
Radiogenomic System for Non-Invasive Identification of Multiple Actionable Mutations and PD-L1 Expression in Non-Small Cell Lung Cancer Based on CT Images
Abstract
:Simple Summary
Abstract
1. Introduction
2. Methods
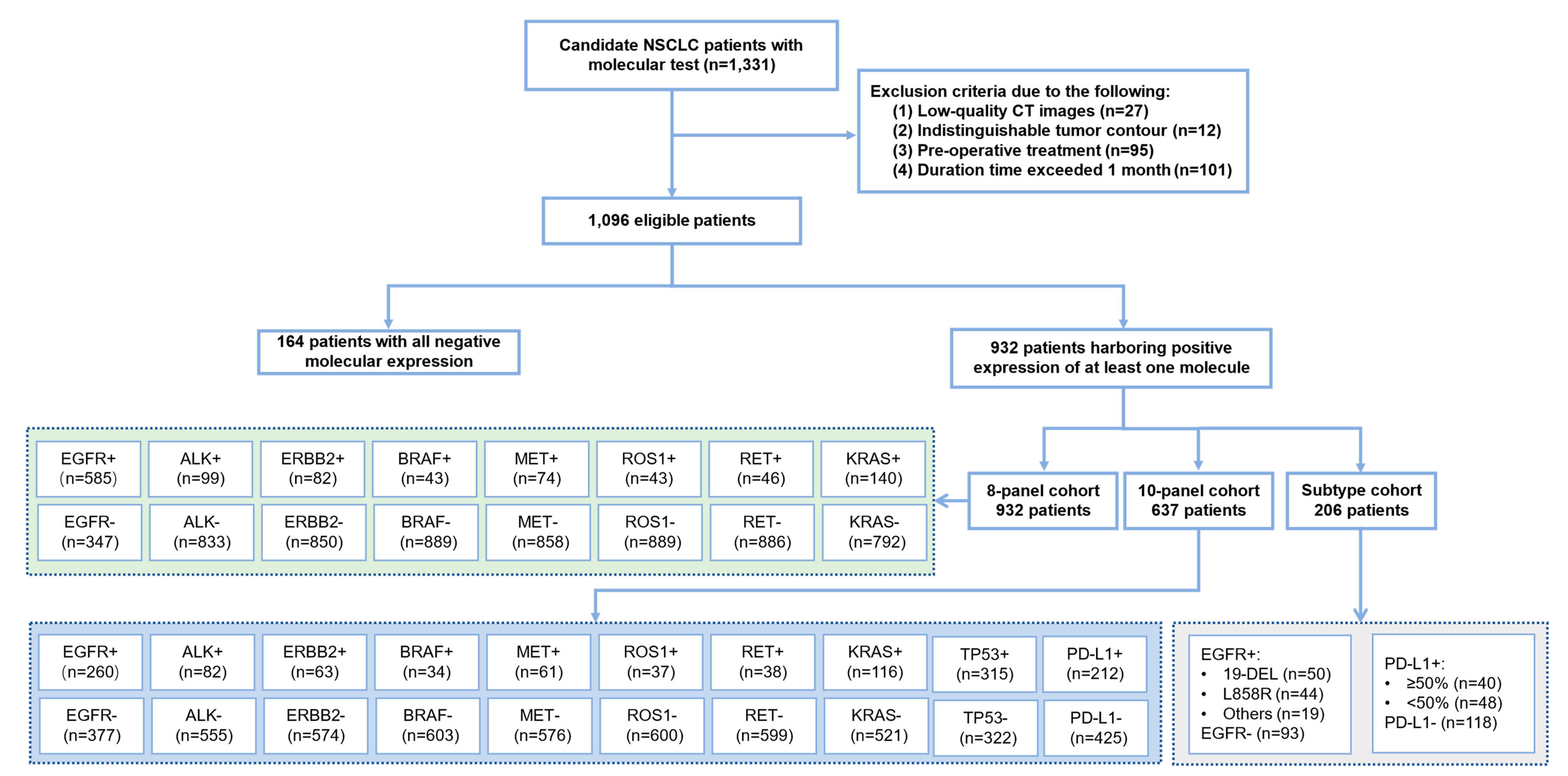
2.1. Study Population
2.2. Imaging Acquisition and Preprocessing
2.3. Radiomics Approach
2.4. Convolutional Neural Network-Based Deep Learning
2.5. Transformer-Based Deep Learning
2.6. Multi-Label Multi-Task Deep Learning (MMDL) System
2.7. Statistical Analysis
3. Results
3.1. Patient Characteristics
3.2. The Performance of the Radiomics Model
3.3. The Performance of the Deep Learning Models
3.4. Performance of the Proposed MMDL Hybrid Model
3.5. Correlation Analysis between Radiomics and Deep Learning Features
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Herbst, R.S.; Morgensztern, D.; Boshoff, C. The biology and management of non-small cell lung cancer. Nature 2018, 553, 446–454. [Google Scholar] [CrossRef]
- Wang, M.; Herbst, R.S.; Boshoff, C. Toward personalized treatment approaches for non-small-cell lung cancer. Nat. Med. 2021, 27, 1345–1356. [Google Scholar] [CrossRef] [PubMed]
- Ettinger, D.S.; Wood, D.E.; Aisner, D.L.; Akerley, W.; Bauman, J.R.; Bharat, A.; Bruno, D.S.; Chang, J.Y.; Chirieac, L.R.; D’Amico, T.A.; et al. NCCN guidelines insights: Non-small cell lung cancer, version 2.2021. J. Natl. Compr. Cancer Netw. 2021, 19, 254–266. [Google Scholar] [CrossRef]
- Luo, W.; Wang, Z.; Zhang, T.; Yang, L.; Xian, J.; Li, Y.; Li, W. Immunotherapy in non-small cell lung cancer: Rationale, recent advances and future perspectives. Precis Clin. Med. 2021, 4, 258–270. [Google Scholar] [CrossRef] [PubMed]
- Canale, M.; Andrikou, K.; Priano, I.; Cravero, P.; Pasini, L.; Urbini, M.; Delmonte, A.; Crinò, L.; Bronte, G.; Ulivi, P. The role of TP53 mutations in EGFR-mutated non-small-cell lung cancer: Clinical significance and implications for therapy. Cancers 2022, 14, 1143. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Ma, J.; Shao, J.; Zhang, S.; Li, J.; Yan, J.; Zhao, Z.; Bai, C.; Yu, Y.; Li, W. Non-invasive measurement using deep learning algorithm based on multi-source features fusion to predict PD-L1 expression and survival in NSCLC. Front. Immunol. 2022, 13, 828560. [Google Scholar] [CrossRef]
- Sanmamed, M.F.; Chen, L. A paradigm shift in cancer immunotherapy: From enhancement to normalization. Cell 2018, 175, 313–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibney, G.T.; Weiner, L.M.; Atkins, M.B. Predictive biomarkers for checkpoint inhibitor-based immunotherapy. Lancet Oncol. 2016, 17, e542–e551. [Google Scholar] [CrossRef] [Green Version]
