
The Expression of the Immunoproteasome Subunit PSMB9 Is Related to Distinct Molecular Subtypes of Uterine Leiomyosarcoma

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Cohort
2.2. RNA-Seq Data Collection
2.3. RNA and Protein Extraction and Library Preparation
2.4. Differential Expression and Enrichment Analysis
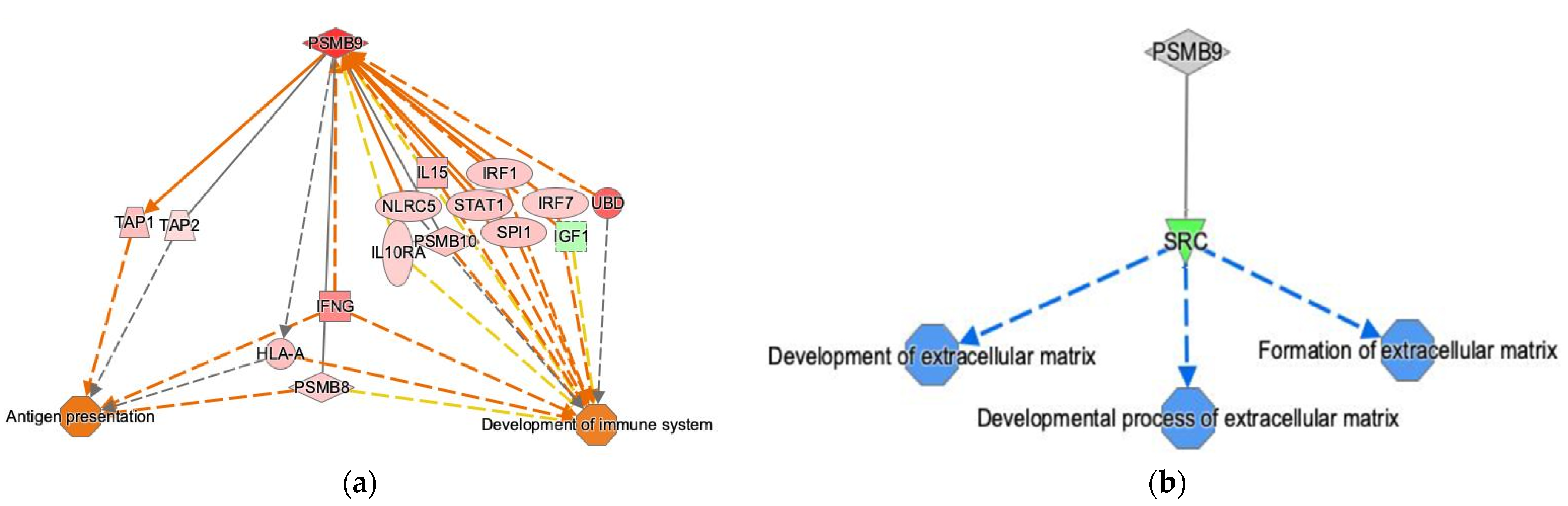
2.5. Ingenuity Pathway Analysis
2.6. Data Integration
2.7. Cell Line Expression
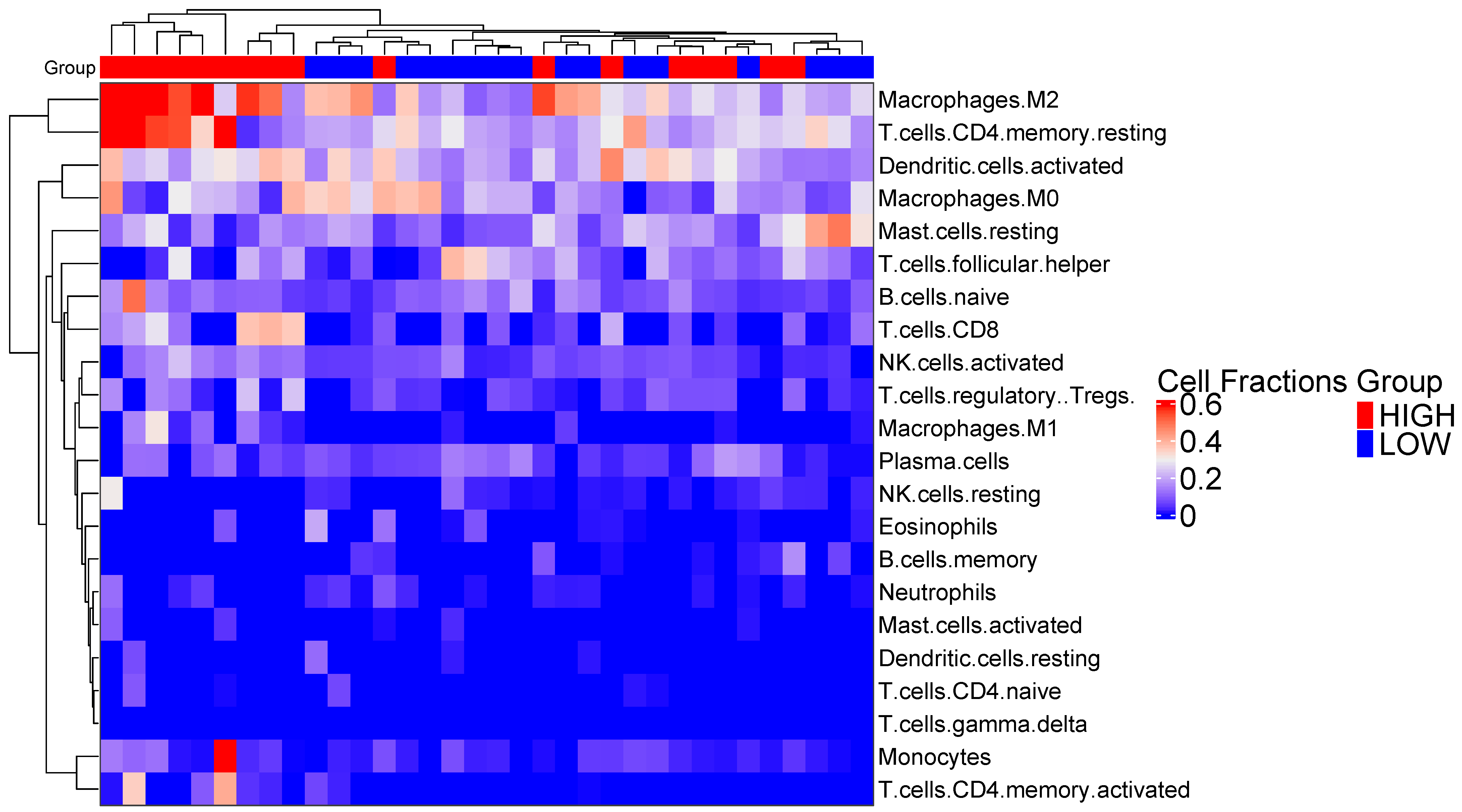
2.8. Immune Cell Subtype Related to Immune Activity
2.9. Survival and Statistical Analysis
3. Results
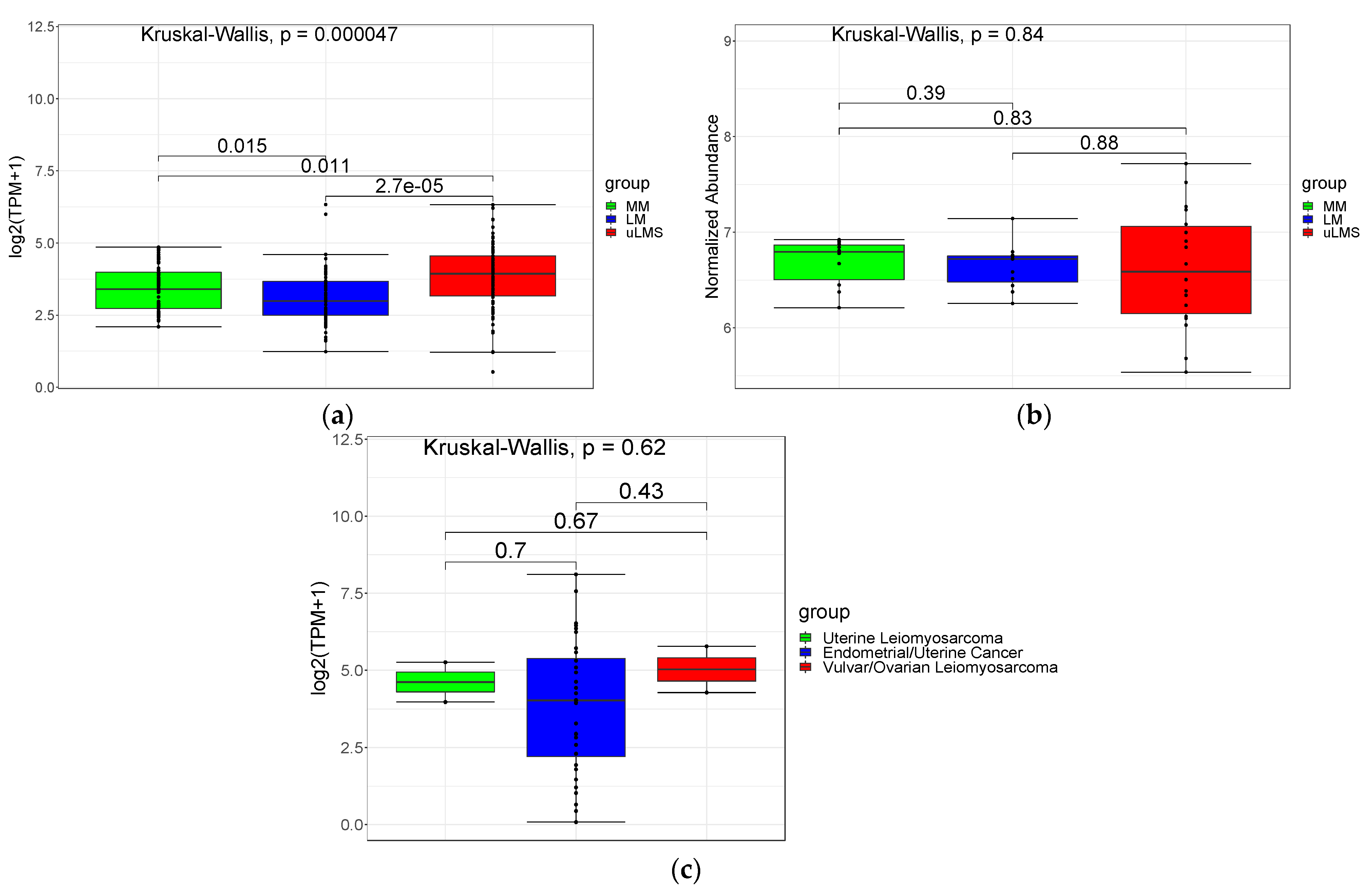
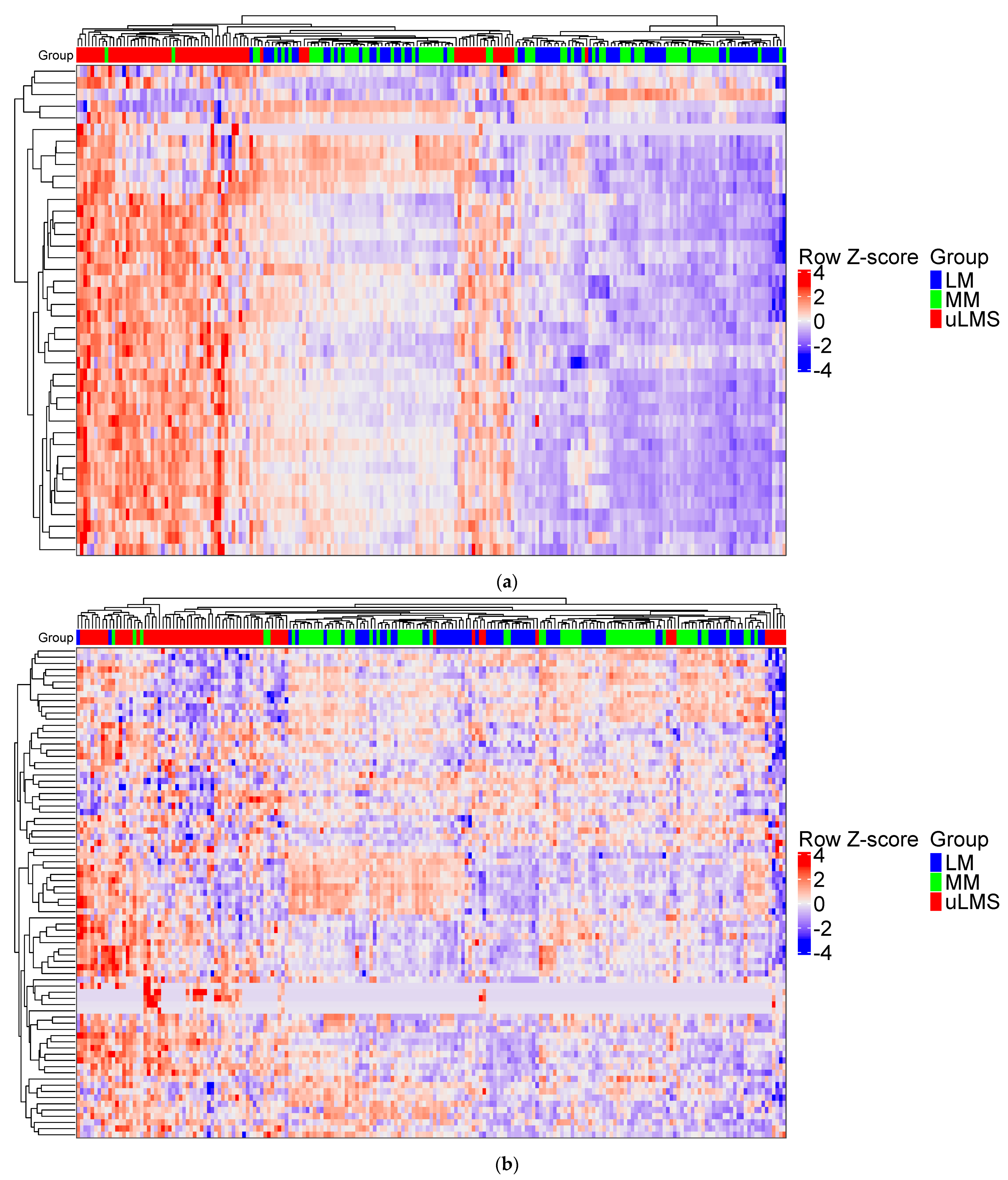
3.1. PSMB9 and the Proteasome Pathway Are Highly Expressed in uLMS, but Not in LM
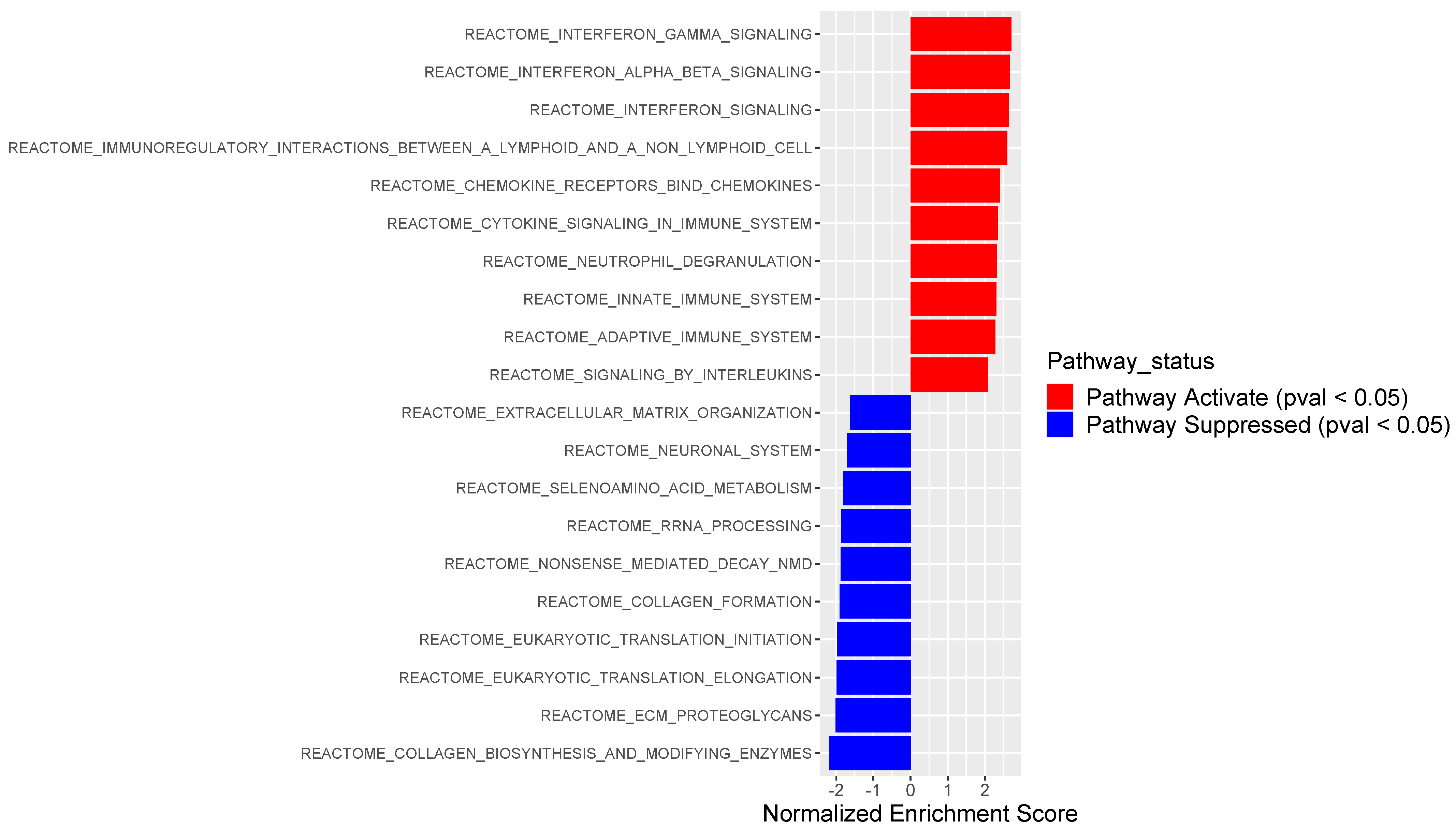
3.2. PSMB9 Expression Level Groups Low or High Are Enriched for ECM and IFNγ Pathways, Respectively
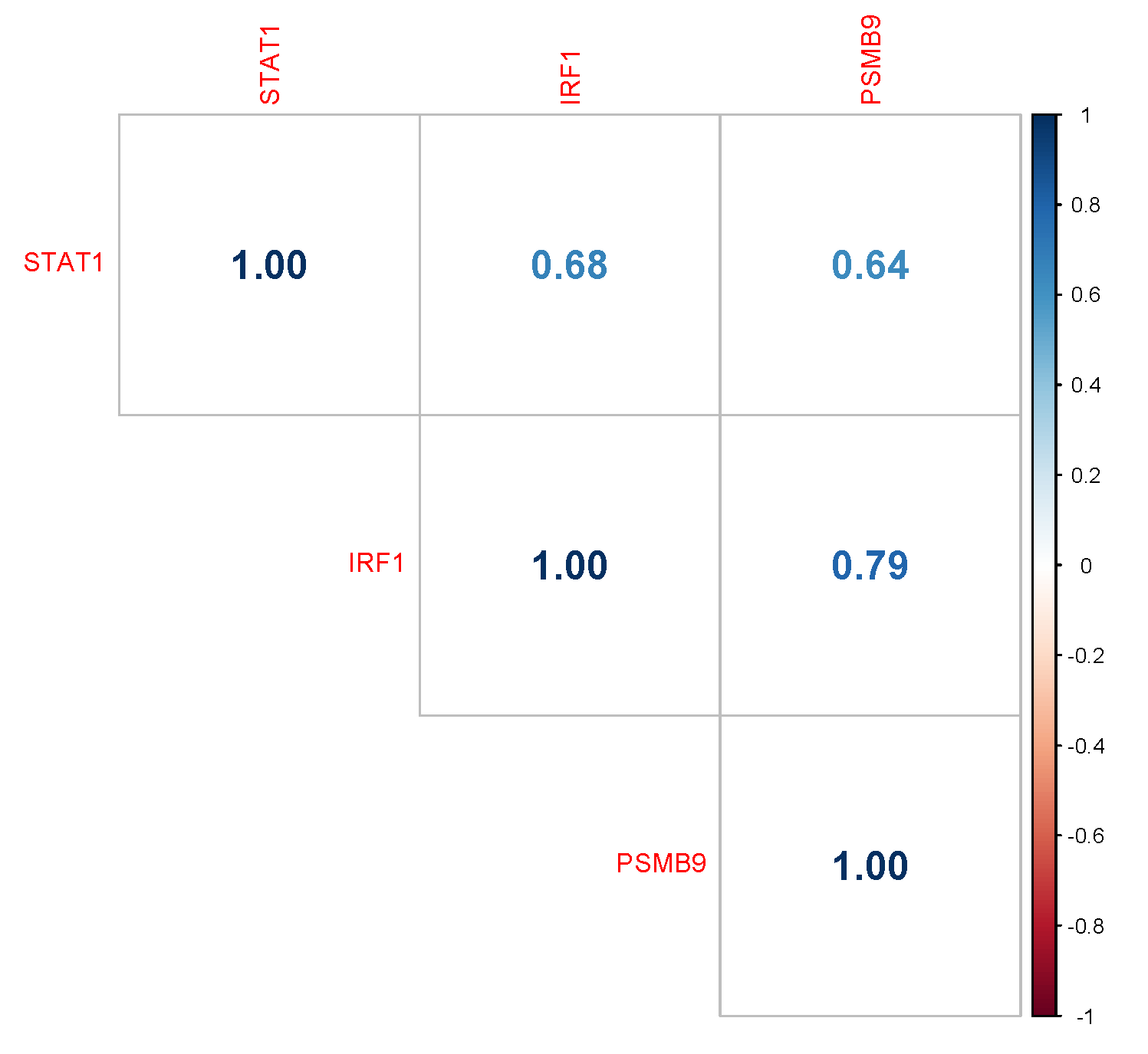
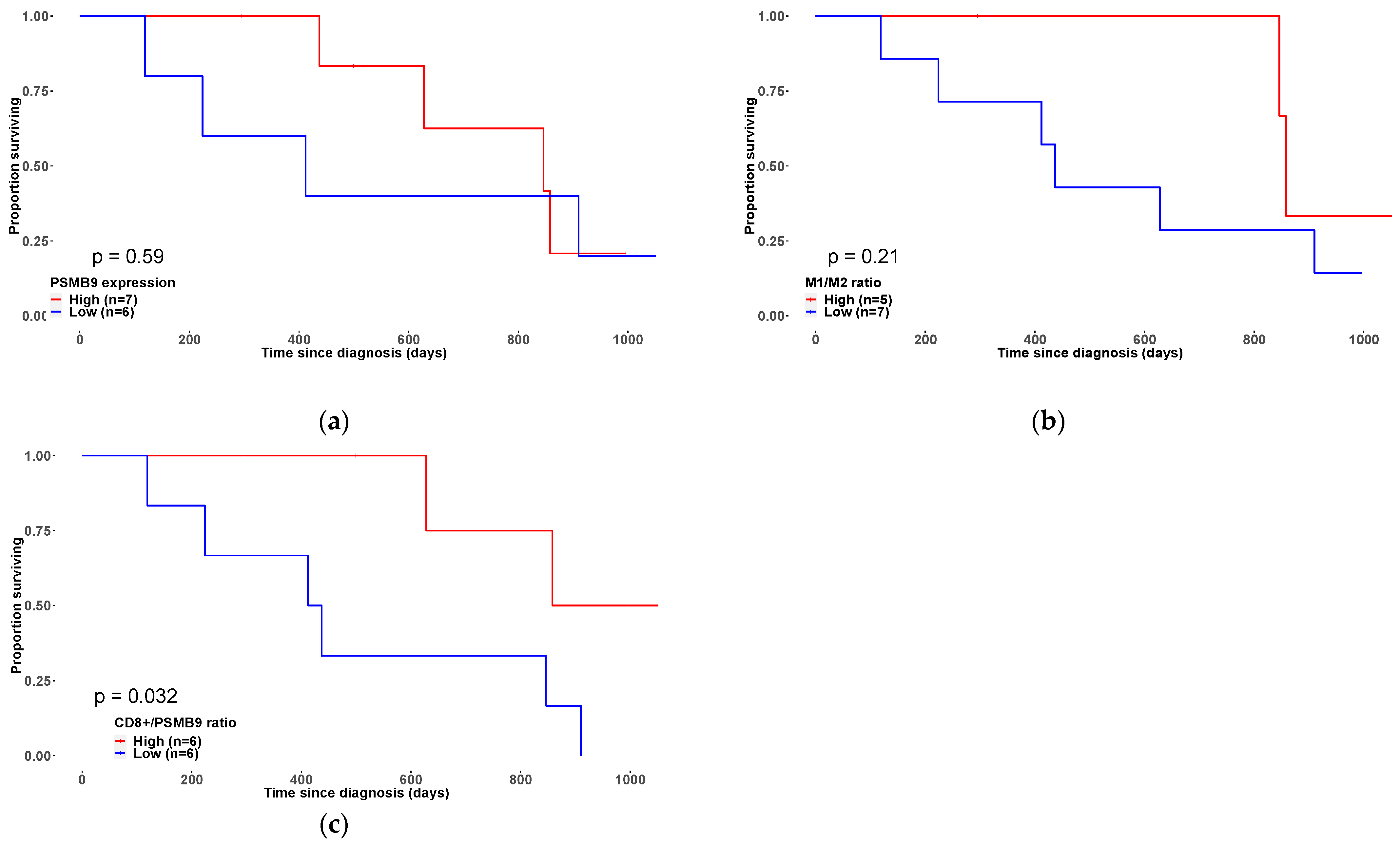
3.3. Overexpression of the Immunoproteasome PSMB9 Subunit Is Correlated with uLMS Patients’ Survival Outcome
