
Epigenetic Alteration of the Cancer-Related Gene TGFBI in B Cells Infected with Epstein–Barr Virus and Exposed to Aflatoxin B1: Potential Role in Burkitt Lymphoma Development

 , ,
, ,  ,
,  , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cases Selection
2.2. Cell Culture and Treatment
2.3. qPCR
2.4. Immunoblotting and Antibodies
2.5. Immunohistochemistry
2.6. Chromatin Immunoprecipitation
2.7. Bisulfite Modification and DNA Methylation Assessment
2.8. DNMTs Assays
3. Results
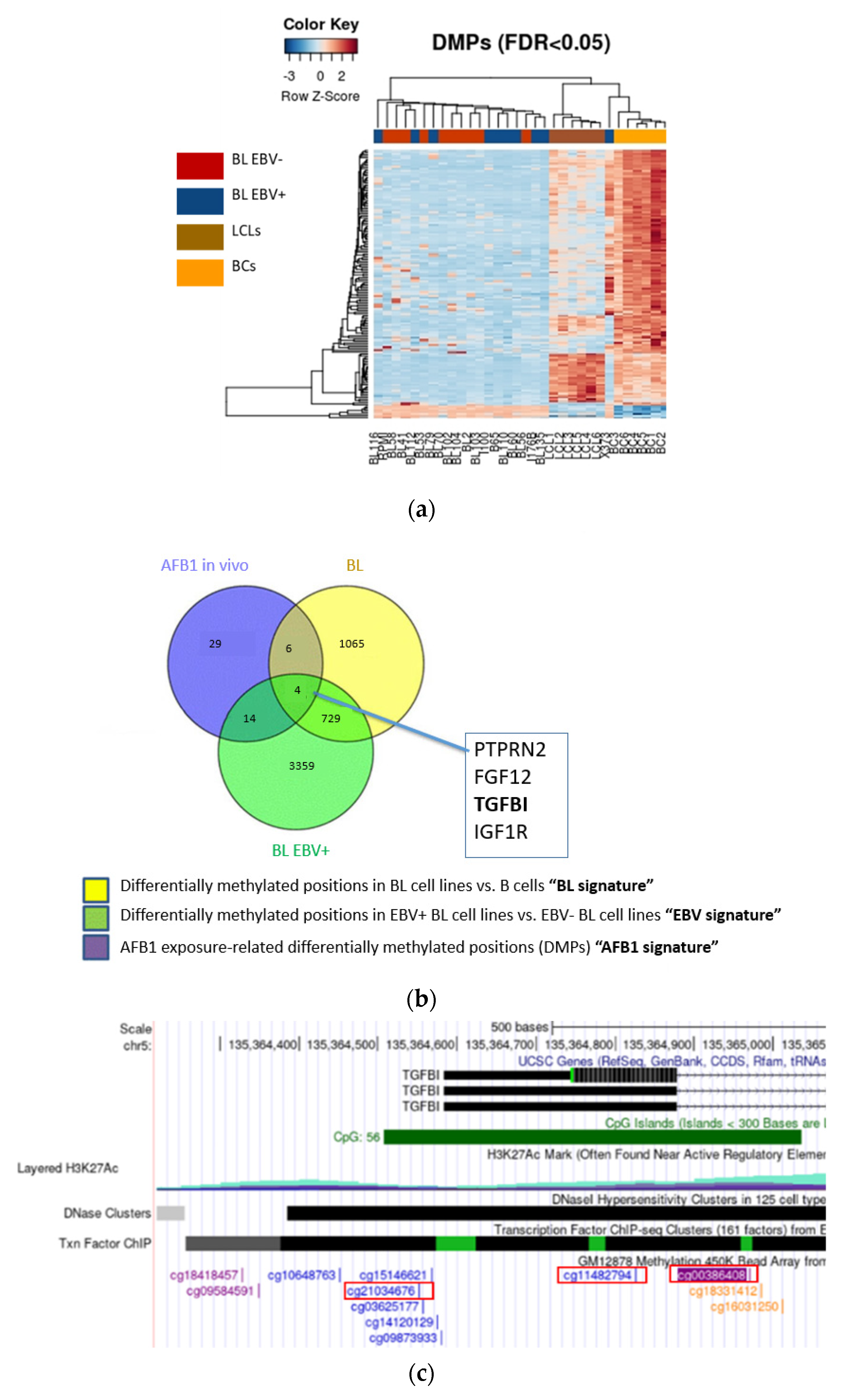
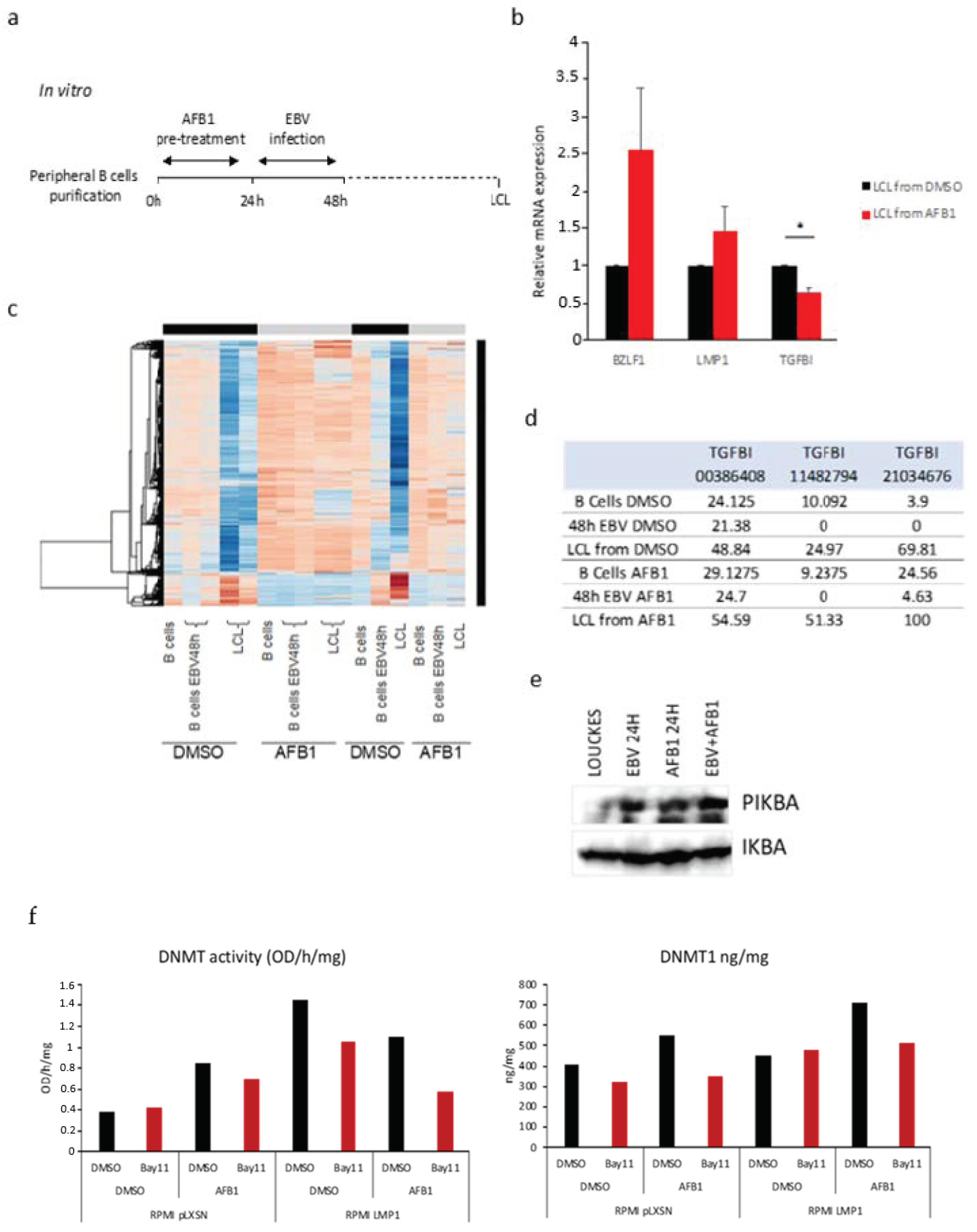
3.1. EBV and AFB1-Related BL Methylome Profiling Identifies Differentially Methylated Genes
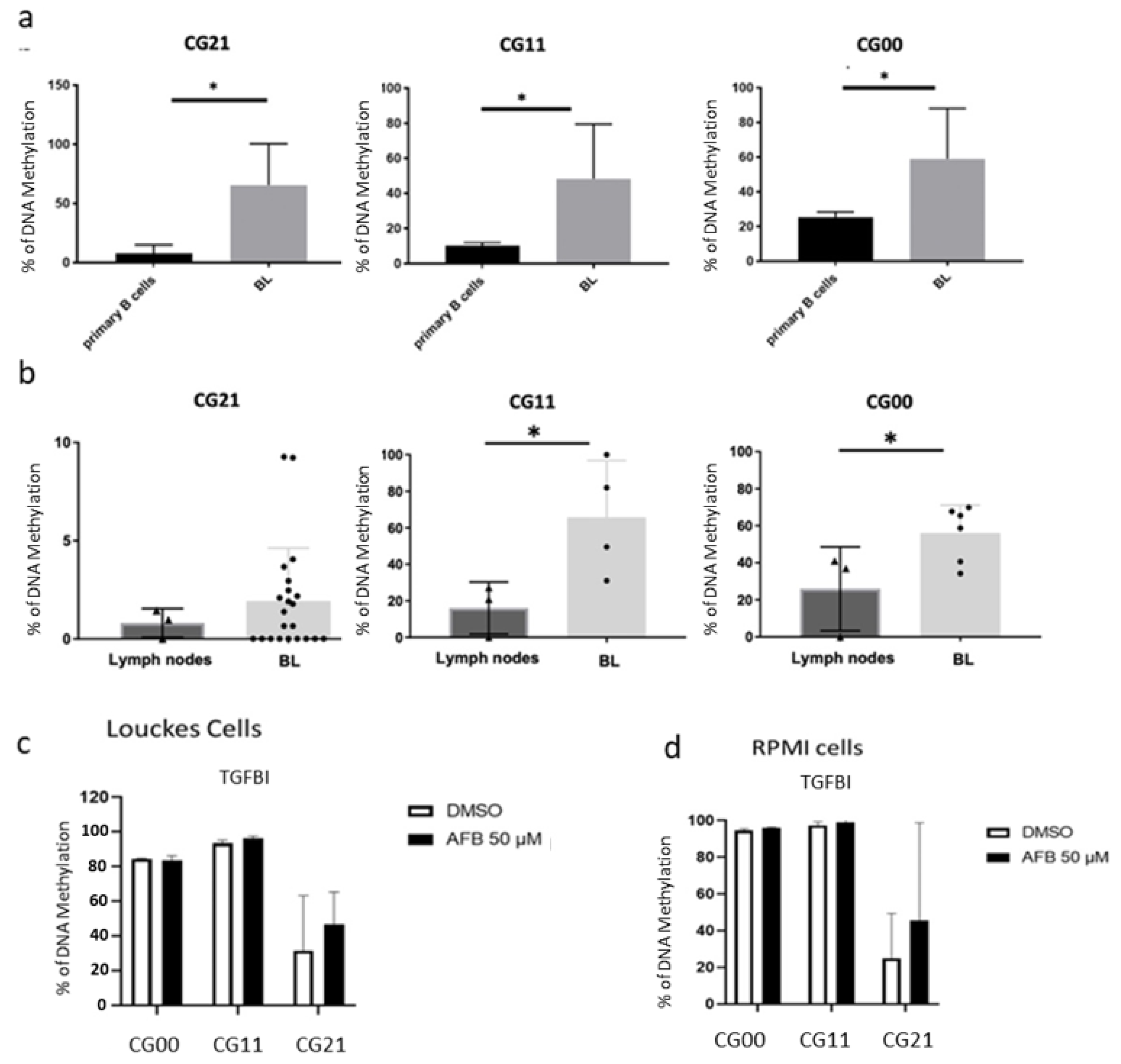
3.2. Validation of the Differential Methylation in the EBV and AFB1-Related BL Cells
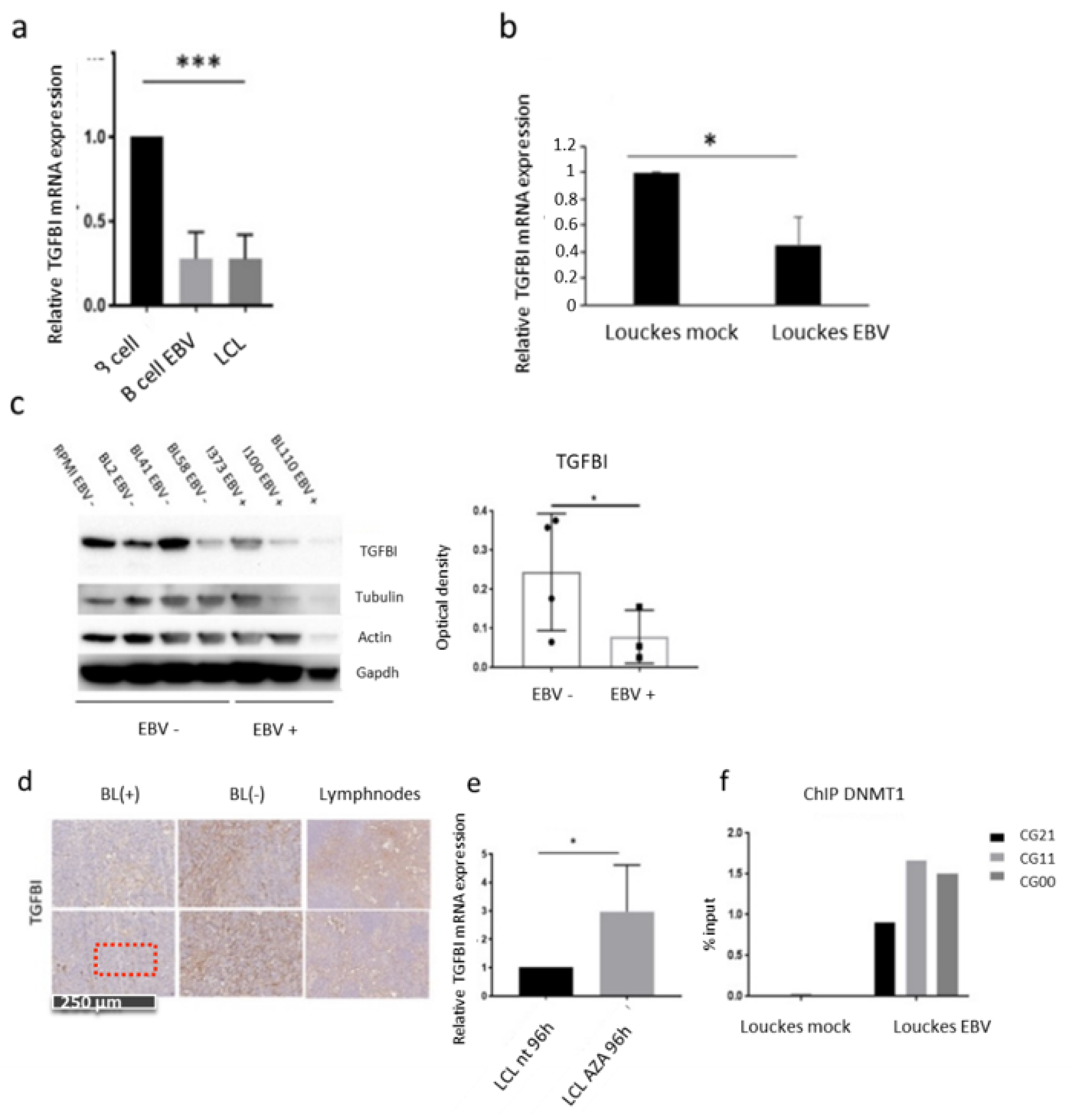
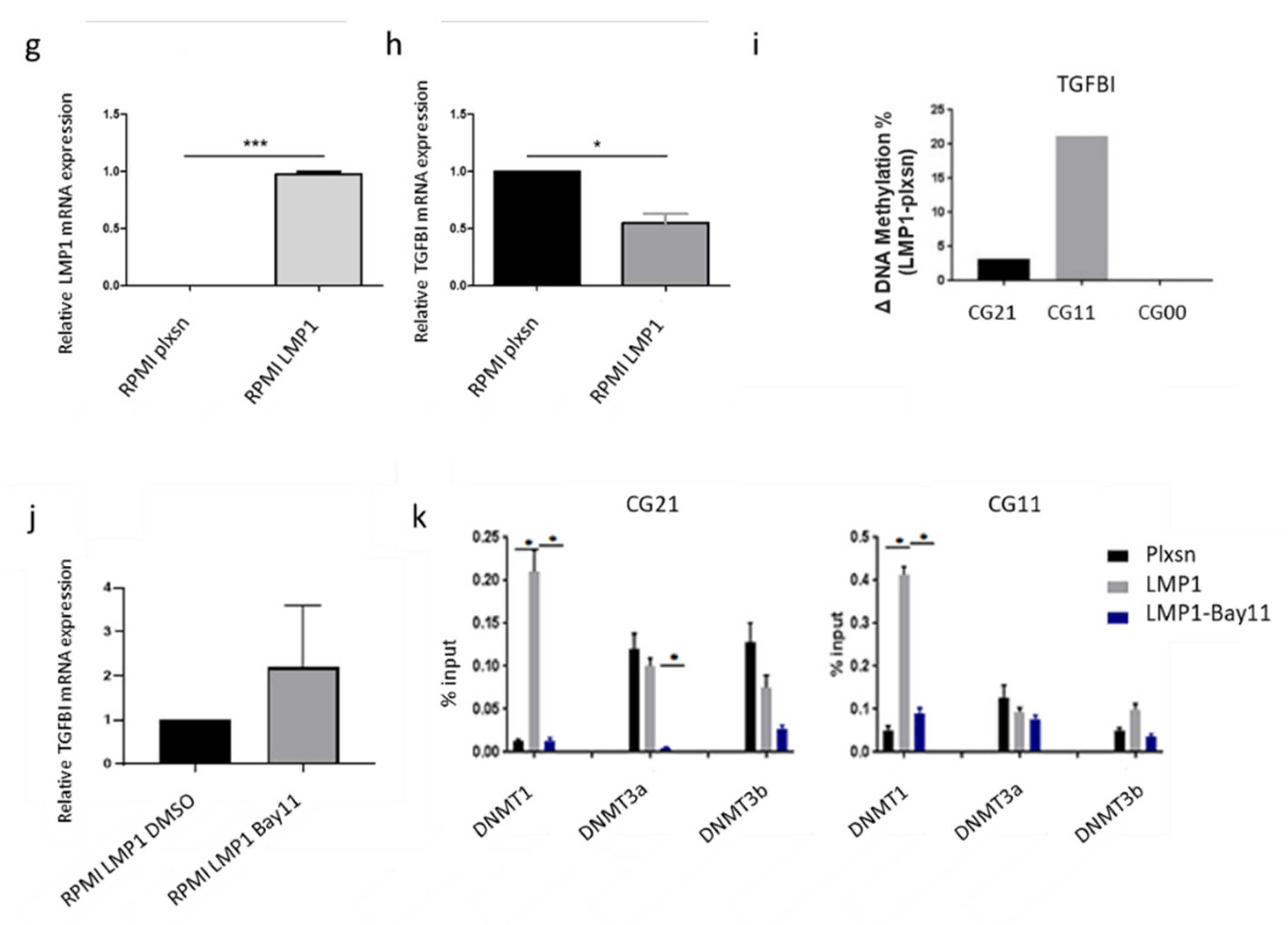
3.3. The Main EBV Oncoprotein LMP1 Downregulates TGFBI Expression during B Cells Infection
3.4. AFB1 Enhances the EBV-Induced TGFBI Downregulation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Disclaimer
References
- Blinder, V.; Fisher, S.G. The Role of Environmental Factors in the Etiology of Lymphoma. Cancer Investig. 2008, 26, 306–316. [Google Scholar] [CrossRef]
- Basso, K.; Dalla-Favera, R. Germinal centres and B cell lymphomagenesis. Nat. Rev. Immunol. 2015, 15, 172–184. [Google Scholar] [CrossRef] [PubMed]
- Herceg, Z.; Vaissière, T. Epigenetic mechanisms and cancer: An interface between the environment and the genome. Epigenetics 2011, 6, 804–819. [Google Scholar] [CrossRef]
- Jaenisch, R.; Bird, A. Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat. Genet. 2003, 33, 245–254. [Google Scholar] [CrossRef]
