

Inhibition of KRAS, MEK and PI3K Demonstrate Synergistic Anti-Tumor Effects in Pancreatic Ductal Adenocarcinoma Cell Lines
 ,
,  , , , ,
, , , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Kinase Inhibitors
2.2. Cell Lines and Cell Culture
2.3. Inhibitor Application Experiments
2.4. Cell Viability Assays
2.4.1. Proliferation
2.4.2. Biomass Quantification
2.5. Apoptosis and Necrosis Analyses
2.6. Evaluation of Combined Inhibitor Application
2.7. Examination of Cell Morphology Changes
2.8. RNA Extraction
2.9. RNA Sequencing Analysis
2.10. Data Pre-Processing and Differentially Expressed Genes (DEGs) Analysis
2.11. Statistical Analyses
3. Results
3.1. KRAS Status of the PDAC Cell Lines
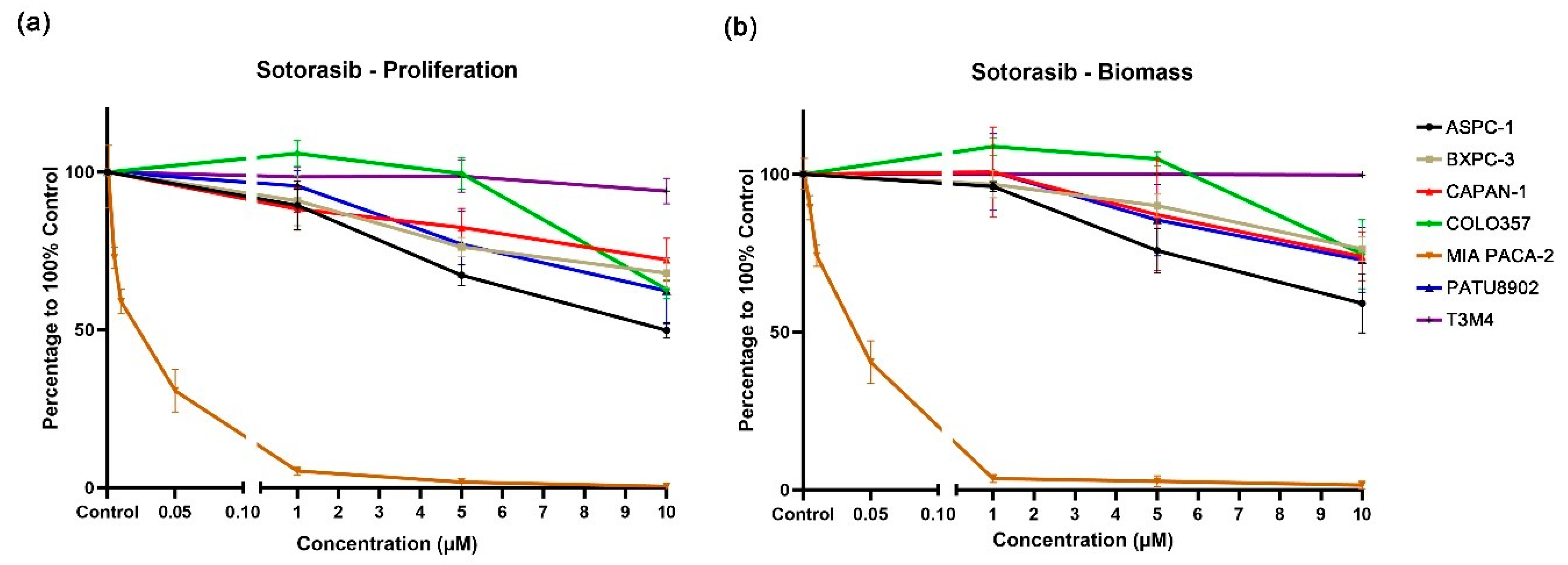
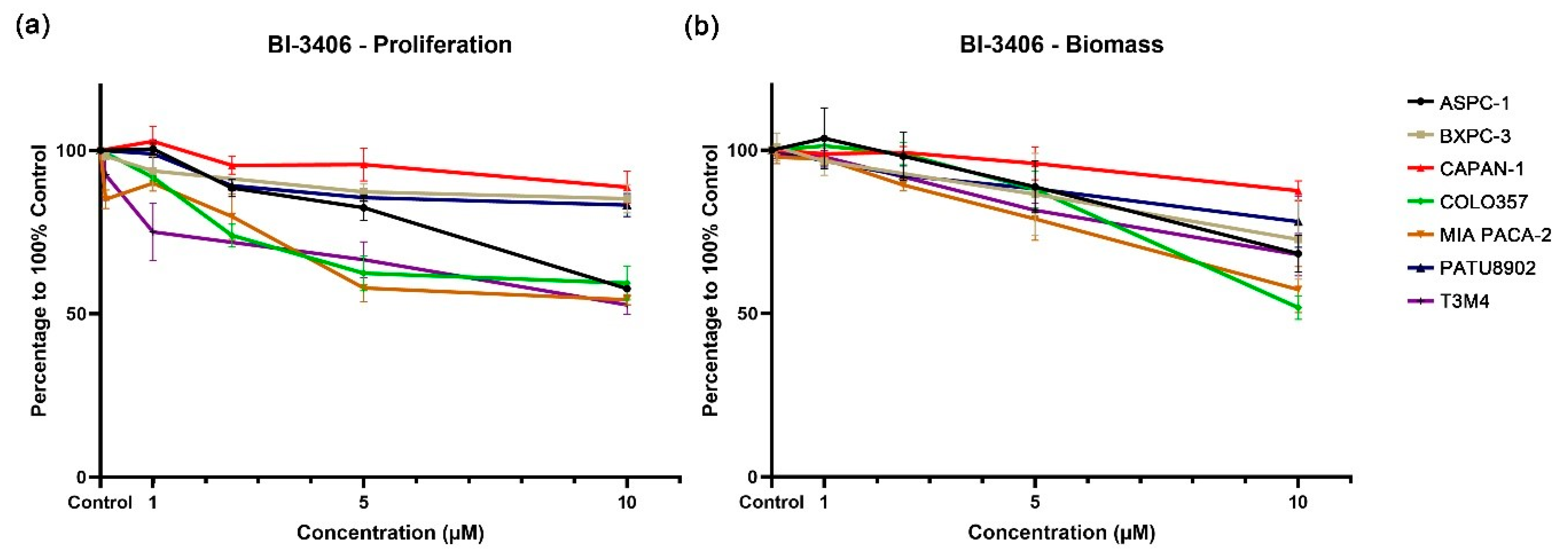
3.2. Single Application of KRAS Inhibitors BI-3406 and Sotorasib to PDAC Cell Lines
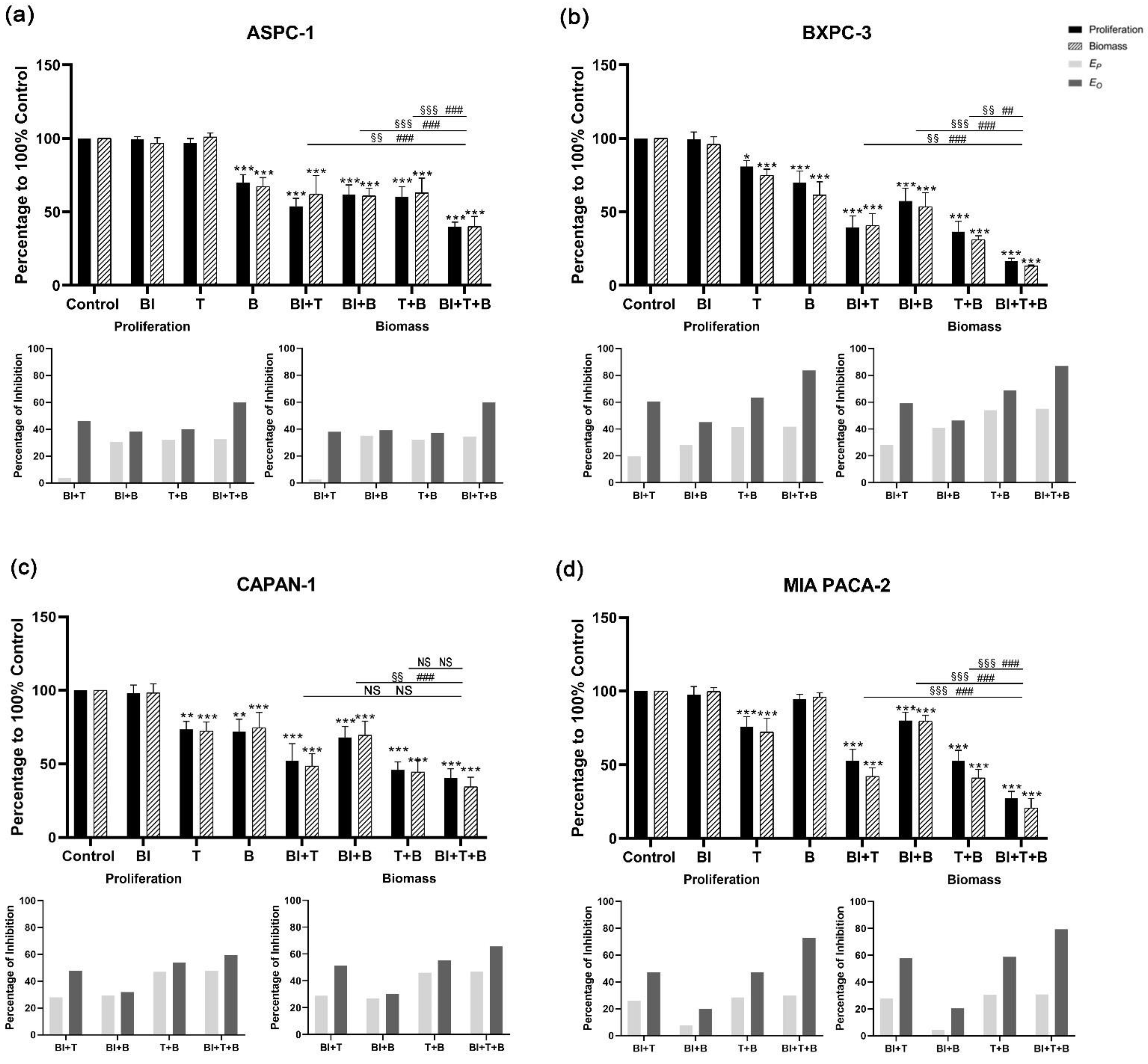
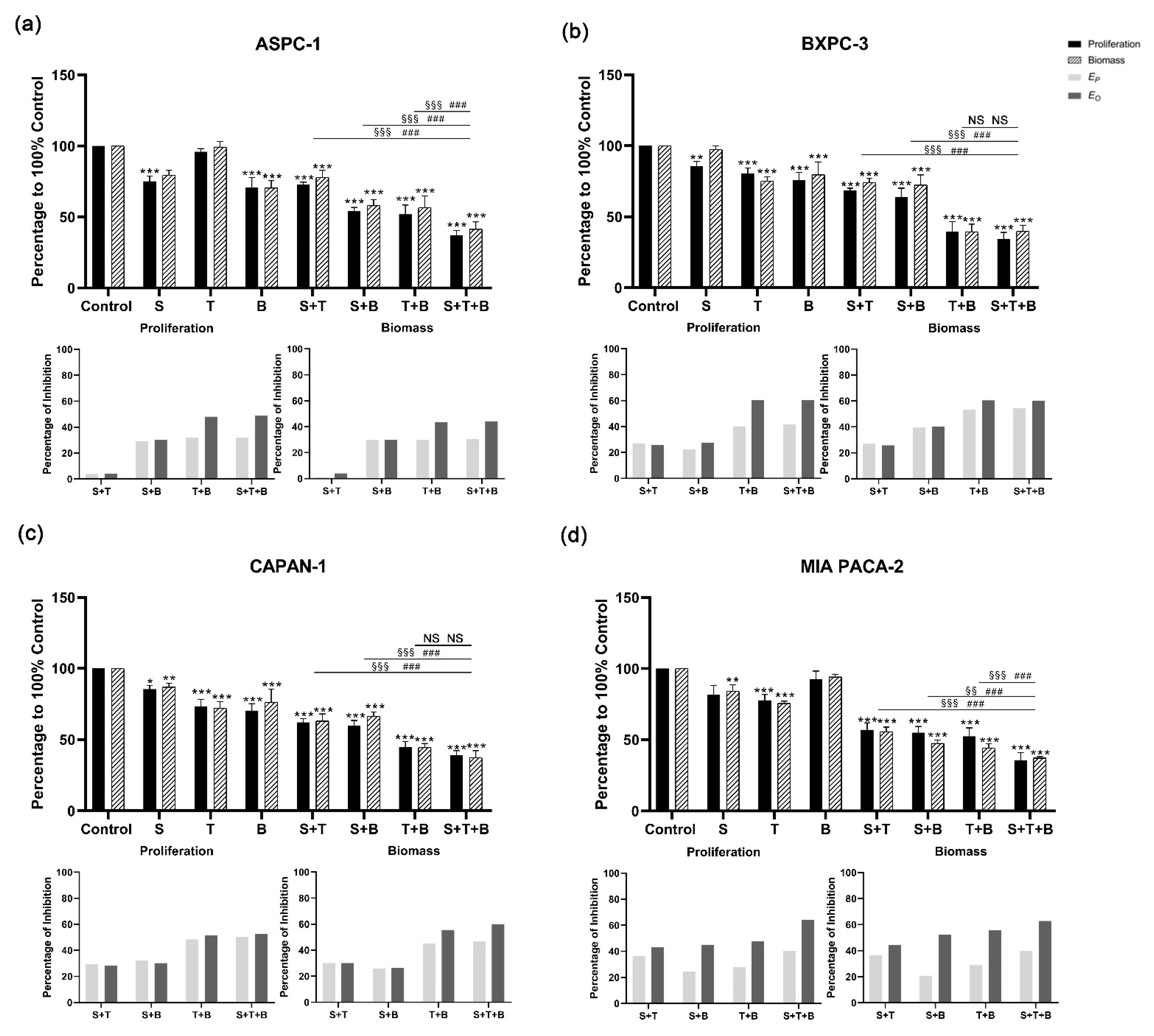
3.3. Combined Applications of KRAS, PI3K, and MEK1/2 Inhibitors Enhance Inhibition of PDAC Cell Lines
3.4. Bliss Analysis Revealed the Synergistic Effects of Double- and Triple-Application
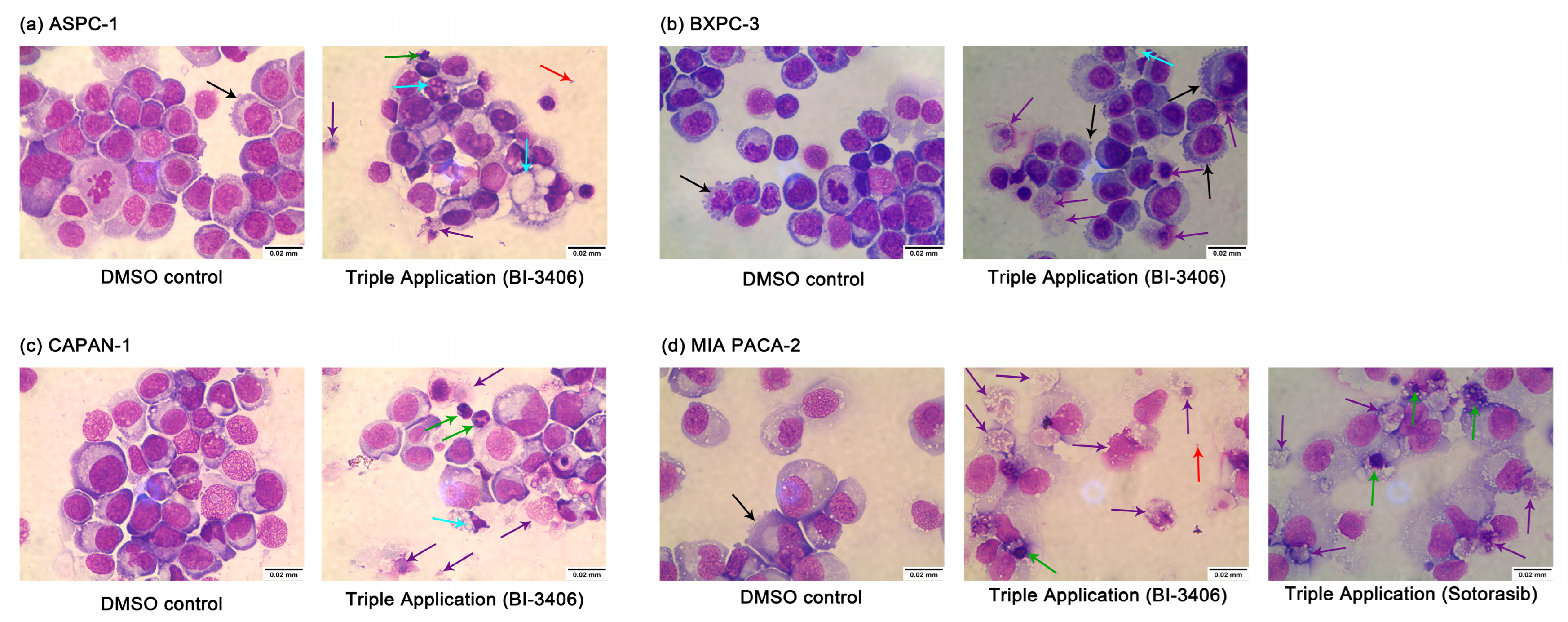
