
The Minimal Residual Disease Using Liquid Biopsies in Hematological Malignancies
by
 , and
, and
Rafael Colmenares
1,
Noemí Álvarez
1,2,
Santiago Barrio
1,2,
Joaquín Martínez-López
1,2,3,4 and
and
Rosa Ayala
1,2,3,4,* 1
Hematology Department, Hospital Universitario 12 de Octubre, Instituto de Investigación Sanitaria Imas12, 28041 Madrid, Spain
2
Hematological Malignancies Clinical Research Unit, CNIO, 28029 Madrid, Spain
3
Department of Medicine, Complutense University of Madrid, 28040 Madrid, Spain
4
Centro de Investigación Biomédica en Red de Cáncer (CIBERONC), Instituto Carlos III, 28029 Madrid, Spain
*
Author to whom correspondence should be addressed.
Cancers 2022, 14(5), 1310; https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14051310
Submission received: 24 January 2022
/
Revised: 23 February 2022
/
Accepted: 27 February 2022
/
Published: 3 March 2022
(This article belongs to the Special Issue Latest Development in B Cell Malignancies)
Abstract
:Simple Summary
Monitoring the response to treatment in hematologic malignancies is essential in defining the best way to optimize patient management. In general, achieving a deeper response has been shown to lead to a better prognosis, and the techniques used to study the minimal residual disease (MRD) are becoming more precise. The use of liquid biopsies, that is, analyzing the presence of alterations in nucleic acids, usually in peripheral blood or other biological fluids, is being studied and optimized with increasingly innovative molecular techniques, such as next-generation sequencing (NGS) in the monitoring of the MRD, avoiding, in many cases, more invasive tests in different hematological neoplasms. Currently, liquid biopsies are not standardized for the MRD monitoring, but there is increasing evidence of its correlation with other techniques to measure responses to treatments and patient outcomes.
Abstract
The study of cell-free DNA (cfDNA) and other peripheral blood components (known as “liquid biopsies”) is promising, and has been investigated especially in solid tumors. Nevertheless, it is increasingly showing a greater utility in the diagnosis, prognosis, and response to treatment of hematological malignancies; in the future, it could prevent invasive techniques, such as bone marrow (BM) biopsies. Most of the studies about this topic have focused on B-cell lymphoid malignancies; some of them have shown that cfDNA can be used as a novel way for the diagnosis and minimal residual monitoring of B-cell lymphomas, using techniques such as next-generation sequencing (NGS). In myelodysplastic syndromes, multiple myeloma, or chronic lymphocytic leukemia, liquid biopsies may allow for an interesting genomic representation of the tumor clones affecting different lesions (spatial heterogeneity). In acute leukemias, it can be helpful in the monitoring of the early treatment response and the prediction of treatment failure. In chronic lymphocytic leukemia, the evaluation of cfDNA permits the definition of clonal evolution and drug resistance in real time. However, there are limitations, such as the difficulty in obtaining sufficient circulating tumor DNA for achieving a high sensitivity to assess the minimal residual disease, or the lack of standardization of the method, and clinical studies, to confirm its prognostic impact. This review focuses on the clinical applications of cfDNA on the minimal residual disease in hematological malignancies.
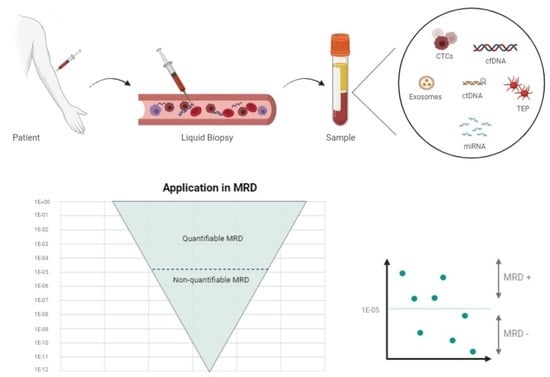
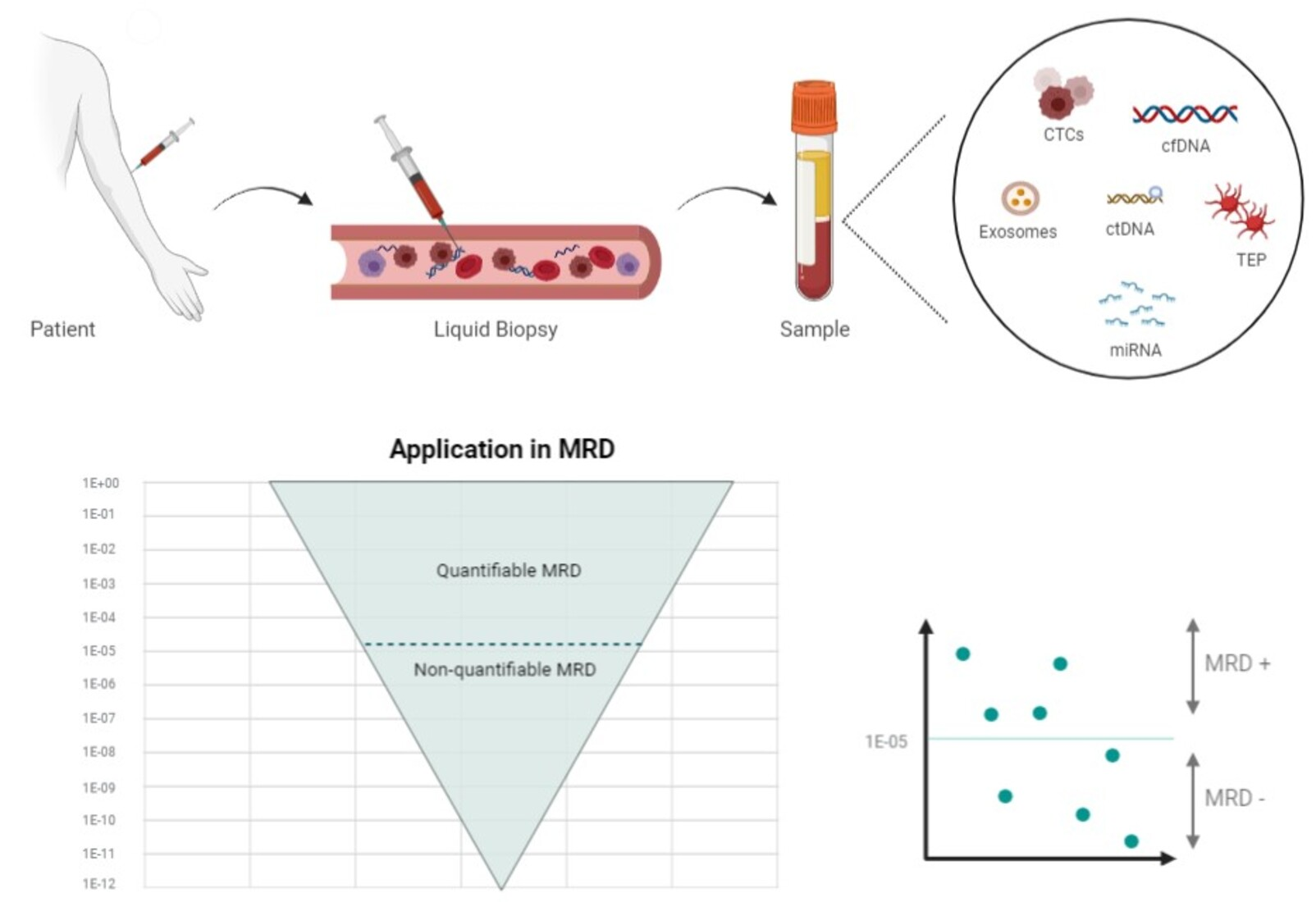
1. Introduction
Hematological malignancies are the result of molecular alterations that affect the genes involved in cell growth and proliferation. Sometimes, the molecular profile of a tumor is obtained only by analyzing a small portion, as in lymphomas, which can lead to the loss of information because of the heterogeneity of the tumor or the presence of multiple tumor sites. In addition, it may, sometimes, be difficult to obtain a good sample to analyze, which involves invasive procedures and increases the risk to the patient. Monitoring the tumor by repeat biopsies is usually not feasible [1].
1.1. Liquid Biopsy Components
Various studies have shown that the blood contains remnants of some tissues, including tumor tissues. The term “liquid biopsy” is an attempt to approximate the molecular profile of a tumor by analyzing the peripheral blood using different methods: circulating tumor cells (CTCs), cell-free circulating nucleic acids (DNA, mRNA, micro-RNA, or non-coding RNA), “tumor-educated platelets” (TEPs), or exosomes [2,3].
- Circulating tumor cells (CTCs): CTCs come from tumors as an early step in blood-borne metastasis. CTCs are transient in blood, with a half-life of 1–2.4 h and are presented in a low abundance in most patients. Various techniques have been defined to isolate and analyze CTCs. These cells can even be used to establish cell line models to carry out therapeutic studies [4]. In hematological malignancies, the analysis of CTCs is possible in acute myeloid leukemia (AML) or acute lymphoblastic leukemia (ALL), myelodysplastic syndromes (MDSs), myeloproliferative neoplasms (MPNs), multiple myelomas (MM), and some lymphomas, such as mantle cell lymphoma (MCL), follicular lymphoma (FL), marginal zone lymphoma, small lymphocytic lymphoma, and a subset of Burkitt lymphoma. In contrast, diffuse large B-cell lymphoma (DLBCL) and classical Hodgkin lymphoma (cHL) do not typically harbor CTCs [5];
- RNA: MicroRNAs (miRNAs) are a class of small molecules of 19–24 nucleotides in length, and they are the most abundant RNA molecules in the blood; they can be carried in the exosomes or TEPs. They have a high stability and play an important role in tumor growth and treatment resistance [6];
- Tumor-educated platelets (TEP): Platelets are circulating anucleated fragments originating from megakaryocytes in the bone marrow, and they participate in hemostasis and the initiation of wound healing. However, they also have a role in systemic and local responses to tumor growth, as tumor cells alter the RNA profile of these platelets. In addition, TEPs can ingest the circulating mRNA released by tumor cells or solubilized tumor-associated proteins [7]. These interactions may signify a potential for cancer diagnosis or monitoring [8];
- Exosomes: Exosomes are a type of extracellular vesicle of endocytic origin, ranging in size between 30 and 100 nm; they are detectable in the blood of patients with some types of cancer, and they carry proteins and nucleic acids. They are analyzed through their RNA content [9]. For example, the ability of HL-specific exosomal microRNAs (miRNAs) to inform a treatment response in HL has been studied. The authors found that the specificity and sensitivity of exosomal miRNAs are superior to protein-bound miRNAs, with regard to HL detection [10];
- DNA: The existence of cell-free DNA (cfDNA) was first described in 1948, and some studies have reported a higher amount of cfDNA in patients with cancer [11,12,13]. Initially, some reports have identified apoptosis, necrosis, or both, as the main source of cfDNA. During apoptosis, the chromosomes are trimmed by DNases into multiple nucleosomal units of 180 bp that are released into the blood stream. These fragments “circulate” for one to three hours before they are ingested by phagocytes and other cells, whereas the cfDNA is completely digested into nucleotides by lysosomes [14]. Necrosis causes the random nonspecific and incomplete digestion of DNA. Some studies have suggested that DNA may be released by living cells [15,16,17]. This review is focused on circulating tumor DNA (ctDNA) and cfDNA, and there are differences between these concepts. cfDNA fragments are longer, with a size range of 160–180 bp, indicating a caspase-dependent apoptotic cleavage, while ctDNA fragments range between 90 and 150 bp, and others range between 250 and 320 bp [18]. The proportion of ctDNA in cfDNA varies, and the most-used methods to detect cfDNA are the real-time quantitative polymerase chain reaction (qRT-PCR) and next-generation sequencing (NGS) [19,20]. Liquid biopsies, by the quantification of cfDNA, can be used for all types of lymphomas, because the plasma from peripheral blood (PB) regularly contains low levels of detectable lymphoma-derived ctDNA, as well as in myeloid pathology (AML, MDS, and MPN) or other lymphoid pathology (MM and ALL) [21]. Therefore, the ctDNA-based liquid biopsy has emerged as a platform to genotype these tumors;
The first methods used to analyze ctDNA were based on PCR, using technologies such as TaqMan, PNA clamps, and the Scorpion Amplification Refractory Mutation System; however, they were limited because of their analytical sensitivity and specificity [22]. In the last decade, other newer techniques have been used: digital PCR (dPCR) and NGS have emerged, increasing the detection thresholds by up to 0.01% for mutant allele abundance [23];
- ○
- dPCR-based methods have a high sensitivity (0.01%) (better than real-time quantitative PCR), but they can only detect a few alterations simultaneously, and they must be optimized for each mutation. In this technique, thousands of individual reactions are analyzed with each sample, calculating whether a PCR end-point is reached, and evaluating the absolute number of molecules in the sample. It is useful in liquid biopsies because it is a precise and sensitive technique, detecting low-abundance targets. [24];
- ○
- In NGS, millions of DNA fragments are generated in a single sequencing process; enrichment can be performed to select the areas of interest. Finally, a massive and parallel sequencing is carried out. NGS-based methods can detect multiple alterations simultaneously. Initially, they had an insufficient sensitivity (>1%), but various groups have been developing methods that allow for a greater sensitivity. Indeed, some of them have tried to use the deep sequencing of a limited number of amplicons for the commonly mutated genes in cancer. The technique, called CAncer Personalized Profiling by deep Sequencing (CAPP-Seq), can detect all major classes of mutations (single nucleotide variants, indels, rearrangements, and copy number alterations). Capture-based NGS methods enrich genomic regions, before sequencing, by a hybridization of target regions to antisense oligonucleotides. With this technique, large portions of the genome can be examined [25].
1.2. Minimal Residual Disease Using Liquid Biopsy
The minimal/measurable residual disease (MRD) is defined as the persistence of a small number of malignant cells after the initial treatment, undetectable by morphologic or conventional screening methods. Table 1 and Table 2 detail the methods used in clinical practice to evaluate the response to treatment in different hematological malignancies.
In myeloid pathology, the MRD-guided approaches have become an attractive therapeutic strategy (e.g., CML, AML, and MDS), allowing for a more individualized therapy, possibly leading to less treatment-related toxicities and better outcomes. Moreover, the treatment, upon molecular relapse, is more effective than upon hematologic relapse (e.g., after allogeneic HCT in AML and MDS) [26]. The monitoring of the clonal evolution of AML identifies the leukemia subtype, clinical outcome, and potential new drug targets for post-remission strategies or relapse [27,28]. However, a universal MRD marker for MDS and AML is unlikely, because of the genotypic and phenotypic heterogeneity of these diseases [29]. Thus, a more effective strategy may be individualized MRD monitoring using a targeted next-generation sequencing panel [30].
In lymphoid pathology, the MRD-guided approaches, mainly based on the monitoring of the clonal rearrangements of Igs or TCR, have also become a therapeutic strategy for ALL and MM. However, the available MRD strategies have not yet been incorporated into the assistance practice for other lymphoid tumors, where the imaging methods are the method of choice to assess responses to treatments.
Liquid biopsies have several advantages over bone marrow for the detection of MRD: They are less invasive, they may provide a more comprehensive molecular overview of tumor heterogeneity, and they make it easier to obtain repeated blood samples over time to understand the dynamics of the response to a specific treatment. Multiple studies have demonstrated a satisfactory positive predictive value of ctDNA detection, as the presence of the MRD identified through ctDNA is associated with a lower disease-free survival (DFS) rate. Nevertheless, several hurdles maintain the negative predictive value of the technique at a low level, namely, the detection methods are not sensitive enough to detect ctDNA at very low amounts, new clones that are not captured by the selected technique can emerge, and the timing of the post-treatment sampling can be inappropriate [31]. Moreover, in the case of cfDNA, the logistics of the sample processing are crucial to avoid white blood cell (WBC) lysis and the subsequent genomic DNA contamination. To avoid this issue, samples need to be processed in less than four hours, or collected in tubes with a cell stabilizer [32].

{kind=link}
Table 1.
Minimal residual disease monitoring methods in myeloid malignancies.
| Method | APL | AML | MDS | MPN | CML |
|---|---|---|---|---|---|
| Image Methods | No | No | No | Yes | No |
| CT or PET/CT MRI | Spleen measurement by CT or MRI (clinical trials) | ||||
| Histologic/morphologic methods | Yes, BM or PB, 10−2 | Yes, BM, 10−2 | Yes, BM, 10−2 | Yes, BM, 10−2 (clinical trials) | No |
| MFC methods | No | Yes, BM, 10−4 or 10−5 | Yes, BM, 10−4 or 10−5 | No recommendations | No |
| Molecular methods | Yes, RQ-PCR (PML/RARA), BM, 10−5 | RQ-PCR *, BM or PB, 10−6 | No recommendations | No recommendations | Yes, PB (BCR/ABL1), 10−5 |
| NGS methods | No | NGS **, BM, 10−6 | Investigational use (clinical trials) | Investigational use (clinical trials) | No |
| Timing of MRD assessment | Post-induction time and PCR every 3 m for 2 years | Upon completion of the initial induction, additional time points should be guided according to the regimen used before allogeneic transplantation | No recommendations | Only in clinical trials *** | PCR every 3 m for one year, then every 6 m |
| References | [33,34] | [34] | [35] | [36,37,38] | [39] |
APL, acute promyelocytic leukemia; AML, acute myeloid leukemia; MDS, myelodysplastic syndrome; MPS, myeloproliferative syndrome; CML, chronic myeloid leukemia; CT, computed tomography; PET, positron emission tomography; MRI, magnetic resonance imaging; CR, complete response; y, year; BM, bone marrow; PB, peripheral blood; FISH, fluorescence in situ hybridization; MFC, multiparametric flow cytometry; RQ-PCR, real quantitative PCR; ASO-PCR, allelic specific oligonucleotide PCR; NGS, next-generation sequencing; MRD, minimal residual disease; m, month. The information in the boxes includes the test used, the sample to be studied, and the sensitivity of the method. * CBFb-MYH11, RUNX1-RUNX1T1, and mutNPM1. ** Excluded DTA mutations. *** 2013 IWG-MRT and ELN guidelines recommend monitoring the response (anemia response, spleen response, and symptom response), as well as signs and symptoms of disease progression every three to six months during the course of treatment. BM should be performed as clinically indicated.