- Molyneux, E.M.; Rochford, R.; Griffin, B.; Newton, R.; Jackson, G.; Menon, G.; Harrison, C.J.; Israels, T.; Bailey, S. Burkitt’s lymphoma. Lancet 2012, 379, 1234–1244. [Google Scholar] [CrossRef] [Green Version]
- Mlynarczyk, C.; Fontán, L.; Melnick, A. Germinal center-derived lymphomas: The darkest side of humoral immunity. Immunol. Rev. 2019, 288, 214–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orem, J.; Mbidde, E.K.; Lambert, B.; de Sanjose, S.; Weiderpass, E. Burkitt’s lymphoma in Africa, a review of the epidemiology and etiology. Afr. Health Sci. 2007, 7, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Magrath, I. Epidemiology: Clues to the pathogenesis of Burkitt lymphoma. Br. J. Haematol. 2012, 156, 744–756. [Google Scholar] [CrossRef] [PubMed]
- Epstein, M.A.; Henle, G.; Achong, B.G.; Barr, Y.M. Morphological and biological studies on a virus in cultured lymphoblasts from burkitt’s lymphoma. J. Exp Med. 1965, 121, 761–770. [Google Scholar] [CrossRef] [Green Version]
- Hatton, O.L.; Harris-Arnold, A.; Schaffert, S.; Krams, S.M.; Martinez, O.M. The interplay between Epstein-Barr virus and B lymphocytes: Implications for infection, immunity, and disease. Immunol. Res. 2014, 58, 268–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, L.S.; Yap, L.F.; Murray, P.G. Epstein–Barr virus: More than 50 years old and still providing surprises. Nat. Rev. Cancer 2016, 16, 789–802. [Google Scholar] [CrossRef]
- Tao, Q.; Robertson, K.D. Stealth technology: How Epstein-Barr virus utilizes DNA methylation to cloak itself from immune detection. Clin. Immunol. 2003, 109, 53–63. [Google Scholar] [CrossRef]
