
Cell-Free DNA–Based Multi-Cancer Early Detection Test in an Asymptomatic Screening Population (NHS-Galleri): Design of a Pragmatic, Prospective Randomised Controlled Trial
 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
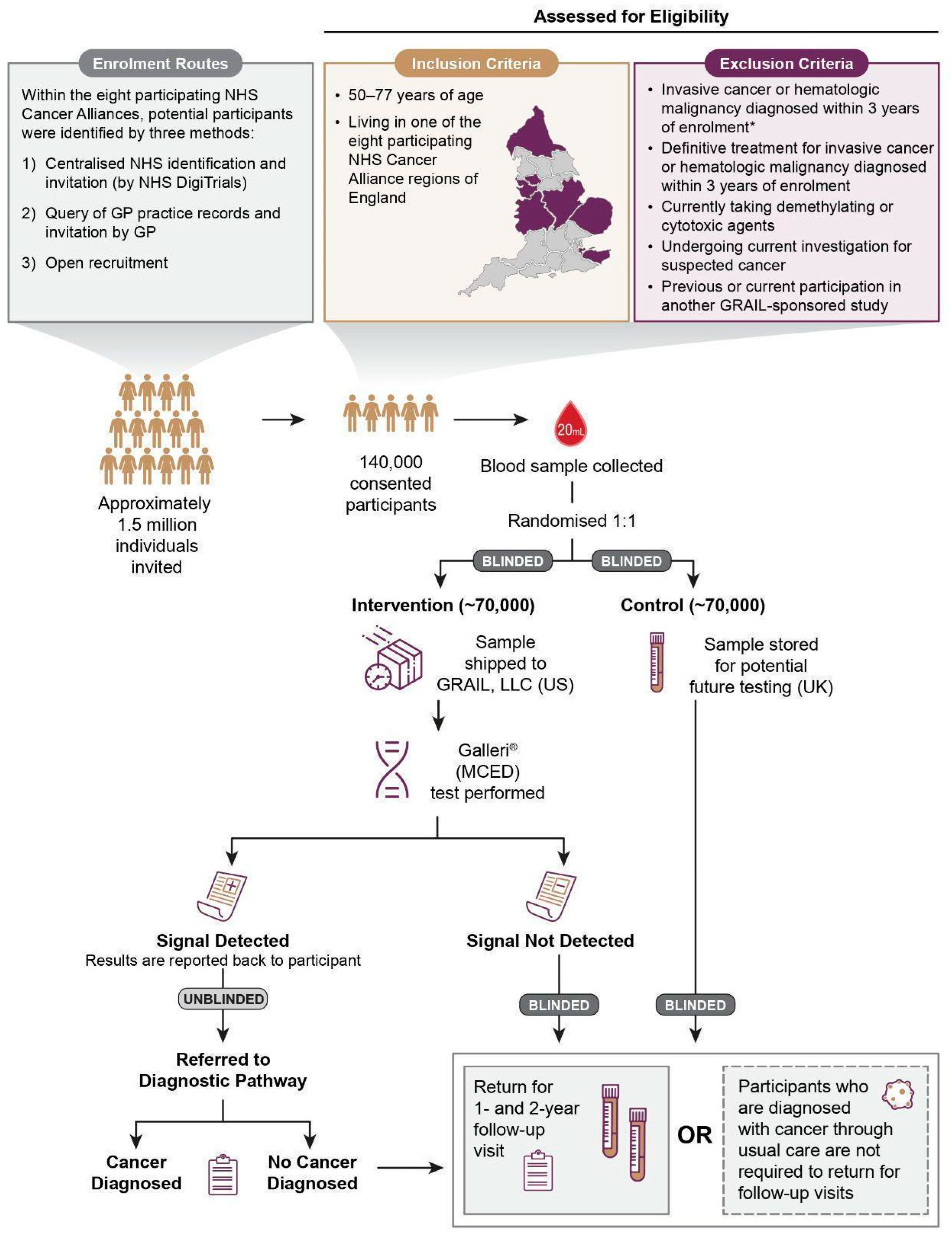
2.1. Trial Design
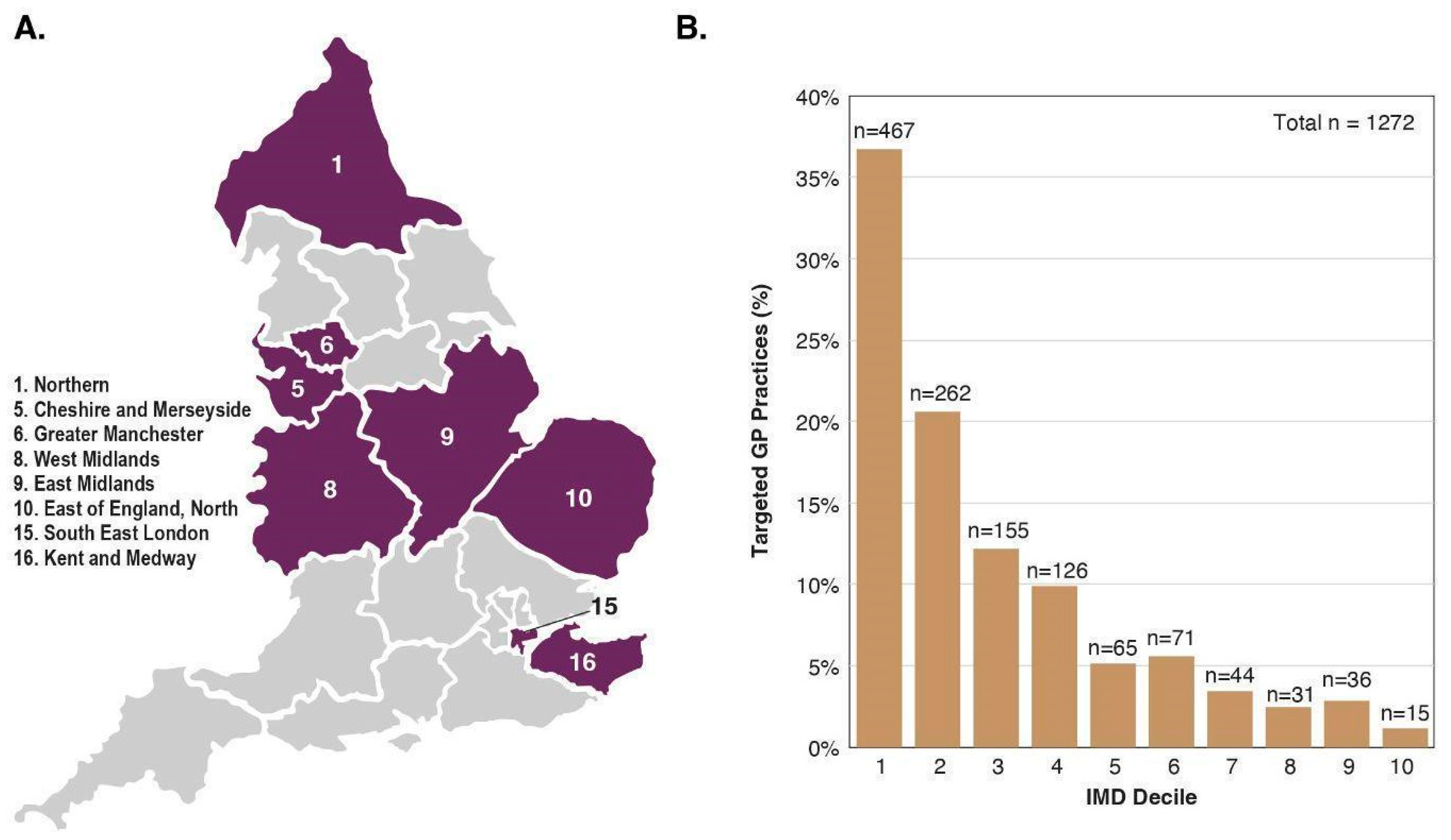
2.2. Participants
2.3. Randomisation and Masking
2.4. Procedures
2.5. Outcomes
2.6. Sample Size Calculation and Statistical Analysis
3. Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Dagenais, G.R.; Leong, D.P.; Rangarajan, S.; Lanas, F.; Lopez-Jaramillo, P.; Gupta, R.; Diaz, R.; Avezum, A.; Oliveira, G.B.F.; Wielgosz, A.; et al. Variations in Common Diseases, Hospital Admissions, and Deaths in Middle-Aged Adults in 21 Countries from Five Continents (PURE): A Prospective Cohort Study. Lancet 2019, 395, 785–794. [Google Scholar] [CrossRef]
