
ER Stress Response and Induction of Apoptosis in Malignant Pleural Mesothelioma: The Achilles Heel Targeted by the Anticancer Ruthenium Drug BOLD-100


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. GRP78 Inhibitor
2.2. Cell Culture and Reagents
2.3. Cytotoxicity Assay
2.4. Colony-Forming Assay
2.5. Activity of Caspase 3 and Caspase 8
2.6. Calpain Activity
2.7. Mitochondrial Permeability Transition Pore Assay
2.8. JC-1 Fluorescence/JC-1 Mitochondrial Membrane Potential Assay
2.9. Measurements of free Cytosolic Ca2+ Concentration ([Ca2+]i)
2.10. Measurements of Intracellular Reactive Oxygen Species (ROS)
2.11. Quantitative Reverse Transcriptase PCR (qRT-PCR)
2.12. Western Immunoblotting
2.13. Cell Surface Enzyme-Linked Immunosorbent Assay (ELISA)
2.14. Statistical Analysis
3. Results
3.1. Cytotoxicity Assay
3.2. Activation of Apoptosis
3.3. ROS Alteration after BOLD-100 Treatment
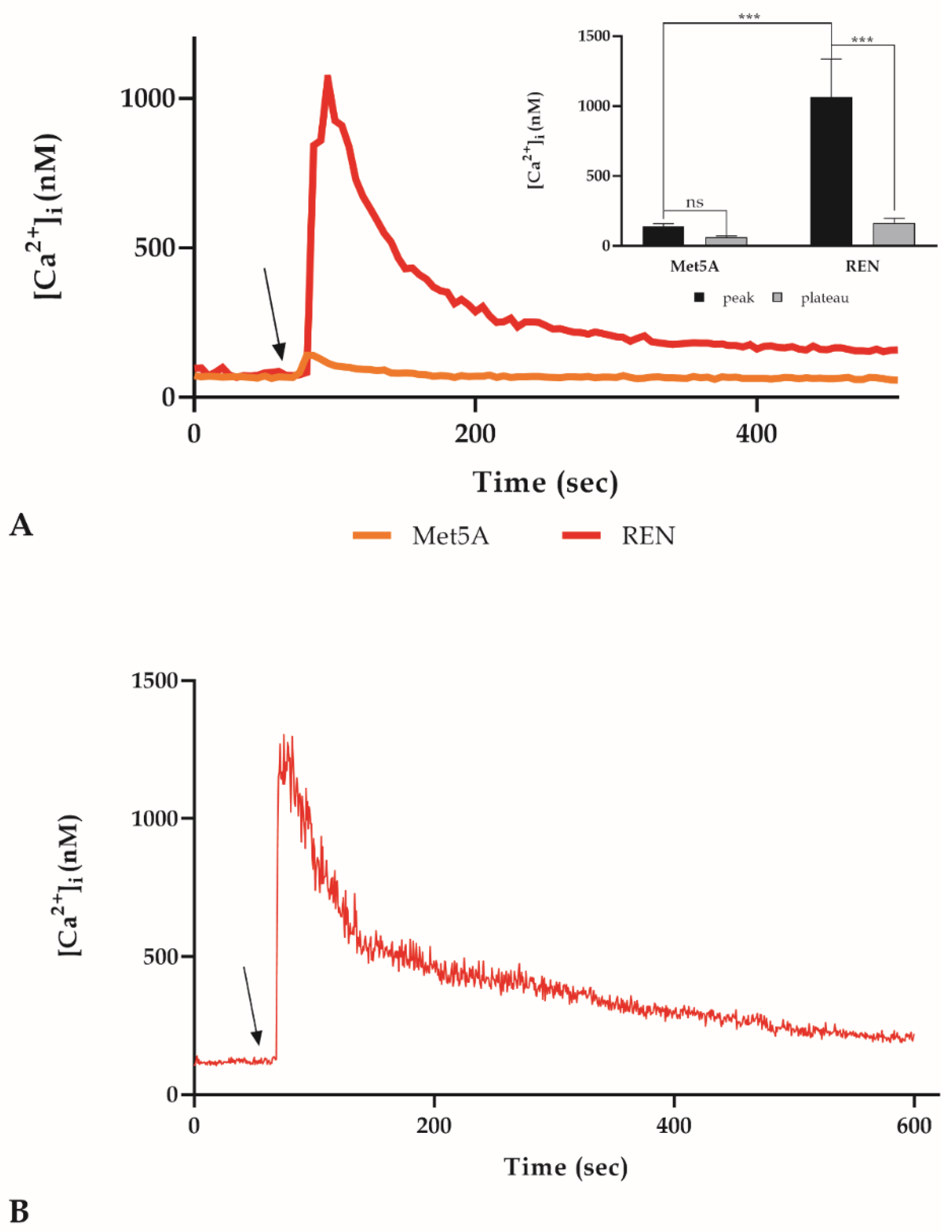
3.4. Intracellular Ca2+ Concentration ([Ca2+]i) Variations
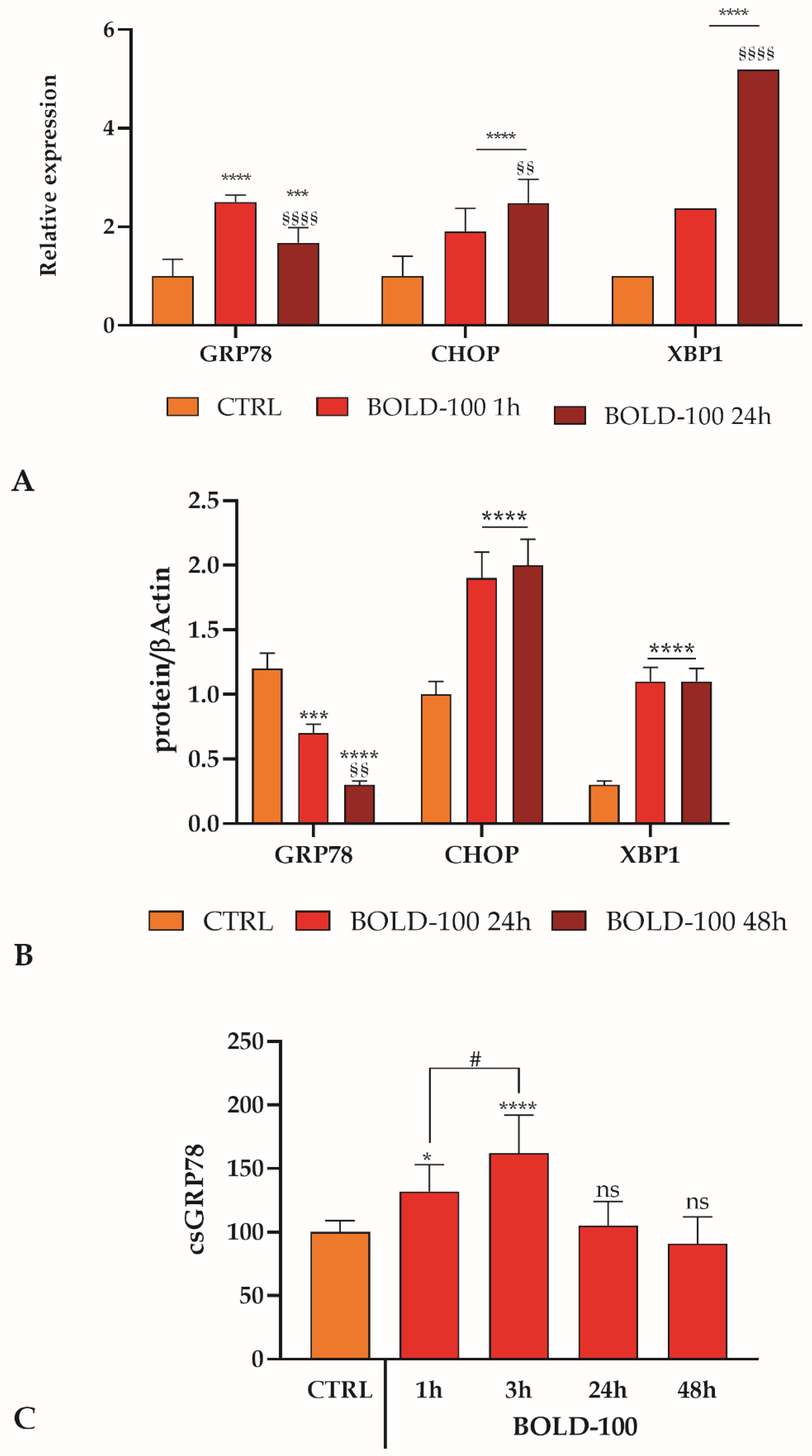
3.5. GRP78 Variation and ER Stress Response
4. Discussion
5. Conclusions
- Treatment with BOLD-100 determines a disruption of ER homeostasis:
- o
- BOLD-100 causes an increase of ROS as a consequence of the induction of ERO1α overexpression;
- o
- The rise in ROS and ERO1α levels destabilize ER calcium regulation, leading to a massive release into the cytosol either through TLC and IP3R leakage, but also a Ca2+ overload into the mitochondria by way of MAM.
- Following ER homeostasis loss, UPR activation starts:
- o
- GRP78 firstly increases and then decreases in a time-dependent manner;
- o
- GRP78 translocates to the plasma membrane in a time-dependent manner immediately after the treatment;
- o
- XBP1 and CHOP increase in a time-dependent manner.
- All these pathways converge in the induction of apoptosis in MPM cells through different ways:
- o
- The activation of caspase3/7 and caspase 8;
- o
- The destabilization of mitochondrial membrane potential;
- o
- The activation of calpain.
