
Deep Learning-Based Pan-Cancer Classification Model Reveals Tissue-of-Origin Specific Gene Expression Signatures

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Gene Expression Data Collection and Pre-Processing
2.2. Unsupervised Clustering
2.3. Implementation Details
2.4. Evaluation of the Model’s Performance
2.5. Model Interpretation Based upon SHAP Values
2.6. ICGC Test Data Set
2.7. Implementation Details of Cloud-Based Web Tool
3. Results
3.1. Unique Gene Expression Signatures Are Associated with Different Cancer Types
3.2. Development and Training of a Deep Neural Network Model
3.3. DNN Model Accurately Predicts Cancer Types Based on Gene Expression Profiles
3.4. Identification of Cancer Tissue-of-Origin Specific Gene Expression Signatures
3.5. Gene Signature Validation Using an Independent Test Data Set
3.6. Expression Profile of Keratins across Cancer Tissues
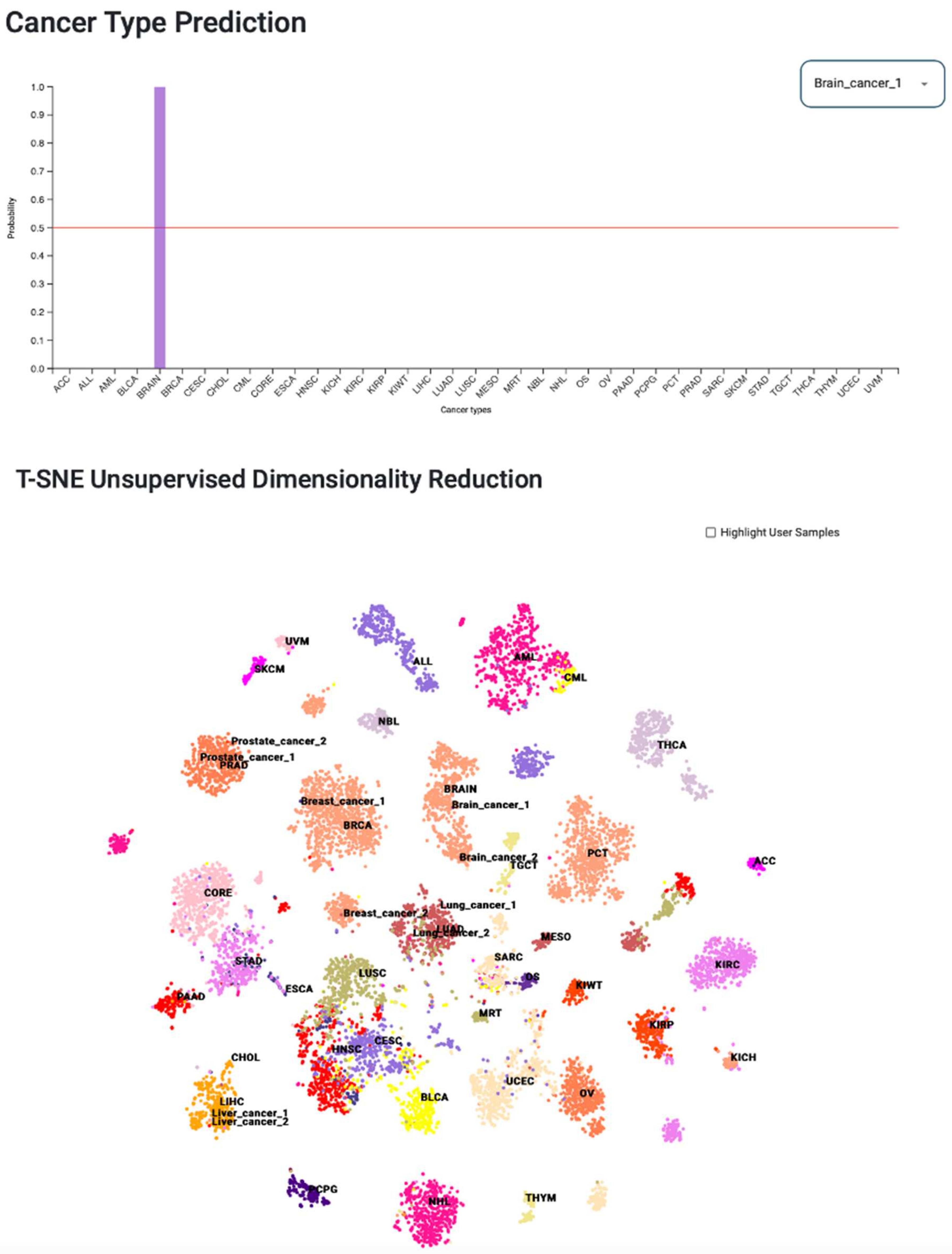
3.7. Web Tool for Cancer-Type Prediction
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hanif, M.; Zaidi, P.; Kamal, S.; Idrees, S.; Rasool, S.A. Significance of prostate specific antigen in prostate cancer patients and in non cancerous prostatic disease patients. J. Pak. Med. Assoc. 2007, 57, 248–251. [Google Scholar] [PubMed]
- Hutter, C.; Zenklusen, J.C. The Cancer Genome Atlas: Creating Lasting Value beyond Its Data. Cell 2018, 173, 283–285. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Baran, J.; Cros, A.; Guberman, J.M.; Haider, S.; Hsu, J.; Liang, Y.; Rivkin, E.; Wang, J.; Whitty, B.; et al. International Cancer Genome Consortium Data Portal—A one-stop shop for cancer genomics data. Database 2011, 2011, bar026. [Google Scholar] [CrossRef] [Green Version]
- LeCun, Y.; Bengio, Y.; Hinton, G. Deep learning. Nature 2015, 521, 436–444. [Google Scholar] [CrossRef]
- Hornik, K.; Stinchcombe, M.; White, H. Multilayer feedforward networks are universal approximators. Neural Netw. 1989, 2, 359–366. [Google Scholar] [CrossRef]
- Cybenko, G. Approximation by superpositions of a sigmoidal function. Math. Control Signals Syst. 1989, 2, 303–314. [Google Scholar] [CrossRef]
- Jiao, W.; Atwal, G.; Polak, P.; Karlic, R.; Cuppen, E.; Danyi, A.; de Ridder, J.; van Herpen, C.; Lolkema, M.P.; Steeghs, N.; et al. A deep learning system accurately classifies primary and metastatic cancers using passenger mutation patterns. Nat. Commun. 2020, 11, 728. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhu, S.; Ma, K.; Liu, W.; Yue, Y.; Hu, G.; Lu, H.; Chen, W. Identification of 12 cancer types through genome deep learning. Sci. Rep. 2019, 9, 17256. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Y.; Shi, Y.; Su, X.; Zou, X.; Luo, Q.; Feng, D.D.; Cai, W.; Han, Z.-G. Cancer type prediction based on copy number aberration and chromatin 3D structure with convolutional neural networks. BMC Genom. 2018, 19, 565. [Google Scholar] [CrossRef] [Green Version]
