
Roles of Protein Disulfide Isomerase in Breast Cancer

 ,
, 
Abstract
:Simple Summary
Abstract
1. Introduction
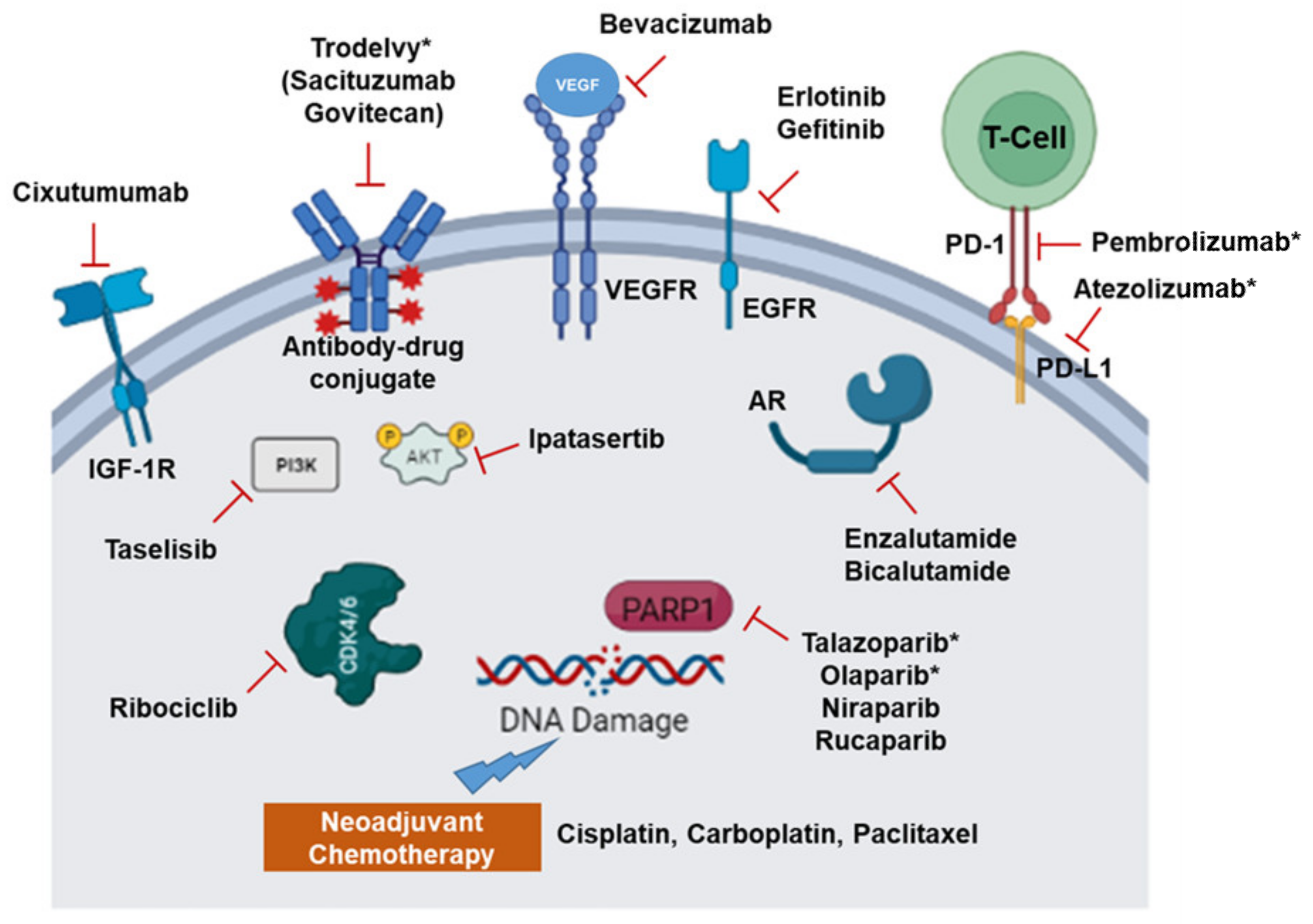
2. Recent Therapeutic Options and Molecular Targets in TNBC
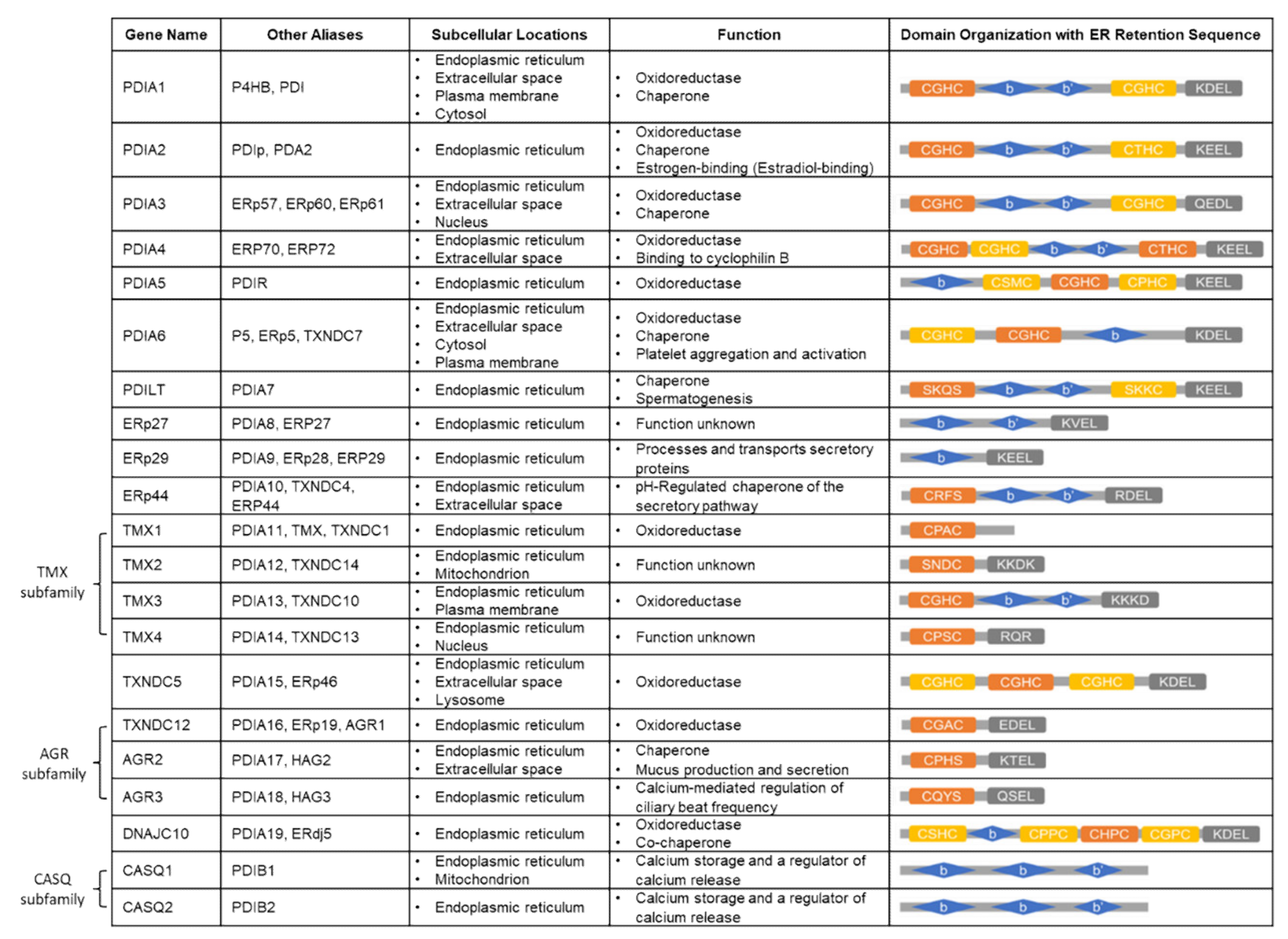
3. Protein Disulfide Isomerase (PDI) Family
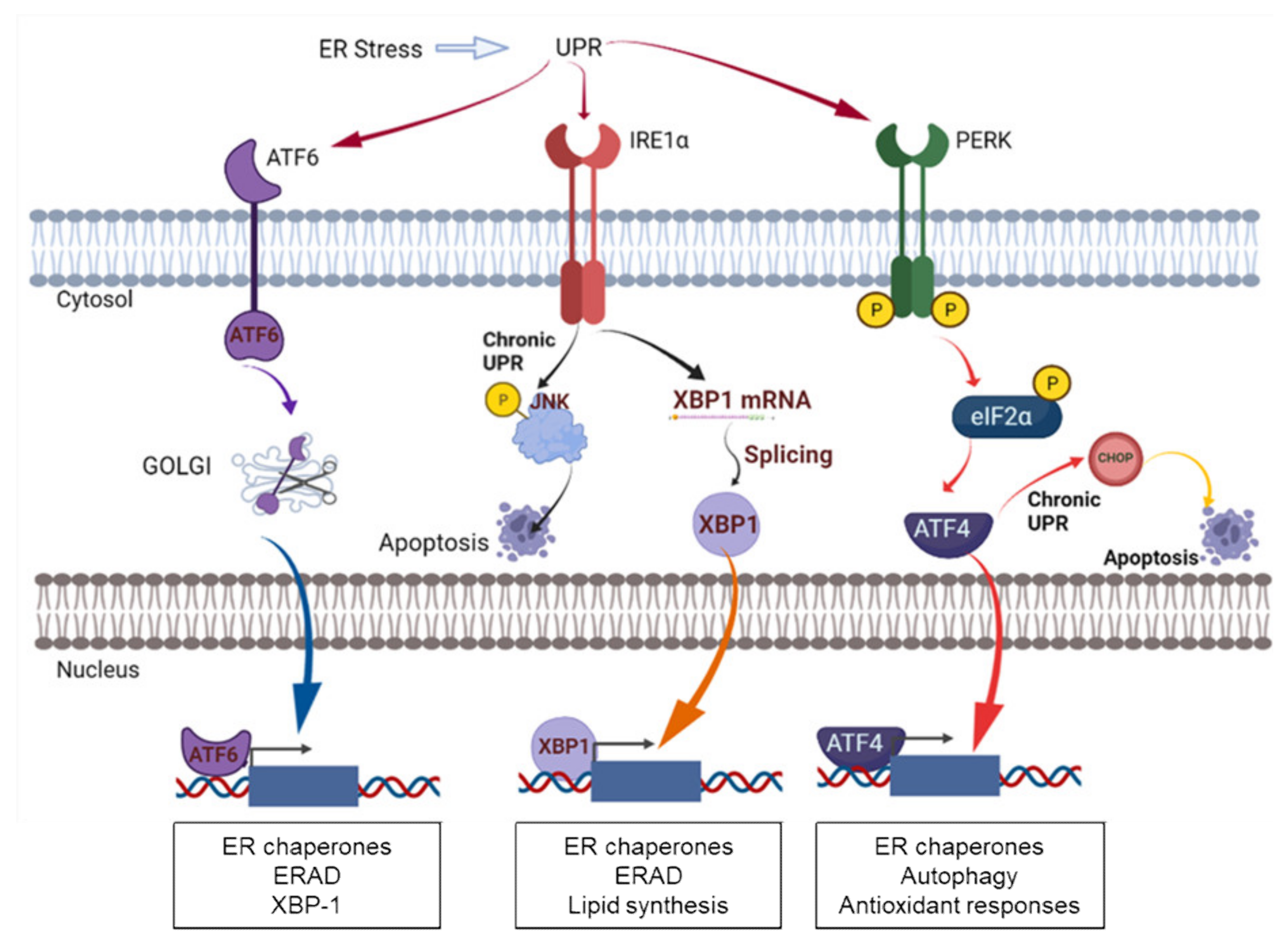
4. PDI in ER Stress and UPR Signaling
5. The Functions/Roles of Specific PDIs in Breast Cancer
5.1. Overexpression of PDIs and the Role of PDI in Breast Cancer Proliferation
5.2. Role of PDI in Breast Cancer Invasion and Metastasis
5.3. Role of PDI in Breast Cancer Chemoresistance and Clinical Outcomes
5.4. Role of PDI as Transcriptional Cofactors
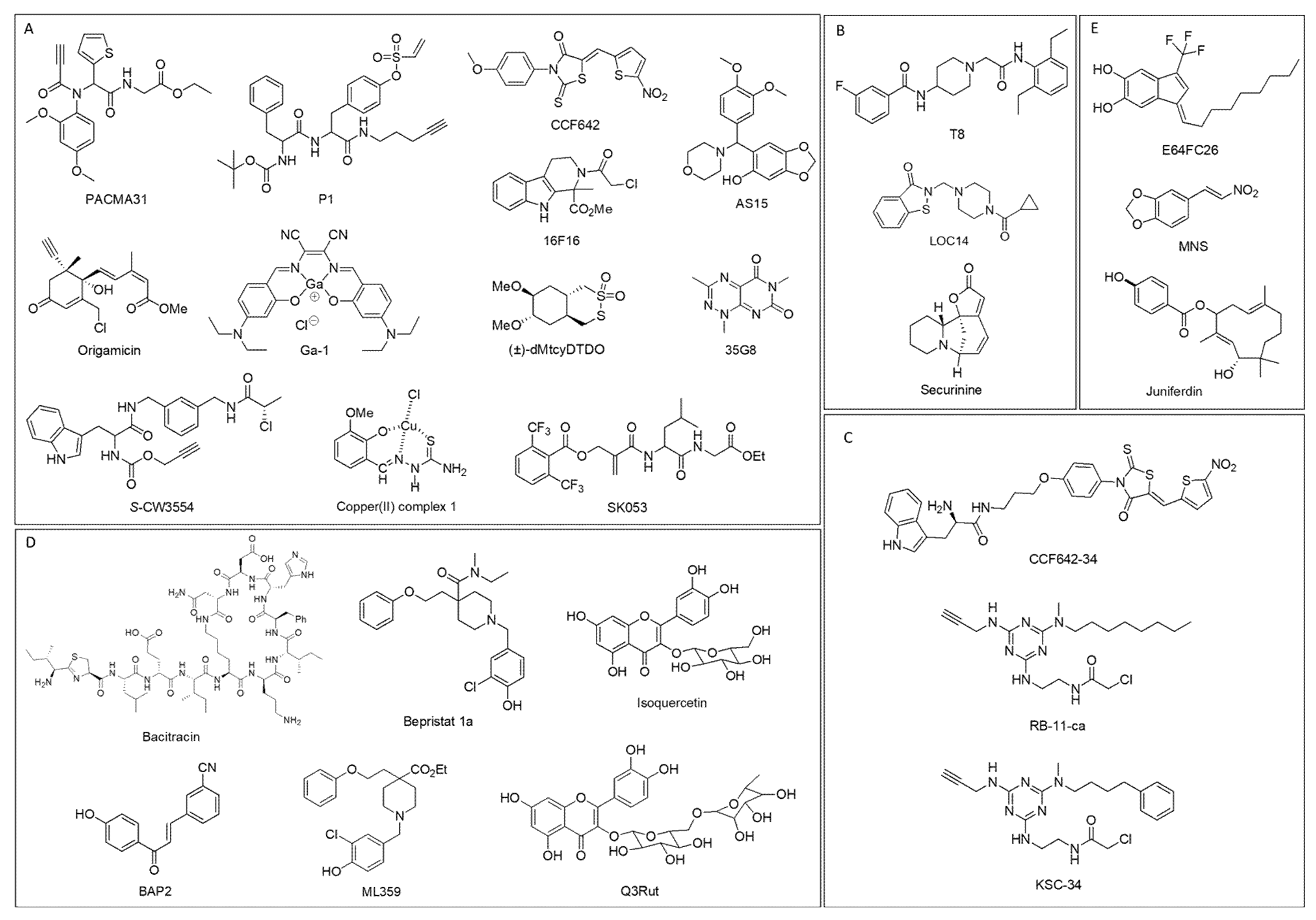
6. PDI Inhibitors
6.1. PDI Inhibitors Categorized Depending on Binding Sites

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDI Inhibitor | Specificity toward PDIs | Mode of Action | Cell-Based and Pre-Clinical Studies | Refs |
|---|---|---|---|---|
| PACMA31 | PDIA1, PDIA2, PDIA3, PDIA4, PDIA6, TXNDC5 |
|
| [81,86,114,126,127] |
| P1 | PDIA1, PDIA4, PDIA6 |
|
| [115] |
| 16F16 | PDIA1, PDIA3 |
|
| [94,116] |
| AS15 | PDIA1 |
|
| [117] |