Table 2.
Minimal residual disease monitoring methods in lymphoid malignancies.
| Method | ALL | DLBCL | FL | HL | CLL | MM |
|---|---|---|---|---|---|---|
| Image Methods | No | Yes | Yes | Yes | Yes | Yes |
| CT or PET/CT MRI | PET-TAC scan or CT scan contrast | PET-TAC scan or CT scan contrast | CR includes PET negative within 3 m posttreatment. Consider body CT with contrast no more often than every 6 m for the first 2 y following completion of therapy | Lymphoid nodes, spleen, and liver evaluation by CT | PET/CT | |
| Histologic/morphologic methods | Yes, BM, 10−2 | BM biopsy (optional) | BM biopsy (optional) | No, unless there is BM involvement at diagnosis | Yes, BM, 10−2 | Yes, BM with FISH, 10−2 |
| MFC methods | Yes, BM, 10−4 | No | No | No | Yes, BM or PB, 10−5 | Yes, BM, 10−5 |
| Molecular methods | RQ-PCR, BM, 10−6 | No | No | No | ASO-PCR, BM or PB, 10−4 | No |
| NGS methods | NGS Igs-TCR, BM, 10−6 | NGS liquid biopsy, investigational use | NGS liquid biopsy, investigational use | Investigational use | NGS Igs, BM or PB, 10−6 | NGS Igs, BM, 10−6 |
| Timing of MRD assessment | Upon completion of initial induction and additional time points; should be guided according to the regimen used | Post-third cycle and every 3 m | Post-third cycle every 3–6 m for 5 years | Post-third cycle. Additional time points should be guided according to the regimen used | Post-third cycle every 3–6 m for 5 years | Post-third cycle. Consider body CT with contrast no more often than every 6 m for the first 2 y following completion of therapy |
| References | [40] | [41] | [41] | [42] | [43] | [44] |
ALL, acute lymphoblastic leukemia; DLBCL, diffuse large B-cell lymphoma; FL, follicular lymphoma; HL, Hodgkin lymphoma; CLL, chronic lymphatic leukemia; MM, multiple myeloma; CT, computed tomography; PET, positron emission tomography; MRI, magnetic resonance imaging; CR, complete response; y, year; BM, bone marrow; PB, peripheral blood; FISH, fluorescence in situ hybridization; MFC, multiparametric flow cytometry; RQ-PCR, real quantitative PCR; ASO-PCR, allelic specific oligonucleotide PCR; NGS, next-generation sequencing; MRD, minimal residual disease; m, month; y; year. The information in the boxes includes the test used, the sample to be studied, and the sensitivity of the method.
2. Methods
References for this review were identified through searches of PUBMED with the search terms “cell free DNA,” “liquid biopsy,” “MDS,” “MPN,” “AML,” “NHL,” “CLL,” “MM,” and “ALL,” published from 1 January 2000 to 31 December 2021. Articles were also identified through searches of the reference authors’ names. Only papers published in English were reviewed. The final reference list was generated on the basis of significance and relevance to the broad scope of this review. Note that when we searched for the term cfDNA and cancer, we found 20,446 papers; however, if we associated these terms with MRD, only 116 results appeared. If we carried out this specific search for leukemia, lymphomas, or myelomas, four, seven, or seven results appeared, respectively. This shows that there are still not many publications with the relevant data on the usefulness of liquid biopsies to assess the MRD. However, there is a great interest in this application and much work is being conducted on different approaches in order to optimize these liquid biopsy techniques to achieve an adequate sensitivity for detecting tumor persistence or relapse.
3. Myeloid Malignancies
3.1. Acute Myeloid Leukemia
Acute myeloid leukemia (AML) is a malignancy of the hematopoietic stem cell precursors of the myeloid lineage, causing an overproduction of neoplastic clonal myeloid stem cells and the infiltration of extramedullary organs [45,46,47,48,49]. A correct diagnosis and classification of the disease are critical for the treatment of patients with AML, and an evaluation of the response to the treatment by the MRD quantification is essential to optimize treatment and select the appropriate method of response consolidation. The quantification of the MRD is very important because it can identify those patients at a high risk of relapse. Currently, studies including the morphologic evaluation on bone marrow (BM) aspirate or biopsies, cytogenetic analyses, and molecular testing using gene panels or NGS are currently being carried out for diagnoses and follow-ups [50]. Nevertheless, several studies have explored the use of peripheral blood (PB) as an input for the detection of residual disease, and the MRD obtained from the PB is already being incorporated into AML protocols for clinical decision making (e.g., CBF and NPM1 AML Pethema protocols with the MRD by RT-qPCR for follow-up RUNX1-RUNX1T1, CBFb-MYH11, and NPM1 mutations) [51,52,53]. Maurillo et al. analyzed BM and PB samples in 50 AML patients, 48 of whom were re-evaluated after consolidation. The preliminary results showed a concordance in the CTC quantification between PB and BM samples, increasing the disease stratification value and improving the MRD monitoring in AML patients. The analysis of the MRD after consolidation revealed useful prognostic information [54].
The MRD in AML is defined as the presence of leukemic blasts at a level lower than the limit of conventional morphologic detection (1:1000–1:106 white blood cells) [55]. The main techniques used, thus far, for the MRD in AML patients are multiparameter flow cytometry (MFC) and molecular techniques using RT-qPCR. The detection of the MRD by MFC uses the leukemia-associated immunophenotypes (LAIPs) approach (LAIPs are defined at diagnosis and their presence is subsequently monitored at follow-up) and the DfN approach (DfN, different from normal, irrespective of the LAIPs at diagnosis) and is applicable in most AML patients, with a sensitivity reported from 10−3 to 10−5. The molecular MRD is more sensitive and specific than flow cytometry, and real-time quantitative PCR (RT-qPCR) can achieve a sensitivity of 10−4–10−6 [51,56,57,58,59]. Droplet digital PCR (ddPCR) can reach a sensitivity of 10−4–10−6 as well [52,58], and NGS can achieve a sensitivity of 10−4–10−5 [27,60,61]. The problem with RT-qPCR is that that its applicability is limited to 30–40% of AMLs. NGS has an acceptable sensitivity, but with a better applicability than RT-qPCR (>85% of AML cases), and it provides new insights into the evolution of subclones that emerge under treatment, or during disease progression. This allows the availability of markers that can be used to measure the MRD to be expanded [62,63,64].
Advances in NGS technology have also enabled the sequencing of increasingly low amounts of DNA, using a variety of sources [65]. Blood contains many types of biological materials, such as circulating cells, mRNA, proteins, and cfDNA. In the blood of patients with AML, a part of the ctDNA is released by the tumor [66], but the tumor cells themselves circulate in the peripheral blood of these AML patients, and are called blastic cells. This CTC quantification has been incorporated into protocols, as previously cited.
However, there is not much reported experience with cfDNA in AML patients (Table 3). Through new technologies, the number of studies carried out in this field is increasing. Short et al. analyzed cfDNA and BM samples in 22 AML patients. They analyzed 28 genes and found that five mutations were detected only in the cfDNA, 15 mutations were detected in the BM, and 19 mutations were detected in both samples. Generally, the mutations detected in only one source had a variant allele frequency (VAF) of less than 10%. This suggests that both methods could miss subclonal populations. In addition, the cfDNA samples detected new or persistent mutations that implied a relapse. The results suggest that cfDNA and BM are complementary in the follow-up and monitoring of diseases in these patients [67].
Nakamura et al. conducted a study in 51 patients (37 with AML and 14 with MDS) to investigate the role of ctDNA in the risk of relapse after undergoing myeloablative allogeneic hematopoietic stem cell transplantation (alloSCT), with a median follow-up of 32 months post-alloSCT, where 16 out of 51 patients relapsed at a median of seven months. They collected ctDNA and BM samples at diagnosis and at follow-up. NGS studies with a 54- or 141-gene panel, or WES, were carried out. They were able to observe an increase in ctDNA in relapsed patients, determining that non-invasive testing has comparable utility in the BM testing, predicting the risk of relapse [68]. In this work, DNMT3A, TET2, and ASXL1 mutations (DTA gene mutations) were prognostic factors of leukemia relapse.
Rausch et al. demonstrated the application of a new method called the double drop-off digital droplet PCR (DDO-ddPCR) for the detection of gene mutations in NPM1, IDH2, and NRAS, which can detect and quantify diverse alterations at two nearby hotspot regions present in these genes. They used this method for disease monitoring, and compared it to qPCR, ddPCR, and NGS. At 38 time points (78%), the results of the cfDNA-ddPCR and BM qPCR were concordant, whereas at 11 time points (22%), during the follow-up, an NPM1 mutation was detected in the BM but not in the PB cfDNA. The cfDNA analysis was found to have a similar sensitivity compared to the quantitative PCR-based analysis of peripheral blood, but a lower sensitivity compared to the BM qPCR [69].
In conclusion, the use of liquid biopsies in AML shows promising results in terms of disease staging, monitoring, prognosis, and treatment [67], as well as in the detection of early relapse [70]. Although CTCs and cfDNA have been shown to be new biomarkers in the blood [25], at the moment, these cannot yet replace BM studies, mainly due to the low concentration of cfDNA in the blood [67]. An additional obstacle in the current molecular MRD approaches, and one that also affects liquid biopsy studies, is to find the molecular variants that represent, and are specific to, leukemic cells, with the capability of relapsing [71]. The determination of the MRD is complicated by the fact that many treated patients have persistent clonal hematopoiesis (CH) that may not reflect residual AML [72]. For this, the ELN group in the consensus document for the MRD in AML 2021 provided the following recommendations: To consider all the detected mutations as potential MRD markers; germline mutations (VAF of ~50% in the genes ANKRD26, CEBPA, DDX41, ETV6, GATA2, RUNX1, and TP53) should be excluded as NGS-MRD markers, as they are non-informative for the MRD; and DTA mutations can be found in age-related clonal hematopoiesis and should be excluded from the MRD analysis, as the mutations associated with clonal hematopoiesis often persist during remission and, thus, may not represent the leukemic clone. If the only detectable mutations are in the DTA genes, it is recommended to use MFC and/or PCR for the MRD assessment; mutations in signaling pathway genes (e.g., FLT3-ITD, FLT3-TKD, KIT, KRAS, and NRAS) likely represent residual AML when detected, but they are often subclonal and have a low negative predictive value. These mutations are best used in combination with additional MRD markers; NGS-MRD analyses in patients treated with targeted agents (FLT3 and IDH1/IDH2 inhibitors) should include the molecular marker that is targeted, but also others that are present in the sample [33].
Table 3.
Liquid biopsies in acute myeloid leukemia.
| Target | Methods | Cohort Size/Disease Stage | Evidence: Key Points | Application | Reference |
|---|---|---|---|---|---|
| IDH1 and IDH2 genes (R140 and R172 mutations) | Sanger, ddPCR, NGS, and qPCR | n = 60 Diagnosis |
| MRD | Grassi et al., 2020 [53] |
| CEBPA mutations and blast cells | RT-qPCR | n = 4 (n = 3 diagnostic, n = 1 relapse) |
| Concordance | Smith et al., 2006 [56] |
| NPM1 mutations | Flow cytometry, RT-qPCR | n = 15 patients n = 45 MRD samples Diagnosis and follow-up |
| MRD | Pettersson et al., 2016 [51] |
| Somatic mutations | ddPCR, RT-qPCR | n = 41 patients with AML-M1/M2 n = 20 healthy volunteers |
| Concordance | Handschuh et al., 2017 [52] |
| Residual leukemic cells | Flow cytometry | n = 135 patients with de novo AML (100 achieving CR after intensive chemotherapy) |
| MRD Response assessment | Buccisano et al., 2006 [55] |
| Somatic mutation | NGS | n = 22 (after remission) post-treatment |
| Concordance MRD | Short et al., 2020 [67] |
| Driver mutations | NGS dPCR | n = 53 (n = 37 AML, n = 14 MDS) (after post-alloSCT) Diagnosis and post-treatment |
| Concordance MRD Response assessment | Nakamura et al., 2019 [68] |
| Somatic mutation | DDO-ddPCR dPCR qPCR NGS | n = 57 samples (cfDNA), n = 28 (PB), n = 53 (BM) Post-treatment |
| Concordance MRD | Rausch et al., 2021 [69] |
| Leukemic cells | Flow cytometry | n = 50 patients with de novo AML |
| Concordance MRD | Maurillo et al., 2007 [54] |
| Primitive blast (CD34+/CD117+/CD133+) | Flow cytometry | n = 114 patients (205 paired BM and PB samples) Diagnosis and post-treatment |
| Concordance MRD Response assessment | Zeijlemaker et al., 2016 [59] |
Ref., reference; ddPCR, droplet digital PCR; NGS, next-generation sequencing; qPCR, quantitative PCR; MRD, minimal residual disease; RT-qPCR, real-time quantitative PCR; FC, flow cytometry; CR, complete response; RFS, relapse-free survival; dPCR, digital PCR; BM, bone marrow; AML, acute myeloid leukemia; MDS, myelodysplastic syndrome; SCT, stem cell transplantation; VAF, variant allele frequency; DDO-ddPCR, D-aspartate oxidase ddPCR, droplet digital PCR; PB, peripheral blood; cfDNA, cell-free DNA.
3.2. Myelodysplastic Syndromes
Myelodysplastic syndromes are a heterogeneous group of hemopathies characterized by a clonal disorder of hematopoietic stem cells, and whose main problems are caused by the morbidities associated with cytopenias, as well as by the potential risk of progression to acute myeloid leukemia. It is a disease typical of older patients, and in most cases, it is not possible to opt for treatments with curative options, so it is not a pathology in which the MRD techniques have been developed, as in others (e.g., AML). However, in recent years, several papers have been published highlighting the usefulness of cfDNA monitoring in patients with MDS (Table 4).
Dawson published a work where the detection of mutations and cytogenetic alterations in cfDNA predicts treatment failure. Targeted deep sequencing (TS) was performed on DNA derived from BM and plasma using a customized panel of 55 genes. The sequencing of the BM samples identified putative driver mutations in 10 out of of 12 patients. A digital polymerase chain reaction (dPCR) was performed to validate the TS results. The quantification of the mutant allele fraction (MAF) showed an excellent correlation between TS and dPCR. Through the analysis of serial bone marrow and the matched plasma samples (n = 75), the authors demonstrated that ctDNA is directly comparable to bone marrow biopsies in representing the genomic heterogeneity of malignant clones in MDS, and the serial monitoring of ctDNA allows the concurrent tracking of mutations throughout therapy, which could be able to anticipate treatment failure. In addition, in the time points with severe neutropenia, the allelic burden by variant allele frequency (VAF) determination was higher in the plasma cfDNA than leucocyte BM [73].
Nakamura published the prognostic impact of the circulating tumor DNA status in a post-allogeneic hematopoietic stem cell transplantation in AML and MDS. Fifty-three patients with MDS/AML were studied by the targeted 37-gene panel NGS at diagnosis, which detected 51 cases (96.2%) with driver mutations, at diagnosis, by NGS with a limit of detection of 0.04%. The follow-up was made by ddPCR in serum samples with one or more markers, with a limit of detection of 0.1–0.01%. They found that ctDNA reflects clonal dynamics, and the persistent molecular MRD status in the post-allogeneic stem cell transplantation (SCT) predicts relapse and survival. Increased ctDNA levels between month 1 and month 3, after the allogeneic transplantation, could be a predictor of relapse. The use of serum ctDNA has a benefit over leukocyte DNA in predicting relapse, with a concordance observed in 8/11 cases; however, in cases with cytopenias, it was only detectable in serum ctDNA [68].
In another study, the peripheral plasma cfDNA samples available from patients with aplastic anemia (AA; n = 25), MDS (n = 27), and a healthy cohort (n = 107) were screened for somatic variants in genes related to the hematologic malignancies by a targeted-panel NGS. The results were further compared to the DNA sequencing of matched blood cells. The authors observed that the concordance between cfDNA and the blood cells was poor for clonal hematopoiesis (CH) detection when variants were at a variant allele frequency <10%, which was mostly observed in the healthy and AA cohort, but not in the MDS group. The use of cfDNA does not offer advantages over CTC for the detection of variants associated with clonal hematopoiesis in diseases with low allele burdens and healthy individuals. They detected numerous new variants with a low number of reads in cfDNA that they considered artifacts, so they recommended strict filtering criteria in these cases. Ultra-sensitive assays with a robust sequencing coverage and error-correction methodology may be required to overcome assay discordance [74].
Table 4.