- Carr, T.H.; McEwen, R.; Dougherty, B.; Johnson, J.H.; Dry, J.R.; Lai, Z.; Ghazoui, Z.; Laing, N.M.; Hodgson, D.R.; Cruzalegui, F.; et al. Defining actionable mutations for oncology therapeutic development. Nat. Rev. Cancer 2016, 16, 319–329. [Google Scholar] [CrossRef]
- Behjati, S.; Tarpey, P.S. What is next generation sequencing? Arch. Dis. Child Educ. Pract. Ed. 2013, 98, 236–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.R.; Schultheis, A.M.; Yu, H.; Mandelker, D.; Ladanyi, M.; Büttner, R. Precision medicine in non-small cell lung cancer: Current applications and future directions. Semin. Cancer Biol. 2022, 84, 184–198. [Google Scholar] [CrossRef]
- Koh, G.; Degasperi, A.; Zou, X.; Momen, S.; Nik-Zainal, S. Mutational signatures: Emerging concepts, caveats and clinical applications. Nat. Rev. Cancer 2021, 21, 619–637. [Google Scholar] [CrossRef]
- Zhou, M.; Leung, A.; Echegaray, S.; Gentles, A.; Shrager, J.B.; Jensen, K.C.; Berry, G.J.; Plevritis, S.K.; Rubin, D.L.; Napel, S.; et al. Non-small cell lung cancer radiogenomics map identifies relationships between molecular and imaging phenotypes with prognostic implications. Radiology 2018, 286, 307–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sacconi, B.; Anzidei, M.; Leonardi, A.; Boni, F.; Saba, L.; Scipione, R.; Anile, M.; Rengo, M.; Longo, F.; Bezzi, M.; et al. Analysis of CT features and quantitative texture analysis in patients with lung adenocarcinoma: A correlation with EGFR mutations and survival rates. Clin. Radiol. 2017, 72, 443–450. [Google Scholar] [CrossRef]
- Rizzo, S.; Petrella, F.; Buscarino, V.; De Maria, F.; Raimondi, S.; Barberis, M.; Fumagalli, C.; Spitaleri, G.; Rampinelli, C.; De Marinis, F.; et al. CT radiogenomic characterization of EGFR, K-RAS, and ALK mutations in non-small cell lung cancer. Eur. Radiol. 2016, 26, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Korn, R.L.; Oklu, R.; Migdal, C.; Gotway, M.B.; Weiss, G.J.; Iafrate, A.J.; Kim, D.W.; Kuo, M.D. ALK molecular phenotype in non-small cell lung cancer: CT radiogenomic characterization. Radiology 2014, 272, 568–576. [Google Scholar] [CrossRef]
- Esteva, A.; Kuprel, B.; Novoa, R.A.; Ko, J.; Swetter, S.M.; Blau, H.M.; Thrun, S. Dermatologist-level classification of skin cancer with deep neural networks. Nature 2017, 542, 115–118. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Liu, X.; Shen, J.; Li, Z.; Sang, Y.; Wu, X.; Zha, Y.; Liang, W.; Wang, C.; Wang, K.; et al. Clinically applicable AI system for accurate diagnosis, quantitative measurements, and prognosis of COVID-19 pneumonia using computed tomography. Cell 2020, 182, 1360. [Google Scholar] [CrossRef]
