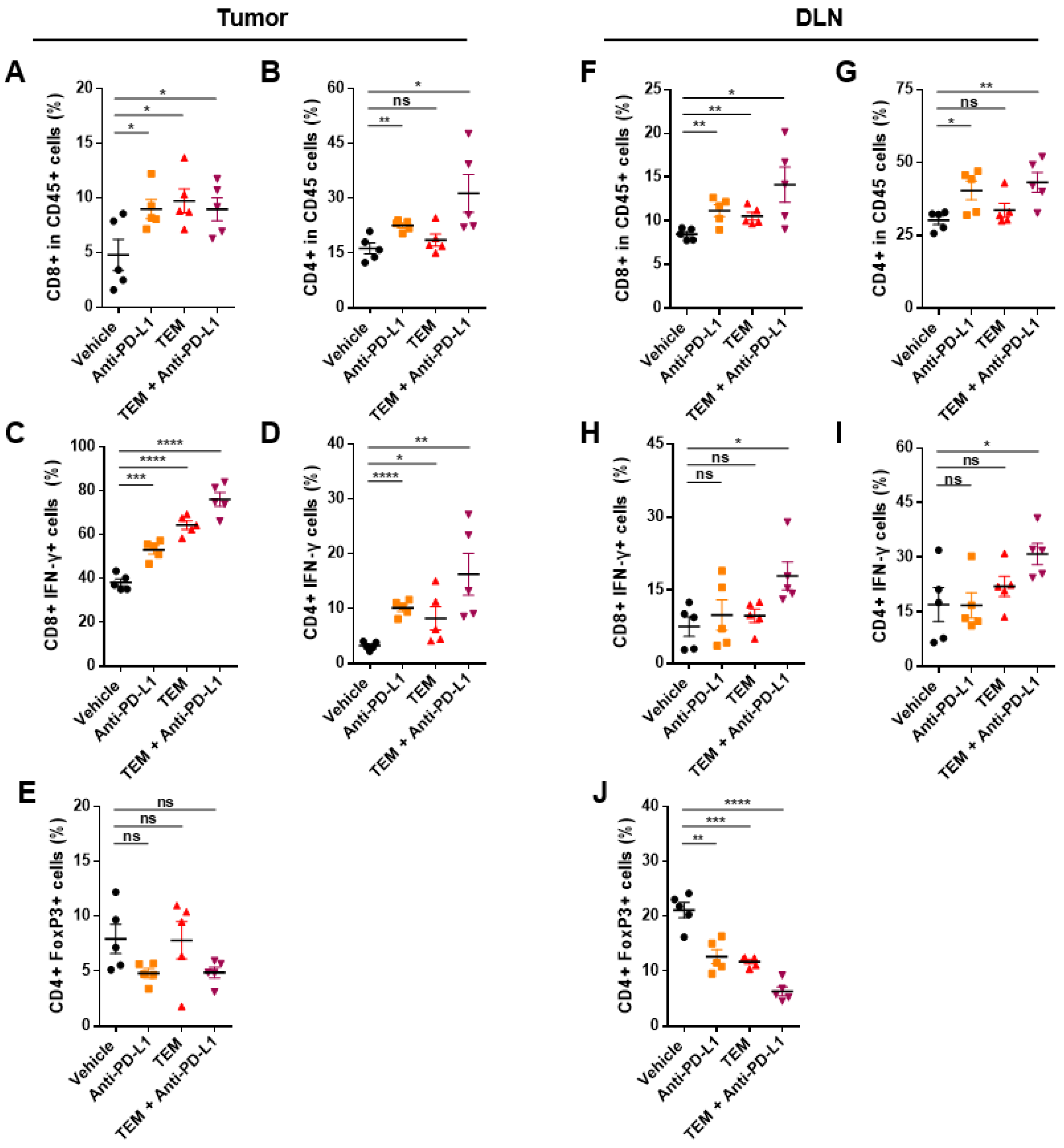
3.5. Combined Administration of TEM and Anti-PD-L1 Boosts Anti-Cancer Immunity
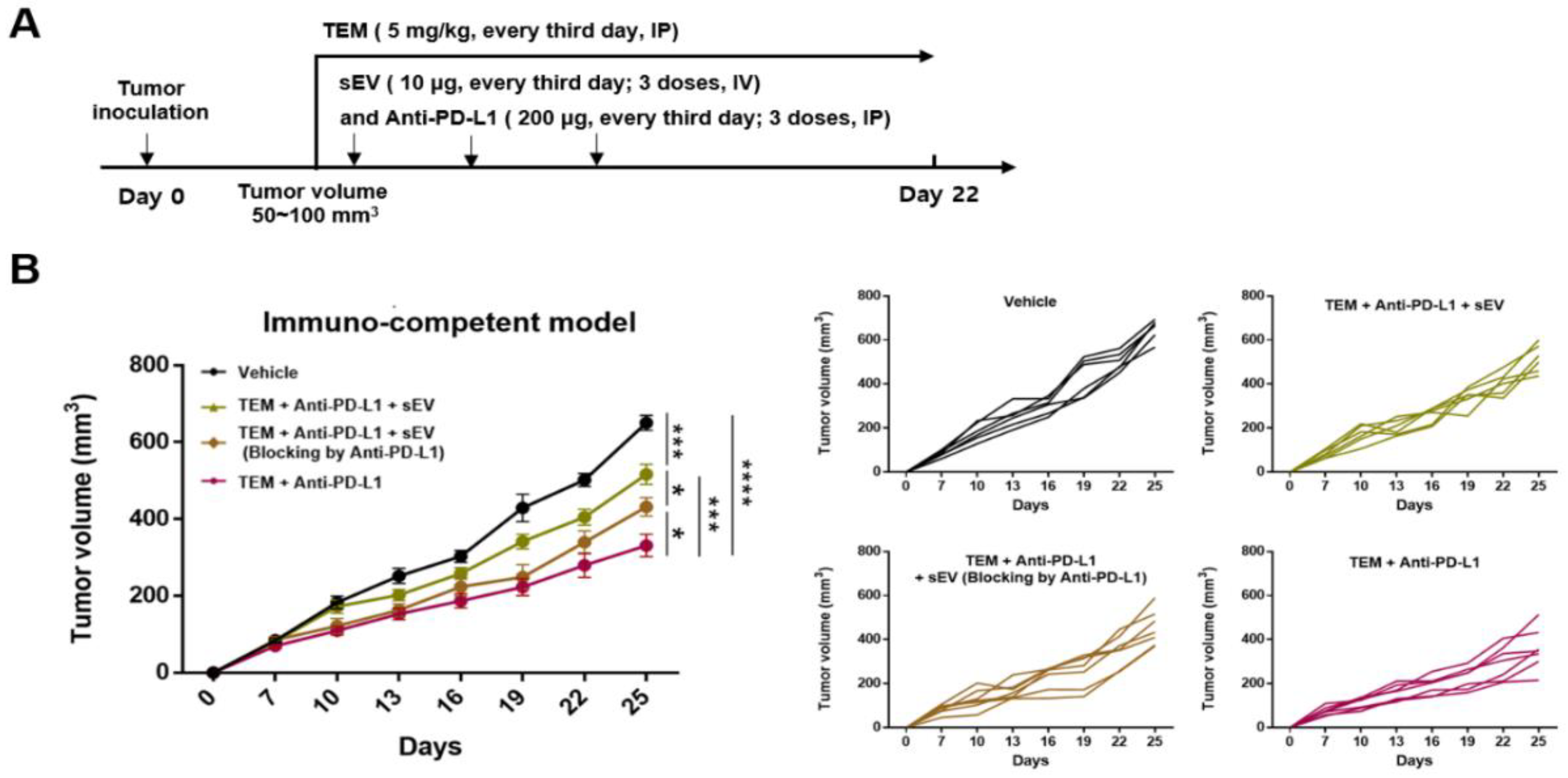
3.6. Combined Administration of TEM and Anti-PD-L1 Antibodies Reverses sEV PD-L1-Mediated Tumor Growth
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jiang, X.; Wang, J.; Deng, X.; Xiong, F.; Ge, J.; Xiang, B.; Wu, X.; Ma, J.; Zhou, M.; Li, X.; et al. Role of the Tumor Microenvironment in PD-L1/PD-1-mediated Tumor Immune Escape. Mol. Cancer 2019, 18, 10. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Mezzadra, R.; Schumacher, T.N. Regulation and Function of the PD-L1 Checkpoint. Immunity 2018, 48, 434–452. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Pan, S.; Chen, X.; Wang, Z.W.; Zhu, X. The Role of lncRNAs and circRNAs in the PD-1/PD-L1 Pathway in Cancer Immunotherapy. Mol. Cancer 2021, 20, 116. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.A.; Drake, V.; Huang, H.S.; Chiu, S.; Zheng, L. Reprogramming the Tumor Microenvironment: Tumor-Induced Immunosuppressive Factors Paralyze T Cells. Oncoimmunology 2015, 4, e1016700. [Google Scholar] [CrossRef]
- Setordzi, P.; Chang, X.; Liu, Z.; Wu, Y.; Zuo, D. The Recent Advances of PD-1 and PD-L1 Checkpoint Signaling Inhibition for Breast Cancer Immunotherapy. Eur. J. Pharmacol. 2021, 895, 173867. [Google Scholar] [CrossRef]
- Sun, J.Y.; Zhang, D.; Wu, S.; Xu, M.; Zhou, X.; Lu, X.J.; Ji, J. Resistance to PD-1/PD-L1 Blockade Cancer Immunotherapy: Mechanisms, Predictive Factors, and Future Perspectives. Biomark. Res. 2020, 8, 35. [Google Scholar] [CrossRef]
- Farkona, S.; Diamandis, E.P.; Blasutig, I.M. Cancer Immunotherapy: The Beginning of the End of Cancer? BMC Med. 2016, 14, 73. [Google Scholar] [CrossRef]
- Tang, H.; Qiao, J.; Fu, Y.X. Immunotherapy and Tumor Microenvironment. Cancer Lett. 2016, 370, 85–90. [Google Scholar] [CrossRef]
- Xing, C.; Li, H.; Li, R.J.; Yin, L.; Zhang, H.F.; Huang, Z.N.; Cheng, Z.; Li, J.; Wang, Z.H.; Peng, H.L. The Roles of Exosomal Immune Checkpoint Proteins in Tumors. Mil. Med. Res. 2021, 8, 56. [Google Scholar] [CrossRef]
- Kalluri, R.; LeBleu, V.S. The Biology, Function, and Biomedical Applications of Exosomes. Science 2020, 367, eaau6977. [Google Scholar] [CrossRef]
