
Specialized Intercellular Communications via Tunnelling Nanotubes in Acute and Chronic Leukemia

 ,
,  , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
1.1. General Considerations on Tunneling Nanotubes
1.2. TNTs and Immune System
2. TNTs and Cancer
3. TNTs and Hematological Malignancies
3.1. TNTs and Acute Lymphoblastic Leukemia (ALL)
3.2. TNTs and T Cell Acute Lymphoblastic Leukaemia (T-ALL)
3.3. TNTs and Chronic Myeloid Leukemia (CML)
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Naus, C.C.; Laird, D.W. Implications and Challenges of Connexin Connections to Cancer. Nat. Rev. Cancer 2010, 10, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Bobrie, A.; Colombo, M.; Raposo, G.; Thery, C. Exosome Secretion: Molecular Mechanisms and Roles in Immune Responses. Traffic 2011, 12, 1659–1668. [Google Scholar] [CrossRef]
- Pap, E.; Pallinger, E.; Falus, A. The Role of Membrane Vesicles in Tumorigenesis. Crit. Rev. Oncol. Hematol. 2011, 79, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Buszczak, M.; Inaba, M.; Yamashita, Y.M. Signaling by cellular protrusion keeping the conversation private. Trends Cell Biol. 2016, 26, 526–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dagar, S.; Pathak, D.; Oza, H.V.; Mylavarapu, S.V.S. Tunneling nanotubes and related structures: Molecular mechanisms of formation and function. Biochem. J. 2021, 22, 3977–3998. [Google Scholar] [CrossRef] [PubMed]
- Cordero Cervantes, D.; Zurzolo, C. Peering into tunneling nanotubes-The path forward. EMBO J. 2021, 40, e105789. [Google Scholar] [CrossRef] [PubMed]
- Zurzolo, C. Tunneling nanotubes: Reshaping connectivity. Curr. Opin. Cell Biol. 2021, 71, 139–147. [Google Scholar] [CrossRef]
- Yamashita, Y.M.; Inaba, M.; Buszczak, M. Specialized Intercellular Communications via Cytonemes and Nanotubes. Annu. Rev. Cell Dev. Biol. 2018, 34, 59–84. [Google Scholar] [CrossRef]
- Ariazi, J.; Benowitz, A.; de Biasi, V.; den Boer, M.L.; Cherqui, S.; Cui, H.; Douillet, N.; Eugenin, E.A.; Favre, D.; Goodman, S.; et al. Tunneling Nanotubes and Gap Junctions-Their Role in Long-Range Intercellular Communication during Development, Health, and Disease Conditions. Front. Mol. Neurosci. 2017, 10, 333. [Google Scholar] [CrossRef]
- Pinto, G.; Brou, C.; Zurzolo, C. Tunneling Nanotubes: The Fuel of Tumor Progression? Trends Cancer 2020, 6, 874–888. [Google Scholar] [CrossRef]
- Dupont, M.; Souriant, S.; Lugo-Villarino, G.; Maridonneau-Parini, I.; Vérollet, C. Tunneling Nanotubes: Intimate Communication between Myeloid Cells. Front. Immunol. 2018, 9, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCoy-Simandle, K.; Hanna, S.J.; Cox, D. Exosomes and Nanotubes: Control of Immune Cell Communication. Int. J. Biochem. Cell Biol. 2016, 71, 44–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, X.; Wang, X. Opportunities and Challenges in Tunneling Nanotubes Research: How Far from Clinical Application? Int. J. Mol. Sci. 2021, 22, 2306. [Google Scholar] [CrossRef] [PubMed]
- Watkins, S.C.; Salter, R.D. Functional connectivity between immune cells mediated by tunneling nanotubules. Immunity 2005, 23, 309–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Gerdes, H.-H. Long-distance electrical coupling via tunneling nanotubes. Biochim. Biophys. Acta. 2012, 1818, 2082–2086. [Google Scholar] [CrossRef] [Green Version]
- Chauveau, A.; Aucher, A.; Eissmann, P.; Vivier, E.; Davis, D.M. Membrane nanotubes facilitate long-distance interactions between natural killer cells and target cells. Proc. Natl. Acad. Sci. USA 2010, 107, 5545–5550. [Google Scholar] [CrossRef] [Green Version]
- Innocenti, M. New insights into the formation and the function of lamellipodia and ruffles in mesenchymal cell migration. Cell Adh. Migr. 2018, 12, 401–416. [Google Scholar] [CrossRef]
- Rustom, A.; Saffrich, R.; Markovic, I.; Walther, P.; Gerdes, H.H. Nanotubular Highways for Intercellular Organelle Transport. Science 2004, 303, 1007–1010. [Google Scholar] [CrossRef] [Green Version]
- Onfelt, B.; Purbhoo, M.A.; Nedvetzki, S.; Sowinski, S.; Davis, D.M. Long-Distance Calls between Cells Connected by Tunneling Nanotubules. Sci. STKE 2005, 2005, 55. [Google Scholar] [CrossRef]
- Veranic, P.; Lokar, M.; Schutz, G.J.; Weghuber, J.; Wieser, S.; Hagerstrand, H.; Kralj-Iglic, V.; Iglic, A. Different Types of Cell-to-Cell Connections Mediated by Nanotubular Structures. Biophys. J. 2008, 95, 4416–4425. [Google Scholar] [CrossRef] [Green Version]
- Gerdes, H.; Rustom, A.; Wang, X. Tunneling Nanotubes, an Emerging Intercellular Communication Route in Development. Mech. Dev. 2013, 130, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Bukoreshtliev, N.V.; Wang, X.; Hodneland, E.; Gurke, S.; Barroso, J.F.; Gerdes, H.H. Selective Block of Tunneling Nanotube (Tnt) Formation Inhibits Intercellular Organelle Transfer between Pc12 Cells. FEBS Lett. 2009, 583, 1481–1488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sowinski, S.; Jolly, C.; Berninghausen, O.; Purbhoo, M.A.; Chauveau, A.; Köhler, K.; Oddos, S.; Eissmann, P.; Brodsky, F.M.; Hopkins, C.; et al. Membrane nanotubes physically connect T cells over long distances presenting a novel route for HIV-1 transmission. Ncb 2008, 10, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Onfelt, B.; Nedvetzki, S.; Benninger, R.K.P.; Purbhoo, M.A.; Sowinski, S.; Hume, A.N.; Seabra, M.C.; Neil, M.A.; French, P.M.; Davis, D.M. Structurally distinct membrane nanotubes between human macrophages support long-distance vesicular traffic or surfing of bacteria. J. Immunol. 2006, 177, 8476–8483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pontes, B.; Viana, N.B.; Campanati, L.; Farina, M.; Neto, V.M.; Nussenzveig, H.M. Structure and elastic properties of tunneling nanotubes. Eur. Biophys. J. Biophys. Lett. 2008, 37, 121–129. [Google Scholar] [CrossRef]
