
Disparities in Lung Cancer: miRNA Isoform Characterization in Lung Adenocarcinoma

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
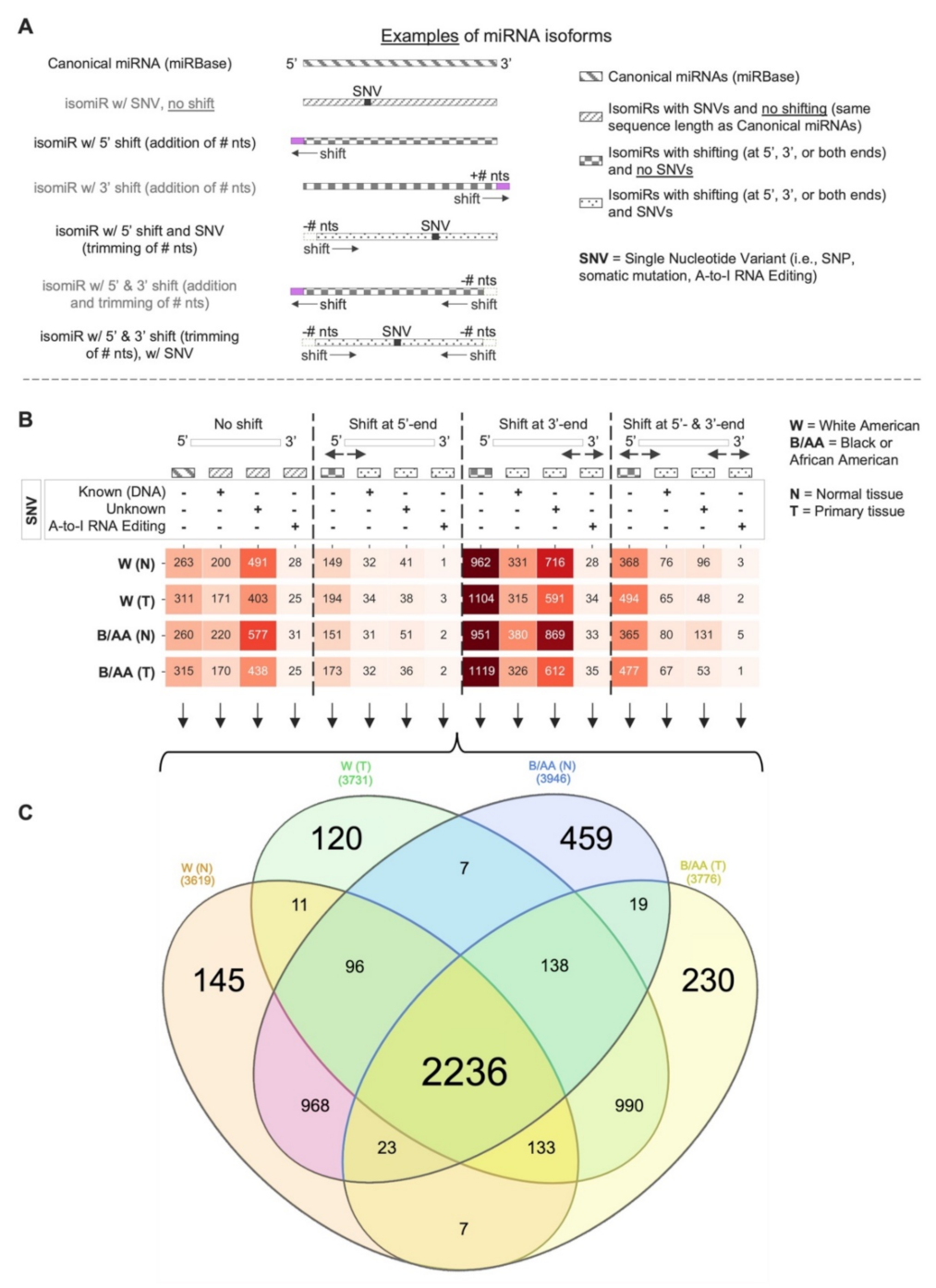
2.1. Systematic miRNA Isoform Characterization in Black or African and White American Lung Adenocarcinoma Patients
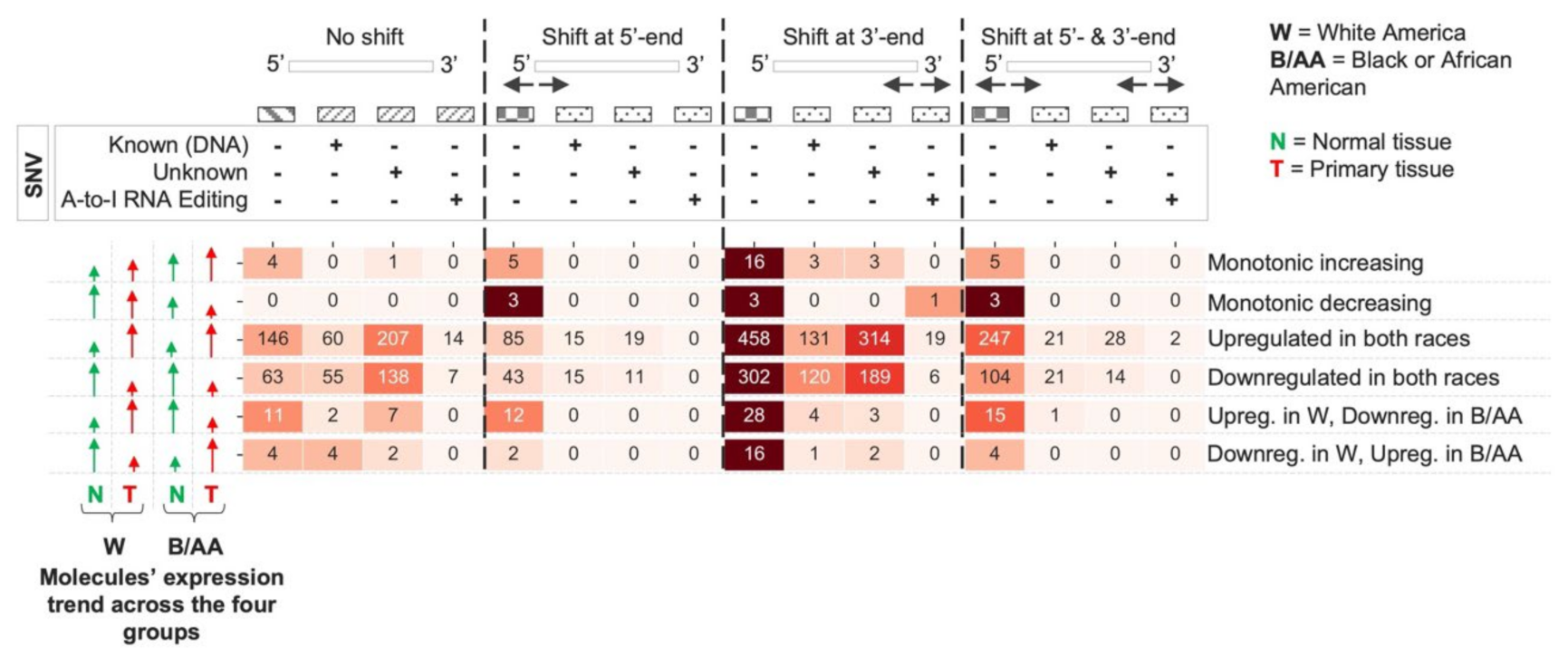
2.2. miRNA Isoform Dysregulation in Lung Adenocarcinoma Race Disparities
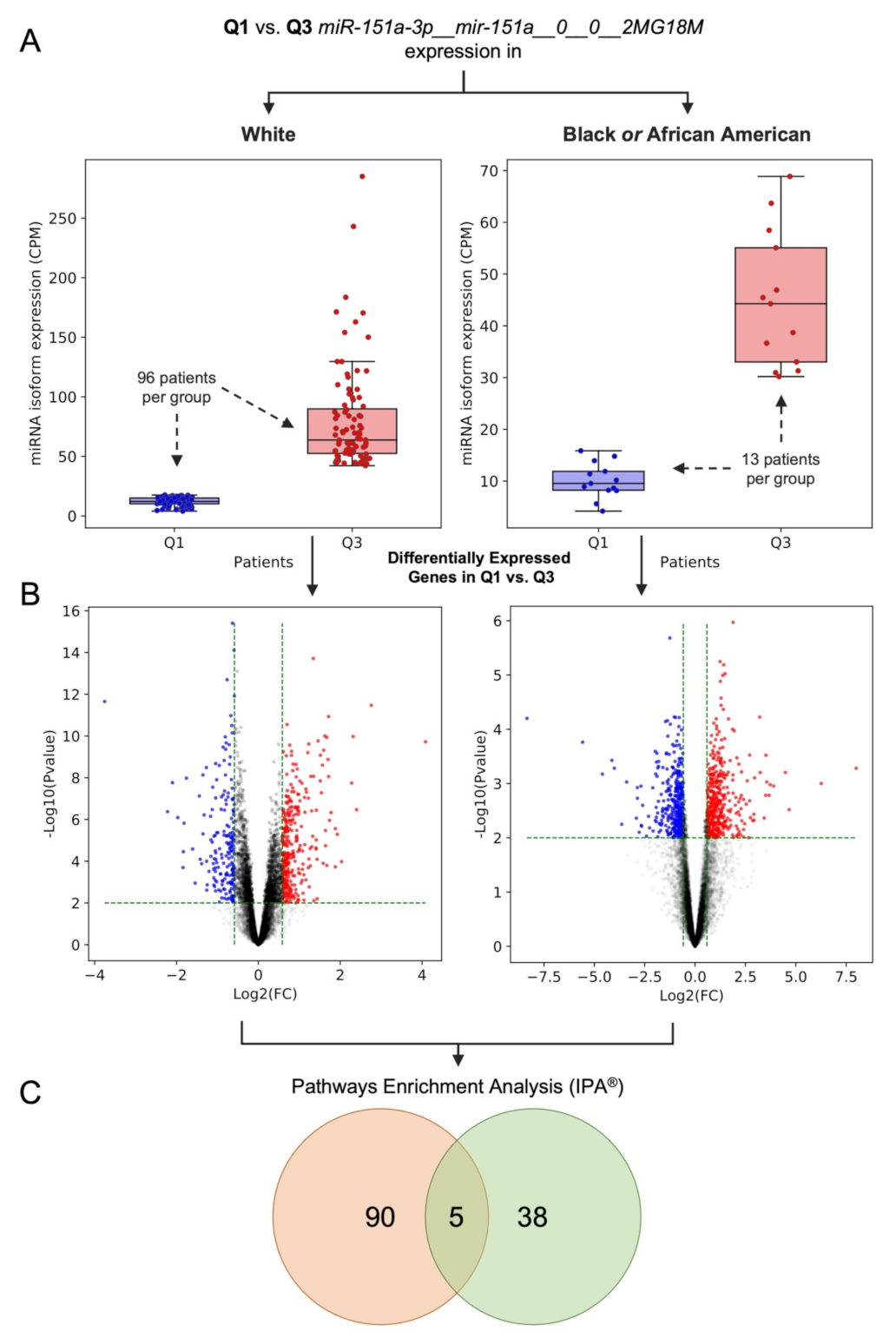
2.3. Dysregulation of Edited miR-151a-3p in Black or African and White American Lung Adenocarcinoma Patients: A Case of Study
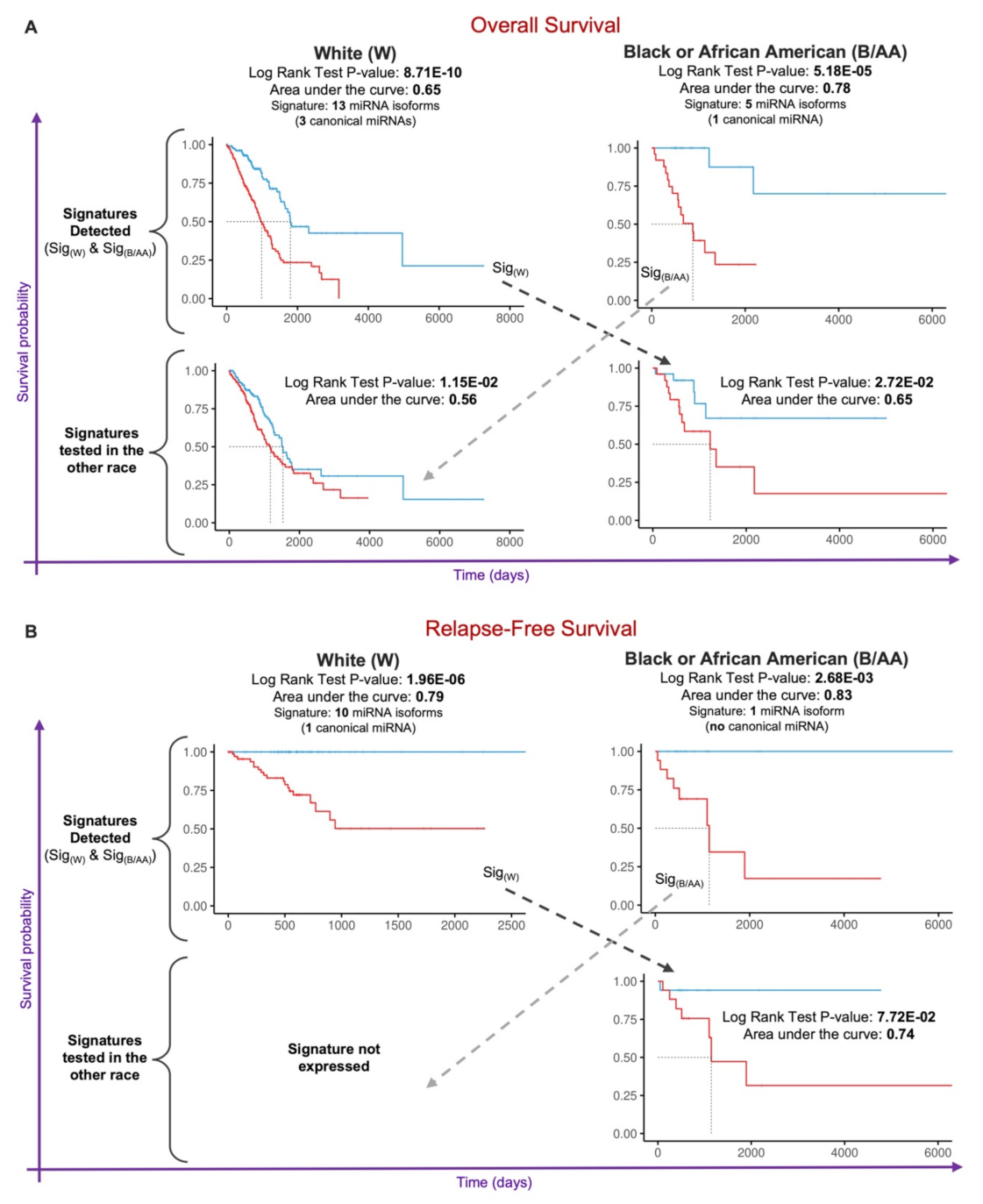
2.4. Prognostic miRNA Isoform Signature in Lung Adenocarcinoma Race Disparities
3. Discussion
4. Materials and Methods
4.1. Data Acquisition and Preparation
4.2. MiRNA Isoforms Mapping, Annotation, and Filtering
4.3. miRNA Isoform Quantification and Multivariate Analysis in LUAD Race Disparities
4.4. Differential Gene Expression and Functional Enrichment Analyses
4.5. Prognostic Signature Identification
4.6. Statistical Analyses and Graphs
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33, Erratum in A Cancer J. Clin. 2021, 71, 359. [Google Scholar] [CrossRef] [PubMed]
- Rivera, M.P.; Katki, H.A.; Tanner, N.T.; Triplette, M.; Sakoda, L.C.; Wiener, R.S.; Cardarelli, R.; Carter-Harris, L.; Crothers, K.; Fathi, J.T.; et al. Addressing Disparities in Lung Cancer Screening Eligibility and Healthcare Access. An Official American Thoracic Society Statement. Am. J. Respir. Crit. Care Med. 2020, 202, e95–e112. [Google Scholar] [CrossRef] [PubMed]