Liquid biopsies in myelodysplastic syndromes.
| Target | Methods | Cohort Size/Disease Stage | Evidence: Key Points | Application | Ref. |
|---|---|---|---|---|---|
| Somatic mutations | NGS (55 genes) | n = 12 patients 75 samples at follow-up |
| MRD Concordance | Yeh et al., 2017 [73] |
| Somatic mutations | NGS diagnostic (37 genes) ddPCR for follow-up | n = 51 (n = 15 novo AML, n = 22 secondary AML, n = 14 MDS) LOD (NGS) = 0.04% LOD (ddPCR) = 0.1–0.01% |
| Concordance MRD | Nakamura et al., 2019 [68] |
| Somatic mutations (CHIP quantification) | NGS (200 genes) | n = 25 AA patients, n = 27 MDS patients, n = 107 healthy controls LOD = 0.96% |
| Concordance | Gutierrez-Rodrigues et al., 2021 [74] |
Ref., reference; NGS, next-generation sequencing; ctDNA, circulant tumor DNA; BM, bone marrow; MRD, minimal residual disease; ddPCR, droplet digital PCR; AML, acute myeloid leukemia; MDS, myelodysplastic syndrome; LOD, limit of detection; alloSCT, allogeneic stem cell transplantation; PB, peripheral blood; CHIP, clonal hematopoiesis of indeterminate potential; AA, aplastic anemia; cfDNA, cell-free DNA.
3.3. Myeloproliferative Neoplasms
Myeloproliferative neoplasms are disorders characterized by stem cell-derived clonal myeloproliferation, with mutually exclusive JAK2, CALR, and MPL mutations as phenotype-driver variants [75,76]. In myeloproliferative neoplasms, the aim of the available treatments is to avoid thrombosis. Good treatments that decrease the disease burden or cure the disease are not available, with the exception of allogeneic transplantation in advanced stage primary myelofibrosis (PMF). To date, drug therapy has not been shown to improve survival or prevent leukemic/fibrotic transformation in either essential thrombocythemia (ET) or polycythemia vera (PV); therefore, treatment is primarily directed at preventing thrombotic complications [77].
Based on these assumptions, the usefulness of markers to quantify residual disease is unclear (Table 5). However, Bellosillo et al. [78] published a work where 107 patients with MPNs were studied by NGS and ddPCR, including 33 PV, 56 ET, 14 PMF, and four unclassifiable MPNs (uMPNs). The aim of the study was to assess the accuracy and reliability of cfDNA analyses in a cohort of MPN patients, comparing this technique to the genotype of peripheral blood granulocytes. The authors showed that the amount of cfDNA in plasma varies among MPN disease phenotypes. A significant positive correlation was observed between the amount of cfDNA and PMF (vs. PV), the leukocyte count, LDH, MPL-mutated group, and those suffering from a thrombotic event at the time of diagnosis or during follow-up. In addition, cfDNA and granulocyte DNA showed an equivalent mutational profile, and the discrepancies were detected with variants at low VAF. MPL-mutated and ASLX1-mutated patients had higher amounts of cfDNA. However, in relation to the role of cfDNA as a way of monitoring responses to treatment, for the PV cases, in those who received hydroxycarbamide, the JAK2V617F VAF remained stable in both the granulocytes and cfDNA during the follow-up. For the ET cases, those who were treated with interferon had a proportional decrease in the JAK2V617F VAF, which was observed in granulocytes and cfDNA [78].
4. Lymphoid Malignancies
4.1. Acute Lymphoblastic Leukemia
Acute lymphoblastic leukemia (ALL) is a disease with an incidence of one to five cases per 100,000 people in the population. Of these, 67% are B-ALL, and 75% are produced in children under six years of age. The prognosis of the disease is defined by age, the leukocyte count, genetic and molecular alterations, and initial responses to treatment, among others [79]. Since this disease affects the pediatric population, it is even more important to avoid invasive tests, so liquid biopsies would represent an important advance.
In recent years, a management that takes into account the result of the MRD has been imposed, such as in adults who may require allogeneic hematopoietic stem cell transplantations. For this, the most commonly used methods, to date, are the multiparametric flow cytometry- (more standardized) and PCR-based tests, such as in ALL with BCR/ABL1 mutations. NGS is not yet routinely used for the MRD testing [80,81,82]. However, guidelines (e.g., NCCN) already recommend the use of NGS to detect IGH and TCR rearrangements as a marker for the MRD. A comparison of the MRD levels in ALL patients, detected by IG/TR RT-qPCR vs. BCR-ABL1 genomic transcript quantification, showed a good correlation, and the sensitivity significantly increased when large numbers of cells were acquired [83]. In adult patients with ALL undergoing cellular therapies (hematopoietic cell transplantation and chimeric antigen receptor T-cell therapies), the authors demonstrated a strong concordance between the NGS-based MRD detected in the PB and BM (r = 0.87; p = 0.001), with a sensitivity and specificity of the MRD detection in the PB of 87% and 90%, respectively, relative to the MRD in the BM [84].
The use of liquid biopsies (cfDNA) in the MRD in ALL has been explored in some studies (Table 6). Van der Velden et al. studied Ig/TCR rearrangements in T-ALL, and its precursor in bone marrow and PB samples, finding a strong correlation in the MRD levels in T-ALL, but not in precursor, B-ALL [85]. Schwarz et al. conducted one of the first studies to see that there was more plasma DNA in patients with ALL than in healthy patients [86]. Cheng et al. compared flow cytometry in bone marrow and peripheral blood plasma PCR quantifying Ig/TCR gene rearrangements. They did not show a correlation, but the peripheral blood could predict a relapse [87]. Another study showed that BCR/ABL1-positive ALL patients had lower pre-transplant cfDNA levels than BCR/ABL1-negative ALL patients [88]. More studies are needed to use cfDNA for monitoring the MRD in ALL.
4.2. Lymphomas and Chronic Lymphocytic Leukemia
Lymphomas are the most common hematologic malignancies, and the gold standard for their diagnosis is tissue or lymph node biopsies, with a pathological examination after surgical resection or lymph node puncture; however, these are invasive examinations (sometimes, as in CNS (central nervous system) lymphoma, the sample may be not accessible) and a biopsy is not useful for monitoring the disease [89]. It is a heterogenous group of neoplasms, with significant differences in the treatment schemes, and non-Hodgkin lymphoma is the most frequent type [90].
Traditionally, imaging scans (including computed tomography (CT) and positron emission tomography (PET)) have been used to assess the responses to treatment of lymphomas, which have several limitations, such as lack of tumor specificity, an inability to detect the disease at the molecular level, an inability to capture dynamic tumor processes, and a radiation risk or cost. However, the study of cfDNA may have advantages (Table 7): A high tumor specificity, a high sensitivity, broad applicability, and the ability to assess tumor heterogeneity [91]. The study of cfDNA in lymphomas includes cfDNA concentrations, DNA methylation, the detection of specific somatic mutations, and IgH gene rearrangement. Although the gold standard method for molecular profiling in lymphomas is based on lymph node or tissue biopsies, they have limitations. Lymphomas are not always expressed in peripheral blood, making it difficult to use the concept of the "minimal residual disease" when employing conventional methods. The use of “liquid biopsies” has obstacles in lymphomas, where it is difficult to identify targets applicable to the diversity of these neoplasms. For example, IGH-BCL2 translocations may be detectable in the blood of patients with FL, transformed FL, and a subset of DLBCL, but there is a variability in the breakpoint regions [92].
4.2.1. Diffuse Large B-Cell Lymphoma
Diffuse large B-cell lymphoma (DLBCL) is the most common type of newly diagnosed lymphoma cases. Three major molecular subtype categories of DLBCL have been defined: the activated B-cell-like (ABC) subtype, the germinal center B-cell-like (GCB) subtype, and the primary mediastinal BCL (PMBL) subtype.
Normal and tumor B-cells express the antigen receptors of B-lymphocytes (BCR), composed of paired heavy and light immunoglobulin chains; there is a wide diversity in the characteristics of the molecule that results from the random rearrangement of the variable diversity joining (VDJ) in the progenitor B-cells, producing unique clonotypes in each lymphoma, which may be useful in anticipating relapses in the peripheral blood, if the relapse comes from the diagnosis clone. However, this technique has disadvantages: There are some DLBCLs with unproductive VDJ rearrangements.
DLBCL is an aggressive and heterogeneous lymphoma, and studies have shown that cfDNA may have a position in the diagnosis of the disease. Various reports have shown a higher amount of cfDNA in patients with this lymphoma, compared to healthy patients, as well as a decrease in the amount of cfDNA when the treatment is effective, and a correlation between the quantification of cfDNA and the adverse prognosis of the disease [21,93,94,95,96].
Digital PCR (dPCR) has been used for detecting common mutations in DLBCL, such as XPO1 EF71K, EZH2 Y641, and MYD88 L265P. Roschewski et al. established the pertinence and the prognostic impact of NGS-based molecular circulating tumor DNA monitoring in a retrospective cohort of 126 patients. The detectable ctDNA in the surveillance serum samples was a strong predictor for disease progression (with an HR of 228) [95,97,98,99,100,101].
Li et al. studied APP gene mutations using RT-PCR in 174 patients (98 DLBCL patients) and concluded that an increase in the cfDNA was associated with the advanced stages of the disease, elevated LDH levels, and a higher prognosis score, with an inferior two-year PFS in patients with high levels of cfDNA [102].
In another study, presented by Rossi et al., a rapid clearance of mutations from cfDNA, among the responsive patients with DLBCL treated with R-CHOP, was demonstrated using CAncer Personalized Profiling by deep Sequencing (CAPPseq) technology, as well as the persistence of basal mutations in the plasma cfDNA of refractory patients [103].
Kurtz et al. studied the dynamics of ctDNA from 217 patients with DLBCL, using CAPPseq, and assessed the prognostic value of ctDNA, regarding risk factors. This study demonstrated that pretreatment ctDNA levels and molecular responses are independently prognostic of outcomes in DLBCL. A two-log drop in ctDNA levels after two chemotherapy courses was associated with an eventual complete response and cure [104]. In another study, Rivas-Delgado et al. reported that high ctDNA levels were associated with a lower response [105,106].
Some studies have demonstrated that ctDNA can be an indicator of the MRD before the lymphoma relapse in DLBCL or T-cell lymphoblastic lymphoma [99,107,108,109]. Shin et al. designed a panel of 66 genes associated with NHLs and analyzed the plasma cfDNA from patients with various subtypes using NGS; this study showed that the level of ctDNA was decreased in patients with a response to therapy, and it increased in patients with disease progression [110]. However, we must be cautious, because some reports have shown that some mutations, such as TP53 or DNMT3A, could have their origin in clonal hematopoiesis of an indeterminate potential (CHIP) [96,111].
4.2.2. Mantle Cell Lymphoma
Mantle cell lymphoma (MCL) is an aggressive lymphoma with a survival median of more than five years. Its management is variable, depending on the risk and patient performance status, and includes chemotherapy, immunotherapy, or autologous stem cell transplantation. The MRD is not very standardized, although some studies have been conducted using the PCR amplification of IgH rearrangements, demonstrating an impact on the clinical outcomes of the patient after autologous transplantation [114,115,116]. Lakhotia et al. used NGS to detect Ig heavy and light chains, and CCND1 and BCL2 gene mutations in ctDNA; they concluded that the baseline ctDNA correlated with the total metabolic tumor volume on the PET scans, and the clearance of ctDNA after one cycle of DA-EPOCH-R + BZ was strongly associated with a superior median PFS (76.4 vs. 20.7 months, p = 0.0037) and a trend toward a superior four-year overall survival (OS) (92.3% vs. 73.0%, p = 0.23). The clearance of ctDNA after two cycles of DA-EPOCH-R + BZ was also associated with a superior median progression-free survival (PFS) (32.4 vs. 21.4 months, p = 0.015) and a trend toward a superior median OS (82.2 vs. 73.2 months, p = 0.15) [117]. Agarwal et al. reported that ctDNA provides valuable prognostic information and enables the real-time assessment of tumor evolution [118].
4.2.3. Follicular Lymphoma
Follicular lymphoma (FL) is the most common indolent lymphoma, and it usually has a prolonged survival (more than 10 years), but the prognosis is variable; treatment consists of immunotherapy (anti CD-20) with or without chemotherapy [119].
Sarkozy et al. reported a study with 133 patients using NGS to detect VDJ mutations, showing that a high ctDNA level is associated with a worse PFS (HR = 6.2, 95% CI 2–162, p = 0.001) (p = 0.14, 0.52, and 0.25 for FLIPI, bone marrow involvement, and the presence of circulating lymphoma cells, respectively) [120].
Delfau-Larue et al. tried to correlate CTCs, cfDNA, and the total metabolic tumor volume (TMTV) with patients’ outcomes using ddPCR and the IgH rearrangements. They reported that CTCs and cfDNA were correlated with TMTV, and that the four-year PFS was lower in patients with a TMTV > 510 cm3 (p = 0.0004), CTCs > 0.0018 PB cells (p = 0.03), and cfDNA > 2550 equivalent-genome/mL (p = 0.04) [121]. Another study using NGS in 29 patients with FL showed that the MRD positivity in the interim, or at the end of treatment, results in significantly inferior PFS (median 12 months vs. not reached, p = 0.009) [122].
4.2.4. Primary Central Nervous System Lymphoma
Primary central nervous system lymphoma (PCNSL) is an aggressive and infrequent lymphoma [123]. In this lymphoma, the MYD88 L265P mutation has been studied in cfDNA using PCR and targeted deep sequencing (TDS). Hattori et al. found this mutation in diagnostics in the majority of patients, but, after chemotherapy, the mutation was undetectable; Yoon et al. concluded that using ctDNA has a limited value for predicting treatment outcomes because it has a low detection efficiency [124,125]. Moreover, for the diagnosis, it may be useful to detect the MYD88 L265P mutation in the cfDNA from cerebrospinal fluid [126,127,128].
4.2.5. T-Cell Lymphomas
In T-cell lymphomas, which is less common than B-cell lymphomas, there is less evidence for the use of liquid biopsy. For the diagnosis, and to determine the mutational landscape, as in an angioimmunoblastic T-cell lymphoma, the detection of HOAG17V/IDH2R172 mutations primarily takes place using an allele-specific polymerase chain reaction (AS-PCR). However, in a previous study with 20 patients, it was not possible to demonstrate an association between mutations and the clinical parameters or survival [129]. Milkjovic et al. used dPCR to detect TCR rearrangements in 34 patients with peripheral T-cell lymphomas (PTCL), and reported a median 2.6-log decrease in their ctDNA levels after the first two cycles of treatment, as well as the early clearance of ctDNA after cycle 2, were not associated with a statistically significant improvement in EFS (median (95% CI), 8.4 (0.1–NR) vs. 2.0 (0.1–NR) years; p = 0.32) or OS (median, 8.4 (0.3–NR) vs. 7.0 (0.5–NR) years; p = 0.44) [130].
4.2.6. Hodgkin Lymphoma
Hodgkin lymphoma (HL) represents 10% of newly diagnosed lymphomas; the standard initial treatment is chemotherapy and/or radiotherapy, with a five-year progression-free survival rate of 65–90% [89,131]. Studies have been conducted in the attempt to detect peripheral blood mutations in cfDNA using NGS, finding that XPO1 E571K, ATM, KMT2D, and TP53 are frequently mutated in HL [132,133].
In 2018, Spina et al. clarified the genetic landscape of classic HL using CAPP-seq on cfDNA, establishing that STAT6, TNFAIP3, ITPKB, GNA13, B2M, ATM, SPEN, and XPO1 are the most commonly mutated genes [134].
Regarding the MRD, Camus et al. reported that the XPO1 E571K mutation may be used as a biomarker in classical HL, using digital PCR, because in their study, patients with a detectable XPO1 mutation at the end of treatment could have a shorter free progression survival, but the results were not statistically significant [135].
Additionally, Spina et al. reported that a two-log reduction in cfDNA, or greater, between the diagnosis and after the two chemotherapy courses, was linked to a complete metabolic response and cure. However, these studies have several limitations, and more evidence is needed to incorporate liquid biopsies into the monitoring of the MRD in lymphomas [130,134].
4.2.7. Chronic Lymphocytic Leukemia
Chronic lymphocytic leukemia (CLL) is the most common leukemia in the Western world, and it has a variable clinical outcome [136]. In CLL, the MRD is not as well implemented as in other hematologic malignancies, but more and more studies reflect its importance in achieving better progression-free and overall survival rates [137,138,139]. The most widely used techniques are MFC and RQ-PCR. Although this is a circulating disease in the peripheral blood, some studies have demonstrated the usefulness of ctDNA, using digital PCR and targeted sequencing, in obtaining a wide mutational landscape of the disease, or when the disease is localized in an organ, as well as to assess the clonal evolution. More studies are needed to implement the MRD monitoring as a complementary method to those currently used [140].
Table 7.