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Yang, H.; Testa, J.R.; Carbone, M. Mesothelioma epidemiology, carcinogenesis, and pathogenesis. Curr. Treat. Options Oncol. 2008, 9, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Fazzo, L.; Minelli, G.; Bruno, C.; Comba, P.; Conti, S.; De Santis, M.; Zona, A.; Binazzi, A.; Magnani, C.; Marinaccio, A.; et al. Early mortality from malignant mesothelioma in Italy as a proxy of environmental exposure to asbestos in children. Ann. Ist. Super. Sanita 2020, 56, 478–486. [Google Scholar] [CrossRef] [PubMed]
- Cavone, D.; Caputi, A.; De Maria, L.; Cannone, E.S.S.; Mansi, F.; Birtolo, F.; Delfino, M.C.; Vimercati, L. Epidemiology of Mesothelioma. Environments 2019, 6, 76. [Google Scholar] [CrossRef]
- Raja, S.; Murthy, S.C.; Mason, D.P. Malignant pleural mesothelioma. Curr. Oncol. Rep. 2011, 13, 259–264. [Google Scholar] [CrossRef]
- Chirieac, L.R.; Corson, J.M. Pathologic evaluation of malignant pleural mesothelioma. Semin. Thorac. Cardiovasc. Surg. 2009, 21, 121–124. [Google Scholar] [CrossRef]
- Vogelzang, N.J.; Rusthoven, J.J.; Symanowski, J.; Denham, C.; Kaukel, E.; Ruffie, P.; Gatzemeier, U.; Boyer, M.; Emri, S.; Manegold, C.; et al. Phase III study of pemetrexed in combination with cisplatin versus cisplatin alone in patients with malignant pleural mesothelioma. J. Clin. Oncol. 2003, 21, 2636–2644. [Google Scholar] [CrossRef]
- Zucali, P.A. Target therapy: New drugs or new combinations of drugs in malignant pleural mesothelioma. J. Thorac. Dis. 2018, 10, S311–S321. [Google Scholar] [CrossRef]
- Baas, P.; Scherpereel, A.; Nowak, A.K.; Fujimoto, N.; Peters, S.; Tsao, A.S.; Mansfield, A.S.; Popat, S.; Jahan, T.; Antonia, S.; et al. First-line nivolumab plus ipilimumab in unresectable malignant pleural mesothelioma (CheckMate 743): A multicentre, randomised, open-label, phase 3 trial. Lancet 2021, 397, 375–386. [Google Scholar] [CrossRef]
- Lee, A.S. Glucose-regulated proteins in cancer: Molecular mechanisms and therapeutic potential. Nat. Rev. Cancer 2014, 14, 263–276. [Google Scholar] [CrossRef]
- Dong, D.; Ni, M.; Li, J.; Xiong, S.; Ye, W.; Virrey, J.J.; Mao, C.; Ye, R.; Wang, M.; Pen, L.; et al. Critical role of the stress chaperone GRP78/BiP in tumor proliferation, survival, and tumor angiogenesis in transgene-induced mammary tumor development. Cancer Res. 2008, 68, 498–505. [Google Scholar] [CrossRef] [Green Version]
- Wey, S.; Luo, B.; Tseng, C.C.; Ni, M.; Zhou, H.; Fu, Y.; Bhojwani, D.; Carroll, W.L.; Lee, A.S. Inducible knockout of GRP78/BiP in the hematopoietic system suppresses Pten-null leukemogenesis and AKT oncogenic signaling. Blood 2012, 119, 817–825. [Google Scholar] [CrossRef] [PubMed]