- Abounit, S.; Zurzolo, C. Wiring through tunneling nanotubes--from electrical signals to organelle transfer. J. Cell Sci. 2012, 125 Pt 5, 1089–1098. [Google Scholar] [CrossRef] [Green Version]
- Gousset, K.; Zurzolo, C. Tunnelling nanotubes: A highway for prion spreading? Prion 2009, 3, 94–98. [Google Scholar] [CrossRef] [Green Version]
- Jansens, R.J.J.; Van den Broeck, W.; De Pelsmaeker, S.; Lamote, J.A.S.; VanWaesberghe, C.; Couck, L.; Favoreel, H.W. Pseudorabies Virus US3-Induced Tunneling Nanotubes Contain Stabilized Microtubules, Interact with Neighboring Cells via Cadherins, and Allow Intercellular Molecular Communication. J. Virol. 2017, 91, e00749-17. [Google Scholar] [CrossRef] [Green Version]
- Sisakhtnezhad, S.; Khosravi, L. Emerging physiological and pathological implications of tunneling nanotubes formation between cells. Eur. J. Cell Biol. 2015, 94, 429–443. [Google Scholar] [CrossRef]
- Wang, X.; Veruki, M.L.; Bukoreshtliev, N.V.; Hartveit, E.; Gerdes, H.H. Animal cells connected by nanotubes can be electrically coupled through interposed gap-junction channels. Proc. Natl. Acad. Sci. USA 2010, 107, 17194–17199. [Google Scholar] [CrossRef] [Green Version]
- Smith, I.F.; Shuai, J.; Parker, I. Active generation and propagation of Ca2+ signals within tunneling membrane nanotubes. Biophys. J. 2011, 100, L37–L39. [Google Scholar] [CrossRef] [Green Version]
- He, K.; Luo, W.; Zhang, Y.; Liu, F.; Liu, D.; Xu, L.; Qin, L.; Xiong, C.; Lu, Z.; Fang, X.; et al. Intercellular Transportation of Quantum Dots Mediated by Membrane Nanotubes. ACS Nano 2010, 4, 3015–3022. [Google Scholar] [CrossRef] [PubMed]
- Daniels, D.R. Transport of solid bodies along tubular membrane tethers. PLoS ONE 2019, 14, e0210259. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Bukoreshtliev, N.V.; Gerdes, H.H. Developing neurons form transient nanotubes facilitating electrical coupling and calcium signaling with distant astrocytes. PLoS ONE 2012, 7, e47429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, D.; Tan, K.S.; Zhang, X.; Sun, A.Y.; Sun, G.Y.; Lee, J.C.-M. Hydrogen peroxide alters membrane and cytoskeleton properties and increases intercellular connections in astrocytes. J. Cell Sci. 2005, 118 Pt 16, 3695–3703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desir, S.; Dickson, E.L.; Vogel, R.I.; Thayanithy, V.; Wong, P.; Teoh, D.; Geller, M.A.; Steer, C.J.; Subramanian, S.; Lou, E. Tunneling nanotube formation is stimulated by hypoxia in ovarian cancer cells. Oncotarget 2016, 7, 43150–43161. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Gerdes, H.-H. Transfer of mitochondria via tunneling nanotubes rescues apoptotic PC12 cells. Cell Death Differ. 2015, 22, 1181–1191. [Google Scholar] [CrossRef] [Green Version]
- Osswald, M.; Jung, E.; Sahm, F.; Solecki, G.; Venkataramani, V.; Blaes, J.; Horstmann, H.; Wiestler, B.; Syed, M.; Huang, L.; et al. Brain tumor cells interconnect to a functional and resistant network. Nature 2015, 528, 93–98. [Google Scholar] [CrossRef]
- Reindl, J.; Shevtsov, M.; Dollinger, G.; Stangl, S.; Multhoff, G. Membrane Hsp70-supported cell-to-cell connections via tunneling nanotubes revealed by live cell STED nanoscopy. Cell Stress Chaperones 2019, 24, 213–221. [Google Scholar] [CrossRef] [Green Version]
- Lou, E.; Fujisawa, S.; Morozov, A.; Barlas, A.; Romin, Y.; Dogan, Y.; Gholami, S.; Moreira, A.L.; Manova-Todorova, K.; Moore, M.A. Tunneling nanotubes provide a unique conduit for intercellular transfer of cellular contents in human malignant pleural mesothelioma. PLoS ONE 2012, 7, e33093. [Google Scholar]
- Kabaso, D.; Lokar, M.; Kralj-Iglič, V.; Veranič, P.; Iglič, A. Temperature and cholera toxin B are factors that influence formation of membrane nanotubes in RT4 and T24 urothelial cancer cell lines. Int. J. Nanomed. 2011, 6, 495–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eugenin, E.A.; Gaskill, P.J.; Berman, J.W. Tunneling nanotubes (TNT) are induced by HIV-infection of macrophages: A potential mechanism for intercellular HIV trafficking. Cell Immunol. 2009, 254, 142–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, S.; Hase, K.; Ohno, H. The molecular basis of induction and formation of tunneling nanotubes. Cell Tissue Res. 2013, 352, 67–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matejka, N.; Reindl, J. Perspectives of cellular communication through tunneling nanotubes in cancer cells and the connection to radiation effects. Radiat. Oncol. 2019, 14, 218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Kazanietz, M.G.; Cooke, M. Rho GTPases and the emerging role of tunneling nanotubes in physiology and disease. Am. J. Physiol. Cell Physiol. 2020, 319, C877–C884. [Google Scholar] [CrossRef]
- Andresen, V.; Wang, X.; Ghimire, S.; Omsland, M.; Gjertsen, B.T.; Gerdes, H.H. Tunneling nanotube (TNT) formation is independent of p53 expression. Cell Death Differ. 2013, 20, 1124. [Google Scholar] [CrossRef] [Green Version]