| CCF642 | PDIA1, PDIA3, PDIA4 |
|
| [118] |
| S-CW3554 | PDIA1 |
|
| [119] |
| Origamicin | PDIA1 |
|
| [120] |
| (±)-dMtcyDTDO | PDIA1, AGR2, AGR3, ERp44 |
|
| [121] |
| Ga-1 | PDIA1, PDIA3, PDIA4, PDIA6 |
|
| [122] |
| 35G8 | PDIA1 |
|
| [123] |
| Copper (II) complex 1 | PDIA1 |
|
| [124] |
| SK053 | PDIA1 |
|
| [125] |
| T8 | PDIA1 |
|
| [128] |
| LOC14 | PDIA1, PDIA3 |
|
| [129,130] |
| Securinine | PDIA1 |
|
| [131] |
| CCF642–34 | PDIA1 |
|
| [132] |
| RB-11-ca | PDIA1 |
|
| [133] |
| KSC-34 | PDIA1 |
|
| [134] |
| Bacitracin | PDIA1 |
|
| [135,136,137,138] |
| BAP2 | PDIA1, PDIA2 |
|
| [66,127] |
| Bepristat 1a | PDIA1 |
|
| [139] |
| Q3Rut | PDIA1 |
|
| [140,141] |
| Isoquercetin | PDIA1 |
|
| [140,141,142] |
| ML359 | PDIA1 |
|
| [143] |
| E64FC26 | PDIA1, PDIA3, PDIA4, PDIA6, TXNDC5 |
|
| [144] |
| MNS | Cell surface PDI |
|
| [95,145,146] |
| Juniferdin | PDIA1 |
|
| [147,148] |
6.2. PDI Inhibitors in Breast Cancer
7. Concluding Remarks and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Breastcancer. U.S. Breast Cancer Statistics. Available online: https://www.breastcancer.org/symptoms/understand_bc/statistics (accessed on 10 November 2021).