- McAndrews, K.M.; Kalluri, R. Mechanisms Associated with Biogenesis of Exosomes in Cancer. Mol. Cancer 2019, 18, 52. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Ji, X.; Liu, J.; Fan, D.; Zhou, Q.; Chen, C.; Wang, W.; Wang, G.; Wang, H.; Yuan, W.; et al. Effects of Exosomes on Pre-Metastatic Niche Formation in Tumors. Mol. Cancer 2019, 18, 39. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Huang, A.C.; Zhang, W.; Zhang, G.; Wu, M.; Xu, W.; Yu, Z.; Yang, J.; Wang, B.; Sun, H.; et al. Exosomal PD-L1 Contributes to Immunosuppression and Is Associated with Anti-PD-1 Response. Nature 2018, 560, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Poggio, M.; Hu, T.; Pai, C.C.; Chu, B.; Belair, C.D.; Chang, A.; Montabana, E.; Lang, U.E.; Fu, Q.; Fong, L.; et al. Suppression of Exosomal PD-L1 Induces Systemic Anti-Tumor Immunity and Memory. Cell 2019, 177, 414–427.e13. [Google Scholar] [CrossRef] [PubMed]
- Ricklefs, F.L.; Alayo, Q.; Krenzlin, H.; Mahmoud, A.B.; Speranza, M.C.; Nakashima, H.; Hayes, J.L.; Lee, K.; Balaj, L.; Passaro, C.; et al. Immune Evasion Mediated by PD-L1 on Glioblastoma-Derived Extracellular Vesicles. Sci. Adv. 2018, 4, eaar2766. [Google Scholar] [CrossRef]
- Im, E.J.; Lee, C.H.; Moon, P.G.; Rangaswamy, G.G.; Lee, B.; Lee, J.M.; Lee, J.C.; Jee, J.G.; Bae, J.S.; Kwon, T.K.; et al. Sulfisoxazole Inhibits the Secretion of Small Extracellular Vesicles by Targeting the Endothelin Receptor A. Nat. Commun. 2019, 10, 1387. [Google Scholar] [CrossRef]
- Shin, J.M.; Lee, C.H.; Son, S.; Kim, C.H.; Lee, J.A.; Ko, H.; Shin, S.; Song, S.H.; Park, S.S.; Bae, J.H.; et al. Sulfisoxazole Elicits Robust Antitumour Immune Response Along with Immune Checkpoint Therapy by Inhibiting Exosomal PD-L1. Adv. Sci. 2022, 9, e2103245. [Google Scholar] [CrossRef]
- Lee, C.H.; Bae, J.H.; Choe, E.J.; Park, J.M.; Park, S.S.; Cho, H.J.; Song, B.J.; Baek, M.C. Macitentan Improves Antitumor Immune Responses by Inhibiting the Secretion of Tumor-Derived Extracellular Vesicle PD-L1. Theranostics 2022, 12, 1971–1987. [Google Scholar] [CrossRef]
- Abdulrahman, B.A.; Abdelaziz, D.H.; Schatzl, H.M. Autophagy Regulates Exosomal Release of Prions in Neuronal Cells. J. Biol. Chem. 2018, 293, 8956–8968. [Google Scholar] [CrossRef]