- Wang, G.; Liu, X.; Shen, J.; Wang, C.; Li, Z.; Ye, L.; Wu, X.; Chen, T.; Wang, K.; Zhang, X.; et al. A deep-learning pipeline for the diagnosis and discrimination of viral, non-viral and COVID-19 pneumonia from chest X-ray images. Nat. Biomed Eng. 2021, 5, 509–521. [Google Scholar] [CrossRef] [PubMed]
- Ardila, D.; Kiraly, A.P.; Bharadwaj, S.; Choi, B.; Reicher, J.J.; Peng, L.; Tse, D.; Etemadi, M.; Ye, W.; Corrado, G.; et al. End-to-end lung cancer screening with three-dimensional deep learning on low-dose chest computed tomography. Nat. Med. 2019, 25, 954–961. [Google Scholar] [CrossRef]
- Wang, C.; Shao, J.; Xu, X.; Yi, L.; Wang, G.; Bai, C.; Guo, J.; He, Y.; Zhang, L.; Yi, Z.; et al. DeepLN: A multi-task ai tool to predict the imaging characteristics, malignancy and pathological subtypes in CT-detected pulmonary nodules. Front. Oncol. 2022, 12, 683792. [Google Scholar] [CrossRef] [PubMed]
- Shao, J.; Wang, G.; Yi, L.; Wang, C.; Lan, T.; Xu, X.; Guo, J.; Deng, T.; Liu, D.; Chen, B.; et al. Deep learning empowers lung cancer screening based on mobile low-dose computed tomography in resource-constrained sites. Front. Biosci. 2022, 27, 212. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Ma, J.; Zhang, S.; Shao, J.; Wang, Y.; Zhou, H.Y.; Song, L.; Zheng, J.; Yu, Y.; Li, W. Development and validation of an abnormality-derived deep-learning diagnostic system for major respiratory diseases. NPJ Digit. Med. 2022, 5, 124. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Yu, H.; Gan, Y.; Wu, Z.; Li, E.; Li, X.; Cao, J.; Zhu, Y.; Wang, L.; Deng, H.; et al. Mining whole-lung information by artificial intelligence for predicting EGFR genotype and targeted therapy response in lung cancer: A multicohort study. Lancet Digit. Health 2022, 4, e309–e319. [Google Scholar] [CrossRef]
- Wang, C.; Xu, X.; Shao, J.; Zhou, K.; Zhao, K.; He, Y.; Li, J.; Guo, J.; Yi, Z.; Li, W. Deep learning to predict EGFR mutation and PD-L1 expression status in non-small-cell lung cancer on computed tomography images. J. Oncol. 2021, 2021, 5499385. [Google Scholar] [CrossRef]
- Wang, C.; Ma, J.; Shao, J.; Zhang, S.; Liu, Z.; Yu, Y.; Li, W. Predicting EGFR and PD-L1 status in NSCLC patients using multitask AI system based on CT images. Front. Immunol. 2022, 13, 813072. [Google Scholar] [CrossRef]
