
Genomic and Epigenomic Landscape of Juvenile Myelomonocytic Leukemia


1
Cancer and Leukemia Epigenetics and Biology Program, Josep Carreras Leukemia Research Institute (IJC), 08916 Badalona, Barcelona, Spain
2
Immuno Procure, Catalan Institute of Oncology (ICO), 08916 Badalona, Barcelona, Spain
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Cancers 2022, 14(5), 1335; https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14051335
Submission received: 6 January 2022
/
Revised: 25 February 2022
/
Accepted: 2 March 2022
/
Published: 4 March 2022
(This article belongs to the Special Issue Genomics of Rare Hematologic Cancers)
Abstract
:Simple Summary
Juvenile myelomonocytic leukemia (JMML) is a rare pediatric myelodysplastic/myeloproliferative neoplasm characterized by the constitutive activation of the RAS pathway. In spite of the recent progresses in the molecular characterization of JMML, this disease is still a clinical challenge due to its heterogeneity, difficult diagnosis, poor prognosis, and the lack of curative treatment options other than hematopoietic stem cell transplantation (HSCT). In this review, we will provide a detailed overview of the genetic and epigenetic alterations occurring in JMML, and discuss their clinical relevance in terms of disease prognosis and risk of relapse after HSCT. We will also present the most recent advances on novel preclinical and clinical therapeutic approaches directed against JMML molecular targets. Finally, we will outline future research perspectives to further explore the oncogenic mechanism driving JMML leukemogenesis and progression, with special attention to the application of single-cell next-generation sequencing technologies.
Abstract
Juvenile myelomonocytic leukemia (JMML) is a rare myelodysplastic/myeloproliferative neoplasm of early childhood. Most of JMML patients experience an aggressive clinical course of the disease and require hematopoietic stem cell transplantation, which is currently the only curative treatment. JMML is characterized by RAS signaling hyperactivation, which is mainly driven by mutations in one of five genes of the RAS pathway, including PTPN11, KRAS, NRAS, NF1, and CBL. These driving mutations define different disease subtypes with specific clinico-biological features. Secondary mutations affecting other genes inside and outside the RAS pathway contribute to JMML pathogenesis and are associated with a poorer prognosis. In addition to these genetic alterations, JMML commonly presents aberrant epigenetic profiles that strongly correlate with the clinical outcome of the patients. This observation led to the recent publication of an international JMML stratification consensus, which defines three JMML clinical groups based on DNA methylation status. Although the characterization of the genomic and epigenomic landscapes in JMML has significantly contributed to better understand the molecular mechanisms driving the disease, our knowledge on JMML origin, cell identity, and intratumor and interpatient heterogeneity is still scarce. The application of new single-cell sequencing technologies will be critical to address these questions in the future.
1. Introduction
Juvenile myelomonocytic leukemia (JMML) is a rare and very heterogeneous myelodysplastic/myeloproliferative neoplasm of early childhood resulting from the malignant transformation of hematopoietic stem/progenitor cells (HSPCs) and characterized by the hyperactivation of the RAS signaling pathway. Children with JMML typically show symptoms related to the infiltration of the bone marrow (BM) and other organs by malignant mature and immature myeloid cells. Formal diagnosis of JMML requires the presence of prominent monocytosis (≥1 × 109/L), a low proportion of blasts in the BM (<20%), splenomegaly, absence of BCR-ABL fusions, and mutations in genes encoding for proteins of the RAS signaling pathway [1]. Granulocyte–macrophage colony-stimulating factor (GM-CSF) in vitro hypersensitivity is a common hallmark in JMML and can be used as a diagnostic criterion in patients in which RAS pathway mutations are not identified [2,3]. Although JMML karyotype is predominantly normal, recurrent cases of monosomy 7 are observed in approximately 25% of the patients, as well as other karyotype abnormalities involving 10% of cases [4]. JMML therapeutic options are scarce, with early allogeneic hematopoietic stem cell transplantation (HSCT) being the only effective therapy for achieving long-term disease control. However, this treatment entails a significant risk of transplant-related mortality and the overall survival at five years in treated patients remains at 64%, largely due to unsuccessful HSCT [3,5,6].
Despite the major advances in the study of the underlying molecular defects in JMML, this disease is still a puzzling disorder with a wide variety of phenotypes and outcomes, ranging from rare self-limiting forms that spontaneously resolve, to aggressive cases prone to relapse and with dismal prognosis. In this context, the characterization of the genomic and epigenomic landscapes in JMML has not only contributed to identify novel oncogenic mechanisms involved in the pathogenesis of the disease, but also provided critical insights in predicting patient prognosis and making clinical decisions.
2. Genetic Alterations in JMML
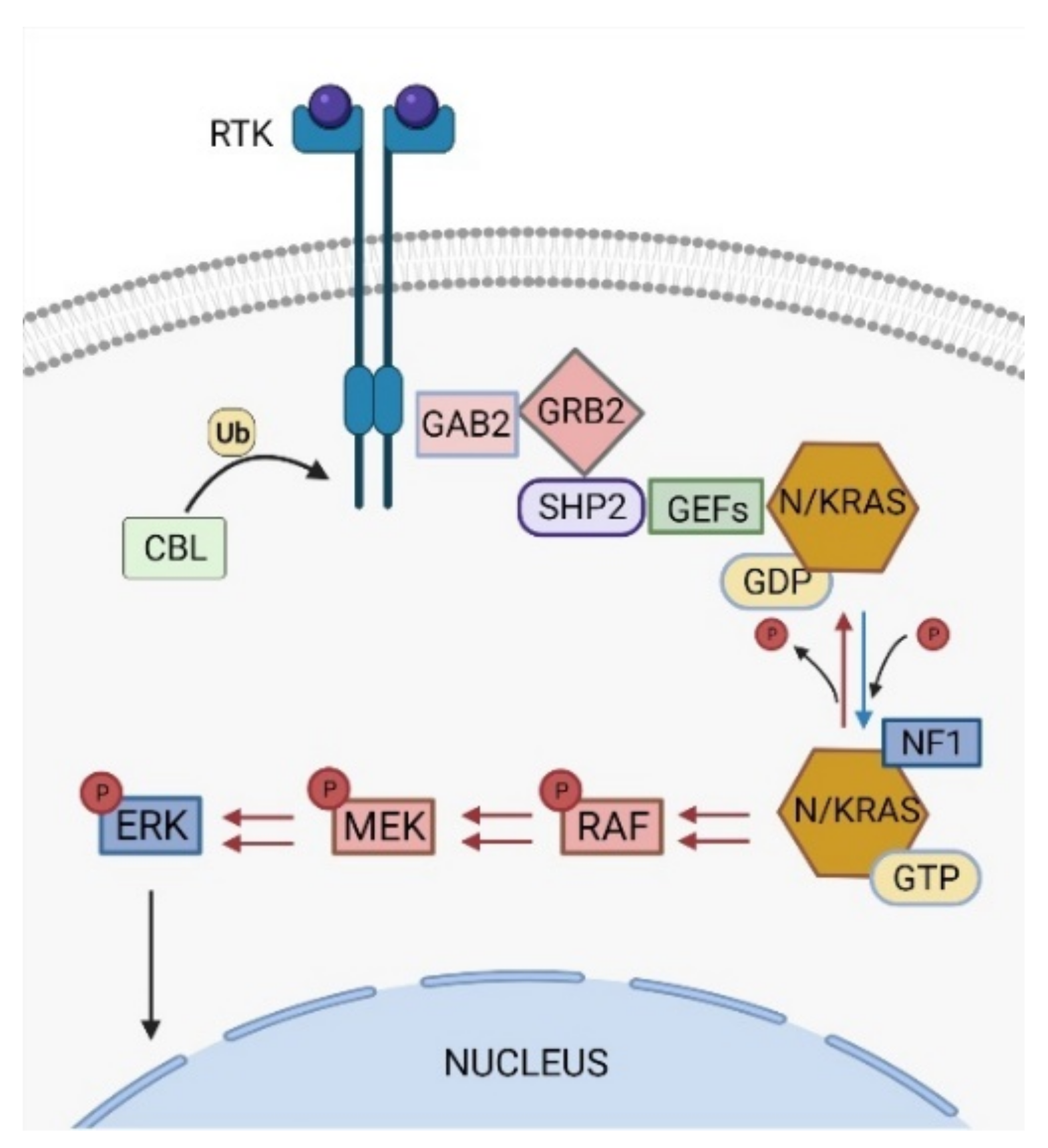
The RAS signaling pathway is one of the most studied pathways in cell biology and its deregulation is widely observed in approximately 40% of cancer patients [7]. Under normal conditions, RAS pathway activation triggers a phosphorylation signaling cascade that ultimately boosts cell proliferation, survival, differentiation, and migration, among other functions (Figure 1) [8].