- Bakewell, S.J.; Rangel, D.F.; Ha, D.P.; Sethuraman, J.; Crouse, R.; Hadley, E.; Costich, T.L.; Zhou, X.; Nichols, P.; Lee, A.S. Suppression of stress induction of the 78-kilodalton glucose regulated protein (GRP78) in cancer by IT-139, an anti-tumor ruthenium small molecule inhibitor. Oncotarget 2018, 9, 29698–29714. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.S. GRP78 induction in cancer: Therapeutic and prognostic implications. Cancer Res. 2007, 67, 3496–3499. [Google Scholar] [CrossRef] [PubMed]
- Li, Z. Glucose regulated protein 78: A critical link between tumor microenvironment and cancer hallmarks. Biochim. Biophys. Acta 2012, 1826, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Ni, M.; Zhang, Y.; Lee, A.S. Beyond the endoplasmic reticulum: Atypical GRP78 in cell viability, signalling and therapeutic targeting. Biochem. J. 2011, 434, 181–188. [Google Scholar] [CrossRef]
- Wang, M.; Wey, S.; Zhang, Y.; Ye, R.; Lee, A.S. Role of the unfolded protein response regulator GRP78/BiP in development, cancer, and neurological disorders. Antioxid. Redox Signal. 2009, 11, 2307–2316. [Google Scholar] [CrossRef]
- Luo, B.; Lee, A.S. The critical roles of endoplasmic reticulum chaperones and unfolded protein response in tumorigenesis and anticancer therapies. Oncogene 2013, 32, 805–818. [Google Scholar] [CrossRef]
- Dalton, L.E.; Clarke, H.J.; Knight, J.; Lawson, M.H.; Wason, J.; Lomas, D.A.; Howat, W.J.; Rintoul, R.C.; Rassl, D.M.; Marciniak, S.J. The endoplasmic reticulum stress marker CHOP predicts survival in malignant mesothelioma. Br. J. Cancer 2013, 108, 1340–1347. [Google Scholar] [CrossRef]
- Martinotti, S.; Ranzato, E.; Burlando, B. (−)-Epigallocatechin-3-gallate induces GRP78 accumulation in the ER and shifts mesothelioma constitutive UPR into proapoptotic ER stress. J. Cell. Physiol. 2018, 233, 7082–7090. [Google Scholar] [CrossRef]
- Burris, H.A.; Bakewell, S.; Bendell, J.C.; Infante, J.; Jones, S.F.; Spigel, D.R.; Weiss, G.J.; Ramanathan, R.K.; Ogden, A.; Von Hoff, D. Safety and activity of IT-139, a ruthenium-based compound, in patients with advanced solid tumours: A first-in-human, open-label, dose-escalation phase I study with expansion cohort. ESMO Open 2016, 1, e000154. [Google Scholar] [CrossRef] [Green Version]