- Zhang, T.; Xu, Z.; Liu, G.; Jiang, B.; de Bock, G.H.; Groen, H.J.M.; Vliegenthart, R.; Xie, X. Simultaneous Identification of EGFR, KRAS, ERBB2, and TP53 mutations in patients with non-small cell lung cancer by machine learning-derived three-dimensional radiomics. Cancers 2021, 13, 1814. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Liu, Z.; Laurens, V.; Weinberger, K.Q. Densely connected convolutional networks. In Proceedings of the IEEE Computer Society, Las Vegas, NV, USA, 27–30 June 2016. [Google Scholar]
- Ronneberger, O.; Fischer, P.; Brox, T. U-Net: Convolutional Networks for Biomedical Image Segmentation; Springer: Berlin/Heidelberg, Germany, 2015. [Google Scholar]
- Zhang, S.; Xu, J.; Chen, Y.-C.; Ma, J.; Li, Z.; Wang, Y.; Yu, Y. Revisiting 3D context modeling with supervised pre-training for universal lesion detection in CT slices. In Proceedings of the Medical Image Computing and Computer Assisted Intervention—MICCAI 2020, Lima, Peru, 4–8 October 2020; pp. 542–551. [Google Scholar]
- Chen, T.; Guestrin, C. XGBoost: A Scalable Tree Boosting System. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, 13–17 August 2016; pp. 785–794. [Google Scholar]
- Liu, Z.; Ning, J.; Cao, Y.; Wei, Y.; Zhang, Z.; Lin, S.; Hu, H. Video swin transformer. arXiv 2022, arXiv:2106.13230. [cs.CV,cs.AI,cs.LG]. [Google Scholar]
- Lundberg, S.; Lee, S.I. A unified approach to interpreting model predictions. Adv. Neural Inf. Process. Syst. 2017, 30. [Google Scholar]
- Coudray, N.; Ocampo, P.S.; Sakellaropoulos, T.; Narula, N.; Snuderl, M.; Fenyö, D.; Moreira, A.L.; Razavian, N.; Tsirigos, A. Classification and mutation prediction from non-small cell lung cancer histopathology images using deep learning. Nat. Med. 2018, 24, 1559–1567. [Google Scholar] [CrossRef] [PubMed]
- Esteva, A.; Robicquet, A.; Ramsundar, B.; Kuleshov, V.; DePristo, M.; Chou, K.; Cui, C.; Corrado, G.; Thrun, S.; Dean, J. A guide to deep learning in healthcare. Nat. Med. 2019, 25, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Boehm, K.M.; Khosravi, P.; Vanguri, R.; Gao, J.; Shah, S.P. Harnessing multimodal data integration to advance precision oncology. Nat. Rev. Cancer 2022, 22, 114–126. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.Y.; Chen, T.Y.; Williamson, D.F.K.; Zhao, M.; Shady, M.; Lipkova, J.; Mahmood, F. AI-based pathology predicts origins for cancers of unknown primary. Nature 2021, 594, 106–110. [Google Scholar] [CrossRef]