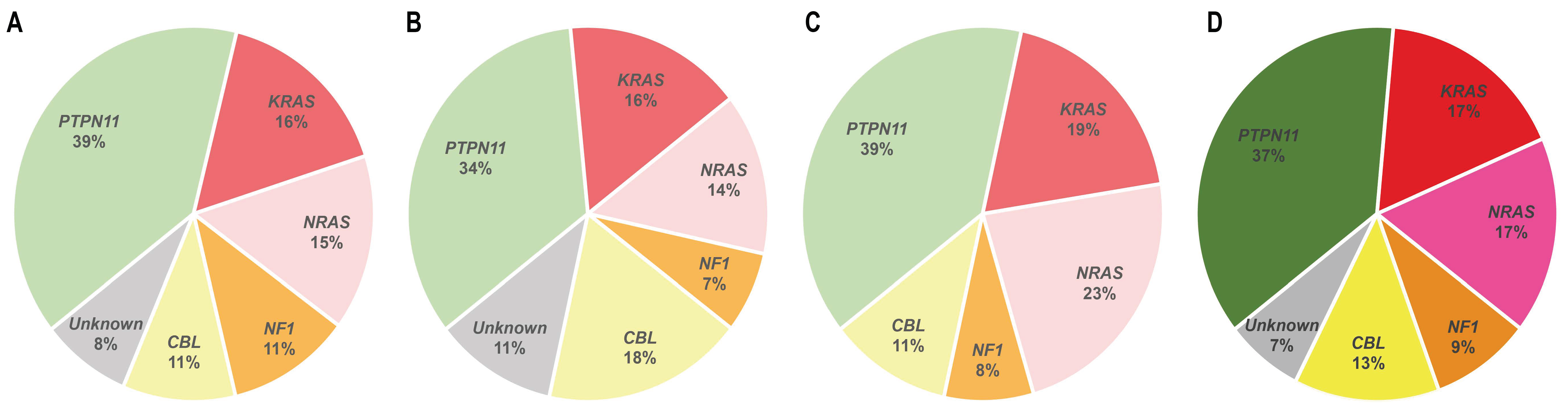
Around 90% of JMML patients carry mutations in one of five genes of the RAS pathway, including PTPN11, NRAS, KRAS, CBL, and NF1. Concomitantly, some other secondary mutations affecting either additional RAS pathway components or external elements have been described in JMML patients [9].
2.1. PTPN11
Activating PTPN11 somatic mutations are the most common genetic drivers of JMML, accounting for approximately 35–40% of the patients (Figure 2), and are associated with an aggressive clinical course and poor disease outcome [10,11,12]. The PTPN11 gene encodes for the protein tyrosine phosphatase SHP2, which acts downstream of various receptor and cytoplasmic tyrosine kinases, and promotes RAS signaling activation (Figure 1) [13].
SHP2 structure consists in two tandem Src homology 2 recognition domains (N-SH2 and C-SH2), followed by a catalytic protein tyrosine phosphatase (PTP) domain, and a C-terminal hydrophilic tail containing phosphorylation sites [14]. In the inactive state, the N-SH2 domain engages the PTP domain, keeping the phosphatase in a close autoinhibited conformation [15]. Under physiological conditions, the binding of tyrosine-phosphorylated ligands to the tandem SH2 domains stabilizes an open SHP2 conformation that renders the active site accessible and allows the dephosphorylation of target substrates [16].
JMML PTPN11 mutations occur mainly in the N-SH2 domain of SHP2, particularly in the residues G60, D61, A72, and E76, which account for more than 70% of PTPN11-mutated patients [12]. These mutations result in ligand-independent forms of the enzyme that constitutively activate downstream effectors of the RAS pathway [17].
Interestingly, germline mutations in PTPN11 are highly prevalent in Noonan Syndrome (NS), a developmental disorder characterized by unusual facial features, a restricted growth, and cardiovascular defects [18]. Approximately 5% of NS patients are affected by a mild myeloproliferative disorder, which is hematologically indistinguishable from JMML, but usually resolves spontaneously without intervention [12,19]. However, a small subset of NS patients (approximately 3%) progress into bona fide JMML and half of them die within the first month of life [19]. The distribution of NS-associated PTPN11 germline mutations differs from the one observed in PTPN11-mutated JMML patients and results in weaker SHP2 forms [12,19].
2.2. NRAS and KRAS
Approximately 25–30% of JMML patients present heterozygous somatic-activating mutations in the RAS paralogs NRAS and KRAS (Figure 2) [20]. The NRAS and KRAS proteins are small GTPases that act as binary molecular switches of the RAS signaling pathway. NRAS and KRAS are active when bound to guanosine triphosphate (GTP) and inactive when bound to guanosine diphosphate (GDP) [20]. This phosphorylation exchange is regulated by the opposing activity of guanine nucleotide-exchange factors (GEFs) and GTPase-activating proteins (GAPs) (Figure 1) and results in allosteric conformational changes at the RAS protein G domain, which is critical for RAS activation [21].
JMML mutations occur mostly in the residues G12, G13, and Q61, which are located at the G domain both in KRAS and NRAS, and render the proteins insensitive to GAP inactivation, stabilize their GTP-bound conformation, and/or affect nucleotide-exchange rate [22,23,24,25,26]. Duplication of NRAS and KRAS oncogenic alleles through acquired uniparental disomy (UPD) by mitotic recombination is observed in some JMML patients and is associated with higher aggressiveness and worst outcomes [27,28]. Interestingly, there is a strong association between KRAS mutations and monosomy 7, being the latter present in approximately 50% of KRAS-mutated JMML patients [29]. This observation suggests an interaction between the oncogenic mechanisms driven by these two genetic alterations.
Germline mutations in RAS proteins have been described in different RASopathies, including NS. However, the distribution of these mutations is different to the one observed in JMML patients harboring NRAS/KRAS somatic mutations, and the evidence pointing to a driver role in JMML leukemogenesis is scarce [30,31,32,33]. A NRAS germline mutation at G13D was found as a possible driver event in a JMML patient, and somatic mosaicism of NRAS mutations, acquired at early developmental stages, has been reported in two JMML patients, who developed a mild clinical form of the disease [34,35]. In addition, a KRAS germline mutation at T58I was identified in a NS patient who presented a JMML-like disorder [36].
2.3. CBL
Germline and somatic CBL loss-of-function mutations account for 10–15% of JMML patients [38,39]. Germline CBL mutations are associated to Noonan-like CBL syndrome, a constitutional disease that is presented as a mild form of NS with a heterogeneous set of clinical features with a variable penetrance (developmental delay, reduced growth, facial dysmorphism, among others) and an increased risk to develop JMML [38,40,41]. Disease progression to JMML normally occurs after loss of heterozygosity of the CBL wild type allele, typically through UPD encompassing the CBL locus [42,43,44]. CBL-mutant JMML patients usually develop indolent forms of the disease that resolve spontaneously, and only a subset of them require HSCT treatment [38,45].
The CBL gene encodes for the RING E3 ubiquitin ligase CBL that acts as negative regulator of activated protein tyrosine kinases by promoting their targeting for degradation by the proteasome (Figure 1) [46]. The CBL protein comprises a highly conserved N-terminal tyrosine kinase-binding (TKB) domain, followed by a central region containing a helical linker and a RING finger domain, which are critical for CBL ubiquitin ligase activity, and a C-terminal proline-rich sequence, that mediates interactions of CBL with SH3 domain-containing proteins [46,47].
Most JMML somatic mutations in CBL occur in the linker region, especially in Y371, or in different residues at the RING finger domain, and lead to the loss of E3 ubiquitin ligase activity against tyrosine kinase substrates [9,38,42]. CBL linker mutants have been shown to enhance LYN- and JAK2-mediated GM-CSF signaling by activating the PI3K/AKT/mTOR and JAK/STAT pathways, respectively [48,49]. Moreover, CBL can also regulate the RAS pathway through an indirect mechanism involving the adaptor protein GRB2 [50]. CBL interaction with GRB2 prevents the binding of this protein to SOS, a GEF that promotes the formation of active RAS-GTP complexes (Figure 1) [51]. CBL loss-of-function mutations impair the binding of CBL to GRB2, indirectly promoting RAS pathway activation by allowing GRB2-SOS interaction and SOS-mediated GDP/GTP nucleotide exchange in RAS proteins [52,53]. Interestingly, GRB2 is a core component of a multiprotein complex that includes SHP2, among other factors (Figure 1) [13,54]. The interaction of this complex with SOS allows SHP2 to dephosphorylate the RAS residue Y32, which increases the binding of RAS to RAF, thus contributing to RAS activation [55]. This adaptor activity of SHP2 and the inhibitory role of CBL in the regulation of GRB2-SOS complex function illustrate how CBL loss-of-function mutations and PTPN11 gain-of-function mutations (through the adaptor activity of SHP2) functionally converge through a similar molecular mechanism to induce RAS activation, which could explain why these two genetic alterations are mutually exclusive in JMML [42].