3.5. Combined Application of KRAS, PI3K, and MEK1/2 Inhibitors Induce Apoptosis and Necrosis of PDAC Cell Lines
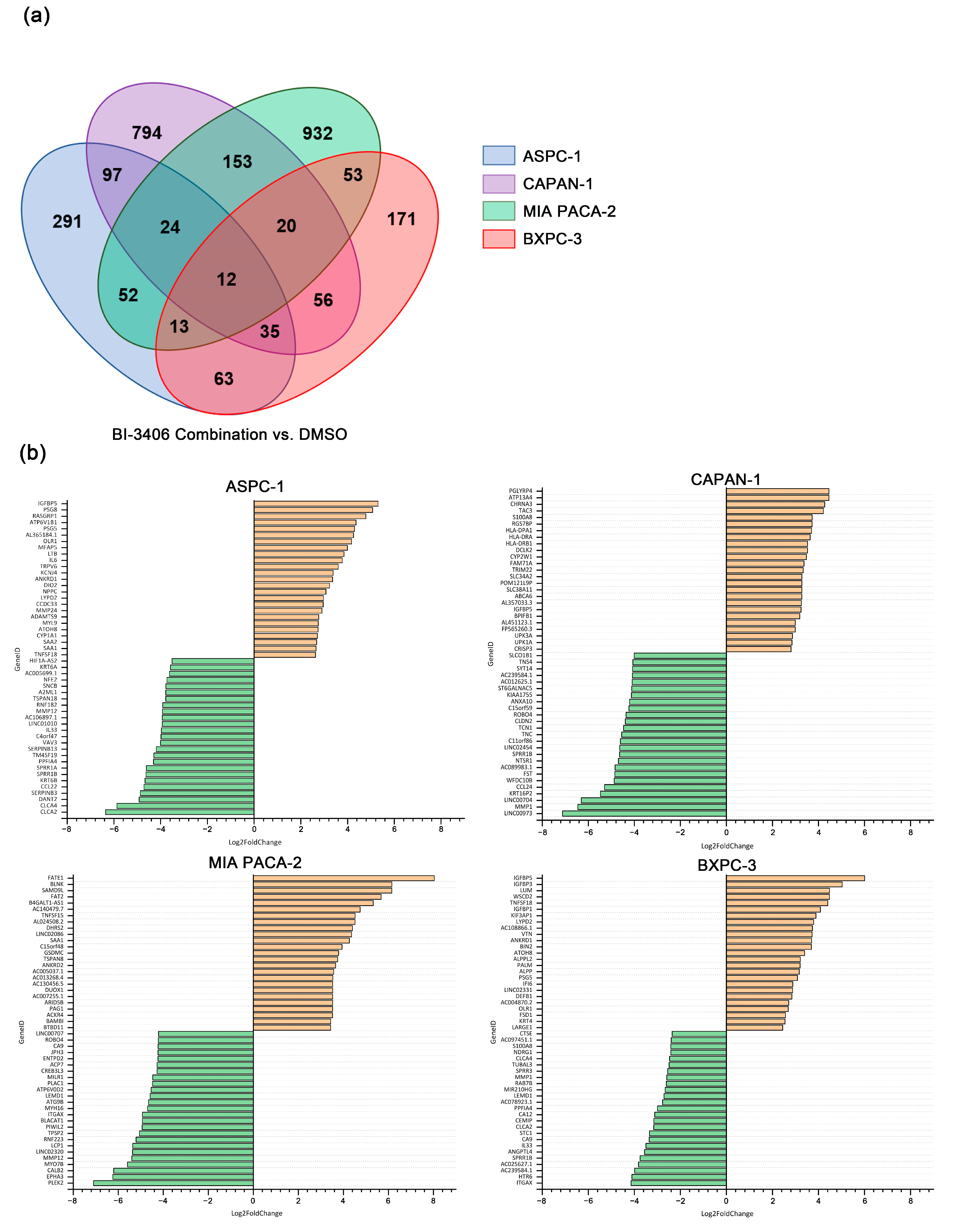
3.6. Comparative Analysis of Differentially Expressed Genes (DEGs) between BI-3406 Combination-Treated and Non-Exposed PDAC Cell Lines
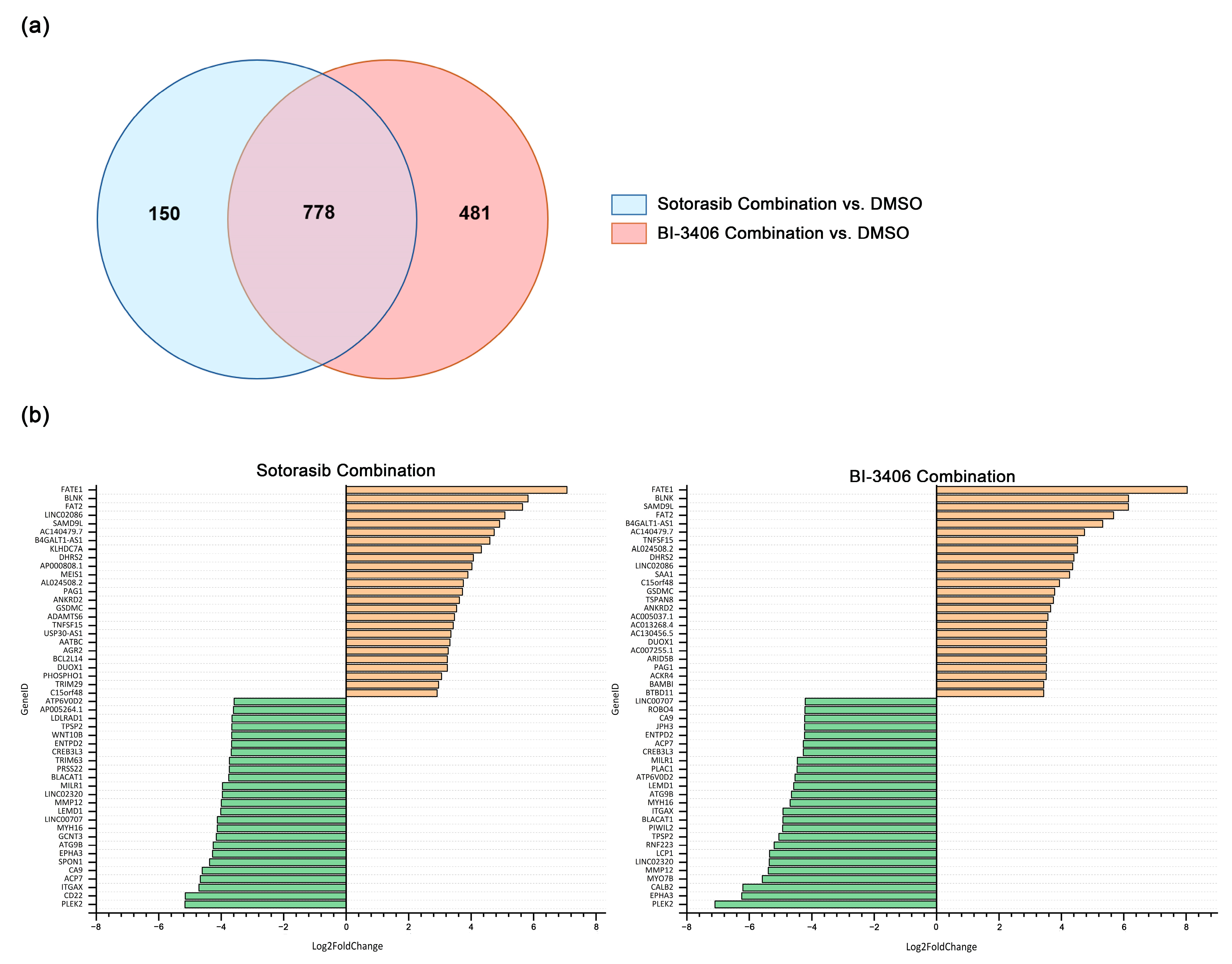
3.7. Comparative Analysis of DEG Changes Induced by BI-3406 Combination-Treated and Sotorasib Combination-Treated in MIA PACA-2 Cell Line
3.8. Functional and Pathway Enrichment Analysis of DEGs Induced by Combination-Treated PDAC Cell Lines
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| A | Alanine |
| AKT | Protein kinase B |
| BP | Biological process |
| BRCA3 | Breast cancer 3 |
| C | Cysteine |
| CC | Cellular components |
| CDKN2A | Cyclin dependent kinase inhibitor 2A |
| CV | Crystal violet |
| D | Aspartic acid |
| DEG | Differentially expressed gene |
| DMSO | Dimethyl sulfoxide |
| EO | Observed inhibition |
| EP | Bliss predicted inhibition |
| ERK | Extracellular signal-regulated kinase |
| FCS | Fetal calf serum |
| G | Glycine |
| GDP | Guanosine diphosphate |
| GO | Gene Ontology |
| GRC2 | Cytosolic glutathione reductase |
| GSEA | Gene set enrichment analysis |
| GTP | Guanosine triphosphate |
