
Everything Comes with a Price: The Toxicity Profile of DNA-Damage Response Targeting Agents


Abstract
:Simple Summary
Abstract
1. Introduction
2. Safety Profile of DDR-Targeting Agents
2.1. Frequent Adverse Events
2.1.1. Hematologic Toxicities
2.1.2. Gastrointestinal Toxicities
2.1.3. Fatigue
2.1.4. Respiratory Toxicities
2.1.5. Neurological and Cardiovascular Toxicities
2.1.6. Secondary Malignancies
2.1.7. Laboratory Alterations
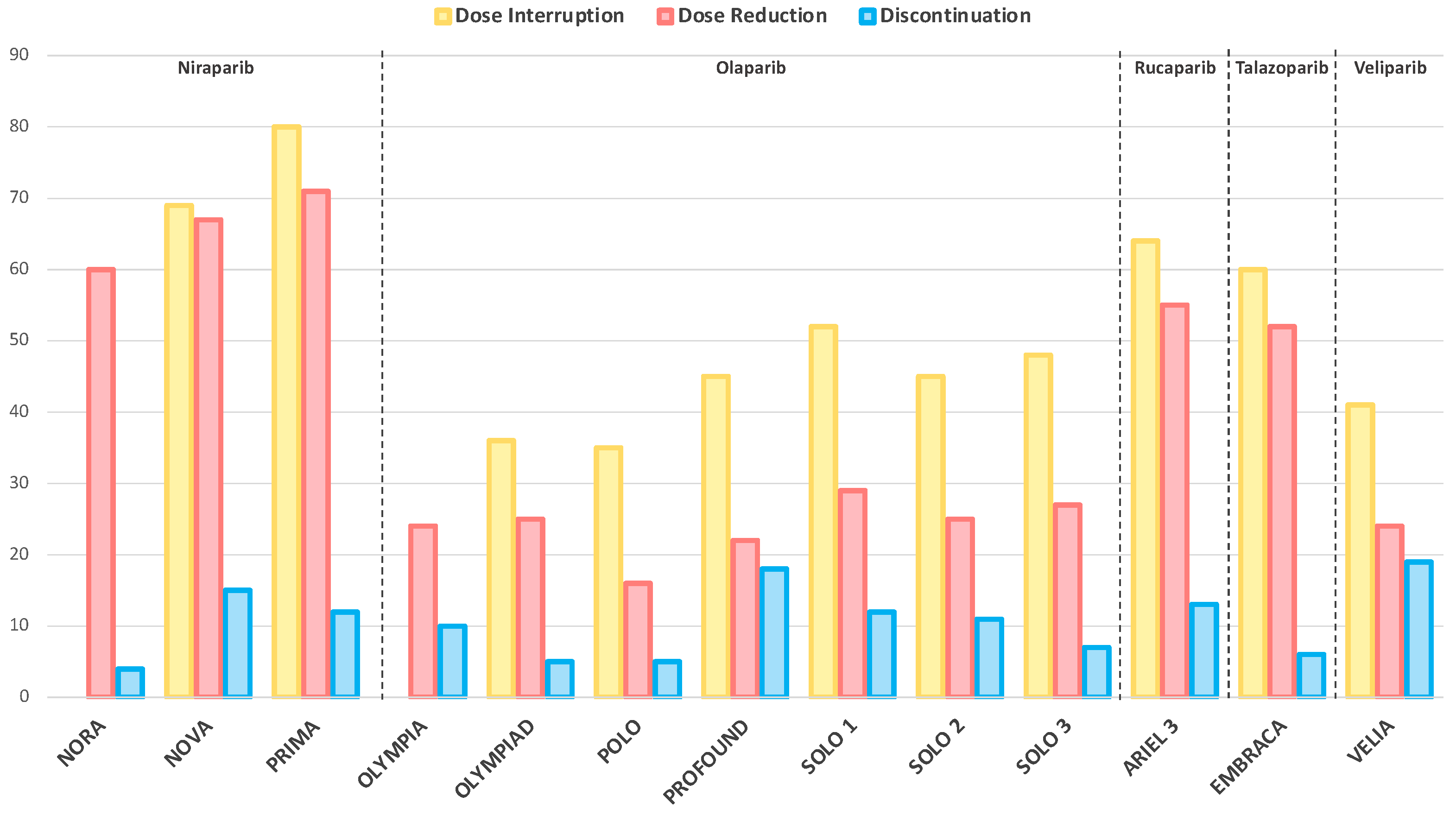
2.2. Dose Interruptions, Reductions and Treatment Discontinuations
3. Combination Regimens
3.1. DDR-Targeting Agents and Chemotherapy
3.2. DDR-Targeting Agents and Anti-Angiogenic Agents
3.3. DDR-Targeting Agents and Immunotherapy
3.4. Other Combination Strategies
4. Future Challenges and Perspectives
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Pilie, P.G.; Tang, C.; Mills, G.B.; Yap, T.A. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat. Rev. Clin. Oncol. 2019, 16, 81–104. [Google Scholar] [CrossRef] [PubMed]
- Roos, W.P.; Thomas, A.D.; Kaina, B. DNA damage and the balance between survival and death in cancer biology. Nat. Rev. Cancer 2016, 16, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- O’Neil, N.J.; Bailey, M.L.; Hieter, P. Synthetic lethality and cancer. Nat. Rev. Genet. 2017, 18, 613–623. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y.; O’Connor, M.J.; de Bono, J. Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci. Transl. Med. 2016, 8, 362ps317. [Google Scholar] [CrossRef] [PubMed]
- Genta, S.; Martorana, F.; Stathis, A.; Colombo, I. Targeting the DNA damage response: PARP inhibitors and new perspectives in the landscape of cancer treatment. Crit. Rev. Oncol. Hematol. 2021, 168, 103539. [Google Scholar] [CrossRef]
- Moore, K.; Colombo, N.; Scambia, G.; Kim, B.G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; Sonke, G.S.; et al. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2018, 379, 2495–2505. [Google Scholar] [CrossRef]
- Pujade-Lauraine, E.; Ledermann, J.A.; Selle, F.; Gebski, V.; Penson, R.T.; Oza, A.M.; Korach, J.; Huzarski, T.; Poveda, A.; Pignata, S.; et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 1274–1284. [Google Scholar] [CrossRef] [Green Version]
- Penson, R.T.; Valencia, R.V.; Cibula, D.; Colombo, N.; Leath, C.A., 3rd; Bidzinski, M.; Kim, J.W.; Nam, J.H.; Madry, R.; Hernandez, C.; et al. Olaparib Versus Nonplatinum Chemotherapy in Patients With Platinum-Sensitive Relapsed Ovarian Cancer and a Germline BRCA1/2 Mutation (SOLO3): A Randomized Phase III Trial. J. Clin. Oncol. 2020, 38, 1164–1174. [Google Scholar] [CrossRef]
- Robson, M.; Im, S.A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.O.; Hochhauser, D.; Arnold, D.; Oh, D.Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef]
- de Bono, J.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Olaparib for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 382, 2091–2102. [Google Scholar] [CrossRef]
- Mirza, M.R.; Monk, B.J.; Herrstedt, J.; Oza, A.M.; Mahner, S.; Redondo, A.; Fabbro, M.; Ledermann, J.A.; Lorusso, D.; Vergote, I.; et al. Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. N. Engl. J. Med. 2016, 375, 2154–2164. [Google Scholar] [CrossRef]
- Moore, K.N.; Secord, A.A.; Geller, M.A.; Miller, D.S.; Cloven, N.; Fleming, G.F.; Wahner Hendrickson, A.E.; Azodi, M.; DiSilvestro, P.; Oza, A.M.; et al. Niraparib monotherapy for late-line treatment of ovarian cancer (QUADRA): A multicentre, open-label, single-arm, phase 2 trial. Lancet Oncol. 2019, 20, 636–648. [Google Scholar] [CrossRef]
- Ray-Coquard, I.; Pautier, P.; Pignata, S.; Perol, D.; Gonzalez-Martin, A.; Berger, R.; Fujiwara, K.; Vergote, I.; Colombo, N.; Maenpaa, J.; et al. Olaparib plus Bevacizumab as First-Line Maintenance in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2416–2428. [Google Scholar] [CrossRef]
- Coleman, R.L.; Oza, A.M.; Lorusso, D.; Aghajanian, C.; Oaknin, A.; Dean, A.; Colombo, N.; Weberpals, J.I.; Clamp, A.; Scambia, G.; et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 390, 1949–1961. [Google Scholar] [CrossRef] [Green Version]
- Swisher, E.M.; Lin, K.K.; Oza, A.M.; Scott, C.L.; Giordano, H.; Sun, J.; Konecny, G.E.; Coleman, R.L.; Tinker, A.V.; O’Malley, D.M.; et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): An international, multicentre, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 75–87. [Google Scholar] [CrossRef] [Green Version]
- Litton, J.K.; Rugo, H.S.; Ettl, J.; Hurvitz, S.A.; Goncalves, A.; Lee, K.H.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.A.; Martin, M.; et al. Talazoparib in Patients with Advanced Breast Cancer and a Germline BRCA Mutation. N. Engl. J. Med. 2018, 379, 753–763. [Google Scholar] [CrossRef]
- Sorensen, C.S.; Syljuasen, R.G. Safeguarding genome integrity: The checkpoint kinases ATR, CHK1 and WEE1 restrain CDK activity during normal DNA replication. Nucleic Acids Res. 2012, 40, 477–486. [Google Scholar] [CrossRef] [Green Version]
- Konstantinopoulos, P.A.; Cheng, S.C.; Wahner Hendrickson, A.E.; Penson, R.T.; Schumer, S.T.; Doyle, L.A.; Lee, E.K.; Kohn, E.C.; Duska, L.R.; Crispens, M.A.; et al. Berzosertib plus gemcitabine versus gemcitabine alone in platinum-resistant high-grade serous ovarian cancer: A multicentre, open-label, randomised, phase 2 trial. Lancet Oncol. 2020, 21, 957–968. [Google Scholar] [CrossRef]
- Yap, T.A.; Krebs, M.G.; Postel-Vinay, S.; El-Khouiery, A.; Soria, J.C.; Lopez, J.; Berges, A.; Cheung, S.A.; Irurzun-Arana, I.; Goldwin, A.; et al. Ceralasertib (AZD6738), an Oral ATR Kinase Inhibitor, in Combination with Carboplatin in Patients with Advanced Solid Tumors: A Phase I Study. Clin. Cancer Res. 2021, 27, 5213–5224. [Google Scholar] [CrossRef]
- Lee, J.-M.; Nair, J.; Zimmer, A.; Lipkowitz, S.; Annunziata, C.M.; Merino, M.J.; Swisher, E.M.; Harrell, M.I.; Trepel, J.B.; Lee, M.-J.; et al. Prexasertib, a cell cycle checkpoint kinase 1 and 2 inhibitor, in BRCA wild-type recurrent high-grade serous ovarian cancer: A first-in-class proof-of-concept phase 2 study. Lancet Oncol. 2018, 19, 207–215. [Google Scholar] [CrossRef]
- Oza, A.M.; Estevez-Diz, M.; Grischke, E.M.; Hall, M.; Marme, F.; Provencher, D.; Uyar, D.; Weberpals, J.I.; Wenham, R.M.; Laing, N.; et al. A Biomarker-enriched, Randomized Phase II Trial of Adavosertib (AZD1775) Plus Paclitaxel and Carboplatin for Women with Platinum-sensitive TP53-mutant Ovarian Cancer. Clin. Cancer Res. 2020, 26, 4767–4776. [Google Scholar] [CrossRef]
- LaFargue, C.J.; Dal Molin, G.Z.; Sood, A.K.; Coleman, R.L. Exploring and comparing adverse events between PARP inhibitors. Lancet Oncol. 2019, 20, e15–e28. [Google Scholar] [CrossRef]
- Madariaga, A.; Bowering, V.; Ahrari, S.; Oza, A.M.; Lheureux, S. Manage wisely: Poly (ADP-ribose) polymerase inhibitor (PARPi) treatment and adverse events. Int. J. Gynecol Cancer 2020, 30, 903–915. [Google Scholar] [CrossRef] [Green Version]