Liquid biopsies in lymphomas and chronic lymphocytic leukemia.
| Target | Methods | Cohort Size/Disease Stage | Evidence: Key Points | Application | Ref. |
|---|---|---|---|---|---|
| β-globin gene | qPCR | n = 142 DLBCL, n = 63 FL, n = 24 MCL, n = 10 HL, n = 45 |
| Concordance Prognosis | Hohaus et al., 2009 [21] |
| IgH gene rearrangements | Locus-specific primer sets por IgH and IgK | n = 17 DLBCL, n = 15 MLBCL, n = 2 Diagnosis and post-treatment |
| Response assessment Prognosis | Armand et al., 2013 [97] |
| IgH/TCR rearrangements | RQ-PCRNGS | n = 68 B-NHL, n = 37 T-NHL, n = 10 HL, n = 5 CLL, n = 16 Pre- and post-HSCT |
| Prognosis MRD | Herrera et al., 2016 [100] |
| Somatic gene | Targeted NGS | n = 32 B-NHL, n = 18 NK or T-NHL, n = 9 Other, n = 5 |
| Concordance | Shin et al., 2019 [110] |
| APP gene | RT-PCR | n = 174 DLBCL, n = 98 HL, n = 18 TCL, n = 9 NK/T cell lymphoma, n = 21 Other B-NHL, n = 28 Diagnosis |
| Prognosis | Li et al., 2017 [102] |
| IgH gene rearrangements | NGS (ctDNA) and PCR vs. CT | DLBCL, n = 126 Diagnosis and post-treatment |
| Response assessment Staging | Roschewski et al., 2015 [99] |
| IgH gene rearrangements | Ig-HTS vs. PET/CT | DLBCL, n = 75 Diagnosis or recurrence |
| Surveillance after complete remission | Kurtz et al., 2015 [98] |
| Somatic mutations | CAPP-seq Ultra-deep targeted NGS | DLBCL, n = 30 Diagnosis and post-treatment |
| Response assessment | Rossi et al., 2017 [103] |
| V(D)J rearrangements | NGS | DLBCL, n = 6 Pre- and post-CAR-T-cell therapy |
| MRD Concordance | Hossain et al., 2019 [112] |
| L1PA2 | qPCR | DLBCL, n = 40 (and 38 controls) Diagnosis |
| Prognosis | Eskandari et al., 2019 [94] |
| Somatic gene | CAPP-seq | DLBCL, n = 217 Diagnosis, relapse, or recurrence |
| Prognosis | Kurtz et al., 2018 [104] |
| Somatic gene | Targeted NGS | DLBCL, n = 79 Diagnosis and post-treatment |
| Concordance Prognosis | Rivas-Delgado et al., 2021 [105] |
| Ig heavy and light chains and CCND1 and BCL2 genes | NGS | MCL, n = 53 Diagnosis and post-treatment |
| Concordance Prognosis | Lakhotia et al., 2018 [117] |
| V(D)J gene | NGS | FL, n = 133 Diagnosis and post-treatment |
| Concordance Prognosis | Sarkozy et al., 2017 [120] |
| IgH gene rearrangements | ddPCR | FL, n = 133 Diagnosis and post-treatment |
| Concordance Prognosis | Delfau-Larue et al., 2018 [121] |
| Somatic mutations | NGS | FL, n = 27 Diagnosis and post-treatment |
| Concordance Follow-up | Jimenez-Ubieto et al., 2020 [122] |
| MYD88 gene | ddPCR TGS | PCNSL, n = 14 Diagnosis and post-treatment |
| Concordance Follow-up | Hattori et al., 2018 [124] |
| MYD88 gene | ddPCR | PCNSL, n = 29 Diagnosis |
| Concordance | Hiemcke-Jiwa et al., 2019 [126] |
| MYD88 gene | ddPCR | PCNSL, n = 11 Diagnosis and relapse |
| Concordance | Rimelen et al., 2019 [127] |
| MYD88 gene | ddPCR | PCNSL, n = 42 Diagnosis |
| Concordance | Yamagishi et al., 2021 [128] |
| RHOAG17V and IDH2R172 mutations | AS-PCR | PCNSL, n = 20 Diagnosis |
| Concordance Response assessment | Hayashida et al., 2020 [129] |
| TCR rearrangements | dPCR | PTCLs, n = 34: ALCL, n = 10 PTCL-NOS, n = 10 Other, n = 14 Diagnosis |
| Response assessment | Miljkovic et al., 2021 [130] |
| XPO1 gene | dPCR | cHL, n = 94 Diagnosis and post-treatment |
| Concordance Response assessment Prognosis MRD | Camus et al., 2016 [135] |
| STAT6 mutations | NGSCAPP-seq | cHl, n = 112 Newly diagnosed, n = 80 Refractory, n = 32 |
| Response assessment Prognosis | Spina et al., 2018 [134] |
| NFKBIE, TNFAIP3, STAT6, PTPN1, B2M, XPO1, ITPKB, GNA13, and SOCS1 genes | NGS | HL, n = 60 Diagnosis and post-treatment |
| Concordance Prognosis | Camus et al., 2021 [133] |
Ref., reference; RQ-PCR, real-time quantitative polymerase chain reaction; MRD, minimal residual disease; PB, peripheral blood; cfDNA, cell-/non-Hodgkin lymphoma; HL, Hodgkin lymphoma; DLBCL, diffuse large B -cell lymphoma; PTCL-NOS: peripheral T-cell lymphoma-not otherwise specified; qPCR, quantitative PCR; FL, follicular lymphoma; MCL, mantle cell lymphoma; LDH, lactate dehydrogenase; ctDNA, circulant tumor DNA; MLBCL, mediastinal large B-cell lymphoma; HSCT, hematopoietic stem cell transplantation; PFS, progression-free survival; NGS, next-generation sequencing; VAF, variant allele frequency; PD, progressive disease; CT, computerized tomography; PET, positron-emission tomography; CAPP-seq, CAncer Personalized Profiling by deep Sequencing; CAR, chimeric antigen receptor; OS, overall survival; MTV, metabolic tumor volume; EMR, early molecular response; MMR, major molecular response; HR, hazard ratio; ddPCR, droplet digital PCR; FLIPI, Follicular Lymphoma International Prognostic Index; TMTV, total MTV; PCNSL, primary central nervous system lymphoma; TDS, target deep sequencing; CSF, cerebrospinal fluid; AS-PCR, allele-specific PCR; dPCR, digital PCR; DS, Deauville score.
4.3. Multiple Myeloma
Multiple myeloma (MM) is a clonal plasma cell proliferative disorder characterized by bone lesions, whose diagnosis is based on the presence of clinical, biochemical, histopathological, and radiological markers of disease. The appearance of new drugs and the improvement of supportive treatment has produced an increase in the median survival, which is currently estimated at five years in developed countries [141,142].
The MRD in MM has been shown to be a predictive factor for PFS and OS; it is performed by multiparametric flow cytometry (MFC) or by allele-specific oligonucleotide PCR (ASO-PCR) sequencing in bone marrow, with a sensitivity of 10−5–10−6 [143,144,145,146], or by NGS, to detect IgH rearrangements with a sensitivity of 10−6 [147].
MM is a disease of complex molecular biology, with different clones of malignant plasma cells in the same patient; these clones evolve in the natural history of the disease. Several genes are involved in many patients, such as KRAS, NRAS, or BRAF [148,149]. In MM, circulating tumor cells are released from the primary tumor into the bloodstream, usually reaching another location in the bone marrow; this process is early on in the carcinogenesis. Different groups have studied circulating tumor cells in MM and other plasma cell dyscrasias; they can be particularly useful in extramedullary, oligosecretory, and non-secretory disease settings [150,151,152,153,154,155].
As in other hematologic neoplasms, circulating tumor DNA can be analyzed in MM through tumor-specific mutations or genetic aberrations. In recent years, the number of articles on this subject has increased (Table 8). In 2015, Sata et al. evaluated the tumor burden in mRNA from peripheral blood cells, whole bone marrow cells, the CD20+CD38L B-cell population in bone marrow, and the cell-free DNA from the sera of patients with MM, and compared them using ASO-PCR. This study, with 30 patients, found statistically significant correlations between the ASO-PCR levels in bone marrow cells and the peripheral blood cells, which suggests that clonogenic plasma cells or MM precursor cells may circulate in peripheral blood, but the ASO-PCR values in the cell-free DNA from the sera did not correlate with those in either the bone marrow or the peripheral blood cells [156].
Oberle et al. conducted a study in 27 MM patients to explore the clonotypic V(D)J rearrangement for monitoring circulating myeloma cells and cell-free myeloma DNA. The positivity for circulating myeloma cells/cell-free myeloma was associated with a conventional remission status, and the majority of non-responders/progressors had evidence of persistent circulating myeloma cells/cell-free myeloma DNA. However, the positivity for circulating myeloma cells and for cell-free myeloma DNA were discordant in 30% of cases, which indicates that cell-free myeloma DNA may not be generated entirely by circulating myeloma cells and may reflect the overall tumor burden [157].
Mithraprabhu et al. analyzed the plasma-derived circulating free tumor DNA as an adjunct to bone marrow biopsies for mutational characterization (for KRAS, NRAS, BRAF, and TP53) and for tracking disease progression in 33 relapsed/refractory and 15 newly diagnosed MM patients, in comparison to 12 healthy donors by NGS, showing a higher amount of cfDNA in MM patients. Some mutations were found only in the cfDNA. Although there were few patients in the study, it may confirm the spatial heterogeneity of MM [158].
Rustad et al. explored the presence of circulating tumor DNA, monitoring recurrent mutations (NRAS, KRAS, and BRAF) using ddPCR, and comparing it to bone marrow plasma cells. They observed a correlation between the concentration of mutated alleles in the serum and the fraction in bone marrow plasma cells, which may reflect mutated cells, the total tumor mass, and the transformation to a more aggressive disease that is complementary to the M protein [159].
Gerber et al. studied the cfDNA in 28 patients, identifying the clonotypic V(D)J rearrangement as a marker, or genotyping a limited set of genes, and designed a panel of NGS, comparing the characteristics of these patients to samples of bone marrow aspirates from patients with plasma cell disorders in different stages. The amount of cfDNA correlated with some parameters that may indicate the tumor burden, such as the percentage of plasma cell infiltration of bone marrow [160].
Biancon et al. analyzed the disease evolution of 25 MM patients receiving second-line therapy, and the study showed that the levels of IgH detected in cfDNA reflected the number of PCs enumerated by MFC, which correlated with clinical outcomes [161].
Other studies have shown that cell-free tumor DNA can be used to obtain the molecular profile of myelomas instead of the bone marrow aspirate, and they have supported the concept of cell-free DNA as a prognostic marker [162,163,164]. In a recent study, after sorting 77 MM patients according to their molecular risk, Deshpande et al. found that cfDNA was higher in high-risk MM, and high cfDNA levels were associated with a worse PFS and OS [165]. In other study, Manzoni et al. analyzed 104 samples from 65 patients (15 monoclonal gammopathy of undetermined significance-MGUS; 33 smoldering multiple myeloma-SMM; and 17 MM) using ultra-deep NGS, concluding that a lower tumor mass correlates with a lower cfDNA tumor fraction, and it could be useful to evaluate MGUS and SMM in the future [166].
Regarding the study of the MRD with cfDNA, there are few published studies that compare it to the currently standardized methods (flow cytometry and DNA sequencing in bone marrow) [167]. Mazzotti et al. demonstrated the absence of a correlation between ctDNA and bone marrow for the MRD by NGS, using only IgH gene rearrangements, although only 37 patients were included [168]. Long et al. analyzed 22 plasma samples from eight extramedullary multiple myelomas (EMM) and 23 plasma samples from 10 MM patients without extramedullary spread, with higher cfDNA concentrations in patients with extramedullary spread. After designing sequencing panels targeting the coding sequence regions of the same 22 recurrently mutated genes, 17 different were detected. The authors concluded that cfDNA can be used to track extramedullary disease progression, including the MRD, when plasmacytomas are inaccessible [169].
In other studies, a significant correlation of the quantity of tumor-specific cell-free DNA levels with clinically meaningful events has been found, but the results in the case of the MRD monitoring were not significant [163,170]. The target sequencing of cfDNA cannot, today, achieve the sensitivity of the MRD detection of flow cytometry or PCR; however, in the future, the MRD detection in cell-free DNA may increase its sensitivity combining parameters (patient-specific mutation panels, methylation patterns, copy number alterations, or IgH rearrangements), which may add further accuracy to progression-free survival prediction and the detection of the false-negative MRD [171,172,173]. At the present time, the isolated use of cfDNA has no clinical applicability in the study of the MRD in multiple myeloma, although there is increasing evidence, which may initially make it a complementary test for the follow-up of the disease and, in the future, with its improvement, into a fundamental tool for the evaluation of the MRD [155].
Table 8.
Liquid biopsies in multiple myeloma.
| Target | Methods | Cohort Size/Disease Stage | Evidence: Key Points | Application | Ref. |
|---|---|---|---|---|---|
| IgH gene | ASO-PCR | n = 30 Diagnosis and post-treatment |
| Concordance | Sata et al., 2015 [156] |
| V(D)J rearrangement | NGS | n = 27 Diagnosis and post-treatment |
| Concordance Prognosis | Oberle et al., 2017 [157] |
| KRAS, NRAS, BRAF, and TP53 mutations | ddPCR | n = 60 New diagnosis, n = 15 Relapse/refractory, n = 33 Diagnosis and post-treatment Normal volunteers, n = 12 |
| Concordance Response assessment | Mithraprabhu et al., 2017 [158] |
| NRAS, KRAS, and BRAF mutations | ddPCR | n = 18 Diagnosis and post-treatment |
| Concordance Response assessment | Rustad et al., 2017 [159] |
| KRAS, NRAS, BRAF, EGFR, and PIK3CA mutations | ddPCR | n = 53 New diagnosis, n = 11 Relapsed, n = 42 |
| Concordance Prognosis | Kis et al., 2017 [162] |
| Somatic mutations | CAPP-seq | n = 28 New diagnosis, n = 25 Relapsed/refractory, n = 3 Diagnosis |
| Concordance | Gerber et al., 2018 [160] |
| IgH gene rearrangements | ddPCR | n = 25 At first relapse |
| Concordance Prognosis | Biancon et al., 2018 [161] |
| IgH gene rearrangements | NGS | n = 37 Post-treatment |
| MRD | Mazzotti et al., 2018 [168] |
| Somatic mutations | WES | n = 163 cfDNA, n = 107 CTCs, n = 56 Diagnosis |
| Concordance | Manier et al., 2018 [170] |
| Somatic mutations | WES | n = 105 MM patients, n = 93 Healthy patients, n = 12 Diagnosis |
| Concordance | Guo et al., 2018 [171] |
| Somatic mutations | Ultra-deep NGS | n = 65 MGUS, n = 15 SMM, n = 33 MM, n = 17 Diagnosis |
| Concordance Prognosis | Manzoni et al., 2020 [166] |
| Somatic mutations | NGS | n =18 EMM patients, n = 8 MM without EM spread, n = 10 Diagnosis |
| Concordance | Long et al., 2020 [169] |
| Somatic mutations | Targeted NGS | n = 77 Newly diagnosed, n = 52 Relapsed, n = 2 Previously treated, n = 23 |
| Concordance Prognosis | Deshpande et al., 2021 [165] |
Ref., reference; ASO-PCR, allele-specific oligonucleotide PCR; NGS, next-generation sequencing; ddPCR, droplet digital PCR; ctDNA, circulant tumor DNA; AF, allele frequency; MM, multiple myeloma; cfDNA, cell-free DNA; BM, bone marrow; CAPPseq, CAncer Personalized Profiling by deep Sequencing; PC, plasmatic cell; HR, hazard ratio; MFC, multiparameter flow cytometry; WES, whole-exome sequencing; CTC, circulant tumor cell; CNV, copy number variation; MGUS, monoclonal gammopathy of undetermined significance; SMM, smoldering multiple myeloma; EM, extramodular; ISS, International Staging System; LDH, lactate dehydrogenase.
5. Current Situation and Preanalytical Recommendations
5.1. Current Situation
In the field of hematology, studies of liquid biopsies, in terms of CTC, have been carried out over the last few years, such as in CML (with a treatment adjustment in terms of BCR/ABL1 peripheral blood leukocyte RNA in CML patients, as well as adjustments in terms of the NPM1-mutated ratio in peripheral blood leukocyte RNA in AML). However, the application of liquid biopsies, in terms of cfDNA, for treatment adjustment (as a biomarker of the MRD) in myeloid pathology has not been highly developed. However, recent publications have shown its usefulness as a complementary technique to CTC or BM studies in the follow-up of the MRD, and they allow an interesting genomic representation of the different tumor clones.
There are several papers on the applicability of cfDNA as a marker for the MRD in B- or T-cell lymphoid malignancies. In these pathologies, there is a lot of information on the usefulness of this approach to evaluate the MRD, where it could be considered a new pathway for the MRD. In chronic lymphocytic leukemia, the evolution of resistance to treatments in real time could be monitored with ctDNA, and in acute leukemias, it can be helpful in the monitoring of early responses to treatments and the prediction of treatment failure.
NGS is the preferred method for liquid biopsy studies to detect the MRD in hematologic pathology, given the great molecular heterogeneity of these tumors. The main limiting factor of these sequencing tools is the error rate, which varies between 1% and 0.01% depending on the sequencing conditions, starting material, and the analysis pipeline. Variant detection with high confidence occurs at a fraction below 1% and, therefore, requires sufficient depth of coverage (i.e., the number of sequences that "read" any nucleotide position) in both patient and control samples, as well as the use of bioinformatics analyses and algorithms that allow for the application of the appropriate quality control criteria [31]. In order to optimize this procedure, the main recommendations are as follows.