- Riaz, M.; van Jaarsveld, M.T.; Hollestelle, A.; Prager-van der Smissen, W.J.; Heine, A.A.; Boersma, A.W.; Liu, J.; Helmijr, J.; Ozturk, B.; Smid, M.; et al. miRNA expression profiling of 51 human breast cancer cell lines reveals subtype and driver mutation-specific miRNAs. Breast Cancer Res. 2013, 15, R33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldhirsch, A.; Wood, W.C.; Coates, A.S.; Gelber, R.D.; Thurlimann, B.; Senn, H.J.; Panel, M. Strategies for subtypes-dealing with the diversity of breast cancer: Highlights of the St. Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2011. Ann. Oncol. 2011, 22, 1736–1747. [Google Scholar] [CrossRef]
- Prat, A.; Adamo, B.; Cheang, M.C.; Anders, C.K.; Carey, L.A.; Perou, C.M. Molecular characterization of basal-like and non-basal-like triple-negative breast cancer. Oncologist 2013, 18, 123–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, N.U.; Claus, E.; Sohl, J.; Razzak, A.R.; Arnaout, A.; Winer, E.P. Sites of distant recurrence and clinical outcomes in patients with metastatic triple-negative breast cancer: High incidence of central nervous system metastases. Cancer 2008, 113, 2638–2645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gluz, O.; Liedtke, C.; Gottschalk, N.; Pusztai, L.; Nitz, U.; Harbeck, N. Triple-negative breast cancer-current status and future directions. Ann. Oncol. 2009, 20, 1913–1927. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Pietenpol, J.A.; Tan, A.R. Triple-negative breast cancer: Molecular subtypes and new targets for therapy. Am. Soc. Clin. Oncol. Educ. Book 2015, 35, e31–e39. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [Green Version]
- Haffty, B.G.; Yang, Q.; Reiss, M.; Kearney, T.; Higgins, S.A.; Weidhaas, J.; Harris, L.; Hait, W.; Toppmeyer, D. Locoregional relapse and distant metastasis in conservatively managed triple negative early-stage breast cancer. J. Clin. Oncol. 2006, 24, 5652–5657. [Google Scholar] [CrossRef]
- Foulkes, W.D.; Smith, I.E.; Reis-Filho, J.S. Triple-negative breast cancer. N. Engl. J. Med. 2010, 363, 1938–1948. [Google Scholar] [CrossRef] [Green Version]
- Carey, L.A.; Perou, C.M.; Livasy, C.A.; Dressler, L.G.; Cowan, D.; Conway, K.; Karaca, G.; Troester, M.A.; Tse, C.K.; Edmiston, S.; et al. Race, breast cancer subtypes, and survival in the Carolina Breast Cancer Study. JAMA 2006, 295, 2492–2502. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.J.; Naidu, S.; Topham, A.K.; Guiles, F.; Xu, Y.; McCue, P.; Schwartz, G.F.; Park, P.K.; Rosenberg, A.L.; Brill, K.; et al. Differences in breast carcinoma characteristics in newly diagnosed African-American and Caucasian patients: A single-institution compilation compared with the National Cancer Institute’s Surveillance, Epidemiology, and End Results database. Cancer 2007, 110, 876–884. [Google Scholar] [CrossRef]
- Prakash, O.; Hossain, F.; Danos, D.; Lassak, A.; Scribner, R.; Miele, L. Racial Disparities in Triple Negative Breast Cancer: A Review of the Role of Biologic and Non-biologic Factors. Front. Public Health 2020, 8, 576964. [Google Scholar] [CrossRef] [PubMed]
- Pogoda, K.; Niwińska, A.; Murawska, M.; Pieńkowski, T. Analysis of pattern, time and risk factors influencing recurrence in triple-negative breast cancer patients. Med. Oncol. 2013, 30, 388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianchini, G.; De Angelis, C.; Licata, L.; Gianni, L. Treatment landscape of triple-negative breast cancer—Expanded options, evolving needs. Nat. Rev. Clin. Oncol. 2021, 19, 91–113. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; O’Shaughnessy, J.; Zhao, J.; Haiderali, A.; Cortés, J.; Ramsey, S.D.; Briggs, A.; Hu, P.; Karantza, V.; Aktan, G.; et al. Association of Pathologic Complete Response with Long-Term Survival Outcomes in Triple-Negative Breast Cancer: A Meta-Analysis. Cancer Res. 2020, 80, 5427–5434. [Google Scholar] [CrossRef]
- Anders, C.K.; Abramson, V.; Tan, T.; Dent, R. The Evolution of Triple-Negative Breast Cancer: From Biology to Novel Therapeutics. Am. Soc. Clin. Oncol. Educ. Book 2016, 36, 34–42. [Google Scholar] [CrossRef]
- Bardia, A.; Hurvitz, S.A.; Tolaney, S.M.; Loirat, D.; Punie, K.; Oliveira, M.; Brufsky, A.; Sardesai, S.D.; Kalinsky, K.; Zelnak, A.B.; et al. Sacituzumab Govitecan in Metastatic Triple-Negative Breast Cancer. N. Engl. J. Med. 2021, 384, 1529–1541. [Google Scholar] [CrossRef]
- Carey, L.A.; Dees, E.C.; Sawyer, L.; Gatti, L.; Moore, D.T.; Collichio, F.; Ollila, D.W.; Sartor, C.I.; Graham, M.L.; Perou, C.M. The triple negative paradox: Primary tumor chemosensitivity of breast cancer subtypes. Clin. Cancer Res. 2007, 13, 2329–2334. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Wu, J.; Zhang, Z.; Tang, Y.; Li, X.; Liu, S.; Cao, S.; Li, X. Association Between BRCA Status and Triple-Negative Breast Cancer: A Meta-Analysis. Front. Pharmacol. 2018, 9, 909. [Google Scholar] [CrossRef]
- Lips, E.H.; Mulder, L.; Oonk, A.; van der Kolk, L.E.; Hogervorst, F.B.; Imholz, A.L.; Wesseling, J.; Rodenhuis, S.; Nederlof, P.M. Triple-negative breast cancer: BRCAness and concordance of clinical features with BRCA1-mutation carriers. Br. J. Cancer 2013, 108, 2172–2177. [Google Scholar] [CrossRef]
- Keung, M.Y.T.; Wu, Y.; Vadgama, J.V. PARP Inhibitors as a Therapeutic Agent for Homologous Recombination Deficiency in Breast Cancers. J. Clin. Med. 2019, 8, 435. [Google Scholar] [CrossRef] [Green Version]
- Litton, J.K.; Rugo, H.S.; Ettl, J.; Hurvitz, S.A.; Gonçalves, A.; Lee, K.-H.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.A.; Martin, M.; et al. Talazoparib in Patients with Advanced Breast Cancer and a Germline BRCA Mutation. N. Engl. J. Med. 2018, 379, 753–763. [Google Scholar] [CrossRef] [PubMed]
- Tutt, A.; Tovey, H.; Cheang, M.C.U.; Kernaghan, S.; Kilburn, L.; Gazinska, P.; Owen, J.; Abraham, J.; Barrett, S.; Barrett-Lee, P.; et al. Carboplatin in BRCA1/2-mutated and triple-negative breast cancer BRCAness subgroups: The TNT Trial. Nat. Med. 2018, 24, 628–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tung, N.; Arun, B.; Hacker, M.R.; Hofstatter, E.; Toppmeyer, D.L.; Isakoff, S.J.; Borges, V.; Legare, R.D.; Isaacs, C.; Wolff, A.C.; et al. TBCRC 031: Randomized Phase II Study of Neoadjuvant Cisplatin Versus Doxorubicin-Cyclophosphamide in Germline BRCA Carriers with HER2-Negative Breast Cancer (the INFORM trial). J. Clin. Oncol. 2020, 38, 1539–1548. [Google Scholar] [CrossRef]
- Osborne, C.; Challagalla, J.D.; Eisenbeis, C.F.; Holmes, F.A.; Neubauer, M.A.; Koutrelakos, N.W.; Taboada, C.A.; Vukelja, S.J.; Wilks, S.T.; Allison, M.A.; et al. Ixabepilone and Carboplatin for Hormone Receptor Positive/HER2-neu Negative and Triple Negative Metastatic Breast Cancer. Clin. Breast Cancer 2018, 18, e89–e95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomssen, C.; Pierga, J.Y.; Pritchard, K.I.; Biganzoli, L.; Cortes-Funes, H.; Petráková, K.; Kaufman, B.; Duenne, A.; Smith, I. First-line bevacizumab-containing therapy for triple-negative breast cancer: Analysis of 585 patients treated in the ATHENA study. Oncology 2012, 82, 218–227. [Google Scholar] [CrossRef]