- Lin, L.; Li, Z.; Yan, L.; Liu, Y.; Yang, H.; Li, H. Global, Regional, and National Cancer Incidence and Death for 29 Cancer Groups in 2019 and Trends Analysis of the Global Cancer Burden, 1990–2019. J. Hematol. Oncol. 2021, 14, 1–24. [Google Scholar] [CrossRef]
- Mortality Statistics-Underlying Cause, Sex and Age. Available online: https://www.nomisweb.co.uk/query/construct/summary.asp?mode=construct&version=0&dataset=161# (accessed on 25 August 2022).
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization Promoting Cancer Early Diagnosis. Available online: https://www.who.int/activities/promoting-cancer-early-diagnosis (accessed on 25 August 2022).
- Routes to Diagnosis-Data Briefing. Available online: http://www.ncin.org.uk/publications/routes_to_diagnosis (accessed on 25 August 2022).
- Stage Breakdown by CCG 2017. Available online: http://www.ncin.org.uk/publications/survival_by_stage (accessed on 25 August 2022).
- Sud, A.; Torr, B.; Jones, M.E.; Broggio, J.; Scott, S.; Loveday, C.; Garrett, A.; Gronthoud, F.; Nicol, D.L.; Jhanji, S.; et al. Effect of Delays in the 2-Week-Wait Cancer Referral Pathway during the COVID-19 Pandemic on Cancer Survival in the UK: A Modelling Study. Lancet Oncol. 2020, 21, 1035–1044. [Google Scholar] [CrossRef]
- National Health Service Long Term Plan 2019. Available online: https://www.longtermplan.nhs.uk/ (accessed on 25 August 2022).
- Cancer Research UK What Is Cancer Screening? Available online: https://www.cancerresearchuk.org/about-cancer/cancer-symptoms/spot-cancer-early/screening/what-is-cancer-screening (accessed on 25 August 2022).
- Cancer Research UK Cancer Mortality for Common Cancers. Available online: https://www.cancerresearchuk.org/health-professional/cancer-statistics/mortality/common-cancers-compared#heading-Zero (accessed on 25 August 2022).
- NHS England Evaluation of the Targeted Lung Health Check Programme. Available online: https://www.england.nhs.uk/contact-us/privacy-notice/how-we-use-your-information/our-services/evaluation-of-the-targeted-lung-health-check-programme/ (accessed on 25 August 2022).