- Hopkins, T.A.; Ainsworth, W.B.; Ellis, P.A.; Donawho, C.K.; DiGiammarino, E.L.; Panchal, S.C.; Abraham, V.C.; Algire, M.A.; Shi, Y.; Olson, A.M.; et al. PARP1 Trapping by PARP Inhibitors Drives Cytotoxicity in Both Cancer Cells and Healthy Bone Marrow. Mol. Cancer Res. 2019, 17, 409–419. [Google Scholar] [CrossRef] [Green Version]
- Farres, J.; Llacuna, L.; Martin-Caballero, J.; Martinez, C.; Lozano, J.J.; Ampurdanes, C.; Lopez-Contreras, A.J.; Florensa, L.; Navarro, J.; Ottina, E.; et al. PARP-2 sustains erythropoiesis in mice by limiting replicative stress in erythroid progenitors. Cell Death Differ. 2015, 22, 1144–1157. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Vandenberg, C.J.; Lieschke, E.; Di Rago, L.; Scott, C.L.; Majewski, I.J. CHK2 Inhibition Provides a Strategy to Suppress Hematologic Toxicity from PARP Inhibitors. Mol. Cancer Res. 2021, 19, 1350–1360. [Google Scholar] [CrossRef]
- Schuler, F.; Afreen, S.; Manzl, C.; Hacker, G.; Erlacher, M.; Villunger, A. Checkpoint kinase 1 is essential for fetal and adult hematopoiesis. EMBO Rep. 2019, 20, e47026. [Google Scholar] [CrossRef]
- Hu, W.; Zong, Q.; John-Baptiste, A.; Jessen, B. Transient knock down of checkpoint kinase 1 in hematopoietic progenitors is linked to bone marrow toxicity. Toxicol. Lett. 2011, 204, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Li, J. Haematologic toxicities with PARP inhibitors in cancer patients: An up-to-date meta-analysis of 29 randomized controlled trials. J. Clin. Pharm. 2021, 46, 571–584. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.L.; Fleming, G.F.; Brady, M.F.; Swisher, E.M.; Steffensen, K.D.; Friedlander, M.; Okamoto, A.; Moore, K.N.; Efrat Ben-Baruch, N.; Werner, T.L.; et al. Veliparib with First-Line Chemotherapy and as Maintenance Therapy in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2403–2415. [Google Scholar] [CrossRef] [PubMed]
- Common Terminology Criteria for Adverse Events (CTCAE). Common Terminology Criteria for Adverse Events (CTCAE). Available online: https://ctep.cancer.gov/ (accessed on 16 January 2022).
- Yap, T.A.; Tan, D.S.P.; Terbuch, A.; Caldwell, R.; Guo, C.; Goh, B.C.; Heong, V.; Haris, N.R.M.; Bashir, S.; Drew, Y.; et al. First-in-Human Trial of the Oral Ataxia Telangiectasia and RAD3-Related (ATR) Inhibitor BAY 1895344 in Patients with Advanced Solid Tumors. Cancer Discov. 2021, 11, 80–91. [Google Scholar] [CrossRef]
- Yap, T.A.; O’Carrigan, B.; Penney, M.S.; Lim, J.S.; Brown, J.S.; de Miguel Luken, M.J.; Tunariu, N.; Perez-Lopez, R.; Rodrigues, D.N.; Riisnaes, R.; et al. Phase I Trial of First-in-Class ATR Inhibitor M6620 (VX-970) as Monotherapy or in Combination With Carboplatin in Patients With Advanced Solid Tumors. J. Clin. Oncol. 2020, 38, 3195–3204. [Google Scholar] [CrossRef]
- Hong, D.S.; Moore, K.; Patel, M.; Grant, S.C.; Burris, H.A., 3rd; William, W.N., Jr.; Jones, S.; Meric-Bernstam, F.; Infante, J.; Golden, L.; et al. Evaluation of Prexasertib, a Checkpoint Kinase 1 Inhibitor, in a Phase Ib Study of Patients with Squamous Cell Carcinoma. Clin. Cancer Res. 2018, 24, 3263–3272. [Google Scholar] [CrossRef] [Green Version]
- Gatti-Mays, M.E.; Karzai, F.H.; Soltani, S.N.; Zimmer, A.; Green, J.E.; Lee, M.J.; Trepel, J.B.; Yuno, A.; Lipkowitz, S.; Nair, J.; et al. A Phase II Single Arm Pilot Study of the CHK1 Inhibitor Prexasertib (LY2606368) in BRCA Wild-Type, Advanced Triple-Negative Breast Cancer. Oncologist 2020, 25, e1013–e1824. [Google Scholar] [CrossRef]
- Byers, L.A.; Navarro, A.; Schaefer, E.; Johnson, M.; Ozguroglu, M.; Han, J.Y.; Bondarenko, I.; Cicin, I.; Dragnev, K.H.; Abel, A.; et al. A Phase II Trial of Prexasertib (LY2606368) in Patients With Extensive-Stage Small-Cell Lung Cancer. Clin. Lung Cancer 2021, 22, 531–540. [Google Scholar] [CrossRef]
- Do, K.; Wilsker, D.; Ji, J.; Zlott, J.; Freshwater, T.; Kinders, R.J.; Collins, J.; Chen, A.P.; Doroshow, J.H.; Kummar, S. Phase I Study of Single-Agent AZD1775 (MK-1775), a Wee1 Kinase Inhibitor, in Patients With Refractory Solid Tumors. J. Clin. Oncol. 2015, 33, 3409–3415. [Google Scholar] [CrossRef] [Green Version]
- Takebe, N.; Naqash, A.R.; O’Sullivan Coyne, G.; Kummar, S.; Do, K.; Bruns, A.; Juwara, L.; Zlott, J.; Rubinstein, L.; Piekarz, R.; et al. Safety, Antitumor Activity, and Biomarker Analysis in a Phase I Trial of the Once-daily Wee1 Inhibitor Adavosertib (AZD1775) in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2021, 27, 3834–3844. [Google Scholar] [CrossRef]
- Liu, J.F.; Xiong, N.; Campos, S.M.; Wright, A.A.; Krasner, C.; Schumer, S.; Horowitz, N.; Veneris, J.; Tayob, N.; Morrissey, S.; et al. Phase II Study of the WEE1 Inhibitor Adavosertib in Recurrent Uterine Serous Carcinoma. J. Clin. Oncol. 2021, 39, 1531–1539. [Google Scholar] [CrossRef]
- Lorusso, D.; Bologna, A.; Cecere, S.C.; De Matteis, E.; Scandurra, G.; Zamagni, C.; Arcangeli, V.; Artioli, F.; Bella, M.; Blanco, G.; et al. Sharing real-world experiences to optimize the management of olaparib toxicities: A practical guidance from an Italian expert panel. Support. Care Cancer 2020, 28, 2435–2442. [Google Scholar] [CrossRef]
- Wu, X.H.; Zhu, J.Q.; Yin, R.T.; Yang, J.X.; Liu, J.H.; Wang, J.; Wu, L.Y.; Liu, Z.L.; Gao, Y.N.; Wang, D.B.; et al. Niraparib maintenance therapy in patients with platinum-sensitive recurrent ovarian cancer using an individualized starting dose (NORA): A randomized, double-blind, placebo-controlled phase III trial(). Ann. Oncol. 2021, 32, 512–521. [Google Scholar] [CrossRef]
- Gonzalez-Martin, A.; Pothuri, B.; Vergote, I.; DePont Christensen, R.; Graybill, W.; Mirza, M.R.; McCormick, C.; Lorusso, D.; Hoskins, P.; Freyer, G.; et al. Niraparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2391–2402. [Google Scholar] [CrossRef] [Green Version]
- Tutt, A.N.J.; Garber, J.E.; Kaufman, B.; Viale, G.; Fumagalli, D.; Rastogi, P.; Gelber, R.D.; de Azambuja, E.; Fielding, A.; Balmana, J.; et al. Adjuvant Olaparib for Patients with BRCA1- or BRCA2-Mutated Breast Cancer. N. Engl. J. Med. 2021, 384, 2394–2405. [Google Scholar] [CrossRef]
- Robson, M.E.; Tung, N.; Conte, P.; Im, S.A.; Senkus, E.; Xu, B.; Masuda, N.; Delaloge, S.; Li, W.; Armstrong, A.; et al. OlympiAD final overall survival and tolerability results: Olaparib versus chemotherapy treatment of physician’s choice in patients with a germline BRCA mutation and HER2-negative metastatic breast cancer. Ann. Oncol. 2019, 30, 558–566. [Google Scholar] [CrossRef]
- Hussain, M.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Survival with Olaparib in Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 383, 2345–2357. [Google Scholar] [CrossRef]