| H | Histidine |
| HIF-1 | Hypoxia inducible factor-1 |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| KRAS | Kirsten rat sarcoma virus |
| MEK | Mitogen-activated protein kinase kinase |
| MF | Molecular function |
| mTOR | Mammalian target of rapamycin |
| NSCLC | Non-small cell lung cancer |
| P- | Phosphorylated- |
| P/S | Penicillin-streptomycin solution |
| PDAC | Pancreatic ductal adenocarcinoma |
| PI | Propidium iodide |
| PI3K | Phosphoinositide 3-kinase |
| Q | Glutamine |
| RAS | Rat sarcoma virus |
| RIN | RNA integrity number |
| SD | Standard deviation |
| SMAD4 | Mothers against decapentaplegic homolog 4 |
| SOS1 | Son of sevenless 1 |
| TP53 | Tumor protein P53 |
| V | Valine |
References
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008, 321, 1801–1806. [Google Scholar] [CrossRef]
- Campbell, P.J.; Yachida, S.; Mudie, L.J.; Stephens, P.J.; Pleasance, E.D.; Stebbings, L.A.; Morsberger, L.A.; Latimer, C.; McLaren, S.; Lin, M.L.; et al. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature 2010, 467, 1109–1113. [Google Scholar] [CrossRef] [PubMed]