- Thomas, P.L.; Madubata, C.J.; Aldrich, M.C.; Lee, M.M.; Owonikoko, T.K.; Minna, J.D.; Rudin, C.M.; Sage, J.; Lovly, C.M. A Call to Action: Dismantling Racial Injustices in Preclinical Research and Clinical Care of Black Patients Living with Small Cell Lung Cancer. Cancer Discov. 2020, 11, 240–244. [Google Scholar] [CrossRef] [PubMed]
- Tanner, N.T.; Gebregziabher, M.; Halbert, C.H.; Payne, E.; Egede, L.E.; Silvestri, G.A. Racial Differences in Outcomes within the National Lung Screening Trial. Implications for Widespread Implementation. Am. J. Respir. Crit. Care Med. 2015, 192, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Ezer, N.; Mhango, G.; Bagiella, E.; Goodman, E.; Flores, R.; Wisnivesky, J.P. Racial Disparities in Resection of Early Stage Non–Small Cell Lung Cancer. Med. Care 2019, 58, 392–398. [Google Scholar] [CrossRef] [PubMed]
- Kitts, A.K.B. The Patient Perspective on Lung Cancer Screening and Health Disparities. J. Am. Coll. Radiol. 2019, 16, 601–606. [Google Scholar] [CrossRef] [PubMed]
- Hastert, T.A.; McDougall, J.A.; Strayhorn, S.M.; Nair, M.; Beebe-Dimmer, J.L.; Schwartz, A.G. Social needs and health-related quality of life among African American cancer survivors: Results from the Detroit Research on Cancer Survivors study. Cancer 2020, 127, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Ryan, B.M. Lung cancer health disparities. Carcinogenesis 2018, 39, 741–751. [Google Scholar] [CrossRef]
- National Lung Screening Trial Research Team; Aberle, D.R.; Adams, A.M.; Berg, C.D.; Black, W.C.; Clapp, J.D.; Fagerstrom, R.M.; Gareen, I.F.; Gatsonis, C.; Marcus, P.M.; et al. Reduced Lung-Cancer Mortality with Low-Dose Computed Tomographic Screening. N. Engl. J. Med. 2011, 365, 395–409. [Google Scholar] [CrossRef] [Green Version]