- Hase, K.; Kimura, S.; Takatsu, H.; Ohmae, M.; Kawano, S.; Kitamura, H.; Ito, M.; Watarai, H.; Hazelett, C.C.; Yeaman, C.; et al. Msec Promotes Membrane Nanotube Formation by Interacting with Ral and the Exocyst Complex. Nat. Cell Biol. 2009, 11, 1427–1432. [Google Scholar] [CrossRef]
- Wang, Y.; Cui, J.; Sun, X.; Zhang, Y. Tunneling-Nanotube Development in Astrocytes Depends on p53 Activation. Cell Death Differ. 2011, 18, 732–742. [Google Scholar] [CrossRef] [Green Version]
- Arkwright, P.D.; Luchetti, F.; Tour, J.; Roberts, C.; Ayub, R.; Morales, A.P.; Rodríguez, J.J.; Gilmore, A.; Canonico, B.; Papa, S.; et al. Fas stimulation of T lymphocytes promotes rapid intercellular exchange of death signals via membrane nanotubes. Cell Res. 2010, 20, 72–88. [Google Scholar] [CrossRef] [Green Version]
- Maus, M.; Medgyesi, D.; Kiss, E.; Schneider, A.E.; Enyedi, A.; Szilagyi, N.; Matko, J.; Sarmay, G. B cell receptor-induced Ca2+ mobilization mediates F-actin rearrangements and is indispensable for adhesion and spreading of B lymphocytes. J. Leukoc. Biol. 2013, 93, 537–547. [Google Scholar] [CrossRef] [Green Version]
- Matula, Z.; Nemeth, A.; Lorincz, P.; Szepesi, A.; Brozik, A.; Buzas, E.I.; Low, P.; Nemet, K.; Uher, F.; Urban, V.S. The Role of Extracellular Vesicle and Tunneling Nanotube-Mediated Intercellular Cross-Talk between Mesenchymal Stem Cells and Human Peripheral T Cells. Stem Cells Dev. 2016, 25, 1818–1832. [Google Scholar] [CrossRef] [PubMed]
- Dhainaut, M.; Moser, M. Regulation of immune reactivity by intercellular transfer. Front. Immunol. 2014, 5, 112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaccard, C.R.; Rinaldo, C.R.; Mailliard, R.B. Linked in: Immunologic membrane nanotube networks. J. Leukoc. Biol. 2016, 100, 81–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakim, L.M.; Bevan, M.J. Cross-dressed dendritic cells drive memory CD8+ T-cell activation after viral infection. Nature 2011, 471, 629–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasquier, J.; Galas, L.; Boulangé-Lecomte, C.; Rioult, D.; Bultelle, F.; Magal, P.; Webb, G.; Le Foll, F. Different modalities of intercellular membrane exchanges mediate cell-to-cell p-glycoprotein transfers in MCF-7 breast cancer cells. J. Biol. Chem. 2012, 287, 7374–7387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiller, C.; Huber, J.E.; Diakopoulos, K.N.; Weiss, E.H. Tunneling nanotubes enable intercellular transfer of MHC class I molecules. Hum. Immunol. 2013, 74, 412–416. [Google Scholar] [CrossRef]
- Bittins, M.; Wang, X. TNT-Induced Phagocytosis: Tunneling Nanotubes Mediate the Transfer of Pro-Phagocytic Signals from Apoptotic to Viable Cells. J. Cell. Physiol. 2017, 232, 2271–2279. [Google Scholar] [CrossRef] [Green Version]
- Rainy, N.; Chetrit, D.; Rouger, V.; Vernitsky, H.; Rechavi, O.; Marguet, D.; Goldstein, I.; Ehrlich, M.; Kloog, Y. H-Ras transfers from B to T cells via tunneling nanotubes. Cell Death Dis. 2013, 4, e726. [Google Scholar] [CrossRef]
- Polak, R.; de Rooij, B.; Pieters, R.; den Boer, M.L. B-Cell Precursor Acute Lymphoblastic Leukemia Cells Use Tunneling Nanotubes to Orchestrate Their Microenvironment. Blood 2015, 126, 2404–2414. [Google Scholar] [CrossRef] [Green Version]
- Venkatesh, V.S.; Lou, E. Tunnelling nanotubes: A bridge for heterogeneity in glioblastoma and a new therapeutic target? Cancer Rep. 2019, 74, e1185. [Google Scholar]
- Osswald, M.; Jung, E.; Wick, W.; Winkler, F. Tunneling nanotube-like structures in brain tumors. Cancer Rep. 2019, 6, 1124. [Google Scholar] [CrossRef] [Green Version]
- Desir, S.; O’Hare, P.; Vogel, R.I.; Sperduto, W.; Sarkari, A.; Dickson, E.L.; Wong, P.; Nelson, A.C.; Fong, Y.; Steer, C.J.; et al. Chemotherapy-induced tunneling nanotubes mediate intercellular drug efflux in pancreatic Cancer. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Thayanithy, V.; Dickson, E.L.; Steer, C.; Subramanian, S.; Lou, E. Tumor-stromal cross talk: Direct cell-to-cell transfer of oncogenic microRNAs via tunneling nanotubes. Transl. Res. 2014, 164, 359–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ady, J.; Thayanithy, V.; Mojica, K.; Wong, P.; Carson, J.; Rao, P.; Fong, Y.; Lou, E. Tunneling nanotubes: An alternate route for propagation of the bystander effect following oncolytic viral infection. Mol. Ther. Oncolytics 2016, 3, 16029. [Google Scholar] [CrossRef] [Green Version]
- Thayanithy, V.; Babatunde, V.; Dickson, E.L.; Wong, P.; Oh, S.; Ke, X.; Barlas, A.; Fujisawa, S.; Romin, Y.; Moreira, A.L.; et al. Tumor exosomes induce tunneling nanotubes in lipid raft-enriched regions of human mesothelioma cells. Exp. Cell Res. 2014, 323, 178–188. [Google Scholar] [CrossRef] [Green Version]
- Ady, J.W.; Desir, S.; Thayanithy, V.; Vogel, R.I.; Moreira, A.L.; Downey, R.J.; Fong, Y.; Manova-Todorova, K.; Moore, M.A.; Lou, E. Intercellular communication in malignant pleural mesothelioma: Properties of tunneling nanotubes. Front. Physiol. 2014, 5, 400. [Google Scholar] [CrossRef]
- Lou, E.; Fujisawa, S.; Barlas, A.; Romin, Y.; Manova-Todorova, K.; Moore, M.A.S.; Subramanian, S. Tunneling Nanotubes: A new paradigm for studying intercellular communication and therapeutics in cancer. Commun. Integr. Biol. 2012, 5, 399–403. [Google Scholar] [CrossRef] [Green Version]
- Desir, S.; Wong, P.; Turbyville, T.; Chen, D.; Shetty, M.; Clark, C.; Zhai, E.; Romin, Y.; Manova-Todorova, K.; Starr, T.K.; et al. Intercellular Transfer of Oncogenic KRAS via Tunneling Nanotubes Introduces Intracellular Mutational Heterogeneity in Colon Cancer Cells. Cancers 2019, 11, 892. [Google Scholar] [CrossRef] [Green Version]