- Croswell, J.M.; Kramer, B.S.; Kreimer, A.R.; Prorok, P.C.; Xu, J.-L.; Baker, S.G.; Fagerstrom, R.; Riley, T.L.; Clapp, J.D.; Berg, C.D.; et al. Cumulative Incidence of False-Positive Results in Repeated, Multimodal Cancer Screening. Ann. Fam. Med. 2009, 7, 212–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rebolj, M.; Parmar, D.; Maroni, R.; Blyuss, O.; Duffy, S.W. Concurrent Participation in Screening for Cervical, Breast, and Bowel Cancer in England. J. Med. Screen. 2019, 27, 9–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hackshaw, A.; Clarke, C.A.; Hartman, A.-R. New Genomic Technologies for Multi-Cancer Early Detection: Rethinking the Scope of Cancer Screening. Cancer Cell 2022, 14, 109–113. [Google Scholar] [CrossRef]
- Klein, E.; Richards, D.; Cohn, A.; Tummala, M.; Lapham, R.; Cosgrove, D.; Chung, G.; Clement, J.; Gao, J.; Hunkapiller, N. Clinical Validation of a Targeted Methylation-Based Multi-Cancer Early Detection Test Using an Independent Validation Set. Ann. Oncol. 2021, 32, 1167–1177. [Google Scholar] [CrossRef]
- Corcoran, R.B.; Chabner, B.A. Application of Cell-Free DNA Analysis to Cancer Treatment. N. Engl. J. Med. 2018, 379, 1754–1765. [Google Scholar] [CrossRef] [Green Version]
- Salvi, S.; Gurioli, G.; Giorgi, U.D.; Conteduca, V.; Tedaldi, G.; Calistri, D.; Casadio, V. Cell-Free DNA as a Diagnostic Marker for Cancer: Current Insights. Oncotargets Ther. 2016, 9, 6549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinsky, P.F.; Gierada, D.S.; Black, W.; Munden, R.; Nath, H.; Aberle, D.; Kazerooni, E. Performance of Lung-RADS in the National Lung Screening Trial: A Retrospective Assessment. Ann. Intern. Med. 2015, 162, 485–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinsky, P.F.; Berg, C.D. Applying the National Lung Screening Trial Eligibility Criteria to the US Population: What Percent of the Population and of Incident Lung Cancers Would Be Covered? J. Med. Screen. 2012, 19, 154–156. [Google Scholar] [CrossRef]
- Lehman, C.D.; Arao, R.F.; Sprague, B.L.; Lee, J.M.; Buist, D.S.M.; Kerlikowske, K.; Henderson, L.M.; Onega, T.; Tosteson, A.N.A.; Rauscher, G.H.; et al. National Performance Benchmarks for Modern Screening Digital Mammography: Update from the Breast Cancer Surveillance Consortium. Radiology 2017, 283, 49–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siu, A.L. Screening for Breast Cancer: US Preventive Services Task Force Recommendation Statement. Ann. Intern. Med. 2016, 164, 279–296. [Google Scholar] [CrossRef] [Green Version]
- Davidson, K.W.; Barry, M.J.; Mangione, C.M.; Cabana, M.; Caughey, A.B.; Davis, E.M.; Donahue, K.E.; Doubeni, C.A.; Krist, A.H.; Kubik, M. Screening for Colorectal Cancer: US Preventive Services Task Force Recommendation Statement. JAMA 2021, 325, 1965–1977. [Google Scholar]
- US Food and Drug Administration. FDA Summary of Safety and Effectiveness Data. PMA P130017; US Dept of Health and Human Services: Silver Spring, MD, USA, 2014. [Google Scholar]
- Kim, J.J.; Burger, E.A.; Regan, C.; Sy, S. Screening for Cervical Cancer in Primary Care: A Decision Analysis for the US Preventive Services Task Force. JAMA 2018, 320, 706. [Google Scholar] [CrossRef]