2.4. NF1
Germline and somatic loss-of-function mutations at the NF1 tumor suppressor gene are found in 10% to 15% of JMML patients (Figure 2) [4]. Germline NF1 mutations are associated to neurofibromatosis type 1, a common autosomal congenital disorder characterized by the presence of café-au-lait macules, skinfold freckling, development of tumors of the nervous system, and overlapping features with other RASopathies, such as NS and Legius syndrome [56]. Although it is not a common complication, NF1-mutated neurofibromatosis type 1 patients have an increased predisposition to develop JMML, with a 200- to 350-fold increased risk compared to their wild type NF1 counterparts [4]. In NF1-mutant patients, JMML progression is triggered by loss of heterozygosity of the wild type NF1 allele, typically by UPD or compound-heterozygous mutations [57].
NF1 encodes for neurofibromin, a GAP that functions as negative regulator of the RAS signaling pathway. Neurofibromin binds to RAS family proteins and stimulates the hydrolysis of active RAS-GTP to RAS-GDP inactive forms (Figure 1) [58,59,60].
Most of JMML reported alterations in NF1 are nonsense or frameshift mutations resulting in a truncated protein due to a premature termination codon [61,62]. NF1 loss-of-function mutations result in a reduced dephosphorylation of RAS-GTP activated proteins and confer sustained activation of the RAS signaling pathway [63].
2.5. Other Driver Genetic Alterations in the RAS Pathway
Although PTPN11, NRAS, KRAS, CBL, and NF1 mutations account for approximately 90% of JMML cases, around 10% of the patients that are clinically diagnosed with JMML do not present mutations in any of these five RAS pathway genes [37]. However, several studies have reported other genetic alterations that can possibly act as molecular drivers of the disease in cases of unknown origin.
Gain-of-function somatic mutations affecting known oncogenic hotspots of the RAS genes were identified in RRAS (Q87L) and RRAS2 (Q72L) in two independent patients that lacked any of the canonical JMML mutations at diagnosis, supporting a driver role of these alterations [64]. In addition, the analysis of a cohort of 16 patients presenting a JMML-like phenotype without mutations in any of the five canonical JMML genes revealed three patients that harbored gain-of-function ALK and ROS1 tyrosine kinase fusions, including RANBP2-ALK, DCTN1-ALK, and TBL1XR1-ROS1 [37]. Fusions involving the tyrosine kinase genes PDGFRB (SPECC1-PDGFRB and NDEL1-PDGFRB) and FLT3 (CCDC88C-FLT3) were also identified in case reports of JMML-diagnosed patients lacking mutations in the classical JMML drivers [65,66,67]. Similar rearrangements involving these tyrosine kinases have been also described in other hematologic malignancies [68,69,70,71,72], supporting the role for these alterations as an alternative oncogenic mechanism of RAS pathway hyperactivation in leukemia transformation. However, although these patients harboring tyrosine kinase fusions recapitulate the clinical features of JMML, it is still a matter of controversy whether they should be diagnosed as JMML or instead represent an as yet undefined category of myelodysplastic/myeloproliferative neoplasms. Further research must be carried out to shed light on this debate.
2.6. Secondary Genetic Alterations in JMML
JMML is characterized by a low mutational rate, suggesting that a limited number of genetic alterations is required to support JMML leukemogenesis [29]. However, secondary mutational events that contribute to the pathogenesis of the disease have been recurrently identified in different cohorts of JMML patients.
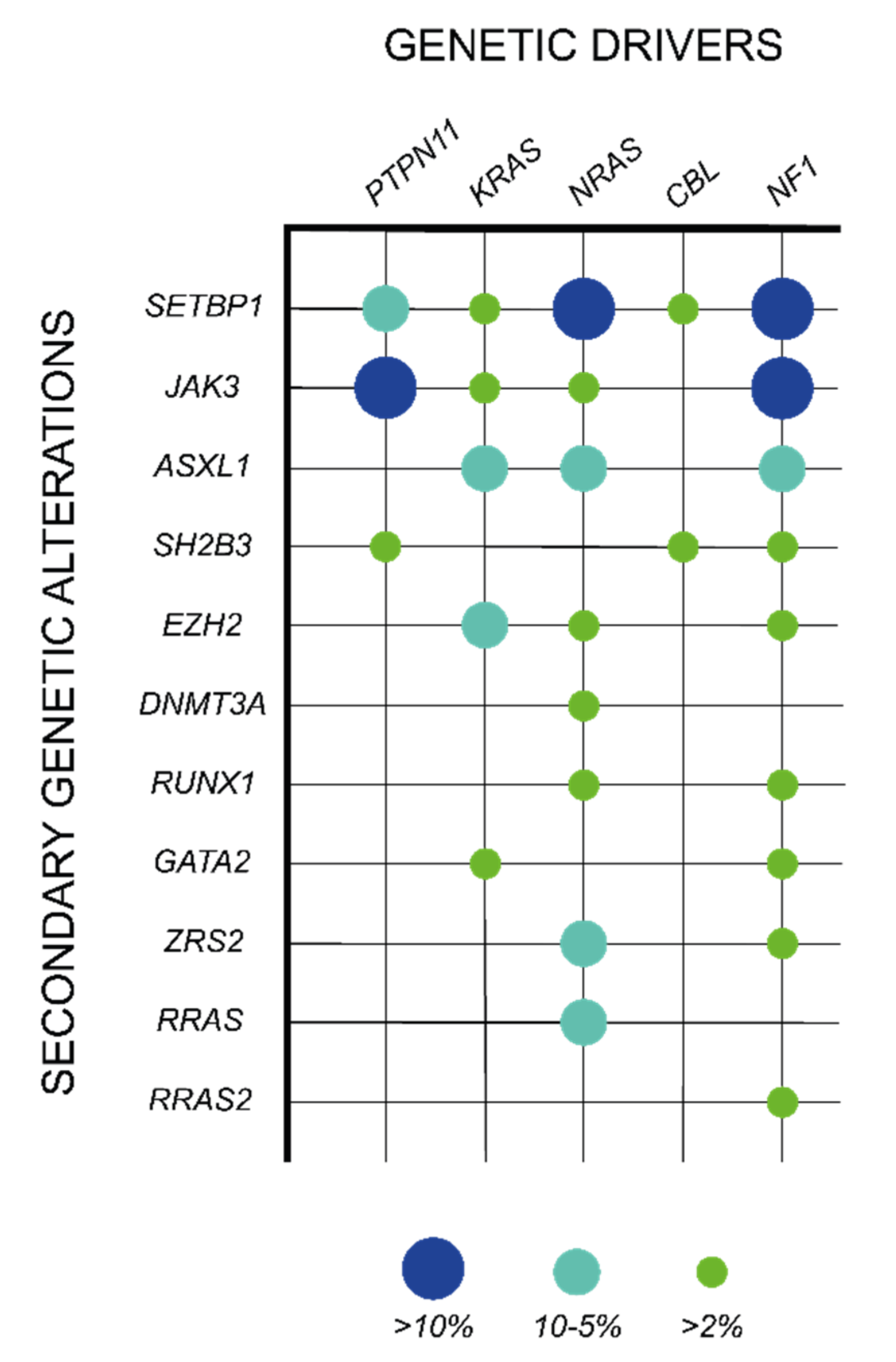
Although the mutations in the five canonical JMML genes are in general mutually exclusive, around 10% of JMML patients harbor co-existing alterations in these genes, being the association of PTPN11 and NF1 mutations the most common co-mutational event [37]. In addition, approximately 50% of the cases present secondary somatic mutations in other genes, which are specifically associated with particular RAS pathway initiating lesions and expand clonally, indicating a cooperative role with the driver event in JMML maintenance (Table 1; Figure 3) [29]. Within the RAS pathway, heterozygous mutations in signaling components (RRAS, RAC2, and SOS1) or RAS regulators (PLXNB2, ABI1, and PDE8A) have been described in JMML in combination with some of the classical driver events and contribute to JMML pathogenesis by enhancing RAS pathway activation (Table 1) [29,37,64]. In addition, other genetic events outside the RAS pathway have been reported as major secondary mutations in some JMML subsets. Among them, SETBP1 activating mutations are the most prevalent genetic events, being present in around 30% of JMML patients and correlating with poorer disease outcomes [73,74]. SETBP1 directly binds to SET, which functions as an inhibitor of the protein phosphatase PP2A, a well-known tumor suppressor in hematopoietic malignancies [75,76]. This interaction protects SET from degradation, potentiating its inhibitory activity over PP2A [77]. SETBP1 mutations disrupt the degron motif of the protein, resulting in an impaired proteasome cleavage and subsequent SETBP1 protein accumulation, which further enhances the inhibitory effects of SET over PP2A and support leukemia cell proliferation [77,78]. In addition to this function, SETBP1 has been shown to directly bind AT-rich promoter regions and contribute to the transcriptional activation a set of target genes that include the hematopoietic master regulators HOXA9 and HOXA10 [79,80]. This effect correlates with an increase in myeloid progenitor self-renewal capacity in Setbp1-overexpressing mouse bone marrow, supporting the existence of additional PP2A-independent oncogenic mechanisms driven by SETBP1 aberrant expression [79]. In JMML, SETBP1 mutations associate with PTPN11 or NRAS somatic mutations [74]. Interestingly, a recently published mouse model combining SETBP1 and NRAS mutations has shed some light on the mechanism of interaction between these two factors by showing that the aberrant expression of SETBP1 enhances both NRAS gene expression signature and NRAS-driven MAPK protein phosphorylation [81].