- Pich, D.; Mrozek-Gorska, P.; Bouvet, M.; Sugimoto, A.; Akidil, E.; Grundhoff, A.; Hamperl, S.; Ling, P.D.; Hammerschmidt, W. First Days in the Life of Naive Human B Lymphocytes Infected with Epstein-Barr Virus. mBio 2019, 10, e01723-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shannon-Lowe, C.; Rickinson, A. The Global Landscape of EBV-Associated Tumors. Front. Oncol. 2019, 9, 713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.W.; Jiang, S.; Gewurz, B.E. Epstein-Barr Virus LMP1-Mediated Oncogenicity. J. Virol. 2017, 91, e01718-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eliopoulos, A.G.; Young, L.S. LMP1 structure and signal transduction. Semin. Cancer Biol. 2001, 11, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Lenardo, M.J.; Baltimore, D. 30 Years of NF-κB: A Blossoming of Relevance to Human Pathobiology. Cell 2017, 168, 37–57. [Google Scholar] [CrossRef] [Green Version]
- Taniguchi, K.; Karin, M. NF-κB, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef] [PubMed]
- Lavorgna, A.; Harhaj, E.W. EBV LMP1: New and shared pathways to NF-κB activation. Proc. Natl. Acad. Sci. USA 2012, 109, 2188–2189. [Google Scholar] [CrossRef] [Green Version]
- Hayden, M.S.; West, A.P.; Ghosh, S. NF-kappaB and the immune response. Oncogene 2006, 25, 6758–6780. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, Y.; Iwai, K. Roles of the NF-κB Pathway in B-Lymphocyte Biology. Curr. Top Microbiol. Immunol. 2016, 393, 177–209. [Google Scholar] [CrossRef]
- Nagel, D.; Vincendeau, M.; Eitelhuber, A.C.; Krappmann, D. Mechanisms and consequences of constitutive NF-κB activation in B-cell lymphoid malignancies. Oncogene 2014, 33, 5655–5665. [Google Scholar] [CrossRef] [Green Version]