- Biankin, A.V.; Waddell, N.; Kassahn, K.S.; Gingras, M.C.; Muthuswamy, L.B.; Johns, A.L.; Miller, D.K.; Wilson, P.J.; Patch, A.M.; Wu, J.; et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 2012, 491, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Waddell, N.; Pajic, M.; Patch, A.M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; Quek, K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015, 518, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Witkiewicz, A.K.; McMillan, E.A.; Balaji, U.; Baek, G.; Lin, W.C.; Mansour, J.; Mollaee, M.; Wagner, K.U.; Koduru, P.; Yopp, A.; et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat. Commun. 2015, 6, 6744. [Google Scholar] [CrossRef]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.M.; Gingras, M.C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef]
- Simanshu, D.K.; Nissley, D.V.; McCormick, F. RAS Proteins and Their Regulators in Human Disease. Cell 2017, 170, 17–33. [Google Scholar] [CrossRef]
- Di Magliano, M.P.; Logsdon, C.D. Roles for KRAS in pancreatic tumor development and progression. Gastroenterology 2013, 144, 1220–1229. [Google Scholar] [CrossRef]
- Hingorani, S.R.; Wang, L.; Multani, A.S.; Combs, C.; Deramaudt, T.B.; Hruban, R.H.; Rustgi, A.K.; Chang, S.; Tuveson, D.A. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 2005, 7, 469–483. [Google Scholar] [CrossRef]