5.2. Preanalytical Recommendations
High-quality cfDNA for liquid biopsies is required to avoid contamination by genomic DNA from white blood cells, and to maintain a sufficient fragment length to allow for the conduction of PCR-based methods. Various preanalytical factors can influence sample quality for ctDNA extraction, such as the centrifugation procedure, or the storage conditions. Then, a blood collection tube, containing a preservative that stabilizes nucleated blood cells for delayed separation, can be used. Recent studies have shown similar ctDNA levels at zero hours, or for up to five days, at room temperature in blood collection tubes with preservative, and a 2 h stability if the blood is drawn in EDTA tubes [174]. A correct centrifugation protocol with the aim of limiting genomic DNA contamination of leukocytes is needed. For cfDNA extraction, the sensitivity of the different assays is driven by a DNA input spin column, and the magnetic bead-based isolation methods are influenced by the size of the DNA fragments. Thus, the selected extraction kit must be adapted to the sample, and the use of the membrane-based method promotes the extraction of a high molecular weight cfDNA fragments (>600 bp), whereas the magnetic bead system yields shorter cfDNA fragments. The subsequent storage should be at −20 or −80 °C for up to nine months with good preservation, without thawing, or with less than three thaws.
6. Discussion
NGS techniques are rapidly evolving, and their sensitivity is improving, allowing their application even in early-stage diseases. Various assays are available, and the PCR-based methods require precise knowledge of the expected alteration, as well as allowing for the detection of SNV with a very low allele frequency, a relatively low time frame, and low costs. Alternatively, the NGS-based assays allow for the detection of non-hotspot mutations and other variations, such as CNV, but they require more time than PCR assays, as well as a thorough bioinformatics pipeline for analyses, and expertise in the technical and complex interpretations of the data read-out.
Basic NGS workflows are designed to detect mutations above a 1% minimum mutant allele fraction. In ctDNA, this mutant allele fraction is generally reached in a metastatic disease context with a high tumor load. For early-stage cancers, a lower detection threshold is needed for ctDNA mutation detection, and the basic NGS workflow needs further adaptation.
Targeted NGS methods have, therefore, been developed to screen a large number of potential mutations in tumor biopsies with an elevated sensitivity (e.g., Guardant Health, which employs the pre-sequencing preparation of a digital library of individually tagged cfDNA molecules, combined with the post-sequencing bioinformatic reconstruction to eliminate nearly all false positives; the Foundation ACT, with hybrid capture-based assays; CAPP-Seq, with limit-sequenced regions by capturing recurrently mutated genomic regions; Oncomine Lung cfTNA, with capture-based methods to assess 12 genes that are frequently mutated in lung cancer; Trusight tumor 15 assay, with capture-based methods to assess 15 genes that are frequently mutated in solid tumors; Safe sequencing, with the assignment of a unique identifier to each DNA template molecule to be analyzed, as well as the amplification of each uniquely tagged template; and the Archer Reveal cDNA 28kit, with capture-based methods to assess 28 genes that are frequently mutated in solid tumors) [175]. However, there are no specifically-targeted NGS methods with an elevated sensitivity oriented towards hematological tumors.
7. Conclusions
The applications of liquid biopsy methods in evaluating the responses to treatment of onco-hematological pathologies have shown their great utility, and they could be imposed in a relatively short time frame, alone or in combination with other methods (e.g., imaging). Most of the studies carried out to date are case reports or small samples, but the results in these, or in larger studies, are encouraging. However, there are still some limitations to overcome, such as improving the obtainment of a sufficient amount of cfDNA and the sensitivity of the techniques used (generally NGS), as well as obtaining validated results in clinical trials.
Author Contributions
Conceptualization, R.C., J.M.-L. and R.A.; investigation, N.Á. and R.C.; writing—original draft preparation, R.C., N.Á., S.B., J.M.-L. and R.A., writing—review and editing, R.C., S.B., J.M.-L. and R.A.; supervision, J.M.-L. and R.A.; funding acquisition, J.M.-L. and R.A. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by the Subdirección General de Investigación Sanitaria (Instituto de Salud Carlos III, Spain), grants PI19/01518, the CRIS against Cancer foundation, grant 2018/001, and by the Instituto de Investigación Hospital 12 de Octubre (IMAS12) (co-financed by FEDER funds).
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
J.M.-L. declares honoraria for lectures from and membership on advisory boards with Janssen, BMS, Sanofi, Novartis, Incyte, Roche, and Amgen. R.A. declares being an advisory board member for Novartis and Incyte and a speaker honorarium from Astellas, Celgene, and Novartis. S.B. is an employee and S.B., J.M.-L. and R.A. are equity shareholders of Altum Sequencing Co. The remaining authors declare no competing financial interests.
References
- Poulet, G.; Massias, J.; Taly, V. Liquid Biopsy: General Concepts. Acta Cytol. 2019, 63, 449–455. [Google Scholar] [CrossRef] [PubMed]
- Alix-Panabières, C.; Schwarzenbach, H.; Pantel, K. Circulating Tumor Cells and Circulating Tumor DNA. Annu. Rev. Med. 2012, 63, 199–215. [Google Scholar] [CrossRef]
- Siravegna, G.; Marsoni, S.; Siena, S.; Bardelli, A. Integrating Liquid Biopsies into the Management of Cancer. Nat. Rev. Clin. Oncol. 2017, 14, 531–548. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Bode, A.M.; Dong, Z. Circulating Tumor Cells: Moving Biological Insights into Detection. Theranostics 2017, 7, 2606–2619. [Google Scholar] [CrossRef]
- Cirillo, M.; Craig, A.F.M.; Borchmann, S.; Kurtz, D.M. Liquid Biopsy in Lymphoma: Molecular Methods and Clinical Applications. Cancer Treat. Rev. 2020, 91, 102106. [Google Scholar] [CrossRef] [PubMed]
- Salehi, M.; Sharifi, M. Exosomal MiRNAs as Novel Cancer Biomarkers: Challenges and Opportunities. J. Cell. Physiol. 2018, 233, 6370–6380. [Google Scholar] [CrossRef]
- Leslie, M. Cell Biology. Beyond Clotting: The Powers of Platelets. Science 2010, 328, 562–564. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, R.J.A.; Balaj, L.; Hulleman, E.; van Rijn, S.; Pegtel, D.M.; Walraven, M.; Widmark, A.; Gerritsen, W.R.; Verheul, H.M.; Vandertop, W.P.; et al. Blood Platelets Contain Tumor-Derived RNA Biomarkers. Blood 2011, 118, 3680–3683. [Google Scholar] [CrossRef]
- Becker, A.; Thakur, B.K.; Weiss, J.M.; Kim, H.S.; Peinado, H.; Lyden, D. Extracellular Vesicles in Cancer: Cell-to-Cell Mediators of Metastasis. Cancer Cell 2016, 30, 836–848. [Google Scholar] [CrossRef] [Green Version]
- Van Eijndhoven, M.A.; Zijlstra, J.M.; Groenewegen, N.J.; Drees, E.E.; van Niele, S.; Baglio, S.R.; Koppers-Lalic, D.; van der Voorn, H.; Libregts, S.F.; Wauben, M.H.; et al. Plasma Vesicle MiRNAs for Therapy Response Monitoring in Hodgkin Lymphoma Patients. JCI Insight 2016, 1, e89631. [Google Scholar] [CrossRef]
- Mandel, P.; Metais, P. Nuclear Acids In Human Blood Plasma. C. R. Seances Soc. Biol. Fil. 1948, 142, 241–243. [Google Scholar] [PubMed]
- Leon, S.A.; Shapiro, B.; Sklaroff, D.M.; Yaros, M.J. Free DNA in the Serum of Cancer Patients and the Effect of Therapy. Cancer Res. 1977, 37, 646–650. [Google Scholar] [PubMed]
- Stroun, M.; Anker, P.; Maurice, P.; Lyautey, J.; Lederrey, C.; Beljanski, M. Neoplastic Characteristics of the DNA Found in the Plasma of Cancer Patients. Oncology 1989, 46, 318–322. [Google Scholar] [CrossRef] [PubMed]
- Khier, S.; Lohan, L. Kinetics of Circulating Cell-Free DNA for Biomedical Applications: Critical Appraisal of the Literature. Future Sci. OA 2018, 4, FSO295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stroun, M.; Lyautey, J.; Lederrey, C.; Olson-Sand, A.; Anker, P. About the Possible Origin and Mechanism of Circulating DNA. Clin. Chim. Acta 2001, 313, 139–142. [Google Scholar] [CrossRef]
- Van der Vaart, M.; Pretorius, P.J. The Origin of Circulating Free DNA. Clin. Chem. 2007, 53, 2215. [Google Scholar] [CrossRef] [Green Version]
- Van der Vaart, M.; Pretorius, P.J. Circulating DNA. Its Origin and Fluctuation. Ann. N. Y. Acad. Sci. 2008, 1137, 18–26. [Google Scholar] [CrossRef]
- Mouliere, F.; Chandrananda, D.; Piskorz, A.M.; Moore, E.K.; Morris, J.; Ahlborn, L.B.; Mair, R.; Goranova, T.; Marass, F.; Heider, K.; et al. Enhanced Detection of Circulating Tumor DNA by Fragment Size Analysis. Sci. Transl. Med. 2018, 10, eaat4921. [Google Scholar] [CrossRef] [Green Version]
- Jahr, S.; Hentze, H.; Englisch, S.; Hardt, D.; Fackelmayer, F.O.; Hesch, R.D.; Knippers, R. DNA Fragments in the Blood Plasma of Cancer Patients: Quantitations and Evidence for Their Origin from Apoptotic and Necrotic Cells. Cancer Res. 2001, 61, 1659–1665. [Google Scholar]
- Corcoran, R.B.; Chabner, B.A. Application of Cell-Free DNA Analysis to Cancer Treatment. N. Engl. J. Med. 2018, 379, 1754–1765. [Google Scholar] [CrossRef] [Green Version]
- Hohaus, S.; Giachelia, M.; Massini, G.; Mansueto, G.; Vannata, B.; Bozzoli, V.; Criscuolo, M.; D’Alò, F.; Martini, M.; Larocca, L.M.; et al. Cell-Free Circulating DNA in Hodgkin’s and Non-Hodgkin’s Lymphomas. Ann. Oncol. 2009, 20, 1408–1413. [Google Scholar] [CrossRef]
- Diaz, L.A.; Bardelli, A. Liquid Biopsies: Genotyping Circulating Tumor DNA. J. Clin. Oncol. 2014, 32, 579–586. [Google Scholar] [CrossRef]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of Circulating Tumor DNA in Early- and Late-Stage Human Malignancies. Sci. Transl. Med. 2014, 6, 224ra24. [Google Scholar] [CrossRef] [Green Version]
- Hindson, C.M.; Chevillet, J.R.; Briggs, H.A.; Gallichotte, E.N.; Ruf, I.K.; Hindson, B.J.; Vessella, R.L.; Tewari, M. Absolute Quantification by Droplet Digital PCR versus Analog Real-Time PCR. Nat. Methods 2013, 10, 1003–1005. [Google Scholar] [CrossRef]
- Bratman, S.V.; Newman, A.M.; Alizadeh, A.A.; Diehn, M. Potential Clinical Utility of Ultrasensitive Circulating Tumor DNA Detection with CAPP-Seq. Expert Rev. Mol. Diagn. 2015, 15, 715–719. [Google Scholar] [CrossRef] [Green Version]
- Schroeder, T.; Rachlis, E.; Bug, G.; Stelljes, M.; Klein, S.; Steckel, N.K.; Wolf, D.; Ringhoffer, M.; Czibere, A.; Nachtkamp, K.; et al. Treatment of Acute Myeloid Leukemia or Myelodysplastic Syndrome Relapse after Allogeneic Stem Cell Transplantation with Azacitidine and Donor Lymphocyte Infusions—A Retrospective Multicenter Analysis from the German Cooperative Transplant Study Group. Biol. Blood Marrow Transplant. 2015, 21, 653–660. [Google Scholar] [CrossRef] [Green Version]
- Onecha, E.; Linares, M.; Rapado, I.; Ruiz-Heredia, Y.; Martinez-Sanchez, P.; Cedena, T.; Pratcorona, M.; Oteyza, J.P.; Herrera, P.; Barragan, E.; et al. A Novel Deep Targeted Sequencing Method for Minimal Residual Disease Monitoring in Acute Myeloid Leukemia. Haematologica 2019, 104, 288–296. [Google Scholar] [CrossRef]
- Onecha, E.; Rapado, I.; Luz Morales, M.; Carreño-Tarragona, G.; Martinez-Sanchez, P.; Gutierrez, X.; Sánchez Pina, J.M.; Linares, M.; Gallardo, M.; Martinez-López, J.; et al. Monitoring of Clonal Evolution of Acute Myeloid Leukemia Identifies the Leukemia Subtype, Clinical Outcome and Potential New Drug Targets for Post-Remission Strategies or Relapse. Haematologica 2021, 106, 2325–2333. [Google Scholar] [CrossRef]
- Shapiro, R.M.; Kim, D.D.H. Next-Generation Sequencing-Based Minimal Residual Disease Monitoring in Patients Receiving Allogeneic Hematopoietic Stem Cell Transplantation for Acute Myeloid Leukemia or Myelodysplastic Syndrome. Curr. Opin. Hematol. 2018, 25, 425–432. [Google Scholar] [CrossRef]
- Platzbecker, U.; Kubasch, A.S.; Homer-Bouthiette, C.; Prebet, T. Current Challenges and Unmet Medical Needs in Myelodysplastic Syndromes. Leukemia 2021, 35, 2182–2198. [Google Scholar] [CrossRef]
- Honoré, N.; Galot, R.; van Marcke, C.; Limaye, N.; Machiels, J.-P. Liquid Biopsy to Detect Minimal Residual Disease: Methodology and Impact. Cancers 2021, 13, 5364. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.; Woo, H.I.; Kim, J.-W.; Md, Y.K.; Lee, K.-A. Clinical Practice Guidelines for Pre-Analytical Procedures of Plasma Epidermal Growth Factor Receptor Variant Testing. Ann. Lab. Med. 2022, 42, 141–149. [Google Scholar] [CrossRef]
- Heuser, M.; Freeman, S.D.; Ossenkoppele, G.J.; Buccisano, F.; Hourigan, C.S.; Ngai, L.L.; Tettero, J.M.; Bachas, C.; Baer, C.; Béné, M.-C.; et al. 2021 Update on MRD in Acute Myeloid Leukemia: A Consensus Document from the European LeukemiaNet MRD Working Party. Blood 2021, 138, 2753–2767. [Google Scholar] [CrossRef]
- National Comprehensive Cancer Network. Acute Myeloid Leukemia (Version 1.2022); National Comprehensive Cancer Network: Plymouth Meeting, PA, USA, 2022. [Google Scholar]
- National Comprehensive Cancer Network. Myelodysplastic Syndromes (Version 3.2022); National Comprehensive Cancer Network: Plymouth Meeting, PA, USA, 2022. [Google Scholar]
- Barosi, G.; Mesa, R.; Finazzi, G.; Harrison, C.; Kiladjian, J.-J.; Lengfelder, E.; McMullin, M.F.; Passamonti, F.; Vannucchi, A.M.; Besses, C.; et al. Revised Response Criteria for Polycythemia Vera and Essential Thrombocythemia: An ELN and IWG-MRT Consensus Project. Blood 2013, 121, 4778–4781. [Google Scholar] [CrossRef]