- Basu, A.; Kopulos, L.; Geissen, N.; Sukhal, S.; Smith, S.B. Analysis of the 30-Pack-Year Smoking Threshold in African Americans From an Underserved Lung Cancer Screening Program. J. Am. Coll. Radiol. 2020, 18, 27–33. [Google Scholar] [CrossRef]
- Haiman, C.A.; Stram, D.O.; Wilkens, L.R.; Pike, M.C.; Kolonel, L.N.; Henderson, B.E.; Le Marchand, L. Ethnic and Racial Differences in the Smoking-Related Risk of Lung Cancer. N. Engl. J. Med. 2006, 354, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Pu, C.Y.; Lusk, C.M.; Neslund-Dudas, C.; Gadgeel, S.; Soubani, A.O.; Schwartz, A.G. Comparison Between the 2021 USPSTF Lung Cancer Screening Criteria and Other Lung Cancer Screening Criteria for Racial Disparity in Eligibility. JAMA Oncol. 2022, e216720. [Google Scholar] [CrossRef] [PubMed]
- Araujo, L.H.; Lammers, P.E.; Matthews-Smith, V.; Eisenberg, R.; Gonzalez, A.; Schwartz, A.G.; Timmers, C.; Shilo, K.; Zhao, W.; Natarajan, T.G.; et al. Somatic Mutation Spectrum of Non–Small-Cell Lung Cancer in African Americans: A Pooled Analysis. J. Thorac. Oncol. 2015, 10, 1430–1436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araujo, L.H.; Timmers, C.; Bell, E.H.; Shilo, K.; Lammers, P.E.; Zhao, W.; Natarajan, T.G.; Miller, C.J.; Zhang, J.; Yilmaz, A.S.; et al. Genomic Characterization of Non–Small-Cell Lung Cancer in African Americans by Targeted Massively Parallel Sequencing. J. Clin. Oncol. 2015, 33, 1966–1973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choudhury, N.J.; Eghtesad, M.; Kadri, S.; Cursio, J.; Ritterhouse, L.; Segal, J.; Husain, A.; Patel, J.D. Fewer actionable mutations but higher tumor mutational burden characterizes NSCLC in black patients at an urban academic medical center. Oncotarget 2019, 10, 5817–5823. [Google Scholar] [CrossRef] [PubMed]