- Bardeesy, N.; Cheng, K.H.; Berger, J.H.; Chu, G.C.; Pahler, J.; Olson, P.; Hezel, A.F.; Horner, J.; Lauwers, G.Y.; Hanahan, D.; et al. Smad4 is dispensable for normal pancreas development yet critical in progression and tumor biology of pancreas cancer. Genes Dev. 2006, 20, 3130–3146. [Google Scholar] [CrossRef]
- Bardeesy, N.; Aguirre, A.J.; Chu, G.C.; Cheng, K.H.; Lopez, L.V.; Hezel, A.F.; Feng, B.; Brennan, C.; Weissleder, R.; Mahmood, U.; et al. Both p16(Ink4a) and the p19(Arf)-p53 pathway constrain progression of pancreatic adenocarcinoma in the mouse. Proc. Natl. Acad. Sci. USA 2006, 103, 5947–5952. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, A.J.; Bardeesy, N.; Sinha, M.; Lopez, L.; Tuveson, D.A.; Horner, J.; Redston, M.S.; DePinho, R.A. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev. 2003, 17, 3112–3126. [Google Scholar] [CrossRef] [PubMed]
- Guerra, C.; Barbacid, M. Genetically engineered mouse models of pancreatic adenocarcinoma. Mol. Oncol. 2013, 7, 232–247. [Google Scholar] [CrossRef] [PubMed]
- Bournet, B.; Buscail, C.; Muscari, F.; Cordelier, P.; Buscail, L. Targeting KRAS for diagnosis, prognosis, and treatment of pancreatic cancer: Hopes and realities. Eur. J. Cancer 2016, 54, 75–83. [Google Scholar] [CrossRef]