- Nakai, K.; Hung, M.C.; Yamaguchi, H. A perspective on anti-EGFR therapies targeting triple-negative breast cancer. Am. J. Cancer Res. 2016, 6, 1609–1623. [Google Scholar]
- Liu, Y.; Zhou, Y.; Huang, K.H.; Li, Y.; Fang, X.; An, L.; Wang, F.; Chen, Q.; Zhang, Y.; Shi, A.; et al. EGFR-specific CAR-T cells trigger cell lysis in EGFR-positive TNBC. Aging 2019, 11, 11054–11072. [Google Scholar] [CrossRef] [PubMed]
- Kwapisz, D. Pembrolizumab and atezolizumab in triple-negative breast cancer. Cancer Immunol. Immunother. 2021, 70, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Schmid, P.; Salgado, R.; Park, Y.H.; Muñoz-Couselo, E.; Kim, S.B.; Sohn, J.; Im, S.A.; Foukakis, T.; Kuemmel, S.; Dent, R.; et al. Pembrolizumab plus chemotherapy as neoadjuvant treatment of high-risk, early-stage triple-negative breast cancer: Results from the phase 1b open-label, multicohort KEYNOTE-173 study. Ann. Oncol. 2020, 31, 569–581. [Google Scholar] [CrossRef]
- Schmid, P.; Rugo, H.S.; Adams, S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Diéras, V.; Henschel, V.; Molinero, L.; Chui, S.Y.; et al. Atezolizumab plus nab-paclitaxel as first-line treatment for unresectable, locally advanced or metastatic triple-negative breast cancer (IMpassion130): Updated efficacy results from a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. Oncol. 2020, 21, 44–59. [Google Scholar] [CrossRef]
- Lyons, T.G. Targeted Therapies for Triple-Negative Breast Cancer. Curr. Treat. Options Oncol. 2019, 20, 82. [Google Scholar] [CrossRef] [PubMed]
- Vinayak, S.; Tolaney, S.M.; Schwartzberg, L.; Mita, M.; McCann, G.; Tan, A.R.; Wahner-Hendrickson, A.E.; Forero, A.; Anders, C.; Wulf, G.M.; et al. Open-label Clinical Trial of Niraparib Combined with Pembrolizumab for Treatment of Advanced or Metastatic Triple-Negative Breast Cancer. JAMA Oncol. 2019, 5, 1132–1140. [Google Scholar] [CrossRef] [Green Version]
- Diamond, J.R.; Eckhardt, S.G.; Pitts, T.M.; van Bokhoven, A.; Aisner, D.; Gustafson, D.L.; Capasso, A.; Sams, S.; Kabos, P.; Zolman, K.; et al. A phase II clinical trial of the Aurora and angiogenic kinase inhibitor ENMD-2076 for previously treated, advanced, or metastatic triple-negative breast cancer. Breast Cancer Res. 2018, 20, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.B.; Dent, R.; Im, S.A.; Espie, M.; Blau, S.; Tan, A.R.; Isakoff, S.J.; Oliveira, M.; Saura, C.; Wongchenko, M.J.; et al. Ipatasertib plus paclitaxel versus placebo plus paclitaxel as first-line therapy for metastatic triple-negative breast cancer (LOTUS): A multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2017, 18, 1360–1372. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Abramson, V.G.; Sanders, M.E.; Mayer, E.L.; Haddad, T.C.; Nanda, R.; Van Poznak, C.; Storniolo, A.M.; Nangia, J.R.; Gonzalez-Ericsson, P.I.; et al. TBCRC 032 IB/II Multicenter Study: Molecular Insights to AR Antagonist and PI3K Inhibitor Efficacy in Patients with AR(+) Metastatic Triple-Negative Breast Cancer. Clin. Cancer Res. 2020, 26, 2111–2123. [Google Scholar] [CrossRef] [PubMed]
- Reddy, T.P.; Choi, D.S.; Anselme, A.C.; Qian, W.; Chen, W.; Lantto, J.; Horak, I.D.; Kragh, M.; Chang, J.C.; Rosato, R.R. Simultaneous targeting of HER family pro-survival signaling with Pan-HER antibody mixture is highly effective in TNBC: A preclinical trial with PDXs. Breast Cancer Res. 2020, 22, 48. [Google Scholar] [CrossRef]
- Rupp, T.; Pelouin, O.; Genest, L.; Legrand, C.; Froget, G.; Castagné, V. Therapeutic potential of Fingolimod in triple negative breast cancer preclinical models. Transl. Oncol. 2021, 14, 100926. [Google Scholar] [CrossRef]
- Zanker, D.J.; Spurling, A.J.; Brockwell, N.K.; Owen, K.L.; Zakhour, J.M.; Robinson, T.; Duivenvoorden, H.M.; Hertzog, P.J.; Mullins, S.R.; Wilkinson, R.W.; et al. Intratumoral administration of the Toll-like receptor 7/8 agonist 3M-052 enhances interferon-driven tumor immunogenicity and suppresses metastatic spread in preclinical triple-negative breast cancer. Clin. Transl. Immunol. 2020, 9, e1177. [Google Scholar] [CrossRef]
- Solomon, V.R.; Alizadeh, E.; Bernhard, W.; Hartimath, S.V.; Hill, W.; Chekol, R.; Barreto, K.M.; Geyer, C.R.; Fonge, H. 111In- and 225Ac-Labeled Cixutumumab for Imaging and α-Particle Radiotherapy of IGF-1R Positive Triple-Negative Breast Cancer. Mol. Pharm. 2019, 16, 4807–4816. [Google Scholar] [CrossRef]
- Galligan, J.J.; Petersen, D.R. The human protein disulfide isomerase gene family. Hum. Genom. 2012, 6, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Depuydt, M.; Messens, J.; Collet, J.F. How proteins form disulfide bonds. Antioxid. Redox Signal. 2011, 15, 49–66. [Google Scholar] [CrossRef] [PubMed]
- Perri, E.R.; Thomas, C.J.; Parakh, S.; Spencer, D.M.; Atkin, J.D. The Unfolded Protein Response and the Role of Protein Disulfide Isomerase in Neurodegeneration. Front. Cell Dev. Biol. 2015, 3, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, E.; Lee, D.H. Emerging roles of protein disulfide isomerase in cancer. BMB Rep. 2017, 50, 401–410. [Google Scholar] [CrossRef] [Green Version]
- Ali Khan, H.; Mutus, B. Protein disulfide isomerase a multifunctional protein with multiple physiological roles. Front. Chem. 2014, 2, 70. [Google Scholar] [CrossRef] [Green Version]
- Kanemura, S.; Matsusaki, M.; Inaba, K.; Okumura, M. PDI Family Members as Guides for Client Folding and Assembly. Int. J. Mol. Sci. 2020, 21, 9351. [Google Scholar] [CrossRef]
- Hatahet, F.; Ruddock, L.W. Protein disulfide isomerase: A critical evaluation of its function in disulfide bond formation. Antioxid. Redox Signal. 2009, 11, 2807–2850. [Google Scholar] [CrossRef]
- Kozlov, G.; Maattanen, P.; Thomas, D.Y.; Gehring, K. A structural overview of the PDI family of proteins. FEBS J. 2010, 277, 3924–3936. [Google Scholar] [CrossRef]
- Klappa, P.; Ruddock, L.W.; Darby, N.J.; Freedman, R.B. The b’ domain provides the principal peptide-binding site of protein disulfide isomerase but all domains contribute to binding of misfolded proteins. EMBO J. 1998, 17, 927–935. [Google Scholar] [CrossRef] [Green Version]
- Powell, L.E.; Foster, P.A. Protein disulphide isomerase inhibition as a potential cancer therapeutic strategy. Cancer Med. 2021, 10, 2812–2825. [Google Scholar] [CrossRef]
- Wang, C.; Li, W.; Ren, J.; Fang, J.; Ke, H.; Gong, W.; Feng, W.; Wang, C.C. Structural insights into the redox-regulated dynamic conformations of human protein disulfide isomerase. Antioxid. Redox Signal. 2013, 19, 36–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okumura, M.; Noi, K.; Kanemura, S.; Kinoshita, M.; Saio, T.; Inoue, Y.; Hikima, T.; Akiyama, S.; Ogura, T.; Inaba, K. Dynamic assembly of protein disulfide isomerase in catalysis of oxidative folding. Nat. Chem. Biol. 2019, 15, 499–509. [Google Scholar] [CrossRef]
- Wang, C.; Yu, J.; Huo, L.; Wang, L.; Feng, W.; Wang, C.C. Human protein-disulfide isomerase is a redox-regulated chaperone activated by oxidation of domain a’. J. Biol. Chem. 2012, 287, 1139–1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okumura, M.; Kadokura, H.; Hashimoto, S.; Yutani, K.; Kanemura, S.; Hikima, T.; Hidaka, Y.; Ito, L.; Shiba, K.; Masui, S.; et al. Inhibition of the functional interplay between endoplasmic reticulum (ER) oxidoreduclin-1alpha (Ero1alpha) and protein-disulfide isomerase (PDI) by the endocrine disruptor bisphenol A. J. Biol. Chem. 2014, 289, 27004–27018. [Google Scholar] [CrossRef] [Green Version]