- Lusk, C.M.; Watza, D.; Dyson, G.; Craig, D.B.; Ratliff, V.; Wenzlaff, A.S.; Lonardo, F.; Bollig-Fischer, A.; Bepler, G.; Purrington, K.S.; et al. Profiling the Mutational Landscape in Known Driver Genes and Novel Genes in African American Non–Small Cell Lung Cancer Patients. Clin. Cancer Res. 2019, 25, 4300–4308. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, K.A.; Zingone, A.; Toulabi, L.; Boeckelman, J.; Ryan, B.M. Comparative Transcriptome Profiling Reveals Coding and Noncoding RNA Differences in NSCLC from African Americans and European Americans. Clin. Cancer Res. 2017, 23, 7412–7425. [Google Scholar] [CrossRef] [Green Version]
- Telonis, A.G.; Rigoutsos, I. Race Disparities in the Contribution of miRNA Isoforms and tRNA-Derived Fragments to Triple-Negative Breast Cancer. Cancer Res. 2017, 78, 1140–1154. [Google Scholar] [CrossRef] [Green Version]
- Lee, L.W.; Zhang, S.; Etheridge, A.; Ma, L.; Martin, D.; Galas, D.; Wang, K. Complexity of the microRNA repertoire revealed by next-generation sequencing. RNA 2010, 16, 2170–2180. [Google Scholar] [CrossRef] [Green Version]
- Esteller, M.; Pandolfi, P.P. The Epitranscriptome of Noncoding RNAs in Cancer. Cancer Discov. 2017, 7, 359–368. [Google Scholar] [CrossRef] [Green Version]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling, H.; Fabbri, M.; Calin, G.A. MicroRNAs and other non-coding RNAs as targets for anticancer drug development. Nat. Rev. Drug Discov. 2013, 12, 847–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glogovitis, I.; Yahubyan, G.; Würdinger, T.; Koppers-Lalic, D.; Baev, V. IsomiRs–Hidden Soldiers in the miRNA Regulatory Army, and How to Find Them? Biomolecules 2020, 11, 41. [Google Scholar] [CrossRef] [PubMed]