- Auger, C.; Christou, N.; Brunel, A.; Perraud, A.; Verdier, M. Autophagy and Extracellular Vesicles in Colorectal Cancer: Interactions and Common Actors? Cancers 2021, 13, 1039. [Google Scholar] [CrossRef]
- Xu, J.; Camfield, R.; Gorski, S.M. The Interplay Between Exosomes and Autophagy—Partners in Crime. J. Cell Sci. 2018, 131, jcs215210. [Google Scholar] [CrossRef] [PubMed]
- Lastwika, K.J.; Wilson, W., 3rd; Li, Q.K.; Norris, J.; Xu, H.; Ghazarian, S.R.; Kitagawa, H.; Kawabata, S.; Taube, J.M.; Yao, S.; et al. Control of PD-L1 Expression by Oncogenic Activation of the AKT-mTOR Pathway in Non-Small Cell Lung Cancer. Cancer Res. 2016, 76, 227–238. [Google Scholar] [CrossRef]
- Wang, X.; Wu, W.K.K.; Gao, J.; Li, Z.; Dong, B.; Lin, X.; Li, Y.; Li, Y.; Gong, J.; Qi, C.; et al. Autophagy Inhibition Enhances PD-L1 Expression in Gastric Cancer. J. Exp. Clin. Cancer Res. 2019, 38, 140. [Google Scholar] [CrossRef] [PubMed]
- Moore, E.C.; Cash, H.A.; Caruso, A.M.; Uppaluri, R.; Hodge, J.W.; Van Waes, C.; Allen, C.T. Enhanced Tumor Control with Combination mTOR and PD-L1 Inhibition in Syngeneic Oral Cavity Cancers. Cancer Immunol. Res. 2016, 4, 611–620. [Google Scholar] [CrossRef] [PubMed]
- Shelke, G.V.; Lässer, C.; Gho, Y.S.; Lötvall, J. Importance of Exosome Depletion Protocols to Eliminate Functional and RNA-Containing Extracellular Vesicles from Fetal Bovine Serum. J. Extracell. Vesicles. 2014, 3, 24783. [Google Scholar] [CrossRef]
- Dong, M.; Wu, S.; Xu, H.; Yu, X.; Wang, L.; Bai, H.; Niu, W. FBS-Derived Exosomes as a Natural Nano-Scale Carrier for Icariin Promote Osteoblast Proliferation. Front. Bioeng. Biotechnol. 2021, 9, 615920. [Google Scholar] [CrossRef]
- Moser, B.; Hochreiter, B.; Herbst, R.; Schmid, J.A. Fluorescence Colocalization Microscopy Analysis Can Be Improved by Combining Object-Recognition with Pixel-Intensity-Correlation. Biotechnol. J. 2017, 12, 1600332. [Google Scholar] [CrossRef]
- Li, C.; Yao, H.; Wang, H.; Fang, J.Y.; Xu, J. Repurposing Screen Identifies Amlodipine as an Inducer of PD-L1 Degradation and Antitumor Immunity. Oncogene 2021, 40, 1128–1146. [Google Scholar] [CrossRef]