- Banerjee, S.; Moore, K.N.; Colombo, N.; Scambia, G.; Kim, B.G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; et al. Maintenance olaparib for patients with newly diagnosed advanced ovarian cancer and a BRCA mutation (SOLO1/GOG 3004): 5-year follow-up of a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2021, 22, 1721–1731. [Google Scholar] [CrossRef]
- Poveda, A.; Floquet, A.; Ledermann, J.A.; Asher, R.; Penson, R.T.; Oza, A.M.; Korach, J.; Huzarski, T.; Pignata, S.; Friedlander, M.; et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): A final analysis of a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2021, 22, 620–631. [Google Scholar] [CrossRef]
- Berek, J.S.; Matulonis, U.A.; Peen, U.; Ghatage, P.; Mahner, S.; Redondo, A.; Lesoin, A.; Colombo, N.; Vergote, I.; Rosengarten, O.; et al. Safety and dose modification for patients receiving niraparib. Ann. Oncol. 2018, 29, 1784–1792. [Google Scholar] [CrossRef]
- Seligmann, J.F.; Fisher, D.J.; Brown, L.C.; Adams, R.A.; Graham, J.; Quirke, P.; Richman, S.D.; Butler, R.; Domingo, E.; Blake, A.; et al. Inhibition of WEE1 Is Effective in TP53- and RAS-Mutant Metastatic Colorectal Cancer: A Randomized Trial (FOCUS4-C) Comparing Adavosertib (AZD1775) With Active Monitoring. J. Clin. Oncol. 2021, 39, 3705–3715. [Google Scholar] [CrossRef]
- Liu, Y.; Meng, J.; Wang, G. Risk of selected gastrointestinal toxicities associated with poly (ADP-ribose) polymerase (PARP) inhibitors in the treatment of ovarian cancer: A meta-analysis of published trials. Drug Des. Devel. 2018, 12, 3013–3019. [Google Scholar] [CrossRef] [Green Version]
- Sun, W.; Li, J.; Zhang, Z.; Su, X. Gastrointestinal events with PARP inhibitors in cancer patients: A meta-analysis of phase II/III randomized controlled trials. J. Clin. Pharm. 2021, 46, 241–255. [Google Scholar] [CrossRef]
- Eakin, C.M.; Norton, T.J.; Monk, B.J.; Chase, D.M. Management of nausea and vomiting from poly(ADP-ribose) polymerase inhibitor therapy for advanced ovarian cancer. Gynecol. Oncol. 2020, 159, 581–587. [Google Scholar] [CrossRef] [PubMed]
- NCCN. Clinical Practice Guidelines in Oncology. Antiemesis. Version 1. 2021. Available online: https://www.nccn.org/professionals/physician_gls/pdf/antiemesis.pdf (accessed on 16 December 2021).
- Colombo, N.; Moore, K.; Scambia, G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; Sonke, G.S.; Gourley, C.; et al. Tolerability of maintenance olaparib in newly diagnosed patients with advanced ovarian cancer and a BRCA mutation in the randomized phase III SOLO1 trial. Gynecol. Oncol. 2021, 163, 41–49. [Google Scholar] [CrossRef]
- Ledermann, J.A.; Oza, A.M.; Lorusso, D.; Aghajanian, C.; Oaknin, A.; Dean, A.; Colombo, N.; Weberpals, J.I.; Clamp, A.R.; Scambia, G.; et al. Rucaparib for patients with platinum-sensitive, recurrent ovarian carcinoma (ARIEL3): Post-progression outcomes and updated safety results from a randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2020, 21, 710–722. [Google Scholar] [CrossRef]
- Durante, M.; Sgambellone, S.; Lanzi, C.; Nardini, P.; Pini, A.; Moroni, F.; Masini, E.; Lucarini, L. Effects of PARP-1 Deficiency and Histamine H4 Receptor Inhibition in an Inflammatory Model of Lung Fibrosis in Mice. Front. Pharm. 2019, 10, 525. [Google Scholar] [CrossRef]
- Dharwal, V.; Naura, A.S. PARP-1 inhibition ameliorates elastase induced lung inflammation and emphysema in mice. Biochem Pharm. 2018, 150, 24–34. [Google Scholar] [CrossRef]
- Zaffini, R.; Gotte, G.; Menegazzi, M. Asthma and poly(ADP-ribose) polymerase inhibition: A new therapeutic approach. Drug Des. Devel. 2018, 12, 281–293. [Google Scholar] [CrossRef] [Green Version]
- Ma, Z.; Sun, X.; Zhao, Z.; Lu, W.; Guo, Q.; Wang, S.; You, J.; Zhang, Y.; Liu, L. Risk of pneumonitis in cancer patients treated with PARP inhibitors: A meta-analysis of randomized controlled trials and a pharmacovigilance study of the FAERS database. Gynecol. Oncol. 2021, 162, 496–505. [Google Scholar] [CrossRef]
- Zhao, H.; Sifakis, E.G.; Sumida, N.; Millan-Arino, L.; Scholz, B.A.; Svensson, J.P.; Chen, X.; Ronnegren, A.L.; Mallet de Lima, C.D.; Varnoosfaderani, F.S.; et al. PARP1- and CTCF-Mediated Interactions between Active and Repressed Chromatin at the Lamina Promote Oscillating Transcription. Mol. Cell 2015, 59, 984–997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerace, E.; Masi, A.; Resta, F.; Felici, R.; Landucci, E.; Mello, T.; Pellegrini-Giampietro, D.E.; Mannaioni, G.; Moroni, F. PARP-1 activation causes neuronal death in the hippocampal CA1 region by increasing the expression of Ca(2+)-permeable AMPA receptors. Neurobiol. Dis. 2014, 70, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Sandhu, D.; Antolin, A.A.; Cox, A.R.; Jones, A.M. Identification of different side effects between PARP inhibitors and their polypharmacological multi-target rationale. Br. J. Clin. Pharm. 2021, 88, 742–752. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.N.; Mirza, M.R.; Matulonis, U.A. The poly (ADP ribose) polymerase inhibitor niraparib: Management of toxicities. Gynecol. Oncol. 2018, 149, 214–220. [Google Scholar] [CrossRef]
- Morice, P.M.; Leary, A.; Dolladille, C.; Chretien, B.; Poulain, L.; Gonzalez-Martin, A.; Moore, K.; O’Reilly, E.M.; Ray-Coquard, I.; Alexandre, J. Myelodysplastic syndrome and acute myeloid leukaemia in patients treated with PARP inhibitors: A safety meta-analysis of randomised controlled trials and a retrospective study of the WHO pharmacovigilance database. Lancet Haematol. 2021, 8, e122–e134. [Google Scholar] [CrossRef]
- Bolton, K.L.; Moukarzel, L.A.; Ptashkin, R.; Gao, T.; Patel, M.; Caltabellotta, N.; Braunstein, L.Z.; Aghajanian, C.; Hyman, D.M.; Berger, M.F.; et al. The impact of poly ADP ribose polymerase (PARP) inhibitors on clonal hematopoiesis. J. Clin. Oncol. 2020, 38, 1513. [Google Scholar] [CrossRef]
- Short, N.J.; Rytting, M.E.; Cortes, J.E. Acute myeloid leukaemia. Lancet 2018, 392, 593–606. [Google Scholar] [CrossRef]
- Ma, Z.; Sun, X.M.; Lu, W.C.; Zhao, Z.X.; Xu, Z.M.; Lyu, J.Y.; Zhao, P.; Liu, L.H. Poly(ADP-ribose) polymerase inhibitor-associated myelodysplastic syndrome/acute myeloid leukemia: A pharmacovigilance analysis of the FAERS database. ESMO Open 2021, 6, 100033. [Google Scholar] [CrossRef]
- Zibetti Dal Molin, G.; Westin, S.N.; Msaouel, P.; Gomes, L.M.; Dickens, A.; Coleman, R.L. Discrepancy in calculated and measured glomerular filtration rates in patients treated with PARP inhibitors. Int. J. Gynecol. Cancer 2020, 30, 89–93. [Google Scholar] [CrossRef]
- Bruin, M.A.C.; Korse, C.M.; van Wijnen, B.; de Jong, V.M.T.; Linn, S.C.; van Triest, B.; Rosing, H.; Beijnen, J.H.; van den Broek, D.; Huitema, A.D.R. A real or apparent decrease in glomerular filtration rate in patients using olaparib? Eur. J. Clin. Pharm. 2021, 77, 179–188. [Google Scholar] [CrossRef]