Along with the mutations in SETBP1, other secondary genetic alterations are also observed as clonal events in JMML patients, including mutations in hematopoietic commitment transcription factors (RUNX1, GATA2, RARA, and HOXA11), spliceosome components (ZRSR2), cAMP pathway components (PDE8A), structural protein components and regulators (WASP, DYNC1H1, TNS3, COL22A1, KRT1, and SMC1A), or JAK/STAT pathway components (JAK3 and SH2B3), among others (Table 1) [29,37,64,73,74,81,84,86,87,88,89]. These secondary mutations are associated with an aggressive clinical course of the disease and an increased risk of relapse after HSCT [64].
Of special relevance are the genetic alterations that affect components of the epigenetic machinery, which are observed in approximately 15% of JMML patients [29,64]. These genetic alterations include mostly mutations at the genes encoding for the polycomb repressive complex 2 (PRC2) core component EZH2 and the PRC2-associated factor ASXL1. Of note, the EZH2 gene is located at chromosome 7 and all JMML-associated EZH2 mutations are hemizygous due to co-occurring monosomy 7 [29,64]. In addition to these mutations, copy number variations (CVN) in genes encoding for other PRC2 complex components and associated factors, such as SUZ12, AEBP2, CDYL, or JARID, have been found in some subsets of JMML patients [29]. Mutations at the DNMT3A gene, encoding for the DNA methyltransferase 3 alpha, have been also described; however, the recurrence of this genetic alterations in JMML is lower than in other hematologic malignancies [64,90].
The PRC2 complex directs the transcriptional repression of target genes by catalyzing histone H3 trimethylation at lysine 27 (H3K27me3) [91]. Interestingly, a recent study demonstrated that JMML patients that present an impaired PRC2 activity show a global decrease in H3K27me3 and a concomitant increase in H3K27 acetylation, suggesting a critical role of PRC2-associated mutations in regulating the JMML transcriptional program at the epigenetic level [29]. These findings illustrate the crosstalk between genetics and epigenetics in JMML and highlight the importance of a better understanding of the oncogenic mechanisms driven by epigenetic dysregulation during JMML leukemogenesis.
3. Epigenetic Alterations in JMML
Although genetic mutational events are generally considered the main drivers of cancer transformation, alterations in the epigenetic landscape of tumor cells have a critical role in cancer pathogenesis, providing additional mechanisms to consolidate specific oncogenic transcriptional programs [92]. Over the last decade, several research groups have explored the epigenetic landscape in JMML and identified important alterations in the methylome of JMML cells that correlate with the severity and prognosis of the disease. This fact highlights the urgent need of a better understanding of the oncogenic mechanisms driven by JMML epigenetic aberrations and postulates the use of epigenetic modifiers as a potential therapeutic strategy for the treatment of this disease.
3.1. Early Studies on DNA Methylation in JMML
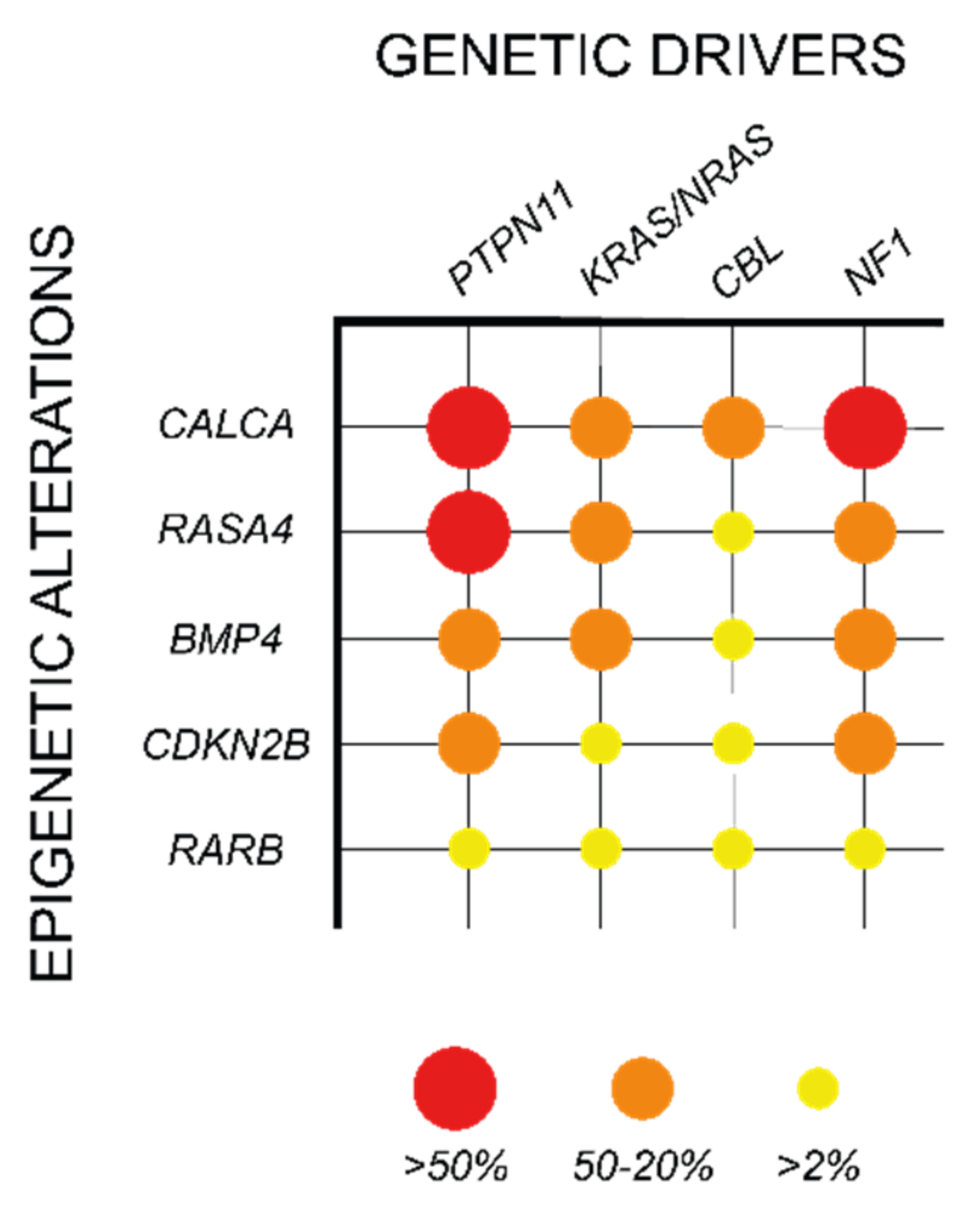
CpG island methylation at gene promoters is an important repressive mechanism of gene expression, which has been shown to play a relevant oncogenic role in different cancers, including myeloid malignancies [93]. In JMML, methylation in CpG islands was first analyzed in a European cohort of 86 patients, in which 14 candidate genes were selected based on their hypermethylation status in other cancer types (CALCA, CDKN1C, CDKN2B, DAPK1, MGMT, MLH1, RARB, RASSF1, SOCS1, and TP73) or their involvement in RAS signaling (BMP4, PAWR, RASA1, and RECK). Among the selected candidates, four genes were found recurrently hypermethylated, including BMP4, CALCA, CDKN2B, and RARB (Table 2; Figure 4). Interestingly, this hypermethylation phenotype correlated with poorer prognosis and a high risk of treatment failure due to relapse after HSCT [94].
These results were further validated in a Japanese cohort of 92 JMML patients, in which the CpG methylation status of 16 genes was analyzed, including nine genes that were common to the European study (CALCA, CDKN2B, DAPK1, MGMT, MLH1, RARB, RASSF1, TP73, and BMP4) and seven new candidate genes (APC, CDH13, CDKN1A, CHFR, ESR1, H19, and IGF2AS). This study not only confirmed the hypermethylation of BMP4, CALCA, CDKN2B, and RARB, but also provided a novel prognostic tool based on the “aberrant methylation score” (AMS). AMS stratifies the patients in three groups based on the number of hypermethylated genes (0, 1–2, or 3–4) and predicts their 5-year overall survival and transplant-free survival, with high AMS patients presenting a dismal prognosis [95].
In addition to the genes identified in these studies, hypermethylation in other genes such as RASA4, CREBBP, and AKAP12 was also observed in JMML patients in correlation with a poor survival and high risk of relapse after HSCT, further supporting the significance of DNA methylation in aggressive JMML phenotypes (Table 2; Figure 4) [96,97,98].