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- George, S.; Serrano, C.; Hensley, M.L.; Ray-Coquard, I. Soft tissue and uterine leiomyosarcoma. J. Clin. Oncol. 2018, 36, 144–150. [Google Scholar] [CrossRef]
- Toro, J.R.; Travis, L.B.; Wu, H.J.; Zhu, K.; Fletcher, C.D.; Devesa, S.S. Incidence patterns of soft tissue sarcomas, regardless of primary site, in the surveillance, epidemiology and end results program, 1978–2001: An analysis of 26,758 cases. Int. J. Cancer 2006, 119, 2922–2930. [Google Scholar] [CrossRef]
- Hosh, M.; Antar, S.; Nazzal, A.; Warda, M.; Gibreel, A.; Refky, B. Uterine sarcoma: Analysis of 13,089 cases based on surveillance, epidemiology, and end results database. Int. J. Gynecol. Cancer 2016, 26, 1098–1104. [Google Scholar] [CrossRef]
- Bosch, T.V.D.; Coosemans, A.; Morina, M.; Timmerman, D.; Amant, F. Screening for uterine tumours. Best Pr. Res. Clin. Obstet. Gynaecol. 2012, 26, 257–266. [Google Scholar] [CrossRef]
- Trope, C.G.; Abeler, V.M.; Kristensen, G.B. Diagnosis and treatment of sarcoma of the uterus. A review. Acta Oncol. 2012, 51, 694–705. [Google Scholar] [CrossRef] [Green Version]