- Liu, W.J.; Ye, L.; Huang, W.F.; Guo, L.J.; Xu, Z.G.; Wu, H.L.; Yang, C.; Liu, H.F. p62 Links the Autophagy Pathway and the ubiqutin-proteasome System upon Ubiquitinated Protein Degradation. Cell. Mol. Biol. Lett. 2016, 21, 29. [Google Scholar] [CrossRef]
- Huang, X.; Qi, Q.; Hua, X.; Li, X.; Zhang, W.; Sun, H.; Li, S.; Wang, X.; Li, B. Beclin 1, an Autophagy-Related Gene, Augments Apoptosis in U87 Glioblastoma Cells. Oncol. Rep. 2014, 31, 1761–1767. [Google Scholar] [CrossRef]
- Mohsen, M.O.; Speiser, D.E.; Michaux, J.; Pak, H.; Stevenson, B.J.; Vogel, M.; Inchakalody, V.P.; de Brot, S.; Dermime, S.; Coukos, G.; et al. Bedside Formulation of a Personalized Multi-Neoantigen Vaccine Against Mammary Carcinoma. J. Immunother. Cancer 2022, 10, e002927. [Google Scholar] [CrossRef] [PubMed]
- Ito, D.; Fujimoto, K.; Mori, T.; Kami, K.; Koizumi, M.; Toyoda, E.; Kawaguchi, Y. In Vivo Antitumor Effect of the mTOR Inhibitor CCI-779 and Gemcitabine in Xenograft Models of Human Pancreatic Cancer. Int. J. Cancer 2006, 118, 2337–2343. [Google Scholar] [CrossRef] [PubMed]
- Geoerger, B.; Kerr, K.; Tang, C.B.; Fung, K.M.; Powell, B.; Sutton, L.N.; Phillips, P.C.; Janss, A.J. Antitumor Activity of the Rapamycin Analog CCI-779 in Human Primitive Neuroectodermal Tumor/Medulloblastoma Models as Single Agent and in Combination Chemotherapy. Cancer Res. 2001, 61, 1527–1532. [Google Scholar] [PubMed]
- Tian, T.; Li, X.; Zhang, J. MTOR Signaling in Cancer and mTOR Inhibitors in Solid Tumor Targeting Therapy. Int. J. Mol. Sci. 2019, 20, 755. [Google Scholar] [CrossRef] [PubMed]
- Hua, H.; Kong, Q.; Zhang, H.; Wang, J.; Luo, T.; Jiang, Y. Targeting mTOR for Cancer Therapy. J. Hematol. Oncol. 2019, 12, 71. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Yu, M.; Ma, T.; Zhang, C.; Huang, S.; Karimzadeh, M.R.; Momtazi-Borojeni, A.A.; Chen, S. Mechanisms Underlying Low-Clinical Responses to PD-1/PD-L1 Blocking Antibodies in Immunotherapy of Cancer: A Key Role of Exosomal PD-L1. J. Immunother. Cancer 2021, 9, e001698. [Google Scholar] [CrossRef] [PubMed]