- Nishikura, K. Functions and Regulation of RNA Editing by ADAR Deaminases. Annu. Rev. Biochem. 2010, 79, 321–349. [Google Scholar] [CrossRef] [Green Version]
- Nishikura, K. A-to-I editing of coding and non-coding RNAs by ADARs. Nat. Rev. Mol. Cell Biol. 2016, 17, 83–96. [Google Scholar] [CrossRef] [Green Version]
- Nigita, G.; Marceca, G.P.; Tomasello, L.; Distefano, R.; Calore, F.; Veneziano, D.; Romano, G.; Nana-Sinkam, S.P.; Acunzo, M.; Croce, C.M. ncRNA Editing: Functional Characterization and Computational Resources. Comput. Biol. Non-Coding RNA 2019, 1912, 133–174. [Google Scholar] [CrossRef]
- Kawahara, Y.; Zinshteyn, B.; Sethupathy, P.; Iizasa, H.; Hatzigeorgiou, A.G.; Nishikura, K. Redirection of Silencing Targets by Adenosine-to-Inosine Editing of miRNAs. Science 2007, 315, 1137–1140. [Google Scholar] [CrossRef] [Green Version]
- Nigita, G.; Acunzo, M.; Romano, G.; Veneziano, D.; Laganà, A.; Vitiello, M.; Wernicke, D.; Ferro, A.; Croce, C.M. microRNA editing in seed region aligns with cellular changes in hypoxic conditions. Nucleic Acids Res. 2016, 44, 6298–6308. [Google Scholar] [CrossRef] [Green Version]
- Pinto, Y.; Buchumenski, I.; Levanon, E.Y.; Eisenberg, E. Human cancer tissues exhibit reduced A-to-I editing of miRNAs coupled with elevated editing of their targets. Nucleic Acids Res. 2017, 46, 71–82. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Wang, Y.; Mojumdar, K.; Zhou, Z.; Jeong, K.J.; Mangala, L.S.; Yu, S.; Tsang, Y.H.; Rodriguez-Aguayo, C.; Lu, Y.; et al. A-to-I–edited miRNA-379-5p inhibits cancer cell proliferation through CD97-induced apoptosis. J. Clin. Investig. 2019, 129, 5343–5356. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Xu, X.; Yu, S.; Jeong, K.J.; Zhou, Z.; Han, L.; Tsang, Y.H.; Li, J.; Chen, H.; Mangala, L.S.; et al. Systematic characterization of A-to-I RNA editing hotspots in microRNAs across human cancers. Genome Res. 2017, 27, 1112–1125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baysal, B.E.; Sharma, S.; Hashemikhabir, S.; Janga, S.C. RNA Editing in Pathogenesis of Cancer. Cancer Res. 2017, 77, 3733–3739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.; Baras, A.S.; Halushka, M.K. miRge 2.0 for comprehensive analysis of microRNA sequencing data. BMC Bioinform. 2018, 19, 275. [Google Scholar] [CrossRef] [PubMed]
- Desvignes, T.; Loher, P.; Eilbeck, K.; Ma, J.; Urgese, G.; Fromm, B.; Sydes, J.; Aparicio-Puerta, E.; Barrera, V.; Espín, R.; et al. Unification of miRNA and isomiR research: The mirGFF3 format and the mirtop API. Bioinformatics 2019, 36, 698–703. [Google Scholar] [CrossRef]
- Marceca, G.P.; Distefano, R.; Tomasello, L.; Lagana, A.; Russo, F.; Calore, F.; Romano, G.; Bagnoli, M.; Gasparini, P.; Ferro, A.; et al. MiREDiBase, a manually curated database of validated and putative editing events in microRNAs. Sci. Data 2021, 8, 199. [Google Scholar] [CrossRef]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [Green Version]
- Sherry, S.T.; Ward, M.-H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef] [Green Version]
- Alon, S.; Mor, E.; Vigneault, F.; Church, G.M.; Locatelli, F.; Galeano, F.; Gallo, A.; Shomron, N.; Eisenberg, E. Systematic identification of edited microRNAs in the human brain. Genome Res. 2012, 22, 1533–1540. [Google Scholar] [CrossRef] [Green Version]