- Bettigole, S.E.; Glimcher, L.H. Endoplasmic reticulum stress in immunity. Annu. Rev. Immunol. 2015, 33, 107–138. [Google Scholar] [CrossRef]
- Eletto, D.; Eletto, D.; Dersh, D.; Gidalevitz, T.; Argon, Y. Protein disulfide isomerase A6 controls the decay of IRE1alpha signaling via disulfide-dependent association. Mol. Cell 2014, 53, 562–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Li, T.; Liu, Y.; Wang, X.; Zhang, J.; Wang, X.; Shi, G.; Lou, J.; Wang, L.; Wang, C.C.; et al. Phosphorylation switches protein disulfide isomerase activity to maintain proteostasis and attenuate ER stress. EMBO J. 2020, 39, e103841. [Google Scholar] [CrossRef] [PubMed]
- Appenzeller-Herzog, C.; Ellgaard, L. The human PDI family: Versatility packed into a single fold. Biochim. Biophys. Acta 2008, 1783, 535–548. [Google Scholar] [CrossRef] [Green Version]
- Kranz, P.; Neumann, F.; Wolf, A.; Classen, F.; Pompsch, M.; Ocklenburg, T.; Baumann, J.; Janke, K.; Baumann, M.; Goepelt, K.; et al. PDI is an essential redox-sensitive activator of PERK during the unfolded protein response (UPR). Cell Death Dis. 2017, 8, e2986. [Google Scholar] [CrossRef] [Green Version]
- Eletto, D.; Eletto, D.; Boyle, S.; Argon, Y. PDIA6 regulates insulin secretion by selectively inhibiting the RIDD activity of IRE1. FASEB J. 2016, 30, 653–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higa, A.; Taouji, S.; Lhomond, S.; Jensen, D.; Fernandez-Zapico, M.E.; Simpson, J.C.; Pasquet, J.M.; Schekman, R.; Chevet, E. Endoplasmic reticulum stress-activated transcription factor ATF6alpha requires the disulfide isomerase PDIA5 to modulate chemoresistance. Mol. Cell. Biol. 2014, 34, 1839–1849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nadanaka, S.; Okada, T.; Yoshida, H.; Mori, K. Role of disulfide bridges formed in the luminal domain of ATF6 in sensing endoplasmic reticulum stress. Mol. Cell. Biol. 2007, 27, 1027–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Kaufman, R.J. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 2016, 529, 326–335. [Google Scholar] [CrossRef]
- Hetz, C.; Bernasconi, P.; Fisher, J.; Lee, A.H.; Bassik, M.C.; Antonsson, B.; Brandt, G.S.; Iwakoshi, N.N.; Schinzel, A.; Glimcher, L.H.; et al. Proapoptotic BAX and BAK modulate the unfolded protein response by a direct interaction with IRE1alpha. Science 2006, 312, 572–576. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Shergalis, A.; Lu, D.; Kyani, A.; Liu, Z.; Ljungman, M.; Neamati, N. Design, Synthesis, and Biological Evaluation of Novel Allosteric Protein Disulfide Isomerase Inhibitors. J. Med. Chem. 2019, 62, 3447–3474. [Google Scholar] [CrossRef]
- Samanta, S.; Tamura, S.; Dubeau, L.; Mhawech-Fauceglia, P.; Miyagi, Y.; Kato, H.; Lieberman, R.; Buckanovich, R.J.; Lin, Y.G.; Neamati, N. Expression of protein disulfide isomerase family members correlates with tumor progression and patient survival in ovarian cancer. Oncotarget 2017, 8, 103543–103556. [Google Scholar] [CrossRef] [PubMed]
- Tufo, G.; Jones, A.W.; Wang, Z.; Hamelin, J.; Tajeddine, N.; Esposti, D.D.; Martel, C.; Boursier, C.; Gallerne, C.; Migdal, C.; et al. The protein disulfide isomerases PDIA4 and PDIA6 mediate resistance to cisplatin-induced cell death in lung adenocarcinoma. Cell Death Differ. 2014, 21, 685–695. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Lee, D.; Ho, A.S.; Pu, J.K.; Zhang, X.Q.; Lee, N.P.; Day, P.J.; Lui, W.M.; Fung, C.F.; Leung, G.K. Inhibition of prolyl 4-hydroxylase, beta polypeptide (P4HB) attenuates temozolomide resistance in malignant glioma via the endoplasmic reticulum stress response (ERSR) pathways. Neuro Oncol. 2013, 15, 562–577. [Google Scholar] [CrossRef]
- Xu, S.; Sankar, S.; Neamati, N. Protein disulfide isomerase: A promising target for cancer therapy. Drug Discov. Today 2014, 19, 222–240. [Google Scholar] [CrossRef]
- Gromov, P.; Gromova, I.; Bunkenborg, J.; Cabezon, T.; Moreira, J.M.; Timmermans-Wielenga, V.; Roepstorff, P.; Rank, F.; Celis, J.E. Up-regulated proteins in the fluid bathing the tumour cell microenvironment as potential serological markers for early detection of cancer of the breast. Mol. Oncol. 2010, 4, 65–89. [Google Scholar] [CrossRef] [Green Version]
- Chahed, K.; Kabbage, M.; Hamrita, B.; Guillier, C.L.; Trimeche, M.; Remadi, S.; Ehret-Sabatier, L.; Chouchane, L. Detection of protein alterations in male breast cancer using two dimensional gel electrophoresis and mass spectrometry: The involvement of several pathways in tumorigenesis. Clin. Chim. Acta 2008, 388, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Chahed, K.; Kabbage, M.; Ehret-Sabatier, L.; Lemaitre-Guillier, C.; Remadi, S.; Hoebeke, J.; Chouchane, L. Expression of fibrinogen E-fragment and fibrin E-fragment is inhibited in the human infiltrating ductal carcinoma of the breast: The two-dimensional electrophoresis and MALDI-TOF-mass spectrometry analyses. Int. J. Oncol. 2005, 27, 1425–1431. [Google Scholar] [CrossRef] [PubMed]
- Thongwatchara, P.; Promwikorn, W.; Srisomsap, C.; Chokchaichamnankit, D.; Boonyaphiphat, P.; Thongsuksai, P. Differential protein expression in primary breast cancer and matched axillary node metastasis. Oncol. Rep. 2011, 26, 185–191. [Google Scholar] [CrossRef]
- Wise, R.; Duhachek-Muggy, S.; Qi, Y.; Zolkiewski, M.; Zolkiewska, A. Protein disulfide isomerases in the endoplasmic reticulum promote anchorage-independent growth of breast cancer cells. Breast Cancer Res. Treat. 2016, 157, 241–252. [Google Scholar] [CrossRef]
- Ramos, F.S.; Serino, L.T.; Carvalho, C.M.; Lima, R.S.; Urban, C.A.; Cavalli, I.J.; Ribeiro, E.M. PDIA3 and PDIA6 gene expression as an aggressiveness marker in primary ductal breast cancer. Genet. Mol. Res. 2015, 14, 6960–6967. [Google Scholar] [CrossRef]
- Salmans, M.L.; Zhao, F.; Andersen, B. The estrogen-regulated anterior gradient 2 (AGR2) protein in breast cancer: A potential drug target and biomarker. Breast Cancer Res. 2013, 15, 204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stojak, M.; Milczarek, M.; Kurpinska, A.; Suraj-Prazmowska, J.; Kaczara, P.; Wojnar-Lason, K.; Banach, J.; Stachowicz-Suhs, M.; Rossowska, J.; Kalvins, I.; et al. Protein Disulphide Isomerase A1 Is Involved in the Regulation of Breast Cancer Cell Adhesion and Transmigration via Lung Microvascular Endothelial Cells. Cancers 2020, 12, 2850. [Google Scholar] [CrossRef]
- Yang, S.; (Charles R. Drew University of Medicine and Science, Los Angeles, CA 90059, USA); Dutta, P.; (Charles R. Drew University of Medicine and Science, Los Angeles, CA 90059, USA); Wu, Y.; (Charles R. Drew University of Medicine and Science, Los Angeles, CA 90059, USA); Wu, Y.; (Charles R. Drew University of Medicine and Science, Los Angeles, CA 90059, USA); Vadgama, J.V.; (Charles R. Drew University of Medicine and Science, Los Angeles, CA 90059, USA). Unpublished work, 2022.