- Lorusso, D.; Garcia-Donas, J.; Sehouli, J.; Joly, F. Management of Adverse Events During Rucaparib Treatment for Relapsed Ovarian Cancer: A Review of Published Studies and Practical Guidance. Target. Oncol. 2020, 15, 391–406. [Google Scholar] [CrossRef]
- Yap, T.A.; Plummer, R.; Azad, N.S.; Helleday, T. The DNA Damaging Revolution: PARP Inhibitors and Beyond. Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 185–195. [Google Scholar] [CrossRef]
- Lu, Y.; Liu, Y.; Pang, Y.; Pacak, K.; Yang, C. Double-barreled gun: Combination of PARP inhibitor with conventional chemotherapy. Pharmacol. Ther. 2018, 188, 168–175. [Google Scholar] [CrossRef]
- Matulonis, U.A.; Monk, B.J. PARP inhibitor and chemotherapy combination trials for the treatment of advanced malignancies: Does a development pathway forward exist? Ann. Oncol. 2017, 28, 443–447. [Google Scholar] [CrossRef]
- Lheureux, S.; Cristea, M.C.; Bruce, J.P.; Garg, S.; Cabanero, M.; Mantia-Smaldone, G.; Olawaiye, A.B.; Ellard, S.L.; Weberpals, J.I.; Wahner Hendrickson, A.E.; et al. Adavosertib plus gemcitabine for platinum-resistant or platinum-refractory recurrent ovarian cancer: A double-blind, randomised, placebo-controlled, phase 2 trial. Lancet 2021, 397, 281–292. [Google Scholar] [CrossRef]
- Moore, K.N.; Hong, D.S.; Patel, M.R.; Pant, S.; Ulahannan, S.V.; Jones, S.; Meric-Bernstam, F.; Wang, J.S.; Aljumaily, R.; Hamilton, E.P.; et al. A Phase 1b Trial of Prexasertib in Combination with Standard-of-Care Agents in Advanced or Metastatic Cancer. Target. Oncol. 2021, 16, 569–589. [Google Scholar] [CrossRef]
- Bang, Y.J.; Xu, R.H.; Chin, K.; Lee, K.W.; Park, S.H.; Rha, S.Y.; Shen, L.; Qin, S.; Xu, N.; Im, S.A.; et al. Olaparib in combination with paclitaxel in patients with advanced gastric cancer who have progressed following first-line therapy (GOLD): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 1637–1651. [Google Scholar] [CrossRef]
- Kalra, M.; Tong, Y.; Jones, D.R.; Walsh, T.; Danso, M.A.; Ma, C.X.; Silverman, P.; King, M.C.; Badve, S.S.; Perkins, S.M.; et al. Cisplatin +/- rucaparib after preoperative chemotherapy in patients with triple-negative or BRCA mutated breast cancer. Jpn. Breast Cancer 2021, 7, 29. [Google Scholar] [CrossRef]
- Byers, L.A.; Bentsion, D.; Gans, S.; Penkov, K.; Son, C.; Sibille, A.; Owonikoko, T.K.; Groen, H.J.M.; Gay, C.M.; Fujimoto, J.; et al. Veliparib in Combination with Carboplatin and Etoposide in Patients with Treatment-Naive Extensive-Stage Small Cell Lung Cancer: A Phase 2 Randomized Study. Clin. Cancer Res. 2021, 27, 3884–3895. [Google Scholar] [CrossRef]
- O’Reilly, E.M.; Lee, J.W.; Zalupski, M.; Capanu, M.; Park, J.; Golan, T.; Tahover, E.; Lowery, M.A.; Chou, J.F.; Sahai, V.; et al. Randomized, Multicenter, Phase II Trial of Gemcitabine and Cisplatin With or Without Veliparib in Patients With Pancreas Adenocarcinoma and a Germline BRCA/PALB2 Mutation. J. Clin. Oncol. 2020, 38, 1378–1388. [Google Scholar] [CrossRef]
- Chiorean, E.G.; Guthrie, K.A.; Philip, P.A.; Swisher, E.M.; Jalikis, F.; Pishvaian, M.J.; Berlin, J.; Noel, M.S.; Suga, J.M.; Garrido-Laguna, I.; et al. Randomized Phase II Study of PARP Inhibitor ABT-888 (Veliparib) with Modified FOLFIRI versus FOLFIRI as Second-line Treatment of Metastatic Pancreatic Cancer: SWOG S1513. Clin. Cancer Res. 2021, 27, 6314–6322. [Google Scholar] [CrossRef] [PubMed]
- Loibl, S.; O’Shaughnessy, J.; Untch, M.; Sikov, W.M.; Rugo, H.S.; McKee, M.D.; Huober, J.; Golshan, M.; von Minckwitz, G.; Maag, D.; et al. Addition of the PARP inhibitor veliparib plus carboplatin or carboplatin alone to standard neoadjuvant chemotherapy in triple-negative breast cancer (BrighTNess): A randomised, phase 3 trial. Lancet Oncol. 2018, 19, 497–509. [Google Scholar] [CrossRef]
- Dieras, V.; Han, H.S.; Kaufman, B.; Wildiers, H.; Friedlander, M.; Ayoub, J.P.; Puhalla, S.L.; Bondarenko, I.; Campone, M.; Jakobsen, E.H.; et al. Veliparib with carboplatin and paclitaxel in BRCA-mutated advanced breast cancer (BROCADE3): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2020, 21, 1269–1282. [Google Scholar] [CrossRef]
- Ramalingam, S.S.; Novello, S.; Guclu, S.Z.; Bentsion, D.; Zvirbule, Z.; Szilasi, M.; Bernabe, R.; Syrigos, K.; Byers, L.A.; Clingan, P.; et al. Veliparib in Combination With Platinum-Based Chemotherapy for First-Line Treatment of Advanced Squamous Cell Lung Cancer: A Randomized, Multicenter Phase III Study. J. Clin. Oncol. 2021, 39, 3633–3644. [Google Scholar] [CrossRef]
- Pal, S.K.; Frankel, P.H.; Mortazavi, A.; Milowsky, M.; Vaishampayan, U.; Parikh, M.; Lyou, Y.; Weng, P.; Parikh, R.; Teply, B.; et al. Effect of Cisplatin and Gemcitabine With or Without Berzosertib in Patients With Advanced Urothelial Carcinoma: A Phase 2 Randomized Clinical Trial. JAMA Oncol. 2021, 7, 1536–1543. [Google Scholar] [CrossRef]
- Mirza, M.R.; Avall Lundqvist, E.; Birrer, M.J.; dePont Christensen, R.; Nyvang, G.B.; Malander, S.; Anttila, M.; Werner, T.L.; Lund, B.; Lindahl, G.; et al. Niraparib plus bevacizumab versus niraparib alone for platinum-sensitive recurrent ovarian cancer (NSGO-AVANOVA2/ENGOT-ov24): A randomised, phase 2, superiority trial. Lancet Oncol. 2019, 20, 1409–1419. [Google Scholar] [CrossRef]
- Liu, J.F.; Barry, W.T.; Birrer, M.; Lee, J.M.; Buckanovich, R.J.; Fleming, G.F.; Rimel, B.; Buss, M.K.; Nattam, S.; Hurteau, J.; et al. Combination cediranib and olaparib versus olaparib alone for women with recurrent platinum-sensitive ovarian cancer: A randomised phase 2 study. Lancet Oncol. 2014, 15, 1207–1214. [Google Scholar] [CrossRef] [Green Version]
- Perez-Fidalgo, J.A.; Cortes, A.; Guerra, E.; Garcia, Y.; Iglesias, M.; Bohn Sarmiento, U.; Calvo Garcia, E.; Manso Sanchez, L.; Santaballa, A.; Oaknin, A.; et al. Olaparib in combination with pegylated liposomal doxorubicin for platinum-resistant ovarian cancer regardless of BRCA status: A GEICO phase II trial (ROLANDO study). ESMO Open 2021, 6, 100212. [Google Scholar] [CrossRef]
- Hanna, C.; Kurian, K.M.; Williams, K.; Watts, C.; Jackson, A.; Carruthers, R.; Strathdee, K.; Cruickshank, G.; Dunn, L.; Erridge, S.; et al. Pharmacokinetics, safety, and tolerability of olaparib and temozolomide for recurrent glioblastoma: Results of the phase I OPARATIC trial. Neuro. Oncol. 2020, 22, 1840–1850. [Google Scholar] [CrossRef]
- Lee, C.K.; Scott, C.; Lindeman, G.J.; Hamilton, A.; Lieschke, E.; Gibbs, E.; Asher, R.; Badger, H.; Paterson, R.; Macnab, L.; et al. Phase 1 trial of olaparib and oral cyclophosphamide in BRCA breast cancer, recurrent BRCA ovarian cancer, non-BRCA triple-negative breast cancer, and non-BRCA ovarian cancer. Br. J. Cancer 2019, 120, 279–285. [Google Scholar] [CrossRef] [Green Version]