- Thorley-Lawson, D.A. EBV Persistence--Introducing the Virus. Curr. Top. Microbiol. Immunol. 2015, 390, 151–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Münz, C. Latency and lytic replication in Epstein–Barr virus-associated oncogenesis. Nat. Rev. Microbiol. 2019, 17, 691–700. [Google Scholar] [CrossRef] [Green Version]
- Buschle, A.; Hammerschmidt, W. Epigenetic lifestyle of Epstein-Barr virus. Semin Immunopathol. 2020, 42, 131–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutcheson, R.L.; Chakravorty, A.; Sugden, B. Burkitt Lymphomas Evolve to Escape Dependencies on Epstein-Barr Virus. Front. Cellul. Infect. Microbiol. 2021, 10, 835. [Google Scholar] [CrossRef] [PubMed]
- Ambinder, R.F. Gammaherpesviruses and "Hit-and-Run" oncogenesis. Am. J. Pathol. 2000, 156, 1–3. [Google Scholar] [CrossRef]
- Vargas-Ayala, R.C.; Jay, A.; Manara, F.; Maroui, M.A.; Hernandez-Vargas, H.; Diederichs, A.; Robitaille, A.; Sirand, C.; Ceraolo, M.G.; Romero-Medina, M.C.; et al. Interplay between the Epigenetic Enzyme Lysine (K)-Specific Demethylase 2B and Epstein-Barr Virus Infection. J. Virol. 2019, 93, e00273-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leong, M.M.L.; Lung, M.L. The Impact of Epstein-Barr Virus Infection on Epigenetic Regulation of Host Cell Gene Expression in Epithelial and Lymphocytic Malignancies. Front. Oncol. 2021, 11, 201. [Google Scholar] [CrossRef]
- Hernandez-Vargas, H.; Gruffat, H.; Cros, M.P.; Diederichs, A.; Sirand, C.; Vargas-Ayala, R.C.; Jay, A.; Durand, G.; Le Calvez-Kelm, F.; Herceg, Z.; et al. Viral driven epigenetic events alter the expression of cancer-related genes in Epstein-Barr-virus naturally infected Burkitt lymphoma cell lines. Sci. Rep. 2017, 7, 5852. [Google Scholar] [CrossRef] [Green Version]