- Hashida, T.; Kotake, Y.; Ohta, S. Protein disulfide isomerase knockdown-induced cell death is cell-line-dependent and involves apoptosis in MCF-7 cells. J. Toxicol. Sci. 2011, 36, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Yamada, R.; Cao, X.; Butkevich, A.N.; Millard, M.; Odde, S.; Mordwinkin, N.; Gundla, R.; Zandi, E.; Louie, S.G.; Petasis, N.A.; et al. Discovery and preclinical evaluation of a novel class of cytotoxic propynoic acid carbamoyl methyl amides (PACMAs). J. Med. Chem. 2011, 54, 2902–2914. [Google Scholar] [CrossRef]
- Fu, X.; Wang, P.; Zhu, B.T. Protein disulfide isomerase is a multifunctional regulator of estrogenic status in target cells. J. Steroid Biochem. Mol. Biol. 2008, 112, 127–137. [Google Scholar] [CrossRef]
- Torpe, N.; Gopal, S.; Baltaci, O.; Rella, L.; Handley, A.; Korswagen, H.C.; Pocock, R. A Protein Disulfide Isomerase Controls Neuronal Migration through Regulation of Wnt Secretion. Cell Rep. 2019, 26, 3183–3190.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Zhang, M.; Xu, F.; Jiang, S. Wnt signaling in breast cancer: Biological mechanisms, challenges and opportunities. Mol. Cancer 2020, 19, 165. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Leftheris, K. Insights into Protein-Ligand Interactions in Integrin Complexes: Advances in Structure Determinations. J. Med. Chem. 2020, 63, 5675–5696. [Google Scholar] [CrossRef] [PubMed]
- Popielarski, M.; Ponamarczuk, H.; Stasiak, M.; Watala, C.; Swiatkowska, M. Modifications of disulfide bonds in breast cancer cell migration and invasiveness. Am. J. Cancer Res. 2019, 9, 1554–1582. [Google Scholar] [PubMed]
- Romagnoli, M.; Mineva, N.D.; Polmear, M.; Conrad, C.; Srinivasan, S.; Loussouarn, D.; Barille-Nion, S.; Georgakoudi, I.; Dagg, A.; McDermott, E.W.; et al. ADAM8 expression in invasive breast cancer promotes tumor dissemination and metastasis. EMBO Mol. Med. 2014, 6, 278–294. [Google Scholar] [CrossRef]
- Willems, S.H.; Tape, C.J.; Stanley, P.L.; Taylor, N.A.; Mills, I.G.; Neal, D.E.; McCafferty, J.; Murphy, G. Thiol isomerases negatively regulate the cellular shedding activity of ADAM17. Biochem. J. 2010, 428, 439–450. [Google Scholar] [CrossRef] [Green Version]
- Mullooly, M.; McGowan, P.M.; Kennedy, S.A.; Madden, S.F.; Crown, J.; O’Donovan, N.; Duffy, M.J. ADAM10: A new player in breast cancer progression? Br. J. Cancer 2015, 113, 945–951. [Google Scholar] [CrossRef] [Green Version]
- Chabottaux, V.; Sounni, N.E.; Pennington, C.J.; English, W.R.; van den Brule, F.; Blacher, S.; Gilles, C.; Munaut, C.; Maquoi, E.; Lopez-Otin, C.; et al. Membrane-type 4 matrix metalloproteinase promotes breast cancer growth and metastases. Cancer Res. 2006, 66, 5165–5172. [Google Scholar] [CrossRef] [Green Version]
- Lorenzen, I.; Eble, J.A.; Hanschmann, E.M. Thiol switches in membrane proteins—Extracellular redox regulation in cell biology. Biol. Chem. 2021, 402, 253–269. [Google Scholar] [CrossRef]
- Liu, D.; Rudland, P.S.; Sibson, D.R.; Platt-Higgins, A.; Barraclough, R. Human homologue of cement gland protein, a novel metastasis inducer associated with breast carcinomas. Cancer Res. 2005, 65, 3796–3805. [Google Scholar] [CrossRef] [Green Version]
- Obacz, J.; Sommerova, L.; Sicari, D.; Durech, M.; Avril, T.; Iuliano, F.; Pastorekova, S.; Hrstka, R.; Chevet, E.; Delom, F.; et al. Extracellular AGR3 regulates breast cancer cells migration via Src signaling. Oncol. Lett. 2019, 18, 4449–4456. [Google Scholar] [CrossRef] [PubMed]
- Young, H.S.; McGowan, L.M.; Jepson, K.A.; Adams, J.C. Impairment of cell adhesion and migration by inhibition of protein disulphide isomerases in three breast cancer cell lines. Biosci. Rep. 2020, 40, BSR20193271. [Google Scholar] [CrossRef]
- Chen, I.H.; Chang, F.R.; Wu, Y.C.; Kung, P.H.; Wu, C.C. 3,4-Methylenedioxy-beta-nitrostyrene inhibits adhesion and migration of human triple-negative breast cancer cells by suppressing beta1 integrin function and surface protein disulfide isomerase. Biochimie 2015, 110, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, H.; Han, B.; Zhang, J.T. Identification of 14-3-3sigma as a contributor to drug resistance in human breast cancer cells using functional proteomic analysis. Cancer Res. 2006, 66, 3248–3255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, M.; Kajino, K.; Abe, M.; Fujimura, T.; Mineki, R.; Ikegami, T.; Ishikawa, T.; Hino, O. NP-1250, an ABCG2 inhibitor, induces apoptotic cell death in mitoxantrone-resistant breast carcinoma MCF7 cells via a caspase-independent pathway. Oncol. Rep. 2013, 29, 1492–1500. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Putti, T.C. Over-expression of ERp29 attenuates doxorubicin-induced cell apoptosis through up-regulation of Hsp27 in breast cancer cells. Exp. Cell Res. 2010, 316, 3522–3531. [Google Scholar] [CrossRef]
- Hrstka, R.; Nenutil, R.; Fourtouna, A.; Maslon, M.M.; Naughton, C.; Langdon, S.; Murray, E.; Larionov, A.; Petrakova, K.; Muller, P.; et al. The pro-metastatic protein anterior gradient-2 predicts poor prognosis in tamoxifen-treated breast cancers. Oncogene 2010, 29, 4838–4847. [Google Scholar] [CrossRef]
- Kutomi, G.; Tamura, Y.; Tanaka, T.; Kajiwara, T.; Kukita, K.; Ohmura, T.; Shima, H.; Takamaru, T.; Satomi, F.; Suzuki, Y.; et al. Human endoplasmic reticulum oxidoreductin 1-alpha is a novel predictor for poor prognosis of breast cancer. Cancer Sci. 2013, 104, 1091–1096. [Google Scholar] [CrossRef]
- Schindl, M.; Schoppmann, S.F.; Samonigg, H.; Hausmaninger, H.; Kwasny, W.; Gnant, M.; Jakesz, R.; Kubista, E.; Birner, P.; Oberhuber, G.; et al. Overexpression of hypoxia-inducible factor 1alpha is associated with an unfavorable prognosis in lymph node-positive breast cancer. Clin. Cancer Res. 2002, 8, 1831–1837. [Google Scholar]
- Jarman, E.J.; Ward, C.; Turnbull, A.K.; Martinez-Perez, C.; Meehan, J.; Xintaropoulou, C.; Sims, A.H.; Langdon, S.P. HER2 regulates HIF-2alpha and drives an increased hypoxic response in breast cancer. Breast Cancer Res. 2019, 21, 10. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Oguro, A.; Hirata, Y.; Imaoka, S. The regulation of Hypoxia-Inducible Factor-1 (HIF-1alpha) expression by Protein Disulfide Isomerase (PDI). PLoS ONE 2021, 16, e0246531. [Google Scholar] [CrossRef] [PubMed]
- Schultz-Norton, J.R.; McDonald, W.H.; Yates, J.R.; Nardulli, A.M. Protein disulfide isomerase serves as a molecular chaperone to maintain estrogen receptor alpha structure and function. Mol. Endocrinol. 2006, 20, 1982–1995. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, T.; Watanabe, Y.; Waga, I. Protein disulfide isomerase suppresses the transcriptional activity of NF-kappaB. Biochem. Biophys. Res. Commun. 2004, 318, 46–52. [Google Scholar] [CrossRef]
- Coppari, S.; Altieri, F.; Ferraro, A.; Chichiarelli, S.; Eufemi, M.; Turano, C. Nuclear localization and DNA interaction of protein disulfide isomerase ERp57 in mammalian cells. J. Cell. Biochem. 2002, 85, 325–333. [Google Scholar] [CrossRef]