- Tomaselli, S.; Galeano, F.; Alon, S.; Raho, S.; Galardi, S.; Polito, V.A.; Presutti, C.; Vincenti, S.; Eisenberg, E.; Locatelli, F.; et al. Modulation of microRNA editing, expression and processing by ADAR2 deaminase in glioblastoma. Genome Biol. 2015, 16, 5. [Google Scholar] [CrossRef]
- Yang, W.; Chendrimada, T.P.; Wang, Q.; Higuchi, M.; Seeburg, P.H.; Shiekhattar, R.; Nishikura, K. Modulation of microRNA processing and expression through RNA editing by ADAR deaminases. Nat. Struct. Mol. Biol. 2006, 13, 13–21. [Google Scholar] [CrossRef] [Green Version]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smeds, L.; Künstner, A. ConDeTri—A Content Dependent Read Trimmer for Illumina Data. PLoS ONE 2011, 6, e26314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fromm, B.; Domanska, D.; Høye, E.; Ovchinnikov, V.; Kang, W.; Aparicio-Puerta, E.; Johansen, M.; Flatmark, K.; Mathelier, A.; Hovig, E.; et al. MirGeneDB 2.0: The metazoan microRNA complement. Nucleic Acids Res. 2019, 48, D132–D141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2013, 42, D68–D73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Hoon, M.J.; Taft, R.J.; Hashimoto, T.; Kanamori-Katayama, M.; Kawaji, H.; Kawano, M.; Kishima, M.; Lassmann, T.; Faulkner, G.J.; Mattick, J.S.; et al. Cross-mapping and the identification of editing sites in mature microRNAs in high-throughput sequencing libraries. Genome Res. 2010, 20, 257–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, M.; Quinton, L.; Blahna, M.T.; Neilson, J.R.; Fu, S.; Ivanov, A.R.; Wolf, D.; Mizgerd, J.P. Zcchc11-dependent uridylation of microRNA directs cytokine expression. Nat. Cell Biol. 2009, 11, 1157–1163. [Google Scholar] [CrossRef]