- Schoenhacker-Alte, B.; Mohr, T.; Pirker, C.; Kryeziu, K.; Kuhn, P.S.; Buck, A.; Hofmann, T.; Gerner, C.; Hermann, G.; Koellensperger, G.; et al. Sensitivity towards the GRP78 inhibitor KP1339/IT-139 is characterized by apoptosis induction via caspase 8 upon disruption of ER homeostasis. Cancer Lett. 2017, 404, 79–88. [Google Scholar] [CrossRef]
- Smythe, W.R.; Hwang, H.C.; Amin, K.M.; Eck, S.L.; Davidson, B.L.; Wilson, J.M.; Kaiser, L.R.; Albelda, S.M. Use of recombinant adenovirus to transfer the herpes simplex virus thymidine kinase (HSVtk) gene to thoracic neoplasms: An effective in vitro drug sensitization system. Cancer Res. 1994, 54, 2055–2059. [Google Scholar] [PubMed]
- Ranzato, E.; Grosso, S.; Patrone, M.; Betta, P.G.; Viarengo, A.; Biffo, S. Spreading of mesothelioma cells is rapamycin-sensitive and requires continuing translation. J. Cell. Biochem. 2009, 108, 867–876. [Google Scholar] [CrossRef]
- Manfredi, M.; Martinotti, S.; Gosetti, F.; Ranzato, E.; Marengo, E. The secretome signature of malignant mesothelioma cell lines. J. Proteom. 2016, 145, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Ranzato, E.; Bonsignore, G.; Patrone, M.; Martinotti, S. Endothelial and Vascular Health: A Tale of Honey, H. Cells 2021, 10, 1071. [Google Scholar] [CrossRef] [PubMed]
- Volta, V.; Ranzato, E.; Martinotti, S.; Gallo, S.; Russo, M.V.; Mutti, L.; Biffo, S.; Burlando, B. Preclinical demonstration of synergistic Active Nutrients/Drug (AND) combination as a potential treatment for malignant pleural mesothelioma. PLoS ONE 2013, 8, e58051. [Google Scholar] [CrossRef]
- Franken, N.A.; Rodermond, H.M.; Stap, J.; Haveman, J.; van Bree, C. Clonogenic assay of cells in vitro. Nat. Protoc. 2006, 1, 2315–2319. [Google Scholar] [CrossRef]
- Belhadj, S.; Hermann, N.S.; Zhu, Y.; Christensen, G.; Strasser, T.; Paquet-Durand, F. Visualizing Cell Death in Live Retina: Using Calpain Activity Detection as a Biomarker for Retinal Degeneration. Int. J. Mol. Sci. 2022, 23, 3892. [Google Scholar] [CrossRef]
- Sivandzade, F.; Bhalerao, A.; Cucullo, L. Analysis of the Mitochondrial Membrane Potential Using the Cationic JC-1 Dye as a Sensitive Fluorescent Probe. Bio-Protocol 2019, 9, e3128. [Google Scholar] [CrossRef]