- Tefferi, A.; Cervantes, F.; Mesa, R.; Passamonti, F.; Verstovsek, S.; Vannucchi, A.M.; Gotlib, J.; Dupriez, B.; Pardanani, A.; Harrison, C.; et al. Revised Response Criteria for Myelofibrosis: International Working Group-Myeloproliferative Neoplasms Research and Treatment (IWG-MRT) and European LeukemiaNet (ELN) Consensus Report. Blood 2013, 122, 1395–1398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- National Comprehensive Cancer Network. Myeloproliferative Neoplasms (Version 2.2021); National Comprehensive Cancer Network: Plymouth Meeting, PA, USA., 2021. [Google Scholar]
- National Comprehensive Cancer Network. Chronic Myeloid Leukemia (Version 2.2022); National Comprehensive Cancer Network: Plymouth Meeting, PA, USA, 2022. [Google Scholar]
- National Comprehensive Cancer Network. Acute Lymphoblastic Leukemia (Version 4.2021); National Comprehensive Cancer Network: Plymouth Meeting, PA, USA, 2021. [Google Scholar]
- National Comprehensive Cancer Network. B-Cell Lymphomas (Version 5.2021); National Comprehensive Cancer Network: Plymouth Meeting, PA, USA, 2021. [Google Scholar]
- National Comprehensive Cancer Network. Hodgkin Lymphoma (Version 1.2022); National Comprehensive Cancer Network: Plymouth Meeting, PA, USA, 2022. [Google Scholar]
- National Comprehensive Cancer Network. Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma (Version 1.2022); National Comprehensive Cancer Network: Plymouth Meeting, PA, USA, 2022. [Google Scholar]
- National Comprehensive Cancer Network. Multiple Myeloma (Version 4.2022); National Comprehensive Cancer Network: Plymouth Meeting, PA, USA, 2022. [Google Scholar]
- Rowe, J.M. Will New Agents Impact Survival in AML? Best Pract. Res. Clin. Haematol. 2019, 32, 101094. [Google Scholar] [CrossRef] [PubMed]
- Pelcovits, A.; Niroula, R. Acute Myeloid Leukemia: A Review. Rhode Isl. Med. J. 2020, 103, 38–40. [Google Scholar]
- De Kouchkovsky, I.; Abdul-Hay, M. Acute Myeloid Leukemia: A Comprehensive Review and 2016 Update. Blood Cancer J. 2016, 6, e441. [Google Scholar] [CrossRef]
- Döhner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute Myeloid Leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitzmaurice, C.; Global Burden of Disease Cancer Collaboration. Global, Regional, and National Cancer Incidence, Mortality, Years of Life Lost, Years Lived with Disability, and Disability-Adjusted Life-Years for 29 Cancer Groups, 2006 to 2016: A Systematic Analysis for the Global Burden of Disease Study. J. Clin. Oncol. 2018, 36, 1568. [Google Scholar] [CrossRef] [Green Version]
- Narayanan, D.; Weinberg, O.K. How I Investigate Acute Myeloid Leukemia. Int. J. Lab. Hematol. 2020, 42, 3–15. [Google Scholar] [CrossRef] [Green Version]
- Pettersson, L.; Levéen, P.; Axler, O.; Dvorakova, D.; Juliusson, G.; Ehinger, M. Improved Minimal Residual Disease Detection by Targeted Quantitative Polymerase Chain Reaction in Nucleophosmin 1 Type a Mutated Acute Myeloid Leukemia: MFC vs. RQ-PCR in MRD Detection in AML. Genes Chromosomes Cancer 2016, 55, 750–766. [Google Scholar] [CrossRef]
- Handschuh, L.; Kaźmierczak, M.; Milewski, M.; Gïralski, M.; Łuczak, M.; Wojtaszewska, M.; Uszczyńska-Ratajczak, B.; Lewandowski, K.; Komarnicki, M.; Figlerowicz, M. Gene Expression Profiling of Acute Myeloid Leukemia Samples from Adult Patients with AML-M1 and -M2 through Boutique Microarrays, Real-Time PCR and Droplet Digital PCR. Int. J. Oncol. 2017, 52, 656–678. [Google Scholar] [CrossRef]
- Grassi, S.; Guerrini, F.; Ciabatti, E.; Puccetti, R.; Salehzadeh, S.; Metelli, M.R.; Di Vita, A.; Domenichini, C.; Caracciolo, F.; Orciuolo, E.; et al. Digital Droplet PCR Is a Specific and Sensitive Tool for Detecting IDH2 Mutations in Acute Myeloid LeuKemia Patients. Cancers 2020, 12, 1738. [Google Scholar] [CrossRef]
- Maurillo, L.; Buccisano, F.; Spagnoli, A.; Del Poeta, G.; Panetta, P.; Neri, B.; Del Principe, M.I.; Mazzone, C.; Consalvo, M.I.; Tamburini, A.; et al. Monitoring of Minimal Residual Disease in Adult Acute Myeloid Leukemia Using Peripheral Blood as an Alternative Source to Bone Marrow. Haematologica 2007, 92, 605–611. [Google Scholar] [CrossRef] [Green Version]
- Buccisano, F.; Maurillo, L.; Gattei, V.; Del Poeta, G.; Del Principe, M.I.; Cox, M.C.; Panetta, P.; Consalvo, M.I.; Mazzone, C.; Neri, B.; et al. The Kinetics of Reduction of Minimal Residual Disease Impacts on Duration of Response and Survival of Patients with Acute Myeloid Leukemia. Leukemia 2006, 20, 1783–1789. [Google Scholar] [CrossRef] [Green Version]
- Smith, L.-L.; Pearce, D.; Smith, M.L.; Jenner, M.; Andrew Lister, T.; Bonnet, D.; Goff, L.; Fitzgibbon, J. Development of a Quantitative Real-Time Polymerase Chain Reaction Method for Monitoring CEBPA Mutations in Normal Karyotype Acute Myeloid Leukaemia. Br. J. Haematol. 2006, 133, 103–105. [Google Scholar] [CrossRef]
- Kern, W.; Bacher, U.; Haferlach, C.; Schnittger, S.; Haferlach, T. The Role of Multiparameter Flow Cytometry for Disease Monitoring in AML. Best Pract. Res. Clin. Haematol. 2010, 23, 379–390. [Google Scholar] [CrossRef]
- Cruz, N.M.; Mencia-Trinchant, N.; Hassane, D.C.; Guzman, M.L. Minimal Residual Disease in Acute Myelogenous Leukemia. Int. J. Lab. Hematol. 2017, 39, 53–60. [Google Scholar] [CrossRef] [Green Version]
- Zeijlemaker, W.; Kelder, A.; Oussoren-Brockhoff, Y.J.M.; Scholten, W.J.; Snel, A.N.; Veldhuizen, D.; Cloos, J.; Ossenkoppele, G.J.; Schuurhuis, G.J. Peripheral Blood Minimal Residual Disease May Replace Bone Marrow Minimal Residual Disease as an Immunophenotypic Biomarker for Impending Relapse in Acute Myeloid Leukemia. Leukemia 2016, 30, 708–715. [Google Scholar] [CrossRef]
- Thol, F.; Kölking, B.; Damm, F.; Reinhardt, K.; Klusmann, J.-H.; Reinhardt, D.; von Neuhoff, N.; Brugman, M.H.; Schlegelberger, B.; Suerbaum, S.; et al. Next-Generation Sequencing for Minimal Residual Disease Monitoring in Acute Myeloid Leukemia Patients with FLT3-ITD or NPM1 Mutations. Genes Chromosomes Cancer 2012, 51, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Vonk, C.M.; Al Hinai, A.S.A.; Hanekamp, D.; Valk, P.J.M. Molecular Minimal Residual Disease Detection in Acute Myeloid Leukemia. Cancers 2021, 13, 5431. [Google Scholar] [CrossRef] [PubMed]
- Grimwade, D.; Freeman, S.D. Defining Minimal Residual Disease in Acute Myeloid Leukemia: Which Platforms Are Ready for “Prime Time”? Hematol. Am. Soc. Hematol. Educ. Program 2014, 2014, 222–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shumilov, E.; Flach, J.; Kohlmann, A.; Banz, Y.; Bonadies, N.; Fiedler, M.; Pabst, T.; Bacher, U. Current Status and Trends in the Diagnostics of AML and MDS. Blood Rev. 2018, 32, 508–519. [Google Scholar] [CrossRef] [PubMed]
- Haferlach, T. Advancing Leukemia Diagnostics: Role of Next Generation Sequencing (NGS) in Acute Myeloid Leukemia. Hematol. Rep. 2020, 12, 8957. [Google Scholar] [CrossRef]
- Flach, J.; Shumilov, E.; Joncourt, R.; Porret, N.; Novak, U.; Pabst, T.; Bacher, U. Current Concepts and Future Directions for Hemato-Oncologic Diagnostics. Crit. Rev. Oncol. Hematol. 2020, 151, 102977. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Zhao, H. Next-Generation Sequencing in Liquid Biopsy: Cancer Screening and Early Detection. Hum. Genom. 2019, 13, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Short, N.J.; Patel, K.P.; Albitar, M.; Franquiz, M.; Luthra, R.; Kanagal-Shamanna, R.; Wang, F.; Assi, R.; Montalban-Bravo, G.; Matthews, J.; et al. Targeted Next-Generation Sequencing of Circulating Cell-Free DNA vs Bone Marrow in Patients with Acute Myeloid Leukemia. Blood Adv. 2020, 4, 1670–1677. [Google Scholar] [CrossRef]
- Nakamura, S.; Yokoyama, K.; Shimizu, E.; Yusa, N.; Kondoh, K.; Ogawa, M.; Takei, T.; Kobayashi, A.; Ito, M.; Isobe, M.; et al. Prognostic Impact of Circulating Tumor DNA Status Post–Allogeneic Hematopoietic Stem Cell Transplantation in AML and MDS. Blood 2019, 133, 2682–2695. [Google Scholar] [CrossRef]
- Rausch, C.; Rothenberg-Thurley, M.; Buerger, S.A.; Tschuri, S.; Dufour, A.; Neusser, M.; Schneider, S.; Spiekermann, K.; Metzeler, K.H.; Ziemann, F. Double Drop-Off Droplet Digital PCR. J. Mol. Diagn. 2021, 23, 975–985. [Google Scholar] [CrossRef]
- Lim, J.K.; Kuss, B.; Talaulikar, D. Role of Cell-Free DNA in Haematological Malignancies. Pathology 2021, 53, 416–426. [Google Scholar] [CrossRef]
- Steensma, D.P. Clinical Implications of Clonal Hematopoiesis. Mayo Clin. Proc. 2018, 93, 1122–1130. [Google Scholar] [CrossRef] [Green Version]
- Hasserjian, R.P.; Steensma, D.P.; Graubert, T.A.; Ebert, B.L. Clonal Hematopoiesis and Measurable Residual Disease Assessment in Acute Myeloid Leukemia. Blood 2020, 135, 1729–1738. [Google Scholar] [CrossRef]
- Yeh, P.; Dickinson, M.; Ftouni, S.; Hunter, T.; Sinha, D.; Wong, S.Q.; Agarwal, R.; Vedururu, R.; Doig, K.; Fong, C.Y.; et al. Molecular Disease Monitoring Using Circulating Tumor DNA in Myelodysplastic Syndromes. Blood 2017, 129, 1685–1690. [Google Scholar] [CrossRef]
- Gutierrez-Rodrigues, F.; Beerman, I.; Groarke, E.M.; Patel, B.A.; Spitofsky, N.; Dillon, L.W.; Raffo, D.Q.; Hourigan, C.S.; Kajigaya, S.; Ferrucci, L.; et al. Utility of Plasma Cell-Free DNA for de Novo Detection and Quantification of Clonal Hematopoiesis. Haematologica 2021. early view. [Google Scholar] [CrossRef]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 Revision to the World Health Organization Classification of Myeloid Neoplasms and Acute Leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Barbui, T.; Thiele, J.; Gisslinger, H.; Kvasnicka, H.M.; Vannucchi, A.M.; Guglielmelli, P.; Orazi, A.; Tefferi, A. The 2016 WHO Classification and Diagnostic Criteria for Myeloproliferative Neoplasms: Document Summary and in-Depth Discussion. Blood Cancer J. 2018, 8, 15. [Google Scholar] [CrossRef]
- Tefferi, A.; Barbui, T. Polycythemia Vera and Essential Thrombocythemia: 2021 Update on Diagnosis, Risk-stratification and Management. Am. J. Hematol. 2020, 95, 1599–1613. [Google Scholar] [CrossRef]
- Garcia-Gisbert, N.; Fernández-Ibarrondo, L.; Fernández-Rodríguez, C.; Gibert, J.; Andrade-Campos, M.; Arenillas, L.; Camacho, L.; Angona, A.; Longarón, R.; Salar, A.; et al. Circulating Cell-Free DNA Improves the Molecular Characterisation of Ph-Negative Myeloproliferative Neoplasms. Br. J. Haematol. 2021, 192, 300–309. [Google Scholar] [CrossRef]
- Redaelli, A.; Laskin, B.L.; Stephens, J.M.; Botteman, M.F.; Pashos, C.L. A Systematic Literature Review of the Clinical and Epidemiological Burden of Acute Lymphoblastic Leukaemia (ALL). Eur. J. Cancer Care 2005, 14, 53–62. [Google Scholar] [CrossRef]
- Nunes, V.; Cazzaniga, G.; Biondi, A. An Update on PCR Use for Minimal Residual Disease Monitoring in Acute Lymphoblastic Leukemia. Expert Rev. Mol. Diagn. 2017, 17, 953–963. [Google Scholar] [CrossRef]
- Theunissen, P.; Mejstrikova, E.; Sedek, L.; van der Sluijs-Gelling, A.J.; Gaipa, G.; Bartels, M.; Sobral da Costa, E.; Kotrová, M.; Novakova, M.; Sonneveld, E.; et al. Standardized Flow Cytometry for Highly Sensitive MRD Measurements in B-Cell Acute Lymphoblastic Leukemia. Blood 2017, 129, 347–357. [Google Scholar] [CrossRef]
- Kruse, A.; Abdel-Azim, N.; Kim, H.N.; Ruan, Y.; Phan, V.; Ogana, H.; Wang, W.; Lee, R.; Gang, E.J.; Khazal, S.; et al. Minimal Residual Disease Detection in Acute Lymphoblastic Leukemia. Int. J. Mol. Sci. 2020, 21, 1054. [Google Scholar] [CrossRef] [Green Version]
- Kotrova, M.; Muzikova, K.; Mejstrikova, E.; Novakova, M.; Bakardjieva-Mihaylova, V.; Fiser, K.; Stuchly, J.; Giraud, M.; Salson, M.; Pott, C.; et al. The Predictive Strength of Next-Generation Sequencing MRD Detection for Relapse Compared with Current Methods in Childhood ALL. Blood 2015, 126, 1045–1047. [Google Scholar] [CrossRef]
- Muffly, L.; Sundaram, V.; Chen, C.; Yurkiewicz, I.; Kuo, E.; Burnash, S.; Spiegel, J.Y.; Arai, S.; Frank, M.J.; Johnston, L.J.; et al. Concordance of Peripheral Blood and Bone Marrow Measurable Residual Disease in Adult Acute Lymphoblastic Leukemia. Blood Adv. 2021, 5, 3147–3151. [Google Scholar] [CrossRef]
- Van der Velden, V.H.J.; Jacobs, D.C.H.; Wijkhuijs, A.J.M.; Comans-Bitter, W.M.; Willemse, M.J.; Hählen, K.; Kamps, W.A.; van Wering, E.R.; van Dongen, J.J.M. Minimal Residual Disease Levels in Bone Marrow and Peripheral Blood Are Comparable in Children with T Cell Acute Lymphoblastic Leukemia (ALL), but Not in Precursor-B-ALL. Leukemia 2002, 16, 1432–1436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, A.K.; Stanulla, M.; Cario, G.; Flohr, T.; Sutton, R.; Möricke, A.; Anker, P.; Stroun, M.; Welte, K.; Bartram, C.R.; et al. Quantification of Free Total Plasma DNA and Minimal Residual Disease Detection in the Plasma of Children with Acute Lymphoblastic Leukemia. Ann. Hematol. 2009, 88, 897–905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, S.H.; Lau, K.M.; Li, C.K.; Chan, N.P.H.; Ip, R.K.L.; Cheng, C.K.; Lee, V.; Shing, M.M.K.; Leung, A.W.K.; Ha, S.Y.; et al. Minimal Residual Disease-Based Risk Stratification in Chinese Childhood Acute Lymphoblastic Leukemia by Flow Cytometry and Plasma DNA Quantitative Polymerase Chain Reaction. PLoS ONE 2013, 8, e69467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yegin, Z.A.; Can, F.; Gökçen, S.; Sadioğlu, R.E.; Özkurt, Z.N.; İlhan, Ç.; Yağcı, M. The Impact of Pre-Transplant Cell-Free DNA Levels on Leukemia Relapse and Transplant-Related Complications in Allogeneic Hematopoietic Stem Cell Transplant Recipients. Balk. Med. J. 2020, 37, 138–143. [Google Scholar] [CrossRef] [PubMed]
- Camus, V.; Jardin, F. Cell-Free DNA for the Management of Classical Hodgkin Lymphoma. Pharm. Basel Switz. 2021, 14, 207. [Google Scholar] [CrossRef]
- Schürch, C.M.; Federmann, B.; Quintanilla-Martinez, L.; Fend, F. Tumor Heterogeneity in Lymphomas: A Different Breed. Pathobiology 2018, 85, 130–145. [Google Scholar] [CrossRef]