- Zhao, H.; Wu, S.; Li, H.; Duan, Q.; Zhang, Z.; Shen, Q.; Wang, C.; Yin, T. ROS/KRAS/AMPK Signaling Contributes to Gemcitabine-Induced Stem-like Cell Properties in Pancreatic Cancer. Mol. Oncolytics 2019, 14, 299–312. [Google Scholar] [CrossRef]
- Kawesha, A.; Ghaneh, P.; Andren-Sandberg, A.; Ograed, D.; Skar, R.; Dawiskiba, S.; Evans, J.D.; Campbell, F.; Lemoine, N.; Neoptolemos, J.P. K-ras oncogene subtype mutations are associated with survival but not expression of p53, p16(INK4A), p21(WAF-1), cyclin D1, erbB-2 and erbB-3 in resected pancreatic ductal adenocarcinoma. Int. J. Cancer 2000, 89, 469–474. [Google Scholar] [CrossRef]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef]
- Strickler, J.H.; Satake, H.; Hollebecque, A.; Sunakawa, Y.; Tomasini, P.; Bajor, D.L.; Schuler, M.H.; Yaeger, R.; George, T.J.; Garrido-Laguna, I.; et al. First data for sotorasib in patients with pancreatic cancer with KRAS p.G12C mutation: A phase I/II study evaluating efficacy and safety. J. Clin. Oncol. 2022, 40, 360490. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Available online: https://www.clinicaltrials.gov/ (accessed on 25 August 2022).