- Swanton, C. Intratumor heterogeneity: Evolution through space and time. Cancer Res. 2012, 72, 4875–4882. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, T.; Horiuchi, A.; Sano, K.; Hiraoka, N.; Kanai, Y.; Shiozawa, T.; Tonegawa, S.; Konishi, I. Molecular approach to uterine leiomyosarcoma: LMP2-deficient mice as an animal model of spontaneous uterine leiomyosarcoma. Sarcoma 2011, 2011, 476498. [Google Scholar] [CrossRef] [Green Version]
- Leone, P.; Shin, E.-C.; Perosa, F.; Vacca, A.; Dammacco, F.; Racanelli, V. MHC Class I Antigen processing and presenting machinery: Organization, function, and defects in tumor cells. JNCI J. Natl. Cancer Inst. 2013, 105, 1172–1187. [Google Scholar] [CrossRef] [Green Version]
- Basler, M.; Groettrup, M. On the role of the immunoproteasome in protein homeostasis. Cells 2021, 10, 3216. [Google Scholar] [CrossRef]
- Angeles, A.; Fung, G.; Luo, H. Immune and non-immune functions of the immunoproteasome. Front. Biosci. 2012, 17, 1904–1916. [Google Scholar] [CrossRef]
- McCarthy, M.K.; Weinberg, J.B. The immunoproteasome and viral infection: A complex regulator of inflammation. Front. Microbiol. 2015, 6, 21. [Google Scholar] [CrossRef] [Green Version]