- Grignani, G.; D’Ambrosio, L.; Pignochino, Y.; Palmerini, E.; Zucchetti, M.; Boccone, P.; Aliberti, S.; Stacchiotti, S.; Bertulli, R.; Piana, R.; et al. Trabectedin and olaparib in patients with advanced and non-resectable bone and soft-tissue sarcomas (TOMAS): An open-label, phase 1b study from the Italian Sarcoma Group. Lancet Oncol. 2018, 19, 1360–1371. [Google Scholar] [CrossRef]
- Bendell, J.; O’Reilly, E.M.; Middleton, M.R.; Chau, I.; Hochster, H.; Fielding, A.; Burke, W.; Burris, H., III. Phase I study of olaparib plus gemcitabine in patients with advanced solid tumours and comparison with gemcitabine alone in patients with locally advanced/metastatic pancreatic cancer. Ann. Oncol. 2015, 26, 804–811. [Google Scholar] [CrossRef]
- Mego, M.; Svetlovska, D.; Reckova, M.; Angelis, D.; Kalavska, K.; Obertova, J.; Palacka, P.; Rejlekova, K.; Sycova-Mila, Z.; Chovanec, M.; et al. Gemcitabine, carboplatin and veliparib in multiple relapsed/refractory germ cell tumours: The GCT-SK-004 phase II trial. Investig. New Drugs 2021, 39, 1664–1670. [Google Scholar] [CrossRef]
- Plummer, R.; Dean, E.; Arkenau, H.T.; Redfern, C.; Spira, A.I.; Melear, J.M.; Chung, K.Y.; Ferrer-Playan, J.; Goddemeier, T.; Locatelli, G.; et al. A phase 1b study evaluating the safety and preliminary efficacy of berzosertib in combination with gemcitabine in patients with advanced non-small cell lung cancer. Lung Cancer 2021, 163, 19–26. [Google Scholar] [CrossRef]
- Middleton, M.R.; Dean, E.; Evans, T.R.J.; Shapiro, G.I.; Pollard, J.; Hendriks, B.S.; Falk, M.; Diaz-Padilla, I.; Plummer, R. Phase 1 study of the ATR inhibitor berzosertib (formerly M6620, VX-970) combined with gemcitabine +/- cisplatin in patients with advanced solid tumours. Br. J. Cancer 2021, 125, 510–519. [Google Scholar] [CrossRef]
- Kim, S.T.; Smith, S.A.; Mortimer, P.; Loembe, A.B.; Cho, H.; Kim, K.M.; Smith, C.; Willis, S.; Irurzun-Arana, I.; Berges, A.; et al. Phase I Study of Ceralasertib (AZD6738), a Novel DNA Damage Repair Agent, in Combination with Weekly Paclitaxel in Refractory Cancer. Clin. Cancer Res. 2021, 27, 4700–4709. [Google Scholar] [CrossRef]
- Keenan, T.E.; Li, T.; Vallius, T.; Guerriero, J.L.; Tayob, N.; Kochupurakkal, B.; Davis, J.; Pastorello, R.; Tahara, R.K.; Anderson, L.; et al. Clinical Efficacy and Molecular Response Correlates of the WEE1 Inhibitor Adavosertib Combined with Cisplatin in Patients with Metastatic Triple-Negative Breast Cancer. Clin. Cancer Res. 2021, 27, 983–991. [Google Scholar] [CrossRef]
- Moore, K.N.; Chambers, S.K.; Hamilton, E.P.; Chen, L.M.; Oza, A.M.; Ghamande, S.A.; Konecny, G.E.; Plaxe, S.C.; Spitz, D.L.; Geenen, J.J.J.; et al. Adavosertib with Chemotherapy in Patients with Primary Platinum-Resistant Ovarian, Fallopian Tube, or Peritoneal Cancer: An Open-Label, Four-Arm, Phase II Study. Clin. Cancer Res. 2021, 1, 36–44. [Google Scholar] [CrossRef]
- Leijen, S.; van Geel, R.M.; Sonke, G.S.; de Jong, D.; Rosenberg, E.H.; Marchetti, S.; Pluim, D.; van Werkhoven, E.; Rose, S.; Lee, M.A.; et al. Phase II Study of WEE1 Inhibitor AZD1775 Plus Carboplatin in Patients With TP53-Mutated Ovarian Cancer Refractory or Resistant to First-Line Therapy Within 3 Months. J. Clin. Oncol. 2016, 34, 4354–4361. [Google Scholar] [CrossRef] [Green Version]
- Alvarez Secord, A.; O’Malley, D.M.; Sood, A.K.; Westin, S.N.; Liu, J.F. Rationale for combination PARP inhibitor and antiangiogenic treatment in advanced epithelial ovarian cancer: A review. Gynecol. Oncol. 2021, 162, 482–495. [Google Scholar] [CrossRef]
- Kim, J.W.; Cardin, D.B.; Vaishampayan, U.N.; Kato, S.; Grossman, S.R.; Glazer, P.M.; Shyr, Y.; Ivy, S.P.; LoRusso, P.M. Clinical Activity and Safety of Cediranib and Olaparib Combination in Patients with Metastatic Pancreatic Ductal Adenocarcinoma without BRCA Mutation. Oncologist 2021, 26, e1104–e1109. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.F.; Tolaney, S.M.; Birrer, M.; Fleming, G.F.; Buss, M.K.; Dahlberg, S.E.; Lee, H.; Whalen, C.; Tyburski, K.; Winer, E.; et al. A Phase 1 trial of the poly(ADP-ribose) polymerase inhibitor olaparib (AZD2281) in combination with the anti-angiogenic cediranib (AZD2171) in recurrent epithelial ovarian or triple-negative breast cancer. Eur. J. Cancer 2013, 49, 2972–2978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bizzaro, F.; Fuso Nerini, I.; Taylor, M.A.; Anastasia, A.; Russo, M.; Damia, G.; Guffanti, F.; Guana, F.; Ostano, P.; Minoli, L.; et al. VEGF pathway inhibition potentiates PARP inhibitor efficacy in ovarian cancer independent of BRCA status. J. Hematol. Oncol. 2021, 14, 186. [Google Scholar] [CrossRef] [PubMed]
- Lheureux, S.; Oaknin, A.; Garg, S.; Bruce, J.P.; Madariaga, A.; Dhani, N.C.; Bowering, V.; White, J.; Accardi, S.; Tan, Q.; et al. EVOLVE: A Multicenter Open-Label Single-Arm Clinical and Translational Phase II Trial of Cediranib Plus Olaparib for Ovarian Cancer after PARP Inhibition Progression. Clin. Cancer Res. 2020, 26, 4206–4215. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.F.; Barry, W.T.; Birrer, M.; Lee, J.M.; Buckanovich, R.J.; Fleming, G.F.; Rimel, B.J.; Buss, M.K.; Nattam, S.R.; Hurteau, J.; et al. Overall survival and updated progression-free survival outcomes in a randomized phase II study of combination cediranib and olaparib versus olaparib in relapsed platinum-sensitive ovarian cancer. Ann. Oncol. 2019, 30, 551–557. [Google Scholar] [CrossRef]
- Stewart, R.A.; Pilie, P.G.; Yap, T.A. Development of PARP and Immune-Checkpoint Inhibitor Combinations. Cancer Res. 2018, 78, 6717–6725. [Google Scholar] [CrossRef] [Green Version]
- Pham, M.M.; Ngoi, N.Y.L.; Peng, G.; Tan, D.S.P.; Yap, T.A. Development of poly(ADP-ribose) polymerase inhibitor and immunotherapy combinations: Progress, pitfalls, and promises. Trends Cancer 2021, 7, 958–970. [Google Scholar] [CrossRef]
- Peyraud, F.; Italiano, A. Combined PARP Inhibition and Immune Checkpoint Therapy in Solid Tumors. Cancers 2020, 12, 1502. [Google Scholar] [CrossRef]
- Jiao, S.; Xia, W.; Yamaguchi, H.; Wei, Y.; Chen, M.K.; Hsu, J.M.; Hsu, J.L.; Yu, W.H.; Du, Y.; Lee, H.H.; et al. PARP Inhibitor Upregulates PD-L1 Expression and Enhances Cancer-Associated Immunosuppression. Clin. Cancer Res. 2017, 23, 3711–3720. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, L.B.; Salama, A.K.S. A review of cancer immunotherapy toxicity. CA Cancer J. Clin. 2020, 70, 86–104. [Google Scholar] [CrossRef] [Green Version]
- Konstantinopoulos, P.A.; Waggoner, S.; Vidal, G.A.; Mita, M.; Moroney, J.W.; Holloway, R.; Van Le, L.; Sachdev, J.C.; Chapman-Davis, E.; Colon-Otero, G.; et al. Single-Arm Phases 1 and 2 Trial of Niraparib in Combination With Pembrolizumab in Patients With Recurrent Platinum-Resistant Ovarian Carcinoma. JAMA Oncol. 2019, 5, 1141–1149. [Google Scholar] [CrossRef] [Green Version]