- Grynkiewicz, G.; Poenie, M.; Tsien, R.Y. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 1985, 260, 3440–3450. [Google Scholar] [CrossRef]
- Martinotti, S.; Patrone, M.; Balbo, V.; Mazzucco, L.; Ranzato, E. Endothelial response boosted by platelet lysate: The involvement of calcium toolkit. Int. J. Mol. Sci. 2020, 21, 808. [Google Scholar] [CrossRef] [PubMed]
- Vidimar, V.; Meng, X.; Klajner, M.; Licona, C.; Fetzer, L.; Harlepp, S.; Hébraud, P.; Sidhoum, M.; Sirlin, C.; Loeffler, J.P.; et al. Induction of caspase 8 and reactive oxygen species by ruthenium-derived anticancer compounds with improved water solubility and cytotoxicity. Biochem. Pharmacol. 2012, 84, 1428–1436. [Google Scholar] [CrossRef]
- Momeni, H.R. Role of calpain in apoptosis. Cell J. 2011, 13, 65–72. [Google Scholar] [PubMed]
- Hartinger, C.G.; Zorbas-Seifried, S.; Jakupec, M.A.; Kynast, B.; Zorbas, H.; Keppler, B.K. From bench to bedside--preclinical and early clinical development of the anticancer agent indazolium trans-[tetrachlorobis(1H-indazole)ruthenate(III)] (KP1019 or FFC14A). J. Inorg. Biochem. 2006, 100, 891–904. [Google Scholar] [CrossRef] [PubMed]
- Chakravarthi, S.; Bulleid, N.J. Glutathione is required to regulate the formation of native disulfide bonds within proteins entering the secretory pathway. J. Biol. Chem. 2004, 279, 39872–39879. [Google Scholar] [CrossRef]
- Ranzato, E.; Biffo, S.; Burlando, B. Selective ascorbate toxicity in malignant mesothelioma: A redox Trojan mechanism. Am. J. Respir. Cell Mol. Biol. 2011, 44, 108–117. [Google Scholar] [CrossRef]
- Kania, E.; Pająk, B.; Orzechowski, A. Calcium homeostasis and ER stress in control of autophagy in cancer cells. BioMed Res. Int. 2015, 2015, 352794. [Google Scholar] [CrossRef]
- Tagliarino, C.; Pink, J.J.; Reinicke, K.E.; Simmers, S.M.; Wuerzberger-Davis, S.M.; Boothman, D.A. Mu-calpain activation in beta-lapachone-mediated apoptosis. Cancer Biol. Ther. 2003, 2, 141–152. [Google Scholar] [CrossRef]
- Eletto, D.; Chevet, E.; Argon, Y.; Appenzeller-Herzog, C. Redox controls UPR to control redox. J. Cell Sci. 2014, 127, 3649–3658. [Google Scholar] [CrossRef]
- Kiviluoto, S.; Vervliet, T.; Ivanova, H.; Decuypere, J.P.; De Smedt, H.; Missiaen, L.; Bultynck, G.; Parys, J.B. Regulation of inositol 1,4,5-trisphosphate receptors during endoplasmic reticulum stress. Biochim. Biophys. Acta 2013, 1833, 1612–1624. [Google Scholar] [CrossRef] [Green Version]