- Lam, W.K.J.; Jiang, P.; Chan, K.C.A.; Peng, W.; Shang, H.; Heung, M.M.S.; Cheng, S.H.; Zhang, H.; Tse, O.Y.O.; Raghupathy, R.; et al. Methylation analysis of plasma DNA informs etiologies of Epstein-Barr virus-associated diseases. Nat. Commun. 2019, 10, 3256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abate, F.; Ambrosio, M.R.; Mundo, L.; Laginestra, M.A.; Fuligni, F.; Rossi, M.; Zairis, S.; Gazaneo, S.; De Falco, G.; Lazzi, S.; et al. Distinct Viral and Mutational Spectrum of Endemic Burkitt Lymphoma. PLoS Pathog. 2015, 11, e1005158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Accardi, R.; Gruffat, H.; Sirand, C.; Fusil, F.; Gheit, T.; Hernandez-Vargas, H.; Le Calvez-Kelm, F.; Traverse-Glehen, A.; Cosset, F.L.; Manet, E.; et al. The mycotoxin aflatoxin B1 stimulates Epstein-Barr virus-induced B-cell transformation in in vitro and in vivo experimental models. Carcinogenesis 2015, 36, 1440–1451. [Google Scholar] [CrossRef] [Green Version]
- Magrath, I. Denis Burkitt and the African lymphoma. Ecancermedicalscience 2009, 3, 159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wild, C.P.; Gong, Y.Y. Mycotoxins and human disease: A largely ignored global health issue. Carcinogenesis 2010, 31, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Claeys, L.; Romano, C.; De Ruyck, K.; Wilson, H.; Fervers, B.; Korenjak, M.; Zavadil, J.; Gunter, M.J.; De Saeger, S.; De Boevre, M.; et al. Mycotoxin exposure and human cancer risk: A systematic review of epidemiological studies. Compr. Rev. Food Sci. Food Saf. 2020, 19, 1449–1464. [Google Scholar] [CrossRef]
- Some traditional herbal medicines, some mycotoxins, naphthalene and styrene. IARC Monogr. Eval. Carcinog. Risks Hum. 2002, 82, 1–556.