- Gao, F.; Wang, W.; Tan, M.; Zhu, L.; Zhang, Y.; Fessler, E.; Vermeulen, L.; Wang, X. DeepCC: A novel deep learning-based framework for cancer molecular subtype classification. Oncogenesis 2019, 8, 44. [Google Scholar] [CrossRef]
- Zhao, Y.; Pan, Z.; Namburi, S.; Pattison, A.; Posner, A.; Balachander, S.; Paisie, C.A.; Reddi, H.V.; Rueter, J.; Gill, A.J.; et al. CUP-AI-Dx: A tool for inferring cancer tissue of origin and molecular subtype using RNA gene-expression data and artificial intelligence. EBioMedicine 2020, 61, 103030. [Google Scholar] [CrossRef]
- Mostavi, M.; Chiu, Y.-C.; Huang, Y.; Chen, Y. Convolutional neural network models for cancer type prediction based on gene expression. BMC Med Genom. 2020, 13, 44. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, J.; Sun, K.; Yang, X.; Dai, C.; Guo, Y. Integrated Multi-omics Analysis Using Variational Autoencoders: Application to Pan-cancer Classification. In Proceedings of the 2019 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), San Diego, CA, USA, 18–21 November 2019; pp. 765–769. [Google Scholar]
- Chaudhary, K.; Poirion, O.B.; Lu, L.; Garmire, L.X. Deep Learning–Based Multi-Omics Integration Robustly Predicts Survival in Liver Cancer. Clin. Cancer Res. 2018, 24, 1248–1259. [Google Scholar] [CrossRef] [Green Version]
- Hassanzadeh, H.R.; Wang, M.D. An Integrated Deep Network for Cancer Survival Prediction Using Omics Data. Front. Big Data 2021, 4, 568352. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Kang, K.; Krahn, J.M.; Croutwater, N.; Lee, K.; Umbach, D.M.; Li, L. A comprehensive genomic pan-cancer classification using The Cancer Genome Atlas gene expression data. BMC Genom. 2017, 18, 508. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Nie, R.; Yu, Z.; Xin, R.; Zheng, C.; Zhang, Z.; Zhang, J.; Cai, J. An interpretable deep-learning architecture of capsule networks for identifying cell-type gene expression programs from single-cell RNA-sequencing data. Nat. Mach. Intell. 2020, 2, 693–703. [Google Scholar] [CrossRef]
- van Hilten, A.; Kushner, S.A.; Kayser, M.; Ikram, M.A.; Adams, H.H.; Klaver, C.C.; Niessen, W.J.; Roshchupkin, G.V. GenNet framework: Interpretable deep learning for predicting phenotypes from genetic data. Commun. Biol. 2021, 4, 1094. [Google Scholar] [CrossRef]
- van der Maaten, L.; Hinton, G. Visualizing Data using t-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2605. [Google Scholar]
- Abadi, M.; Agarwal, A.; Barham, P.; Brevdo, E.; Chen, Z.; Citro, C.; Corrado, G.S.; Davis, A.; Dean, J.; Devin, M.; et al. TensorFlow: Large-Scale Machine Learning on Heterogeneous Distributed Systems. arXiv 2016, arXiv:1603.04467. [Google Scholar]
- Kingma, D.P.; Ba, J. Adam: A Method for Stochastic Optimization. arXiv 2014, arXiv:1412.6980. [Google Scholar]