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Hammadi, M.; Oulidi, A.; Gackière, F.; Katsogiannou, M.; Slomianny, C.; Roudbaraki, M.; Dewailly, E.; Delcourt, P.; Lepage, G.; Lotteau, S.; et al. Modulation of ER stress and apoptosis by endoplasmic reticulum calcium leak via translocon during unfolded protein response: Involvement of GRP78. FASEB J. 2013, 27, 1600–1609. [Google Scholar] [CrossRef] [PubMed]
- Flourakis, M.; Van Coppenolle, F.; Lehen’kyi, V.; Beck, B.; Skryma, R.; Prevarskaya, N. Passive calcium leak via translocon is a first step for iPLA2-pathway regulated store operated channels activation. FASEB J. 2006, 20, 1215–1217. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, I.M.; Abdelmalek, D.H.; Elfiky, A.A. GRP78: A cell’s response to stress. Life Sci. 2019, 226, 156–163. [Google Scholar] [CrossRef]
- Gonzalez-Gronow, M.; Gopal, U.; Austin, R.C.; Pizzo, S.V. Glucose-regulated protein (GRP78) is an important cell surface receptor for viral invasion, cancers, and neurological disorders. IUBMB Life 2021, 73, 843–854. [Google Scholar] [CrossRef]
- Bibby, A.C.; Tsim, S.; Kanellakis, N.; Ball, H.; Talbot, D.C.; Blyth, K.G.; Maskell, N.A.; Psallidas, I. Malignant pleural mesothelioma: An update on investigation, diagnosis and treatment. Eur. Respir. Rev. 2016, 25, 472–486. [Google Scholar] [CrossRef]
- Popat, S.; Baas, P.; Faivre-Finn, C.; Girard, N.; Nicholson, A.G.; Nowak, A.K.; Opitz, I.; Scherpereel, A.; Reck, M. Malignant pleural mesothelioma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2022, 33, 129–142. [Google Scholar] [CrossRef]
- Zhang, F.; Chen, F.; Yang, C.; Wang, L.; Hu, H.; Li, X.; Zheng, X.; Wang, Z.; Chang, Z.; Li, T.; et al. Coordination and Redox Dual-Responsive Mesoporous Organosilica Nanoparticles Amplify Immunogenic Cell Death for Cancer Chemoimmunotherapy. Small 2021, 17, e2100006. [Google Scholar] [CrossRef]
- Bakewell, S.; Conde, I.; Fallah, Y.; McCoy, M.; Jin, L.; Shajahan-Haq, A.N. Inhibition of DNA Repair Pathways and Induction of ROS Are Potential Mechanisms of Action of the Small Molecule Inhibitor BOLD-100 in Breast Cancer. Cancers 2020, 12, 2647. [Google Scholar] [CrossRef]