- Ayala-Mar, S.; Donoso-Quezada, J.; González-Valdez, J. Clinical Implications of Exosomal PD-L1 in Cancer Immunotherapy. J. Immunol. Res. 2021, 2021, 8839978. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Li, C.W.; Chan, L.C.; Wei, Y.; Hsu, J.M.; Xia, W.; Cha, J.H.; Hou, J.; Hsu, J.L.; Sun, L.; et al. Exosomal PD-L1 Harbors Active Defense Function to Suppress T Cell Killing of Breast Cancer Cells and Promote Tumor Growth. Cell Res. 2018, 28, 862–864. [Google Scholar] [CrossRef]
- Kim, D.H.; Kim, H.; Choi, Y.J.; Kim, S.Y.; Lee, J.E.; Sung, K.J.; Sung, Y.H.; Pack, C.G.; Jung, M.K.; Han, B.; et al. Exosomal PD-L1 Promotes Tumor Growth through Immune Escape in Non-Small Cell Lung Cancer. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef]
- Greenberg, J.W.; Kim, H.; Ahn, M.; Moustafa, A.A.; Zhou, H.; Barata, P.C.; Boulares, A.H.; Abdel-Mageed, A.B.; Krane, L.S. Combination of Tipifarnib and Sunitinib Overcomes Renal Cell Carcinoma Resistance to Tyrosine Kinase Inhibitors via Tumor-Derived Exosome and T Cell Modulation. Cancers 2022, 14, 903. [Google Scholar] [CrossRef]
- Hao, M.; Yeo, S.K.; Turner, K.; Harold, A.; Yang, Y.; Zhang, X.; Guan, J.L. Autophagy Blockade Limits HER2+ Breast Cancer Tumorigenesis by Perturbing HER2 Trafficking and Promoting Release via Small Extracellular Vesicles. Dev. Cell 2021, 56, 341–355.e5. [Google Scholar] [CrossRef] [PubMed]
- Zou, W.; Lai, M.; Zhang, Y.; Zheng, L.; Xing, Z.; Li, T.; Zou, Z.; Song, Q.; Zhao, X.; Xia, L.; et al. Exosome Release Is Regulated by mTORC1. Adv. Sci. 2018, 6, 1801313. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.C.; Lin, Y.C.; Liao, Y.H.; Liou, J.P.; Chen, C.H. MPT0G612, a Novel HDAC6 Inhibitor, Induces Apoptosis and Suppresses IFN-Gamma-Induced Programmed Death-Ligand 1 in Human Colorectal Carcinoma Cells. Cancers 2019, 11, 1617. [Google Scholar] [CrossRef] [PubMed]
- Langdon, S.; Hughes, A.; Taylor, M.A.; Kuczynski, E.A.; Mele, D.A.; Delpuech, O.; Jarvis, L.; Staniszewska, A.; Cosulich, S.; Carnevalli, L.S.; et al. Combination of Dual mTORC1/2 Inhibition and Immune-Checkpoint Blockade Potentiates Anti-Tumour Immunity. Oncoimmunology 2018, 7, e1458810. [Google Scholar] [CrossRef]
- Chapman, N.M.; Chi, H. MTOR Signaling, Tregs and Immune Modulation. Immunotherapy 2014, 6, 1295–1311. [Google Scholar] [CrossRef] [PubMed]