- Jiang, Y.; Jolly, P.E.; Preko, P.; Wang, J.S.; Ellis, W.O.; Phillips, T.D.; Williams, J.H. Aflatoxin-related immune dysfunction in health and in human immunodeficiency virus disease. Clin. Dev. Immunol. 2008, 2008, 790309. [Google Scholar] [CrossRef]
- Gong, Y.; Hounsa, A.; Egal, S.; Turner, P.C.; Sutcliffe, A.E.; Hall, A.J.; Cardwell, K.; Wild, C.P. Postweaning exposure to aflatoxin results in impaired child growth: A longitudinal study in Benin, West Africa. Environ. Health Perspect. 2004, 112, 1334–1338. [Google Scholar] [CrossRef] [PubMed]
- Dhakal, A.; Sbar, E. Aflatoxin Toxicity. In StatPearls; StatPearls Publishing LLC: Treasure Island, FL, USA, 2021. [Google Scholar]
- McCullough, A.K.; Lloyd, R.S. Mechanisms underlying aflatoxin-associated mutagenesis-Implications in carcinogenesis. DNA Repair 2019, 77, 76–86. [Google Scholar] [CrossRef]
- Hernandez-Vargas, H.; Castelino, J.; Silver, M.J.; Dominguez-Salas, P.; Cros, M.-P.; Durand, G.; Le Calvez-Kelm, F.; Prentice, A.M.; Wild, C.P.; Moore, S.E.; et al. Exposure to aflatoxin B1 in utero is associated with DNA methylation in white blood cells of infants in The Gambia. Int. J. Epidemiol. 2015, 44, 1238–1248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, P.C.; Collinson, A.C.; Cheung, Y.B.; Gong, Y.; Hall, A.J.; Prentice, A.M.; Wild, C.P. Aflatoxin exposure in utero causes growth faltering in Gambian infants. Int. J. Epidemiol. 2007, 36, 1119–1125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Granai, M.; Mundo, L.; Akarca, A.U.; Siciliano, M.C.; Rizvi, H.; Mancini, V.; Onyango, N.; Nyagol, J.; Abinya, N.O.; Maha, I.; et al. Immune landscape in Burkitt lymphoma reveals M2-macrophage polarization and correlation between PD-L1 expression and non-canonical EBV latency program. Infect. Agent Cancer 2020, 15, 28. [Google Scholar] [CrossRef] [PubMed]
- Mundo, L.; Del Porro, L.; Granai, M.; Siciliano, M.C.; Mancini, V.; Santi, R.; Marcar, L.; Vrzalikova, K.; Vergoni, F.; Di Stefano, G.; et al. Correction: Frequent traces of EBV infection in Hodgkin and non-Hodgkin lymphomas classified as EBV-negative by routine methods: Expanding the landscape of EBV-related lymphomas. Mod. Pathol. 2020, 33, 2637. [Google Scholar] [CrossRef]
- Delecluse, H.J.; Hilsendegen, T.; Pich, D.; Zeidler, R.; Hammerschmidt, W. Propagation and recovery of intact, infectious Epstein-Barr virus from prokaryotic to human cells. Proc. Natl. Acad. Sci. USA 1998, 95, 8245–8250. [Google Scholar] [CrossRef] [Green Version]
- Giarrè, M.; Caldeira, S.; Malanchi, I.; Ciccolini, F.; Leão, M.J.; Tommasino, M. Induction of pRb degradation by the human papillomavirus type 16 E7 protein is essential to efficiently overcome p16INK4a-imposed G1 cell cycle Arrest. J. Virol. 2001, 75, 4705–4712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez-Vargas, H.; Lambert, M.P.; Le Calvez-Kelm, F.; Gouysse, G.; McKay-Chopin, S.; Tavtigian, S.V.; Scoazec, J.Y.; Herceg, Z. Hepatocellular carcinoma displays distinct DNA methylation signatures with potential as clinical predictors. PLoS ONE 2010, 5, e9749. [Google Scholar] [CrossRef] [PubMed]
- Karpf, A.R.; Jones, D.A. Reactivating the expression of methylation silenced genes in human cancer. Oncogene 2002, 21, 5496–5503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, C.L.; Li, H.P.; Lu, Y.J.; Hsueh, C.; Liang, Y.; Chen, C.L.; Tsao, S.W.; Tse, K.P.; Yu, J.S.; Chang, Y.S. Activation of DNA methyltransferase 1 by EBV LMP1 Involves c-Jun NH(2)-terminal kinase signaling. Cancer Res. 2006, 66, 11668–11676. [Google Scholar] [CrossRef] [Green Version]
- Niller, H.H.; Wolf, H.; Minarovits, J. Viral hit and run-oncogenesis: Genetic and epigenetic scenarios. Cancer Lett. 2011, 305, 200–217. [Google Scholar] [CrossRef]
- da Silva, J.V.B.; de Oliveira, C.A.F.; Ramalho, L.N.Z. Effects of Prenatal Exposure to Aflatoxin B1: A Review. Molecules 2021, 26, 7312. [Google Scholar] [CrossRef] [PubMed]
- Hjalgrim, H.; Friborg, J.; Melbye, M. The epidemiology of EBV and its association with malignant disease. In Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis; Arvin, A., Campadelli-Fiume, G., Mocarski, E., Moore, P.S., Roizman, B., Whitley, R., Yamanishi, K., Eds.; Cambridge University Press: Cambridge, MA, USA, 2007. [Google Scholar]