- Melani, C.; Wilson, W.H.; Roschewski, M. Monitoring Clinical Outcomes in Aggressive B-Cell Lymphoma: From Imaging Studies to Circulating Tumor DNA. Best Pract. Res. Clin. Haematol. 2018, 31, 285–292. [Google Scholar] [CrossRef]
- Camus, V.; Jardin, F.; Tilly, H. The Value of Liquid Biopsy in Diagnosis and Monitoring of Diffuse Large B-Cell Lymphoma: Recent Developments and Future Potential. Expert Rev. Mol. Diagn. 2017, 17, 557–566. [Google Scholar] [CrossRef]
- Li, M.; Xu, C. Circulating Cell-Free DNA Utility for the Surveillance of Patients with Treated Diffuse Large B-Cell Lymphoma. Clin. Oncol. R. Coll. Radiol. G. B. 2017, 29, 637–638. [Google Scholar] [CrossRef]
- Eskandari, M.; Manoochehrabadi, S.; Pashaiefar, H.; Zaimy, M.A.; Ahmadvand, M. Clinical Significance of Cell-Free DNA as a Prognostic Biomarker in Patients with Diffuse Large B-Cell Lymphoma. Blood Res. 2019, 54, 114–119. [Google Scholar] [CrossRef] [Green Version]
- Hur, J.Y.; Kim, Y.J.; Yoon, S.E.; Son, D.-S.; Park, W.-Y.; Kim, S.J.; Park, D.; Kim, W.S. Plasma Cell-Free DNA Is a Prognostic Biomarker for Survival in Patients with Aggressive Non-Hodgkin Lymphomas. Ann. Hematol. 2020, 99, 1293–1302. [Google Scholar] [CrossRef]
- Lv, L.; Liu, Y. Clinical Application of Liquid Biopsy in Non-Hodgkin Lymphoma. Front. Oncol. 2021, 11, 658234. [Google Scholar] [CrossRef]
- Armand, P.; Oki, Y.; Neuberg, D.S.; Faham, M.; Cummings, C.; Klinger, M.; Weng, L.; Bhattar, S.; Lacasce, A.S.; Jacobsen, E.D.; et al. Detection of Circulating Tumour DNA in Patients with Aggressive B-Cell Non-Hodgkin Lymphoma. Br. J. Haematol. 2013, 163, 123–126. [Google Scholar] [CrossRef]
- Kurtz, D.M.; Green, M.R.; Bratman, S.V.; Scherer, F.; Liu, C.L.; Kunder, C.A.; Takahashi, K.; Glover, C.; Keane, C.; Kihira, S.; et al. Noninvasive Monitoring of Diffuse Large B-Cell Lymphoma by Immunoglobulin High-Throughput Sequencing. Blood 2015, 125, 3679–3687. [Google Scholar] [CrossRef] [Green Version]
- Roschewski, M.; Dunleavy, K.; Pittaluga, S.; Moorhead, M.; Pepin, F.; Kong, K.; Shovlin, M.; Jaffe, E.S.; Staudt, L.M.; Lai, C.; et al. Circulating Tumour DNA and CT Monitoring in Patients with Untreated Diffuse Large B-Cell Lymphoma: A Correlative Biomarker Study. Lancet Oncol. 2015, 16, 541–549. [Google Scholar] [CrossRef] [Green Version]
- Herrera, A.F.; Kim, H.T.; Kong, K.A.; Faham, M.; Sun, H.; Sohani, A.R.; Alyea, E.P.; Carlton, V.E.; Chen, Y.-B.; Cutler, C.S.; et al. Next-Generation Sequencing-Based Detection of Circulating Tumour DNA After Allogeneic Stem Cell Transplantation for Lymphoma. Br. J. Haematol. 2016, 175, 841–850. [Google Scholar] [CrossRef]
- Camus, V.; Jardin, F. Cell-Free DNA and the Monitoring of Lymphoma Treatment. Pharmacogenomics 2019, 20, 1271–1282. [Google Scholar] [CrossRef]
- Li, M.; Jia, Y.; Xu, J.; Cheng, X.; Xu, C. Assessment of the Circulating Cell-Free DNA Marker Association with Diagnosis and Prognostic Prediction in Patients with Lymphoma: A Single-Center Experience. Ann. Hematol. 2017, 96, 1343–1351. [Google Scholar] [CrossRef] [Green Version]
- Rossi, D.; Diop, F.; Spaccarotella, E.; Monti, S.; Zanni, M.; Rasi, S.; Deambrogi, C.; Spina, V.; Bruscaggin, A.; Favini, C.; et al. Diffuse Large B-Cell Lymphoma Genotyping on the Liquid Biopsy. Blood 2017, 129, 1947–1957. [Google Scholar] [CrossRef]
- Kurtz, D.M.; Scherer, F.; Jin, M.C.; Soo, J.; Craig, A.F.M.; Esfahani, M.S.; Chabon, J.J.; Stehr, H.; Liu, C.L.; Tibshirani, R.; et al. Circulating Tumor DNA Measurements As Early Outcome Predictors in Diffuse Large B-Cell Lymphoma. J. Clin. Oncol. 2018, 36, 2845–2853. [Google Scholar] [CrossRef] [PubMed]
- Rivas-Delgado, A.; Nadeu, F.; Enjuanes, A.; Casanueva-Eliceiry, S.; Mozas, P.; Magnano, L.; Castrejón de Anta, N.; Rovira, J.; Dlouhy, I.; Martín, S.; et al. Mutational Landscape and Tumor Burden Assessed by Cell-Free DNA in Diffuse Large B-Cell Lymphoma in a Population-Based Study. Clin. Cancer Res. 2021, 27, 513–521. [Google Scholar] [CrossRef]
- Kurtz, D.M.; Soo, J.; Co Ting Keh, L.; Alig, S.; Chabon, J.J.; Sworder, B.J.; Schultz, A.; Jin, M.C.; Scherer, F.; Garofalo, A.; et al. Enhanced Detection of Minimal Residual Disease by Targeted Sequencing of Phased Variants in Circulating Tumor DNA. Nat. Biotechnol. 2021, 39, 1537–1547. [Google Scholar] [CrossRef]
- Scherer, F.; Kurtz, D.M.; Newman, A.M.; Stehr, H.; Craig, A.F.M.; Esfahani, M.S.; Lovejoy, A.F.; Chabon, J.J.; Klass, D.M.; Liu, C.L.; et al. Distinct Biological Subtypes and Patterns of Genome Evolution in Lymphoma Revealed by Circulating Tumor DNA. Sci. Transl. Med. 2016, 8, 364ra155. [Google Scholar] [CrossRef] [Green Version]
- Arzuaga-Mendez, J.; Prieto-Fernández, E.; Lopez-Lopez, E.; Martin-Guerrero, I.; García-Ruiz, J.C.; García-Orad, A. Cell-Free DNA as a Biomarker in Diffuse Large B-Cell Lymphoma: A Systematic Review. Crit. Rev. Oncol. Hematol. 2019, 139, 7–15. [Google Scholar] [CrossRef]
- Chen, F.; Pang, D.; Guo, H.; Jiang, X.; Liu, S.; Huang, L.; Wei, X.; Liang, Z.; Wang, X.; Li, W. Clinicopathological Characteristics and Mutational Profiling of Adult T-Cell Lymphoblastic Lymphoma in a Chinese Population. Cancer Manag. Res. 2020, 12, 3003–3012. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.-H.; Kim, Y.J.; Lee, D.; Cho, D.; Ko, Y.H.; Cho, J.; Park, W.-Y.; Park, D.; Kim, S.J.; Kim, W.S. Analysis of Circulating Tumor DNA by Targeted Ultra-Deep Sequencing across Various Non-Hodgkin Lymphoma Subtypes. Leuk. Lymphoma 2019, 60, 2237–2246. [Google Scholar] [CrossRef] [PubMed]
- Suehara, Y.; Sakata-Yanagimoto, M.; Hattori, K.; Kusakabe, M.; Nanmoku, T.; Sato, T.; Noguchi, M.; Chiba, S. Mutations Found in Cell-free DNAs of Patients with Malignant Lymphoma at Remission Can Derive from Clonal Hematopoiesis. Cancer Sci. 2019, 110, 3375–3381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hossain, N.M.; Dahiya, S.; Le, R.; Abramian, A.M.; Kong, K.A.; Muffly, L.S.; Miklos, D.B. Circulating Tumor DNA Assessment in Patients with Diffuse Large B-Cell Lymphoma Following CAR T-Cell Therapy. Leuk. Lymphoma 2019, 60, 503–506. [Google Scholar] [CrossRef] [PubMed]
- Galimberti, S.; Genuardi, E.; Mazziotta, F.; Iovino, L.; Morabito, F.; Grassi, S.; Ciabatti, E.; Guerrini, F.; Petrini, M. The Minimal Residual Disease in Non-Hodgkin’s Lymphomas: From the Laboratory to the Clinical Practice. Front. Oncol. 2019, 9, 528. [Google Scholar] [CrossRef]
- Cowan, A.J.; Stevenson, P.A.; Cassaday, R.D.; Graf, S.A.; Fromm, J.R.; Wu, D.; Holmberg, L.A.; Till, B.G.; Chauncey, T.R.; Smith, S.D.; et al. Pretransplantation Minimal Residual Disease Predicts Survival in Patients with Mantle Cell Lymphoma Undergoing Autologous Stem Cell Transplantation in Complete Remission. Biol. Blood Marrow Transplant. 2016, 22, 380–385. [Google Scholar] [CrossRef]
- Kolstad, A.; Pedersen, L.B.; Eskelund, C.W.; Husby, S.; Grønbæk, K.; Jerkeman, M.; Laurell, A.; Räty, R.; Elonen, E.; Andersen, N.S.; et al. Molecular Monitoring after Autologous Stem Cell Transplantation and Preemptive Rituximab Treatment of Molecular Relapse; Results from the Nordic Mantle Cell Lymphoma Studies (MCL2 and MCL3) with Median Follow-Up of 8.5 Years. Biol. Blood Marrow Transplant. 2017, 23, 428–435. [Google Scholar] [CrossRef] [Green Version]
- Jung, D.; Jain, P.; Yao, Y.; Wang, M. Advances in the Assessment of Minimal Residual Disease in Mantle Cell Lymphoma. J. Hematol. Oncol. 2020, 13, 127. [Google Scholar] [CrossRef]
- Lakhotia, R.; Melani, C.; Pittaluga, S.; Dunleavy, K.; Saba, N.S.; Lucas, A.N.; Jacob, A.; Yusko, E.; Steinberg, S.M.; Jaffe, E.S.; et al. Circulating Tumor DNA Dynamics during Therapy Predict Outcomes in Mantle Cell Lymphoma. Blood 2018, 132, 147. [Google Scholar] [CrossRef]
- Agarwal, R.; Chan, Y.-C.; Tam, C.S.; Hunter, T.; Vassiliadis, D.; Teh, C.E.; Thijssen, R.; Yeh, P.; Wong, S.Q.; Ftouni, S.; et al. Dynamic Molecular Monitoring Reveals That SWI–SNF Mutations Mediate Resistance to Ibrutinib plus Venetoclax in Mantle Cell Lymphoma. Nat. Med. 2019, 25, 119–129. [Google Scholar] [CrossRef]
- Bachy, E.; Houot, R.; Morschhauser, F.; Sonet, A.; Brice, P.; Belhadj, K.; Cartron, G.; Audhuy, B.; Fermé, C.; Feugier, P.; et al. Long-Term Follow up of the FL2000 Study Comparing CHVP-Interferon to CHVP-Interferon plus Rituximab in Follicular Lymphoma. Haematologica 2013, 98, 1107–1114. [Google Scholar] [CrossRef]
- Sarkozy, C.; Huet, S.; Carlton, V.E.H.; Fabiani, B.; Delmer, A.; Jardin, F.; Delfau-Larue, M.-H.; Hacini, M.; Ribrag, V.; Guidez, S.; et al. The Prognostic Value of Clonal Heterogeneity and Quantitative Assessment of Plasma Circulating Clonal IG-VDJ Sequences at Diagnosis in Patients with Follicular Lymphoma. Oncotarget 2017, 8, 8765–8774. [Google Scholar] [CrossRef] [Green Version]
- Delfau-Larue, M.-H.; van der Gucht, A.; Dupuis, J.; Jais, J.-P.; Nel, I.; Beldi-Ferchiou, A.; Hamdane, S.; Benmaad, I.; Laboure, G.; Verret, B.; et al. Total Metabolic Tumor Volume, Circulating Tumor Cells, Cell-Free DNA: Distinct Prognostic Value in Follicular Lymphoma. Blood Adv. 2018, 2, 807–816. [Google Scholar] [CrossRef]
- Jimenez-Ubieto, A.I.; Heredia, Y.; de la Rosa, J.M.; Rodriguez-Izquierdo, A.; Rufian, L.; Carrillo, J.; Sanchez, R.; Onecha, E.; Wang, C.; Sarandeses, P.; et al. Minimal Residual Disease Monitoring from Liquid Biopsy By Next Generation Sequencing in Follicular Lymphoma Patients. In Proceedings of the 62nd ASH Annual Meeting and Exposition, Virtual, 5–8 December 2020. 282. [Google Scholar]
- Zeremski, V.; Koehler, M.; Fischer, T.; Schalk, E. Characteristics and Outcome of Patients with Primary CNS Lymphoma in a “Real-Life” Setting Compared to a Clinical Trial. Ann. Hematol. 2016, 95, 793–799. [Google Scholar] [CrossRef]
- Hattori, K.; Sakata-Yanagimoto, M.; Suehara, Y.; Yokoyama, Y.; Kato, T.; Kurita, N.; Nishikii, H.; Obara, N.; Takano, S.; Ishikawa, E.; et al. Clinical Significance of Disease-Specific MYD88 Mutations in Circulating DNA in Primary Central Nervous System Lymphoma. Cancer Sci. 2018, 109, 225–230. [Google Scholar] [CrossRef] [Green Version]
- Yoon, S.E.; Kim, Y.J.; Shim, J.H.; Park, D.; Cho, J.; Ko, Y.H.; Park, W.-Y.; Mun, Y.-C.; Lee, K.E.; Cho, D.; et al. Plasma Circulating Tumor DNA in Patients with Primary Central Nervous System Lymphoma. Cancer Res. Treat. 2021. Epub ahead of print. [Google Scholar] [CrossRef]
- Hiemcke-Jiwa, L.S.; Leguit, R.J.; Snijders, T.J.; Bromberg, J.E.C.; Nierkens, S.; Jiwa, N.M.; Minnema, M.C.; Huibers, M.M.H. MYD88 p.(L265P) Detection on Cell-Free DNA in Liquid Biopsies of Patients with Primary Central Nervous System Lymphoma. Br. J. Haematol. 2019, 185, 974–977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rimelen, V.; Ahle, G.; Pencreach, E.; Zinniger, N.; Debliquis, A.; Zalmaï, L.; Harzallah, I.; Hurstel, R.; Alamome, I.; Lamy, F.; et al. Tumor Cell-Free DNA Detection in CSF for Primary CNS Lymphoma Diagnosis. Acta Neuropathol. Commun. 2019, 7, 43. [Google Scholar] [CrossRef]
- Yamagishi, Y.; Sasaki, N.; Nakano, Y.; Matushita, Y.; Omura, T.; Shimizu, S.; Saito, K.; Kobayashi, K.; Narita, Y.; Kondo, A.; et al. Liquid Biopsy of Cerebrospinal Fluid for MYD88 L265P Mutation Is Useful for Diagnosis of Central Nervous System Lymphoma. Cancer Sci. 2021, 112, 4702–4710. [Google Scholar] [CrossRef]
- Hayashida, M.; Maekawa, F.; Chagi, Y.; Iioka, F.; Kobashi, Y.; Watanabe, M.; Ohno, H. Combination of Multicolor Flow Cytometry for Circulating Lymphoma Cells and Tests for the RHOAG17V and IDH2R172 Hot-Spot Mutations in Plasma Cell-Free DNA as Liquid Biopsy for the Diagnosis of Angioimmunoblastic T-Cell Lymphoma. Leuk. Lymphoma 2020, 61, 2389–2398. [Google Scholar] [CrossRef]
- Miljkovic, M.D.; Melani, C.; Pittaluga, S.; Lakhotia, R.; Lucas, N.; Jacob, A.; Yusko, E.; Jaffe, E.S.; Wilson, W.H.; Roschewski, M. Next-Generation Sequencing-Based Monitoring of Circulating Tumor DNA Reveals Clonotypic Heterogeneity in Untreated PTCL. Blood Adv. 2021, 5, 4198–4210. [Google Scholar] [CrossRef]
- Diehl, V.; Thomas, R.K.; Re, D. Part II: Hodgkin’s Lymphoma--Diagnosis and Treatment. Lancet Oncol. 2004, 5, 19–26. [Google Scholar] [CrossRef]
- Bessi, L.; Viailly, P.-J.; Bohers, E.; Ruminy, P.; Maingonnat, C.; Bertrand, P.; Vasseur, N.; Beaussire, L.; Cornic, M.; Etancelin, P.; et al. Somatic Mutations of Cell-Free Circulating DNA Detected by Targeted next-Generation Sequencing and Digital Droplet PCR in Classical Hodgkin Lymphoma. Leuk. Lymphoma 2019, 60, 498–502. [Google Scholar] [CrossRef] [PubMed]
- Camus, V.; Viennot, M.; Lequesne, J.; Viailly, P.-J.; Bohers, E.; Bessi, L.; Marcq, B.; Etancelin, P.; Dubois, S.; Picquenot, J.-M.; et al. Targeted Genotyping of Circulating Tumor DNA for Classical Hodgkin Lymphoma Monitoring: A Prospective Study. Haematologica 2021, 106, 154–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spina, V.; Bruscaggin, A.; Cuccaro, A.; Martini, M.; Di Trani, M.; Forestieri, G.; Manzoni, M.; Condoluci, A.; Arribas, A.; Terzi-Di-Bergamo, L.; et al. Circulating Tumor DNA Reveals Genetics, Clonal Evolution, and Residual Disease in Classical Hodgkin Lymphoma. Blood 2018, 131, 2413–2425. [Google Scholar] [CrossRef] [Green Version]
- Camus, V.; Stamatoullas, A.; Mareschal, S.; Viailly, P.-J.; Sarafan-Vasseur, N.; Bohers, E.; Dubois, S.; Picquenot, J.M.; Ruminy, P.; Maingonnat, C.; et al. Detection and Prognostic Value of Recurrent Exportin 1 Mutations in Tumor and Cell-Free Circulating DNA of Patients with Classical Hodgkin Lymphoma. Haematologica 2016, 101, 1094–1101. [Google Scholar] [CrossRef]
- Baumann, T.; Delgado, J.; Santacruz, R.; Martínez-Trillos, A.; Royo, C.; Navarro, A.; Pinyol, M.; Rozman, M.; Pereira, A.; Villamor, N.; et al. Chronic Lymphocytic Leukemia in the Elderly: Clinico-Biological Features, Outcomes, and Proposal of a Prognostic Model. Haematologica 2014, 99, 1599–1604. [Google Scholar] [CrossRef] [Green Version]