- Deng, K.; Wang, L.; Liu, Y.; Li, X.; Hou, Q.; Cao, M.; Ng, N.N.; Wang, H.; Chen, H.; Yeom, K.W.; et al. A deep learning-based system for survival benefit prediction of tyrosine kinase inhibitors and immune checkpoint inhibitors in stage IV non-small cell lung cancer patients: A multicenter, prognostic study. EClinicalMedicine 2022, 51, 101541. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Prediction Task | Dataset | Sensitivity (95%CI) | Specificity (95%CI) | Accuracy (95%CI) | AUC (95%CI) |
|---|---|---|---|---|---|
| Binary | Validation | 0.680 (0.629–0.739) | 0.840 (0.758–0.912) | 0.836 (0.803–0.881) | 0.818 (0.773–0.871) |
| Testing | 0.856 (0.805–0.904) | 0.722 (0.607–0.853) | 0.829 (0.789–0.874) | 0.807 (0.738–0.884) | |
| 8-panel | Validation | 0.814 (0.625–0.980) | 0.833 (0.802–0.868) | 0.951 (0.933–0.971) | 0.831 (0.702–0.949) |
| Testing | 0.691 (0.504–0.888) | 0.882 (0.839–0.921) | 0.928 (0.894–0.959) | 0.809 (0.692–0.927) | |
| 10-panel | Validation | 0.796 (0.656–0.933) | 0.852 (0.810–0.896) | 0.901 (0.869–0.933) | 0.847 (0.762–0.936) |
| Testing | 0.705 (0.496–0.918) | 0.880 (0.836–0.918) | 0.876 (0.836–0.915) | 0.821 (0.703–0.936) | |
| Subtype | Validation | 0.820 (0.640–0.961) | 0.769 (0.642–0.887) | 0.754 (0.646–0.861) | 0.771 (0.606–0.900) |
| Testing | 0.741 (0.443–0.968) | 0.793 (0.654–0.914) | 0.783 (0.682–0.894) | 0.732 (0.536–0.925) |
| Deep Learning Algorithm | Prediction Task | Dataset | Sensitivity (95% CI) | Specificity (95% CI) | Accuracy (95% CI) | AUC (95% CI) |
|---|---|---|---|---|---|---|
| CNN-Based | Binary | Validation | 0.919 (0.879–0.955) | 0.724 (0.621–0.857) | 0.884 (0.854–0.933) | 0.836 (0.777–0.911) |
| Testing | 0.960 (0.924–0.982) | 0.611 (0.464–0.743) | 0.611 (0.464–0.743) | 0.825 (0.682–0.891) | ||
| 8-panel | Validation | 0.767 (0.636–0.883) | 0.906 (0.879–0.933) | 0.943 (0.922–0.963) | 0.869 (0.745–0.926) | |
| Testing | 0.721 (0.588–0.864) | 0.932 (0.907–0.954) | 0.946 (0.926–0.966) | 0.839 (0.757–0.931) | ||
| 10-panel | Validation | 0.743 (0.592–0.902) | 0.932 (0.905–0.956) | 0.937 (0.914–0.960) | 0.848 (0.732–0.921) | |
| Testing | 0.706 (0.563–0.844) | 0.906 (0.877–0.933) | 0.924 (0.900–0.948) | 0.829 (0.724–0.888) | ||
| Subtype | Validation | 0.858 (0.692–0.973) | 0.830 (0.700–0.939) | 0.840 (0.742–0.923) | 0.839 (0.673–0.933) | |
| Testing | 0.881 (0.765–0.972) | 0.764 (0.622–0.885) | 0.786 (0.684–0.884) | 0.810 (0.648–0.915) | ||
| Transformer-Based | Binary | Validation | 0.967 (0.943–0.984) | 0.710 (0.579–0.840) | 0.930 (0.906–0.953) | 0.857 (0.782–0.931) |
| Testing | 0.979 (0.964–0.995) | 0.632 (0.467–0.826) | 0.944 (0.920–0.967) | 0.847 (0.763–0.942) | ||
| 8-panel | Validation | 0.758 (0.598–0.917) | 0.962 (0.940–0.978) | 0.950 (0.927–0.973) | 0.872 (0.774–0.969) | |