- Esteller, M. CpG island hypermethylation and tumor suppressor genes: A booming present, a brighter future. Oncogene 2002, 21, 5427–5440. [Google Scholar] [CrossRef] [Green Version]
- Corona, A.; Blobe, G.C. The role of the extracellular matrix protein TGFBI in cancer. Cell Signal. 2021, 84, 110028. [Google Scholar] [CrossRef] [PubMed]
- Thapa, N.; Lee, B.H.; Kim, I.S. TGFBIp/betaig-h3 protein: A versatile matrix molecule induced by TGF-beta. Int. J. Biochem. Cell Biol. 2007, 39, 2183–2194. [Google Scholar] [CrossRef]
- Moses, H.L.; Coffey, R.J., Jr.; Leof, E.B.; Lyons, R.M.; Keski-Oja, J. Transforming growth factor beta regulation of cell proliferation. J. Cell Physiol. Suppl. 1987, 133 (Suppl. S5), 1–7. [Google Scholar] [CrossRef]
- Velapasamy, S.; Dawson, C.W.; Young, L.S.; Paterson, I.C.; Yap, L.F. The Dynamic Roles of TGF-β Signalling in EBV-Associated Cancers. Cancers 2018, 10, 247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palomeras, S.; Diaz-Lagares, Á.; Viñas, G.; Setien, F.; Ferreira, H.J.; Oliveras, G.; Crujeiras, A.B.; Hernández, A.; Lum, D.H.; Welm, A.L.; et al. Epigenetic silencing of TGFBI confers resistance to trastuzumab in human breast cancer. Breast Cancer Res. 2019, 21, 79. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Wen, G.; Zhao, Y.; Tong, J.; Hei, T.K. The role of TGFBI in mesothelioma and breast cancer: Association with tumor suppression. BMC Cancer 2012, 12, 239. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.; Dong, S.M.; Park, N.H. Frequent promoter hypermethylation of TGFBI in epithelial ovarian cancer. Gynecol. Oncol. 2010, 118, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.; Liu, J.; Guo, D.; Liu, P.; Zhao, Y. Epigenetic regulation of putative tumor suppressor TGFBI in human leukemias. Chin. Med. J. 2014, 127, 1645–1650. [Google Scholar] [PubMed]
- Lai, M.W.; Liang, K.H.; Yeh, C.T. Hepatitis B Virus preS/S Truncation Mutant rtM204I/sW196* Increases Carcinogenesis through Deregulated HIF1A, MGST2, and TGFbi. Int. J. Mol. Sci. 2020, 21, 6366. [Google Scholar] [CrossRef]
- Jiang, S.S.; Huang, S.F.; Huang, M.S.; Chen, Y.T.; Jhong, H.J.; Chang, I.C.; Chen, Y.T.; Chang, J.W.; Chen, W.L.; Lee, W.C.; et al. Dysregulation of the TGFBI gene is involved in the oncogenic activity of the nonsense mutation of hepatitis B virus surface gene sW182*. Biochim. Biophys. Acta 2014, 1842, 1080–1087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takegawa, S.; Jin, Z.; Nakayama, T.; Oyama, T.; Hieshima, K.; Nagakubo, D.; Shirakawa, A.K.; Tsuzuki, T.; Nakamura, S.; Yoshie, O. Expression of CCL17 and CCL22 by latent membrane protein 1-positive tumor cells in age-related Epstein-Barr virus-associated B-cell lymphoproliferative disorder. Cancer Sci. 2008, 99, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Nakagomi, H.; Dolcetti, R.; Bejarano, M.T.; Pisa, P.; Kiessling, R.; Masucci, M.G. The Epstein-Barr virus latent membrane protein-1 (LMP1) induces interleukin-10 production in Burkitt lymphoma lines. Int. J. Cancer 1994, 57, 240–244. [Google Scholar] [CrossRef]
- Cahir-McFarland, E.D.; Carter, K.; Rosenwald, A.; Giltnane, J.M.; Henrickson, S.E.; Staudt, L.M.; Kieff, E. Role of NF-kappa B in cell survival and transcription of latent membrane protein 1-expressing or Epstein-Barr virus latency III-infected cells. J. Virol. 2004, 78, 4108–4119. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Brooks, L.; Busson, P.; Wang, F.; Charron, D.; Kieff, E.; Rickinson, A.B.; Tursz, T. Epstein-Barr virus (EBV) latent membrane protein 1 increases HLA class II expression in an EBV-negative B cell line. Eur. J. Immunol. 1994, 24, 1467–1470. [Google Scholar] [CrossRef]
- Campbell, K.J.; Rocha, S.; Perkins, N.D. Active repression of antiapoptotic gene expression by RelA(p65) NF-kappa B. Mol. Cell 2004, 13, 853–865. [Google Scholar] [CrossRef]