- Allegra, A.; Alonci, A.; Campo, S.; Penna, G.; Petrungaro, A.; Gerace, D.; Musolino, C. Circulating microRNAs: New biomarkers in diagnosis, prognosis and treatment of cancer (review). Int. J. Oncol. 2012, 41, 1897–1912. [Google Scholar] [CrossRef] [Green Version]
- Alonci, A.; Allegra, A.; Bellomo, G.; Penna, G.; D’Angelo, A.; Quartarone, E.; Musolino, C. Evaluation of circulating endothelial cells, VEGF and VEGFR2 serum levels in patients with chronic myeloproliferative diseases. Hematol. Oncol. 2008, 26, 235–239. [Google Scholar] [CrossRef]
- Quartarone, E.; Alonci, A.; Allegra, A.; Bellomo, G.; Calabrò, L.; D’Angelo, A.; Del Fabro, V.; Grasso, A.; Cincotta, M.; Muso-lino, C. Differential levels of soluble angiopoietin-2 and Tie-2 in patients with haematological malignancies. Eur. J. Haematol. 2006, 77, 480–485. [Google Scholar] [CrossRef] [PubMed]
- Errede, M.; Mangieri, D.; Longo, G.; Girolamo, F.; de Trizio, I.; Vimercati, A.; Serio, G.; Frei, K.; Perris, R.; Virgintino, D. Tunneling Nanotubes Evoke Pericyte/Endothelial Communication During Normal and Tumoral Angiogenesis. Fluids Barriers CNS 2018, 15, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Astanina, K.; Koch, M.; Jungst, C.; Zumbusch, A.; Kiemer, A.K. Lipid Droplets as a Novel Cargo of Tunnelling Nanotubes in Endothelial Cells. Sci. Rep. 2015, 5, 11453. [Google Scholar] [CrossRef] [Green Version]
- Connor, Y.; Tekleab, S.; Nandakumar, S.; Walls, C.; Tekleab, Y.; Husain, A.; Gadish, O.; Sabbisetti, V.; Kaushik, S.; Sehrawat, S.; et al. Physical Nanoscale Conduit-Mediated Communication between Tumor Cells and the Endothelium Modulates Endothelial Phenotype. Nat. Commun. 2015, 6, 8671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno-Sanchez, R.; Rodriguez-Enriquez, S.; Marin-Hernandez, A.; Saavedra, E. Energy metabolism in tumor cells. FEBS J. 2007, 274, 1393–1418. [Google Scholar] [CrossRef] [PubMed]
- Ashton, T.M.; McKenna, W.G.; Kunz-Schughart, L.A.; Higgins, G.S. Oxidative Phosphorylation as an Emerging Target in Cancer Therapy. Clin. Cancer Res. 2018, 24, 2482–2491. [Google Scholar] [CrossRef] [Green Version]
- Roh, J.S.; Sohn, D.H. Damage-associated molecular patterns in inflammatory diseases. Immune Netw. 2018, 18, 1–14. [Google Scholar] [CrossRef]
- Rubartelli, A.; Lotze, M.T. Inside, outside, upside down: Damage-associated molecular-pattern molecules (DAMPs) and redox. Trends Immunol. 2007, 28, 429–436. [Google Scholar] [CrossRef]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef] [Green Version]
- Hernandez, C.; Huebener, P.; Schwabe, R. Damage-associated molecular patterns in cancer: A double edged sword. Oncogene 2016, 35, 5931–5941. [Google Scholar] [CrossRef]
- Krysko, O.; Aaes, T.L.; Bachert, C.; Vandenabeele, P.; Krysko, D.V. Many faces of DAMPs in cancer therapy. Cell Death Dis. 2013, 4, e631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsvetkov, P.; Detappe, A.; Cai, K.; Keys, H.R.; Brune, Z.; Ying, W.; Thiru, P.; Reidy, M.; Kugener, G.; Rossen, J.; et al. Mitochondrial metabolism promotes adaptation to proteotoxic stress. Nat. Chem. Biol. 2019, 15, 681–689. [Google Scholar] [CrossRef] [PubMed]
- Song, I.S.; Kim, H.K.; Lee, S.R.; Jeong, S.H.; Kim, N.; Ko, K.S.; Rhee, B.D.; Han, J. Mitochondrial modulation decreases the bortezomib-resistance in multiple myeloma cells. Int. J. Cancer 2013, 133, 1357–1367. [Google Scholar] [CrossRef] [PubMed]
- Marlein, C.R.; Zaitseva, L.; Piddock, R.E.; Robinson, S.D.; Edwards, D.R.; Shafat, M.S.; Zhou, Z.; Lawes, M.; Bowles, K.M.; Rushworth, S.A. NADPH oxidase-2 derived superoxide drives mitochondrial transfer from bone marrow stromal cells to leukemic blasts. Blood 2017, 130, 1649–1660. [Google Scholar] [CrossRef]
- Gousset, K.; Marzo, L.; Commere, P.; Zurzolo, C. Myo10 is a key regulator of TNT formation in neuronal cells. J. Cell Sci. 2013, 126, 4424–4435. [Google Scholar] [CrossRef] [Green Version]
- Tardivel, M.; Bégard, S.; Bousset, L.; Dujardin, S.; Coens, A.; Melki, R. Tunneling nanotube (TNT)-mediated neuron-to neuron transfer of pathological Tau protein assemblies. Acta Neuropathol. Commun. 2016, 4, 117. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Yu, Z.; Jiang, D.; Liang, X.; Liao, S.; Zhang, Z.; Yue, W.; Li, X.; Chiu, S.M.; Chai, Y.H.; et al. iPSC-MSCs with High Intrinsic MIRO1 and Sensitivity to TNF-_ Yield Efficacious Mitochondrial Transfer to Rescue Anthracycline-Induced Cardiomyopathy. Stem Cell Rep. 2016, 7, 749–763. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, T.; Mukherjee, S.; Pattnaik, B.; Kumar, M.; Singh, S.; Rehman, R.; Tiwari, B.K.; Jha, K.A.; Barhanpurkar, A.P.; Wani, M.R.; et al. Miro1 regulates intercellular mitochondrial transport & enhances mesenchymal stem cell rescue efficacy. EMBO J. 2014, 33, 994–1010. [Google Scholar]
- López-Doménech, G.; Covill-Cooke, C.; Ivankovic, D.; Hal, E.F.; Sheehan, D.F.; Norkett, R.; Birsa, N.; Kittler, J.T. Miro proteins coordinate microtubule- and actin-dependent mitochondrial transport and distribution. EMBO J. 2018, 37, 321–336. [Google Scholar] [CrossRef]
- Tan, A.S.; Baty, J.W.; Dong, L.F.; Bezawork-Geleta, A.; Endaya, B.; Goodwin, J.; Bajzikova, M.; Kovarova, J.; Peterka, M.; Yan, B.; et al. Mitochondrial Genome Acquisition Restores Respiratory Function and Tumorigenic Potential of Cancer Cells without Mitochondrial DNA. Cell Metab. 2015, 21, 81–94. [Google Scholar] [CrossRef] [Green Version]