3.2. The JMML Methylation Landscape
Early methylation studies in JMML focused on the analysis of specific gene subsets. However, a global view on the DNA methylation landscape in JMML was not obtained until the first genome-wide CpG methylation analyses were performed [25,37,99]. Based on these studies, patients were clustered according to their global methylation status in three groups: low methylation (LM), intermediate methylation (IM), and high methylation (HM). These groups were not only associated with particular outcomes [99], but also to specific molecular profiles [25,37]. Thus, LM patients showed enrichment of somatic NRAS and CBL mutations and presented high survival rates, the IM group was associated to somatic KRAS mutations and monosomy 7, and the HM patients showed an enrichment in PTPN11 mutations and were characterized by a poor clinical outcome [25]. Interestingly, the analysis of HM samples showed an upregulation in the expression of the genes encoding for the DNA methyltransferases DNMT1 and DNMT3B, suggesting an association between activation of DNA methylation mechanisms and specific JMML mutational profiles [25]. Overall, these data further supported the diagnostic value of DNA methylation in JMML patients.
3.3. DNA Methylation as a Prognostic Tool in JMML
The accumulating evidences on the role of DNA methylation in the pathogenesis of JMML has led to the recent publication of an international JMML stratification consensus, which has defined the parameters and characteristics of the different DNA methylation subgroups in JMML [100]. In here, the Illumina Infinium 450 k/EPIC array technology was applied to develop and validate a machine learning classifier for prospective patient classification.
To complement this novel prognostic tool, a new technique called Digital Restriction Enzyme Analysis of Methylation (DREAM) was developed to provide an easy and robust method to evaluate DNA methylation in JMML clinical samples [101]. In this method, DNA is sequentially digested with a methylation-sensitive restriction enzyme (SmaI), followed by a restriction enzyme that is tolerant to DNA methylation (XmaI). This digestion results in the generation of two DNA fragment types, carrying either a CCGGG tag in their 5′ end in methylated DNA sites, or a GGG tag in unmethylated DNA sites. These fragments are then analyzed by Next-Generation Sequencing (NGS), allowing a quantitative whole-genome evaluation of DNA methylation [102]. DREAM represents a promising and cost-efficient technology for the evaluation of DNA methylation in clinical settings; however, further research will be required to address its implementation and determine the robustness of the approach in different laboratories.
The standardized use of DNA methylation as a biomarker in JMML and the incorporation of techniques such as Illumina Infinium 450 k/EPIC arrays or new techniques such as DREAM as prognostic methods represent a unique breakthrough for the stratification and management of JMML patients. These tools will not only improve clinical decision-making, but also contribute to optimize the inclusion criteria of specific JMML subsets in future clinical trials.
4. Genetic and Epigenetic Therapeutic Targets for the Treatment of JMML
JMML has historically represented a clinical challenge mainly due to the limited number of therapeutic options for its treatment and the inevitable fatal outcome in children with the most aggressive forms of the disease [103,104]. HSCT is currently the only effective therapy for achieving long-term disease control in JMML [103]. However, the advances in the characterization of the molecular mechanisms driving and supporting the progression of JMML have provided new potential genetic and epigenetic therapeutic targets that are currently being explored in different preclinical assays and clinical trials as alternative treatments for JMML (Table 3). These studies hold promise for an improvement in the pre- and post-HSCT management of the disease, and might have a direct impact on the prognosis and survival of JMML patients in the future.
4.1. JMML Therapeutic Targets on Signaling Pathways
4.1.1. RAS Pathway Targeting
Although RAS hyperactivation is a hallmark in JMML, the therapeutic targeting of factors involved in RAS signaling has been shown to provide limited benefits due to the frequent treatment-associated toxicities, and the functional redundancy and complexity of the pathway [105,106,107]. However, some studies have explored the use of different tyrosine kinase (TK) and mitogen-activated protein kinase (MEK) inhibitors to target RAS signaling, as a therapeutic alternative for JMML treatment.
A small subset of clinically-diagnosed JMML patients harbor fusions involving diverse RAS components, which result in the abnormal activation of specific TKs, such as ALK, ROS1, or FLT3 [37,67]. Some examples illustrate how drug repurposing of TK inhibitors (TKIs) that are approved for the treatment of other pathologies could have a potential therapeutic benefit on this atypical subset of JMML patients. Such is the case of crizotinib, a potent inhibitor of ALK and ROS1 that is approved for the treatment of non-small cell lung cancer [108]. Current evidences on the benefits of crizotinib treatment on JMML are limited to the case of a patient carrying RANBP2-ALK fusion that was refractory to conventional cytotoxic chemotherapy. The addition of crizotinib to the treatment resulted in complete molecular remission and allowed successful HSCT in this patient [37]. In the same study, another two patients who harbored ALK or ROS1 fusions were not treated with crizotinib and succumbed due to tumor progression [37].
Similar to this case, the use of another TKI, sorafenib, has been also explored in the context of JMML [67]. Sorafenib, is currently approved for the treatment of different types of cancer, including acute myeloid leukemia patients that carry an activating internal tandem duplication mutation on FLT3 (FLT3-ITD) [109]. A JMML patient harboring a CCDC88C-FLT3 fusion who was refractory to conventional chemotherapy, was treated with sorafenib, resulting in FLT3 inhibition and cytogenetic remission, which also in this case allowed successful HSCT [67].
In the case of PTPN11-mutant JMML, a recent study has identified the non-receptor tyrosine kinase ACK1 (encoded by the TNK2 gene) as a potential therapeutic target for these patients [110]. In vitro assays showed that the ACK1-specific inhibitors AIM-100 and XMD8-87 can reduce the transforming potential of JMML-associated PTPN11 mutations [110]. Clinically, the use of dasatinib, a TKI that is approved for the treatment of different types of leukemia and can target ACK1 (among other TKs), was shown to reduce disease burden and provide extended survival in a PTPN11-mutant JMML patient [110].
Together, these data postulate the use of TKIs as a potential therapeutic approach to achieve disease remission and facilitate HSCT in some JMML patient subsets; however, further research is required to validate these observations.
In addition to TKIs, the use of MEK inhibitors (MEKi) in JMML has also been explored. Especially remarkable is the case of trametinib, a MAP2K1/MAP2K2 inhibitor that is currently approved for the treatment of metastatic melanoma [111]. The first evidences on the potential therapeutic effect of trametinib in JMML were obtained in in vitro assays using induced pluripotent stem cells (iPSCs) derived from PTPN11- and CBL-mutant JMML cells [112]. In this experimental setting, trametinib treatment resulted in efficient RAS signaling and cell growth inhibition in PTPN11-mutant iPSCs [112]. More recently, trametinib has also been shown to provide a survival benefit in a mouse model in which leukemogenesis is driven by the combined expression of JMML-associated NRAS and SETBP1 mutations [81]. The application of trametinib for the treatment of JMML is currently being explored in a phase II clinical trial, which aims to determine the safety and efficacy of this drug in refractory or relapsed JMML patients (ClinicalTrials.gov Identifier: NCT03190915).
4.1.2. Targeting of Other Signaling Pathways
The PI3K/AKT/mTOR pathway is one of the main downstream effectors of the RAS pathway and it is also affected by RAS hyperactivation [106]. In JMML, some studies have provided evidence of the beneficial effects of PI3K/AKT/mTOR inhibition. The use of rapamycin, a specific and potent mTOR inhibitor, has been shown to reduce signaling and proliferation of JMML-derived PTPN11-mutant iPSCs in vitro [112]. Similar results were obtained in the same experimental setting upon treatment with idelalisib, a PI3Kδ inhibitor [112]. Moreover, the use of idelalisib on primary JMML cells resulted in a dose-dependent reduction in GM-CSF hypersensitivity in two of the three samples analyzed [113]. Finally, both rapamycin and idelalisib have also shown a therapeutic effect in in vivo leukemia mouse models driven by the expression of JMML-associated PTPN11 mutations [114,115].
Secondary mutations affecting components of the JAK/STAT pathway, such as JAK3 or SH2B3, are recurrently found in JMML patients, indicating a relevant role of this signaling pathway in the pathogenesis of JMML [29,64,73,74]. Therefore, targeting the JAK/STAT pathway could potentially report beneficial effects in some JMML patient subsets. In this context, a recent in vitro study revealed that JMML-derived CBL-mutant iPSCs are sensitive to the JAK inhibitors momelotinib and ruxolitinib, reducing both cell proliferation and aberrant signaling [112].
Although clinical data on the use of these inhibitors in JMML are not available, the promising results obtained in in vitro and in vivo preclinical models support a potential therapeutic benefit of these treatments in JMML patients. However, further research will be required to explore this possibility in a clinical setting.