- Bottou, L. Stochastic Gradient Learning in Neural Networks. Proc. Neuro-Nımes 1991, 91, 12. [Google Scholar]
- Hinton, G.; Srivastava, N.; Swersky, K. RMSprop: Divide the gradient by a running average of its recent magnitude. COURSERA Neural Netw. Mach. Learn 2012. [Google Scholar]
- Lundberg, S.M.; Lee, S.-I. A unified approach to interpreting model predictions. In Proceedings of the 31st International Conference on Neural Information Processing Systems, Long Beach, CA, USA, 4–9 December 2017; pp. 4768–4777. [Google Scholar]
- Sha, Y.; Phan, J.H.; Wang, M.D. Effect of low-expression gene filtering on detection of differentially expressed genes in RNA-seq data. In Proceedings of the 2015 37th Annual International Conference of the IEEE Engineering in Medicine and Biology Society (EMBC), Milan, Italy, 25–29 August 2015; pp. 6461–6464. [Google Scholar]
- Angermueller, C.; Pärnamaa, T.; Parts, L.; Stegle, O. Deep learning for computational biology. Mol. Syst. Biol. 2016, 12, 878. [Google Scholar] [CrossRef]
- Karantza, V. Keratins in health and cancer: More than mere epithelial cell markers. Oncogene 2010, 30, 127–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moll, R.; Divo, M.; Langbein, L. The human keratins: Biology and pathology. Histochem. Cell Biol. 2008, 129, 705–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moll, R.; Franke, W.W.; Schiller, D.L.; Geiger, B.; Krepler, R. The catalog of human cytokeratins: Patterns of expression in normal epithelia, tumors and cultured cells. Cell 1982, 31, 11–24. [Google Scholar] [CrossRef]
- Chu, P.G.; Weiss, L.M. Keratin expression in human tissues and neoplasms. Histopathology 2002, 40, 403–439. [Google Scholar] [CrossRef]
- Divate, M.; Tyagi, A.; Richard, D.J.; Prasad, P.A.; Gowda, H.; Nagaraj, S.H. Deep learning-based pan-cancer classification model reveals cancer-specific gene expression signatures. bioRxiv 2021. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Divate, M.; Tyagi, A.; Richard, D.J.; Prasad, P.A.; Gowda, H.; Nagaraj, S.H. Deep Learning-Based Pan-Cancer Classification Model Reveals Tissue-of-Origin Specific Gene Expression Signatures. Cancers 2022, 14, 1185. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14051185
Divate M, Tyagi A, Richard DJ, Prasad PA, Gowda H, Nagaraj SH. Deep Learning-Based Pan-Cancer Classification Model Reveals Tissue-of-Origin Specific Gene Expression Signatures. Cancers. 2022; 14(5):1185. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14051185
Chicago/Turabian StyleDivate, Mayur, Aayush Tyagi, Derek J. Richard, Prathosh A. Prasad, Harsha Gowda, and Shivashankar H. Nagaraj. 2022. "Deep Learning-Based Pan-Cancer Classification Model Reveals Tissue-of-Origin Specific Gene Expression Signatures" Cancers 14, no. 5: 1185. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14051185