- Dong, L.F.; Kovarova, J.; Bajzikova, M.; Bezawork-Geleta, A.; Svec, D.; Endaya, B.; Sachaphibulkij, K.; Coelho, A.R.; Sebkova, N.; Ruzickova, A.; et al. Horizontal Transfer of Whole Mitochondria Restores Tumorigenic Potential in Mitochondrial DNA-Deficient Cancer Cells. Elife 2017, 6, e22187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allegra, A.; Ettari, R.; Innao, V.; Bitto, A. Potential Role of microRNAs in inducing Drug Resistance in Patients with Multiple Myeloma. Cells 2021, 10, 448. [Google Scholar] [CrossRef] [PubMed]
- Innao, V.; Rizzo, V.; Allegra, A.G.; Musolino, C.; Allegra, A. Promising Anti-Mitochondrial Agents for Overcoming Acquired Drug Resistance in Multiple Myeloma. Cells 2021, 10, 439. [Google Scholar] [CrossRef] [PubMed]
- Pasquier, J.; Guerrouahen, B.S.; Al Thawadi, H.; Ghiabi, P.; Maleki, M.; Abu-Kaoud, N.; Jacob, A.; Mirshahi, M.; Galas, L.; Rafii, S.; et al. Preferential Transfer of Mitochondria from Endothelial to Cancer Cells Through Tunneling Nanotubes Modulates Chemoresistance. J. Transl. Med. 2013, 11, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.; Zheng, X.; Li, F.; Yu, Y.; Chen, Z.; Liu, Z.; Wang, Z.; Xu, H.; Yang, W. Tunneling Nanotubes Promote Intercellular Mitochondria Transfer Followed by Increased Invasiveness in Bladder Cancer Cells. Oncotarget 2017, 8, 15539–15552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desouky, O.; Ding, N.; Zhou, G. Targeted and non-targeted effects of ionizing radiation. J. Radiat. Res. Appl. Sci. 2015, 8, 247–254. [Google Scholar] [CrossRef] [Green Version]
- Prise, K.M.; O’Sullivan, J.M. Radiation-induced bystander signalling in cancer therapy. Nat. Rev. Cancer 2009, 9, 351–360. [Google Scholar] [CrossRef]
- Yahyapour, R.; Motevaseli, E.; Rezaeyan, A.; Abdollahi, H.; Farhood, B.; Cheki, M.; Najafi, M.; Villa, V. Mechanisms of radiation bystander and non-targeted effects: Implications to radiation carcinogenesis and radiotherapy. Curr. Radiopharm. 2018, 11, 34–45. [Google Scholar] [CrossRef]
- Marín, A.; Martín, M.; Liñán, O.; Alvarenga, F.; López, M.; Fernández, L.; Büchser, D.; Cerezo, L. Bystander effects and radiotherapy. Rep. Pract. Oncol. Radiother. 2015, 20, 12–21. [Google Scholar] [CrossRef] [Green Version]
- Ariyoshi, K.; Miura, T.; Kasai, K.; Fujishima, Y.; Nakata, A.; Yoshida, M. Radiation-induced bystander effect is mediated by mitochondrial DNA in exosome-like vesicles. Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Gerdes, H.H.; Carvalho, R.N. Intercellular transfer mediated by tunneling nanotubes. Curr. Opin. Cell Biol. 2008, 20, 470–475. [Google Scholar] [CrossRef] [PubMed]
- Allegra, A.; Speciale, A.; Molonia, M.S.; Guglielmo, L.; Musolino, C.; Ferlazzo, G.; Costa, G.; Saija, A.; Cimino, F. Curcumin ameliorates the in vitro efficacy of carfilzomib in human multiple myeloma U266 cells targeting p53 and NF-κB pathways. Toxicol In Vitro 2018, 47, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Ettari, R.; Zappalà, M.; Grasso, S.; Musolino, C.; Innao, V.; Allegra, A. Immunoproteasome-selective and non-selective inhibitors: A promising approach for the treatment of multiple myeloma. Pharmacol. Ther. 2018, 182, 176–192. [Google Scholar] [CrossRef] [PubMed]
- Allegra, A.; Innao, V.; Gerace, D.; Vaddinelli, D.; Musolino, C. Adoptive immunotherapy for hematological malignancies: Current status and new insights in chimeric antigen receptor T cells. Blood Cells Mol. Dis. 2016, 62, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Allegra, A.; Penna, G.; Alonci, A.; Russo, S.; Greve, B.; Innao, V.; Minardi, V.; Musolino, C. Monoclonal antibodies: Potential new therapeutic treatment against multiple myeloma. Eur. J. Haematol. 2013, 90, 441–468. [Google Scholar] [CrossRef] [Green Version]
- Allegra, A.; Sant’Antonio, E.; Penna, G.; Alonci, A.; D’Angelo, A.; Russo, S.; Cannavò, A.; Gerace, D.; Musolino, C. Novel therapeutic strategies in multiple myeloma: Role of the heat shock protein inhibitors. Eur. J. Haematol. 2011, 86, 93–110. [Google Scholar] [CrossRef]
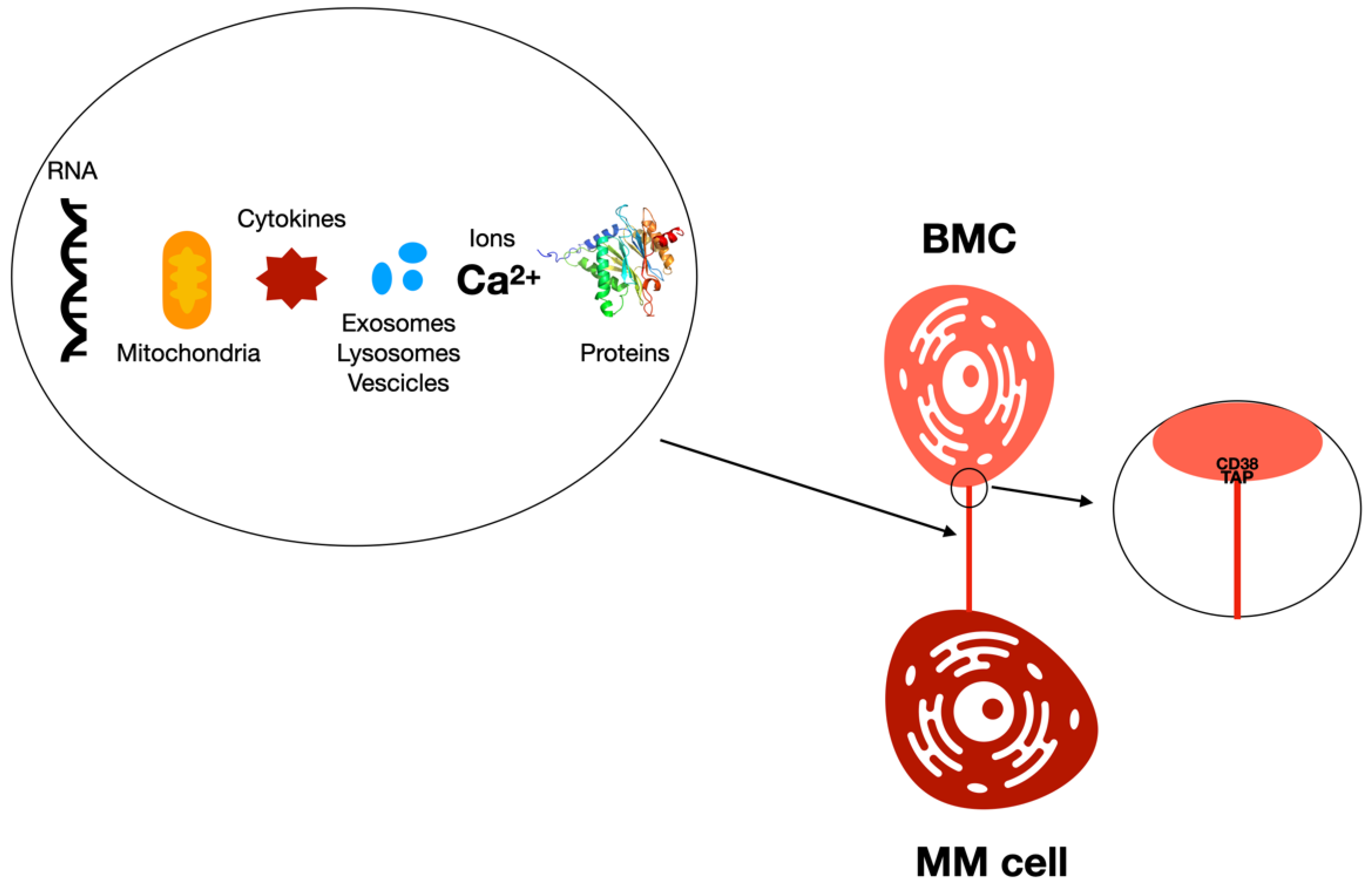
- Marlein, C.R.; Piddock, R.E.; Mistry, J.J.; Zaitseva, L.; Hellmich, C.; Horton, R.H.; Zhou, Z.; Auger, M.J.; Bowles, K.M.; Rushworth, S.A. CD38-driven mitochondrial trafficking promotes bioenergetic plasticity in multiple myeloma. Cancer Res. 2019, 79, 2285–2297. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, A.; Kukita, A.; Li, Y.J.; Zhang, J.Q.; Nomiyama, H.; Yamaza, T.; Ayukawa, Y.; Koyano, K.; Kukita, T. Tunneling nanotube formation is essential for the regulation of osteoclastogenesis. J Cell Biochem. 2013, 114, 1238–1247. [Google Scholar] [CrossRef]
- Dohner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute myeloid leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef] [Green Version]
- Mitroulis, I.; Kalafati, L.; Hajishengallis, G.; Chavakis, T. Myelopoiesis in the Context of Innate Immunity. J. Innate. Immun. 2018, 10, 365–372. [Google Scholar] [CrossRef]
