
Targeting TET2 as a Therapeutic Approach for Angioimmunoblastic T Cell Lymphoma

1
Sun Yat-sen University Cancer Center, State Key Laboratory of Oncology in South China, Collaborative Innovation Center for Cancer Medicine, Guangzhou 510060, China
2
Department of Pathology, Sun Yat-sen University Cancer Center, Guangzhou 510060, China
3
Department of Medical Oncology, Sun Yat-sen University Cancer Center, Guangzhou, China
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Cancers 2022, 14(22), 5699; https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14225699
Submission received: 25 October 2022
/
Revised: 18 November 2022
/
Accepted: 19 November 2022
/
Published: 20 November 2022
(This article belongs to the Special Issue Advances in Cancer Therapeutics)
Abstract
:Simple Summary
Recent advances in exome sequencing and comprehensive genomic studies have shed new light on the pathogenesis and mutational landscape of Angioimmunoblastic T-cell lymphoma (AITL). The essential pathogenic role of recurrent TET2, DNMT3A, IDH2, and RHOA mutations in AITL has been identified. Notably, the detection of TET2 mutations in bystander B cells in AITL samples has allowed new mechanistic advances in AITL combined with B-cell lymphoma. In this review, we highlight the current role of TET2 mutations in the pathogenesis of AITL and how mutational crosstalk in TET2 and other partner genes (RHOA, DNMT3A, and IDH2) cooperate to contribute to the disease. In addition, we elaborate on recent advances in AITL frequently comprising B-cell lymphoma and discuss the potential prospects of targeting TET2 as an anti-AITL agent.
Abstract
Angioimmunoblastic T-cell lymphoma (AITL), a type of malignant lymphoma with unique genomic aberrations, significant clinicopathological features, and poor prognosis, is characterized by immune system dysregulation. Recent sequencing studies have identified recurrent mutations and interactions in tet methylcytosine dioxygenase 2 (TET2), ras homology family member A (RHOA), DNA methyltransferase 3 alpha (DNMT3A), and mitochondrial isocitrate dehydrogenase II (IDH2). Notably, since B-cell lymphomas are frequently observed along with AITL, this review first summarizes its controversial mechanisms based on traditional and recent views. Epigenetic regulation represented by TET2 plays an increasingly important role in understanding the multi-step and multi-lineage tumorigenesis of AITL, providing new research directions and treatment strategies for patients with AITL. Here, we review the latest advances in our understanding of AITL and highlight relevant issues that have yet to be addressed in clinical practice.
1. Introduction
Angioimmunoblastic T-cell lymphoma (AITL) is a malignant hematologic tumor derived from T follicular helper (TFH) cells. AITL is among the most aggressive of non-Hodgkin lymphomas with a poor prognosis [1]. Only a small proportion of AITLs are detected at an early stage, which is amenable to the current treatment. Nonetheless, AITLs are mostly diagnosed at an advanced stage, and patients with advanced AITLs show a limited response to intensified chemotherapy along with high relapse rates, mainly due to a lack of effective targeted therapies. Therefore, an in-depth understanding of the pathogenesis of AITL will provide a mechanistic basis for the design of effective therapeutic regimens.
Recent exome sequencing and comprehensive genomic studies of AITL samples have led to the discovery of frequent mutations in three genes (TET2, DNMT3A, and IDH2) that are directly or indirectly involved in the regulation of DNA methylation or hydroxymethylation [2,3,4]. Mutations in TET2, which is a member of the ten-eleven translocation (TET) family of 2-oxoglutarate (2OG)-dependent dioxygenases, were more frequent than the other mutations. Moreover, TET2 mutations frequently coexist with RHOA or DNMT3A in AITL [5,6,7]. Loss of TET2 function leads to the defect in successively oxidizing 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC), and 5-carboxylcytosine (5caC) in the mammalian genome [4]. Thus, TET2 plays a crucial role in the pathogenesis of AITL through active cytosine demethylation.
In this review, we focus on exploring the current role of TET2 mutations in the pathogenesis of AITL and how mutations in TET2 as well as other genes (RHOA, DNMT3A, and IDH2) cooperatively contribute to disease pathogenesis. Recently, TET2 mutations have been detected in the AITL tumor clones of bystander B cells in AITL samples [8,9,10]. Therefore, it is likely that genetic lesions of TET2 are critical in the co-occurrence of T and B-cell lymphomas in the same patient. Here, we elaborate a summary of TET2 mutations in the frequent co-occurrence of AITL and B-cell lymphomas (e.g., diffuse large B-cell lymphoma and classic Hodgkin lymphoma) and discuss the potential of targeting TET2 as an anti-AITL agent.
2. AITL and Other Lymphomas with a TFH-Cell Phenotype
According to a review of the upcoming fifth edition of the World Health Organization Classification of Haematolymphoid Tumors update on mature T-and natural killer (NK) cell neoplasms, AITL and other lymphomas with a TFH-cell phenotype have been regarded as entities in the family of nodal T-follicular helper cell lymphomas (nTFHLs) [11]. This family includes three entities of nodal T-cell lymphomas that arise from TFH cells, which are defined by the expression of at least two (ideally three) TFH markers: CD10, BCL-6, PD1, CXCL13, and ICOS. Diseases previously referred to as “angioimmunoblastic T-cell lymphoma” “follicular T-cell lymphoma” and “peripheral T-cell lymphoma with TFH phenotype” have been renamed as nTFHL angioimmunoblastic-type (nTFHL-AI), nTFHL follicular-type (nTFHL-F) and nTFHL not otherwise specified (nTFHL-NOS), respectively. Cutaneous T-cell lymphomas with a TFH-cell phenotype were excluded.
The prototype of these lymphomas is nTFHL-AI (referred to as AITL in this paper), which has distinct clinical and histological features. AITL is typically manifested by lymphadenopathy, hepatosplenomegaly, polyclonal hyperglobulinemia, and systemic symptoms. Skin rashes are frequently observed, as is pruritus. Other common findings are pleural effusion, arthritis, and ascites [12,13]. The histology of AITL is very unique: (1) AITL tumor cells which are small to medium in size typically have abundant pale cytoplasms with round and slightly irregular nuclei; (2) AITL tumor cells show the cell-of-origin of TFH cells with expression of five TFHcell-associated genes (CD10, BCL-6, PD1, CXCL13, and ICOS) in most cases. (3) A polymorphous inflammatory background that contains variable numbers of reactive lymphocytes, histiocytes, plasma cells, and eosinophils; (4) Arborization of high endothelial venules (HEVs) and follicular dendritic cells (FDCs) are very prominent; (5) Variable numbers of B immunoblasts are usually present in the paracortex and may be positive or negative for EBV. More recently, studies based on next-generation sequencing have determined frequent mutations in genes encoding epigenetic modifiers, such as TET2, DNMT3A, and IDH2, as well as the small GTPase RHOA. Among these, IDH2 R172 mutation apparently has specificity for AITL [2,7,8], while others are visible in other peripheral T-cell lymphomas, particularly those with a TFH-cell phenotype. This suggests a consistent oncogenic course in these related types of lymphomas.
nTFHL follicular-type (nTFHL-F) is a rare subtype of nTFHLs, with a predominantly follicular growth pattern that distinguish from AITL in the histological features [10]. Patients with nTFHL-F resemble AITL, characterized by lymphadenopathy, hepatosplenomegaly, systemic symptoms, polyclonal hypergammaglobulinemia, and autoimmune manifestations. nTFHL-F and AITL have very similar genetic profiles with mutations in TET2, DNMT3A, and IDH2, as well as the small GTPase RHOA [14,15]; however, nTFHL-F has specific genetic alterations due to the translocation t (5;9) (q32; q22) leading to ITK-SYK fusion, which is rarely seen in AITL [16,17].
nTFHL-NOS, previously classified as peripheral T-cell lymphomas, not otherwise specified (PTCL-NOS), is now included in the family of nodal T-follicular helper cell lymphomas. This lymphoma lacks the characteristic histological features (e.g., inflammatory infiltration, vascular hyperplasia, or dilation of follicular dendritic cell meshwork) of AITL but presents with a TFH-cell phenotype and genetic abnormalities similar to AITL.
3. The Highly Recurrent TET2 Mutation in AITL
TET proteins were named due to the rare “ten-eleven translocation” and included TET1, TET2, and TET3 [4]. TET1 and TET3 are virtually mutation-free in hematological malignancies. However, the gene located in the 4q24 region of human chromosome 4, which encodes TET2, frequently undergoes microdeletions and copy number-neutral loss-of-heterozygosity in various hematological malignancies [4,18]. Most missense mutations in TET2 impair enzyme activity and induce aberrant DNA methylation [4,18].
More recently, TET2 mutations have been frequently observed in T-cell lymphomas, particularly in AITL and other lymphomas with a TFH-cell phenotype. Table 1 summarizes the studies that have included TFH cell lymphoma and PTCL-NOS. The techniques used for the detection of TET2 mutations are listed in Table 1. According to previous studies, TET2 mutations have been detected in 47–100% of AITL cases [8,18,19,20,21,22,23,24,25,26,27]. Among patients with TET2 mutations, over 57% harbored two or more mutations. Mutations in TET2 were identified through the entire coding region, while most of them were insertions/deletions generating frameshift and nonsense mutations. Only one study assessed TET2 mutations in nTFHL-F, with a mutation rate of 75% [10]. Up to 58–73% of nTFHL-NOS had TET2 mutations [10,18,20]. The mutation rate of TET2 in PTCL-NOS ranged from 14.6% to 38% [10,18,20]. In conclusion, TET2 has the highest mutation rate in AITL (up to 100%), which is more common than that in other tumors.
In AITL, TET2 mutations are associated with advanced-stage disease [18,22], thrombocytopenia [18], high International Prognostic Index (IPI) scores [18,28], an increased number of involved extranodal sites [18], the presence of B symptoms [22], and elevated lactate dehydrogenase (LDH) levels [22]. Elevated LDH levels and positive B symptoms are considered indicators of tumor load and play an overwhelming role in tumor maintenance [29], thereby suggesting that TET2 mutations can lead to increased tumor burden.
Whether TET2 mutations affect the prognosis of AITL patients is unclear. Two studies have shown that TET2 mutations are correlated with shorter progression-free survival (PFS) [6,18], whereas another study found no significant difference in OS or PFS when compared between the group of wild-type TET2 and the mutant group; the only individual mutation that significantly reduced survival was IDH2 [22]. TET2 and IDH2 co-occurrence possessed better PFS than TET2 mutations alone, which also demonstrated the emphasis on epigenetic modifications [22]. In conclusion, the prognostic value of TET2 mutations in AITL remains controversial and is still being explored.
4. Crosstalk between TET2 and Other Genes in AITL
4.1. Interaction between TET2 and RHOA Mutations
Ras homology family member A (RHOA) functions as a small GTPase, which is activated by a specific guanine nucleotide exchange factor (GEF), to promote the exchange of GTP and GDP [33,34]. Missense mutations in the pG17V substitution encoding the RHOA GTPase were detected in 50–70% of AITL patients [6,7,35,36]. As a result, RHOA G17V loses its GTP-binding activity to inhibit RHO signaling by isolating the GEF protein [6,7,37]. Thus, RHOA mutants promote the development of PTCL by dominantly and negatively inhibiting the binding of wild-type RHOA protein to GTP [6,7]. Notably, the RHOA mutation is specific to T-cell lymphoma and absent in B-cell lymphoma [37].