- Flocke, L.S.; Trondl, R.; Jakupec, M.A.; Keppler, B.K. Molecular mode of action of NKP-1339-a clinically investigated ruthenium-based drug-involves ER- and ROS-related effects in colon carcinoma cell lines. Investig. New Drugs 2016, 34, 261–268. [Google Scholar] [CrossRef] [Green Version]
- Rizzuto, R.; Brini, M.; Murgia, M.; Pozzan, T. Microdomains with high Ca2+ close to IP3-sensitive channels that are sensed by neighboring mitochondria. Science 1993, 262, 744–747. [Google Scholar] [CrossRef] [PubMed]
- Rizzuto, R.; Pinton, P.; Carrington, W.; Fay, F.S.; Fogarty, K.E.; Lifshitz, L.M.; Tuft, R.A.; Pozzan, T. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 1998, 280, 1763–1766. [Google Scholar] [CrossRef] [PubMed]
- De Stefani, D.; Raffaello, A.; Teardo, E.; Szabò, I.; Rizzuto, R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 2011, 476, 336–340. [Google Scholar] [CrossRef]
- Rapizzi, E.; Pinton, P.; Szabadkai, G.; Wieckowski, M.R.; Vandecasteele, G.; Baird, G.; Tuft, R.A.; Fogarty, K.E.; Rizzuto, R. Recombinant expression of the voltage-dependent anion channel enhances the transfer of Ca2+ microdomains to mitochondria. J. Cell Biol. 2002, 159, 613–624. [Google Scholar] [CrossRef] [PubMed]
- Deniaud, A.; el Dein, O.S.; Maillier, E.; Poncet, D.; Kroemer, G.; Lemaire, C.; Brenner, C. Endoplasmic reticulum stress induces calcium-dependent permeability transition, mitochondrial outer membrane permeabilization and apoptosis. Oncogene 2008, 27, 285–299. [Google Scholar] [CrossRef]
- Higo, T.; Hattori, M.; Nakamura, T.; Natsume, T.; Michikawa, T.; Mikoshiba, K. Subtype-specific and ER lumenal environment-dependent regulation of inositol 1,4,5-trisphosphate receptor type 1 by ERp44. Cell 2005, 120, 85–98. [Google Scholar] [CrossRef]
- Szlufcik, K.; Missiaen, L.; Parys, J.B.; Callewaert, G.; De Smedt, H. Uncoupled IP3 receptor can function as a Ca2+-leak channel: Cell biological and pathological consequences. Biol. Cell 2006, 98, 1–14. [Google Scholar] [CrossRef]
- Sadafi, F.Z.; Massai, L.; Bartolommei, G.; Moncelli, M.R.; Messori, L.; Tadini-Buoninsegni, F. Anticancer ruthenium(III) complex KP1019 interferes with ATP-dependent Ca2+ translocation by sarco-endoplasmic reticulum Ca2+-ATPase (SERCA). ChemMedChem 2014, 9, 1660–1664. [Google Scholar] [CrossRef]