4.2. Epigenetic Therapeutic Targets in JMML
DNA hypermethylation has been showed to be a hallmark in the most aggressive cases of JMML [25,37,99]. For that reason, the use of the DNA methyltransferase inhibitor azacitidine has been explored as a potential therapeutic agent for the treatment of this disease, alone or in combination with conventional cytotoxic chemotherapy [116]. Azacitidine use in JMML was first reported on a patient that achieved complete clinical and genetic remission of the disease after eight cycles of treatment prior to HSCT [117]. Later on, a retrospective study showed that low-dose azacitidine was effective and tolerable in JMML, and documented another three JMML cases in which this drug induced complete remission before HSCT [118]. Finally, the beneficial effects of pre-HSCT azacitidine treatment were formally validated in a phase II clinical trial (NCT02447666) that demonstrated that the use of this drug as a single agent is a suitable option for newly diagnosed JMML patients, independent of their methylation status [116]. In addition, the histone deacetylase inhibitor vorinostat, is currently being explored in a phase I clinical trial (NCT03843528) in combination with low dose azacitidine for the treatment of pediatric myeloid malignancies, including JMML. Further clinical studies will be required to determine long-term safety and efficacy of these treatments in JMML.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 3.
Experimental therapeutic strategies for JMML treatment.
| Pathway | Target | Inhibitor | Status | References | |
|---|---|---|---|---|---|
| Signaling pathway inhibitors | RAS | ACK1 | Dasatinib | In vitro | [110] |
| ALK/ROS1/MET | Crizotinib | In vitro | [37] | ||
| MEK | Trametinib | Phase II clinical trial | [81,112,119,120] | ||
| FLT3 | Sorafenib | Clinical use | [67] | ||
| PI3K | mTOR | Rapamycin | Preclinical in vivo | [112,114,115] | |
| PI3Kδ | Idelalisib | Preclinical in vivo | [112,113,114,115] | ||
| JAK/STAT | JAK1/JAK2 | Momelotinib | In vitro | [112] | |
| JAK1/JAK2 | Ruxolitinib | In vitro | [112] | ||
| Epigenetic inhibitors | Methylation | DNMTs | Azacitidine | Clinical use | [37,116,117,118,121,122] |
| Acetylation | HDACs | Vorinostat | Phase I clinical trial |
5. Current Challenges and Future Perspectives in JMML Research
Despite of the advances in the genetic and epigenetic characterization of JMML, there are still several experimental challenges and clinically relevant open questions that remain to be addressed.
5.1. JMML Experimental Models
JMML research has been traditionally hindered due to its low incidence (1.2 cases per million children under 14 years of age), and the impossibility of maintaining primary JMML cells in vitro for extended periods of time or establishing immortalized JMML cells lines [123,124]. In this context, the development of methods to generate JMML patient-derived xenografts (PDX) and iPSCs has been instrumental to overcome low sample number difficulties and provide an unlimited source of JMML cells for experimental purposes.
JMML PDXs have been established in immunodeficient NSG (NOD/SCID/IL2rγ−/−) and NSG-S (NOD/SCID/IL2rγ−/−/IL-3/GM/SF) mice, and in both cases these models present not only the immunophenotypical features of the primary sample, but also maintain the clonal diversity of the original tumor [84,125]. On the other hand, different reprogramming techniques have been successfully applied towards the generation of JMML-derived iPSCs, providing a unique experimental tool to perform high-throughput analysis, such as drug or CRISPR screens [112,126,127,128]. However, the lack of a physiological bone marrow microenvironment in iPSC models gives rise to important concerns regarding whether these systems can faithfully recapitulate the molecular and functional features of the tumor. Future JMML modeling efforts should focus on the establishment of biomimetic 3D culture systems that replicate the molecular and cellular complexity of the bone marrow microenvironment [129,130,131]. The development of these approaches might help to overcome the limitations of primary JMML cultures and provide new physiologically relevant in vitro models to study the oncogenic mechanism driving JMML pathogenesis and resistance to therapy.
5.2. State-of-the-Art Methods to Explore JMML Tumor Origin, Heterogeneity and Evolution
Although the phenotype of JMML patients is dominated by the expansion of myeloid cells, there is evidence that also indicates the involvement of other hematopoietic lineages [4,20,132]. For this reason, JMML is considered a disease of the HSPC compartment [1,84,133]. However, very little is known about the cell hierarchies involved in leukemia progression, the intratumor heterogeneity, the specific identity of the leukemia initiating cells, or the clonal evolution of the disease. However, all these features might have an important impact in the prognosis of the disease and risk of relapse after HSCT.
Over the last years, the rapid development of new technologies based on the use of next generation sequencing has changed the paradigms of cancer research. More specifically, the analysis of genomic, epigenomic, and/or transcriptomic information at single-cell resolution, and the comprehensive integration of these data can provide critical information on tumor origin, progression, and cellular heterogeneity.
In JMML, these approaches have only been applied to the transcriptomic analysis of HSPCs from two JMML patient samples by single-cell RNA sequencing (scRNA-seq) [134]. In this study, different JMML-specific HSPC clusters were identified, each of them showing upregulation of particular sets of genes, including myeloid genes, stem cell, and fetal genes, or genes associated to proliferation, leukemia, and erythroid differentiation [134]. These results demonstrated a broad heterogeneity within the JMML HSPC compartment, with the expression of aberrant transcriptional signatures that are not found in control cord blood HSPCs [134]. However, the collection of only transcriptional data and the low number of samples analyzed, limited the conclusions that could be extracted regarding the role of each of these clusters in tumor progression or maintenance.
Although not yet applied to JMML research, single-cell DNA-sequencing (scDNA-seq) combined with scRNA-seq has been successfully applied to the analysis of different types of cancers [135,136]. In these analyses, scRNA-seq data are used to identify specific cell clusters based on their transcriptomic profile. Then, this information is integrated with the scDNA-seq results to correlate different cell subpopulations with a specific mutational status and assess tumor clonal evolution during the differentiation process. In addition, transcriptomic data can be used to infer cell lineage differentiation between the annotated cell subpopulations by trajectory-based differential expression analysis using pseudotime analytical tools [137,138,139]. These bioinformatic approaches could be applied to the study of JMML tumoral heterogeneity and clonal evolution by tracking “first hit” mutations to a cell of origin, and studying their differentiation trajectories and acquisition of mutations.
Similarly, JMML epigenomic intratumoral heterogeneity could be studied using methods that allow the analysis of DNA methylation at single-cell level. Simultaneous profiling of the transcriptome and DNA methylome from individual cells (scMT-seq) provides both a functional annotation of different cell subsets and a methylation profile [140]. A similar approach integrating scRNA-seq with methylome microarray data has been recently used to track the cell of origin in chronic lymphocytic leukemia [141]. The application of these technologies to the study of JMML could be critical to decipher the molecular mechanisms driving JMML leukemogenesis and progression.
6. Conclusions
The characterization of the genetic and epigenetic landscapes of JMML has significantly improved our understanding of the oncogenic pathways controlling the pathogenesis of this disease. Moreover, these analyses have turned the spotlight on DNA hypermethylation and secondary mutations as critical alterations that cooperate with canonical RAS pathway mutations and have an important prognostic value in the clinic. However, key questions regarding JMML origin, tumor cell identity, and intratumor and interpatient heterogeneity, remain open and must be addressed in the future. The use of new technologies allowing single-cell molecular profiling will be instrumental to achieve this aim and provide new relevant information on JMML pathobiology. This will in turn improve JMML diagnostic and prognostic criteria and contribute to identify potential therapeutic targets for the treatment of this disease.
Author Contributions
Conceptualization: L.B. Literature review: C.F. and N.G.-M. Manuscript preparation: C.F., N.G.-M. and L.B. Figure preparation: C.F., N.G.-M. and S.A.-M. Supervision: L.B. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by Spanish Ministry of Science and Innovation (PID2020-117645RB-I00/AEI/10.13039/501100011033 and RYC2020-029400-I/AEI/10.13039/501100011033), the FERO Foundation (BFero2020.03), and the Government of Andorra (ATC025-AND-2020).