VAV1 (Vav Guanine Nucleotide Exchange Factor 1) has been characterized as a G17V RHOA-specific binding companion [38]. This protein not only acts as a GEF (Guanine Nucleotide Exchange Factor) to activate small GTPases in the T-cell receptor (TCR) signaling pathway but also acts as an adapter to promote the formation or function of the TCR signaling complex [38,39]. The G17V RHOA-VAV1 axis activated in AITL accelerates TCR signal transduction by enhancing phosphorylation at 174Tyr [38,40]. Genes with the highest mutation frequency in the TCR signaling pathway include PLCG1, CD28, PIK3, GTF2I, and CTNNB1 [35], which are related to cell activation and TFH-derived lymphoma [35]. In addition, RHOA G17V expression in CD4+ T cells promotes Tfh lineage specification and AITL transformation through a mechanism dependent on the ICOS-PI3K-mTOR signaling pathway [40,41]. The blocking of the ICOS-PI3K-mTOR signaling pathway provides an innovative and possible strategy for targeting RHOA G17V therapy for AITL. As a costimulatory molecule and migration receptor for Tfh cells, ICOS co-receptor signaling phosphorylates transcription factor FOXO1 (forkhead box O1) and decreases nuclear transcription factor KIF2 [42,43], which is essential for driving Tfh lineage differentiation and maintaining the Tfh phenotype. Therefore, RHOA G17V induces Tfh lineage differentiation and drives AITL transformation by accelerating TCR signal transduction and enhancing the ICOS-PI3K signaling pathway.
Notably, TET2 mutation and RHOA G17V have a synergistic effect on human AITL pathogenesis, with co-mutations of TET2 and RHOA observed in 60–70% of AITL cases [6,7,36], leading to AITL-like disease with high penetrance [41,43,44]. In multiple mouse models reported, Tet2 loss and RHOA G17V expression jointly led to AITL, which supports a multi-step model of AITL development (Figure 1) [41,43,44,45]. TET2 deletion with RHOA G17V expression can lead to abnormal activation of CD4+ T cells and the imbalance of T cell homeostasis in the peripheral blood, which results in a reduction of Treg cells and an increase of Tfh cells [43]. In addition, the loss of function of RHOA and TET2 together interfere with FOXO1 function, which is manifested by inhibiting FOXO1 transcription and promoting its phosphorylation, thereby inducing the differentiation of Tfh cells and greatly reducing the need for ICOS signal transduction [42,43]. In most cases, RHOA mutations are specifically identified only in tumor cells, whereas TET2 mutations are found in both tumor and non-tumor hematopoietic cells [44]. The allele frequencies of TET2 were distinctly higher when compared to the frequencies of RHOA and IDH2. DNMT3A and TET2 mutations have a large degree of overlap and similar allele burden, whereas RHOA and IDH2 mutations share similar allele frequencies [7]. This indicates that TET2 and/or DNMT3A mutations precede RHOA and/or IDH2 mutations in the majority of cases [7], while RHOA G17V is the secondary strike of AITL pathogenic mechanism relay TET2 in early hematopoietic progenitor cell mutation.
4.2. Interaction between TET2 and DNMT3A Mutation
In AITL, DNMT3A mutations occur at a frequency of 20–38.5% [6,7,24,30]. The majority of DNMT3A mutations are concentrated in the MTase-binding domain, which can lead to loss-of-function. Notably, Arg-882 (R882), located in the MTase binding domain, was the most common mutation site [24]. DNMT3A deficiency progressively impairs hematopoietic stem cell (HSC) differentiation while amplifying the number of HSCs in the bone marrow [46], thereby, making hematopoietic stem cells prone to malignant transformation [47].
DNMT3A is a de novo DNA methyltransferase that mediates DNA methylation [48], whereas the TET protein family can oxidize 5-methylcytosine (5-mc) to 5-hydroxymethylcytosine (5-hmc) [49] during DNA demethylation. Thus, the interaction between TET2 and DNMT3A co-regulates the addition or removal of DNA methyl groups [48,49]. The DNMT3A R882H mutant combined with TET2 inactivation plays a role in DNA methylation dysregulation and induces malignant tumors in the mouse lymphatic system [5].
Interestingly, TET2 and DNMT3A mutations co-occur significantly, particularly in AITL [3,5,6,7,30]. DNMT3A and TET2 mutations have a large degree of overlap and similar allele burden. These mutations have been found not only in tumor cells from patients with T-cell lymphoma [3,4,5,8,30,48,50] but also in multiple lineages of normal bone marrow and blood cells [3,4,7,51,52], including B cells, myeloid cells, and myeloid progenitor cells [7,8], whereas RHOA and IDH2 mutations are confined only to tumor cells [7,8]. These data provide further confirmation of the hypothesis that TET2 and DNMT3A mutations may exist in precancerous cells.
4.3. Interaction between TET2 and IDH2 Mutations
Mitochondrial isocitrate dehydrogenase II (IDH2) is an NADP+-dependent enzyme that is critical for cell proliferation and catalyzes the conversion of isocitrate to α-ketoglutarate (αKG) in the mitochondria [53]. However, the IDH2 R172 mutant lacks this ability and catalyzes the NADPH-dependent reduction of αKG to R(−)-2-hydroxyglutarate(2HG) [53,54,55], contributing to the malignant transformation [54,55]. Since 2HG is a structural analog of αKG, mutant IDH-induced increases in 2HG levels may interfere with the normal cycle of DNA methylation and demethylation by inhibiting the activity of α-KG-dependent dioxygenases, such as TET family proteins [55,56]. IDH2 R172 mutant-mediated DNA hypermethylation inhibits T-cell receptor signal transduction and T-cell differentiation, thereby supporting the idea that 2HG produced by the mutant enzyme is responsible for driving tumorigenesis and progression [24,57].
In acute myeloid leukemia (AML), the IDH1/2 and TET2 mutations repel each other. IDH1/2 mutations mediate hypermethylation and suppress TET2-mediated demethylation, impelling myeloid differentiation [55]. IDH2 mutations are seemingly specific events in AITL, with a mutation frequency of 20–45% [2,7,8,24,30,58], most of which coexist with TET2 mutations [7,24,30,58]. It is interesting to note that the IDH2 mutation in AITL is hypothesized to constitute a second strike and may refine the differentiation of precancerous clones [7,19]. Cases with IDH2 and TET2 double mutations showed the upregulation of TFH-related genes (IL21 and ICOS) and an increased TFH cell-like phenotype, thereby defining a distinct subgroup of AITL with unique follicular T helper gene expression signatures [24].
Therefore, IDH2 R172 mutations can not only affect epigenetic regulation by competitively inhibiting TET2 demethylation but also enhance the expression profile of TFH and promote AITL development.
5. Role of TET2 Mutations in B-Cell Lymphoma Observed in AITL
In clinical practice, the incidence of B-cell lymphoma in the setting of AITL is as high as 10% [59,60,61,62]. However, the reason for this unique-recurring, frequent concomitant appearance of B- and T-cell lymphomas is not known.
5.1. Traditional View: AITL Precedes B-Cell Lymphoma
B-cell lymphoma has traditionally been regarded as a follow-up to AITL, as indicated by this model (Figure 2). Latent Epstein-Barr virus (EBV) infection is a common feature of sporadic non-neoplastic bystander B cells, showing that B-cells are usually positive for EBV-coding RNA in 66–97% of companion/secondary B-cell lymphomas [10,59,60,61,62,63]. EBV has a significant effect on B cell differentiation, leading to the survival and clonal expansion of Ig-less B cells, and promoting the progression of B-cell lymphoma [59,60,62,64]. Zhou et al. suggested that the reduction in antiviral immune surveillance in AITL may be conducive to the proliferation of potential EBV-infected B lymphocytes and the subsequent progression of EBV+ B cell malignancies [59]. Furthermore, the B cell-stimulating properties of TFH tumor cells may promote B cell expansion [9,65]. Although EBV infection may contribute to the oncogenic mechanism of EBV-positive B-cell lymphoma, a considerable proportion of patients with B-cell lymphoma are EBV-negative [60,66,67], suggesting that other factors besides EBV may be responsible for the occurrence of B-cell lymphoma.
5.2. Recent View: AITL Occurs Concurrently with B Cell Lymphoma
TET2 mutations represent early genetic lesions that occur in upstream hematopoietic stem/progenitor cells (HSC/HPC) and have been detected in both tumor tissue and normal blood cells of patients with AITL [3,4,7,30]. Between 10% and 60% of polyclonal B cells in AITL lymph nodes carry the same TET2 mutation in their corresponding T-cell lymphoma clones [9]. Furthermore, premalignant cells with TET2 mutations may differentiate into not only T-lineage tumor cells but also B cells [8,9]. Single-cell analysis revealed that 7–66% of B cells were derived from the differentiation of TET2-mutated HSC rather than through clonal expansion of a minority of B cells with TET2 mutations [9,64]. Notably, all NOTCH1 mutations in B-cell lymphomas secondary to AITL were detected only in B cells, whereas RHOA G17V mutations were specific to all tumor T cells [8], showing that NOTCH1 mutation and RHOA G17V mutation play a role in B-cell lymphoma and AITL, respectively, and both are secondary mutations following TET2 mutation. Consequently, AITL with B-cell lymphoma is a multistep alteration, with AITL and B-cell lymphoma occurring in the T-cell zone and germinal center, following the differentiation of hematopoietic stem cells carrying the TET2 mutation into premalignant T-cell and B-cell lineages, respectively.
Taken together, these studies suggest that clonal hematopoietic cells carrying the TET2 mutation may acquire the NOTCH1 mutation when they differentiate into B cells, which may subsequently enhance the effects of the TET2 mutation. The co-occurrence of AITL and B-cell lymphoma was mediated by B cells harboring TET2 and NOTCH1 mutations, and Tfh cells carrying TET2 and RHOA mutations, as indicated by the model outlined in Figure 3.
6. New Drugs Targeting TET2 for Treatment of AITL
Most peripheral T-cell lymphomas (PTCLs) have a high degree of malignancy, rapid disease progression, poor clinical prognosis, low remission rates, and high recurrence rates after first-line treatment. Despite several novel drugs currently being investigated [68], there has been little improvement in the prognosis of AITL patients over the past 20 years [69]. According to the 2022 NCCN guidelines, anthracycline-based chemotherapy regimens [e.g., CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone), CHOP + etoposide (CHOEP), or dose-adjusted EPOCH (etoposide, prednisone, vincristine, cyclophosphamide, and doxorubicin)] are the most commonly used first-line therapy regimens, although with a high failure rate and frequent relapse in PTCL. Additionally, epigenetic drugs have been applied in clinical trials (Table 2) and recommended as second-line therapy by NCCN clinical practice guidelines [70], providing new research directions to promote promising clinical outcomes as a focus of research in PTCL [71].
6.1. Histone Deacetylase Inhibitor (HDACis)
Histone deacetylase (HDAC) is an enzyme engaged in chromatin remodeling and regulates the epigenetics of gene expression [82]. HDAC inhibitors, such as romidepsin and belinostat, have previously been demonstrated to induce acetylation of histones and other proteins [83,84], producing antitumor activity by inhibiting gene transcription, regulating cell cycle, and increasing tumor cell apoptosis [83,85].
A pivotal, open-label phase II clinical trial of romidepsin for relapsed or refractory PTCL involving 130 patients with an objective response rate (ORR) of 25% and a complete response/unconfirmed complete response rate (CR/CRu) of 15% demonstrated that single-agent romidepsin induced complete and durable responses with manageable toxicity [72]. These results contributed to the FDA’s (Food and Drug Administration) approval of romidepsin in June 2011. Since then, several pivotal multicenter phase II studies have shown the activity of HDAC inhibitors as monotherapy in patients with relapsed or refractory PTCL (ALCL, ALK-negative, PCTL-NOS, and AITL) [73,74]. The combination of romidepsin and conventional CHOP chemotherapy did not seem to have the desired effect. A non-randomized phase 1b/2 study of 37 previously untreated patients with PTCL showed that the combination of romidepsin was more toxic than expected with CHOP alone (panel) [77]. Another homogeneous phase 3 combination trial involving 421 previously untreated PTCL patients also showed that the combination did not improve OS, PFS, or response rates, but instead increased the frequency of grade ≥3 therapeutic adverse events [79]. In contrast, the combination of romidepsin, duvelisib (a PI3K-δ,γ Inhibitor), and 5-azacytidine showed good efficacy in clinical trials of T-cell lymphoma, with the highest ORR and CR of 61% and 48%, respectively [41,78,80].