- Nagasaka, M.; Li, Y.; Sukari, A.; Ou, S.I.; Al-Hallak, M.N.; Azmi, A.S. KRAS G12C Game of Thrones, which direct KRAS inhibitor will claim the iron throne? Cancer Treat Rev. 2020, 84, 101974. [Google Scholar] [CrossRef]
- Hallin, J.; Engstrom, L.D.; Hargis, L.; Calinisan, A.; Aranda, R.; Briere, D.M.; Sudhakar, N.; Bowcut, V.; Baer, B.R.; Ballard, J.A.; et al. The KRAS(G12C) Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov. 2020, 10, 54–71. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, 1. [Google Scholar] [CrossRef] [PubMed]
- Cherniack, A.D.; Klarlund, J.K.; Conway, B.R.; Czech, M.P. Disassembly of Son-of-sevenless proteins from Grb2 during p21ras desensitization by insulin. J. Biol. Chem. 1995, 270, 1485–1488. [Google Scholar] [CrossRef] [PubMed]
- Waters, S.B.; Yamauchi, K.; Pessin, J.E. Insulin-stimulated disassociation of the SOS-Grb2 complex. Mol. Cell. Biol. 1995, 15, 2791–2799. [Google Scholar] [CrossRef] [PubMed]
- Jeng, H.H.; Taylor, L.J.; Bar-Sagi, D. Sos-mediated cross-activation of wild-type Ras by oncogenic Ras is essential for tumorigenesis. Nat. Commun. 2012, 3, 1168. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, M.H.; Gmachl, M.; Ramharter, J.; Savarese, F.; Gerlach, D.; Marszalek, J.R.; Sanderson, M.P.; Kessler, D.; Trapani, F.; Arnhof, H.; et al. BI-3406, a Potent and Selective SOS1-KRAS Interaction Inhibitor, Is Effective in KRAS-Driven Cancers through Combined MEK Inhibition. Cancer Discov. 2021, 11, 142–157. [Google Scholar] [CrossRef]
- Ma, Y.; Sender, S.; Sekora, A.; Kong, W.; Bauer, P.; Ameziane, N.; Al-Ali, R.; Krake, S.; Radefeldt, M.; Weiss, F.U.; et al. The Inhibitory Response to PI3K/AKT Pathway Inhibitors MK-2206 and Buparlisib Is Related to Genetic Differences in Pancreatic Ductal Adenocarcinoma Cell Lines. Int. J. Mol. Sci. 2022, 23, 4295. [Google Scholar] [CrossRef]
- Goldoni, M.; Johansson, C. A mathematical approach to study combined effects of toxicants in vitro: Evaluation of the Bliss independence criterion and the Loewe additivity model. Toxicol. Vitr. 2007, 21, 759–769. [Google Scholar] [CrossRef]
- Babraham Bioinformatics. Available online: http://www.bioinformatics.babraham.ac.uk/ (accessed on 1 May 2022).