- Katoh, T.; Sakaguchi, Y.; Miyauchi, K.; Suzuki, T.; Kashiwabara, S.-I.; Baba, T.; Suzuki, T. Selective stabilization of mammalian microRNAs by 3′ adenylation mediated by the cytoplasmic poly(A) polymerase GLD-2. Genes Dev. 2009, 23, 433–438. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2009, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- McCarthy, D.J.; Chen, Y.; Smyth, G.K. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012, 40, 4288–4297. [Google Scholar] [CrossRef] [Green Version]
- Gentleman, R.C.; Carey, V.J.; Bates, D.M.; Bolstad, B.; Dettling, M.; Dudoit, S.; Ellis, B.; Gautier, L.; Ge, Y.; Gentry, J.; et al. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol. 2004, 5, R80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Heberle, H.; Meirelles, G.V.; Da Silva, F.R.; Telles, G.P.; Minghim, R. InteractiVenn: A web-based tool for the analysis of sets through Venn diagrams. BMC Bioinform. 2015, 16, 169. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Distefano, R.; Nigita, G.; Le, P.; Romano, G.; Acunzo, M.; Nana-Sinkam, P. Disparities in Lung Cancer: miRNA Isoform Characterization in Lung Adenocarcinoma. Cancers 2022, 14, 773. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14030773
Distefano R, Nigita G, Le P, Romano G, Acunzo M, Nana-Sinkam P. Disparities in Lung Cancer: miRNA Isoform Characterization in Lung Adenocarcinoma. Cancers. 2022; 14(3):773. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14030773
Chicago/Turabian StyleDistefano, Rosario, Giovanni Nigita, Patricia Le, Giulia Romano, Mario Acunzo, and Patrick Nana-Sinkam. 2022. "Disparities in Lung Cancer: miRNA Isoform Characterization in Lung Adenocarcinoma" Cancers 14, no. 3: 773. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14030773