- Vinayak, S.; Tolaney, S.M.; Schwartzberg, L.; Mita, M.; McCann, G.; Tan, A.R.; Wahner-Hendrickson, A.E.; Forero, A.; Anders, C.; Wulf, G.M.; et al. Open-label Clinical Trial of Niraparib Combined With Pembrolizumab for Treatment of Advanced or Metastatic Triple-Negative Breast Cancer. JAMA Oncol. 2019, 5, 1132–1140. [Google Scholar] [CrossRef] [Green Version]
- Ramalingam, S.S.; Thara, E.; Awad, M.M.; Dowlati, A.; Haque, B.; Stinchcombe, T.E.; Dy, G.K.; Spigel, D.R.; Lu, S.; Iyer Singh, N.; et al. JASPER: Phase 2 trial of first-line niraparib plus pembrolizumab in patients with advanced non-small cell lung cancer. Cancer 2022, 128, 65–74. [Google Scholar] [CrossRef]
- Domchek, S.M.; Postel-Vinay, S.; Im, S.A.; Park, Y.H.; Delord, J.P.; Italiano, A.; Alexandre, J.; You, B.; Bastian, S.; Krebs, M.G.; et al. Olaparib and durvalumab in patients with germline BRCA-mutated metastatic breast cancer (MEDIOLA): An open-label, multicentre, phase 1/2, basket study. Lancet Oncol. 2020, 21, 1155–1164. [Google Scholar] [CrossRef]
- Karzai, F.; VanderWeele, D.; Madan, R.A.; Owens, H.; Cordes, L.M.; Hankin, A.; Couvillon, A.; Nichols, E.; Bilusic, M.; Beshiri, M.L.; et al. Activity of durvalumab plus olaparib in metastatic castration-resistant prostate cancer in men with and without DNA damage repair mutations. J. Immunother Cancer 2018, 6, 141. [Google Scholar] [CrossRef]
- Lampert, E.J.; Zimmer, A.; Padget, M.; Cimino-Mathews, A.; Nair, J.R.; Liu, Y.; Swisher, E.M.; Hodge, J.W.; Nixon, A.B.; Nichols, E.; et al. Combination of PARP Inhibitor Olaparib, and PD-L1 Inhibitor Durvalumab, in Recurrent Ovarian Cancer: A Proof-of-Concept Phase II Study. Clin. Cancer Res. 2020, 26, 4268–4279. [Google Scholar] [CrossRef]
- Thomas, A.; Vilimas, R.; Trindade, C.; Erwin-Cohen, R.; Roper, N.; Xi, L.; Krishnasamy, V.; Levy, E.; Mammen, A.; Nichols, S.; et al. Durvalumab in Combination with Olaparib in Patients with Relapsed SCLC: Results from a Phase II Study. J. Thorac. Oncol. 2019, 14, 1447–1457. [Google Scholar] [CrossRef]
- Kim, R.; Kwon, M.; An, M.; Kim, S.T.; Smith, S.A.; Loembe, A.B.; Mortimer, P.G.S.; Armenia, J.; Lukashchuk, N.; Shah, N.; et al. Phase II study of ceralasertib (AZD6738) in combination with durvalumab in patients with advanced/metastatic melanoma who have failed prior anti-PD-1 therapy. Ann. Oncol. 2021. [Google Scholar] [CrossRef]
- Martorana, F.; Motta, G.; Pavone, G.; Motta, L.; Stella, S.; Vitale, S.R.; Manzella, L.; Vigneri, P. AKT Inhibitors: New Weapons in the Fight Against Breast Cancer? Front. Pharm. 2021, 12, 662232. [Google Scholar] [CrossRef]
- Vitale, S.R.; Martorana, F.; Stella, S.; Motta, G.; Inzerilli, N.; Massimino, M.; Tirro, E.; Manzella, L.; Vigneri, P. PI3K inhibition in breast cancer: Identifying and overcoming different flavors of resistance. Crit Rev. Oncol. Hematol. 2021, 162, 103334. [Google Scholar] [CrossRef]
- van der Ploeg, P.; Uittenboogaard, A.; Thijs, A.M.J.; Westgeest, H.M.; Boere, I.A.; Lambrechts, S.; van de Stolpe, A.; Bekkers, R.L.M.; Piek, J.M.J. The effectiveness of monotherapy with PI3K/AKT/mTOR pathway inhibitors in ovarian cancer: A meta-analysis. Gynecol. Oncol. 2021, 163, 433–444. [Google Scholar] [CrossRef]
- Ibrahim, Y.H.; Garcia-Garcia, C.; Serra, V.; He, L.; Torres-Lockhart, K.; Prat, A.; Anton, P.; Cozar, P.; Guzman, M.; Grueso, J.; et al. PI3K inhibition impairs BRCA1/2 expression and sensitizes BRCA-proficient triple-negative breast cancer to PARP inhibition. Cancer Discov. 2012, 2, 1036–1047. [Google Scholar] [CrossRef] [Green Version]
- Juvekar, A.; Burga, L.N.; Hu, H.; Lunsford, E.P.; Ibrahim, Y.H.; Balmana, J.; Rajendran, A.; Papa, A.; Spencer, K.; Lyssiotis, C.A.; et al. Combining a PI3K inhibitor with a PARP inhibitor provides an effective therapy for BRCA1-related breast cancer. Cancer Discov. 2012, 2, 1048–1063. [Google Scholar] [CrossRef] [Green Version]
- Matulonis, U.A.; Wulf, G.M.; Barry, W.T.; Birrer, M.; Westin, S.N.; Farooq, S.; Bell-McGuinn, K.M.; Obermayer, E.; Whalen, C.; Spagnoletti, T.; et al. Phase I dose escalation study of the PI3kinase pathway inhibitor BKM120 and the oral poly (ADP ribose) polymerase (PARP) inhibitor olaparib for the treatment of high-grade serous ovarian and breast cancer. Ann. Oncol. 2017, 28, 512–518. [Google Scholar] [CrossRef] [PubMed]
- Konstantinopoulos, P.A.; Barry, W.T.; Birrer, M.; Westin, S.N.; Cadoo, K.A.; Shapiro, G.I.; Mayer, E.L.; O’Cearbhaill, R.E.; Coleman, R.L.; Kochupurakkal, B.; et al. Olaparib and alpha-specific PI3K inhibitor alpelisib for patients with epithelial ovarian cancer: A dose-escalation and dose-expansion phase 1b trial. Lancet Oncol. 2019, 20, 570–580. [Google Scholar] [CrossRef]
- Yap, T.A.; Kristeleit, R.; Michalarea, V.; Pettitt, S.J.; Lim, J.S.J.; Carreira, S.; Roda, D.; Miller, R.; Riisnaes, R.; Miranda, S.; et al. Phase I Trial of the PARP Inhibitor Olaparib and AKT Inhibitor Capivasertib in Patients with BRCA1/2- and Non-BRCA1/2-Mutant Cancers. Cancer Discov. 2020, 10, 1528–1543. [Google Scholar] [CrossRef]
- Chiappa, M.; Guffanti, F.; Bertoni, F.; Colombo, I.; Damia, G. Overcoming PARPi resistance: Preclinical and clinical evidence in ovarian cancer. Drug Resist. Update 2021, 55, 100744. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Lheureux, S.; Moore, K.N. PARP Inhibitors for Ovarian Cancer: Current Indications, Future Combinations, and Novel Assets in Development to Target DNA Damage Repair. Am. Soc. Clin. Oncol. Educ. Book 2020, 40, 1–16. [Google Scholar] [CrossRef]
- Do, K.T.; Kochupurakkal, B.; Kelland, S.; de Jonge, A.; Hedglin, J.; Powers, A.; Quinn, N.; Gannon, C.; Vuong, L.; Parmar, K.; et al. Phase 1 Combination Study of the CHK1 Inhibitor Prexasertib and the PARP Inhibitor Olaparib in High-grade Serous Ovarian Cancer and Other Solid Tumors. Clin. Cancer Res. 2021, 27, 4710–4716. [Google Scholar] [CrossRef]
- Mahdi, H.; Hafez, N.; Doroshow, D.; Sohal, D.; Keedy, V.; Do, K.T.; LoRusso, P.; Jurgensmeier, J.; Avedissian, M.; Sklar, J.; et al. Ceralasertib-Mediated ATR Inhibition Combined With Olaparib in Advanced Cancers Harboring DNA Damage Response and Repair Alterations (Olaparib Combinations). JCO Precis Oncol. 2021, 5, 1432–1442. [Google Scholar] [CrossRef]
- Shah, P.D.; Wethington, S.L.; Pagan, C.; Latif, N.; Tanyi, J.; Martin, L.P.; Morgan, M.; Burger, R.A.; Haggerty, A.; Zarrin, H.; et al. Combination ATR and PARP Inhibitor (CAPRI): A phase 2 study of ceralasertib plus olaparib in patients with recurrent, platinum-resistant epithelial ovarian cancer. Gynecol. Oncol. 2021, 163, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Landrum, L.M.; Brady, W.E.; Armstrong, D.K.; Moore, K.N.; DiSilvestro, P.A.; O’Malley, D.M.; Tenney, M.E.; Rose, P.G.; Fracasso, P.M. A phase I trial of pegylated liposomal doxorubicin (PLD), carboplatin, bevacizumab and veliparib in recurrent, platinum-sensitive ovarian, primary peritoneal, and fallopian tube cancer: An NRG Oncology/Gynecologic Oncology Group study. Gynecol. Oncol. 2016, 140, 204–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drew, Y.; Penson, R.T.; O’Malley, D.M.; Kim, J.W.; Zimmermann, S.; Roxburgh, P.; Sohn, J.; Stemmer, S.M.; Bastian, S.; Ferguson, M.; et al. 814MO Phase II study of olaparib (O) plus durvalumab (D) and bevacizumab (B) (MEDIOLA): Initial results in patients (pts) with non-germline BRCA-mutated (non-gBRCAm) platinum sensitive relapsed (PSR) ovarian cancer (OC). Ann. Oncol. 2020, 31, S615–S616. [Google Scholar] [CrossRef]
