
GOF Mutant p53 in Cancers: A Therapeutic Challenge


1
CRUK Manchester, University of Manchester, Alderley Park, Manchester SK10 4TG, UK
2
Department of Biosciences, Durham University, Stockton Road, Durham DH1 3LE, UK
*
Author to whom correspondence should be addressed.
Cancers 2022, 14(20), 5091; https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14205091
Submission received: 24 September 2022
/
Revised: 13 October 2022
/
Accepted: 14 October 2022
/
Published: 18 October 2022
(This article belongs to the Special Issue Targeting Therapies for the p53 Protein in Cancer Treatments)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
In normal cells, p53 is a protein which regulates the cell cycle progression to ensure normal cell division, growth, and development. However, in cancer, changes in the p53 DNA sequence, called genetic mutation, results in the protein either losing its normal function or exhibiting advanced pro-tumorigenic functions that lead to cancer. Importantly, cancers with mutations in the p53 protein often represent ones which are more aggressive and more resistant to chemotherapy. As a result, many studies have and continue to investigate multiple ways to target mutant p53-bearing cancer using targeted therapy, gene therapy, immunotherapy, and combination therapies. Knowledge of these strategies is important in improving the overall therapeutic response of cancers with mutant p53. This review highlights new strategies and discusses the progression of such therapies.
Abstract
TP53 is mutated in the majority of human cancers. Mutations can lead to loss of p53 expression or expression of mutant versions of the p53 protein. These mutant p53 proteins have oncogenic potential. They can inhibit any remaining WTp53 in a dominant negative manner, or they can acquire new functions that promote tumour growth, invasion, metastasis and chemoresistance. In this review we explore some of the mechanisms that make mutant p53 cells resistant to chemotherapy. As mutant p53 tumours are resistant to many traditional chemotherapies, many have sought to explore new ways of targeting mutant p53 tumours and reinstate chemosensitivity. These approaches include targeting of mutant p53 stability, mutant p53 binding partners and downstream pathways, p53 vaccines, restoration of WTp53 function, and WTp53 gene delivery. The current advances and challenges of these strategies are discussed.
1. p53 and Mutations in Cancers
p53 is a tumour suppressor protein and nuclear transcription factor (53 kDa) regulating target genes and involved in apoptosis, senescence, cell cycle arrest, and DNA repair. In response to low doses of genomic stress, both extrinsic (e.g., UV-induced DNA damage) and intrinsic (e.g., chromosomal aberrations), p53 regulates cell cycle arrest to allow for DNA repair [1,2,3]. In response to high doses of stress, p53 is more likely to promote apoptosis. Importantly, many chemotherapeutics act by inducing this stress-induced cell death function of p53 to destroy tumour cells.
In the absence of stress, p53 protein expression is kept at low levels [4]. This is facilitated by the E3 ubiquitin ligase MDM-2 (mouse double minute-2) that ubiquitinates p53 leading to its degradation. In response to DNA damage, p53 is released from MDM2 suppression allowing for p53-mediated transcription. MDM-2 limits p53 expression whilst p53 directly promotes MDM-2 expression. This creates an autofeedback loop that allows for a fast and dynamic signalling response to react to differences in stress quickly (Figure 1) [5,6].
TP53 mutation occurs in ~50–60% of all human cancers and can result in both the absence of protein expression or the expression of a mutated protein [7]. p53 mutational status within tumours is heterogeneous and the onset of TP53 mutations can vary greatly in different cancers. As an example, in colorectal [8], breast [9], and pancreatic [10] cancers, TP53 mutation is marked as a late stage tumourigenic event aiding more with tumour progression than with tumour initiation, while in pre-malignant breast lesions [11], hepatocellular carcinoma [12], and in astrocytoma [13] TP53 mutations present during the early stages of tumorigenesis.
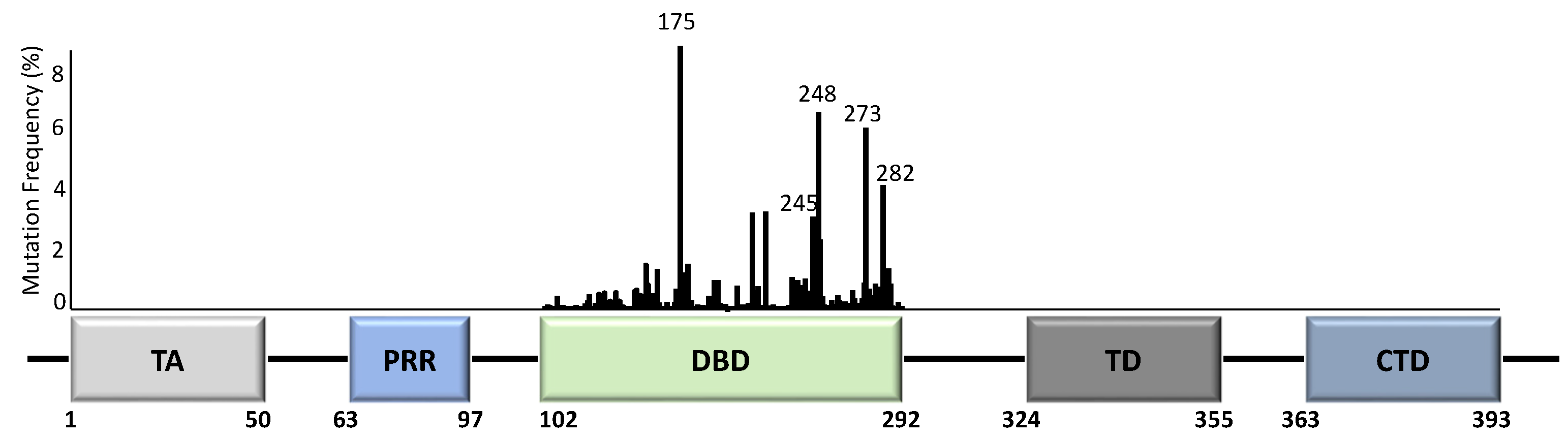
Unlike most other tumour suppressor genes, TP53 mutations often affect a single allele with loss of expression from the remaining allele [14]. This occurs via deletion of part of chromosome 17p [15], methylation of the second allele [16], or through additional mutations [17]. Principally, whilst the presence of TP53 mutations span across almost all of its 393 aa residues (Figure 2), the specificity and frequency of the >25,000 registered TP53 mutations can be differential based on the tumour type, with individual mutants often showing different phenotypical changes [18,19]. Importantly, most mutations are found in the DNA-binding domain (DBD) with six hotspot mutations at codons 175, 245, 248, 249, 273, and 282 (Figure 2) [14,20].
TP53 mutations can cause truncations or frameshifts in TP53 that almost always result in loss of p53 expression. Missense mutations generally result in expression of mutant proteins with one amino acid variation from WTp53 [14,18]. This generates a stable mutant p53 protein with longer half-life, seen as increased expression in human cancers [21]. These mutant proteins, including all hotspots, can have alterations in the protein’s structure such as unfolding of the DBD (conformational/structural mutants) [22] or a decreased DNA binding ability (contact mutants) [18].
Mutant p53 proteins often lose some or all of p53’s tumour suppressive function (loss-of-function, LOF) but may also acquire gain-of-function (GOF). This GOF resembles an oncogenic phenotype and is independent of WTp53 [18]. We and others have shown that mutant p53 promotes invasion and metastasis, tumour growth, genomic instability and chemoresistance [23,24,25,26], via a multitude of different mechanisms (reviewed in [27,28,29,30]). Mutant p53 proteins can further have dominant-negative effects over the remaining WT protein [20,28]. This was attributed to mutp53’s ability to form hetero–tetramer complexes with the WTp53 protein [20], causing multimer inactivation [31]. This was seen for both contact and conformational p53 mutants [20].
2. Mutant p53 and Chemoresistance
Mutp53 forms a challenging anti-cancer therapeutic target, mainly due to its lack of druggable allosteric sites, the occurrence of thermodynamically disrupted states as well as its intrinsic ability to confer drug resistance [24,32,33].
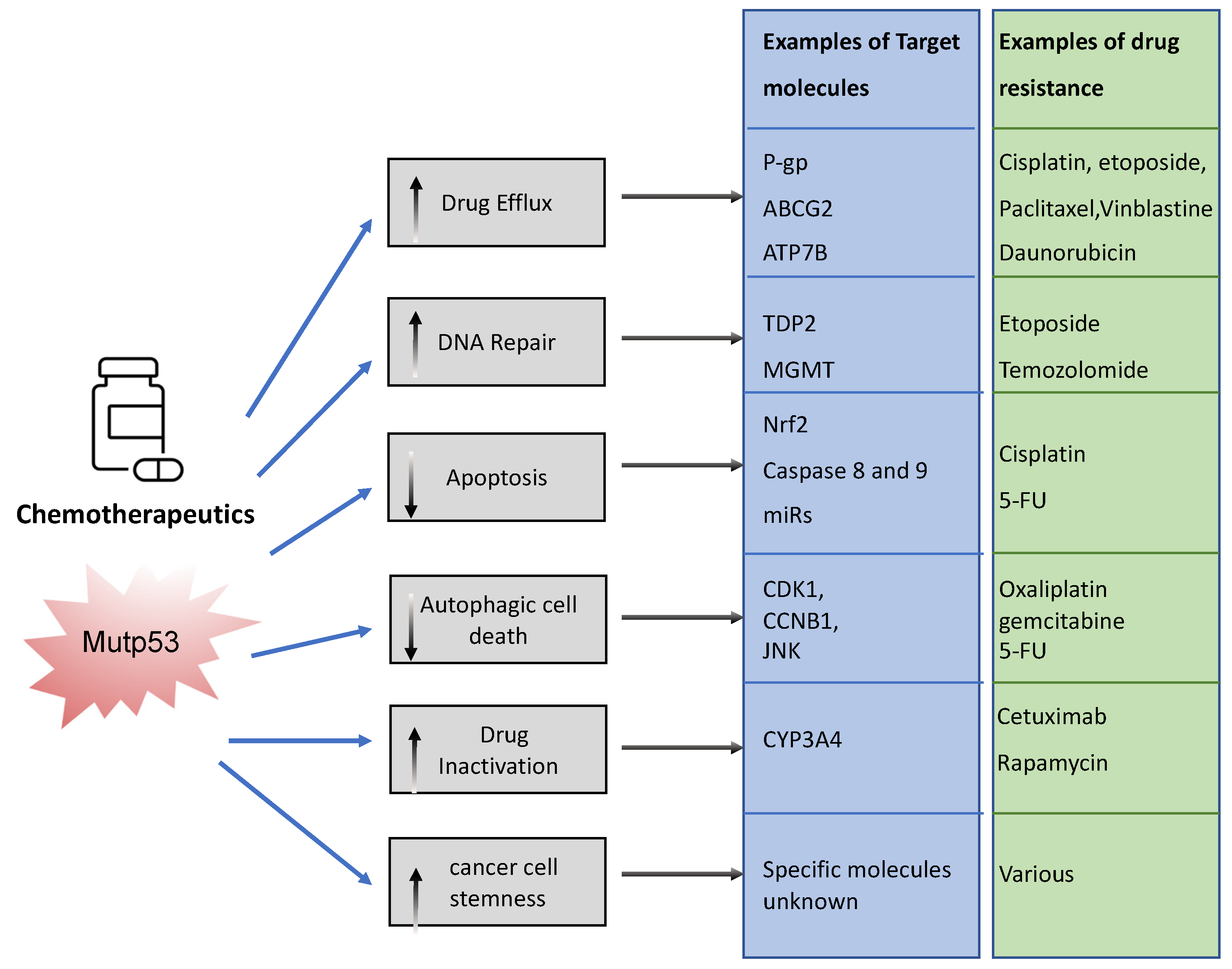
The association between mutp53 expression and decreased chemosensitivity is seen in various primary cancers, including breast [34], ovarian [35], lung [36], and hematopoietic [37]. Loss of WTp53 expression can underlie this chemoresistance, but there are also ways in which mutant p53 acquires chemoresistance via its GOF [33]. Mechanisms include, but are not limited to, upregulation of drug transporters, enhanced DNA repair, activation of stemness, apoptosis avoidance, and drug inactivation (Figure 3).
To limit toxicity of drugs, mutp53 can directly act on drug availability by regulating drug efflux or drug stability. Mutp53 promotes expression of the (MDR1) gene encoding for the ATP-binding cassette (ABC) transmembrane transporter ABCB1/P-glycoprotein (P-gp) [38]. P-gp extrudes xenobiotic substances/toxic compounds and chemotherapeutic drugs [39]. The transcriptional upregulation of P-gp in cancer is driven by GOF-mutp53’s direct binding to MDR1′s promoter [40]. This enhances drug efflux, reduces drug absorption, and minimizes drug retention/accumulation, causing resistance to anti-cancer drugs, such as taxanes (paclitaxel), vinca alkaloids (vinblastine), and anthracyclines (daunorubicin) [41]. Of note, other ABC transporters such as ABCG2 can also be upregulated by mutant p53 to enhance secretion of 5-flouracil (5-FU) [42]. Interestingly, mutant p53 does not only regulate the gene expression of such transporters. We recently discovered that mutp53 also specifically enhances plasma membrane expression of P-gp and ATP7B in response to cisplatin and etoposide to enhance efflux, perhaps working in concert with transcriptional regulation of such transporters [24,26]. GOF-mutp53 (R248W and R282W) can also directly inactivate chemotoxic drugs by upregulating cytochrome P450 enzyme 3A4 (CYP3A4) that help neutralize these drugs [43].
In order to limit toxicity of drugs, mutp53 intercepts in many downstream signalling pathways. In response to drug-induced DNA damage, mutant p53 promotes DNA repair. As an example, GOF-mutp53 (R175H and R248Q) promoted etoposide resistance by enhancing tyrosyl-DNA phosphodiesterase 2′s (TDP2) expression in lung cancer cells in an Ets2 dependent manner [44]. TDP2 in turn repaired etoposide induced double-strand DNA breaks [45], resulting in chemoresistance. Likewise, GOF-mutp53 upregulated the expression of O(6)-methylguanine-DNA-methyltransferase (MGMT) in glioblastoma, enabling the repair of alkylation induced DNA damage by temozolomide [46]. Mutp53 can also directly or indirectly prevent apoptosis. Transcriptionally, it can upregulate Nrf2 (nuclear factor erythroid 2-related factor 2) in response to cisplatin to induce expression of the anti-apoptotic mitochondrial genes: Bcl2 and Bcl-xL [47]. Alternatively, GOF-mutp53’s apoptotic resistance can also occur via direct inhibition of caspases 8 and 9 [48,49,50] or through transcriptional upregulation of miRs that target the apoptosis machinery [51]. Many of the chemotherapeutics are known to cause autophagic cell death through apoptosis. Mutp53 can avoid apoptosis by inducing autophagy via the mTor/AMPK signalling pathway [52], although autophagy itself also regulates mutp53 expression (see Section 3.1.2).
It is likely that in an actual cancer, mutant p53 employs one or more of these mechanisms to combat chemotherapeutics, resulting in selection for p53 mutations. In fact, selection of mutp53 is driven by the fact that mutp53 actively promotes stemness [53]. This could be seen as promoting chemoresistance because cancer stem cells are relatively quiescent and therefore less vulnerable to chemotherapy that predominantly acts on highly proliferative cells [53].
3. Strategies That Exploit Mutant p53 Expression
Numerous current and previous studies have explored targeted treatment strategies that either directly target mutp53 or exploit the cancers’ dependence on pathways that rely on mutp53 expression [54]. However, so far, very few of these have progressed to pre-clinical/clinical trial studies and many have resorted to trialling combinations of various treatments.
In this review, we provide an updated review on potential therapeutic strategies, both current and new, that can be employed in mutp53-bearing cancers. The reviewed strategies are grouped under the three main types of p53 specific anti-cancer treatment approaches: mutant p53-targeted therapy, gene delivery therapy, and immunotherapy (Figure 4). We also discuss the option to combine these therapies.
3.1. Mutant p53-Specific Targeted Therapy
Most mutant p53 targeting strategies in cancers have focused on incorporating one of three key mechanisms: reactivation of mutp53 into a WTp53-like state [55,56,57], degradation of mutp53 [58,59,60], or perturbation of mutp53’s function while reactivating WT function [61]. These mechanisms are appealing because expression of mutant p53 in cancers is often high, whereas WTp53 expression in normal tissue is low [62], allowing for specific targeting of mutant p53 with minimal side effects.
3.1.1. Reactivating Mutant p53 to Behave like a WT Molecule
Most common p53 missense mutations, including all hotspots have a complete or partial loss of WT function [63,64]. Many mutants exist in a distorted conformation that prevents them from binding to the DNA and exert p53 function. The p53 hotspot mutant R175H, a conformational mutant, has been most extensively studied. Murine studies showed that expressing WTp53 or restoring WTp53 expression causes tumour regression [56], tumour clearance [57], senescence [55,57] and increased survival [56], suggesting that reactivation therapies in which mutants are converted to a WT molecule could work in vivo.
The earliest discovery of a small molecule compound that was reported to mediate mutp53 reactivation was CP31398 [65,66]. The compound was initially suggested to bind to mutp53’s core domain [66], although a later study contested this binding and instead suggested the compound functioned as a DNA intercalating agent that exhibits both p53-dependent and -independent anti-tumour activity [67]. Subsequent studies then discovered molecules such as PRIMA-1, APR-246, MIRA-1, and STIMA-1, which are called Michael acceptors due to their ability to bind covalently with p53’s cysteine residues [68,69]. This results in an enhanced thermal stability of reactivated mutp53 in a WTp53-like folding state [69] and tumour growth inhibition [65,70,71]. APR-246, the methylated form of PRIMA-1 is one of the few p53 specific targeted therapies which has progressed to clinical trials [65,70,72]. Another p53-specific targeted therapy in clinical trials is COTI-2, a thiosemicarbazone compound, which primarily induces Zn+2 chelation-mediated p53 refolding, restoring p53’s DNA binding capacity [73,74]. However, the molecule’s pro-apoptotic effect was reported to be both p53-dependent and -independent in pre-clinical models [73,75]. More recently, arsenic trioxide, a cysteine reactive compound stabilizes the DNA-binding loop–sheet–helix motif alongside the overall β-sandwich fold of p53 structural mutants through covalent binding [76]. This drug restored p53’s transcriptional activity both in vitro and in vivo and is currently in clinical trial for patients with AML (acute myeloid leukaemia) [76,77].
Compounds that target specific p53 mutants using covalent binding have also been developed. PK083 and PK7088 were synthesized to target the base substitution-induced cavity in hotspot Y220C p53 mutants [71]. In particular, PK7088 was found to promote re-folding of the Y220C p53 mutant with subsequent induction of p53 target genes’ (p21 and NOXA) expression [71].
Compounds such as SCH529074 [78] and peptides like CDB3 [79] were developed to act as chaperones by non-covalently binding to mutp53’s DBD and consequently restoring WTp53 activity. Interestingly, a similar non-covalent binding could also be demonstrated by using peptides such as pCAPs, that change the equilibrium between unfolded and folded p53 states with their stronger binding affinity (non-covalent) to WTp53 over mutp53 (R175H and R273H) [80].
The interest of researchers in almost all of these drugs originates from the notion that mutp53 expression in cancers is so high that, upon conversion into a WTp53-like molecule, a death response is likely initiated, making such compounds likely to act even in the absence of additional therapies. However, they are generally explored in conjunction with conventional chemotherapy to further enhance WTp53-dependent cell death. Although these compounds were designed to target structural mutants, DNA contact mutants as well as WTp53 can under certain conditions unfold (e.g., in hypoxia) [81]. This would suggest that these compounds could target other mutants, dependent on the tumour environment.
In many tumours, instead of missense mutations, p53 expression is lost due to nonsense TP53 mutations, such as Q192X and E298X, resulting in truncated p53 mRNA expression, nonsense mediated mRNA decay (NMD), and subsequent loss of protein expression. Compounds like Ataluren [82] and aminoglycoside Geneticin (G418) [83] were reported to induce enhanced translational readthrough of the p53 mutants, resulting in the translation of full-length p53 protein that was functionally active. However, the potential high cytotoxicity of read-through-inducing treatments such as G418 remains a major challenge for long-term clinical application [84].
3.1.2. Induction of Mutp53 Degradation
Although counterintuitive at first glance, mutant p53 degradation strategies are also explored as a mutp53 targeting strategy in the clinic [60,77,85,86,87,88,89]. The idea behind removal strategies is that mutp53 causes genetic changes in the cancer cells that make them dependent on mutp53 expression [90]. Many cell lines in which mutp53 expression is severely reduced are impaired in growth and do not survive when xenografted in mice [91,92]. Importantly, loss of mutp53 expression in vivo also resulted in decreased tumour growth and tumour regression [93,94].
As the use of CRISPR or RNAi in the clinic is still mainly exploratory, many researchers have focused on decreasing mutp53 stability with drugs. In particular, the inhibition of Heat-shock protein 90 (Hsp90) and Histone deacetylase 6 (HDAC6) chaperone complex which stabilizes mutp53 remains the most studied method for mutp53 degradation. Mechanistically, the Hsp90 protein conceals the ARF-binding site of MDM2 protein, preventing p53 degradation [58]. The earliest use of an Hsp90 inhibitor, Geldanamycin, in mutp53 cancer cell lines reduced mutant p53 expression and concomitantly refolded mutp53 into a more WT-like conformation [95]. Similarly, Hsp90 inhibition by 17AAG (17-allylamino-17-demethoxygeldanamycin) activated MDM-2 and another mutp53 targeting E3 ubiquitin ligase, CHIP (carboxy-terminus of Hsp70-interacting protein), to degrade mutp53 [60]. Notably, Ganetespib, a more potent Hsp90 inhibitor was in a Phase III clinical trial GALAXY-2 for patients with advanced NSCLC, but has been terminated early due to lack of significant improvement in overall survival [96].
HDAC6 is believed to activate Hsp90 and promote Hsp90′s inhibition of MDM2 and CHIP [97] and is therefore also of interest to destabilize mutp53. Indeed, inhibitors such as FR901228, the FDA approved drug SAHA [59], A542 and trichostatin A caused Hsp90-dependent mutp53 depletion [98]. In contrast to proteasomal degradation, SAHA was shown to cause mutp53 degradation through autophagy [99]. Spautin-1′s inhibition of macro autophagy and consequent activation of chaperone-mediated autophagy (CMA) caused lysosomal uptake and degradation of mutp53 upon nutrient deprivation [100].
In 2012, Freed-Pastor et al. reported that GOF-mutp53 regulated the mevalonate pathway to promote tumorigenesis [101]. Statins inhibit this pathway and were studied as agents that block mutp53 downstream signalling. More recently, statins were also found to destabilize mutant p53 through DNAJA1 and CHIP-mediated ubiquitination [102]. A retrospective study investigating statin use in lung cancer, found that the usage of statins reduced the 5-year mortality [103]. It would be interesting to see if this changes dependent on p53 status in these cancers.
Numerous other compounds promote degradation of mutp53, including, but not limited to, disulfiram [104], YK-3-237 [105], arsenic trioxide [106], NSC59984 [107], and gold nanoparticles [108], although the specificity, the exact mechanism of degradation and/or the clinical use of such compounds needs further investigation.
3.1.3. Disruption of Mutp53 Function
Mutp53 function often relies on binding partner proteins. These include other transcription factors, the p53 family members p63 and p73, ETS1, SREBP, but also other proteins such as Pin1 [109]. Binding of mutp53 to these proteins either inhibits their function (e.g., p53 family members) or potentiates their function (e.g., ETS1). Mutp53 treatment strategies aim to prevent these interactions or disturb downstream signalling.
p63 and p73 are p53 homologs and exist in different isoforms in various tissues and tumours [110,111]. In humans, p63 is important for embryonic development, differentiation and for epithelial cell maintenance [112,113,114,115]. Likewise, p73 regulates cytoskeletal rearrangement [116], cell adhesion [117,118], ciliogenesis [119], and planar cell polarity [120]. The full-length versions of these proteins, TAp63 and TAp73, are generally thought to have tumour suppressive function [110,111]. Mutp53 proteins inhibit TAp63 and TAp73 function and so promote tumorigenesis, metastasis, and chemoresistance [14]. Most strategies focus on disruption of mutp53’s inhibitory interaction with p73 [121]. Prodigiosin facilitated p73 upregulation by disrupting mutp53’s interaction with p73. This induced WTp53-like transcriptional activity of p73 with p21 activation and anti-tumour potential [122]. Compounds such as RETRA [61] and short interfering mutant p53 peptides (SIMPs) [123], also act by disrupting mutp53’s interaction with p73 and restoring its function. Interestingly, a compound called 1-carbaldehyde-3,4-dimethoxyxanthone (LEM2) was found to prevent mutp53’s inhibition of TAp73α (a C-terminal splice variant with tumour suppressive function) by disrupting both mutp53 and MDM2 binding to p73 in neuroblastoma, further enhancing p73 function [124].
The above approaches are only a handful of approaches that are currently being explored to tackle mutp53 cancers. Other approaches target mutp53 downstream signalling pathways including the EGFR signalling pathway with drugs such as NA20 [125] or cetuximab [126] use synthetic lethal approaches to find vulnerabilities of mutant p53 cancer cells that can be targeted with drugs [127,128] or target the capacity of mutp53 to form aggregates using compounds such as ReAcp53 [129,130].
Importantly, novel discoveries on the function and consequence of mutp53 expression in cancers are still being made on a fairly regular basis. It is likely that effective strategies that disrupt mutp53 function rely on a much better understanding of the mechanisms underlying all of mutp53’s actions.
3.2. Mutant p53-Specific Gene Therapy
Re-expressing p53 using p53 gene therapy is a very appealing strategy to allow for restoration of a p53-mediated cell-death response upon chemotherapeutic challenge in cancer cells. However, restoring p53 expression in tumours remains challenging and has mainly been approached using viral delivery or nanotherapeutics/lipid particle delivery of p53.
p53 viral gene delivery research started around 1994 and used replication-deficient recombinant adenovirus in tumour cell lines, in xenografts, and in orthotopic murine models. In all cases, a p53-dependent growth inhibition and marked apoptotic response could be detected when viruses successfully delivered p53 to the target cells [55,57,131,132,133,134].
The Onyx company developed a tumour-restricted adenovirus for WTp53 gene delivery and reported effective replication of the virus in cells with p53 mutants but not WTp53 cells [135]. Likewise, in China, Gendicine is an approved recombinant p53 adenovirus gene therapy product that was initially administered in combination with radiotherapy to treat head and neck cancer, but it has now also been reported effective in other cancers [136].
A major limitation of replication-deficient viruses is efficiency with not all tumour cells being targeted and effects likely to be transient [133]. Frequent re-dosing to ensure a long-term effect might therefore be necessary. Replication competent viruses or oncolytic viruses, known as CRAdp53 vectors [137], including ONYX15 [138], SG600-p53 [139], AdDelta24-p53 [137], and H101 [140] were therefore developed and could negate some of these problems, although the safety of using such viruses needs further testing. Interestingly, even though some of the p53 gene therapy’s WT-p53 protein expression in only some cells, secondary effects in inducing systemic immunological response led to long-lasting effects on tumour regression [55,57]. In a hepatocarcinoma mouse model, re-expression of p53 using a doxycycline model induced a cooperative mechanism between tumour cell senescence and the innate immune system leading to complete tumour regression [57]. These data suggest that expression of p53 in only part of the tumour cells might be sufficient to trigger an immune response to eliminate more than just the infected cells.
As an alternative to viral delivery, liposome-mediated delivery of WTp53 protein was studied in head and neck cancer [141]. By targeting the liposomes with transferrin (a ligand recognized by the transferrin receptor that is expressed to high levels on cancer cells), it was possible to deliver WTp53 and cause tumour regression [141]. This strategy was further developed into a clinical nano-therapy treatment, SGT-53, that is currently in advanced clinical trial stage for various solid cancers and even for COVID-19, in which p53 is thought to play a role in viral infection [142,143,144].
p53 restoration gene therapy has mostly been studied in tumours without p53. It seems plausible that the potential ability of GOF-p53 mutants to induce a dominant negative effect on reintroduced WTp53 would negate the effect of the p53 gene therapy. However, in one of the earliest studies, the dominant negative p53 mutants did not abrogate adenoviral transmitted WTp53 protein function [132]. Some reports suggest that the ONYX-15 p53 gene therapy actually relies on deregulated p53 signalling and works best in mutp53 cells [145], although others demonstrated enhanced oncolytic activity of ONYX-15 in WTp53 cells [146] or activity that was independent on p53 status [147,148]. More recently, parvovirus targeting of glioblastoma stem cells with a cytotoxic protein NS1 showed that this virus is more effective in mutp53 cells [149]. These studies likely suggest disease/cell specificity and raise the question on how p53 is involved in viral infection, viral replication, and which patient would benefit most from such therapies.
Although technically not a p53 gene delivery strategy, a more recently CRISPR/Cas9-based therapeutic vector (inducible and tumour specific) has been proposed to restore p53 function in mutp53 cells [150,151]. This technique could replace the mutated p53 locus with a functional p53 copy through homologous recombination. Just like gene delivery, delivery of the CRISPR/Cas9 system could be done via viral vectors or lipid particles, although functionality still needs to be demonstrated in cells [150,151]. Notably, CRISPR/Cas has recently been shown to select for p53 mutations in p53 WT cells, which could indicate that there are restrictions associated with this approach [152].
In conclusion, p53 gene delivery could be an effective strategy as long as the hurdles to effective delivery, side-effects, and selectivity can be addressed in the future.
3.3. Mutant p53 Specific Immunotherapy
Immunotherapy is one of the most prominent methods that can provide long-term tumour regression [153], as has been demonstrated with therapy targeted against K-RAS and other mutations [153,154]. In contrast to K-RAS, p53 is mutated on many more amino acids, making it challenging to find a specific ‘mutp53’ targeting region for immunotherapy. Preliminary studies and trials have shown promising data corresponding to the immunogenicity of mutp53-derived peptide epitopes. These peptides were used to generate activated T-cell response in vitro for in vivo delivery via vaccination [155,156,157,158]. Likewise, a high-throughput screening of p53 hotspot mutants (R175H, Y220C, G245S, R248Q, R248W, and R282W) found neoantigens in metastatic epithelial cancers. A mutp53-specific T-cell response was elicited when co-cultured with autologous APCs (antigen-presenting cells) that then recognized mutant p53 [153]. Of note, cells with different p53 mutants had different capacities to present immunogenic epitopes [153], suggesting a potential link between the types of p53 mutants and their immunogenicity in different cancers.
As WTp53 is expressed at low levels in normal cells and mutp53 accumulates in tumour cells, immunotherapy using WTp53 peptide has also been explored [159]. The efficacy of this approach is supported by the observation that a WTp53 peptide induced a p53-specific cytotoxic T-cell response against mutp53/WTp53 in both mice [160,161] and in cancer patients [159,162,163,164]. One study also showed selective killing of tumour cells over normal cells [159]. Importantly, this method would bypass the requirement of mutp53-specific immunogenicity.
Over 20 different clinical trials have been conducted using p53 vaccination as a strategy to combat cancers. Although a p53 response is seen and vaccines are generally considered safe, a phase II trial did not show enough benefit to warrant progression to phase III trial [165]. Therefore, further research is needed to enhance immune strategies for mutant p53 in the future.
3.4. Mutant p53 Specific Combination Therapy
For future anti-cancer therapeutic application, the effect of a single strategy application might not be sufficient to cause long-term tumoricidal effects or to overcome potential resistance. This could be resolved by combining the different methods discussed above or by coupling of p53 specific therapeutic strategies with other therapies that target relevant pathways to exploit synergistic effects for improved therapeutic benefit.
Past and ongoing trials like to combine p53 immunotherapy with p53 gene therapy. As an example, one strategy includes priming the autologous lymphocytes with anti-p53 genes in vitro, followed by their in administration in patients [166]. The use of this strategy has rendered promising therapeutic potential in xenograft studies [167]. However, the clinical application of this strategy in humans, so far, has not shown evidences of objective tumour response [167] due to lack of p53-specific self-tolerance and the presence of immunogenicity against the viral vectors.
Another strategy combines p53-specific gene therapy with conventional immunotherapy by direct administration of adenoviral p53 (Ad-p53) gene therapy with immune checkpoint inhibitors such as anti-PD1 or agonists of cytokines such as CD122. This is to stimulate a WT-p53 specific immune response using the Ad-p53 and at the same time, bypass the immune checkpoint blockade normally found in cancer with inhibitors such as anti-PD1. This strategy has been investigated in recurrent and metastatic cancers [168] and was reported to cause effective tumour remission in murine tumour models [169].
p53-specific gene or immunotherapy with conventional chemotherapy in more advanced and often chemo-resistant cancers has also been explored. As an example, in platinum resistant ovarian cancer patients (phase I), the combined therapy of modified vaccinia Ankara vaccine delivering wild-type human p53 (p53MVA) with gemcitabine chemotherapy yielded immunological response in some patients with durable disease control [170]. This study also proposed that the combination therapy of p53MVA with immunotherapeutic agents that have immunomodulatory effects, such as anti-CTLA-4, would improve therapeutic effect [170].
Numerous combinations of p53-specific targeted therapies have been trialled with conventional chemotherapies or cancer-specific treatments in different types of cancers. In particular, APR246 has been shown to synergize with DNA-damaging anti-cancer drugs cisplatin and adriamycin [171,172]. This was attributed to the synergistic crosstalk between APR-246′s reactivation of mutp53 sensitizing the tumour cells to the DNA damaging agents [65]. APR-246 was also noted to demonstrate enhanced synergy with inhibitors of other cancer signalling, including proteosome inhibitor carfilzomib [173], BRAF inhibitor vemurafenib [174], Poly (ADP-ribose) polymerase (PARP) inhibitor Olaparib [175], etc. Other mutant p53 reactivating strategies also show promise for combination therapy. As an example, a phase II clinical trial for myelodysplastic syndrome (MDS)/acute myeloid leukaemia (AML) patients is currently being conducted combining arsenic trioxide with decitabine and cytarabine to treat MDS and AML, respectively [77].
Finally, completely novel vulnerabilities created by mutant p53 expression are explored for synthetic lethality. An example of this is the acetylation of codon 158 in mutp53 cancers. Acetylation of this mutant by a variety of acetylators, including HDAC, JQ1, and topotecan makes this mutant vulnerable to cisplatin-induced cell death [176].
Taken together, it is likely that given the role of p53 and the mutant form of p53 in many different cell processes, not one single therapy will be totally successful in eliminating all mutp53-bearing tumour cells. Combining current chemotherapy with new therapy is often the way in which new drugs are trialled in cancer and therefore would form the easiest and most cost-effective combination to explore.
4. Conclusions and Future Perspectives
The search for mutp53-specific treatment strategies began almost two decades ago. However, very few have advanced to clinical trial stages, with none being currently approved for patient treatments (except for Gendicine in China). This is in part caused by the elusiveness of p53 mutants existing in different states/forms (e.g., conformational or contact mutants) bearing different targetabilities as well as the intrinsic ability of mutp53 to overcome its dependence on multiple pathways and allow for resistance to various treatment regimes.
However, with p53-specific targeted therapy, the development of small molecules that directly or indirectly target mutp53 has proven to be highly promising particularly with discoveries such as APR-246 and COTI-2, currently in clinical trials. Likewise, targeted inhibition of mutp53’s molecular chaperones such as HDAC-6 and hsp90 has progressed to pre-clinical and phase I, II and III clinical trial studies, respectively. Yet, the key challenge involving p53-specific targeted therapy remains selectivity and specificity of the compound ensuring the delivery works in the targeted cells with minimal effect on normal cells.
For p53-specific immunotherapy, the data shown so far reflect the important knowledge that both mutp53 and WTp53 are immunogenic and can elicit tumoricidal immunogenic reactions from immune cells in vivo. However, whilst adoptive cell therapy against mutp53 appears to be promising, it potentially could facilitate a more effective and durable objective clinical response when combined sequentially with conventional chemotherapy or simultaneously with gene therapy.
p53-specific gene delivery therapy has a highly encouraging therapeutic potential but requires further optimization to improve efficacy and reduce toxicity. Long-term validation within in vivo models with delivery systems capable of ensuring uniform systemic transfections/infections will be pivotal. More importantly, the combination of p53-specific gene therapy in the form of p53 cancer vaccines with targeted immunotherapy could be one of the more important strategies that bears significant potential and necessitates further investigations.
Author Contributions
L.D. wrote the manuscript and produced the figures. P.A.J.M. edited the manuscript and managed the project. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bertoli, C.; Skotheim, J.M.; de Bruin, R.A. Control of cell cycle transcription during G1 and S phases. Nat. Rev. Mol. Cell Biol. 2013, 14, 518–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, S.L.; Levine, A.J. The p53 pathway: Positive and negative feedback loops. Oncogene 2005, 24, 2899–2908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senturk, E.; Manfredi, J.J. p53 and cell cycle effects after DNA damage. Methods Mol. Biol. 2013, 962, 49–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espinosa, J.N.M.; Verdun, R.E.; Emerson, B.M. p53 Functions through Stress- and Promoter-Specific Recruitment of Transcription Initiation Components before and after DNA Damage. Mol. Cell 2003, 12, 1015–1027. [Google Scholar] [CrossRef]
- Sullivan, K.D.; Galbraith, M.D.; Andrysik, Z.; Espinosa, J.M. Mechanisms of transcriptional regulation by p53. Cell Death Differ. 2018, 25, 133–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moll, U.M.; Petrenko, O. The MDM2-p53 Interaction. Mol. Cancer Res. 2003, 1, 1001. [Google Scholar]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Olivier, M.; Langerød, A.; Carrieri, P.; Bergh, J.; Klaar, S.; Eyfjord, J.; Theillet, C.; Rodriguez, C.; Lidereau, R.; Bièche, I.; et al. The clinical value of somatic TP53 gene mutations in 1,794 patients with breast cancer. Clin. Cancer Res. 2006, 12, 1157–1167. [Google Scholar] [CrossRef] [Green Version]
- Hruban, R.H.; Goggins, M.; Parsons, J.; Kern, S.E. Progression model for pancreatic cancer. Clin. Cancer Res. 2000, 6, 2969–2972. [Google Scholar] [PubMed]
- Aubele, M.; Werner, M.; Höfler, H. Genetic alterations in presumptive precursor lesions of breast carcinomas. Anal. Cell. Pathol. 2002, 24, 69–76. [Google Scholar] [CrossRef]
- Iakova, P.; Timchenko, L.; Timchenko, N.A. Intracellular signaling and hepatocellular carcinoma. Semin. Cancer Biol. 2011, 21, 28–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nozaki, M.; Tada, M.; Kobayashi, H.; Zhang, C.L.; Sawamura, Y.; Abe, H.; Ishii, N.; Van Meir, E.G. Roles of the functional loss of p53 and other genes in astrocytoma tumorigenesis and progression. Neuro. Oncol. 1999, 1, 124–137. [Google Scholar] [CrossRef]
- Muller, P.A.; Vousden, K.H. Mutant p53 in cancer: New functions and therapeutic opportunities. Cancer Cell 2014, 25, 304–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, S.J.; Preisinger, A.C.; Jessup, J.M.; Paraskeva, C.; Markowitz, S.; Willson, J.K.; Hamilton, S.; Vogelstein, B. p53 gene mutations occur in combination with 17p allelic deletions as late events in colorectal tumorigenesis. Cancer Res. 1990, 50, 7717–7722. [Google Scholar]
- Teoh, P.J.; Chung, T.H.; Sebastian, S.; Choo, S.N.; Yan, J.; Ng, S.B.; Fonseca, R.; Chng, W.J. p53 haploinsufficiency and functional abnormalities in multiple myeloma. Leukemia 2014, 28, 2066–2074. [Google Scholar] [CrossRef] [PubMed]
- Donehower, L.A.; Soussi, T.; Korkut, A.; Liu, Y.; Schultz, A.; Cardenas, M.; Li, X.; Babur, O.; Hsu, T.K.; Lichtarge, O.; et al. Integrated Analysis of TP53 Gene and Pathway Alterations in The Cancer Genome Atlas. Cell. Rep. 2019, 28, 1370–1384.e1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soussi, T.; Wiman, K.G. TP53: An oncogene in disguise. Cell Death Differ. 2015, 22, 1239–1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, X. P53: A Target and a Biomarker of Cancer Therapy? Recent Adv. Cancer Res. Ther. 2012, 12, 197–213. [Google Scholar] [CrossRef]
- Willis, A.; Jung, E.J.; Wakefield, T.; Chen, X. Mutant p53 exerts a dominant negative effect by preventing wild-type p53 from binding to the promoter of its target genes. Oncogene 2004, 23, 2330–2338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iggo, R.; Gatter, K.; Bartek, J.; Lane, D.; Harris, A.L. Increased expression of mutant forms of p53 oncogene in primary lung cancer. Lancet 1990, 335, 675–679. [Google Scholar] [CrossRef]
- Gannon, J.V.; Greaves, R.; Iggo, R.; Lane, D.P. Activating mutations in p53 produce a common conformational effect. A monoclonal antibody specific for the mutant form. Embo J. 1990, 9, 1595–1602. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.; Caswell, P.T.; Doyle, B.; Iwanicki, M.P.; Tan, E.H.; Karim, S.; Lukashchuk, N.; Gillespie, D.A.; Ludwig, R.L.; Gosselin, P.; et al. Mutant p53 drives invasion by promoting integrin recycling. Cell 2009, 139, 1327–1341. [Google Scholar] [CrossRef] [PubMed]
- Phatak, V.; von Grabowiecki, Y.; Janus, J.; Officer, L.; Behan, C.; Aschauer, L.; Pinon, L.; Mackay, H.; Zanivan, S.; Norman, J.C.; et al. Mutant p53 promotes RCP-dependent chemoresistance coinciding with increased delivery of P-glycoprotein to the plasma membrane. Cell Death Dis. 2021, 12, 207. [Google Scholar] [CrossRef] [PubMed]
- Mackay, H.L.; Moore, D.; Hall, C.; Birkbak, N.J.; Jamal-Hanjani, M.; Karim, S.A.; Phatak, V.M.; Pinon, L.; Morton, J.P.; Swanton, C.; et al. Genomic instability in mutant p53 cancer cells upon entotic engulfment. Nat. Commun. 2018, 9, 3070. [Google Scholar] [CrossRef] [Green Version]
- von Grabowiecki, Y.; Phatak, V.; Aschauer, L.; Muller, P.A.J. Rab11-FIP1/RCP Functions as a Major Signalling Hub in the Oncogenic Roles of Mutant p53 in Cancer. Front. Oncol. 2021, 11, 804107. [Google Scholar] [CrossRef]
- Kennedy, M.C.; Lowe, S.W. Mutant p53: It’s not all one and the same. Cell Death Differ. 2022, 29, 983–987. [Google Scholar] [CrossRef] [PubMed]
- Stein, Y.; Aloni-Grinstein, R.; Rotter, V. Mutant p53 oncogenicity: Dominant-negative or gain-of-function? Carcinogenesis 2020, 41, 1635–1647. [Google Scholar] [CrossRef] [PubMed]
- Alvarado-Ortiz, E.; de la Cruz-López, K.G.; Becerril-Rico, J.; Sarabia-Sánchez, M.A.; Ortiz-Sánchez, E.; García-Carrancá, A. Mutant p53 Gain-of-Function: Role in Cancer Development, Progression, and Therapeutic Approaches. Front. Cell Dev. Biol. 2020, 8, 607670. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, L.; Singh, A.; Kaur, P.; Ethayathulla, A.S. Comprehensive omics studies of p53 mutants in human cancer. Brief Funct. Genom. 2022, elac015. [Google Scholar] [CrossRef]
- Friedlander, P.; Haupt, Y.; Prives, C.; Oren, M. A mutant p53 that discriminates between p53-responsive genes cannot induce apoptosis. Mol. Cell. Biol. 1996, 16, 4961–4971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomes, A.S.; Ramos, H.; Inga, A.; Sousa, E.; Saraiva, L. Structural and Drug Targeting Insights on Mutant p53. Cancers 2021, 13, 3344. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Li, L.; Guan, X.; Xiong, L.; Miao, X. Mutant p53 Gain of Function and Chemoresistance: The Role of Mutant p53 in Response to Clinical Chemotherapy. Chemotherapy 2017, 62, 43–53. [Google Scholar] [CrossRef]
- Bergh, J.; Norberg, T.; Sjögren, S.; Lindgren, A.; Holmberg, L. Complete sequencing of the p53 gene provides prognostic information in breast cancer patients, particularly in relation to adjuvant systemic therapy and radiotherapy. Nat. Med. 1995, 1, 1029–1034. [Google Scholar] [CrossRef] [PubMed]
- Righetti, S.C.; Della Torre, G.; Pilotti, S.; Ménard, S.; Ottone, F.; Colnaghi, M.I.; Pierotti, M.A.; Lavarino, C.; Cornarotti, M.; Oriana, S.; et al. A comparative study of p53 gene mutations, protein accumulation, and response to cisplatin-based chemotherapy in advanced ovarian carcinoma. Cancer Res. 1996, 56, 689–693. [Google Scholar]
- Horio, Y.; Takahashi, T.; Kuroishi, T.; Hibi, K.; Suyama, M.; Niimi, T.; Shimokata, K.; Yamakawa, K.; Nakamura, Y.; Ueda, R.; et al. Prognostic significance of p53 mutations and 3p deletions in primary resected non-small cell lung cancer. Cancer Res. 1993, 53, 1–4. [Google Scholar]
- Wattel, E.; Preudhomme, C.; Hecquet, B.; Vanrumbeke, M.; Quesnel, B.; Dervite, I.; Morel, P.; Fenaux, P. p53 mutations are associated with resistance to chemotherapy and short survival in hematologic malignancies. Blood 1994, 84, 3148–3157. [Google Scholar] [CrossRef] [Green Version]
- Chin, K.V.; Ueda, K.; Pastan, I.; Gottesman, M.M. Modulation of activity of the promoter of the human MDR1 gene by Ras and p53. Science 1992, 255, 459–462. [Google Scholar] [CrossRef]
- Cascorbi, I. P-glycoprotein: Tissue distribution, substrates, and functional consequences of genetic variations. Handb. Exp. Pharmacol. 2011, 2011, 261–283. [Google Scholar] [CrossRef]
- Sampath, J.; Sun, D.; Kidd, V.J.; Grenet, J.; Gandhi, A.; Shapiro, L.H.; Wang, Q.; Zambetti, G.P.; Schuetz, J.D. Mutant p53 cooperates with ETS and selectively up-regulates human MDR1 not MRP1. J. Biol. Chem. 2001, 276, 39359–39367. [Google Scholar] [CrossRef] [Green Version]
- Sharom, F.J. ABC multidrug transporters: Structure, function and role in chemoresistance. Pharmacogenomics 2008, 9, 105–127. [Google Scholar] [CrossRef]
- Alam, S.K.; Yadav, V.K.; Bajaj, S.; Datta, A.; Dutta, S.K.; Bhattacharyya, M.; Bhattacharya, S.; Debnath, S.; Roy, S.; Boardman, L.A.; et al. DNA damage-induced ephrin-B2 reverse signaling promotes chemoresistance and drives EMT in colorectal carcinoma harboring mutant p53. Cell Death Differ. 2016, 23, 707–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Wang, J.; Hu, Y.; Qian, J.; Xu, B.; Chen, H.; Zou, W.; Fang, J.Y. Unequal prognostic potentials of p53 gain-of-function mutations in human cancers associate with drug-metabolizing activity. Cell Death Dis. 2014, 5, e1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Do, P.M.; Varanasi, L.; Fan, S.; Li, C.; Kubacka, I.; Newman, V.; Chauhan, K.; Daniels, S.R.; Boccetta, M.; Garrett, M.R.; et al. Mutant p53 cooperates with ETS2 to promote etoposide resistance. Genes Dev. 2012, 26, 830–845. [Google Scholar] [CrossRef] [PubMed]
- Ledesma, F.C.; El Khamisy, S.F.; Zuma, M.C.; Osborn, K.; Caldecott, K.W. A human 5′-tyrosyl DNA phosphodiesterase that repairs topoisomerase-mediated DNA damage. Nature 2009, 461, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chen, J.X.; Liu, Y.H.; You, C.; Mao, Q. Mutant TP53 enhances the resistance of glioblastoma cells to temozolomide by up-regulating O(6)-methylguanine DNA-methyltransferase. Neurol. Sci. 2013, 34, 1421–1428. [Google Scholar] [CrossRef] [PubMed]
- Tung, M.C.; Lin, P.L.; Wang, Y.C.; He, T.Y.; Lee, M.C.; Yeh, S.D.; Chen, C.Y.; Lee, H. Mutant p53 confers chemoresistance in non-small cell lung cancer by upregulating Nrf2. Oncotarget 2015, 6, 41692–41705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chee, J.L.; Saidin, S.; Lane, D.P.; Leong, S.M.; Noll, J.E.; Neilsen, P.M.; Phua, Y.T.; Gabra, H.; Lim, T.M. Wild-type and mutant p53 mediate cisplatin resistance through interaction and inhibition of active caspase-9. Cell Cycle 2013, 12, 278–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehrhardt, H.; Häcker, S.; Wittmann, S.; Maurer, M.; Borkhardt, A.; Toloczko, A.; Debatin, K.M.; Fulda, S.; Jeremias, I. Cytotoxic drug-induced, p53-mediated upregulation of caspase-8 in tumor cells. Oncogene 2008, 27, 783–793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, H.; Schroering, A.; Ding, H.F. p53 mediates DNA damaging drug-induced apoptosis through a caspase-9-dependent pathway in SH-SY5Y neuroblastoma cells. Mol. Cancer Ther. 2002, 1, 679–686. [Google Scholar]
- Donzelli, S.; Fontemaggi, G.; Fazi, F.; Di Agostino, S.; Padula, F.; Biagioni, F.; Muti, P.; Strano, S.; Blandino, G. MicroRNA-128-2 targets the transcriptional repressor E2F5 enhancing mutant p53 gain of function. Cell Death Differ. 2012, 19, 1038–1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morselli, E.; Tasdemir, E.; Maiuri, M.C.; Galluzzi, L.; Kepp, O.; Criollo, A.; Vicencio, J.M.; Soussi, T.; Kroemer, G. Mutant p53 protein localized in the cytoplasm inhibits autophagy. Cell Cycle 2008, 7, 3056–3061. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J. Cancer stem cells and chemoresistance: The smartest survives the raid. Pharmacol. Ther. 2016, 160, 145–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, P.A.; Vousden, K.H. p53 mutations in cancer. Nat. Cell Biol. 2013, 15, 2–8. [Google Scholar] [CrossRef]
- Ventura, A.; Kirsch, D.G.; McLaughlin, M.E.; Tuveson, D.A.; Grimm, J.; Lintault, L.; Newman, J.; Reczek, E.E.; Weissleder, R.; Jacks, T. Restoration of p53 function leads to tumour regression in vivo. Nature 2007, 445, 661–665. [Google Scholar] [CrossRef]
- Martins, C.P.; Brown-Swigart, L.; Evan, G.I. Modeling the Therapeutic Efficacy of p53 Restoration in Tumors. Cell 2006, 127, 1323–1334. [Google Scholar] [CrossRef] [Green Version]
- Xue, W.; Zender, L.; Miething, C.; Dickins, R.A.; Hernando, E.; Krizhanovsky, V.; Cordon-Cardo, C.; Lowe, S.W. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007, 445, 656–660. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.; Chen, L.; Li, C.; Lu, W.; Chen, J. Inhibition of MDM2 by hsp90 contributes to mutant p53 stabilization. J. Biol. Chem. 2001, 276, 40583–40590. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Marchenko, N.D.; Moll, U.M. SAHA shows preferential cytotoxicity in mutant p53 cancer cells by destabilizing mutant p53 through inhibition of the HDAC6-Hsp90 chaperone axis. Cell Death Differ. 2011, 18, 1904–1913. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Marchenko, N.D.; Schulz, R.; Fischer, V.; Velasco-Hernandez, T.; Talos, F.; Moll, U.M. Functional inactivation of endogenous MDM2 and CHIP by HSP90 causes aberrant stabilization of mutant p53 in human cancer cells. Mol. Cancer Res. 2011, 9, 577–588. [Google Scholar] [CrossRef] [Green Version]
- Kravchenko, J.E.; Ilyinskaya, G.V.; Komarov, P.G.; Agapova, L.S.; Kochetkov, D.V.; Strom, E.; Frolova, E.I.; Kovriga, I.; Gudkov, A.V.; Feinstein, E.; et al. Small-molecule RETRA suppresses mutant p53-bearing cancer cells through a p73-dependent salvage pathway. Proc. Natl. Acad. Sci. USA 2008, 105, 6302–6307. [Google Scholar] [CrossRef] [Green Version]
- Bártek, J.; Bártková, J.; Vojtĕsek, B.; Stasková, Z.; Lukás, J.; Rejthar, A.; Kovarík, J.; Midgley, C.A.; Gannon, J.V.; Lane, D.P. Aberrant expression of the p53 oncoprotein is a common feature of a wide spectrum of human malignancies. Oncogene 1991, 6, 1699–1703. [Google Scholar] [PubMed]
- Klimovich, B.; Merle, N.; Neumann, M.; Elmshäuser, S.; Nist, A.; Mernberger, M.; Kazdal, D.; Stenzinger, A.; Timofeev, O.; Stiewe, T. p53 partial loss-of-function mutations sensitize to chemotherapy. Oncogene 2022, 41, 1011–1023. [Google Scholar] [CrossRef] [PubMed]
- Baugh, E.H.; Ke, H.; Levine, A.J.; Bonneau, R.A.; Chan, C.S. Why are there hotspot mutations in the TP53 gene in human cancers? Cell Death Differ. 2018, 25, 154–160. [Google Scholar] [CrossRef]
- Bykov, V.J.N.; Eriksson, S.E.; Bianchi, J.; Wiman, K.G. Targeting mutant p53 for efficient cancer therapy. Nat. Rev. Cancer 2018, 18, 89–102. [Google Scholar] [CrossRef]
- Foster, B.A.; Coffey, H.A.; Morin, M.J.; Rastinejad, F. Pharmacological rescue of mutant p53 conformation and function. Science 1999, 286, 2507–2510. [Google Scholar] [CrossRef] [Green Version]
- Rippin, T.M.; Bykov, V.J.N.; Freund, S.M.V.; Selivanova, G.; Wiman, K.G.; Fersht, A.R. Characterization of the p53-rescue drug CP-31398 in vitro and in living cells. Oncogene 2002, 21, 2119–2129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bykov, V.J.N.; Issaeva, N.; Zache, N.; Shilov, A.; Hultcrantz, M.; Bergman, J.; Selivanova, G.; Wiman, K.G. Reactivation of Mutant p53 and Induction of Apoptosis in Human Tumor Cells by Maleimide Analogs*. J. Biol. Chem. 2005, 280, 30384–30391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bykov, V.J.; Issaeva, N.; Shilov, A.; Hultcrantz, M.; Pugacheva, E.; Chumakov, P.; Bergman, J.; Wiman, K.G.; Selivanova, G. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat. Med. 2002, 8, 282–288. [Google Scholar] [CrossRef]
- Zache, N.; Lambert, J.M.; Wiman, K.G.; Bykov, V.J. PRIMA-1MET inhibits growth of mouse tumors carrying mutant p53. Cell Oncol. 2008, 30, 411–418. [Google Scholar] [CrossRef]
- Bykov, V.J.N.; Wiman, K.G. Mutant p53 reactivation by small molecules makes its way to the clinic. FEBS Lett. 2014, 588, 2622–2627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, J.M.R.; Gorzov, P.; Veprintsev, D.B.; Söderqvist, M.; Segerbäck, D.; Bergman, J.; Fersht, A.R.; Hainaut, P.; Wiman, K.G.; Bykov, V.J.N. PRIMA-1 Reactivates Mutant p53 by Covalent Binding to the Core Domain. Cancer Cell 2009, 15, 376–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindemann, A.; Patel, A.A.; Silver, N.L.; Tang, L.; Liu, Z.; Wang, L.; Tanaka, N.; Rao, X.; Takahashi, H.; Maduka, N.K.; et al. COTI-2, A Novel Thiosemicarbazone Derivative, Exhibits Antitumor Activity in HNSCC through p53-dependent and -independent Mechanisms. Clin. Cancer Res. 2019, 25, 5650–5662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Synnott, N.C.; O’Connell, D.; Crown, J.; Duffy, M.J. COTI-2 reactivates mutant p53 and inhibits growth of triple-negative breast cancer cells. Breast Cancer Res. Treat. 2020, 179, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Salim, K.Y.; Maleki Vareki, S.; Danter, W.R.; Koropatnick, J. COTI-2, a novel small molecule that is active against multiple human cancer cell lines in vitro and in vivo. Oncotarget 2016, 7, 41363–41379. [Google Scholar] [CrossRef]
- Chen, S.; Wu, J.L.; Liang, Y.; Tang, Y.G.; Song, H.X.; Wu, L.L.; Xing, Y.F.; Yan, N.; Li, Y.T.; Wang, Z.Y.; et al. Arsenic Trioxide Rescues Structural p53 Mutations through a Cryptic Allosteric Site. Cancer Cell 2021, 39, 225–239.e228. [Google Scholar] [CrossRef]
- Decitabine, Cytarabine and Arsenic Trioxide for Acute Myeloid Leukemia with p53 Mutations. Available online: https://ClinicalTrials.gov/show/NCT03381781 (accessed on 1 January 2022).
- Demma, M.; Maxwell, E.; Ramos, R.; Liang, L.; Li, C.; Hesk, D.; Rossman, R.; Mallams, A.; Doll, R.; Liu, M.; et al. SCH529074, a small molecule activator of mutant p53, which binds p53 DNA binding domain (DBD), restores growth-suppressive function to mutant p53 and interrupts HDM2-mediated ubiquitination of wild type p53. J. Biol. Chem. 2010, 285, 10198–10212. [Google Scholar] [CrossRef] [Green Version]
- Friedler, A.; Hansson, L.O.; Veprintsev, D.B.; Freund, S.M.; Rippin, T.M.; Nikolova, P.V.; Proctor, M.R.; Rüdiger, S.; Fersht, A.R. A peptide that binds and stabilizes p53 core domain: Chaperone strategy for rescue of oncogenic mutants. Proc. Natl. Acad. Sci. USA 2002, 99, 937–942. [Google Scholar] [CrossRef] [Green Version]
- Tal, P.; Eizenberger, S.; Cohen, E.; Goldfinger, N.; Pietrokovski, S.; Oren, M.; Rotter, V. Cancer therapeutic approach based on conformational stabilization of mutant p53 protein by small peptides. Oncotarget 2016, 7, 11817–11837. [Google Scholar] [CrossRef] [Green Version]
- Gogna, R.; Madan, E.; Kuppusamy, P.; Pati, U. Chaperoning of mutant p53 protein by wild-type p53 protein causes hypoxic tumor regression. J. Biol. Chem. 2012, 287, 2907–2914. [Google Scholar] [CrossRef] [Green Version]
- Welch, E.M.; Barton, E.R.; Zhuo, J.; Tomizawa, Y.; Friesen, W.J.; Trifillis, P.; Paushkin, S.; Patel, M.; Trotta, C.R.; Hwang, S.; et al. PTC124 targets genetic disorders caused by nonsense mutations. Nature 2007, 447, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Floquet, C.; Deforges, J.; Rousset, J.P.; Bidou, L. Rescue of non-sense mutated p53 tumor suppressor gene by aminoglycosides. Nucleic Acids Res. 2011, 39, 3350–3362. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Feng, J.; Song, W.; Wang, J.; Tsai, B.; Zhang, Y.; Scaringe, W.A.; Hill, K.A.; Margaritis, P.; High, K.A.; et al. A mouse model for nonsense mutation bypass therapy shows a dramatic multiday response to geneticin. Proc. Natl. Acad. Sci. USA 2007, 104, 15394–15399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- GANNET53: Ganetespib in Metastatic, p53-Mutant, Platinum-Resistant Ovarian Cancer. 2013. Available online: https://ClinicalTrials.gov/show/NCT02012192 (accessed on 1 January 2022).
- Mutant p53-Based Personalized Trial Using Decitabine and Arsenic Trioxide on AML/MDS. 2019. Available online: https://ClinicalTrials.gov/show/NCT03855371 (accessed on 1 January 2022).
- Vorinostat in Treating Patients with Metastatic or Unresectable Melanoma. 2015. Available online: https://ClinicalTrials.gov/show/NCT00121225 (accessed on 1 September 2022).
- Vorinostat and Alvocidib in Treating Patients with Advanced Solid Tumors. 2006. Available online: https://ClinicalTrials.gov/show/NCT00324480 (accessed on 1 September 2022).
- MLN9708 and Vorinostat in Patients with Advanced p53 Mutant Malignancies. 2014. Available online: https://ClinicalTrials.gov/show/NCT02042989 (accessed on 1 September 2022).
- Alexandrova, E.M.; Yallowitz, A.R.; Li, D.; Xu, S.; Schulz, R.; Proia, D.A.; Lozano, G.; Dobbelstein, M.; Moll, U.M. Improving survival by exploiting tumour dependence on stabilized mutant p53 for treatment. Nature 2015, 523, 352–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turrell, F.K.; Kerr, E.M.; Gao, M.; Thorpe, H.; Doherty, G.J.; Cridge, J.; Shorthouse, D.; Speed, A.; Samarajiwa, S.; Hall, B.A.; et al. Lung tumors with distinct p53 mutations respond similarly to p53 targeted therapy but exhibit genotype-specific statin sensitivity. Genes Dev. 2017, 31, 1339–1353. [Google Scholar] [CrossRef]
- Lang, G.A.; Iwakuma, T.; Suh, Y.A.; Liu, G.; Rao, V.A.; Parant, J.M.; Valentin-Vega, Y.A.; Terzian, T.; Caldwell, L.C.; Strong, L.C.; et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell 2004, 119, 861–872. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Cao, J.; Topatana, W.; Juengpanich, S.; Li, S.; Zhang, B.; Shen, J.; Cai, L.; Cai, X.; Chen, M. Targeting mutant p53 for cancer therapy: Direct and indirect strategies. J. Hematol. Oncol. 2021, 14, 157. [Google Scholar] [CrossRef]
- Zhu, H.-B.; Yang, K.; Xie, Y.-Q.; Lin, Y.-W.; Mao, Q.-Q.; Xie, L.-P. Silencing of mutant p53 by siRNA induces cell cycle arrest and apoptosis in human bladder cancer cells. World J. Surg. Oncol. 2013, 11, 22. [Google Scholar] [CrossRef] [Green Version]
- Blagosklonny, M.V.; Toretsky, J.; Neckers, L. Geldanamycin selectively destabilizes and conformationally alters mutated p53. Oncogene 1995, 11, 933–939. [Google Scholar]
- A Phase 3 Study of Ganetespib in Combination with Docetaxel versus Docetaxel alone in Patients with Advanced NSCLC. Available online: https://clinicaltrials.gov/ct2/show/NCT01798485 (accessed on 24 September 2022).
- Zhang, C.; Liu, J.; Xu, D.; Zhang, T.; Hu, W.; Feng, Z. Gain-of-function mutant p53 in cancer progression and therapy. J. Mol. Cell Biol. 2020, 12, 674–687. [Google Scholar] [CrossRef]
- Blagosklonny, M.V.; Trostel, S.; Kayastha, G.; Demidenko, Z.N.; Vassilev, L.T.; Romanova, L.Y.; Bates, S.; Fojo, T. Depletion of Mutant p53 and Cytotoxicity of Histone Deacetylase Inhibitors. Cancer Res. 2005, 65, 7386–7392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foggetti, G.; Ottaggio, L.; Russo, D.; Mazzitelli, C.; Monti, P.; Degan, P.; Miele, M.; Fronza, G.; Menichini, P. Autophagy induced by SAHA affects mutant P53 degradation and cancer cell survival. Biosci. Rep. 2019, 39, BSR20181345. [Google Scholar] [CrossRef] [Green Version]
- Vakifahmetoglu-Norberg, H.; Kim, M.; Xia, H.G.; Iwanicki, M.P.; Ofengeim, D.; Coloff, J.L.; Pan, L.; Ince, T.A.; Kroemer, G.; Brugge, J.S.; et al. Chaperone-mediated autophagy degrades mutant p53. Genes Dev. 2013, 27, 1718–1730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freed-Pastor, W.A.; Mizuno, H.; Zhao, X.; Langerød, A.; Moon, S.H.; Rodriguez-Barrueco, R.; Barsotti, A.; Chicas, A.; Li, W.; Polotskaia, A.; et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell 2012, 148, 244–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parrales, A.; Thoenen, E.; Iwakuma, T. The interplay between mutant p53 and the mevalonate pathway. Cell Death Differ. 2018, 25, 460–470. [Google Scholar] [CrossRef]
- Chou, C.W.; Lin, C.H.; Hsiao, T.H.; Lo, C.C.; Hsieh, C.Y.; Huang, C.C.; Sher, Y.P. Therapeutic effects of statins against lung adenocarcinoma via p53 mutant-mediated apoptosis. Sci. Rep. 2019, 9, 20403. [Google Scholar] [CrossRef]
- Paranjpe, A.; Srivenugopal, K.S. Degradation of NF-κB, p53 and other regulatory redox-sensitive proteins by thiol-conjugating and -nitrosylating drugs in human tumor cells. Carcinogenesis 2013, 34, 990–1000. [Google Scholar] [CrossRef] [Green Version]
- Yi, Y.W.; Kang, H.J.; Kim, H.J.; Kong, Y.; Brown, M.L.; Bae, I. Targeting mutant p53 by a SIRT1 activator YK-3-237 inhibits the proliferation of triple-negative breast cancer cells. Oncotarget 2013, 4, 984–994. [Google Scholar] [CrossRef] [Green Version]
- Yan, W.; Jung, Y.S.; Zhang, Y.; Chen, X. Arsenic trioxide reactivates proteasome-dependent degradation of mutant p53 protein in cancer cells in part via enhanced expression of Pirh2 E3 ligase. PLoS ONE 2014, 9, e103497. [Google Scholar] [CrossRef]
- Zhang, S.; Zhou, L.; El-Deiry, W.S. Small-Molecule NSC59984 Induces Mutant p53 Degradation through a ROS-ERK2-MDM2 Axis in Cancer Cells. Mol. Cancer Res. 2022, 20, 622–636. [Google Scholar] [CrossRef]
- García-Garrido, E.; Cordani, M.; Somoza, Á. Modified Gold Nanoparticles to Overcome the Chemoresistance to Gemcitabine in Mutant p53 Cancer Cells. Pharmaceutics 2021, 13, 2067. [Google Scholar] [CrossRef] [PubMed]
- Girardini, J.E.; Napoli, M.; Piazza, S.; Rustighi, A.; Marotta, C.; Radaelli, E.; Capaci, V.; Jordan, L.; Quinlan, P.; Thompson, A.; et al. A Pin1/mutant p53 axis promotes aggressiveness in breast cancer. Cancer Cell 2011, 20, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Flores, E.R.; Tsai, K.Y.; Crowley, D.; Sengupta, S.; Yang, A.; McKeon, F.; Jacks, T. p63 and p73 are required for p53-dependent apoptosis in response to DNA damage. Nature 2002, 416, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Pflaum, J.; Schlosser, S.; Muller, M. p53 Family and Cellular Stress Responses in Cancer. Front. Oncol. 2014, 4, 285. [Google Scholar] [CrossRef] [Green Version]
- Rinne, T.; Brunner, H.G.; van Bokhoven, H. p63-Associated Disorders. Cell Cycle 2007, 6, 262–268. [Google Scholar] [CrossRef] [Green Version]
- Pozzi, S.; Zambelli, F.; Merico, D.; Pavesi, G.; Robert, A.; Maltère, P.; Gidrol, X.; Mantovani, R.; Vigano, M.A. Transcriptional Network of p63 in Human Keratinocytes. PLoS ONE 2009, 4, e5008. [Google Scholar] [CrossRef]
- Koster, M.I.; Dai, D.; Roop, D.R. Conflicting Roles for p63 in Skin Development and Carcinogenesis. Cell Cycle 2007, 6, 269–273. [Google Scholar] [CrossRef] [Green Version]
- Crum, C.P.; McKeon, F.D. p63in Epithelial Survival, Germ Cell Surveillance, and Neoplasia. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 349–371. [Google Scholar] [CrossRef]
- Maeso-Alonso, L.; López-Ferreras, L.; Marques, M.M.; Marin, M.C. p73 as a Tissue Architect. Front. Cell Dev. Biol. 2021, 9, 716957. [Google Scholar] [CrossRef]
- Santos Guasch, G.L.; Beeler, J.S.; Marshall, C.B.; Shaver, T.M.; Sheng, Q.; Johnson, K.N.; Boyd, K.L.; Venters, B.J.; Cook, R.S.; Pietenpol, J.A. p73 Is Required for Ovarian Follicle Development and Regulates a Gene Network Involved in Cell-to-Cell Adhesion. iScience 2018, 8, 236–249. [Google Scholar] [CrossRef] [Green Version]
- Holembowski, L.; Kramer, D.; Riedel, D.; Sordella, R.; Nemajerova, A.; Dobbelstein, M.; Moll, U.M. TAp73 is essential for germ cell adhesion and maturation in testis. J. Cell Biol. 2014, 204, 1173–1190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marshall, C.B.; Mays, D.J.; Beeler, J.S.; Rosenbluth, J.M.; Boyd, K.L.; Santos Guasch, G.L.; Shaver, T.M.; Tang, L.J.; Liu, Q.; Shyr, Y.; et al. p73 Is Required for Multiciliogenesis and Regulates the Foxj1-Associated Gene Network. Cell Rep. 2016, 14, 2289–2300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuertes-Alvarez, S.; Maeso-Alonso, L.; Villoch-Fernandez, J.; Wildung, M.; Martin-Lopez, M.; Marshall, C.; Villena-Cortes, A.J.; Diez-Prieto, I.; Pietenpol, J.A.; Tissir, F.; et al. p73 regulates ependymal planar cell polarity by modulating actin and microtubule cytoskeleton. Cell Death Dis. 2018, 9, 1183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Como, C.J.; Gaiddon, C.; Prives, C. p73 function is inhibited by tumor-derived p53 mutants in mammalian cells. Mol. Cell Biol. 1999, 19, 1438–1449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, B.; Prabhu, V.V.; Zhang, S.; van den Heuvel, A.P.; Dicker, D.T.; Kopelovich, L.; El-Deiry, W.S. Prodigiosin rescues deficient p53 signaling and antitumor effects via upregulating p73 and disrupting its interaction with mutant p53. Cancer Res. 2014, 74, 1153–1165. [Google Scholar] [CrossRef] [Green Version]
- Di Agostino, S.; Cortese, G.; Monti, O.; Dell’Orso, S.; Sacchi, A.; Eisenstein, M.; Citro, G.; Strano, S.; Blandino, G. The disruption of the protein complex mutantp53/p73 increases selectively the response of tumor cells to anticancer drugs. Cell Cycle 2008, 7, 3440–3447. [Google Scholar] [CrossRef]
- Gomes, S.; Raimundo, L.; Soares, J.; Loureiro, J.B.; Leão, M.; Ramos, H.; Monteiro, M.N.; Lemos, A.; Moreira, J.; Pinto, M.; et al. New inhibitor of the TAp73 interaction with MDM2 and mutant p53 with promising antitumor activity against neuroblastoma. Cancer Lett. 2019, 446, 90–102. [Google Scholar] [CrossRef]
- Zhang, G.; Li, L.; Bi, J.; Wu, Y.; Li, E. Targeting DNA and mutant p53 by a naphthalimide derivative, NA20, exhibits selective inhibition in gastric tumorigenesis by blocking mutant p53-EGFR signaling pathway. Eur. J. Pharmacol. 2020, 887, 173584. [Google Scholar] [CrossRef]
- Yang, M.; Schell, M.J.; Loboda, A.; Nebozhyn, M.; Li, J.; Teer, J.K.; Pledger, W.J.; Yeatman, T.J. Repurposing EGFR Inhibitor Utility in Colorectal Cancer in Mutant APC and TP53 Subpopulations. Cancer Epidemiol. Biomarkers Prev. 2019, 28, 1141–1152. [Google Scholar] [CrossRef] [Green Version]
- Rokudai, S. High-Throughput RNA Interference Screen Targeting Synthetic-Lethal Gain-of-Function of Oncogenic Mutant TP53 in Triple-Negative Breast Cancer. Methods Mol. Biol. 2020, 2108, 297–303. [Google Scholar] [CrossRef]
- Moser, R.; Gurley, K.E.; Nikolova, O.; Qin, G.; Joshi, R.; Mendez, E.; Shmulevich, I.; Ashley, A.; Grandori, C.; Kemp, C.J. Synthetic lethal kinases in Ras/p53 mutant squamous cell carcinoma. Oncogene 2022, 41, 3355–3369. [Google Scholar] [CrossRef] [PubMed]
- Soragni, A.; Janzen, D.M.; Johnson, L.M.; Lindgren, A.G.; Thai-Quynh Nguyen, A.; Tiourin, E.; Soriaga, A.B.; Lu, J.; Jiang, L.; Faull, K.F.; et al. A Designed Inhibitor of p53 Aggregation Rescues p53 Tumor Suppression in Ovarian Carcinomas. Cancer cell 2016, 29, 90–103. [Google Scholar] [CrossRef] [Green Version]
- Pradhan, M.R.; Siau, J.W.; Kannan, S.; Nguyen, M.N.; Ouaray, Z.; Kwoh, C.K.; Lane, D.P.; Ghadessy, F.; Verma, C.S. Simulations of mutant p53 DNA binding domains reveal a novel druggable pocket. Nucleic Acids Res. 2019, 47, 1637–1652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, L.L.; Dell, J.; Maxwell, E.; Armstrong, L.; Maneval, D.; Catino, J.J. Efficacy of p53 adenovirus-mediated gene therapy against human breast cancer xenografts. Cancer Gene Ther. 1997, 4, 129–138. [Google Scholar] [PubMed]
- Harris, M.P.; Sutjipto, S.; Wills, K.N.; Hancock, W.; Cornell, D.; Johnson, D.E.; Gregory, R.J.; Shepard, H.M.; Maneval, D.C. Adenovirus-mediated p53 gene transfer inhibits growth of human tumor cells expressing mutant p53 protein. Cancer Gene Ther. 1996, 3, 121–130. [Google Scholar]
- Lane, D.P.; Cheok, C.F.; Lain, S. p53-based cancer therapy. Cold Spring Harb. Perspect. Biol. 2010, 2, a001222. [Google Scholar] [CrossRef] [Green Version]
- Fujiwara, T.; Cai, D.W.; Georges, R.N.; Mukhopadhyay, T.; Grimm, E.A.; Roth, J.A. Therapeutic effect of a retroviral wild-type p53 expression vector in an orthotopic lung cancer model. J. Natl. Cancer Inst. 1994, 86, 1458–1462. [Google Scholar] [CrossRef]
- Bischoff, J.R.; Kirn, D.H.; Williams, A.; Heise, C.; Horn, S.; Muna, M.; Ng, L.; Nye, J.A.; Sampson-Johannes, A.; Fattaey, A.; et al. An adenovirus mutant that replicates selectively in p53-deficient human tumor cells. Science 1996, 274, 373–376. [Google Scholar] [CrossRef]
- Zhang, W.; Li, L.; Li, D.; Liu, J.; Li, X.; Li, W.; Xu, X.; Zhang, M.J.; Chandler, L.A.; Lin, H.; et al. The First Approved Gene Therapy Product for Cancer Ad-p53 (Gendicine): 12 Years in the Clinic. Hum. Gene Ther. 2018, 29, 160–179. [Google Scholar] [CrossRef] [Green Version]
- van Beusechem, V.W.; van den Doel, P.B.; Grill, J.; Pinedo, H.M.; Gerritsen, W.R. Conditionally replicative adenovirus expressing p53 exhibits enhanced oncolytic potency. Cancer Res. 2002, 62, 6165–6171. [Google Scholar]
- O’Shea, C.C.; Soria, C.; Bagus, B.; McCormick, F. Heat shock phenocopies E1B-55K late functions and selectively sensitizes refractory tumor cells to ONYX-015 oncolytic viral therapy. Cancer Cell 2005, 8, 61–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Su, C.; Cao, H.; Li, K.; Chen, J.; Jiang, L.; Zhang, Q.; Wu, X.; Jia, X.; Liu, Y.; et al. A novel triple-regulated oncolytic adenovirus carrying p53 gene exerts potent antitumor efficacy on common human solid cancers. Mol. Cancer Ther. 2008, 7, 1598–1603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, W.; Fang, H. Clinical trials with oncolytic adenovirus in China. Curr. Cancer Drug. Targets 2007, 7, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Pirollo, K.F.; Tang, W.H.; Rait, A.; Chang, E.H. Transferrin-liposome-mediated systemic p53 gene therapy in combination with radiation results in regression of human head and neck cancer xenografts. Hum. Gene Ther. 1999, 10, 2941–2952. [Google Scholar] [CrossRef]
- Senzer, N.; Nemunaitis, J.; Nemunaitis, D.; Bedell, C.; Edelman, G.; Barve, M.; Nunan, R.; Pirollo, K.F.; Rait, A.; Chang, E.H. Phase I study of a systemically delivered p53 nanoparticle in advanced solid tumors. Mol. Ther. 2013, 21, 1096–1103. [Google Scholar] [CrossRef] [Green Version]
- Pirollo, K.F.; Nemunaitis, J.; Leung, P.K.; Nunan, R.; Adams, J.; Chang, E.H. Safety and Efficacy in Advanced Solid Tumors of a Targeted Nanocomplex Carrying the p53 Gene Used in Combination with Docetaxel: A Phase 1b Study. Mol.Ther. 2016, 24, 1697–1706. [Google Scholar] [CrossRef] [Green Version]
- Harford, J.B.; Kim, S.S.; Pirollo, K.F.; Chang, E.H. TP53 Gene Therapy as a Potential Treatment for Patients with COVID-19. Viruses 2022, 14, 739. [Google Scholar] [CrossRef]
- Hann, B.; Balmain, A. Replication of an E1B 55-kilodalton protein-deficient adenovirus (ONYX-015) is restored by gain-of-function rather than loss-of-function p53 mutants. J. Virol. 2003, 77, 11588–11595. [Google Scholar] [CrossRef]
- Royds, J.A.; Hibma, M.; Dix, B.R.; Hananeia, L.; Russell, I.A.; Wiles, A.; Wynford-Thomas, D.; Braithwaite, A.W. p53 promotes adenoviral replication and increases late viral gene expression. Oncogene 2006, 25, 1509–1520. [Google Scholar] [CrossRef] [Green Version]
- Geoerger, B.; Grill, J.; Opolon, P.; Morizet, J.; Aubert, G.; Terrier-Lacombe, M.J.; Bressac De-Paillerets, B.; Barrois, M.; Feunteun, J.; Kirn, D.H.; et al. Oncolytic activity of the E1B-55 kDa-deleted adenovirus ONYX-015 is independent of cellular p53 status in human malignant glioma xenografts. Cancer Res. 2002, 62, 764–772. [Google Scholar]
- Edwards, S.J.; Dix, B.R.; Myers, C.J.; Dobson-Le, D.; Huschtscha, L.; Hibma, M.; Royds, J.; Braithwaite, A.W. Evidence that replication of the antitumor adenovirus ONYX-015 is not controlled by the p53 and p14(ARF) tumor suppressor genes. J. Virol. 2002, 76, 12483–12490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gil-Ranedo, J.; Gallego-García, C.; Almendral, J.M. Viral targeting of glioblastoma stem cells with patient-specific genetic and post-translational p53 deregulations. Cell Rep. 2021, 36, 109673. [Google Scholar] [CrossRef] [PubMed]
- Chira, S.; Gulei, D.; Hajitou, A.; Berindan-Neagoe, I. Restoring the p53 ‘Guardian’ Phenotype in p53-Deficient Tumor Cells with CRISPR/Cas9. Trends Biotechnol. 2018, 36, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Mirgayazova, R.; Khadiullina, R.; Chasov, V.; Mingaleeva, R.; Miftakhova, R.; Rizvanov, A.; Bulatov, E. Therapeutic Editing of the TP53 Gene: Is CRISPR/Cas9 an Option? Genes 2020, 11, 704. [Google Scholar] [CrossRef]
- Enache, O.M.; Rendo, V.; Abdusamad, M.; Lam, D.; Davison, D.; Pal, S.; Currimjee, N.; Hess, J.; Pantel, S.; Nag, A.; et al. Cas9 activates the p53 pathway and selects for p53-inactivating mutations. Nat. Genet 2020, 52, 662–668. [Google Scholar] [CrossRef]
- Malekzadeh, P.; Pasetto, A.; Robbins, P.F.; Parkhurst, M.R.; Paria, B.C.; Jia, L.; Gartner, J.J.; Hill, V.; Yu, Z.; Restifo, N.P.; et al. Neoantigen screening identifies broad TP53 mutant immunogenicity in patients with epithelial cancers. J. Clin. Investig. 2021, 129, 1109–1114. [Google Scholar] [CrossRef]
- Tran, E.; Robbins, P.F.; Lu, Y.C.; Prickett, T.D.; Gartner, J.J.; Jia, L.; Pasetto, A.; Zheng, Z.; Ray, S.; Groh, E.M.; et al. T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N. Engl. J. Med. 2016, 375, 2255–2262. [Google Scholar] [CrossRef] [Green Version]
- Couch, M.E.; Ferris, R.L.; Brennan, J.A.; Koch, W.M.; Jaffee, E.M.; Leibowitz, M.S.; Nepom, G.T.; Erlich, H.A.; Sidransky, D. Alteration of cellular and humoral immunity by mutant p53 protein and processed mutant peptide in head and neck cancer. Clin. Cancer Res. 2007, 13, 7199–7206. [Google Scholar] [CrossRef] [Green Version]
- Ito, D.; Visus, C.; Hoffmann, T.K.; Balz, V.; Bier, H.; Appella, E.; Whiteside, T.L.; Ferris, R.L.; DeLeo, A.B. Immunological characterization of missense mutations occurring within cytotoxic T cell-defined p53 epitopes in HLA-A*0201+ squamous cell carcinomas of the head and neck. Int. J. Cancer 2007, 120, 2618–2624. [Google Scholar] [CrossRef]
- Carbone, D.P.; Ciernik, I.F.; Kelley, M.J.; Smith, M.C.; Nadaf, S.; Kavanaugh, D.; Maher, V.E.; Stipanov, M.; Contois, D.; Johnson, B.E.; et al. Immunization with mutant p53- and K-ras-derived peptides in cancer patients: Immune response and clinical outcome. J. Clin. Oncol. 2005, 23, 5099–5107. [Google Scholar] [CrossRef] [Green Version]
- Deniger, D.C.; Pasetto, A.; Robbins, P.F.; Gartner, J.J.; Prickett, T.D.; Paria, B.C.; Malekzadeh, P.; Jia, L.; Yossef, R.; Langhan, M.M.; et al. T-cell Responses to TP53 “Hotspot” Mutations and Unique Neoantigens Expressed by Human Ovarian Cancers. Clin. Cancer Res. 2018, 24, 5562–5573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonia, S.J.; Mirza, N.; Fricke, I.; Chiappori, A.; Thompson, P.; Williams, N.; Bepler, G.; Simon, G.; Janssen, W.; Lee, J.-H.; et al. Combination of p53 Cancer Vaccine with Chemotherapy in Patients with Extensive Stage Small Cell Lung Cancer. Clin. Cancer Res. 2006, 12, 878–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayordomo, J.I.; Loftus, D.J.; Sakamoto, H.; De Cesare, C.M.; Appasamy, P.M.; Lotze, M.T.; Storkus, W.J.; Appella, E.; DeLeo, A.B. Therapy of murine tumors with p53 wild-type and mutant sequence peptide-based vaccines. J. Exp. Med. 1996, 183, 1357–1365. [Google Scholar] [CrossRef] [Green Version]
- Ishida, T.; Chada, S.; Stipanov, M.; Nadaf, S.; Ciernik, F.I.; Gabrilovich, D.I.; Carbone, D.P. Dendritic cells transduced with wild-type p53 gene elicit potent anti-tumour immune responses. Clin. Exp. Immunol. 1999, 117, 244–251. [Google Scholar] [CrossRef]
- Nikitina, E.Y.; Clark, J.I.; van Beynen, J.; Chada, S.; Virmani, A.K.; Carbone, D.P.; Gabrilovich, D.I. Dendritic Cells Transduced with Full-Length Wild-Type p53 Generate Antitumor Cytotoxic T Lymphocytes from Peripheral Blood of Cancer Patients1. Clin. Cancer Res. 2001, 7, 127–135. [Google Scholar]
- Vaccine Therapy in Treating Patients with Head and Neck Cancer. 2006. Available online: https://ClinicalTrials.gov/show/NCT00404339 (accessed on 1 January 2022).
- Rahma, O.E.; Ashtar, E.; Czystowska, M.; Szajnik, M.E.; Wieckowski, E.; Bernstein, S.; Herrin, V.E.; Shams, M.A.; Steinberg, S.M.; Merino, M.; et al. A gynecologic oncology group phase II trial of two p53 peptide vaccine approaches: Subcutaneous injection and intravenous pulsed dendritic cells in high recurrence risk ovarian cancer patients. Cancer Immunol. Immunother. CII 2012, 61, 373–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, S.; Fan, C.; Zeng, Z.; Young, K.H.; Li, Y. Clinical and Immunological Effects of p53-Targeting Vaccines. Front. Cell Dev. Biol. 2021, 9, 762796. [Google Scholar] [CrossRef]
- Rosenberg, S.A. Phase II Study of Metastatic Cancer That Overexpresses P53 Using Lymphodepleting Conditioning Followed by Infusion of Anti-P53 TCR-Gene Engineered Lymphocytes. 2006. Available online: https://clinicaltrials.gov/ct2/show/NCT00393029 (accessed on 22 April 2022).
- Kuball, J.; Schuler, M.; Antunes Ferreira, E.; Herr, W.; Neumann, M.; Obenauer-Kutner, L.; Westreich, L.; Huber, C.; Wölfel, T.; Theobald, M. Generating p53-specific cytotoxic T lymphocytes by recombinant adenoviral vector-based vaccination in mice, but not man. Gene Ther. 2002, 9, 833–843. [Google Scholar] [CrossRef]
- Sellman, B.H.; Menander, K.B. Safety and Efficacy of p53 Gene Therapy Combined with Immune Checkpoint Inhibitors in Solid Tumors. 2018. Available online: https://clinicaltrials.gov/ct2/show/NCT03544723 (accessed on 24 April 2022).
- Chada, S.; Wiederhold, D.; Menander, K.B.; Sellman, B.; Talbott, M.; Nemunaitis, J.J.; Ahn, H.M.; Jung, B.-K.; Yun, C.-O.; Sobol, R.E. Tumor suppressor immune gene therapy to reverse immunotherapy resistance. Cancer Gene Ther. 2021, 29, 825–834. [Google Scholar] [CrossRef]
- Hardwick, N.R.; Frankel, P.; Ruel, C.; Kilpatrick, J.; Tsai, W.; Kos, F.; Kaltcheva, T.; Leong, L.; Morgan, R.; Chung, V.; et al. p53-Reactive T Cells Are Associated with Clinical Benefit in Patients with Platinum-Resistant Epithelial Ovarian Cancer After Treatment with a p53 Vaccine and Gemcitabine Chemotherapy. Clin. Cancer Res. 2018, 24, 1315–1325. [Google Scholar] [CrossRef]
- Bykov, V.J.; Zache, N.; Stridh, H.; Westman, J.; Bergman, J.; Selivanova, G.; Wiman, K.G. PRIMA-1(MET) synergizes with cisplatin to induce tumor cell apoptosis. Oncogene 2005, 24, 3484–3491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohell, N.; Alfredsson, J.; Fransson, Å.; Uustalu, M.; Byström, S.; Gullbo, J.; Hallberg, A.; Bykov, V.J.N.; Björklund, U.; Wiman, K.G. APR-246 overcomes resistance to cisplatin and doxorubicin in ovarian cancer cells. Cell Death Dis. 2015, 6, e1794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walerych, D.; Lisek, K.; Sommaggio, R.; Piazza, S.; Ciani, Y.; Dalla, E.; Rajkowska, K.; Gaweda-Walerych, K.; Ingallina, E.; Tonelli, C.; et al. Proteasome machinery is instrumental in a common gain-of-function program of the p53 missense mutants in cancer. Nat. Cell Biol. 2016, 18, 897–909. [Google Scholar] [CrossRef]
- Krayem, M.; Journe, F.; Wiedig, M.; Morandini, R.; Najem, A.; Salès, F.; van Kempen, L.C.; Sibille, C.; Awada, A.; Marine, J.C.; et al. p53 Reactivation by PRIMA-1(Met) (APR-246) sensitises (V600E/K)BRAF melanoma to vemurafenib. Eur. J. Cancer 2016, 55, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Deben, C.; Lardon, F.; Wouters, A.; Op de Beeck, K.; Van den Bossche, J.; Jacobs, J.; Van Der Steen, N.; Peeters, M.; Rolfo, C.; Deschoolmeester, V.; et al. APR-246 (PRIMA-1(MET)) strongly synergizes with AZD2281 (olaparib) induced PARP inhibition to induce apoptosis in non-small cell lung cancer cell lines. Cancer Lett. 2016, 375, 313–322. [Google Scholar] [CrossRef]
- Kong, L.R.; Ong, R.W.; Tan, T.Z.; Mohamed Salleh, N.A.B.; Thangavelu, M.; Chan, J.V.; Koh, L.Y.J.; Periyasamy, G.; Lau, J.A.; Le, T.B.U.; et al. Targeting codon 158 p53-mutant cancers via the induction of p53 acetylation. Nat. Commun. 2020, 11, 2086. [Google Scholar] [CrossRef]
Figure 1.
p53 and mouse double minute-2 (MDM2) auto-feedback loop. DNA damage and cellular stress increase p53 expression and facilitate its nuclear import. This allows for p53’s transcriptional activation of target genes, including MDM2. p53-induced MDM2 activation then results in p53 binding to MDM2 and its proteasomal degradation.
Figure 1.
p53 and mouse double minute-2 (MDM2) auto-feedback loop. DNA damage and cellular stress increase p53 expression and facilitate its nuclear import. This allows for p53’s transcriptional activation of target genes, including MDM2. p53-induced MDM2 activation then results in p53 binding to MDM2 and its proteasomal degradation.

Figure 2.
TP53 structure and mutation distribution (%) within the DNA-binding domain (DBD). The frequency of each mutation in all cancers based on the p53 database (www.p53.fr) is indicated for the DBD of TP53. Amino acid positions are indicated below the domains. Five TP53 hotspot mutation sites are further indicated with codon numbers above the bars. TA = transactivation domain; PRR = proline-rich region; DBD = DNA-binding domain; TD = tetramerization domain; CTD = carboxyl terminal regulatory domain.
Figure 2.
TP53 structure and mutation distribution (%) within the DNA-binding domain (DBD). The frequency of each mutation in all cancers based on the p53 database (www.p53.fr) is indicated for the DBD of TP53. Amino acid positions are indicated below the domains. Five TP53 hotspot mutation sites are further indicated with codon numbers above the bars. TA = transactivation domain; PRR = proline-rich region; DBD = DNA-binding domain; TD = tetramerization domain; CTD = carboxyl terminal regulatory domain.

Figure 3.
Examples of ways in which mutant p53 promotes chemoresistance. Mutant p53 can impact various cellular processes to prevent chemotherapeutics drugs from working. It can neutralize chemotherapy, promote efflux and impair the way in which these drugs promote cancer cell death. This figure shows examples of these pathways.
Figure 3.
Examples of ways in which mutant p53 promotes chemoresistance. Mutant p53 can impact various cellular processes to prevent chemotherapeutics drugs from working. It can neutralize chemotherapy, promote efflux and impair the way in which these drugs promote cancer cell death. This figure shows examples of these pathways.

Figure 4.
Mutant p53-targeting therapies. Examples of three treatment strategies specifically targeting mutant p53 using immunotherapy, targeted therapy, or gene therapy are shown.
Figure 4.
Mutant p53-targeting therapies. Examples of three treatment strategies specifically targeting mutant p53 using immunotherapy, targeted therapy, or gene therapy are shown.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Dolma, L.; Muller, P.A.J. GOF Mutant p53 in Cancers: A Therapeutic Challenge. Cancers 2022, 14, 5091. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14205091
AMA Style
Dolma L, Muller PAJ. GOF Mutant p53 in Cancers: A Therapeutic Challenge. Cancers. 2022; 14(20):5091. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14205091
Chicago/Turabian StyleDolma, Lobsang, and Patricia A. J. Muller. 2022. "GOF Mutant p53 in Cancers: A Therapeutic Challenge" Cancers 14, no. 20: 5091. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14205091
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.