- Wolf, A.M.D.; Wender, R.C.; Etzioni, R.B.; Thompson, I.M.; D’Amico, A.V.; Volk, R.J.; Brooks, D.D.; Dash, C.; Guessous, I.; Andrews, K.; et al. American Cancer Society Guideline for the Early Detection of Prostate Cancer: Update 2010. Cancer J. Clin. 2010, 60, 70–98. [Google Scholar] [CrossRef] [Green Version]
- Hubbell, E.; Clarke, C.A.; Aravanis, A.M.; Berg, C.D. Modeled Reductions in Late-Stage Cancer with a Multi-Cancer Early Detection Test. Cancer Epidemiol. Biomark. Amp. Prev. 2021, 30, 460. [Google Scholar] [CrossRef]
- Liu, M.; Cummings, S.; Vachon, C.; Kerlikowske, K.; Couch, F.; Morris, E.; Olson, J.; Polley, E.; Conners, A.; Ellis, R.; et al. Abstract OT3-02-01: Development of Cell-Free Nucleic Acid-Based Tests for Early Detection of Breast Cancer: The STRIVE Study. Cancer Res. 2018, 78, OT3-02. [Google Scholar] [CrossRef]
- Chen, X.; Gole, J.; Gore, A.; He, Q.; Lu, M.; Min, J.; Yuan, Z.; Yang, X.; Jiang, Y.; Zhang, T.; et al. Non-Invasive Early Detection of Cancer Four Years before Conventional Diagnosis Using a Blood Test. Nat. Commun. 2020, 11, 3475. [Google Scholar] [CrossRef]
- Lennon, A.M.; Buchanan, A.H.; Kinde, I.; Warren, A.; Honushefsky, A.; Cohain, A.T.; Ledbetter, D.H.; Sanfilippo, F.; Sheridan, K.; Rosica, D.; et al. Feasibility of Blood Testing Combined with PET-CT to Screen for Cancer and Guide Intervention. Science 2020, 369, eabb9601. [Google Scholar] [CrossRef]
- Nadauld, L.D.; McDonnell, C.H.; Beer, T.M.; Liu, M.C.; Klein, E.A.; Hudnut, A.; Whittington, R.A.; Taylor, B.; Oxnard, G.R.; Lipson, J.; et al. The PATHFINDER Study: Assessment of the Implementation of an Investigational Multi-Cancer Early Detection Test into Clinical Practice. Cancers 2021, 13, 3501. [Google Scholar] [CrossRef]
- Beer, T.M.; McDonnell, C.H.; Nadauld, L.; Liu, M.C.; Klein, E.A.; Reid, R.L.; Marinac, C.; Chung, K.; Lopatin, M.; Fung, E.T.; et al. Interim Results of PATHFINDER, a Clinical Use Study Using a Methylation-Based Multi-Cancer Early Detection Test. J. Clin. Oncol. 2021, 39, 3010. [Google Scholar] [CrossRef]
- Beer, T.M.; McDonnell, C.H.; Nadauld, L.; Liu, M.C.; Klein, E.A.; Reid, R.L.; Chung, K.; Lopatin, M.; Fung, E.T.; Schrag, D. A Prespecified Interim Analysis of the PATHFINDER Study: Performance of a Multicancer Early Detection Test in Support of Clinical Implementation. J. Clin. Oncol. 2021, 39, 3070. [Google Scholar] [CrossRef]
- Pinsky, P.; Miller, A.; Kramer, B.; Church, T.; Reding, D.; Prorok, P.; Gelmann, E.; Schoen, R.; Buys, S.; Hayes, R.; et al. Evidence of a Healthy Volunteer Effect in the Prostate, Lung, Colorectal, and Ovarian Cancer Screening Trial. Am. J. Epidemiol. 2007, 165, 874–881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menon, U.; Gentry-Maharaj, A.; Burnell, M.; Singh, N.; Ryan, A.; Karpinskyj, C.; Carlino, G.; Taylor, J.; Massingham, S.K.; Raikou, M. Ovarian Cancer Population Screening and Mortality after Long-Term Follow-up in the UK Collaborative Trial of Ovarian Cancer Screening (UKCTOCS): A Randomised Controlled Trial. Lancet 2021, 397, 2182–2193. [Google Scholar] [CrossRef]
- Ministry of Housing, Communities & Local Government The English Indices of Deprivation 2019 Frequently Asked Questions. Available online: https://www.gov.uk/government/statistics/english-indices-of-deprivation-2019 (accessed on 25 August 2022).