- Chi, H. Regulation and Function of MTOR Signaling in T Cell Fate Decisions. Nat. Rev. Immunol. 2012, 12, 325–338. [Google Scholar] [CrossRef]
- Chapman, N.M.; Zeng, H.; Nguyen, T.M.; Wang, Y.; Vogel, P.; Dhungana, Y.; Liu, X.; Neale, G.; Locasale, J.W.; Chi, H. mTOR Coordinates Transcriptional Programs and Mitochondrial Metabolism of Activated Treg Subsets to Protect Tissue Homeostasis. Nat. Commun. 2018, 9, 2095. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Yamada, D.; Kawai, T.; Sato, Y.; Teshima, T.; Yamada, Y.; Nakamura, M.; Suzuki, M.; Matsumoto, A.; Nakagawa, T.; et al. Different Immunological Effects of the Molecular Targeted Agents Sunitinib, Everolimus and Temsirolimus in Patients with Renal Cell Carcinoma. Int. J. Oncol. 2020, 56, 999–1013. [Google Scholar] [CrossRef]
- Rong, L.; Li, R.; Li, S.; Luo, R. Immunosuppression of Breast Cancer Cells Mediated by Transforming Growth Factor-Beta in Exosomes from Cancer Cells. Oncol. Lett. 2016, 11, 500–504. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, P.; Li, J.; Kulkarni, A.B.; Perruche, S.; Chen, W. A Critical Function for TGF-Beta Signaling in the Development of Natural CD4+CD25+Foxp3+ Regulatory T Cells. Nat. Immunol. 2008, 9, 632–640. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, S.-S.; Kim, J.-I.; Lee, C.-H.; Bae, J.-H.; Park, J.-M.; Choe, E.-J.; Baek, M.-C. Temsirolimus Enhances Anti-Cancer Immunity by Inducing Autophagy-Mediated Degradation of the Secretion of Small Extracellular Vesicle PD-L1. Cancers 2022, 14, 4081. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14174081
Park S-S, Kim J-I, Lee C-H, Bae J-H, Park J-M, Choe E-J, Baek M-C. Temsirolimus Enhances Anti-Cancer Immunity by Inducing Autophagy-Mediated Degradation of the Secretion of Small Extracellular Vesicle PD-L1. Cancers. 2022; 14(17):4081. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14174081
Chicago/Turabian StylePark, Seong-Sik, Jong-In Kim, Chan-Hyeong Lee, Ju-Hyun Bae, Ju-Mi Park, Eun-Ji Choe, and Moon-Chang Baek. 2022. "Temsirolimus Enhances Anti-Cancer Immunity by Inducing Autophagy-Mediated Degradation of the Secretion of Small Extracellular Vesicle PD-L1" Cancers 14, no. 17: 4081. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14174081