Chidamide is a novel selective HDAC inhibitor characterized by a unique mechanism of action. Two chidamide monotherapies and two combination therapies were administered in PTCL [75,76,81]. Among them, combination therapy has shown more promising results than monotherapy. The ORR and CR of chidamide combined with 5-azacytidine were 68.8% and 31.2%, respectively, which had the best results. Panobinostat is a potent oral pan-deacetylase inhibitor as well. In a clinical trial, the combination of panobinostat and the proteasome inhibitor bortezomib was shown to be effective in patients with relapsed or refractory peripheral T-cell lymphoma [76].
6.2. Hypomethylating Agents (HMAs)
Hypomethylating agents (HMAs), for instance 5-azacytidine (Aza) and decitabine, can balance the hypermethylated state of DNA induced by mutations in epigenetic regulators such as TET2, DNMT3A, and IDH2. HMAs have been authorized for the treatment of myelodysplastic syndromes (MDS) in many countries, and MDS patients with TET2 mutations show an increased response rate to HMAs [86].
One study showed that TET2 mutations are associated with a better response to azacytidine and revealed that the TET2 gene is the strongest biomarker of clinical response [87]. Since TET2 mutations are occurring more frequently in AITL than in MDS, the therapeutic effect of Aza was expected to be more in the case of AITL. In a retrospective study, Aza et al. showed promising results, where out of the 12 AITL patients with TET2 mutations, nine responded to Aza, including six complete responses (CRs) and three partial responses (PRs), resulting in an ORR of 75% [31]. Two case reports showed complete remission in AITL patients with TET2 mutations after the administration of Aza [28,88], suggesting that targeting aberrant DNA methylation in AITL may be as a potential strategy to alternate previous chemotherapy regimens. Furthermore, in a phase I clinical trial investigating the efficacy of Aza combined with romidepsin in 33 patients with PTCL, 10 (32%) obtained an objective response, and seven (23%) achieved a complete response. These results indicate that the combination of epigenetic modification drugs exhibits promising therapeutic activity in patients with PTCL [89].
Decitabine (5-aza-2’deoxycytidine) is a more effective methyltransferase inhibitor than 5-azacytidine (Aza), and its combination with HDAC inhibitors has a synergistic effect on reactivating gene expression [90]. Decitabine has been demonstrated to have potent antineoplastic activity in anaplastic cell lymphoma (ALCL) and MDS [91,92]. A study showed that the combination of the PARPi niraparib (Npb), HDACi romidepsin (Rom), and HMA decitabine (DAC) or panobinostat (Pano) was synergistically cytotoxic to leukemia and lymphoma cells and inhibited cell proliferation by up to 70% through activating the ATM pathway [93]. To date, no study has combined AITL therapy with decitabine.
6.3. Preclinical Trials of TET2 Targeting Agents
DNA methylation changes resulting from the silencing or activation of genes by TET2 mutations play a key role in AITL pathogenesis. At present, there are no clinically approved drugs capable of reactivating TET function; however, many preclinical trials have been conducted on TET2-related drugs (Table 3), thereby representing potential therapeutic opportunities.
6.3.1. TET Enzyme Inhibitors
Recently, novel cytosine-based TETase suppressors have been identified. A promising cytosine-based lead compound, Bobcat339, reduced the abundance of DNA 5hmC by inhibiting TETase activity in cells [94]. Furthermore, a TET-selective small-molecule inhibitor (TETi76) was effective in limiting the clonal growth of TET2 mutants and reduced cellular hydroxymethylation both in vitro and in vivo [93]. However, the limitations of these TET2 inhibitors restrict their clinical application. Bobcat339 has not been tested in animal models and clinical trials, and its effects and toxicities among humans are unknown. Furthermore, while TETi76 decreases cytosine hydroxymethylation levels and limits clonal growth of TET2 mutant cells in vitro and in vivo, a potential drawback of TETi is that it may replicate the TET2 mutation and drive the malignant transformation of normal hematopoietic cells. More validation and discussion are needed for their future application.
6.3.2. TET Enzyme Cofactors
Recent studies on embryonic stem cells have shown that ascorbic acid (AA)/Vitamin C is a cofactor for TET. Ascorbic acid is capable of binding to the catalytic domain of TET to promote the oxidation of 5-methylcytosine and DNA demethylation [96,103,104,105]. AA can exert beneficial anti-proliferative effects on AML cells carrying co-existing TET2 and TP53 mutations without interfering with targeted cytotoxic therapies [97]. Additionally, the combination of AA and a class I/II histone deacetylase inhibitor (HDACis) can prevent TET2 mutant myeloid neoplasia (MN), thereby providing a preclinical theoretical basis for further research [98]. Furthermore, AA can combine with 5-AZA to further upregulate methylated genes and human endogenous retroviruses (HERVs) [99]. In addition, vitamin C also promotes decitabine- or azacitidine-induced DNA hydroxymethylation and reactivation of the tumor suppressor CDKN1A [100]. The dual use of AA and vitamin C may improve the response to epigenetic therapy.
The limited early studies suggest that AA levels may vary in different tumor patients, suggesting the need to clarify who to administer, the route of administration, and the dose. Therefore, prospective studies are urgently required to elucidate the incidence of AA deficiency according to tumor type and stage, and whether blood levels of AA correlate with the methylation landscape of tumors to maximize patient benefit.
6.3.3. Other Possibilities
Transcriptomic profiling showed that a four antibiotic cocktail therapy inhibited the differentiation and proliferation of TET2-loss myeloid and lymphoid tumor cells in vivo by mediating genetic alterations in the TNF-α signaling pathway [101]. However, how TNF-α signaling is activated and how antibiotic treatment inhibits pathways associated with TNF remain unresolved. Ginkgo biloba extract can also inhibit cell expansion and invasion by increasing TET2 expression in colorectal cancer cells through miR-29a [102]. These findings suggest that new molecular biology tools will provide effective insights into the field of epigenetics and that TET inhibitors may prove to be promising targeted drugs in TET2 mutational tumorigenesis.
7. Conclusions
In this review, we have summarized the synergistic effects of TET2 mutations and additional genes (RHOA, DNMT3A, and IDH2) in the pathogenesis of AITL. Evidence of “multi-step”and“multi-lineage” genetic events in AITL can provide insights and ideas for the origin and occurrence of this unique subtype of T-cell lymphoma. Identification of novel therapeutic vulnerabilities has induced a significant shift in the clinical management of AITL. The success of epigenetic drugs for AITL, particularly HDAC inhibitors and hypomethylation drugs, offers new hope for patients with refractory AITL. Continued progress in preclinical experiments based on cell lines and animal models will help improve the survival rate of patients with TET2 mutant malignant tumors. Furthermore, challenges and opportunities coexist, and many opportunities for improvement remain, including the identification of biomarkers for targeted treatment response, the description of drug resistance mechanisms, and the development of successful AITL combination therapies.
Author Contributions
Conceptualization, S.Z.; validation, L.H. and X.Z.; data curation, L.H. and H.L.; writing—original draft preparation, L.H.; writing—review and editing, S.Z. and L.H.; supervision, S.Z. and S.L.; project administration, S.Z.; funding acquisition, S.Z. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by Natural Science Foundation of Guangdong Province (Grant No. 2019A1515011354).
Acknowledgments
We acknowledge the many colleagues and trainees who have contributed to the study of AITL.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Grogg, K.L.; Attygalle, A.D.; Macon, W.R.; Remstein, E.D.; Kurtin, P.J.; Dogan, A. Angioimmunoblastic T-cell lymphoma: A neoplasm of germinal-center T-helper cells? Blood 2005, 106, 1501–1502. [Google Scholar] [CrossRef]
- Rob, A.C.; Javeed, I.; François, L.; Can, K.; de Laurence, L.; Jean-Philippe, J.; Marie, P.; Antoine, M.; Luc, X.; Pierre, B.; et al. IDH2 mutations are frequent in angioimmunoblastic T-cell lymphoma. Blood 2012, 119, 1901–1903. [Google Scholar] [CrossRef] [Green Version]
- Lucile, C.; Christian, B.; Olivier, A.B. TET2 and DNMT3A mutations in human T-cell lymphoma. N. Engl. J. Med. 2012, 366, 95–96. [Google Scholar] [CrossRef]
- Quivoron, C.; Couronne, L.; Della Valle, V.; Lopez, C.K.; Plo, I.; Wagner-Ballon, O.; Do Cruzeiro, M.; Delhommeau, F.; Arnulf, B.; Stern, M.H.; et al. TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer Cell 2011, 20, 25–38. [Google Scholar] [CrossRef] [Green Version]
- Scourzic, L.; Couronne, L.; Pedersen, M.T.; Della Valle, V.; Diop, M.; Mylonas, E.; Calvo, J.; Mouly, E.; Lopez, C.K.; Martin, N.; et al. DNMT3A(R882H) mutant and Tet2 inactivation cooperate in the deregulation of DNA methylation control to induce lymphoid malignancies in mice. Leukemia 2016, 30, 1388–1398. [Google Scholar] [CrossRef] [Green Version]
- Palomero, T.; Couronné, L.; Khiabanian, H.; Kim, M.-Y.; Ambesi-Impiombato, A.; Perez-Garcia, A.; Carpenter, Z.; Abate, F.; Allegretta, M.; Haydu, J.E.; et al. Recurrent mutations in epigenetic regulators, RHOA and FYN kinase in peripheral T cell lymphomas. Nat. Genet. 2014, 46, 166–170. [Google Scholar] [CrossRef]
- Sakata-Yanagimoto, M.; Enami, T.; Yoshida, K.; Shiraishi, Y.; Ishii, R.; Miyake, Y.; Muto, H.; Tsuyama, N.; Sato-Otsubo, A.; Okuno, Y.; et al. Somatic RHOA mutation in angioimmunoblastic T cell lymphoma. Nat. Genet. 2014, 46, 171–175. [Google Scholar] [CrossRef]
- Nguyen, T.B.; Sakata-Yanagimoto, M.; Asabe, Y.; Matsubara, D.; Kano, J.; Yoshida, K.; Shiraishi, Y.; Chiba, K.; Tanaka, H.; Miyano, S.; et al. Identification of cell-type-specific mutations in nodal T-cell lymphomas. Blood Cancer J. 2017, 7, e516. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, F.H.; Cai, Q.; Fellmann, E.; Hartmann, S.; Mayranpaa, M.I.; Karjalainen-Lindsberg, M.L.; Sundstrom, C.; Scholtysik, R.; Hansmann, M.L.; Kuppers, R. TET2 mutations in B cells of patients affected by angioimmunoblastic T-cell lymphoma. J. Pathol. 2017, 242, 129–133. [Google Scholar] [CrossRef]
- Dobay, M.P.; Lemonnier, F.; Missiaglia, E.; Bastard, C.; Vallois, D.; Jais, J.-P.; Scourzic, L.; Dupuy, A.; Fataccioli, V.; Pujals, A.; et al. Integrative clinicopathological and molecular analyses of angioimmunoblastic T-cell lymphoma and other nodal lymphomas of follicular helper T-cell origin. Haematologica 2017, 102, e148–e151. [Google Scholar] [CrossRef]
- Alaggio, R.; Amador, C.; Anagnostopoulos, I.; Attygalle, A.D.; Araujo, I.B.O.; Berti, E.; Bhagat, G.; Borges, A.M.; Boyer, D.; Calaminici, M.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms. Leukemia 2022, 36, 1720–1748. [Google Scholar] [CrossRef]
- Byeong-Bae, P.; Baek-Yeol, R.; Jae, H.L.; Hyuck, C.K.; Sung, H.Y.; Hye, J.K.; Hyo, J.K.; Sung, Y.O.; Young, H.K.; Joo, R.H.; et al. Clinical features and treatment outcomes of angioimmunoblastic T-cell lymphoma. Leuk. Lymphoma 2007, 48, 716–722. [Google Scholar] [CrossRef]
- Chittima, S.; Kanchana, C.; Arnuparp, L.; Jakrawadee, J.; Kitsada, W.; Udomsak, B.; Lalita, N.; Weerasak, N.; Archrob, K.; Supachai, E.; et al. Clinical Features and Treatment Outcomes of Angioimmunoblastic T-Cell Lymphoma: An Analysis from a Nationwide Multicenter Registry, Thailand. Blood 2015, 126, 5064. [Google Scholar] [CrossRef]
- Jenni, K.; Pyry, U.; Kirsi-Maria, H.; Saila, K.; Hanna-Riikka, T.; Milla, E.L.K.; Siria, L.; Taina, T.-H.; Martine, V.; Mine, E.; et al. Incidence and clinicopathological features of Follicular T-cell lymphoma in Finland: A population-based immunohistochemical study. Hum. Pathol. 2021, 117, 79–87. [Google Scholar] [CrossRef]
- Hiroaki, M.; Mamiko, S.Y.; Joji, S.; Noriaki, Y.; Keiichiro, H.; Fumiko, A.; Eriko, Y.; Mai, T.; Kyohei, Y.; Takaharu, S.; et al. RHOA mutation in follicular T-cell lymphoma: Clinicopathological analysis of 16 cases. Pathol. Int. 2020, 70, 653–660. [Google Scholar] [CrossRef]
- Huang, Y.; Moreau, A.; Dupuis, J.; Streubel, B.; Petit, B.; Le Gouill, S.; Martin-Garcia, N.; Copie-Bergman, C.; Gaillard, F.; Qubaja, M.; et al. Peripheral T-cell lymphomas with a follicular growth pattern are derived from follicular helper T cells (TFH) and may show overlapping features with angioimmunoblastic T-cell lymphomas. Am. J. Surg. Pathol. 2009, 33, 682–690. [Google Scholar] [CrossRef]
- Liang, P.I.; Chang, S.T.; Lin, M.Y.; Hsieh, Y.C.; Chu, P.Y.; Chen, C.J.; Lin, K.J.; Jung, Y.C.; Hwang, W.S.; Huang, W.T.; et al. Angioimmunoblastic T-cell lymphoma in Taiwan shows a frequent gain of ITK gene. Int. J. Clin. Exp. Pathol. 2014, 7, 6097–6107. [Google Scholar]
- Lemonnier, F.; Couronne, L.; Parrens, M.; Jais, J.P.; Travert, M.; Lamant, L.; Tournillac, O.; Rousset, T.; Fabiani, B.; Cairns, R.A.; et al. Recurrent TET2 mutations in peripheral T-cell lymphomas correlate with TFH-like features and adverse clinical parameters. Blood 2012, 120, 1466–1469. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Zhang, S.; Chuang, S.; Ashton-Key, M.; Ochoa, E.; Bolli, N.; Vassiliou, G.; Gao, Z.; Du, M. Angioimmunoblastic T cell lymphoma: Novel molecular insights by mutation profiling. Oncotarget 2017, 8, 17763–17770. [Google Scholar] [CrossRef] [Green Version]
- Yao, W.Q.; Wu, F.; Zhang, W.; Chuang, S.S.; Thompson, J.S.; Chen, Z.; Zhang, S.W.; Clipson, A.; Wang, M.; Liu, H.; et al. Angioimmunoblastic T-cell lymphoma contains multiple clonal T-cell populations derived from a common TET2 mutant progenitor cell. J. Pathol. 2020, 250, 346–357. [Google Scholar] [CrossRef] [Green Version]
- Alexandra, B.; Kaushik, S.; Diwash, J.; Jyoti, K.; Malaya, K.S.; Nahid, S.; Roger, A.W.; Elumalai, R.; Benjamin, A.P.; Robert, S.O. A Comprehensive Analysis of Rhoa Mutation Positive and Negative Angioimmunoblastic T-Cell Lymphomas By Targeted Deep Sequencing, Expression Profiling, and Single Cell Digital Image Analysis. Blood 2019, 134, 5228. [Google Scholar] [CrossRef]
- Yingying, Y.; Ning, D.; Lan, M.; Yunfei, S.; Weiping, L.; Yuqin, S.; Shaokun, S.; Jun, Z. Correlation of mutational landscape and survival outcome of peripheral T-cell lymphomas. Exp. Hematol. Oncol. 2021, 10, 9. [Google Scholar] [CrossRef]
- Nguyen, T.B.; Sakata-Yanagimoto, M.; Fujisawa, M.; Nuhat, S.T.; Miyoshi, H.; Nannya, Y.; Hashimoto, K.; Fukumoto, K.; Bernard, O.A.; Kiyoki, Y.; et al. Dasatinib Is an Effective Treatment for Angioimmunoblastic T-cell Lymphoma. Cancer Res. 2020, 80, 1875–1884. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; McKeithan, T.W.; Gong, Q.; Zhang, W.; Bouska, A.; Rosenwald, A.; Gascoyne, R.D.; Wu, X.; Wang, J.; Muhammad, Z.; et al. IDH2R172 mutations define a unique subgroup of patients with angioimmunoblastic T-cell lymphoma. Blood 2015, 126, 1741–1752. [Google Scholar] [CrossRef] [Green Version]
- François, L.; Violaine, S.; Asma, B.-F.; Anne-Ségolène, C.; Emmanuel, B.; Guillaume, C.; Virginie, F.; Laura, P.; Cyrielle, R.; Audrey, L.; et al. Integrative analysis of a phase 2 trial combining lenalidomide with CHOP in angioimmunoblastic T-cell lymphoma. Blood Adv. 2021, 5, 539–548. [Google Scholar] [CrossRef]
- Steinhilber, J.; Mederake, M.; Bonzheim, I.; Serinsoz-Linke, E.; Muller, I.; Fallier-Becker, P.; Lemonnier, F.; Gaulard, P.; Fend, F.; Quintanilla-Martinez, L. The pathological features of angioimmunoblastic T-cell lymphomas with IDH2(R172) mutations. Mod. Pathol. 2019, 32, 1123–1134. [Google Scholar] [CrossRef]
- Marta, R.; Ruth, A.-A.; Laura, T.-R.; Socorro, M.R.-P.; Rebeca, M.-A.; Laura, C.; Jennifer, B.; Teresa, V.; Raúl, C.; Margarita, S.-B.; et al. Peripheral T-cell lymphoma: Molecular profiling recognizes subclasses and identifies prognostic markers. Blood Adv. 2021, 5, 5588–5598. [Google Scholar] [CrossRef]
- Saillard, C.; Guermouche, H.; Derrieux, C.; Bruneau, J.; Frenzel, L.; Couronne, L.; Asnafi, V.; Macintyre, E.; Trinquand, A.; Lhermitte, L.; et al. Response to 5-azacytidine in a patient with TET2-mutated angioimmunoblastic T-cell lymphoma and chronic myelomonocytic leukaemia preceded by an EBV-positive large B-cell lymphoma. Hematol. Oncol. 2017, 35, 864–868. [Google Scholar] [CrossRef]
- Fadela, B.; Jocelyne, D.; Jacques, B.; Catherine, T.; Daniel, E.; Gilles, S.; Bertrand, C. Profiles and prognostic values of serum LDH isoenzymes in patients with haematopoietic malignancies. Bull. Cancer 2004, 91, E229–E240. [Google Scholar]
- Odejide, O.; Weigert, O.; Lane, A.A.; Toscano, D.; Lunning, M.A.; Kopp, N.; Kim, S.; van Bodegom, D.; Bolla, S.; Schatz, J.H.; et al. A targeted mutational landscape of angioimmunoblastic T-cell lymphoma. Blood 2014, 123, 1293–1296. [Google Scholar] [CrossRef]
- François, L.; Jehan, D.; Pierre, S.; Olivier, T.; Morgane, C.; Clémentine, S.; Laura, P.; Ambroise, M.; Cyrielle, R.; Virginie, F.; et al. Treatment with 5-azacytidine induces a sustained response in patients with angioimmunoblastic T-cell lymphoma. Blood 2018, 132, 2305–2309. [Google Scholar] [CrossRef] [Green Version]
- Sebastian, F.-P.; Lisa, M.; Rohan, P.J.; Daniel, A.A. A Survey of Somatic Mutations in 41 Genes in a Cohort of T-Cell Lymphomas Identifies Frequent Mutations in Genes Involved in Epigenetic Modification. Appl. Immunohistochem. Mol. Morphol. 2019, 27, 416–422. [Google Scholar] [CrossRef]
- Bar-Sagi, D.; Hall, A. Ras and Rho GTPases: A family reunion. Cell 2000, 103, 227–238. [Google Scholar] [CrossRef] [Green Version]
- Sandrine, E.-M.; Alan, H. Rho GTPases in cell biology. Nature 2002, 420, 629–635. [Google Scholar] [CrossRef]
- Vallois, D.; Dobay, M.P.; Morin, R.D.; Lemonnier, F.; Missiaglia, E.; Juilland, M.; Iwaszkiewicz, J.; Fataccioli, V.; Bisig, B.; Roberti, A.; et al. Activating mutations in genes related to TCR signaling in angioimmunoblastic and other follicular helper T-cell-derived lymphomas. Blood 2016, 128, 1490–1502. [Google Scholar] [CrossRef] [Green Version]
- Hae Yong, Y.; Min Kyung, S.; Seung Ho, L.; Sangok, K.; Haeseung, L.; Seongjin, P.; Sang Cheol, K.; Byungwook, L.; Kyoohyoung, R.; Jong-Eun, L.; et al. A recurrent inactivating mutation in RHOA GTPase in angioimmunoblastic T cell lymphoma. Nat. Genet. 2014, 46, 371–375. [Google Scholar] [CrossRef]
- Fujisawa, M.; Sakata-Yanagimoto, M.; Nishizawa, S.; Komori, D.; Gershon, P.; Kiryu, M.; Tanzima, S.; Fukumoto, K.; Enami, T.; Muratani, M.; et al. Activation of RHOA-VAV1 signaling in angioimmunoblastic T-cell lymphoma. Leukemia 2018, 32, 694–702. [Google Scholar] [CrossRef] [Green Version]
- Tybulewicz, V.L. Vav-family proteins in T-cell signalling. Curr. Opin. Immunol. 2005, 17, 267–274. [Google Scholar] [CrossRef]
- Komori, D.; Sakata-Yanagimoto, M.; Nuhat, S.T.; Fukumoto, K.; Fujisawa, M.; Nishizawa, S.; Matsue, K.; Izutsu, K.; Nakamura, N.; Yoshida, K.; et al. Recurrent VAV1 Abnormalities in Angioimmunoblastic T Cell Lymphoma. Blood 2016, 128, 4104. [Google Scholar] [CrossRef]
- Samuel, Y.N.; Leon, B.; Kristen, S.; Tiffany, d.; Jon, C.A.; Abner, L.; David, M.W. RhoA G17V is sufficient to induce autoimmunity and promotes T cell lymphomagenesis in mice. Blood 2018, 132, 935–947. [Google Scholar] [CrossRef] [Green Version]
- Horwitz, S.M.; Moskowitz, A.J.; Jacobsen, E.D.; Mehta-Shah, N.; Khodadoust, M.S.; Fisher, D.C.; Myskowski, P.; Wang, E.B.K.; Tawa, M.; Davey, T.; et al. The Combination of Duvelisib, a PI3K-δ,γ Inhibitor, and Romidepsin Is Highly Active in Relapsed/Refractory Peripheral T-Cell Lymphoma with Low Rates of Transaminitis: Results of Parallel Multicenter, Phase 1 Combination Studies with Expansion Cohorts. Blood 2018, 132, 683. [Google Scholar] [CrossRef]
- Stone, E.L.; Pepper, M.; Katayama, C.D.; Kerdiles, Y.M.; Lai, C.Y.; Emslie, E.; Lin, Y.C.; Yang, E.; Goldrath, A.W.; Li, M.O.; et al. ICOS coreceptor signaling inactivates the transcription factor FOXO1 to promote Tfh cell differentiation. Immunity 2015, 42, 239–251. [Google Scholar] [CrossRef]
- Zang, S.; Li, J.; Yang, H.; Zeng, H.; Han, W.; Zhang, J.; Lee, M.; Moczygemba, M.; Isgandarova, S.; Yang, Y.; et al. Mutations in 5-methylcytosine oxidase TET2 and RhoA cooperatively disrupt T cell homeostasis. J. Clin. Investig. 2017, 127, 2998–3012. [Google Scholar] [CrossRef] [Green Version]
- Cortes, J.R.; Ambesi-Impiombato, A.; Couronne, L.; Quinn, S.A.; Kim, C.S.; da Silva Almeida, A.C.; West, Z.; Belver, L.; Martin, M.S.; Scourzic, L.; et al. RHOA G17V Induces T Follicular Helper Cell Specification and Promotes Lymphomagenesis. Cancer Cell 2018, 33, 259–273. [Google Scholar] [CrossRef] [Green Version]
- Mhaidly, R.; Krug, A.; Gaulard, P.; Lemonnier, F.; Ricci, J.-E.; Verhoeyen, E. New preclinical models for angioimmunoblastic T-cell lymphoma: Filling the GAP. Oncogenesis 2020, 9, 73. [Google Scholar] [CrossRef]
- Challen, G.A.; Sun, D.; Jeong, M.; Luo, M.; Jelinek, J.; Berg, J.S.; Bock, C.; Vasanthakumar, A.; Gu, H.; Xi, Y.; et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat. Genet. 2011, 44, 23–31. [Google Scholar] [CrossRef] [Green Version]
- Allison, M.; Liubin, Y.; Benjamin, R.; Ting, Z.; Edmund, C.; Choladda, V.C.; Grant, A.C.; Wei, L.; David, W.; Vivienne, I.R.; et al. Dnmt3a loss predisposes murine hematopoietic stem cells to malignant transformation. Blood 2015, 125, 629–638. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Su, J.; Jeong, M.; Ko, M.; Huang, Y.; Park, H.J.; Guzman, A.; Lei, Y.; Huang, Y.H.; Rao, A.; et al. DNMT3A and TET2 compete and cooperate to repress lineage-specific transcription factors in hematopoietic stem cells. Nat. Genet. 2016, 48, 1014–1023. [Google Scholar] [CrossRef] [Green Version]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-Methylcytosine to 5-Hydroxymethylcytosine in Mammalian DNA by MLL Partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef] [Green Version]
- Sakata-Yanagimoto, M. Multistep tumorigenesis in peripheral T cell lymphoma. Int. J. Hematol. 2015, 102, 523–527. [Google Scholar] [CrossRef] [Green Version]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef] [Green Version]
- Xie, M.; Lu, C.; Wang, J.; McLellan, M.D.; Johnson, K.J.; Wendl, M.C.; McMichael, J.F.; Schmidt, H.K.; Yellapantula, V.; Miller, C.A.; et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat. Med. 2014, 20, 1472–1478. [Google Scholar] [CrossRef]
- Ward, P.S.; Patel, J.; Wise, D.R.; Abdel-Wahab, O.; Bennett, B.D.; Coller, H.A.; Cross, J.R.; Fantin, V.R.; Hedvat, C.V.; Perl, A.E.; et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 2010, 17, 225–234. [Google Scholar] [CrossRef] [Green Version]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744. [Google Scholar] [CrossRef] [Green Version]
- Maria, E.F.; Omar, A.-W.; Chao, L.; Patrick, S.W.; Jay, P.; Alan, S.; Yushan, L.; Neha, B.; Aparna, V.; Hugo, F.F.; et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef] [Green Version]
- Cairns, R.A.; Mak, T.W. Oncogenic isocitrate dehydrogenase mutations: Mechanisms, models, and clinical opportunities. Cancer Discov. 2013, 3, 730–741. [Google Scholar] [CrossRef] [Green Version]
- François, L.; Rob, A.C.; Satoshi, I.; Wanda, Y.L.; Aurélie, D.; Sophie, B.; Nadine, M.; Virginie, F.; Romain, P.; Andrew, W.; et al. The IDH2 R172K mutation associated with angioimmunoblastic T-cell lymphoma produces 2HG in T cells and impacts lymphoid development [Medical Sciences]. Proc. Natl. Acad. Sci. USA 2016, 113, 15084–15089. [Google Scholar] [CrossRef] [Green Version]
- Tayla, B.H.; Alyssa, B.; Jiayu, Y.; Waseem, L.; Catalina, A.; Qiang, G.; Weiwei, Z.; Yuping, L.; Bhavana, J.D.; Maarja-Liisa, N.; et al. Genetic drivers of oncogenic pathways in molecular subgroups of peripheral T-cell lymphoma. Blood 2019, 133, 1664–1676. [Google Scholar] [CrossRef] [Green Version]
- Mario, L.M.-P.; Yessenia, I.S.; Carlos, P.; Renato, B.-H.; Francisco, V.; Roberto, N.M. Epstein–Barr virus-associated B-cell lymphoproliferative disorders and lymphomas: A review. Pathology 2020, 52, 40–52. [Google Scholar] [CrossRef] [Green Version]
- Ho, J.W.; Ho, F.C.; Chan, A.C.; Liang, R.H.; Srivastava, G. Frequent detection of Epstein-Barr virus-infected B cells in peripheral T-cell lymphomas. J. Pathol. 1998, 185, 79–85. [Google Scholar] [CrossRef]
- Willenbrock, K.; Brauninger, A.; Hansmann, M.L. Frequent occurrence of B-cell lymphomas in angioimmunoblastic T-cell lymphoma and proliferation of Epstein-Barr virus-infected cells in early cases. Br. J. Haematol. 2007, 138, 733–739. [Google Scholar] [CrossRef]
- Bräuninger, A.; Spieker, T.; Willenbrock, K.; Gaulard, P.; Wacker, H.H.; Rajewsky, K.; Hansmann, M.L.; Küppers, R. Survival and clonal expansion of mutating "forbidden" (immunoglobulin receptor-deficient) epstein-barr virus-infected b cells in angioimmunoblastic t cell lymphoma. J. Exp. Med. 2001, 194, 927–940. [Google Scholar] [CrossRef]
- Marie, P.; Antoine, M.; Laurence, L.; Richard, D.; Corinne, H.; Olivier, T.; Francoise, B.; Céline, B.; Brigitte, B.h.; Josette, B.; et al. Angioimmunoblastic T-Cell Lymphoma (AITL) Is the Most Prevalent T-Cell Lymphoma Entity in Western Europe. Blood 2012, 120, 1607. [Google Scholar] [CrossRef]
- Lemonnier, F.; Mak, T.W. Angioimmunoblastic T-cell lymphoma: More than a disease of T follicular helper cells. J. Pathol. 2017, 242, 387–390. [Google Scholar] [CrossRef] [Green Version]
- Nicolae, A.; Pittaluga, S.; Venkataraman, G.; Vijnovich-Baron, A.; Xi, L.; Raffeld, M.; Jaffe, E.S. Peripheral T-cell lymphomas of follicular T-helper cell derivation with Hodgkin/Reed-Sternberg cells of B-cell lineage: Both EBV-positive and EBV-negative variants exist. Am. J. Surg. Pathol. 2013, 37, 816–826. [Google Scholar] [CrossRef]
- Suefuji, N.; Niino, D.; Arakawa, F.; Karube, K.; Kimura, Y.; Kiyasu, J.; Takeuchi, M.; Miyoshi, H.; Yoshida, M.; Ichikawa, A.; et al. Clinicopathological analysis of a composite lymphoma containing both T- and B-cell lymphomas. Pathol. Int. 2012, 62, 690–698. [Google Scholar] [CrossRef]
- Ayoma Deepthi, A.; Charalampia, K.; Jehan, D.; Karen Lynne, G.; Timothy Charles, D.; Andrew Charles, W.; Shih Sung, C.; José, C.; Peter Gershon, I.; Ming-Qing, D.; et al. Histologic evolution of angioimmunoblastic T-cell lymphoma in consecutive biopsies: Clinical correlation and insights into natural history and disease progression. Am. J. Surg. Pathol. 2007, 31, 1077–1088. [Google Scholar] [CrossRef]
- Krug, A.; Tari, G.; Saidane, A.; Gaulard, P.; Ricci, J.-E.; Lemonnier, F.; Verhoeyen, E. Novel T Follicular Helper-like T-Cell Lymphoma Therapies: From Preclinical Evaluation to Clinical Reality. Cancers 2022, 14, 2392. [Google Scholar] [CrossRef]
- James, O.A. The aggressive peripheral T-cell lymphomas: 2017. Am. J. Hematol. 2017, 92, 706–715. [Google Scholar] [CrossRef] [Green Version]
- Horwitz, S.M.; Ansell, S.; Ai, W.Z.; Barnes, J.; Barta, S.K.; Brammer, J.; Clemens, M.W.; Dogan, A.; Foss, F.; Ghione, P.; et al. T-Cell Lymphomas, Version 2.2022, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. JNCCN 2022, 20, 285–308. [Google Scholar] [CrossRef]
- Tari, G.; Lemonnier, F.; Morschhauser, F. Epigenetic focus on angioimmunoblastic T-cell lymphoma: Pathogenesis and treatment. Curr. Opin. Oncol. 2021, 33, 400–405. [Google Scholar] [CrossRef]
- Coiffier, B.; Pro, B.; Prince, H.M.; Foss, F.; Sokol, L.; Greenwood, M.; Caballero, D.; Borchmann, P.; Morschhauser, F.; Wilhelm, M.; et al. Results From a Pivotal, Open-Label, Phase II Study of Romidepsin in Relapsed or Refractory Peripheral T-Cell Lymphoma After Prior Systemic Therapy. J. Clin. Oncol. 2012, 30, 631–636. [Google Scholar] [CrossRef]
- Foss, F.; Advani, R.; Duvic, M.; Hymes, K.B.; Intragumtornchai, T.; Lekhakula, A.; Shpilberg, O.; Lerner, A.; Belt, R.J.; Jacobsen, E.D.; et al. A Phase II trial of Belinostat (PXD101) in patients with relapsed or refractory peripheral or cutaneous T-cell lymphoma. Br. J. Haematol. 2014, 168, 811–819. [Google Scholar] [CrossRef]
- O’Connor, O.A.; Horwitz, S.; Masszi, T.; Van Hoof, A.; Brown, P.; Doorduijn, J.; Hess, G.; Jurczak, W.; Knoblauch, P.; Chawla, S.; et al. Belinostat in Patients With Relapsed or Refractory Peripheral T-Cell Lymphoma: Results of the Pivotal Phase II BELIEF (CLN-19) Study. J. Clin. Oncol. 2015, 33, 2492–2499. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Dong, M.; Hong, X.; Zhang, W.; Feng, J.; Zhu, J.; Yu, L.; Ke, X.; Huang, H.; Shen, Z.; et al. Results from a multicenter, open-label, pivotal phase II study of chidamide in relapsed or refractory peripheral T-cell lymphoma. Ann. Oncol. 2015, 26, 1766–1771. [Google Scholar] [CrossRef]
- Shi, Y.; Jia, B.; Xu, W.; Li, W.; Liu, T.; Liu, P.; Zhao, W.; Zhang, H.; Sun, X.; Yang, H.; et al. Chidamide in relapsed or refractory peripheral T cell lymphoma: A multicenter real-world study in China. J. Hematol. Oncol. 2017, 10, 69. [Google Scholar] [CrossRef] [Green Version]
- Dupuis, J.; Morschhauser, F.; Ghesquières, H.; Tilly, H.; Casasnovas, O.; Thieblemont, C.; Ribrag, V.; Bossard, C.; Bras, F.L.; Bachy, E.; et al. Combination of romidepsin with cyclophosphamide, doxorubicin, vincristine, and prednisone in previously untreated patients with peripheral T-cell lymphoma: A non-randomised, phase 1b/2 study. Lancet Haematol. 2015, 2, e160–e165. [Google Scholar] [CrossRef]
- Moskowitz, A.J.; Koch, R.; Mehta-Shah, N.; Myskowski, P.; Kheterpal, M.; Dogan, A.; Davey, T.; Galasso, N.; Evan, M.; Shah, M.; et al. In Vitro, In Vivo, and Parallel Phase I Evidence Support the Safety and Activity of Duvelisib, a PI3K-δ,γ Inhibitor, in Combination with Romidepsin or Bortezomib in Relapsed/Refractory T-Cell Lymphoma. Blood 2017, 130, 819. [Google Scholar] [CrossRef]
- Bachy, E.; Camus, V.; Thieblemont, C.; Sibon, D.; Casasnovas, R.-O.; Ysebaert, L.; Damaj, G.; Guidez, S.; Pica, G.M.; Kim, W.S.; et al. Romidepsin Plus CHOP Versus CHOP in Patients With Previously Untreated Peripheral T-Cell Lymphoma: Results of the Ro-CHOP Phase III Study (Conducted by LYSA). J. Clin. Oncol. 2021, 40, 242–251. [Google Scholar] [CrossRef]
- Falchi, L.; Ma, H.; Klein, S.; Lue, J.K.; Montanari, F.; Marchi, E.; Deng, C.; Kim, H.A.; Rada, A.; Jacob, A.T.; et al. Combined oral 5-azacytidine and romidepsin are highly effective in patients with PTCL: A multicenter phase 2 study. Blood 2021, 137, 2161–2170. [Google Scholar] [CrossRef]
- Ding, K.; Shi, X.; Yang, H.; Cao, L.; Zhao, X.; Liu, H.; Wu, W.; Zhang, X.; Wang, L.; Xu, W.; et al. The Interim Efficacy of Epigenetic Priming Regimen with Azacytidine and Chidamide in Patients with Relapsed or Refractory Peripheral T Cell Lymphoma. Blood 2021, 138, 2461. [Google Scholar] [CrossRef]
- Jessica, E.B.; Melissa, J.P.; Ricky, W.J. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discov. 2006, 5, 769–784. [Google Scholar] [CrossRef]
- Ricky, W.J.; Jonathan, D.L. Histone deacetylase inhibitors in cancer therapy: Is transcription the primary target? Cancer Cell 2003, 4, 13–18. [Google Scholar] [CrossRef] [Green Version]
- Peart, M.J.; Smyth, G.K.; Laar, R.K.v.; Bowtell, D.D.; Richon, V.M.; Marks, P.A.; Holloway, A.J.; Johnstone, R.W. Identification and functional significance of genes regulated by structurally different histone deacetylase inhibitors. Proc. Natl. Acad. Sci. USA 2005, 102, 3697–3702. [Google Scholar] [CrossRef] [Green Version]
- Walid, R.; Mark, B.; Ricky, W.J.; Prince, H.M. Histone deacetylase inhibitors in lymphoma and solid malignancies. Expert Rev. Anticancer Ther. 2008, 8, 413–432. [Google Scholar] [CrossRef]
- Yun, L.; Zhijuan, L.; Kun, C.; Zhihong, F.; Zhifeng, L.; Yiming, L.; Bing, X. Prognostic role of TET2 deficiency in myelodysplastic syndromes: A meta-analysis. Oncotarget 2017, 8, 43295–43305. [Google Scholar] [CrossRef] [Green Version]
- Cedena, M.T.; Inmaculada, R.; Alejandro, S.-L.; Rosa, A.; Esther, O.; María, A.; Esperanza, S.; Fernando, R.; José, C.; María, D.-C.; et al. Mutations in the DNA methylation pathway and number of driver mutations predict response to azacitidine in myelodysplastic syndromes. Oncotarget 2017, 8, 106948–106961. [Google Scholar] [CrossRef] [Green Version]
- Morgane, C.; Julie, B.; Olivier, K.; François, L.; Richard, D.; Philippe, G.; Isabelle, R.; Coralie, D.; Olivier, H.; François, L. Efficacy of 5-azacytidine in a TET2 mutated angioimmunoblastic T cell lymphoma. Br. J. Haematol. 2014, 168, 913–916. [Google Scholar] [CrossRef]
- O’Connor, O.A.; Falchi, L.; Lue, J.K.; Marchi, E.; Kinahan, C.; Sawas, A.; Deng, C.; Montanari, F.; Amengual, J.E.; Kim, H.A.; et al. Oral 5-azacytidine and romidepsin exhibit marked activity in patients with PTCL: A multicenter phase 1 study. Blood 2019, 134, 1395–1405. [Google Scholar] [CrossRef]
- Elizabeth, E.C.; Kurtis, E.B.; Sanna, M.; James, G.H.; Stephen, B.B. Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat. Genet. 1999, 21, 103–107. [Google Scholar] [CrossRef]
- Melanie, R.H.; Aleksandra, K.; Karoline, K.; Irene, S.; Martin, B.; Ana-Iris, S.; Veronika, S.; Gerda, E. Antineoplastic activity of the DNA methyltransferase inhibitor 5-aza-2’-deoxycytidine in anaplastic large cell lymphoma. Biochimie 2012, 94, 2297–2307. [Google Scholar] [CrossRef] [Green Version]
- Jabbour, E.; Issa, J.P.; Garcia-Manero, G.; Kantarjian, H. Evolution of decitabine development: Accomplishments, ongoing investigations, and future strategies. Cancer 2008, 112, 2341–2351. [Google Scholar] [CrossRef]
- Benigno, C.V.; Yang, L.; David, M.; Yan, L.; Yago, N.; Richard, E.C.; Borje, S.A. Combination of a hypomethylating agent and inhibitors of PARP and HDAC traps PARP1 and DNMT1 to chromatin, acetylates DNA repair proteins, down-regulates NuRD and induces apoptosis in human leukemia and lymphoma cells. Oncotarget 2017, 9, 3908–3921. [Google Scholar] [CrossRef] [Green Version]
- Chua, G.N.L.; Wassarman, K.L.; Sun, H.; Alp, J.A.; Jarczyk, E.I.; Kuzio, N.J.; Bennett, M.J.; Malachowsky, B.G.; Kruse, M.; Kennedy, A.J. Cytosine-Based TET Enzyme Inhibitors. ACS Med. Chem. Lett. 2019, 10, 180–185. [Google Scholar] [CrossRef]
- Guan, Y.; Tiwari, A.D.; Phillips, J.G.; Hasipek, M.; Grabowski, D.R.; Pagliuca, S.; Gopal, P.; Kerr, C.M.; Adema, V.; Radivoyevitch, T.; et al. A Therapeutic Strategy for Preferential Targeting of TET2 Mutant and TET-dioxygenase Deficient Cells in Myeloid Neoplasms. Blood Cancer Discov. 2020, 2, 146–161. [Google Scholar] [CrossRef]
- Shenoy, N.; Bhagat, T.; Nieves, E.; Stenson, M.; Lawson, J.; Choudhary, G.S.; Habermann, T.; Nowakowski, G.; Singh, R.; Wu, X.; et al. Upregulation of TET activity with ascorbic acid induces epigenetic modulation of lymphoma cells. Blood Cancer J. 2017, 7, e587. [Google Scholar] [CrossRef] [Green Version]
- Smith-Díaz, C.C.; Magon, N.J.; McKenzie, J.L.; Hampton, M.B.; Vissers, M.C.M.; Das, A.B. Ascorbate Inhibits Proliferation and Promotes Myeloid Differentiation in TP53-Mutant Leukemia. Front. Oncol. 2021, 11, 709543. [Google Scholar] [CrossRef]
- Guan, Y.; Greenberg, E.F.; Hasipek, M.; Chen, S.; Liu, X.; Kerr, C.M.; Gackowski, D.; Zarakowska, E.; Radivoyevitch, T.; Gu, X.; et al. Context dependent effects of ascorbic acid treatment in TET2 mutant myeloid neoplasia. Commun. Biol. 2020, 3, 493. [Google Scholar] [CrossRef]
- Bensberg, M.; Rundquist, O.; Selimović, A.; Lagerwall, C.; Benson, M.; Gustafsson, M.; Vogt, H.; Lentini, A.; Nestor, C.E. TET2 as a tumor suppressor and therapeutic target in T-cell acute lymphoblastic leukemia [Genetics]. Proc. Natl. Acad. Sci. USA 2021, 118, e2110758118. [Google Scholar] [CrossRef]
- Gerecke, C.; Schumacher, F.; Edlich, A.; Wetzel, A.; Yealland, G.; Neubert, L.K.; Scholtka, B.; Homann, T.; Kleuser, B. Vitamin C promotes decitabine or azacytidine induced DNA hydroxymethylation and subsequent reactivation of the epigenetically silenced tumour suppressor CDKN1A in colon cancer cells. Oncotarget 2018, 9, 32822–32840. [Google Scholar] [CrossRef] [Green Version]
- Zeng, H.; He, H.; Guo, L.; Li, J.; Lee, M.; Han, W.; Guzman, A.G.; Zang, S.; Zhou, Y.; Zhang, X.; et al. Antibiotic treatment ameliorates Ten-eleven translocation 2 (TET2) loss-of-function associated hematological malignancies. Cancer Lett. 2019, 467, 1–8. [Google Scholar] [CrossRef]
- Li, C.; Peng, C.; Jiang, Z.; Hu, H.; Lin, C.; Gao, Y.; Liu, D.; Sun, B.; Wang, D. Ginkgo biloba Extract Inhibited Cell Proliferation and Invasion by Stimulating TET2 Expression Through miR-29a in Colorectal Carcinoma Cells. DNA Cell Biol. 2022, 41. [Google Scholar] [CrossRef]
- Kawahori, K.; Kondo, Y.; Yuan, X.; Kawasaki, Y.; Hanzawa, N.; Tsujimoto, K.; Wada, F.; Kohda, T.; Ishigami, A.; Yamada, T.; et al. Ascorbic acid during the suckling period is required for proper DNA demethylation in the liver. Sci. Rep. 2020, 10, 21228. [Google Scholar] [CrossRef]
- Yin, R.; Mao, S.-Q.; Zhao, B.; Chong, Z.; Yang, Y.; Zhao, C.; Zhang, D.; Huang, H.; Gao, J.; Li, Z.; et al. Ascorbic Acid Enhances Tet-Mediated 5-Methylcytosine Oxidation and Promotes DNA Demethylation in Mammals. J. Am. Chem. Soc. 2013, 35, 10396–10403. [Google Scholar] [CrossRef]
- Blaschke, K.; Ebata, K.T.; Karimi, M.M.; Zepeda-Martínez, J.A.; Goyal, P.; Mahapatra, S.; Tam, A.; Laird, D.J.; Hirst, M.; Rao, A.; et al. Vitamin C induces Tet-dependent DNA demethylation and a blastocyst-like state in ES cells. Nature 2013, 500, 222–226. [Google Scholar] [CrossRef]
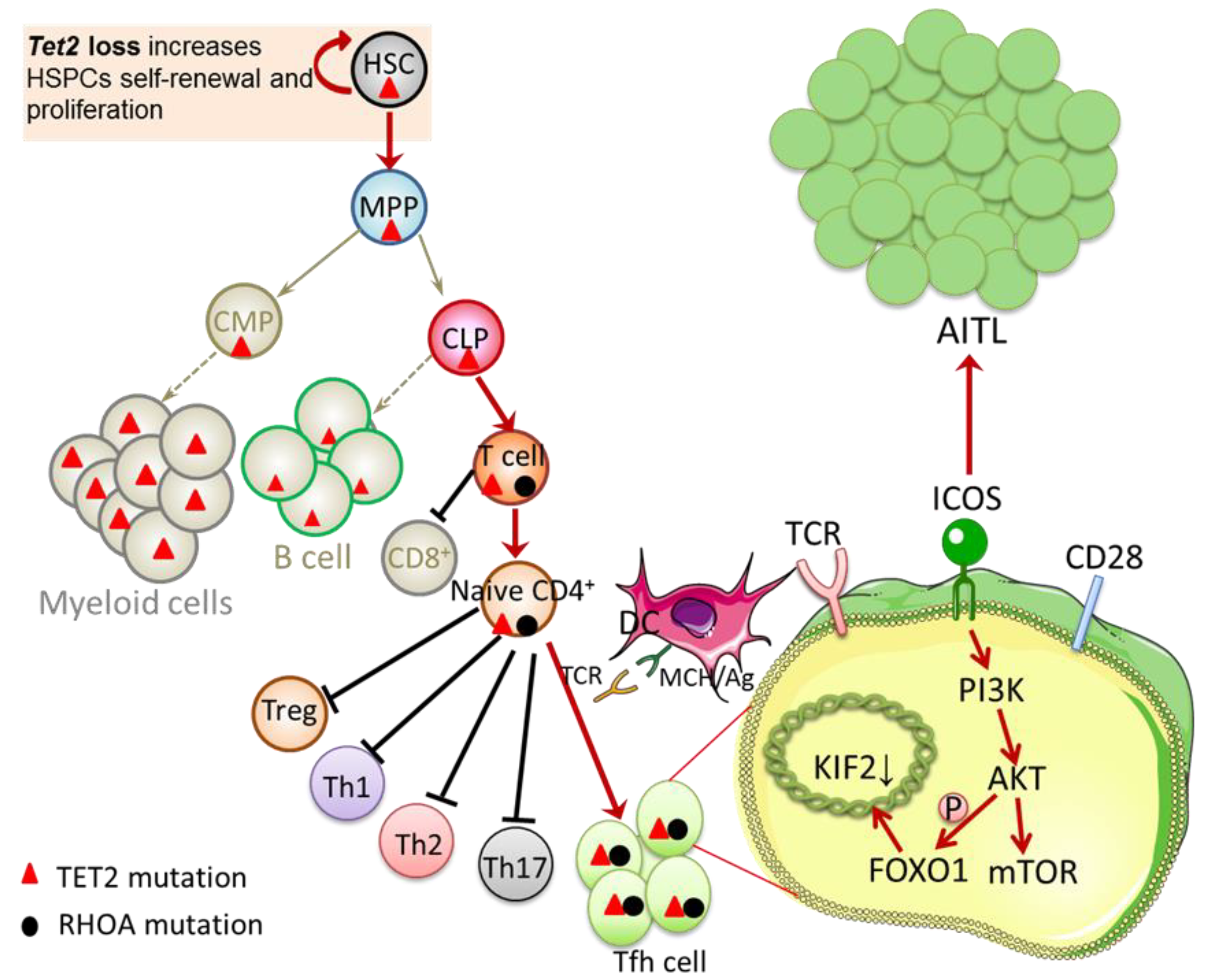
Figure 1.
Interaction between TET2 and RHOA mutations. TET2 loss increases HSPCs self-renewal and proliferation. Cooperativity between TET2 loss and RHOA G17V in T cell lineage suppresses the CD8+ T cell differentiation and endows the naïve CD4+ T cells with a competitive advantage, skews cells differentiation toward Tfh cells, and induces abnormal Tfh cell activation and transformation. ICOS exerts its costimulatory function via the ICOS-PI3K-mTOR signaling pathway, which is essential in driving Tfh lineage differentiation and maintaining the Tfh phenotype. In addition, ICOS activates AKT via PI3K. By phosphorylating the transcription factor FOXO1, AKT reduces the transcription factor KIF2, leading to a significant increase in the number of Tfh cells. Both of these pathways play an essential role in promoting Tfh lineage specification and AITL transformation. HSC, hematopoietic stem cells; MPP, multipotent blood progenitors; CLP, common lymphoid progenitors; CMP, common myeloid progenitors; Treg, regulatory T cell; Th1, T helper 1 cell; Th2, T helper 2 cell; Th17, T helper 17 cell; Tfh cell, follicular helper T cell; DC, dendritic cell; MHC/Ag, antigen presented on major histocompatibility complex; TCR, T-cell receptor; ICOS, inducible T-cell co-stimulator; PI3K, phosphatidylinositol-3-kinase; AKT, protein kinase B; mTOR, mammalian target of rapamycin; FOXO1, forkhead box O1; AITL, angioimmunoblastic T-cell lymphoma.
Figure 1.
Interaction between TET2 and RHOA mutations. TET2 loss increases HSPCs self-renewal and proliferation. Cooperativity between TET2 loss and RHOA G17V in T cell lineage suppresses the CD8+ T cell differentiation and endows the naïve CD4+ T cells with a competitive advantage, skews cells differentiation toward Tfh cells, and induces abnormal Tfh cell activation and transformation. ICOS exerts its costimulatory function via the ICOS-PI3K-mTOR signaling pathway, which is essential in driving Tfh lineage differentiation and maintaining the Tfh phenotype. In addition, ICOS activates AKT via PI3K. By phosphorylating the transcription factor FOXO1, AKT reduces the transcription factor KIF2, leading to a significant increase in the number of Tfh cells. Both of these pathways play an essential role in promoting Tfh lineage specification and AITL transformation. HSC, hematopoietic stem cells; MPP, multipotent blood progenitors; CLP, common lymphoid progenitors; CMP, common myeloid progenitors; Treg, regulatory T cell; Th1, T helper 1 cell; Th2, T helper 2 cell; Th17, T helper 17 cell; Tfh cell, follicular helper T cell; DC, dendritic cell; MHC/Ag, antigen presented on major histocompatibility complex; TCR, T-cell receptor; ICOS, inducible T-cell co-stimulator; PI3K, phosphatidylinositol-3-kinase; AKT, protein kinase B; mTOR, mammalian target of rapamycin; FOXO1, forkhead box O1; AITL, angioimmunoblastic T-cell lymphoma.

Figure 2.
Traditional view: AITL precedes B-cell lymphoma. Early and subsequent oncogenic events, such as TET2 loss and the acquisition of RHOA G17V synergize to induce the cause of AITL. On this basis, EBV infection of sporadic non-neoplastic bystander B cells has a significant impact on the differentiation of B cells, allowing its survival and clonal expansion, and promoting the occurrence of B cell lymphoma. HSC, hematopoietic stem cells; HEV, high endothelial venule; FDC, follicular dendritic cell; EBV, Epstein-Barr virus.
Figure 2.
Traditional view: AITL precedes B-cell lymphoma. Early and subsequent oncogenic events, such as TET2 loss and the acquisition of RHOA G17V synergize to induce the cause of AITL. On this basis, EBV infection of sporadic non-neoplastic bystander B cells has a significant impact on the differentiation of B cells, allowing its survival and clonal expansion, and promoting the occurrence of B cell lymphoma. HSC, hematopoietic stem cells; HEV, high endothelial venule; FDC, follicular dendritic cell; EBV, Epstein-Barr virus.

Figure 3.
Schematic model of co-occurrence of AITL and B cell lymphoma. This multistep and multilineage model shows that TET2/DNMT3A mutation in HSCs will be an initiating event in the process of transformation, inducing the generation of multipotent premalignant T and B cell lines. In the T cell zone, naive CD4+ T cells contact with myeloid DC cells and migrate to the T-B border, activating B cells. Upon acquisition of the second hit in RHOA/IDH2 mutation, Tfh cells will be abnormally activated and proliferate, eventually inducing the development of AITL. B cells that already carry the Tet2/DNMT3A mutation are derived from activated B cells. Correspondingly, subsequent acquisition of NOTCH1 mutation/EBV infection in B cells may lead to the transformation of B cell lymphoma. Eventually, AITL and B Cell lymphoma develop simultaneously. HSC, hematopoietic stem cells; mDC, myeloid dendritic cell; MHC/Ag, antigen presented on major histocompatibility complex; TCR, T-cell receptor; ICOS, inducible T-cell co-stimulator; ICOS-L, ICOS ligand; AITL, angioimmunoblastic T-cell lymphoma.
Figure 3.
Schematic model of co-occurrence of AITL and B cell lymphoma. This multistep and multilineage model shows that TET2/DNMT3A mutation in HSCs will be an initiating event in the process of transformation, inducing the generation of multipotent premalignant T and B cell lines. In the T cell zone, naive CD4+ T cells contact with myeloid DC cells and migrate to the T-B border, activating B cells. Upon acquisition of the second hit in RHOA/IDH2 mutation, Tfh cells will be abnormally activated and proliferate, eventually inducing the development of AITL. B cells that already carry the Tet2/DNMT3A mutation are derived from activated B cells. Correspondingly, subsequent acquisition of NOTCH1 mutation/EBV infection in B cells may lead to the transformation of B cell lymphoma. Eventually, AITL and B Cell lymphoma develop simultaneously. HSC, hematopoietic stem cells; mDC, myeloid dendritic cell; MHC/Ag, antigen presented on major histocompatibility complex; TCR, T-cell receptor; ICOS, inducible T-cell co-stimulator; ICOS-L, ICOS ligand; AITL, angioimmunoblastic T-cell lymphoma.


{kind=link}
{kind=link}
{kind=link}
Table 1.
Frequencies of TET2 mutation in AITL.
| Diagnosis | Experiment Type | AITL Cases | Mutation Cases | Mutation Rate | Mutation Sites | Mutation Type (Rate) | Amino Acid Change | Mult-Mutation Rate | Reference |
|---|---|---|---|---|---|---|---|---|---|
| AITL& PTCL-NOS | Microarray | 30 | 10 | AITL: 33.3%; PTCL-NOS: 20% | NA | NA | NA | NA | Cyril Quivoron [4] et al. (2011) |
| AITL& PTCL-NOS | direct sequencing | 86 | 40 | AITL: 47%; PTCL-NOS: 38% | NA | NA | NA | NA | Lemonnier [18] et al. (2012) |
| AITL | NGS | 85 | 65 | 76% | 115 | Missense: 20/115 (17%); Nonsense: 38/115 (33%); Splice site: 3/115 (3%); Frameshift: 54/115 (47%); | p.Q673*, p.Q727*, p.Q765*, p.R1486*, p.Y1148* | 43/65 (66%) | Odejide [30] et al. (2014) |
| AITL& PTCL-NOS | Targeted resequencing | 46 | 38 | AITL: 82.6%; PTCL-NOS: 48.5% | 70 | Frameshift: 29/70 (42%); Missense: 19/70 (27%); Nonframeshift: 1/70 (1%); Nonsense: 16/70 (23%); Splice site: 5/70 (7%) | p.E1318 splice; | 28/38 (74%) | Sakata-Yanagimoto [7] et al. (2014) |
| AITL& PTCL-NOS | Targeted resequencing | 39 | 32 | AITL: 82.1%; PTCL-NOS: 46.3% | 48 | NA | NA | 13/32 (41%) | Wang [24] et al. (2015) |
| AITL | WES | 9 | 9 | 100% | 15 | frameshift or nonsense changes: 14/15 (93%) | NA | 6/9 (67%) | Wang [19] et al. (2017) |
| AITL | Sanger | 13 | 12 | 92% | 15 | premature stop codons or deletions:11/15 (73%);replacement:4/15 (27%) | NA | NA | Schwartz [9] et al. (2017) |
| AITL& Nodal PTCL with TFH phenotype& PTCL-NOS | Targeted resequencing | 48 | 36 | AITL:75%; Nodal PTCL with TFH phenotype:100% PTCL-NOS: 55.9% | NA | NA | NA | NA | T B Nguyen [8] et al. (2017) |
| AITL& PTCL-NOS & PTCL-TFH&FTCL | NA | 64 | 31 | AITL: 48%; PTCL-NOS:17% PTCL-TFH: 64% FTCL:75% | NA | NA | NA | NA | Dobay [10] et al. (2017) |
| AITL | Targeted resequencing | 12 | 12 | 100% | 17 | NA | NA | 7/12 (58%) | Lemonnier [31] et al. (2018) |
| AITL& PTCL-NOS | Targeted Exon Sequencing | 13 | 5 | AITL:38%; PTCL-NOS:31% | 65 | NA | R126C, G1869W(2/13); N202K; D302Y; Y620; A893T; W1291; | NA | Fernandez-Pol [32] et al. (2019) |
| AITL | NGS | 44 | 38 | 86% | 60 | NA | NA | 23/38 (61%) | Julia Steinhilber [26] et al. (2019) |
| AITL& PTCL-TFH | Fluidigm Access Array& Illumina MiSeq | 94 | NA | AITL: 72%; PTCL-TFH: 73% | 154 | frameshift indels or Nonsense changes: 118 (77%) | NA | 57% | Yao [20] et al. (2020) |
| AITL | Targeted sequencing | 10 | 6 | 60% | 6 | NA | R550; Q1274; G422Efs’ 5; L34F; Q909; G422Efs’ 5 | 0/6 | Butzmann [21] et al. (2020) |
| AITL | Targeted sequencing | 5 | 4 | 80% | 8 | Frameshift insertion: 2/8 (25%); Nonsilent: 3/8 (37.5%);Frameshift deletion:3/8 (37.5%) | NA | 4/5 (80%) | Tran B. Nguyen [23] et al. (2020) |
| AITL | NGS | 28 | 25 | 85% | 75 | Missense: 26/75 (34.7%);Nosens: 22/75 (29.3%);Frameshift: 22/75 (29.3%);Splice: 4/75 (5.3%);CDS-indel: 1/75 (1.3%) | NA | 22/28 (79%) | Ye [22] et al. (2021) |
| AITL | NGS | 44 | NA | NA | 49 | Frameshift: 18/49 (36.7%);Missense: 12/49 (24.5%);Splice: 3/49 (6%);Stop_gained: 12/49 (24.5%);Synonymous: 3/49 (6%);3_prime_UTR: 1/49 (2%) | NA | NA | Marta Rodríguez [27] et al. (2021) |
| AITL& PTCL-TFH | Targeted resequencing | 63 | 49 | AITL: 78%; PTCL-TFH: 58% | NA | NA | NA | 28/49 (57%) | Lemonnier [25] et al. (2021) |
Abbreviations: AITL, angioimmunoblastic T-cell lymphoma; PTCL-NOS, peripheral T-cell lymphoma, not otherwise specified; FTLC, follicular T-cel1 lymphoma; PTCL-TFH, peripheral T-cell lymphoma with TFH phenotype.
Table 2.
Treatment outcomes of novel monotherapy and combination therapy for refractory and relapsed T-cell lymphoma including AITL.
Table 2.
Treatment outcomes of novel monotherapy and combination therapy for refractory and relapsed T-cell lymphoma including AITL.
| 5 | PTCL Subtype | Design | Primary Endpoint | ORR | CR | PR | Median PFS (Months) | Median OS (Months) | Reference |
|---|---|---|---|---|---|---|---|---|---|
| Romidepsin | PTCL n = 130 (PTCL-NOS n = 67, AITL n = 27) | Phase II, Open-Label | CR/Cru | 25% | 15% | 11% | 4 | NA | Coiffier [72] et al. (2012) |
| Belinostat | PTCL n = 24 (PTCL-NOS n = 13, AITL n = 3) | Phase II | ORR | 25% | 8.30% | 16.70% | NA | NA | Foss [73] et al. (2015) |
| Belinostat | PTCL n = 120 (PTCL-NOS n = 77, AITL n = 22) | Phase II, Open-Label, multicenter | ORR | 26% | 11% | 15% | 1.6 | 7.9 | O’Connor [74] et al. (2015) |
| Chidamide | PTCL n = 79 (PTCL-NOS n = 27, AITL n = 10) | Phase II, Open-Label, multicenter | ORR | 28% | 9% | 14% | 2.1 | 21.4 | Shi [75] et al. (2015) |
| Chidamide | PTCL n = 256 | Phase II, multicenter | ORR | 39.06% (PTCL 37.3% AITL 49.23%) | PTCL 8.73% AITL 9.23% | PTCL 28.57% AITL 40% | 4.3 | NA | Shi [76] et al. (2017) |
| Romidepsin + CHOP | PTCL n = 37 (PTCL-NOS n = 9, AITL n = 15) | phase 1b/2 | ORR | 69% | 51% | 17% | 21.3 | NA | Dupuis [77] et al. (2015) |
| Panobinostat + bortezomib | PTCL n = 25 (PTCL-NOS n = 9, AITL n = 8) | Phase II, Open-Label, multicenter | ORR | 43% (PTCL 22% AITL 50%) | 21.5% (PTCL 11% AITL 25%) | 21.5% (PTCL 11% AITL 25%) | NA | NA | Tan [76] et al. (2015) |
| Chidamide + chemotherapy | PTCL n = 127 | Phase II, multicenter | ORR | 51.18% | NA | NA | 5.4 | NA | Shi [76] et al. (2017) |
| Duvelisib + Romidepsin | T-cell lymphoma n = 12 | Phase I | ORR | 50% | NA | NA | NA | NA | Moskowitz [78] et al. (2017) |
| Duvelisib + bortezomib | T-cell lymphoma n = 17 | Phase I | ORR | 53% | 20% | 23% | NA | NA | Moskowitz [78] et al. (2017) |
| 5-Azacytidine | AITL n = 12 PTCL n = 37 | Clinic trial | ORR | 75% | 50% | 25% | 15 | 21 | Lemonnier [31] et al. (2018) |
| Duvelisib + Romidepsin | T-cell lymphoma n = 39 (PTCL n = 22) | Phase I, Parallel Multicenter | ORR | 51% (PTCL 55%) | 17% (PTCL 27%) | 34% | 8.8 (PTCL) | NA | Horwitz [41] et al. (2018) |
| Romidepsin + CHOP | PTCL n = 421 (Ro-CHOP n = 211) | Phase III | PFS | 63% | 41% | 22% | 12 | 51.8 | Bachy [79] et al. (2021) |
| 5-Azacytidine + romidepsin | PTCL n = 25 (PTCL-NOS n = 4, AITL n = 14) | Phase II, multicenter | ORR | 61% | 48% | 13% | 8 | Not reached | Falchi [80] et al. (2021) |
| 5-Azacytidine + Chidamide | PTCL n = 24 (PTCL-NOS n = 4, AITL n = 15) | Phase II | ORR | 68.8% (AITL 72.7%) | 31.2% (AITL 36.4%) | 37.5% (AITL 36.4%) | NA | NA | Ding [81] et al. (2021) |
Abbreviation: PTCL, peripheral T-cell lymphoma; PTCL-NOS, PTCL not otherwise specified; AITL, angioimmunoblastic T-cell lymphoma; CHOP, cyclophosphamide, doxorubicin, vincristine, prednisone; ORR, overall response rate; CR, complete response; Cru, complete response unconfirmed; PR, partial response; PFS, progression-free survival; OS, overall survival; NA, not analyzed.
Table 3.
Preclinical trials of TET2 targeting agents.
| Drugs | Disease of Study | Models | Mechanism | Limitations | Reference |
|---|---|---|---|---|---|
| Bobcat339 (TET enzyme inhibitors) | NA | HT-22 cells | Reduce DNA 5hmC levels in hippocampal | No testing in animal model and clinical trial | Gabriella [94] et al. (2019) |
| TET-specific inhibitors (TETi76) | MDS | Cell-permeable diethyl ester of TETi76 and different human leukemia cell lines (K562, MEG-01, SIG-M5, OCI-AML5, and MOLM13) | Decrease cytosine hydroxymethylation and restrict clonal out-growth of TET2 mutant | Potential to replicate the TET2 mutation | Guan [95] et al. (2020) |
| Ascorbic acid (AA) | DLBCL | Lymphoma cell lines LY-1 (DLBCL), Karpas 299 (T-cell NHL), and Jeko (mantle cell NHL) | Enhance TET activity and an increase in the hydroxymethylcytosine fraction; reactivate SMAD1 | The target, route of administration, and dose are unclear | N Shenoy [96] et al. (2017) |
| Ascorbate | AML | SKM-1 cells | Increase TET activity | The target, route of administration, and dose are unclear | Carlos [97] et al. (2021) |
| Ascorbic acid (AA) | Myeloid neoplasia (MN) | TET2−/− mice | facilitate Fe(III)/Fe(II) redox reaction | The target, route of administration, and dose are unclear | Guan [98] et al. (2020) |
| Ascorbic acid (AA) + 5-Azacytidine (5-aza) | Pediatric T-cell acute lymphoblastic leukemia (T-ALL) | TET2-silenced T-ALL cells | Stable re-expression of the TET2 gene; up-regulation of methylated genes and human endogenous retroviruses (HERVs) | The target, route of administration, and dose are unclear | Maike [99] et al. (2021) |
| Vitamin C | Colorectal cancer (CRC) | HCT 116 cells | Increase expression of CDKN1A | The target, route of administration, and dose are unclear | Christian [100] et al. (2018) |
| Four-antibiotic cocktail | CMML | TET2 KO mice | Supress TNF-α signaling | How antibiotics inhibit the pathways associated with TNF is unclear | Zeng [101] et al. (2019) |
| Ginkgo biloba extract (GBE) | Colorectal cancer (CRC) | SW480 cells | Reduce expression of miR-29a | No testing in the clinical trial | Li [102] et al. (2022) |
Abbreviation: DLBCL, Diffuse large B-cell lymphoma; CMML, chronic myelocytic leukemia; MDS, myelodysplastic syndromes; AML, acute myelocytic leukemia; NA, Not available; TET, ten-eleven translocation protein; CDKN1A, tumor suppressor p21; 5hmc, 5-Hydroxymethylcytosine.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Hu, L.; Zhang, X.; Li, H.; Lin, S.; Zang, S. Targeting TET2 as a Therapeutic Approach for Angioimmunoblastic T Cell Lymphoma. Cancers 2022, 14, 5699. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14225699
AMA Style
Hu L, Zhang X, Li H, Lin S, Zang S. Targeting TET2 as a Therapeutic Approach for Angioimmunoblastic T Cell Lymphoma. Cancers. 2022; 14(22):5699. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14225699
Chicago/Turabian StyleHu, Lina, Xuanye Zhang, Huifeng Li, Suxia Lin, and Shengbing Zang. 2022. "Targeting TET2 as a Therapeutic Approach for Angioimmunoblastic T Cell Lymphoma" Cancers 14, no. 22: 5699. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14225699
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.