- Liu, J.; Gaillard, S.; Hendrickson, A.W.; Moroney, J.; Yeku, O.; Diver, E.; Gunderson, C.; Arend, R.; Ratner, E.; Samnotra, V.; et al. An open-label phase II study of dostarlimab (TSR-042), bevacizumab (bev), and niraparib combination in patients (pts) with platinum-resistant ovarian cancer (PROC): Cohort A of the OPAL trial. In Proceedings of the Society of Gynecologic Oncology 2021 Virtual Annual Meeting on Women’s Cancer, Phoenix, Arizona, 19–25 March 2021; Virtual Abstract 23. [Google Scholar]
- Gillen, J.; Miller, A.; Bell-McGuinn, K.M.; Schilder, R.J.; Walker, J.L.; Mathews, C.A.; Duska, L.R.; Guntupalli, S.R.; O’Cearbhaill, R.; Hays, J.; et al. Post hoc analyses of GOG 9923: Does BRCA status affect toxicities?: An NRG oncology study. Gynecol. Oncol. 2021, 161, 512–515. [Google Scholar] [CrossRef]
- Churpek, J.E.; Marquez, R.; Neistadt, B.; Claussen, K.; Lee, M.K.; Churpek, M.M.; Huo, D.; Weiner, H.; Bannerjee, M.; Godley, L.A.; et al. Inherited mutations in cancer susceptibility genes are common among survivors of breast cancer who develop therapy-related leukemia. Cancer 2016, 122, 304–311. [Google Scholar] [CrossRef]
- McNerney, M.E.; Godley, L.A.; Le Beau, M.M. Therapy-related myeloid neoplasms: When genetics and environment collide. Nat. Rev. Cancer 2017, 17, 513–527. [Google Scholar] [CrossRef]
- Kwan, T.T.; Oza, A.M.; Tinker, A.V.; Ray-Coquard, I.; Oaknin, A.; Aghajanian, C.; Lorusso, D.; Colombo, N.; Dean, A.; Weberpals, J.; et al. Preexisting TP53-Variant Clonal Hematopoiesis and Risk of Secondary Myeloid Neoplasms in Patients with High-grade Ovarian Cancer Treated with Rucaparib. JAMA Oncol. 2021, 7, 1772–1781. [Google Scholar] [CrossRef]
- Morton, L.M.; Dores, G.M.; Schonfeld, S.J.; Linet, M.S.; Sigel, B.S.; Lam, C.J.K.; Tucker, M.A.; Curtis, R.E. Association of Chemotherapy for Solid Tumors With Development of Therapy-Related Myelodysplastic Syndrome or Acute Myeloid Leukemia in the Modern Era. JAMA Oncol. 2019, 5, 318–325. [Google Scholar] [CrossRef] [Green Version]
- Antolin, A.A.; Ameratunga, M.; Banerji, U.; Clarke, P.A.; Workman, P.; Al-Lazikani, B. The kinase polypharmacology landscape of clinical PARP inhibitors. Sci. Rep. 2020, 10, 2585. [Google Scholar] [CrossRef] [Green Version]
- Thorsell, A.G.; Ekblad, T.; Karlberg, T.; Low, M.; Pinto, A.F.; Tresaugues, L.; Moche, M.; Cohen, M.S.; Schuler, H. Structural Basis for Potency and Promiscuity in Poly(ADP-ribose) Polymerase (PARP) and Tankyrase Inhibitors. J. Med. Chem. 2017, 60, 1262–1271. [Google Scholar] [CrossRef]




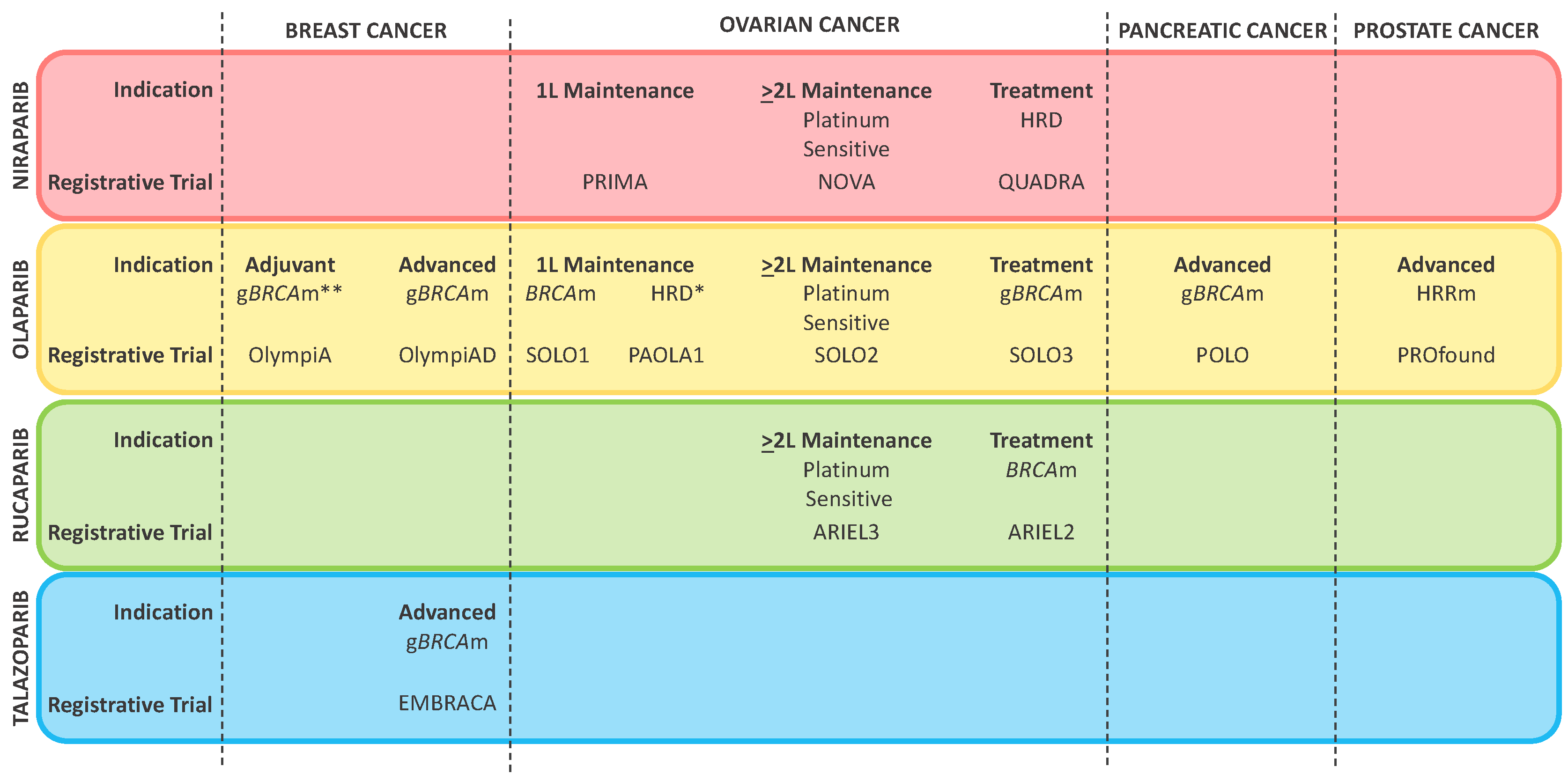{kind=link}
{kind=link}
{kind=link}
| Class | Molecule | Trial | Ref. | Anemia | Thrombocytopenia | Neutropenia | Nausea | Vomiting | Diarrhea | Constipation | Dyspepsia | Dysgeusia | Decreased Appetite | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Any G | G ≥ 3 | Any G | G ≥ 3 | Any G | G ≥ 3 | Any G | G ≥ 3 | Any G | G ≥ 3 | Any G | G ≥ 3 | Any G | G ≥ 3 | Any G | G ≥ 3 | Any G | G ≥ 3 | Any G | G ≥ 3 | ||||
| PARPi | NIRAPARIB | NORA | [44] | 53% | 15% | 55% | 11% | 59% | 20% | 53% | 0% | 32% | 2% | 14% | 0% | 30% | <1% | NR | NR | NR | NR | 18% | 0% |
| NOVA | [14] | 50% | 25% | 61% | 34% | 30% | 20% | 74% | 3% | 34% | 2% | 19% | <1% | 40% | <1% | 11% | 0% | 10% | 0% | 25% | <1% | ||
| PRIMA | [45] | 63% | 31% | 73% | 42% | 43% | 20% | 57% | 1% | 22% | <1% | 19% | <1% | 39% | <1% | NR | NR | NR | NR | 19% | 1% | ||
| OLAPARIB | OlympiA | [46] | 24% | 9% | NR | NR | 16% | 5% | 57% | 1% | 23% | 1% | 18% | <1% | NR | NR | NR | NR | 12% | 0% | 13% | <1% | |
| OlympiAD | [47] | 40% | 16% | NR | NR | 27% | 9% | 58% | 0% | 32% | 0% | 21% | <1% | 13% | <1% | NR | NR | NR | NR | 17% | 0% | ||
| POLO | [12] | 27% | 11% | NR | NR | NR | NR | 45% | 0% | 20% | 1% | 29% | 1% | 23% | 0% | NR | NR | NR | NR | 25% | 3% | ||
| PROfound | [48] | 50% | 23% | NR | 4% | NR | 4% | 43% | 2% | 20% | 2% | 21% | <1% | 19% | 0% | NR | NR | NR | NR | 31% | 2% | ||
| SOLO 1 | [49] | 40% | 22% | 11% | 1% | 24% | 9% | 78% | 1% | 40% | <1% | 35% | 3% | 28% | 0% | 17% | 0% | 22% | 0% | 20% | 0% | ||
| SOLO 2 | [50] | 46% | 21% | 17% | 3% | 24% | 8% | 76% | 3% | 40% | 3% | 34% | 1% | 24% | 0% | 15% | 0% | 19% | 0% | 23% | 1% | ||
| SOLO 3 | [10] | 51% | 21% | 12% | 4% | 23% | 10% | 65% | 1% | 38% | 1% | 28% | 0% | 12% | 0% | 11% | 0% | NR | NR | NR | NR | ||
| RUCAPARIB | ARIEL 3 | [17] | 37% | 19% | 28% | 5% | 18% | 7% | 75% | 4% | 37% | 4% | 32% | 1% | 37% | 2% | 15% | <1% | 39% | 0% | 23% | 1% | |
| TALAZOPARIB | EMBRACA | [19] | 53% | 39% | 27% | 15% | 35% | 21% | 49% | <1% | 25% | 2% | 22% | <1% | 22% | <1% | NR | NR | NR | NR | 21% | <1% | |
| VELIPARIB | VELIA | [33] | 17% | 7% | 20% | 7% | 17% | 5% | 56% | 5% | 34% | 2% | 19% | <1% | 12% | 0% | NR | NR | NR | NR | 11% | <1% | |
| ATRi | ELIMUSERTIB | NCT03188965 | [35] | 82% | 82% | 45% | 18% | 73% | 55% | 50% | 9% | 19% | 5% | 23% | 5% | 14% | 0% | NR | NR | NR | NR | 14% | 0% |
| CHEK1i | PREXASERTIB | NCT02203513 | [23] | 93% | 11% | 82% | 25% | 97% | 93% | 64% | 0% | 29% | 4% | 39% | 7% | 11% | 0% | 4% | 0% | NR | NR | NR | 0% |
| NCT02203513 | [38] | 99% | 33% | 89% | 11% | 89% | 89% | 33% | 0% | 0% | 0% | 44% | 0% | 0% | 0% | 0% | 0% | NR | NR | NR | NR | ||
| NCT01115790 | [37] | 33% | 14% | 46% | 16% | 92% | 89% | 15% | 0% | NR | NR | 8% | 0% | NR | NR | NR | NR | NR | NR | NR | NR | ||
| NCT02735980 | [39] | 40% | 15% | 54% | 27% | 72% | 66% | 23% | 0 | 13% | <1% | 14% | <1% | 10% | 0% | NR | NR | NR | NR | 25% | 3% | ||
| WEE1i | ADAVOSERTIB | NCT03668340 | [42] | 68% | 24% | 62% | 17% | 44% | 32% | 62% | 9% | 41% | 6% | 85% | 6% | 38% | 0% | NR | NR | 21% | 0% | 32% | 3% |
| NCT01748825 | [41] | 68% | 21% | 45% | 13% | 34% | 22% | 81% | 7% | 69% | 12% | 65% | 5% | NR | NR | NR | NR | NR | NR | NR | NR | ||
| Class | Molecule | Trial | Ref. | Fatigue | Increased AST/ALT | Increased Creatinine | Cough | Dyspnea | Headache | Insomnia | Hypertension | Tachycardia | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Any G | G ≥ 3 | Any G | G ≥ 3 | Any G | G ≥ 3 | Any G | G ≥ 3 | Any G | G ≥ 3 | Any G | G ≥ 3 | Any G | G ≥ 3 | Any G | G ≥ 3 | Any G | G ≥ 3 | ||||