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstrale, M.; Laurila, E.; et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Brouwer, C. Pathview: An R/Bioconductor package for pathway-based data integration and visualization. Bioinformatics 2013, 29, 1830–1831. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- Hunter, J.C.; Manandhar, A.; Carrasco, M.A.; Gurbani, D.; Gondi, S.; Westover, K.D. Biochemical and Structural Analysis of Common Cancer-Associated KRAS Mutations. Mol. Cancer Res. 2015, 13, 1325–1335. [Google Scholar] [CrossRef]
- Hong, D.S.; Fakih, M.G.; Strickler, J.H.; Desai, J.; Durm, G.A.; Shapiro, G.I.; Falchook, G.S.; Price, T.J.; Sacher, A.; Denlinger, C.S.; et al. KRAS(G12C) Inhibition with Sotorasib in Advanced Solid Tumors. N. Engl. J. Med. 2020, 383, 1207–1217. [Google Scholar] [CrossRef]
- Rodriguez-Viciana, P.; Warne, P.H.; Dhand, R.; Vanhaesebroeck, B.; Gout, I.; Fry, M.J.; Waterfield, M.D.; Downward, J. Phosphatidylinositol-3-OH kinase as a direct target of Ras. Nature 1994, 370, 527–532. [Google Scholar] [CrossRef]
- Rodriguez-Viciana, P.; Warne, P.H.; Vanhaesebroeck, B.; Waterfield, M.D.; Downward, J. Activation of phosphoinositide 3-kinase by interaction with Ras and by point mutation. EMBO J. 1996, 15, 2442–2451. [Google Scholar] [CrossRef]
- Rubio, I.; Rodriguez-Viciana, P.; Downward, J.; Wetzker, R. Interaction of Ras with phosphoinositide 3-kinase gamma. Biochem. J. 1997, 326 Pt 3, 891–895. [Google Scholar] [CrossRef]
- Vanhaesebroeck, B.; Welham, M.J.; Kotani, K.; Stein, R.; Warne, P.H.; Zvelebil, M.J.; Higashi, K.; Volinia, S.; Downward, J.; Waterfield, M.D. P110delta, a novel phosphoinositide 3-kinase in leukocytes. Proc. Natl. Acad. Sci. USA 1997, 94, 4330–4335. [Google Scholar] [CrossRef]
- Semenza, G.L. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene 2010, 29, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Laughner, E.; Taghavi, P.; Chiles, K.; Mahon, P.C.; Semenza, G.L. HER2 (neu) signaling increases the rate of hypoxia-inducible factor 1alpha (HIF-1alpha) synthesis: Novel mechanism for HIF-1-mediated vascular endothelial growth factor expression. Mol. Cell. Biol. 2001, 21, 3995–4004. [Google Scholar] [CrossRef] [PubMed]
- Pages, G.; Pouyssegur, J. Transcriptional regulation of the Vascular Endothelial Growth Factor gene—A concert of activating factors. Cardiovasc. Res. 2005, 65, 564–573. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Lines | BI-3406 | Sotorasib | Trametinib | Buparlisib |
|---|---|---|---|---|
| ASPC-1 | 4 μM | 4 μM | 0.001 μM | 0.3 μM |
| BXPC-3 | 4 μM | 4 μM | 0.001 μM | 1 μM |
| CAPAN-1 | 4 μM | 4 μM | 0.005 μM | 0.3 μM |
| MIA PACA-2 | 4 μM | 0.005 μM | 0.0025 μM | 0.6 μM |
| Cell Line | #Chr | Start | End | Ref | Obs | Zygosities | VAF | Gene | Base Change | AA Change |
|---|---|---|---|---|---|---|---|---|---|---|
| BXPC-3 | chr12 | 25398284 | 25398284 | G | G | hom | 100 | KRAS | - | - |
| ASPC-1 | chr12 | 25398284 | 25398284 | G | A | hom | 100 | KRAS | NM_033360.2:c.35G>A | G12D |
| COLO357 | chr12 | 25398284 | 25398284 | G | A | het | 23.8 | KRAS | NM_033360.2:c.35G>A | G12D |
| CAPAN-1 | chr12 | 25398284 | 25398284 | G | T | hom | 97.1 | KRAS | NM_033360.2:c.35G>T | G12V |
| PATU8902 | chr12 | 25398284 | 25398284 | G | T | hom | 100 | KRAS | NM_033360.2:c.35G>T | G12V |
| MIA PACA-2 | chr12 | 25398285 | 25398285 | G | T | hom | 99.6 | KRAS | NM_004985.5:c.34G>T | G12C |
| T3M4 | chr12 | 25380275 | 25380275 | A | C | het | 32.6 | KRAS | NM_033360.2:c.183A>C | Q61H |
| PDAC Cell Line | KRAS Mutation | GO Term | KEGG Pathway | ||
|---|---|---|---|---|---|
| Biological Process | Cellular Components | Molecular Functions | |||
| BXPC-3 | Wild Type | 847 | 49 | 96 | 24 |
| ASPC-1 | KRAS G12D | 744 | 80 | 96 | 48 |
| CAPAN-1 | KRAS G12V | 1447 | 116 | 168 | 59 |
| MIA PACA-2 | KRAS G12C | 1053 | 76 | 120 | 66 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, Y.; Schulz, B.; Trakooljul, N.; Al Ammar, M.; Sekora, A.; Sender, S.; Hadlich, F.; Zechner, D.; Weiss, F.U.; Lerch, M.M.; et al. Inhibition of KRAS, MEK and PI3K Demonstrate Synergistic Anti-Tumor Effects in Pancreatic Ductal Adenocarcinoma Cell Lines. Cancers 2022, 14, 4467. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14184467
Ma Y, Schulz B, Trakooljul N, Al Ammar M, Sekora A, Sender S, Hadlich F, Zechner D, Weiss FU, Lerch MM, et al. Inhibition of KRAS, MEK and PI3K Demonstrate Synergistic Anti-Tumor Effects in Pancreatic Ductal Adenocarcinoma Cell Lines. Cancers. 2022; 14(18):4467. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14184467
Chicago/Turabian StyleMa, Yixuan, Benjamin Schulz, Nares Trakooljul, Moosheer Al Ammar, Anett Sekora, Sina Sender, Frieder Hadlich, Dietmar Zechner, Frank Ulrich Weiss, Markus M. Lerch, and et al. 2022. "Inhibition of KRAS, MEK and PI3K Demonstrate Synergistic Anti-Tumor Effects in Pancreatic Ductal Adenocarcinoma Cell Lines" Cancers 14, no. 18: 4467. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14184467