- Schajnovitz, A.; Itkin, T.; D’Uva, G.; Kalinkovich, A.; Golan, K.; Ludin, A.; Cohen, D.; Shulman, Z.; Avigdor, A.; Nagler, A.; et al. CXCL12 secretion by bone marrow stromal cells is dependent on cell contact and mediated by connexin-43 and connexin-45 gap junctions. Nat. Immunol. 2011, 12, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.H.; Nayak, R.C.; Roy, S.; Perumbeti, A.; Wellendorf, A.M.; Bezold, K.Y.; Pirman, M.; Hill, S.E.; Starnes, J.; Loberg, A.; et al. Vasculopathy-associated hyperangiotensinemia mobilizes haematopoietic stem cells/progenitors through endothelial AT(2)R and cytoskeletal dysregulation. Nat. Commun. 2015, 6, 5914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Omsland, M.; Bruserud, O.; Gjertsen, B.T.; Andresen, V. Tunneling nanotube (TNT) formation is downregulated by cytarabine and NF- κ B inhibition in acute myeloid leukemia (AML). Oncotarget 2017, 8, 7946–7963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moschoi, R.; Imbert, V.; Nebout, M.; Chiche, J.; Mary, D.; Prebet, T.; Saland, E.; Castellano, R.; Pouyet, L.; Collette, Y.; et al. Protective mitochondrial transfer from bone marrow stromal cells to acute myeloid leukemic cells during chemotherapy. Blood 2016, 128, 253–264. [Google Scholar] [CrossRef] [Green Version]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Allegra, A.; Innao, V.; Gerace, D.; Bianco, O.; Musolino, C. The metabolomic signature of hematologic malignancies. Leuk. Res. 2016, 49, 22–35. [Google Scholar] [CrossRef]
- Duvel, K.; Yecies, J.L.; Menon, S.; Raman, P.; Lipovsky, A.I.; Souza, A.L.; Triantafellow, E.; Ma, Q.; Gorski, R.; Cleaver, S.; et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell. 2010, 39, 171–183. [Google Scholar] [CrossRef] [Green Version]
- Suganuma, K.; Miwa, H.; Imai, N.; Shikami, M.; Gotou, M.; Goto, M.; Mizuno, S.; Takahashi, M.; Yamamoto, H.; Hiramatsu, A.; et al. Energy metabolism of leukemia cells: Glycolysis versus oxidative phosphorylation. Leuk. Lymphoma 2010, 51, 2112–2119. [Google Scholar] [CrossRef]
- Boultwood, J.; Fidler, C.; Mills, K.I.; Frodsham, P.M.; Kusec, R.; Gaiger, A.; Gale, R.E.; Linch, D.C.; Littlewood, T.J.; Moss, P.A.; et al. Amplification of mitochondrial DNA in acute myeloid leukaemia. Br. J. Haematol. 1996, 95, 426–431. [Google Scholar] [CrossRef]
- Skrtic, M.; Sriskanthadevan, S.; Jhas, B.; Gebbia, M.; Wang, X.; Wang, Z.; Hurren, R.; Jitkova, Y.; Gronda, M.; Maclean, N.; et al. Inhibition of mitochondrial translation as a therapeutic strategy for human acute myeloid leukemia. Cancer Cell. 2011, 20, 674–688. [Google Scholar] [CrossRef] [Green Version]
- Whiteside, T. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008, 27, 5904–5912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griessinger, E.; Moschoi, R.; Biondani, G.; Peyron, J.F. Mitochondrial transfer in the leukemia microenvironment. Trends Cancer 2017, 3, 828–839. [Google Scholar] [CrossRef] [PubMed]
- Musolino, C.; Allegra, A.; Saija, A.; Alonci, A.; Russo, S.; Spatari, G.; Penna, G.; Gerace, D.; Cristani, M.; David, A.; et al. Changes in advanced oxidation protein products, advanced glycation end products, and s-nitrosylated proteins, in patients affected by polycythemia vera and essential thrombocythemia. Clin. Biochem. 2012, 45, 1439–1443. [Google Scholar] [CrossRef] [PubMed]
- Imbesi, S.; Musolino, C.; Allegra, A.; Saija, A.; Morabito, F.; Calapai, G.; Gangemi, S. Oxidative stress in oncohematologic diseases: An update. Expert Rev. Hematol. 2013, 6, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Gangemi, S.; Allegra, A.; Alonci, A.; Cristani, M.; Russo, S.; Speciale, A.; Penna, G.; Spatari, G.; Cannavò, A.; Bellomo, G.; et al. Increase of novel biomarkers for oxidative stress in patients with plasma cell disorders and in multiple myeloma patients with bone lesions. Inflamm. Res. 2012, 61, 1063–1067. [Google Scholar] [CrossRef] [PubMed]
- Saito, K.; Zhang, Q.; Yang, H.; Yamatani, K.; Ai, T.; Ruvolo, V.; Baran, N.; Cai, T.; Ma, H.; Jacamo, R.; et al. Exogenous mitochondrial transfer and endogenous mitochondrial fission facilitate AML resistance to OxPhos inhibition. Blood Adv. 2021, 5, 4233–4255. [Google Scholar] [CrossRef]
- Allegra, A.; Innao, V.; Allegra, A.G.; Musolino, C. Relationship between mitofusin 2 and cancer. Adv. Protein Chem. Struct. Biol. 2019, 116, 209–236. [Google Scholar] [CrossRef]
- Rosilio, C.; Lounnas, N.; Nebout, M.; Imbert, V.; Hagenbeek, T.; Spits, H.; Asnafi, V.; Pontier-Bres, R.; Reverso, J.; Michiels, J.F.; et al. The metabolic perturbators metformin, phenformin and AICAR interfere with the growth and survival of murine PTEN-deficient T cell lymphomas and human T-ALL/T-LL cancer cells. Cancer Lett. 2013, 336, 114–126. [Google Scholar] [CrossRef]
- Caicedo, A.; Fritz, V.; Brondello, J.M.; Ayala, M.; Dennemont, I.; Abdellaoui, N.; de Fraipont, F.; Moisan, A.; Prouteau, C.A.; Boukhaddaoui, H.; et al. MitoCeption as a new tool to assess the effects of mesenchymal stem/stromal cell mitochondria on cancer cell metabolism and function. Sci. Rep. 2015, 5, 9073. [Google Scholar] [CrossRef] [Green Version]
- Colmone, A.; Amorim, M.; Pontier, A.L.; Wang, S.; Jablonski, E.; Sipkins, D.A. Leukemic cells create bone marrow niches that disrupt the behavior of normal hematopoietic progenitor cells. Science 2008, 322, 1861–1865. [Google Scholar] [CrossRef] [Green Version]
- Burt, R.; Dey, A.; Aref, S.; Aguiar, M.; Akarca, A.; Bailey, K.; Day, W.; Hooper, S.; Kirkwood, A.; Kirschner, K.; et al. Activated stromal cells transfer mitochondria to rescue acute lymphoblastic leukemia cells from oxidative stress. Blood 2019, 134, 1415–1429. [Google Scholar] [CrossRef] [PubMed]