Acknowledgments
We thank the CERCA Program (Generalitat de Catalunya), the Josep Carreras Leukemia Research Institute and the Catalan Institute of Oncology for their institutional support. LB is supported by the Spanish Ministry of Science and Innovation (MCI) (PID2020-117645RB-I00/AEI/10.13039/501100011033 and RYC2020-029400-I/AEI/10.13039/501100011033), and the FERO Foundation (BFero2020.03). CF is supported by a PhD grant from the Government of Andorra (ATC025-AND-2020).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Emanuel, P.; Bates, L.; Castleberry, R.; Gualtieri, R.; Zuckerman, K. Selective Hypersensitivity to Granulocyte-Macrophage Colony-Stimulating Factor by Juvenile Chronic Myeloid Leukemia Hematopoietic Progenitors. Blood 1991, 77, 925–929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Locatelli, F.; Niemeyer, C.M. How I treat juvenile myelomonocytic leukemia. Blood 2015, 125, 1083–1090. [Google Scholar] [CrossRef] [PubMed]
- Niemeyer, C.M.; Aricó, M.; Basso, G.; Biondi, A.; Cantú Rajnoldi, A.; Creutzig, U.; Haas, O.; Harbott, J.; Hasle, H.; Kerndrup, G.; et al. Chronic Myelomonocytic Leukemia in Childhood: A Retrospective Analysis of 110 Cases. Blood 1997, 89, 3534–3543. [Google Scholar] [CrossRef]
- Yabe, M.; Ohtsuka, Y.; Watanabe, K.; Inagaki, J.; Yoshida, N.; Sakashita, K.; Kakuda, H.; Yabe, H.; Kurosawa, H.; Kudo, K.; et al. Transplantation for juvenile myelomonocytic leukemia: A retrospective study of 30 children treated with a regimen of busulfan, fludarabine, and melphalan. Int. J. Hematol. 2014, 101, 184–190. [Google Scholar] [CrossRef]
- Dvorak, C.C.; Satwani, P.; Stieglitz, E.; Cairo, M.S.; Dang, H.; Pei, Q.; Gao, Y.; Wall, D.; Mazor, T.; Olshen, A.B.; et al. Disease Burden and Conditioning Regimens in ASCT1221, A Randomized Phase II Trial in Children with Juvenile Myelomonocytic Leukemia: A Children’s Oncology Group Study. Pediatr. Blood Cancer 2018, 65, e27034. [Google Scholar] [CrossRef]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337. [Google Scholar] [CrossRef] [Green Version]
- Chung, E.; Kondo, M. Role of Ras/Raf/MEK/ERK signaling in physiological hematopoiesis and leukemia development. Immunol. Res. 2011, 49, 248–268. [Google Scholar] [CrossRef]
- Niemeyer, C.M. RAS diseases in children. Haematologica 2014, 99, 1653–1662. [Google Scholar] [CrossRef]
- Tartaglia, M.; Niemeyer, C.M.; Fragale, A.; Song, X.; Buechner, J.; Jung, A.; Hählen, K.; Hasle, H.; Licht, J.D.; Gelb, B.D.; et al. Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nat. Genet. 2003, 34, 148–150. [Google Scholar] [CrossRef]
- Loh, M.L.; Vattikuti, S.; Schubbert, S.; Reynolds, M.G.; Carlson, E.; Lleuw, K.H.; Cheng, J.W.; Lee, C.M.; Stokoe, D.; Bonifas, J.M.; et al. Mutations in PTPN11 implicate the SHP-2 phosphatase in leukemogenesis. Blood 2004, 103, 2325–2331. [Google Scholar] [CrossRef]
- Kratz, C.P.; Niemeyer, C.M.; Castleberry, R.P.; Cetin, M.; Bergsträsser, E.; Emanuel, P.D.; Hasle, H.; Kardos, G.; Klein, C.; Kojima, S.; et al. The mutational spectrum of PTPN11 in juvenile myelomonocytic leukemia and Noonan syndrome/myeloproliferative disease. Blood 2005, 106, 2183–2185. [Google Scholar] [CrossRef] [Green Version]
- Dance, M.; Montagner, A.; Salles, J.P.; Yart, A.; Raynal, P. The molecular functions of Shp2 in the Ras/Mitogen-activated protein kinase (ERK1/2) pathway. Cell. Signal. 2008, 20, 453–459. [Google Scholar] [CrossRef]
- Neel, B.G.; Gu, H.; Pao, L. The ’Shp’ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem. Sci. 2003, 28, 284–293. [Google Scholar] [CrossRef]
- Hof, P.; Pluskey, S.; Dhe-Paganon, S.; Eck, M.J.; Shoelson, S.E. Crystal Structure of the Tyrosine Phosphatase SHP-2. Cell 1998, 92, 441–450. [Google Scholar] [CrossRef] [Green Version]
- Pluskey, S.; Wandless, T.J.; Walsh, C.T.; Shoelson, S.E. Potent stimulation of SH-PTP2 phosphatase activity by simultaneous occupancy of both SH2 domains. J. Biol. Chem. 1995, 270, 2897–2900. [Google Scholar] [CrossRef] [Green Version]
- LaRochelle, J.R.; Fodor, M.; Xu, X.; Durzynska, I.; Fan, L.; Stams, T.; Chan, H.M.; LaMarche, M.J.; Chopra, R.; Wang, P.; et al. Structural and Functional Consequences of Three Cancer-Associated Mutations of the Oncogenic Phosphatase SHP2. Biochemistry 2016, 55, 2269–2277. [Google Scholar] [CrossRef] [Green Version]
- Tartaglia, M.; Kalidas, K.; Shaw, A.; Song, X.; Musat, D.L.; van der Burgt, I.; Brunner, H.G.; Bertola, D.R.; Crosby, A.; Ion, A.; et al. PTPN11 Mutations in Noonan Syndrome: Molecular Spectrum, Genotype-Phenotype Correlation, and Phenotypic Heterogeneity. Am. J. Hum. Genet. 2002, 70, 1555. [Google Scholar] [CrossRef] [Green Version]
- Strullu, M.; Caye, A.; Lachenaud, J.; Cassinat, B.; Gazal, S.; Fenneteau, O.; Pouvreau, N.; Pereira, S.; Baumann, C.; Contet, A.; et al. Juvenile myelomonocytic leukaemia and Noonan syndrome. J. Med. Genet. 2014, 51, 689–697. [Google Scholar] [CrossRef]
- Flotho, C.; Valcamonica, S.; Mach-Pascual, S.; Schmahl, G.; Corral, L.; Ritterbach, J.; Hasle, H.; Aricò, M.; Biondi, A.; Niemeyer, C.; et al. RAS mutations and clonality analysis in children with juvenile myelomonocytic leukemia (JMML). Leukemia 1999, 13, 32–37. [Google Scholar] [CrossRef] [Green Version]
- Shima, F.; Ijiri, Y.; Muraoka, S.; Liao, J.; Ye, M.; Araki, M.; Matsumoto, K.; Yamamoto, N.; Sugimoto, T.; Yoshikawa, Y.; et al. Structural Basis for Conformational Dynamics of GTP-bound Ras Protein. J. Biol. Chem. 2010, 285, 22696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuda, K.; Shimada, A.; Yoshida, N.; Ogawa, A.; Watanabe, A.; Yajima, S.; Iizuka, S.; Koike, K.K.; Yanai, F.; Kawasaki, K.; et al. Spontaneous improvement of hematologic abnormalities in patients having juvenile myelomonocytic leukemia with specific RAS mutations. Blood 2007, 109, 5477–5480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraoua, L.; Journel, H.; Bonnet, P.; Amiel, J.; Pouvreau, N.; Baumann, C.; Verloes, A.; Cavé, H. Constitutional NRAS mutations are rare among patients with Noonan syndrome or juvenile myelomonocytic leukemia. Am. J. Med. Genet. Part A 2012, 158A, 2407–2411. [Google Scholar] [CrossRef] [PubMed]
- Hobbs, G.A.; Der, C.J.; Rossman, K.L. RAS isoforms and mutations in cancer at a glance. J. Cell Sci. 2016, 129, 1287. [Google Scholar] [CrossRef] [Green Version]
- Lipka, D.B.; Witte, T.; Toth, R.; Yang, J.; Wiesenfarth, M.; Nöllke, P.; Fischer, A.; Brocks, D.; Gu, Z.; Park, J.; et al. RAS-pathway mutation patterns define epigenetic subclasses in juvenile myelomonocytic leukemia. Nat. Commun. 2017, 8, 2126. [Google Scholar] [CrossRef] [Green Version]
- Simanshu, D.K.; Nissley, D.V.; Mccormick, F. Leading Edge Review RAS Proteins and Their Regulators in Human Disease. Cell 2017, 170, 17–33. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, K.; Nakazawa, Y.; Sakashita, K.; Shiohara, M.; Yamauchi, K.; Koike, K. Acquisition of loss of the wild-type NRAS locus with aggressive disease progression in a patient with juvenile myelomonocytic leukemia and a heterozygous NRAS mutation. Haematologica 2007, 92, 1576–1578. [Google Scholar] [CrossRef]
- Kato, M.; Yasui, N.; Seki, M.; Kishimoto, H.; Sato-Otsubo, A.; Hasegawa, D.; Kiyokawa, N.; Hanada, R.; Ogawa, S.; Manabe, A.; et al. Aggressive Transformation of Juvenile Myelomonocytic Leukemia Associated with Duplication of Oncogenic KRAS due to Acquired Uniparental Disomy. J. Pediatr. 2013, 162, 1285–1288.e1. [Google Scholar] [CrossRef]