- Gaucci, E.; Altieri, F.; Turano, C.; Chichiarelli, S. The protein ERp57 contributes to EGF receptor signaling and internalization in MDA-MB-468 breast cancer cells. J. Cell. Biochem. 2013, 114, 2461–2470. [Google Scholar] [CrossRef] [PubMed]
- Bakker, E.Y.; Fujii, M.; Krstic-Demonacos, M.; Demonacos, C.; Alhammad, R. Protein disulfide isomerase A1associated pathways in the development of stratified breast cancer therapies. Int. J. Oncol. 2022, 60, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Pokkunuri, I.D.; Lokhandwala, M.F.; Banday, A.A. Protein disulfide isomerase inhibition impairs Keap1/Nrf2 signaling and mitochondrial function and induces apoptosis in renal proximal tubular cells. Am. J. Physiol. Ren. Physiol. 2020, 319, F686–F696. [Google Scholar] [CrossRef] [PubMed]
- Alhammad, R.; Khunchai, S.; Tongmuang, N.; Limjindaporn, T.; Yenchitsomanus, P.T.; Mutti, L.; Krstic-Demonacos, M.; Demonacos, C. Protein disulfide isomerase A1 regulates breast cancer cell immunorecognition in a manner dependent on redox state. Oncol. Rep. 2020, 44, 2406–2418. [Google Scholar] [CrossRef]
- Tanaka, T.; Kutomi, G.; Kajiwara, T.; Kukita, K.; Kochin, V.; Kanaseki, T.; Tsukahara, T.; Hirohashi, Y.; Torigoe, T.; Okamoto, Y.; et al. Cancer-associated oxidoreductase ERO1-alpha promotes immune escape through up-regulation of PD-L1 in human breast cancer. Oncotarget 2017, 8, 24706–24718. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, T.; Kajiwara, T.; Torigoe, T.; Okamoto, Y.; Sato, N.; Tamura, Y. Cancer-associated oxidoreductase ERO1-alpha drives the production of tumor-promoting myeloid-derived suppressor cells via oxidative protein folding. J. Immunol. 2015, 194, 2004–2010. [Google Scholar] [CrossRef] [Green Version]
- Park, B.; Lee, S.; Kim, E.; Cho, K.; Riddell, S.R.; Cho, S.; Ahn, K. Redox regulation facilitates optimal peptide selection by MHC class I during antigen processing. Cell 2006, 127, 369–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, S.; Butkevich, A.N.; Yamada, R.; Zhou, Y.; Debnath, B.; Duncan, R.; Zandi, E.; Petasis, N.A.; Neamati, N. Discovery of an orally active small-molecule irreversible inhibitor of protein disulfide isomerase for ovarian cancer treatment. Proc. Natl. Acad. Sci. USA 2012, 109, 16348–16353. [Google Scholar] [CrossRef] [Green Version]
- Ge, J.; Zhang, C.J.; Li, L.; Chong, L.M.; Wu, X.; Hao, P.; Sze, S.K.; Yao, S.Q. Small molecule probe suitable for in situ profiling and inhibition of protein disulfide isomerase. ACS Chem. Biol. 2013, 8, 2577–2585. [Google Scholar] [CrossRef] [PubMed]
- Hoffstrom, B.G.; Kaplan, A.; Letso, R.; Schmid, R.S.; Turmel, G.J.; Lo, D.C.; Stockwell, B.R. Inhibitors of protein disulfide isomerase suppress apoptosis induced by misfolded proteins. Nat. Chem. Biol. 2010, 6, 900–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shergalis, A.; Xue, D.; Gharbia, F.Z.; Driks, H.; Shrestha, B.; Tanweer, A.; Cromer, K.; Ljungman, M.; Neamati, N. Characterization of Aminobenzylphenols as Protein Disulfide Isomerase Inhibitors in Glioblastoma Cell Lines. J. Med. Chem. 2020, 63, 10263–10286. [Google Scholar] [CrossRef]
- Vatolin, S.; Phillips, J.G.; Jha, B.K.; Govindgari, S.; Hu, J.; Grabowski, D.; Parker, Y.; Lindner, D.J.; Zhong, F.; Distelhorst, C.W.; et al. Novel Protein Disulfide Isomerase Inhibitor with Anticancer Activity in Multiple Myeloma. Cancer Res. 2016, 76, 3340–3350. [Google Scholar] [CrossRef] [Green Version]
- Allimuthu, D.; Adams, D.J. 2-Chloropropionamide as a Low-Reactivity Electrophile for Irreversible Small-Molecule Probe Identification. ACS Chem. Biol. 2017, 12, 2124–2131. [Google Scholar] [CrossRef]
- Ozcelik, D.; Pezacki, J.P. Small Molecule Inhibition of Protein Disulfide Isomerase in Neuroblastoma Cells Induces an Oxidative Stress Response and Apoptosis Pathways. ACS Chem. Neurosci. 2019, 10, 4068–4075. [Google Scholar] [CrossRef]
- Law, M.E.; Yaaghubi, E.; Ghilardi, A.F.; Davis, B.J.; Ferreira, R.B.; Koh, J.; Chen, S.; DePeter, S.F.; Schilson, C.M.; Chiang, C.-W.; et al. Inhibitors of ERp44, PDIA1, and AGR2 induce disulfide-mediated oligomerization of Death Receptors 4 and 5 and cancer cell death. bioRxiv 2021. [Google Scholar] [CrossRef]
- Yin, H.Y.; Gao, J.J.; Chen, X.; Ma, B.; Yang, Z.S.; Tang, J.; Wang, B.W.; Chen, T.; Wang, C.; Gao, S.; et al. A Gallium(III) Complex that Engages Protein Disulfide Isomerase A3 (PDIA3) as an Anticancer Target. Angew. Chem. Int. Ed. Engl. 2020, 59, 20147–20153. [Google Scholar] [CrossRef]
- Kyani, A.; Tamura, S.; Yang, S.; Shergalis, A.; Samanta, S.; Kuang, Y.; Ljungman, M.; Neamati, N. Discovery and Mechanistic Elucidation of a Class of Protein Disulfide Isomerase Inhibitors for the Treatment of Glioblastoma. ChemMedChem 2018, 13, 164–177. [Google Scholar] [CrossRef]
- Carcelli, M.; Tegoni, M.; Bartoli, J.; Marzano, C.; Pelosi, G.; Salvalaio, M.; Rogolino, D.; Gandin, V. In vitro and in vivo anticancer activity of tridentate thiosemicarbazone copper complexes: Unravelling an unexplored pharmacological target. Eur. J. Med. Chem. 2020, 194, 112266. [Google Scholar] [CrossRef] [PubMed]
- Chlebowska-Tuz, J.; Sokolowska, O.; Gaj, P.; Lazniewski, M.; Firczuk, M.; Borowiec, K.; Sas-Nowosielska, H.; Bajor, M.; Malinowska, A.; Muchowicz, A.; et al. Inhibition of protein disulfide isomerase induces differentiation of acute myeloid leukemia cells. Haematologica 2018, 103, 1843–1852. [Google Scholar] [CrossRef] [PubMed]
- Won, J.K.; Yu, S.J.; Hwang, C.Y.; Cho, S.H.; Park, S.M.; Kim, K.; Choi, W.M.; Cho, H.; Cho, E.J.; Lee, J.H.; et al. Protein disulfide isomerase inhibition synergistically enhances the efficacy of sorafenib for hepatocellular carcinoma. Hepatology 2017, 66, 855–868. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.; Liu, Y.; Yang, K.; Wang, H.; Shergalis, A.; Kyani, A.; Bankhead, A., III; Tamura, S.; Yang, S.; Wang, X.; et al. Inhibition of protein disulfide isomerase in glioblastoma causes marked downregulation of DNA repair and DNA damage response genes. Theranostics 2019, 9, 2282–2298. [Google Scholar] [CrossRef] [PubMed]
- Eirich, J.; Braig, S.; Schyschka, L.; Servatius, P.; Hoffmann, J.; Hecht, S.; Fulda, S.; Zahler, S.; Antes, I.; Kazmaier, U.; et al. A small molecule inhibits protein disulfide isomerase and triggers the chemosensitization of cancer cells. Angew. Chem. Int. Ed. Engl. 2014, 53, 12960–12965. [Google Scholar] [CrossRef]