- Xu, D.; Yang, H.; Yang, Z.; Berezowska, S.; Gao, Y.; Liang, S.Q.; Marti, T.M.; Hall, S.R.R.; Dorn, P.; Kocher, G.J.; et al. Endoplasmic Reticulum Stress Signaling as a Therapeutic Target in Malignant Pleural Mesothelioma. Cancers 2019, 11, 1502. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, L.; Zhou, L.; Lei, Y.; Zhang, Y.; Huang, C. Redox signaling and unfolded protein response coordinate cell fate decisions under ER stress. Redox Biol. 2019, 25, 101047. [Google Scholar] [CrossRef]
- Hu, H.; Tian, M.; Ding, C.; Yu, S. The C/EBP Homologous Protein (CHOP) Transcription Factor Functions in Endoplasmic Reticulum Stress-Induced Apoptosis and Microbial Infection. Front. Immunol. 2018, 9, 3083. [Google Scholar] [CrossRef] [PubMed]
- Ge, R.; Kao, C. Cell Surface GRP78 as a Death Receptor and an Anticancer Drug Target. Cancers 2019, 11, 1787. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Sequences |
|---|---|
| GRP78 | TGTTCAACCAATTATCAGCAAACTC TTCTGCTGTATCCTCTTCACCAGT |
| CHOP | AGAACCAGGAAACGGAAACAGA TCTCCTTCATGCGCTGCTTT |
| sXBP1 | CTGAGTCCGAATCAGGTGCAG ATCCATGGGGAGATGTTCTGG |
| usXBP1 | CAGCACTCAGACTACGTGCA ATCCATGGGGAGATGTTCTGG |
| β-actin | TCCCTGGAGAAGAGCTACGA AGCACTGTGTTGGCGTACAG |
| Cell Line | EC50 at 24 h | EC50 at 48 h |
|---|---|---|
| REN | 111 (81–152) | 71 (50–102) |
| MM98 | 219 (176–273) | 90 (70–116) |
| Met5A | 572 (460–712) | 399 (249–636) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ranzato, E.; Bonsignore, G.; Martinotti, S. ER Stress Response and Induction of Apoptosis in Malignant Pleural Mesothelioma: The Achilles Heel Targeted by the Anticancer Ruthenium Drug BOLD-100. Cancers 2022, 14, 4126. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14174126
Ranzato E, Bonsignore G, Martinotti S. ER Stress Response and Induction of Apoptosis in Malignant Pleural Mesothelioma: The Achilles Heel Targeted by the Anticancer Ruthenium Drug BOLD-100. Cancers. 2022; 14(17):4126. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14174126
Chicago/Turabian StyleRanzato, Elia, Gregorio Bonsignore, and Simona Martinotti. 2022. "ER Stress Response and Induction of Apoptosis in Malignant Pleural Mesothelioma: The Achilles Heel Targeted by the Anticancer Ruthenium Drug BOLD-100" Cancers 14, no. 17: 4126. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14174126