- Böttcher, S.; Ritgen, M.; Fischer, K.; Stilgenbauer, S.; Busch, R.M.; Fingerle-Rowson, G.; Fink, A.M.; Bühler, A.; Zenz, T.; Wenger, M.K.; et al. Minimal Residual Disease Quantification Is an Independent Predictor of Progression-Free and Overall Survival in Chronic Lymphocytic Leukemia: A Multivariate Analysis from the Randomized GCLLSG CLL8 Trial. J. Clin. Oncol. 2012, 30, 980–988. [Google Scholar] [CrossRef]
- Ghia, P. A Look into the Future: Can Minimal Residual Disease Guide Therapy and Predict Prognosis in Chronic Lymphocytic Leukemia? Hematol. Am. Soc. Hematol. Educ. Program 2012, 2012, 97–104. [Google Scholar] [CrossRef] [Green Version]
- Strati, P.; Keating, M.J.; O’Brien, S.M.; Burger, J.; Ferrajoli, A.; Jain, N.; Tambaro, F.P.; Estrov, Z.; Jorgensen, J.; Challagundla, P.; et al. Eradication of Bone Marrow Minimal Residual Disease May Prompt Early Treatment Discontinuation in CLL. Blood 2014, 123, 3727–3732. [Google Scholar] [CrossRef] [Green Version]
- Del Giudice, I.; Raponi, S.; Della Starza, I.; De Propris, M.S.; Cavalli, M.; De Novi, L.A.; Cappelli, L.V.; Ilari, C.; Cafforio, L.; Guarini, A.; et al. Minimal Residual Disease in Chronic Lymphocytic Leukemia: A New Goal? Front. Oncol. 2019, 9, 689. [Google Scholar] [CrossRef] [Green Version]
- Landgren, O.; Kyle, R.A.; Pfeiffer, R.M.; Katzmann, J.A.; Caporaso, N.E.; Hayes, R.B.; Dispenzieri, A.; Kumar, S.; Clark, R.J.; Baris, D.; et al. Monoclonal Gammopathy of Undetermined Significance (MGUS) Consistently Precedes Multiple Myeloma: A Prospective Study. Blood 2009, 113, 5412–5417. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.K.; Dispenzieri, A.; Lacy, M.Q.; Gertz, M.A.; Buadi, F.K.; Pandey, S.; Kapoor, P.; Dingli, D.; Hayman, S.R.; Leung, N.; et al. Continued Improvement in Survival in Multiple Myeloma: Changes in Early Mortality and Outcomes in Older Patients. Leukemia 2014, 28, 1122–1128. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Paiva, B.; Anderson, K.C.; Durie, B.; Landgren, O.; Moreau, P.; Munshi, N.; Lonial, S.; Bladé, J.; Mateos, M.-V.; et al. International Myeloma Working Group Consensus Criteria for Response and Minimal Residual Disease Assessment in Multiple Myeloma. Lancet Oncol. 2016, 17, e328–e346. [Google Scholar] [CrossRef]
- Flores-Montero, J.; Sanoja-Flores, L.; Paiva, B.; Puig, N.; García-Sánchez, O.; Böttcher, S.; van der Velden, V.H.J.; Pérez-Morán, J.-J.; Vidriales, M.-B.; García-Sanz, R.; et al. Next Generation Flow for Highly Sensitive and Standardized Detection of Minimal Residual Disease in Multiple Myeloma. Leukemia 2017, 31, 2094–2103. [Google Scholar] [CrossRef] [Green Version]
- Anderson, K.C.; Auclair, D.; Kelloff, G.J.; Sigman, C.C.; Avet-Loiseau, H.; Farrell, A.T.; Gormley, N.J.; Kumar, S.K.; Landgren, O.; Munshi, N.C.; et al. The Role of Minimal Residual Disease Testing in Myeloma Treatment Selection and Drug Development: Current Value and Future Applications. Clin. Cancer Res. 2017, 23, 3980–3993. [Google Scholar] [CrossRef] [Green Version]
- Kubaczkova, V.; Vrabel, D.; Sedlarikova, L.; Besse, L.; Sevcikova, S. Cell-Free DNA—Minimally Invasive Marker of Hematological Malignancies. Eur. J. Haematol. 2017, 99, 291–299. [Google Scholar] [CrossRef] [Green Version]
- Wong, S.W.; Shah, N.; Ledergor, G.; Martin, T.; Wolf, J.; Shui, A.M.; Huang, C.-Y.; Martinez-Lopez, J. Early Dynamics and Depth of Response in Multiple Myeloma Patients Treated With BCMA CAR-T Cells. Front. Oncol. 2021, 11, 783703. [Google Scholar] [CrossRef]
- Bolli, N.; Avet-Loiseau, H.; Wedge, D.C.; Van Loo, P.; Alexandrov, L.B.; Martincorena, I.; Dawson, K.J.; Iorio, F.; Nik-Zainal, S.; Bignell, G.R.; et al. Heterogeneity of Genomic Evolution and Mutational Profiles in Multiple Myeloma. Nat. Commun. 2014, 5, 2997. [Google Scholar] [CrossRef] [Green Version]
- Walker, B.A.; Wardell, C.P.; Melchor, L.; Brioli, A.; Johnson, D.C.; Kaiser, M.F.; Mirabella, F.; Lopez-Corral, L.; Humphray, S.; Murray, L.; et al. Intraclonal Heterogeneity Is a Critical Early Event in the Development of Myeloma and Precedes the Development of Clinical Symptoms. Leukemia 2014, 28, 384–390. [Google Scholar] [CrossRef] [Green Version]
- Ghobrial, I.M. Myeloma as a Model for the Process of Metastasis: Implications for Therapy. Blood 2012, 120, 20–30. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Beasley, S.; Prigozhina, N.L.; Higgins, R.; Ikeda, S.; Lee, F.Y.; Marrinucci, D.; Jia, S. Detection and Characterization of Circulating Tumour Cells in Multiple Myeloma. J. Circ. Biomark. 2016, 5, 10. [Google Scholar] [CrossRef] [Green Version]
- Lohr, J.G.; Kim, S.; Gould, J.; Knoechel, B.; Drier, Y.; Cotton, M.J.; Gray, D.; Birrer, N.; Wong, B.; Ha, G.; et al. Genetic Interrogation of Circulating Multiple Myeloma Cells at Single-Cell Resolution. Sci. Transl. Med. 2016, 8, 363ra147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foulk, B.; Schaffer, M.; Gross, S.; Rao, C.; Smirnov, D.; Connelly, M.C.; Chaturvedi, S.; Reddy, M.; Brittingham, G.; Mata, M.; et al. Enumeration and Characterization of Circulating Multiple Myeloma Cells in Patients with Plasma Cell Disorders. Br. J. Haematol. 2018, 180, 71–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcés, J.-J.; Simicek, M.; Vicari, M.; Brozova, L.; Burgos, L.; Bezdekova, R.; Alignani, D.; Calasanz, M.-J.; Growkova, K.; Goicoechea, I.; et al. Transcriptional Profiling of Circulating Tumor Cells in Multiple Myeloma: A New Model to Understand Disease Dissemination. Leukemia 2020, 34, 589–603. [Google Scholar] [CrossRef] [PubMed]
- Ntanasis-Stathopoulos, I.; Gavriatopoulou, M.; Terpos, E.; Fotiou, D.; Kastritis, E.; Dimopoulos, M.A. Monitoring Plasma Cell Dyscrasias with Cell-Free DNA Analysis. Clin. Lymphoma Myeloma Leuk. 2020, 20, e905–e909. [Google Scholar] [CrossRef] [PubMed]
- Sata, H.; Shibayama, H.; Maeda, I.; Habuchi, Y.; Nakatani, E.; Fukushima, K.; Fujita, J.; Ezoe, S.; Tadokoro, S.; Maeda, T.; et al. Quantitative Polymerase Chain Reaction Analysis with Allele-Specific Oligonucleotide Primers for Individual IgH VDJ Regions to Evaluate Tumor Burden in Myeloma Patients. Exp. Hematol. 2015, 43, 374–381.e2. [Google Scholar] [CrossRef]
- Oberle, A.; Brandt, A.; Voigtlaender, M.; Thiele, B.; Radloff, J.; Schulenkorf, A.; Alawi, M.; Akyüz, N.; März, M.; Ford, C.T.; et al. Monitoring Multiple Myeloma by Next-Generation Sequencing of V(D)J Rearrangements from Circulating Myeloma Cells and Cell-Free Myeloma DNA. Haematologica 2017, 102, 1105–1111. [Google Scholar] [CrossRef]
- Mithraprabhu, S.; Khong, T.; Ramachandran, M.; Chow, A.; Klarica, D.; Mai, L.; Walsh, S.; Broemeling, D.; Marziali, A.; Wiggin, M.; et al. Circulating Tumour DNA Analysis Demonstrates Spatial Mutational Heterogeneity That Coincides with Disease Relapse in Myeloma. Leukemia 2017, 31, 1695–1705. [Google Scholar] [CrossRef]
- Rustad, E.H.; Coward, E.; Skytøen, E.R.; Misund, K.; Holien, T.; Standal, T.; Børset, M.; Beisvag, V.; Myklebost, O.; Meza-Zepeda, L.A.; et al. Monitoring Multiple Myeloma by Quantification of Recurrent Mutations in Serum. Haematologica 2017, 102, 1266–1272. [Google Scholar] [CrossRef] [Green Version]
- Gerber, B.; Manzoni, M.; Spina, V.; Bruscaggin, A.; Lionetti, M.; Fabris, S.; Barbieri, M.; Ciceri, G.; Pompa, A.; Forestieri, G.; et al. Circulating Tumor DNA as a Liquid Biopsy in Plasma Cell Dyscrasias. Haematologica 2018, 103, e245–e248. [Google Scholar] [CrossRef] [Green Version]
- Biancon, G.; Gimondi, S.; Vendramin, A.; Carniti, C.; Corradini, P. Noninvasive Molecular Monitoring in Multiple Myeloma Patients Using Cell-Free Tumor DNA. J. Mol. Diagn. 2018, 20, 859–870. [Google Scholar] [CrossRef] [Green Version]
- Kis, O.; Kaedbey, R.; Chow, S.; Danesh, A.; Dowar, M.; Li, T.; Li, Z.; Liu, J.; Mansour, M.; Masih-Khan, E.; et al. Circulating Tumour DNA Sequence Analysis as an Alternative to Multiple Myeloma Bone Marrow Aspirates. Nat. Commun. 2017, 8, 15086. [Google Scholar] [CrossRef]
- Vrabel, D.; Sedlarikova, L.; Besse, L.; Rihova, L.; Bezdekova, R.; Almasi, M.; Kubaczkova, V.; Brožová, L.; Jarkovsky, J.; Plonkova, H.; et al. Dynamics of Tumor-specific CfDNA in Response to Therapy in Multiple Myeloma Patients. Eur. J. Haematol. 2020, 104, 190–197. [Google Scholar] [CrossRef]
- Mithraprabhu, S.; Morley, R.; Khong, T.; Kalff, A.; Bergin, K.; Hocking, J.; Savvidou, I.; Bowen, K.M.; Ramachandran, M.; Choi, K.; et al. Monitoring Tumour Burden and Therapeutic Response through Analysis of Circulating Tumour DNA and Extracellular RNA in Multiple Myeloma Patients. Leukemia 2019, 33, 2022–2033. [Google Scholar] [CrossRef]
- Deshpande, S.; Tytarenko, R.G.; Wang, Y.; Boyle, E.M.; Ashby, C.; Schinke, C.D.; Thanendrarajan, S.; Zangari, M.; Zhan, F.; Davies, F.E.; et al. Monitoring Treatment Response and Disease Progression in Myeloma with Circulating Cell-free DNA. Eur. J. Haematol. 2021, 106, 230–240. [Google Scholar] [CrossRef]
- Manzoni, M.; Pompa, A.; Fabris, S.; Pelizzoni, F.; Ciceri, G.; Seia, M.; Ziccheddu, B.; Bolli, N.; Corradini, P.; Baldini, L.; et al. Limits and Applications of Genomic Analysis of Circulating Tumor DNA as a Liquid Biopsy in Asymptomatic Forms of Multiple Myeloma. HemaSphere 2020, 4, e402. [Google Scholar] [CrossRef]
- Pugh, T.J. Circulating Tumour DNA for Detecting Minimal Residual Disease in Multiple Myeloma. Semin. Hematol. 2018, 55, 38–40. [Google Scholar] [CrossRef]
- Mazzotti, C.; Buisson, L.; Maheo, S.; Perrot, A.; Chretien, M.-L.; Leleu, X.; Hulin, C.; Manier, S.; Hébraud, B.; Roussel, M.; et al. Myeloma MRD by Deep Sequencing from Circulating Tumor DNA Does Not Correlate with Results Obtained in the Bone Marrow. Blood Adv. 2018, 2, 2811–2813. [Google Scholar] [CrossRef] [Green Version]
- Long, X.; Xu, Q.; Lou, Y.; Li, C.; Gu, J.; Cai, H.; Wang, D.; Xu, J.; Li, T.; Zhou, X.; et al. The Utility of Non-invasive Liquid Biopsy for Mutational Analysis and Minimal Residual Disease Assessment in Extramedullary Multiple Myeloma. Br. J. Haematol. 2020, 189, e45–e48. [Google Scholar] [CrossRef] [Green Version]
- Manier, S.; Park, J.; Capelletti, M.; Bustoros, M.; Freeman, S.S.; Ha, G.; Rhoades, J.; Liu, C.J.; Huynh, D.; Reed, S.C.; et al. Whole-Exome Sequencing of Cell-Free DNA and Circulating Tumor Cells in Multiple Myeloma. Nat. Commun. 2018, 9, 1691. [Google Scholar] [CrossRef]
- Guo, G.; Raje, N.S.; Seifer, C.; Kloeber, J.; Isenhart, R.; Ha, G.; Yee, A.J.; O’Donnell, E.K.; Tai, Y.-T.; Richardson, P.G.; et al. Genomic Discovery and Clonal Tracking in Multiple Myeloma by Cell-Free DNA Sequencing. Leukemia 2018, 32, 1838–1841. [Google Scholar] [CrossRef] [PubMed]
- Waldschmidt, J.M.; Vijaykumar, T.; Knoechel, B.; Lohr, J.G. Tracking Myeloma Tumor DNA in Peripheral Blood. Best Pract. Res. Clin. Haematol. 2020, 33, 101146. [Google Scholar] [CrossRef]
- Mithraprabhu, S.; Spencer, A. Circulating Tumour DNA Analysis in Multiple Myeloma. Oncotarget 2017, 8, 90610–90611. [Google Scholar] [CrossRef] [PubMed]
- Medina Diaz, I.; Nocon, A.; Mehnert, D.H.; Fredebohm, J.; Diehl, F.; Holtrup, F. Performance of Streck CfDNA Blood Collection Tubes for Liquid Biopsy Testing. PLoS ONE 2016, 11, e0166354. [Google Scholar] [CrossRef] [PubMed]
- Franczak, C.; Filhine-Tresarrieu, P.; Gilson, P.; Merlin, J.-L.; Au, L.; Harlé, A. Technical Considerations for Circulating Tumor DNA Detection in Oncology. Expert Rev. Mol. Diagn. 2019, 19, 121–135. [Google Scholar] [CrossRef] [PubMed]
Table 5.
Liquid biopsies in myeloproliferative neoplasms.
| Target | Methods | Cohort Size/Disease Stage | Evidence: Key Points | Application | Ref. |
|---|---|---|---|---|---|
| Somatic mutations | NGS ddPCR | n = 107 patients (33 PV, 56 ET, 14 PMF, and 4 uMPNs) |
| Concordance Response assessment | Garcia-Gisbert, et al., 2021 [78] |
Ref., reference; NGS, next-generation sequencing; ddPCR, digital droplet PCR; PV, polycythemia vera; ET, essential thrombocythemia; PMF, primary myelofibrosis; uMPNs, unclassifiable myeloproliferative neoplasms; cfDNA, cell-free DNA; LDH, lactate dehydrogenase; IFN, interferon; VAF, variant allele frequency.
Table 6.
Liquid biopsies in lymphoblastic acute leukemia.
| Target | Methods | Cohort Size/Disease Stage | Evidence: Key Points | Application | Ref. |
|---|---|---|---|---|---|
| IgH/TCR rearrangements | RQ-PCR | Precursor B-ALL, n = 62 T-ALL, n = 22 Diagnosis and post-treatment |
| Concordance Response assessment | van der Velden et al., 2002 [85] |
| IgH/TCR rearrangements | RQ-PCR in plasma vs. leucocytes | n = 21 (2–16 years) Diagnosis and post-treatment |
| Concordance | Schwarz et al., 2009 [86] |
| IgH/TCR rearrangements | RQ-PCR vs. flow cytometry | n = 206 Diagnosis and post-treatment |
| Response assessment by MRD Prognostication | Cheng et al., 2013 [87] |
Ref., reference; ALL, acute lymphoblastic leukemia; RQ-PCR, real-time quantitative polymerase chain reaction; MRD, minimal residual disease; PB, peripheral blood; cfDNA, cell-free DNA; rs, Spearman’s rank correlation coefficient.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Colmenares, R.; Álvarez, N.; Barrio, S.; Martínez-López, J.; Ayala, R. The Minimal Residual Disease Using Liquid Biopsies in Hematological Malignancies. Cancers 2022, 14, 1310. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14051310
AMA Style
Colmenares R, Álvarez N, Barrio S, Martínez-López J, Ayala R. The Minimal Residual Disease Using Liquid Biopsies in Hematological Malignancies. Cancers. 2022; 14(5):1310. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14051310
Chicago/Turabian StyleColmenares, Rafael, Noemí Álvarez, Santiago Barrio, Joaquín Martínez-López, and Rosa Ayala. 2022. "The Minimal Residual Disease Using Liquid Biopsies in Hematological Malignancies" Cancers 14, no. 5: 1310. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14051310
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.