- Henson, K.E.; Elliss-Brookes, L.; Coupland, V.H.; Payne, E.; Vernon, S.; Rous, B.; Rashbass, J. Data Resource Profile: National Cancer Registration Dataset in England. Int. J. Epidemiol. 2020, 49, 16–16h. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marteau, T.M.; Bekker, H. The Development of a Six-item Short-form of the State Scale of the Spielberger State—Trait Anxiety Inventory (STAI). Br. J. Clin. Psychol. 1992, 31, 301–306. [Google Scholar] [CrossRef]
- Cockburn, J.; Luise, T.D.; Hurley, S.; Clover, K. Development and Validation of the PCQ: A Questionnaire to Measure the Psychological Consequences of Screening Mammography. Soc. Sci. Med. 1992, 34, 1129–1134. [Google Scholar] [CrossRef]
- Herbert, A.; Wijlaars, L.; Zylbersztejn, A.; Cromwell, D.; Hardelid, P. Data Resource Profile: Hospital Episode Statistics Admitted Patient Care (HES APC). Int. J. Epidemiol. 2017, 46, 1093–1093i. [Google Scholar] [CrossRef] [Green Version]
- National Health Service Diagnostic Imaging Data Set 2021. Available online: https://www.england.nhs.uk/statistics/statistical-work-areas/diagnostic-imaging-dataset/diagnostic-imaging-dataset-2020-21-data/ (accessed on 25 August 2022).
- Office for National Statistics Mortality Statistics in England and Wales QMI 2022. Available online: https://www.ons.gov.uk/peoplepopulationandcommunity/birthsdeathsandmarriages/deaths/methodologies/mortalitystatisticsinenglandandwalesqmi (accessed on 25 August 2022).
- Zhang, J.; Braun, J.; Simon, N.; Hubbell, E.; Zhang, N. Stage Shift and Mortality Reduction by Screening with a Multi-Cancer Early-Detection Test: Microsimulation Modeling of a Randomized Controlled Trial. Available online: https://ww2.amstat.org/meetings/jsm/2021/onlineprogram/AbstractDetails.cfm?abstractid=319050 (accessed on 8 August 2021).
- Liu, M.C.; Oxnard, G.R.; Klein, E.A.; Swanton, C.; Seiden, M.V.; Cummings, S.R.; Absalan, F.; Alexander, G.; Allen, B.; Amini, H.; et al. Sensitive and Specific Multi-Cancer Detection and Localization Using Methylation Signatures in Cell-Free DNA. Ann. Oncol. 2020, 31, 745–759. [Google Scholar] [CrossRef]
- Zelen, M. A New Design for Randomized Clinical Trials. N. Engl. J. Med. 1979, 300, 1242–1245. [Google Scholar] [CrossRef]
- Zelen, M. Randomized Consent Designs for Clinical Trials: An Update. Stat. Med. 1990, 9, 645–656. [Google Scholar] [CrossRef]
- Spencer, K.; Jones, C.M.; Girdler, R.; Roe, C.; Sharpe, M.; Lawton, S.; Miller, L.; Lewis, P.; Evans, M.; Sebag-Montefiore, D. The Impact of the COVID-19 Pandemic on Radiotherapy Services in England, UK: A Population-Based Study. Lancet Oncol. 2021, 22, 309–320. [Google Scholar] [CrossRef]
- Quinn-Scoggins, H.D.; Cannings-John, R.; Moriarty, Y.; Whitelock, V.; Whitaker, K.L.; Grozeva, D.; Hughes, J.; Townson, J.; Osborne, K.; Goddard, M. Cancer Symptom Experience and Help-Seeking Behaviour during the COVID-19 Pandemic in the UK: A Cross-Sectional Population Survey. BMJ Open 2021, 11, e053095. [Google Scholar] [CrossRef]
- DeGregori, J.; Pharoah, P.; Sasieni, P.; Swanton, C. Cancer Screening, Surrogates of Survival, and the Soma. Cancer Cell 2020, 38, 433–437. [Google Scholar] [CrossRef]
- Sasieni, P.D.; Wald, N.J. Should a Reduction in All-Cause Mortality Be the Goal When Assessing Preventive Medical Therapies? Circulation 2017, 135, 1985–1987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Autier, P.; Héry, C.; Haukka, J.; Boniol, M.; Byrnes, G. Advanced Breast Cancer and Breast Cancer Mortality in Randomized Controlled Trials on Mammography Screening. J. Clin. Oncol. 2009, 27, 5919–5923. [Google Scholar] [CrossRef] [PubMed]