| Testing | 0.746 (0.573–0.926) | 0.970 (0.951–0.987) | 0.956 (0.936–0.978) | 0.863 (0.752–0.968) | ||
| 10-panel | Validation | 0.785 (0.597–0.947) | 0.918 (0.886–0.948) | 0.941 (0.913–0.965) | 0.864 (0.743–0.935) | |
| Testing | 0.733 (0.559–0.910) | 0.925 (0.898–0.949) | 0.941 (0.914–0.967) | 0.842 (0.690–0.917) | ||
| Subtype | Validation | 0.749 (0.553–0.958) | 0.941 (0.886–0.988) | 0.883 (0.814–0.957) | 0.855 (0.701–0.912) | |
| Testing | 0.760 (0.592–0.924) | 0.932 (0.877–0.975) | 0.862 (0.796–0.936) | 0.843 (0.718–0.924) |
| Prediction Task | Dataset | Sensitivity (95% CI) | Specificity (95% CI) | Accuracy (95% CI) | AUC (95% CI) |
|---|---|---|---|---|---|
| Binary | Validation | 0.918 (0.891–0.952) | 0.774 (0.667–0.903) | 0.930 (0.906–0.958) | 0.894 (0.837–0.954) |
| Testing | 0.990 (0.979–1.000) | 0.722 (0.550–0.905) | 0.962 (0.939–0.986) | 0.877 (0.794–0.961) | |
| 8-panel | Validation | 0.829 (0.669–0.986) | 0.927 (0.900–0.955) | 0.956 (0.934–0.978) | 0.896 (0.802–0.983) |
| Testing | 0.759 (0.591–0.933) | 0.948 (0.922–0.973) | 0.954 (0.933–0.977) | 0.862 (0.758–0.969) | |
| 10-panel | Validation | 0.827 (0.678–0.945) | 0.914 (0.881–0.947) | 0.948 (0.923–0.972) | 0.891 (0.756–0.952) |
| Testing | 0.797 (0.623–0.947) | 0.953 (0.929–0.975) | 0.953 (0.928–0.976) | 0.856 (0.663–0.948) | |
| Subtype | Validation | 0.870 (0.689–0.987) | 0.858 (0.761–0.952) | 0.842 (0.748–0.921) | 0.879 (0.761–0.962) |
| Testing | 0.850 (0.642–0.977) | 0.902 (0.794–0.976) | 0.876 (0.778–0.951) | 0.868 (0.641–0.972) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shao, J.; Ma, J.; Zhang, S.; Li, J.; Dai, H.; Liang, S.; Yu, Y.; Li, W.; Wang, C. Radiogenomic System for Non-Invasive Identification of Multiple Actionable Mutations and PD-L1 Expression in Non-Small Cell Lung Cancer Based on CT Images. Cancers 2022, 14, 4823. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14194823
Shao J, Ma J, Zhang S, Li J, Dai H, Liang S, Yu Y, Li W, Wang C. Radiogenomic System for Non-Invasive Identification of Multiple Actionable Mutations and PD-L1 Expression in Non-Small Cell Lung Cancer Based on CT Images. Cancers. 2022; 14(19):4823. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14194823
Chicago/Turabian StyleShao, Jun, Jiechao Ma, Shu Zhang, Jingwei Li, Hesen Dai, Shufan Liang, Yizhou Yu, Weimin Li, and Chengdi Wang. 2022. "Radiogenomic System for Non-Invasive Identification of Multiple Actionable Mutations and PD-L1 Expression in Non-Small Cell Lung Cancer Based on CT Images" Cancers 14, no. 19: 4823. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14194823