- Caye, A.; Strullu, M.; Guidez, F.; Cassinat, B.; Gazal, S.; Fenneteau, O.; Lainey, E.; Nouri, K.; Nakhaei-Rad, S.; Dvorsky, R.; et al. Juvenile myelomonocytic leukemia displays mutations in components of the RAS pathway and the PRC2 network. Nat. Genet. 2015, 47, 1334–1340. [Google Scholar] [CrossRef]
- Schubbert, S.; Zenker, M.; Rowe, S.L.; Böll, S.; Klein, C.; Bollag, G.; van der Burgt, I.; Musante, L.; Kalscheuer, V.; Wehner, L.-E.E.; et al. Germline KRAS mutations cause Noonan syndrome. Nat. Genet. 2006, 38, 331–336. [Google Scholar] [CrossRef]
- Zenker, M.; Lehmann, K.; Schulz, A.L.; Barth, H.; Hansmann, D.; Koenig, R.; Korinthenberg, R.; Kreiss-Nachtsheim, M.; Meinecke, P.; Morlot, S.; et al. Expansion of the genotypic and phenotypic spectrum in patients with KRAS germline mutations. J. Med. Genet. 2007, 44, 131–135. [Google Scholar] [CrossRef] [Green Version]
- Ekvall, S.; Wilbe, M.; Dahlgren, J.; Legius, E.; van Haeringen, A.; Westphal, O.; Annerén, G.; Bondeson, M.L. Mutation in NRAS in familial Noonan syndrome-case report and review of the literature. BMC Med. Genet. 2015, 16, 95. [Google Scholar] [CrossRef] [Green Version]
- Altmüller, F.; Lissewski, C.; Bertola, D.; Flex, E.; Stark, Z.; Spranger, S.; Baynam, G.; Buscarilli, M.; Dyack, S.; Gillis, J.; et al. Genotype and phenotype spectrum of NRAS germline variants. Eur. J. Hum. Genet. 2017, 25, 823–831. [Google Scholar] [CrossRef]
- De Filippi, P.; Zecca, M.; Lisini, D.; Rosti, V.; Cagioni, C.; Carlo-Stella, C.; Radi, O.; Veggiotti, P.; Mastronuzzi, A.; Acquaviva, A.; et al. Germ-line mutation of the NRAS gene may be responsible for the development of juvenile myelomonocytic leukaemia. Br. J. Haematol. 2009, 147, 706–709. [Google Scholar] [CrossRef]
- Doisaki, S.; Muramatsu, H.; Shimada, A.; Takahashi, Y.; Mori-Ezaki, M.; Sato, M.; Kawaguchi, H.; Kinoshita, A.; Sotomatsu, M.; Hayashi, Y.; et al. Somatic mosaicism for oncogenic NRAS mutations in juvenile myelomonocytic leukemia. Blood 2012, 120, 1485–1488. [Google Scholar] [CrossRef] [Green Version]
- Kratz, C.P.; Schubbert, S.; Bollag, G.; Niemeyer, C.M.; Shannon, K.M.; Zenker, M. Germline Mutations in Components of the Ras Signaling Pathway in Noonan Syndrome and Related Disorders. Cell Cycle 2006, 5, 1607–1611. [Google Scholar] [CrossRef] [Green Version]
- Murakami, N.; Okuno, Y.; Yoshida, K.; Shiraishi, Y.; Nagae, G.; Suzuki, K.; Narita, A.; Sakaguchi, H.; Kawashima, N.; Wang, X.; et al. Integrated molecular profiling of juvenile myelomonocytic leukemia. Blood 2018, 131, 1576–1586. [Google Scholar] [CrossRef]
- Niemeyer, C.M.; Kang, M.W.; Shin, D.H.; Furlan, I.; Erlacher, M.; Bunin, N.J.; Bunda, S.; Finklestein, J.Z.; Gorr, T.A.; Mehta, P.; et al. Germline CBL mutations cause developmental abnormalities and predispose to juvenile myelomonocytic leukemia. Nat. Genet. 2010, 42, 794. [Google Scholar] [CrossRef]
- Hecht, A.; Meyer, J.A.; Behnert, A.; Wong, E.; Chehab, F.; Olshen, A.; Hechmer, A.; Aftandilian, C.; Bhat, R.; Choi, S.W.; et al. Molecular and phenotypic diversity of CBL-mutated juvenile myelomonocytic leukemia. Haematologica 2022, 107, 178–186. [Google Scholar] [CrossRef]
- Pérez, B.; Mechinaud, F.; Galambrun, C.; Ben Romdhane, N.; Isidor, B.; Philip, N.; Derain-Court, J.; Cassinat, B.; Lachenaud, J.; Kaltenbach, S.; et al. Germline mutations of the CBL gene define a new genetic syndrome with predisposition to juvenile myelomonocytic leukaemia. J. Med. Genet. 2010, 47, 686–691. [Google Scholar] [CrossRef]
- Martinelli, S.; De Luca, A.; Stellacci, E.; Rossi, C.; Checquolo, S.; Lepri, F.; Caputo, V.; Silvano, M.; Buscherini, F.; Consoli, F.; et al. Heterozygous germline mutations in the CBL tumor-suppressor gene cause a Noonan syndrome-like phenotype. Am. J. Hum. Genet. 2010, 87, 250–257. [Google Scholar] [CrossRef] [Green Version]
- Loh, M.L.; Sakai, D.S.; Flotho, C.; Kang, M.; Fliegauf, M.; Archambeault, S.; Mullighan, C.G.; Chen, L.; Bergstraesser, E.; Bueso-Ramos, C.E.; et al. Mutations in CBL occur frequently in juvenile myelomonocytic leukemia. Blood 2009, 114, 1859–1863. [Google Scholar] [CrossRef] [Green Version]
- Muramatsu, H.; Makishima, H.; Jankowska, A.M.; Cazzolli, H.; O’Keefe, C.; Yoshida, N.; Xu, Y.; Nishio, N.; Hama, A.; Yagasaki, H.; et al. Mutations of an E3 ubiquitin ligase c-Cbl but not TET2 mutations are pathogenic in juvenile myelomonocytic leukemia. Blood 2010, 115, 1969–1975. [Google Scholar] [CrossRef] [Green Version]
- Pérez, B.; Kosmider, O.; Cassinat, B.; Renneville, A.; Lachenaud, J.; Kaltenbach, S.; Bertrand, Y.; Baruchel, A.; Chomienne, C.; Fontenay, M.; et al. Genetic typing of CBL, ASXL1, RUNX1, TET2 and JAK2 in juvenile myelomonocytic leukaemia reveals a genetic profile distinct from chronic myelomonocytic leukaemia. Br. J. Haematol. 2010, 151, 460–468. [Google Scholar] [CrossRef]
- Matsuda, K.; Taira, C.; Sakashita, K.; Saito, S.; Tanaka-Yanagisawa, M.; Yanagisawa, R.; Nakazawa, Y.; Shiohara, M.; Fukushima, K.; Oda, M.; et al. Long-term survival after nonintensive chemotherapy in some juvenile myelomonocytic leukemia patients with CBL mutations, and the possible presence of healthy persons with the mutations. Blood 2010, 115, 5429–5431. [Google Scholar] [CrossRef]
- Mohapatra, B.; Ahmad, G.; Nadeau, S.; Zutshi, N.; An, W.; Scheffe, S.; Dong, L.; Feng, D.; Goetz, B.; Arya, P.; et al. Protein tyrosine kinase regulation by ubiquitination: Critical roles of Cbl-family ubiquitin ligases. Biochim. Biophys. Acta 2013, 1833, 122–139. [Google Scholar] [CrossRef] [Green Version]
- Zheng, N.; Wang, P.; Jeffrey, P.D.; Pavletich, N.P. Structure of a c-Cbl–UbcH7 Complex: RING Domain Function in Ubiquitin-Protein Ligases. Cell 2000, 102, 533–539. [Google Scholar] [CrossRef] [Green Version]
- Javadi, M.; Richmond, T.D.; Huang, K.; Barber, D.L. CBL Linker Region and RING Finger Mutations Lead to Enhanced Granulocyte-Macrophage Colony-stimulating Factor (GM-CSF) Signaling via Elevated Levels of JAK2 and LYN. J. Biol. Chem. 2013, 288, 19459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belizaire, R.; Koochaki, S.H.J.; Udeshi, N.D.; Vedder, A.; Sun, L.; Svinkina, T.; Hartigan, C.; McConkey, M.; Kovalcik, V.; Bizuayehu, A.; et al. CBL mutations drive PI3K/AKT signaling via increased interaction with LYN and PIK3R1. Blood 2021, 137, 2209–2220. [Google Scholar] [CrossRef] [PubMed]
- Park, R.K.; Kyono, W.T.; Liu, Y.; Durden, D.L. CBL-GRB2 Interaction in Myeloid Immunoreceptor Tyrosine Activation Motif Signaling. J. Immunol. 1998, 160, 5018–5027. [Google Scholar] [PubMed]
- Schmidt, M.H.H.; Dikic, I. The Cbl interactome and its functions. Nat. Rev. Mol. Cell Biol. 2005, 6, 907–919. [Google Scholar] [CrossRef]
- Budays, L.; Egan, S.E.; Rodriguez Viciana, P.; Cantrello, D.A.; Downward1, J. A Complex of Grb2 Adaptor Protein, Sos Exchange Factor, and a 36-kDa Membrane-bound Tyrosine Phosphoprotein Is Implicated in Ras Activation in T Cells. J. Biol. Chem. 1994, 269, 9019–9023. [Google Scholar] [CrossRef]
- Buday, L.; Khwaja, A.; Sipeki, S.; Faragó, A.; Downward, J. Interactions of Cbl with two adapter proteins, Grb2 and Crk, upon T cell activation. J. Biol. