
Increased Activity of a NK-Specific CAR-NK Framework Targeting CD3 and CD5 for T-Cell Leukemias



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. CAR Construct Design and Characterization
2.2. Mice and Treatment
2.3. Flow Cytometry
2.4. Patient Samples and Cell Lines
2.5. Cytotoxic Assay
2.6. Cytokine Assay
2.7. Statistics
3. Results
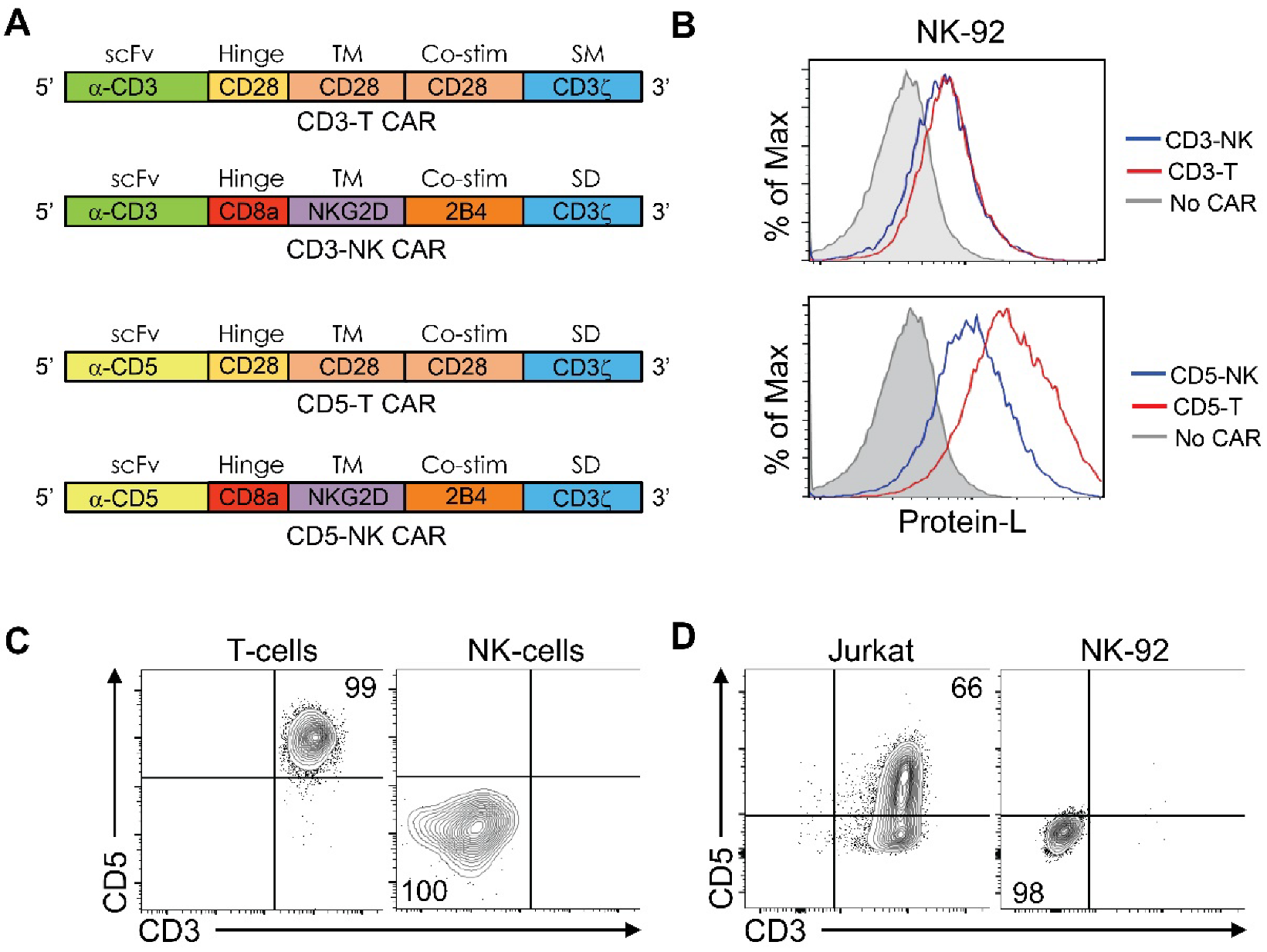
3.1. Design of CAR Constructs
3.2. Activity of the CAR Constructs In Vitro
3.3. Efficacy of CAR Constructs to Eliminate Peripheral T-Cell Lymphoma and Downregulation of the Target Antigen
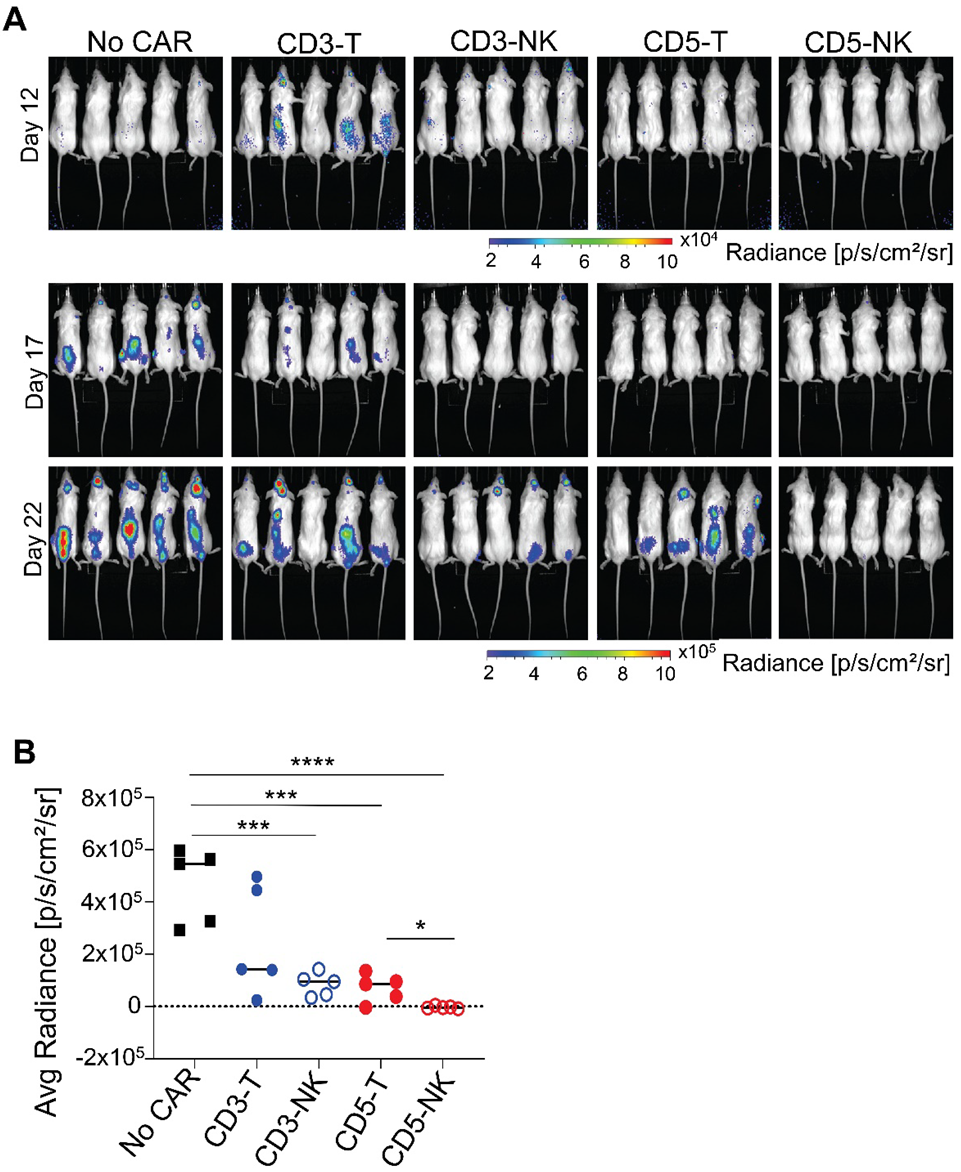
3.4. Activity of the CAR Constructs In Vivo
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Westin, J.R.; Kersten, M.J.; Salles, G.; Abramson, J.S.; Schuster, S.J.; Locke, F.L.; Andreadis, C. Efficacy and safety of CD19-directed CAR-T cell therapies in patients with relapsed/refractory aggressive B-cell lymphomas: Observations from the JULIET, ZUMA-1, and TRANSCEND trials. Am. J. Hematol. 2021, 96, 1295–1312. [Google Scholar] [CrossRef]
- Ma, G.; Shen, J.; Pinz, K.; Wada, M.; Park, J.; Kim, S.; Togano, T.; Tse, W. Targeting T Cell Malignancies Using CD4CAR T-Cells and Implementing a Natural Safety Switch. Stem Cell. Rev. Rep. 2019, 15, 443–447. [Google Scholar] [CrossRef]
- Mamonkin, M.; Rouce, R.H.; Tashiro, H.; Brenner, M.K. A T-cell-directed chimeric antigen receptor for the selective treatment of T-cell malignancies. Blood 2015, 126, 983–992. [Google Scholar] [CrossRef] [Green Version]
- Mamonkin, M.; Mukherjee, M.; Srinivasan, M.; Sharma, S.; Gomes-Silva, D.; Mo, F.; Krenciute, G.; Orange, J.S.; Brenner, M.K. Reversible Transgene Expression Reduces Fratricide and Permits 4-1BB Costimulation of CAR T Cells Directed to T-cell Malignancies. Cancer Immunol. Res. 2018, 6, 47–58. [Google Scholar] [CrossRef] [Green Version]
- Png, Y.T.; Vinanica, N.; Kamiya, T.; Shimasaki, N.; Coustan-Smith, E.; Campana, D. Blockade of CD7 expression in T cells for effective chimeric antigen receptor targeting of T-cell malignancies. Blood Adv. 2017, 1, 2348–2360. [Google Scholar] [CrossRef] [Green Version]
- Rasaiyaah, J.; Georgiadis, C.; Preece, R.; Mock, U.; Qasim, W. TCRalphabeta/CD3 disruption enables CD3-specific antileukemic T cell immunotherapy. JCI Insight 2018, 3, e99442. [Google Scholar] [CrossRef] [Green Version]
- Gomes-Silva, D.; Srinivasan, M.; Sharma, S.; Lee, C.M.; Wagner, D.L.; Davis, T.H.; Rouce, R.H.; Bao, G.; Brenner, M.K.; Mamonkin, M. CD7-edited T cells expressing a CD7-specific CAR for the therapy of T-cell malignancies. Blood 2017, 130, 285–296. [Google Scholar] [CrossRef]
- Arai, S.; Meagher, R.; Swearingen, M.; Myint, H.; Rich, E.; Martinson, J.; Klingemann, H. Infusion of the allogeneic cell line NK-92 in patients with advanced renal cell cancer or melanoma: A phase I trial. Cytotherapy 2008, 10, 625–632. [Google Scholar] [CrossRef]
- Tonn, T.; Schwabe, D.; Klingemann, H.G.; Becker, S.; Esser, R.; Koehl, U.; Suttorp, M.; Seifried, E.; Ottmann, O.G.; Bug, G. Treatment of patients with advanced cancer with the natural killer cell line NK-92. Cytotherapy 2013, 15, 1563–1570. [Google Scholar] [CrossRef]
- Tang, X.; Yang, L.; Li, Z.; Nalin, A.P.; Dai, H.; Xu, T.; Yin, J.; You, F.; Zhu, M.; Shen, W.; et al. First-in-man clinical trial of CAR NK-92 cells: Safety test of CD33-CAR NK-92 cells in patients with relapsed and refractory acute myeloid leukemia. Am. J. Cancer Res. 2018, 8, 1083–1089. [Google Scholar]
- McCauley, S.M.; Kim, K.; Nowosielska, A.; Dauphin, A.; Yurkovetskiy, L.; Diehl, W.E.; Luban, J. Intron-containing RNA from the HIV-1 provirus activates type I interferon and inflammatory cytokines. Nat. Commun. 2018, 9, 5305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermans, I.F.; Silk, J.D.; Yang, J.; Palmowski, M.J.; Gileadi, U.; McCarthy, C.; Salio, M.; Ronchese, F.; Cerundolo, V. The VITAL assay: A versatile fluorometric technique for assessing CTL- and NKT-mediated cytotoxicity against multiple targets in vitro and in vivo. J. Immunol. Methods 2004, 285, 25–40. [Google Scholar] [CrossRef] [PubMed]
- Studnicka, G.M.; Soares, S.; Better, M.; Williams, R.E.; Nadell, R.; Horwitz, A.H. Human-engineered monoclonal antibodies retain full specific binding activity by preserving non-CDR complementarity-modulating residues. Protein Eng. 1994, 7, 805–814. [Google Scholar] [CrossRef]
- Li, Y.; Hermanson, D.L.; Moriarity, B.S.; Kaufman, D.S. Human iPSC-Derived Natural Killer Cells Engineered with Chimeric Antigen Receptors Enhance Anti-tumor Activity. Cell Stem Cell 2018, 23, 181–192. [Google Scholar] [CrossRef] [Green Version]
- Safarzadeh Kozani, P.; Safarzadeh Kozani, P.; Rahbarizadeh, F. CAR-T cell therapy in T-cell malignancies: Is success a low-hanging fruit? Stem Cell Res. Ther. 2021, 12, 527. [Google Scholar] [CrossRef]
- Xie, G.; Dong, H.; Liang, Y.; Ham, J.D.; Rizwan, R.; Chen, J. CAR-NK cells: A promising cellular immunotherapy for cancer. EBioMedicine 2020, 59, 102975. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.H.; Wada, M.; Firor, A.E.; Pinz, K.G.; Jares, A.; Liu, H.; Salman, H.; Golightly, M.; Lan, F.; Jiang, X.; et al. Novel anti-CD3 chimeric antigen receptor targeting of aggressive T cell malignancies. Oncotarget 2016, 7, 56219–56232. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.H.; Wada, M.; Pinz, K.G.; Liu, H.; Lin, K.W.; Jares, A.; Firor, A.E.; Shuai, X.; Salman, H.; Golightly, M.; et al. Preclinical targeting of aggressive T-cell malignancies using anti-CD5 chimeric antigen receptor. Leukemia 2017, 31, 2151–2160. [Google Scholar] [CrossRef] [Green Version]
- McNerney, M.E.; Lee, K.M.; Kumar, V. 2B4 (CD244) is a non-MHC binding receptor with multiple functions on natural killer cells and CD8+ T cells. Mol. Immunol. 2005, 42, 489–494. [Google Scholar] [CrossRef]
- Brown, M.H.; Boles, K.; van der Merwe, P.A.; Kumar, V.; Mathew, P.A.; Barclay, A.N. 2B4, the natural killer and T cell immunoglobulin superfamily surface protein, is a ligand for CD48. J. Exp. Med. 1998, 188, 2083–2090. [Google Scholar] [CrossRef]
- Meinke, S.; Watzl, C. NK cell cytotoxicity mediated by 2B4 and NTB-A is dependent on SAP acting downstream of receptor phosphorylation. Front. Immunol. 2013, 4, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanier, L.L. Up on the tightrope: Natural killer cell activation and inhibition. Nat. Immunol. 2008, 9, 495–502. [Google Scholar] [CrossRef]
- Upshaw, J.L.; Arneson, L.N.; Schoon, R.A.; Dick, C.J.; Billadeau, D.D.; Leibson, P.J. NKG2D-mediated signaling requires a DAP10-bound Grb2-Vav1 intermediate and phosphatidylinositol-3-kinase in human natural killer cells. Nat. Immunol. 2006, 7, 524–532. [Google Scholar] [CrossRef]
- Wu, J.; Song, Y.; Bakker, A.B.; Bauer, S.; Spies, T.; Lanier, L.L.; Phillips, J.H. An activating immunoreceptor complex formed by NKG2D and DAP10. Science 1999, 285, 730–732. [Google Scholar] [CrossRef]
- Xu, Y.; Liu, Q.; Zhong, M.; Wang, Z.; Chen, Z.; Zhang, Y.; Xing, H.; Tian, Z.; Tang, K.; Liao, X.; et al. 2B4 costimulatory domain enhancing cytotoxic ability of anti-CD5 chimeric antigen receptor engineered natural killer cells against T cell malignancies. J. Hematol. Oncol. 2019, 12, 49. [Google Scholar] [CrossRef] [PubMed]
- Majzner, R.G.; Mackall, C.L. Tumor Antigen Escape from CAR T-cell Therapy. Cancer Discov. 2018, 8, 1219–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, J.; Paret, C.; El Malki, K.; Alt, F.; Wingerter, A.; Neu, M.A.; Kron, B.; Russo, A.; Lehmann, N.; Roth, L.; et al. CD19 Isoforms Enabling Resistance to CART-19 Immunotherapy Are Expressed in B-ALL Patients at Initial Diagnosis. J. Immunother. 2017, 40, 187–195. [Google Scholar] [CrossRef] [Green Version]
- Fry, T.J.; Shah, N.N.; Orentas, R.J.; Stetler-Stevenson, M.; Yuan, C.M.; Ramakrishna, S.; Wolters, P.; Martin, S.; Delbrook, C.; Yates, B.; et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat. Med. 2018, 24, 20–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Rhodes, M.; Wiest, D.L.; Vignali, D.A. On the dynamics of TCR:CD3 complex cell surface expression and downmodulation. Immunity 2000, 13, 665–675. [Google Scholar] [CrossRef] [Green Version]
- Sgro, C. Side-effects of a monoclonal antibody, muromonab CD3/orthoclone OKT3: Bibliographic review. Toxicology 1995, 105, 23–29. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Voynova, E.; Hawk, N.; Flomerfelt, F.A.; Telford, W.G.; Gress, R.E.; Kanakry, J.A.; Kovalovsky, D. Increased Activity of a NK-Specific CAR-NK Framework Targeting CD3 and CD5 for T-Cell Leukemias. Cancers 2022, 14, 524. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14030524
Voynova E, Hawk N, Flomerfelt FA, Telford WG, Gress RE, Kanakry JA, Kovalovsky D. Increased Activity of a NK-Specific CAR-NK Framework Targeting CD3 and CD5 for T-Cell Leukemias. Cancers. 2022; 14(3):524. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14030524
Chicago/Turabian StyleVoynova, Elisaveta, Nga Hawk, Francis A. Flomerfelt, William G. Telford, Ronald E. Gress, Jennifer A. Kanakry, and Damian Kovalovsky. 2022. "Increased Activity of a NK-Specific CAR-NK Framework Targeting CD3 and CD5 for T-Cell Leukemias" Cancers 14, no. 3: 524. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14030524