- Manshouri, T.; Estrov, Z.; Quint’as-Cardama, A.; Burger, J.; Zhang, Y.; Livun, A.; Knez, L.; Harris, D.; Creighton, C.J.; Kantarjian, H.M.; et al. Bone marrow stroma-secreted cytokines protect JAK2(V617F)-mutated cells from the effects of a JAK2 inhibitor. Cancer Res. 2011, 71, 3831–3840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernardo, M.E.; Fibbe, W.E. Mesenchymal stromal cells: Sensors and switchers of inflammation. Cell Stem Cell. 2013, 13, 392–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgess, M.; Cheung, C.; Chambers, L.; Ravindranath, K.; Minhas, G.; Knop, L.; Mollee, P.; McMillan, N.A.; Gill, D. CCL2 and CXCL2 enhance survival of primary chronic lymphocytic leukemia cells in vitro. Leuk. Lymphoma 2012, 53, 1988–1998. [Google Scholar] [CrossRef] [Green Version]
- Francia di Celle, P.; Mariani, S.; Riera, L.; Stacchini, A.; Reato, G.; Foa, R. Interleukin-8 induces the accumulation of B-cell chronic lymphocytic leukemia cells by prolonging survival in an autocrine fashion. Blood 1996, 87, 4382–4389. [Google Scholar] [CrossRef] [Green Version]
- Harris, J. Autophagy and cytokines. Cytokine 2011, 56, 140–144. [Google Scholar] [CrossRef]
- Mathew, R.; Karantza--Wadsworth, V.; White, E. Role of autophagy in cancer. Nat. Rev. Cancer 2007, 7, 961–967. [Google Scholar] [CrossRef]
- Mi, N.; Chen, Y.; Wang, S.; Chen, M.; Zhao, M.; Yang, G.; Ma, M.; Su, Q.; Luo, S.; Shi, J.; et al. CapZ regulates autophagosomal membrane shaping by promoting actin assembly inside the isolation membrane. Nat. Cell Biol. 2015, 17, 1112–1123. [Google Scholar] [CrossRef]
- Hubbard, A.K.; Rothlein, R. Intercellular adhesion molecule--1 (ICAM--1) expression and cell signaling cascades. Free Radic. Biol. Med. 2000, 128, 1379-–1386. [Google Scholar] [CrossRef]
- De Rooij, B.; Polak, R.; Stalpers, F.; Pieters, R.; den Boer, M.L. Tunneling nanotubes facilitate autophagosome transfer in the leukemic niche. Leukemia 2017, 31, 1651–1654. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Liu, X.; Qiu, Y.; Shi, Y.; Cai, J.; Wang, B.; Wei, X.; Ke, Q.; Sui, X.; Wang, Y.; et al. Cell adhesion-mediated mitochondria transfer contributes to mesenchymal stem cell-induced chemoresistance on T cell acute lymphoblastic leukemia cells. J. Hematol. Oncol. 2018, 11, 1. [Google Scholar] [CrossRef] [PubMed]
- Kogure, Y.; Kameda, T.; Koya, J.; Yoshimitsu, M.; Nosaka, K.; Yasunaga, J.I.; Imaizumi, Y.; Watanabe, M.; Saito, Y.; Ito, Y.; et al. Whole-genome landscape of adult T-cell leukemia/lymphoma. Blood 2021. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.S.; Petrow-Sadowski, C.; Huang, Y.K.; Bertolette, D.C.; Ruscetti, F.W. Cell-free HTLV-1 infects dendritic cells leading to transmission and transformation of CD4(+) T cells. Nat. Med. 2008, 14, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Pique, C.; Jones, K.S. Pathways of cell-cell transmission of HTLV-1. Front. Microbiol. 2012, 3, 378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizkallah, G.; Alais, S.; Futsch, N.; Tanaka, Y.; Journo, C.; Mahieux, R.; Dutartre, H. Dendritic cell maturation, but not type I interferon exposure, restricts infection by HTLV-1, and viral transmission to T-cells. PLoS Pathog. 2017, 13, e1006353. [Google Scholar] [CrossRef]
- Igakura, T.; Stinchcombe, J.C.; Goon, P.K.; Taylor, G.P.; Weber, J.N.; Griffiths, G.M.; Tanaka, Y.; Osame, M.; Bangham, C.R. Spread of HTLV-I between lymphocytes by virus-induced polarization of the cytoskeleton. Science 2003, 299, 1713–1716. [Google Scholar] [CrossRef] [Green Version]
- Van Prooyen, N.; Gold, H.; Andresen, V.; Schwartz, O.; Jones, K.; Ruscetti, F.; Lockett, S.; Gudla, P.; Venzon, D.; Franchini, G. Human T-cell leukemia virus type 1p8 protein increases cellular conduits and virus transmission. Proc. Natl. Acad. Sci. USA 2010, 107, 20738–20743. [Google Scholar] [CrossRef] [Green Version]
- Omsland, M.; Pise-Masison, C.; Fujikawa, D.; Galli, V.; Fenizia, C.; Parks, R.W.; Gjertsen, B.T.; Franchini, G.; Andresen, V. Inhibition of Tunneling Nanotube (TNT) Formation and Human T-cell Leukemia Virus Type 1 (HTLV-1) Transmission by Cytarabine. Sci. Rep. 2018, 8, 11118. [Google Scholar] [CrossRef]
- Sant’Antonio, E.; Camerini, C.; Rizzo, V.; Musolino, C.; Allegra, A. Genetic Heterogeneity in Chronic Myeloid Leukemia: How Clonal Hematopoiesis and Clonal Evolution May Influence Prognosis, Treatment Outcome, and Risk of Cardiovascular Events. Clin. Lymphoma Myeloma Leuk. 2021, 21, 573–579. [Google Scholar] [CrossRef]
- Krause, D.S.; Scadden, D.T. A hostel for the hostile: The bone marrow niche in hematologic neoplasms. Haematologica 2015, 100, 1376–1387. [Google Scholar] [CrossRef]
- Duarte, D.; Hawkins, E.D.; Lo Celso, C. The interplay of leukemia cells and the bone marrow microenvironment. Blood 2018, 131, 1507–1511. [Google Scholar] [CrossRef] [PubMed]
- Podszywalow-Bartnicka, P.; Cmoch, A.; Wolczyk, M.; Bugajski, L.; Tkaczyk, M.; Dadlez, M.; Nieborowska-Skorska, M.; Koromilas, A.E.; Skorski, T.; Piwocka, K. Increased phosphorylation of eIF2α in chronic myeloid leukemia cells stimulates secretion of matrix modifying enzymes. Oncotarget 2016, 7, 79706–79721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittal, R.; Karhu, E.; Wang, J.S.; Delgado, S.; Zukerman, R.; Mittal, J.; Jhaveri, V.M. Cell communication by tunneling nanotubes: Implications in disease and therapeutic applications. J. Cell Physiol. 2019, 234, 1130–1146. [Google Scholar] [CrossRef] [PubMed]