| PARPi | NIRAPARIB | NORA | [44] | 25% | <1% | 24% | 1% | NR | NR | 12% | 0% | NR | NR | 18% | <1% | 29% | <1% | 11% | 1% | 18% | <1% |
| NOVA | [14] | 59% | 8% | NR | NR | NR | NR | 15% | 0% | 19% | 1% | 26% | <1% | 24% | <1% | 19% | 8% | 10% | 0% | ||
| PRIMA | [45] | 34% | 2% | NR | NR | 11% | <1% | 15% | 0% | 18% | <1% | 26% | <1% | 25% | <1% | 17% | 6% | NR | NR | ||
| OLAPARIB | OLIMPIA | [46] | 40% | 2% | NR | NR | NR | NR | NR | NR | NR | NR | 20% | <1% | NR | NR | NR | NR | NR | NR | |
| OLIMPIAD | [47] | 30% | 3% | 12% | 2% | NR | NR | 17% | 0% | NR | NR | 20% | 1% | NR | NR | NR | NR | NR | NR | ||
| POLO | [12] | 60% | 5% | NR | NR | NR | NR | NR | NR | NR | NR | NR | NR | NR | NR | NR | NR | NR | NR | ||
| PROFOUND | [48] | 42% | 8% | NR | NR | NR | NR | 11% | 0% | 11% | 2% | NR | NR | NR | NR | NR | NR | NR | NR | ||
| SOLO 1 | [49] | 64% | 4% | NR | NR | NR | NR | 17% | 0% | 15% | 0% | 23% | <1% | 10% | 0% | 3% | <1% | NR | NR | ||
| SOLO 2 | [50] | 67% | 6% | NR | NR | 11% | 0% | 19% | 1% | 12% | 1% | 26% | 1% | 7% | 0% | 4% | 0% | NR | NR | ||
| SOLO 3 | [10] | 52% | 4% | NR | NR | <1% | <1% | NR | NR | NR | NR | 16% | 0% | NR | NR | NR | NR | NR | NR | ||
| RUCAPARIB | ARIEL 3 | [17] | 71% | 7% | 34% | 10% | 15% | <1% | 15% | 0% | 14% | 0% | 19% | <1% | 15% | 0% | 9% | 2% | NR | NR | |
| TALAZOPARIB | EMBRACA | [19] | 50% | 2% | NR | NR | NR | NR | NR | NR | 18% | 2% | 33% | 2% | NR | NR | NR | NR | NR | NR | |
| VELIPARIB | VELIA | [33] | 23% | 6% | NR | NR | NR | NR | NR | NR | NR | NR | 10% | <1% | 13% | 1% | NR | NR | NR | NR | |
| ATRi | ELIMUSERTIB | NCT03188965 | [35] | 68% | 9% | NR | NR | NR | NR | 14% | 0% | 14% | 0% | 23% | 0% | NR | NR | NR | NR | NR | NR |
| CHEK1i | PREXASERTIB | NCT02203513 | [23] | 53% | 7% | NR | NR | NR | NR | NR | NR | NR | NR | 4% | 0% | NR | NR | NR | NR | NR | NR |
| NCT02203513 | [38] | 67% | 0% | NR | NR | NR | NR | NR | NR | 22% | 0% | 11% | 0% | NR | NR | NR | NR | NR | NR | ||
| NCT01115790 | [37] | 28% | 2% | NR | NR | NR | NR | NR | NR | NR | NR | 12% | 1% | NR | NR | NR | NR | NR | NR | ||
| NCT02735980 | [39] | 39% | 8% | NR | NR | NR | NR | 22% | 0% | 23% | 8% | NR | NR | NR | NR | NR | NR | NR | NR | ||
| WEE1i | ADAVOSERTIB | NCT03668340 | [42] | 65% | 24% | 38% | 9% | NR | NR | 21% | 0% | 32% | 0% | NR | NR | 27% | 0% | NR | NR | NR | NR |
| NCT01748825 | [41] | 52% | 7% | 26% | 0% | NR | NR | NR | NR | NR | NR | NR | NR | NR | NR | NR | NR | NR | NR | ||
| Partner Agent | DDR Class | DDR Molecule | Treatment Arms | Population | Trial Phase | AEs More Frequent with Combination | Ref. | |
|---|---|---|---|---|---|---|---|---|
| Any Grade (Δ ≥ 5%) | Grade ≥ 3 (Δ ≥ 5%) | |||||||
| Chemotherapy | PARP-i | Olaparib | Pacli + Ola vs. Plcb + Ola | Advanced Gastric Cancer | III | Anemia; Diarrhea | Anemia; Neutropenia | [79] |
| Rucaparib | CDDP + Ruca vs. CDDP | Triple Negative Brest Cancer (Adjuvant) | II | Fatigue; Nausea | Fatigue; Nausea Neutropenia; Vomiting | [80] | ||
| Veliparib | CBDCA + VP16 + Veli vs. CBDCA + VP16 | Small-Cell Lung Cancer | II | Anemia; Fatigue; Headache; Hypokalemia; Hyponatremia; Nausea; Neutropenia; Thrombocytopenia | Anemia; Febrile neutropenia; Hypokalemia; Hyponatremia; Neutropenia; Thrombocytopenia | [81] | ||
| CDDP + Gem + Veli CDDP + Gem + Plcb | Stage III-IV Pancreatic Carcinoma gBRCA/PALB2 mut | II * | Nausea | Anemia; Neutropenia; Thrombocytopenia | [82] | |||
| mFOLFIRI + Veli vs. FOLFIRI | Advanced Pancreatic Carcinoma | II | NR | Dehydration; Diarrhea; Fatigue; Nausea; Neutropenia; Vomiting | [83] | |||
| CBDCA + Pacli + Veli vs. CBDCA + Pacli + Plcb | Epithelial Ovarian Cancer (First Line) | III | Anemia; Constipation; Insomnia; Neutropenia; Thrombocytopenia | Anemia; Neutropenia; Thrombocytopenia | [33] | |||
| CBDCA + Pacli + Veli vs. CBDCA + Pacli + Plcb | Triple Negative Brest Cancer (Neoadjuvant) | III | Diarrhea; Nausea; Neutropenia; Stomatitis; Thrombocytopenia; Vomiting | Anemia | [84] | |||
| CBDCA + Pacli + Veli vs. CBDCA + Pacli + Plcb | Advanced Triple Negative Brest Cancer gBRCA mut | III | Anemia; Back pain; Cough; Diarrhea; Hypomagnesemia; Nausea; Peripheral edema | Thrombocytopenia | [85] | |||
| CBDCA + Pacli + Veli vs. CBDCA + Pacli + Plcb | Advanced Squamous NSCLC | III | No differences ≥5% | No differences ≥5% | [86] | |||
| ATR-i | Berzosertib | CDDP + Gem + Berzo vs. CDDP + Gem | Urothelial Cancer | II | Emesis; Fatigue; Peripheral edema | Neutropenia; Thrombocytopenia | [87] | |
| Gem + Berzo vs. Gem | Platinum Resistant Ovarian Cancer | II | Anemia; AST/ALT increase; Headache; Nausea; Neutropenia; Thrombocytopenia; Vomiting | Neutropenia; Thrombocytopenia | [21] | |||
| WEE1-i | Adavosertib | Gem + Ada vs. Gem+ Plcb | Platinum Resistant Ovarian Cancer | II | Abdominal painAlopecia; Diarrhea; Constipation; Fatigue; Fever; Hypertension; Hypokalemia; Hypomagnesemia; Hyponatremia Insomnia; Nausea; Neutropenia; Thrombocytopenia; Vomiting | Anemia; Febrile neutropenia; Hypertension; Hypokalemia; Hypomagnesemia; Neutropenia; Rash Thrombocytopenia; Vomiting | [77] | |
| CBDCA + Pacli + Ada vs. CBDCA + Pacli + Plcb | Platinum Sensitive Ovarian Cancer TP53 mut | II | Abdominal pain; Anemia Constipation; Diarrhea; Dyspnea; Myalgia; Nausea; Neutropenia Thrombocytopenia; Vomiting | Anemia, Diarrhea; Febrile Neutropenia; Neutropenia; Thrombocytopenia; Vomiting | [24] | |||
| Anti--angiogenics | PARP-i | Niraparib | Beva + Nira vs. Nira | Platinum Sensitive Ovarian Cancer | II | Anemia; Anorexia; Cough; Headache; Hypertension; Myalgia; Nausea; Proteinuria; Vomiting | Hypertension | [88] |
| Olaparib | Beva + Ola vs. Beva + Plcb | Platinum Sensitive Ovarian Cancer | III | Anemia; Fatigue; Hypertension; Nausea; Thrombocytopenia; Vomiting | Anemia | [16] | ||
| Cedi + Ola vs. Ola | Platinum Resistan Ovarian Cancer | II | Abdominal pain; Anorexia; Constipation; Diarrhea; Fatigue; Headache; Hypertension; Hypothyroidism; Mucositis; Proteinuria; Thrombocytopenia | Diarrhea; Fatigue; Hypertension | [89] | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martorana, F.; Da Silva, L.A.; Sessa, C.; Colombo, I. Everything Comes with a Price: The Toxicity Profile of DNA-Damage Response Targeting Agents. Cancers 2022, 14, 953. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14040953
Martorana F, Da Silva LA, Sessa C, Colombo I. Everything Comes with a Price: The Toxicity Profile of DNA-Damage Response Targeting Agents. Cancers. 2022; 14(4):953. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14040953
Chicago/Turabian StyleMartorana, Federica, Leandro Apolinario Da Silva, Cristiana Sessa, and Ilaria Colombo. 2022. "Everything Comes with a Price: The Toxicity Profile of DNA-Damage Response Targeting Agents" Cancers 14, no. 4: 953. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14040953