- Tabar, L.; Fagerberg, G.; Chen, H.H.; Duffy, S.W.; Smart, C.R.; Gad, A.; Smith, R.A. Efficacy of Breast Cancer Screening by Age. New Results from the Swedish Two-County Trial. Cancer 1995, 75, 2507–2517. [Google Scholar] [CrossRef]
- McPhail, S.; Johnson, S.; Greenberg, D.; Peake, M.; Rous, B. Stage at Diagnosis and Early Mortality from Cancer in England. Br. J. Cancer 2015, 112, S108–S115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welch, H.G.; Black, W.C. Overdiagnosis in Cancer. JNCI J. Natl. Cancer Inst. 2010, 102, 605–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Dong, Z.; Hubbell, E.; Kurtzman, K.N.; Oxnard, G.R.; Venn, O.; Melton, C.; Clarke, C.A.; Shaknovich, R.; Ma, T.; et al. Prognostic Significance of Blood-Based Multi-Cancer Detection in Plasma Cell-Free DNA. Clin. Cancer Res. 2021, 27, 4221–4229. [Google Scholar] [CrossRef] [PubMed]
- Chabon, J.J.; Hamilton, E.G.; Kurtz, D.M.; Esfahani, M.S.; Moding, E.J.; Stehr, H.; Schroers-Martin, J.; Nabet, B.Y.; Chen, B.; Chaudhuri, A.A.; et al. Integrating Genomic Features for Non-Invasive Early Lung Cancer Detection. Nature 2020, 580, 245–251. [Google Scholar] [CrossRef]
- Surveillance Research Program; National Cancer Institute SEER*Stat Software (Www.Seer.Cancer.Gov/Seerstat), Version 8.3.8; Data: Surveillance, Epidemiology, and End Results (SEER) Program (Www.Seer.Cancer.Gov) SEER*Stat Database: Incidence-SEER Research Data, 9 Registries, Nov 2019 Sub (1975–2017)-Linked To County Attributes-Time Dependent (1990–2017) Income/Rurality, 1969-2018 Counties, National Cancer Institute, DCCPS, Surveillance Research Program; National Cancer Institute: Bethesda, MD, USA, 2020.
- Gale, D.; Heider, K.; Perry, M.; Marsico, G.; Ruiz-Valdepeñas, A.; Rundell, V.; Wulff, J.; Sharma, G.; Howarth, K.; Gilligan, D.; et al. Residual CtDNA after Treatment Predicts Early Relapse in Patients with Early-Stage NSCLC. J. Clin. Oncol. 2021, 39, 8517. [Google Scholar] [CrossRef]
- Hackshaw, A.; Berg, C.D. An Efficient Randomised Trial Design for Multi-Cancer Screening Blood Tests: Nested Enhanced Mortality Outcomes of Screening Trial. Lancet Oncol. 2021, 22, 1360–1362. [Google Scholar] [CrossRef]
- Jacobs, I.J.; Menon, U.; Ryan, A.; Gentry-Maharaj, A.; Burnell, M.; Kalsi, J.K.; Amso, N.N.; Apostolidou, S.; Benjamin, E.; Cruickshank, D.; et al. Ovarian Cancer Screening and Mortality in the UK Collaborative Trial of Ovarian Cancer Screening (UKCTOCS): A Randomised Controlled Trial. Lancet 2016, 387, 945–956. [Google Scholar] [CrossRef] [Green Version]
- Field, J.K.; Vulkan, D.; Davies, M.P.A.; Baldwin, D.R.; Brain, K.E.; Devaraj, A.; Eisen, T.; Gosney, J.; Green, B.A.; Holemans, J.A.; et al. Lung Cancer Mortality Reduction by LDCT Screening: UKLS Randomised Trial Results and International Meta-Analysis. Lancet Reg. Health Eur. 2021, 10, 100179. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Objective/Endpoint | First Screening Round * | Second Screening Round * | Third Screening Round * | Three Screening Rounds Aggregated * | 3–4 Years after Randomisation † | 3 Years after Final Visit | 6 Years after Final Visit | |
|---|---|---|---|---|---|---|---|---|
| Primary objective/endpoint | Demonstrate a significant reduction in the incidence rate of stage III and IV cancers diagnosed in the intervention arm compared with the control arm | X | ||||||
| Key secondary objectives/endpoints | Demonstrate a significant reduction in the incidence rate of stage IV cancers diagnosed in the intervention arm compared with the control arm (excluding cancers identified by the test performed at the second visit) | X | ||||||
| Evaluate the MCED test performance (overall sensitivity, specificity, PPV, NPV, and cancer signal origin accuracy) in the intervention arm | X | X | X | X | ||||