- Van Kaer, L.; Ashton-Rickardt, P.G.; Eichelberger, M.; Gaczynska, M.; Nagashima, K.; Rock, K.L.; Goldberg, A.L.; Doherty, P.C.; Tonegawa, S. Altered Peptidase and Viral-Specific T Cell Response in LMP2 Mutant Mice. Immunity 1994, 1, 533–541. [Google Scholar]
- Ferrington, D.A.; Gregerson, D.S. Immunoproteasomes: Structure, function, and antigen presentation. Prog. Mol. Biol. Transl. Sci. 2012, 109, 75–112. [Google Scholar] [CrossRef] [Green Version]
- Brucet, M.; Marqués, L.; Sebastián, C.; Lloberas, J.; Celada, A. Regulation of murine Tap1 and Lmp2 genes in macrophages by interferon gamma is mediated by STAT1 and IRF-1. Genes Immun. 2004, 5, 26–35. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, T.; Horiuchi, A.; Sano, K.; Hiraoka, N.; Kasai, M.; Ichimura, T.; Sudo, T.; Tagawa, Y.-I.; Nishimura, R.; Ishiko, O.; et al. Potential role of PSMB9 as tumor-suppressor defines new targets for uterine leiomyosarcoma therapy. Sci. Rep. 2011, 1, 180. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, N.; Taniguchi, T. The interferon regulatory factors and oncogenesis. Semin. Cancer Biol. 2000, 10, 73–81. [Google Scholar] [CrossRef]
- Hayashi, T.; Horiuchi, A.; Sano, K.; Yaegashi, N.; Konishi, I. Uterine leiomyosarcoma tumorigenesis in PSMB9-deficient mice: Involvement of impaired anti-oncogenic factor IRF1. Anticancer Res. 2015, 35, 4665–4679. [Google Scholar] [PubMed]
- Newman, A.M.; Steen, C.B.; Liu, C.L.; Gentles, A.J.; Chaudhuri, A.A.; Scherer, F.; Khodadoust, M.S.; Esfahani, M.S.; Luca, B.A.; Steiner, D.; et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 2019, 37, 773–782. [Google Scholar] [CrossRef]
- Kalaora, S.; Lee, J.S.; Barnea, E.; Levy, R.; Greenberg, P.; Alon, M.; Yagel, G.; Bar Eli, G.; Oren, R.; Peri, A.; et al. Immunoproteasome expression is associated with better prognosis and response to checkpoint therapies in melanoma. Nat. Commun. 2020, 11, 896. [Google Scholar] [CrossRef] [Green Version]
- Shan, W.; Akinfenwa, P.Y.; Savannah, K.B.; Kolomeyevskaya, N.; Laucirica, R.; Thomas, D.G.; Odunsi, K.; Creighton, C.J.; Lev, D.C.; Anderson, M.L. A small-molecule inhibitor targeting the mitotic spindle checkpoint impairs the growth of uterine leiomyosarcoma. Clin. Cancer Res. 2012, 18, 3352–3365. [Google Scholar] [CrossRef] [Green Version]
- Beck, A.H.; Lee, C.-H.; Witten, D.M.; Gleason, B.C.; Edris, B.; Espinosa, I.; Zhu, S.; Li, R.; Montgomery, K.D.; Marinelli, R.J.; et al. Discovery of molecular subtypes in leiomyosarcoma through integrative molecular profiling. Oncogene 2009, 29, 845–854. [Google Scholar] [CrossRef]
- Skubitz, K.M.; Skubitz, A. Differential gene expression in leiomyosarcoma. Cancer 2003, 98, 1029–1038. [Google Scholar] [CrossRef]
- Barlin, J.N.; Zhou, Q.C.; Leitao, M.M.; Bisogna, M.; Olvera, N.; Shih, K.K.; Jacobsen, A.; Schultz, N.; Tap, W.D.; Hensley, M.L.; et al. Molecular subtypes of uterine leiomyosarcoma and correlation with clinical outcome. Neoplasia 2015, 17, 183–189. [Google Scholar] [CrossRef] [Green Version]
- Zang, Y.; Gu, L.; Zhang, Y.; Wang, Y.; Xue, F. Identification of key genes and pathways in uterine leiomyosarcoma through bioinformatics analysis. Oncol. Lett. 2018, 15, 9361–9368. [Google Scholar] [CrossRef]
- Binzer-Panchal, A.; Hardell, E.; Viklund, B.; Ghaderi, M.; Bosse, T.; Nucci, M.R.; Lee, C.-H.; Hollfelder, N.; Corcoran, P.; Gonzalez-Molina, J.; et al. Integrated molecular analysis of undifferentiated uterine sarcomas reveals clinically relevant molecular subtypes. Clin. Cancer Res. 2019, 25, 2155–2165. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, S.C.; Vedpathak, D.; Ostrin, E.J. The functional and mechanistic roles of immunoproteasome subunits in cancer. Cells 2021, 10, 3587. [Google Scholar] [CrossRef]
- Hayashi, T.; Faustman, D.L. Development of spontaneous uterine tumors in low molecular mass polypeptide-2 knockout mice. Cancer Res. 2002, 62, 24–27. [Google Scholar]
- McBurney, M.W.; Sutherland, L.C.; Adra, C.N.; LeClair, B.; Rudnicki, M.A.; Jardine, K. The mouse Pgk-1 gene promoter contains an upstream activator sequence. Nucleic Acids Res. 1991, 19, 5755–5761. [Google Scholar] [CrossRef] [Green Version]
- Valera, A.; Perales, J.C.; Hatzoglou, M.; Bosch, F. Expression of the neomycin-resistance (neo) gene induces alterations in gene expression and metabolism. Hum. Gene Ther. 1994, 5, 449–456. [Google Scholar]
- Bhat, M.Y.; Solanki, H.S.; Advani, J.; Khan, A.A.; Prasad, T.S.K.; Gowda, H.; Thiyagarajan, S.; Chatterjee, A. Comprehensive network map of interferon gamma signaling. J. Cell Commun. Signal. 2018, 12, 745–751. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maia Falcão, R.; Kokaraki, G.; De Wispelaere, W.; Amant, F.; De Souza, G.A.; de Souza, J.E.S.; Carlson, J.W.; Petta, T.B. The Expression of the Immunoproteasome Subunit PSMB9 Is Related to Distinct Molecular Subtypes of Uterine Leiomyosarcoma. Cancers 2022, 14, 5007. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14205007
Maia Falcão R, Kokaraki G, De Wispelaere W, Amant F, De Souza GA, de Souza JES, Carlson JW, Petta TB. The Expression of the Immunoproteasome Subunit PSMB9 Is Related to Distinct Molecular Subtypes of Uterine Leiomyosarcoma. Cancers. 2022; 14(20):5007. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14205007
Chicago/Turabian StyleMaia Falcão, Raul, Georgia Kokaraki, Wout De Wispelaere, Frédéric Amant, Gustavo Antônio De Souza, Jorge Estefano Santana de Souza, Joseph Woodward Carlson, and Tirzah Braz Petta. 2022. "The Expression of the Immunoproteasome Subunit PSMB9 Is Related to Distinct Molecular Subtypes of Uterine Leiomyosarcoma" Cancers 14, no. 20: 5007. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14205007