- Wehbe, H.; Henson, R.; Meng, F.; Mize-Berge, J.; Patel, T. Interleukin-6 contributes to growth in cholangiocarcinoma cells by aberrant promoter methylation and gene expression. Cancer Res. 2006, 66, 10517–10524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Mayo, M.W.; Nagji, A.S.; Smith, P.W.; Ramsey, C.S.; Li, D.; Jones, D.R. Phosphorylation of RelA/p65 promotes DNMT-1 recruitment to chromatin and represses transcription of the tumor metastasis suppressor gene BRMS1. Oncogene 2012, 31, 1143–1154. [Google Scholar] [CrossRef]
- Iannetti, A.; Ledoux, A.C.; Tudhope, S.J.; Sellier, H.; Zhao, B.; Mowla, S.; Moore, A.; Hummerich, H.; Gewurz, B.E.; Cockell, S.J.; et al. Regulation of p53 and Rb links the alternative NF-κB pathway to EZH2 expression and cell senescence. PLoS Genet. 2014, 10, e1004642. [Google Scholar] [CrossRef] [Green Version]
- Winkler, J.; Abisoye-Ogunniyan, A.; Metcalf, K.J.; Werb, Z. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat. Commun. 2020, 11, 5120. [Google Scholar] [CrossRef] [PubMed]
- Menter, T.; Tzankov, A.; Dirnhofer, S. The tumor microenvironment of lymphomas: Insights into the potential role and modes of actions of checkpoint inhibitors. Hematol. Oncol. 2021, 39, 3–10. [Google Scholar] [CrossRef]
- Girigoswami, K.; Saini, D.; Girigoswami, A. Extracellular Matrix Remodeling and Development of Cancer. Stem Cell Rev. Rep. 2021, 17, 739–747. [Google Scholar] [CrossRef]
- Liu, Y.; Zhou, X.; Wang, X. Targeting the tumor microenvironment in B-cell lymphoma: Challenges and opportunities. J. Hematol. Oncol. 2021, 14, 125. [Google Scholar] [CrossRef]
- Autio, M.; Leivonen, S.K.; Brück, O.; Mustjoki, S.; Mészáros Jørgensen, J.; Karjalainen-Lindsberg, M.L.; Beiske, K.; Holte, H.; Pellinen, T.; Leppä, S. Immune cell constitution in the tumor microenvironment predicts the outcome in diffuse large B-cell lymphoma. Haematologica 2021, 106, 718–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Use and Gene | Forward Primer (5′ to 3′) | Reverse Primer (5′ to 3′) |
|---|---|---|
| qPCR | ||
| TGFBI N.1 | GTCCACAGCCATTGACCTTT | GAGTTTCCAGGGTCTGTCCA |
| TGFBI N.2 | GCCCTACCACTCTCAAACCT | GTTGACATTGCTGACCAGGG |
| LMP1 | CCAGTCCAGTCACTCATAACG | CCTACATAAGCCTCTCACACT |
| EBNA1 | GGTCGTGGACGTGGAGAAAA | GGTGGAGACCCGGATGATG |
| BZLF1 | AATGCCGGGCCAAGTTTAAGCA | TTGGGCACATCTGCTTCAACAGGA |
| β2 microglobulin | CTCACGTCATCCAGCAGAGA | CGGCAGGCATACTCATCTTT |
| ACTIN | CGGCAGGCATACTCATCTTT | TCAACTGGTCTCAAGTCAGTG |
| GAPDH | GCCAAAAGGGTCATCATC | TGCCAGTGAGCTTCCCGTTC |
| ChIP-qPCR | ||
| CG11482794 | CTCCATGGCCGCTCTCGT | CCCCGACTACCTGACCTTC |
| CG21034676 | AAGGGCTGGGAAAACTGAG | GGCTCCAGGGAAGTGAGAG |
| CG00386408 | CTGCGGAAGGTCAGGTAGTC | AACTCCCTCCCTCTCTCCTT |
| Pyrosequencing Primers | Sequence | |||
|---|---|---|---|---|
| Forward Primer (5′ to 3′) | Reverse Primer (5′ to 3′) | Sequencing Primer | Sequence to Analyse | |
| CG11482794 | GTTTTGGTTTTGGTTTTGGG | TCCCTCCCTCTCTCCTTCC | GGGTTTYGTTAAG | TYGTTTTATTAG |
| CG21034676 | TGGGTGTTTAGGGTAGTTAGGG | CCCAAAACCAAAACCAAAAC | TAGGGTAGTTAGG | GGYGTAYGGGT |
| CG00386408 | GTTTTGGTTTTGGTTTTGGG | TCCCTCCCTCTCTCCTTCC | TGAATTGGGTTGGG | GGYGTAGGGGA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manara, F.; Jay, A.; Odongo, G.A.; Mure, F.; Maroui, M.A.; Diederichs, A.; Sirand, C.; Cuenin, C.; Granai, M.; Mundo, L.; et al. Epigenetic Alteration of the Cancer-Related Gene TGFBI in B Cells Infected with Epstein–Barr Virus and Exposed to Aflatoxin B1: Potential Role in Burkitt Lymphoma Development. Cancers 2022, 14, 1284. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14051284
Manara F, Jay A, Odongo GA, Mure F, Maroui MA, Diederichs A, Sirand C, Cuenin C, Granai M, Mundo L, et al. Epigenetic Alteration of the Cancer-Related Gene TGFBI in B Cells Infected with Epstein–Barr Virus and Exposed to Aflatoxin B1: Potential Role in Burkitt Lymphoma Development. Cancers. 2022; 14(5):1284. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14051284
Chicago/Turabian StyleManara, Francesca, Antonin Jay, Grace Akinyi Odongo, Fabrice Mure, Mohamed Ali Maroui, Audrey Diederichs, Cecilia Sirand, Cyrille Cuenin, Massimo Granai, Lucia Mundo, and et al. 2022. "Epigenetic Alteration of the Cancer-Related Gene TGFBI in B Cells Infected with Epstein–Barr Virus and Exposed to Aflatoxin B1: Potential Role in Burkitt Lymphoma Development" Cancers 14, no. 5: 1284. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14051284