| Evaluate the safety, including harms, in the intervention arm among participants with a cancer signal detected result by assessing the number of complications and deaths resulting from confirmatory diagnostic procedures, estimated radiation exposure per participant due to test result-directed evaluations, and participant-reported psychological impact among participants with a cancer signal detected result | X | X | X | X | X | |||
| Assess the impact of the use of the MCED test across three annual timepoints on healthcare resource utilisation for cancer diagnosis and treatment, by measuring the number of follow-up procedures and number of invasive procedures needed to achieve diagnostic resolution among participants with a cancer signal detected result, and the number and type(s) of medical encounters and cancer-specific confirmatory diagnostic and treatment procedures among participants with a cancer signal detected result | X | |||||||
| Compare cancer-specific mortality in the intervention and control arms using a retrospective nested analysis | X (for 12 prespecified cancer types ‡) | X | X | |||||
| Assess the potential impact of overdiagnosis by studying the excess in cancers diagnosed after a baseline cancer signal detected result in the intervention arm compared with the control arm (retrospectively testing baseline samples from all participants diagnosed with cancer in the control arm) | X | |||||||
| Key exploratory objectives | Retrospectively test the participants in the control arm who were diagnosed with a cancer of unknown primary and report the cancer signal origin detected by the MCED test | X | ||||||
| Assess the primary and secondary objectives in clinically meaningful subsets (e.g., by age, gender, ethnicity, socio-economic groups, risk factors at enrolment, prior cancer history at enrolment) | X | |||||||
| Assess the potential for avoidance/postponement of cancer death by comparing the cancer-specific mortality rates among participants with a baseline cancer signal detected result in the intervention arm versus the control arm (retrospectively testing baseline samples in the control arm) | X | |||||||
| Assess any potential impact of a baseline cancer signal detected result on non-cancer and all-cause mortality by comparing cancer signal detected non-cancer deaths and cancer signal detected all-cause mortality in the intervention arm compared with the control arm (retrospectively testing baseline samples in the control arm) | X | |||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neal, R.D.; Johnson, P.; Clarke, C.A.; Hamilton, S.A.; Zhang, N.; Kumar, H.; Swanton, C.; Sasieni, P. Cell-Free DNA–Based Multi-Cancer Early Detection Test in an Asymptomatic Screening Population (NHS-Galleri): Design of a Pragmatic, Prospective Randomised Controlled Trial. Cancers 2022, 14, 4818. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14194818
Neal RD, Johnson P, Clarke CA, Hamilton SA, Zhang N, Kumar H, Swanton C, Sasieni P. Cell-Free DNA–Based Multi-Cancer Early Detection Test in an Asymptomatic Screening Population (NHS-Galleri): Design of a Pragmatic, Prospective Randomised Controlled Trial. Cancers. 2022; 14(19):4818. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14194818
Chicago/Turabian StyleNeal, Richard D., Peter Johnson, Christina A. Clarke, Stephanie A. Hamilton, Nan Zhang, Harpal Kumar, Charles Swanton, and Peter Sasieni. 2022. "Cell-Free DNA–Based Multi-Cancer Early Detection Test in an Asymptomatic Screening Population (NHS-Galleri): Design of a Pragmatic, Prospective Randomised Controlled Trial" Cancers 14, no. 19: 4818. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14194818