- Kaplan, A.; Gaschler, M.M.; Dunn, D.E.; Colligan, R.; Brown, L.M.; Palmer, A.G., III; Lo, D.C.; Stockwell, B.R. Small molecule-induced oxidation of protein disulfide isomerase is neuroprotective. Proc. Natl. Acad. Sci. USA 2015, 112, E2245–E2252. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Li, G.; Kaplan, A.; Gaschler, M.M.; Zhang, X.; Hou, Z.; Jiang, M.; Zott, R.; Cremers, S.; Stockwell, B.R.; et al. Small molecule modulator of protein disulfide isomerase attenuates mutant huntingtin toxicity and inhibits endoplasmic reticulum stress in a mouse model of Huntington’s disease. Hum. Mol. Genet. 2018, 27, 1545–1555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaplan, A.; Stockwell, B.R. Structural Elucidation of a Small Molecule Inhibitor of Protein Disulfide Isomerase. ACS Med. Chem. Lett. 2015, 6, 966–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasipek, M.; Grabowski, D.; Guan, Y.; Alugubelli, R.R.; Tiwari, A.D.; Gu, X.; DeAvila, G.A.; Silva, A.S.; Meads, M.B.; Parker, Y.; et al. Therapeutic Targeting of Protein Disulfide Isomerase PDIA1 in Multiple Myeloma. Cancers 2021, 13, 2649. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, R.; Pace, N.J.; Brown, D.R.; Weerapana, E. 1,3,5-Triazine as a modular scaffold for covalent inhibitors with streamlined target identification. J. Am. Chem. Soc. 2013, 135, 2497–2500. [Google Scholar] [CrossRef] [PubMed]
- Cole, K.S.; Grandjean, J.M.D.; Chen, K.; Witt, C.H.; O’Day, J.; Shoulders, M.D.; Wiseman, R.L.; Weerapana, E. Characterization of an A-Site Selective Protein Disulfide Isomerase A1 Inhibitor. Biochemistry 2018, 57, 2035–2043. [Google Scholar] [CrossRef] [PubMed]
- Dickerhof, N.; Kleffmann, T.; Jack, R.; McCormick, S. Bacitracin inhibits the reductive activity of protein disulfide isomerase by disulfide bond formation with free cysteines in the substrate-binding domain. FEBS J. 2011, 278, 2034–2043. [Google Scholar] [CrossRef]
- Karala, A.R.; Ruddock, L.W. Bacitracin is not a specific inhibitor of protein disulfide isomerase. FEBS J. 2010, 277, 2454–2462. [Google Scholar] [CrossRef]
- Lovat, P.E.; Corazzari, M.; Armstrong, J.L.; Martin, S.; Pagliarini, V.; Hill, D.; Brown, A.M.; Piacentini, M.; Birch-Machin, M.A.; Redfern, C.P. Increasing melanoma cell death using inhibitors of protein disulfide isomerases to abrogate survival responses to endoplasmic reticulum stress. Cancer Res. 2008, 68, 5363–5369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goplen, D.; Wang, J.; Enger, P.O.; Tysnes, B.B.; Terzis, A.J.; Laerum, O.D.; Bjerkvig, R. Protein disulfide isomerase expression is related to the invasive properties of malignant glioma. Cancer Res. 2006, 66, 9895–9902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bekendam, R.H.; Bendapudi, P.K.; Lin, L.; Nag, P.P.; Pu, J.; Kennedy, D.R.; Feldenzer, A.; Chiu, J.; Cook, K.M.; Furie, B.; et al. A substrate-driven allosteric switch that enhances PDI catalytic activity. Nat. Commun. 2016, 7, 12579. [Google Scholar] [CrossRef] [PubMed]
- Jasuja, R.; Passam, F.H.; Kennedy, D.R.; Kim, S.H.; van Hessem, L.; Lin, L.; Bowley, S.R.; Joshi, S.S.; Dilks, J.R.; Furie, B.; et al. Protein disulfide isomerase inhibitors constitute a new class of antithrombotic agents. J. Clin. Investig. 2012, 122, 2104–2113. [Google Scholar] [CrossRef]
- Lin, L.; Gopal, S.; Sharda, A.; Passam, F.; Bowley, S.R.; Stopa, J.; Xue, G.; Yuan, C.; Furie, B.C.; Flaumenhaft, R.; et al. Quercetin-3-rutinoside Inhibits Protein Disulfide Isomerase by Binding to Its b’x Domain. J. Biol. Chem. 2015, 290, 23543–23552. [Google Scholar] [CrossRef] [Green Version]
- Zwicker, J.I.; Schlechter, B.L.; Stopa, J.D.; Liebman, H.A.; Aggarwal, A.; Puligandla, M.; Caughey, T.; Bauer, K.A.; Kuemmerle, N.; Wong, E.; et al. Targeting protein disulfide isomerase with the flavonoid isoquercetin to improve hypercoagulability in advanced cancer. JCI Insight 2019, 4, e125851. [Google Scholar] [CrossRef] [Green Version]
- Khodier, C.; VerPlank, L.; Nag, P.P.; Pu, J.; Wurst, J.; Pilyugina, T.; Dockendorff, C.; Galinski, C.N.; Scalise, A.A.; Passam, F.; et al. Identification of ML359 as a Small Molecule Inhibitor of Protein Disulfide Isomerase. In Probe Reports from the NIH Molecular Libraries Program; National Center for Biotechnology Information (US): Bethesda, MD, USA, 2010. [Google Scholar]
- Robinson, R.M.; Reyes, L.; Duncan, R.M.; Bian, H.; Reitz, A.B.; Manevich, Y.; McClure, J.J.; Champion, M.M.; Chou, C.J.; Sharik, M.E.; et al. Inhibitors of the protein disulfide isomerase family for the treatment of multiple myeloma. Leukemia 2019, 33, 1011–1022. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.Y.; Wu, Y.C.; Wu, C.C. Prevention of platelet glycoprotein IIb/IIIa activation by 3,4-methylenedioxy-beta-nitrostyrene, a novel tyrosine kinase inhibitor. Mol. Pharmacol. 2006, 70, 1380–1389. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, P.W.; Chang, Y.T.; Chuang, W.Y.; Shih, H.C.; Chiang, S.Z.; Wu, C.C. The synthesis and biologic evaluation of anti-platelet and cytotoxic beta-nitrostyrenes. Bioorganic Med. Chem. 2010, 18, 7621–7627. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Chang, K.O. Protein disulfide isomerases as potential therapeutic targets for influenza A and B viruses. Virus Res. 2018, 247, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.M.; Simizu, S.; Lai, N.S.; Kawatani, M.; Shimizu, T.; Osada, H. Discovery of a small molecule PDI inhibitor that inhibits reduction of HIV-1 envelope glycoprotein gp120. ACS Chem. Biol. 2011, 6, 245–251. [Google Scholar] [CrossRef]
- Chitambar, C.R.; Al-Gizawiy, M.M.; Alhajala, H.S.; Pechman, K.R.; Wereley, J.P.; Wujek, R.; Clark, P.A.; Kuo, J.S.; Antholine, W.E.; Schmainda, K.M. Gallium Maltolate Disrupts Tumor Iron Metabolism and Retards the Growth of Glioblastoma by Inhibiting Mitochondrial Function and Ribonucleotide Reductase. Mol. Cancer Ther. 2018, 17, 1240–1250. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.W.; Ryu, H.H.; Li, S.Y.; Li, C.H.; Lim, S.H.; Jang, W.Y.; Jung, S. PDIA6 regulation of ADAM17 shedding activity and EGFR-mediated migration and invasion of glioblastoma cells. J. Neurosurg. 2017, 126, 1829–1838. [Google Scholar] [CrossRef] [Green Version]
- Kaufman, R.J.; Malhotra, J.D. Calcium trafficking integrates endoplasmic reticulum function with mitochondrial bioenergetics. Biochim. Biophys. Acta 2014, 1843, 2233–2239. [Google Scholar] [CrossRef] [Green Version]





| PDI Inhibitor | Findings in Breast Cancer Research (Cell-Based and Pre-Clinical Studies) | Mechanism of Action of PDI Inhibitors (Proposed Pathways) |
|---|---|---|
| PACMA31 |
|
|
| P1 |
|
|
| 16F16 |
|
|
| DDAs |
|
|
| Ga-1 |
|
|
| T8 |
|
|
| MNS |
|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, S.; Jackson, C.; Karapetyan, E.; Dutta, P.; Kermah, D.; Wu, Y.; Wu, Y.; Schloss, J.; Vadgama, J.V. Roles of Protein Disulfide Isomerase in Breast Cancer. Cancers 2022, 14, 745. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14030745
Yang S, Jackson C, Karapetyan E, Dutta P, Kermah D, Wu Y, Wu Y, Schloss J, Vadgama JV. Roles of Protein Disulfide Isomerase in Breast Cancer. Cancers. 2022; 14(3):745. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14030745
Chicago/Turabian StyleYang, Suhui, Chanel Jackson, Eduard Karapetyan, Pranabananda Dutta, Dulcie Kermah, Yong Wu, Yanyuan Wu, John Schloss, and Jaydutt V. Vadgama. 2022. "Roles of Protein Disulfide Isomerase in Breast Cancer" Cancers 14, no. 3: 745. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14030745