- Kolba, M.D.; Dudka, W.; Zaręba-Kozioł, M.; Kominek, A.; Ronchi, P.; Turos, L.; Chroscicki, P.; Wlodarczyk, J.; Schwab, Y.; Klejman, A.; et al. Tunneling nanotube-mediated intercellular vesicle and protein transfer in the stroma-provided imatinib resistance in chronic myeloid leukemia cells. Cell Death Dis. 2019, 10, 817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Omsland, M.; Andresen, V.; Gullaksen, S.E.; Ayuda-Durán, P.; Popa, M.; Hovland, R.; Brendehaug, A.; Enserink, J.; McCormack, E.; Gjertsen, B.T. Tyrosine kinase inhibitors and interferon-α increase tunneling nanotube (TNT) formation and cell adhesion in chronic myeloid leukemia (CML) cell lines. FASEB J. 2020, 34, 3773–3791. [Google Scholar] [CrossRef] [Green Version]
- Dubois, F.; Bénard, M.; Jean-Jacques, B.; Schapman, D.; Roberge, H.; Lebon, A.; Goux, D.; Monterroso, B.; Elie, N.; Komuro, H.; et al. Investigating Tunneling Nanotubes in Cancer Cells: Guidelines for Structural and Functional Studies through Cell Imaging. Biomed Res Int. 2020, 2020, 2701345. [Google Scholar] [CrossRef]
- Sáenz-de-Santa-María, I.; Bernardo-Castiñeira, C.; Enciso, E.; García-Moreno, I.; Chiara, J.L.; Suarez, C.; Chiara, M.D. Control of long-distance cell-to-cell communication and autophagosome transfer in squamous cell carcinoma via tunneling nanotubes. Oncotarget 2017, 8, 20939–20960. [Google Scholar] [CrossRef] [Green Version]
- Whitehead, J.; Zhang, J.; Harvestine, J.N.; Kothambawala, A.; Liu, G.Y.; Leach, J.K. Tunneling nanotubes mediate the expression of senescence markers in mesenchymal stem/stromal cell spheroids. Stem Cells. 2020, 38, 80–89. [Google Scholar] [CrossRef] [Green Version]
- Dilsizoglu Senol, A.; Pepe, A.; Grudina, C.; Sassoon, N.; Reiko, U.; Bousset, L.; Melki, R.; Piel, J.; Gugger, M.; Zurzolo, C. Effect of tolytoxin on tunneling nanotube formation and function. Sci. Rep. 2019, 9, 5741. [Google Scholar] [CrossRef]
- Fortemaison, N.; Blancquaert, S.; Dumont, J.E.; Maenhaut, C.; Aktories, K.; Roger, P.P.; Dremier, S. Differential involvement of the actin cytoskeleton in differentiation and mitogenesis of thyroid cells: Inactivation of rho proteins contributes to cyclic adenosine monophosphate-dependent gene expression but prevents mitogenesis. Endocrinology 2005, 146, 5485–5495. [Google Scholar] [CrossRef] [Green Version]
- Susanto, O.; Stewart, S.E.; Voskoboinik, I.; Brasacchio, D.; Hagn, M.; Ellis, S.; Asquith, S.; Sedelies, K.A.; Bird, P.I.; Waterhouse, N.J.; et al. Mouse granzyme A induces a novel death with writhing morphology that is mechanistically distinct from granzyme B-induced apoptosis. Cell Death Differ. 2013, 20, 1183–1193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Souriant, S.; Balboa, L.; Dupont, M.; Pingris, K.; Kviatcovsky, D.; Cougoule, C.; Lastrucci, C.; Bah, A.; Gasser, R.; Poincloux, R.; et al. Tuberculosis Exacerbates HIV-1 Infection through IL-10/STAT3-Dependent Tunneling Nanotube Formation in Macrophages. Cell Rep. 2019, 26, 3586–3599.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vargas, J.Y.; Loria, F.; Wu, Y.J.; Cordova, G.; Nonaka, T.; Bellow, S.; Syan, S.; Hasegawa, M.; van Woerden, G.M.; Trollet, C.; et al. The Wnt/Ca2+ pathway is involved in interneuronal communication mediated by tunneling nanotubes. EMBO J. 2019, 38, e101230. [Google Scholar] [CrossRef] [PubMed]
- Sarma, V.; Wolf, F.W.; Marks, R.M.; Shows, T.B.; Dixit, V.M. Cloning of a novel tumor necrosis factor-alpha-inducible primary response gene that is differentially expressed in development and capillary tube-like formation in vitro. J. Immunol. 1992, 148, 3302–3312. [Google Scholar] [PubMed]
- Jackson, M.V.; Morrison, T.J.; Doherty, D.F.; McAuley, D.F.; Matthay, M.A.; Kissenpfennig, A.; O’Kane, C.M.; Krasnodembskaya, A.D. Mitochondrial Transfer Via Tunneling Nanotubes is an Important Mechanism by Which Mesenchymal Stem Cells Enhance Macrophage Phagocytosis in the In Vitro and In Vivo Models of ARDS. Stem Cells 2016, 34, 2210–2223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vignais, M.L.; Caicedo, A.; Brondello, J.M.; Jorgensen, C. Cell Connections by Tunneling Nanotubes: Effects of Mitochondrial Trafficking on Target Cell Metabolism, Homeostasis, and Response to Therapy. Stem Cells Int. 2017, 2017, 6917941. [Google Scholar] [CrossRef] [Green Version]
- Dewhirst, M.W.; Secomb, T.W. Transport of drugs from blood vessels to tumor tissue. Nat. Rev. Cancer 2017, 17, 738–750. [Google Scholar] [CrossRef]
- Ljubojevic, N.; Henderson, J.M.; Zurzolo, C. The Ways of Actin: Why Tunneling Nanotubes Are Unique Cell Protrusions. Trends Cell Biol. 2021, 31, 130–142. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Allegra, A.; Di Gioacchino, M.; Cancemi, G.; Casciaro, M.; Petrarca, C.; Musolino, C.; Gangemi, S. Specialized Intercellular Communications via Tunnelling Nanotubes in Acute and Chronic Leukemia. Cancers 2022, 14, 659. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14030659
Allegra A, Di Gioacchino M, Cancemi G, Casciaro M, Petrarca C, Musolino C, Gangemi S. Specialized Intercellular Communications via Tunnelling Nanotubes in Acute and Chronic Leukemia. Cancers. 2022; 14(3):659. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14030659
Chicago/Turabian StyleAllegra, Alessandro, Mario Di Gioacchino, Gabriella Cancemi, Marco Casciaro, Claudia Petrarca, Caterina Musolino, and Sebastiano Gangemi. 2022. "Specialized Intercellular Communications via Tunnelling Nanotubes in Acute and Chronic Leukemia" Cancers 14, no. 3: 659. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14030659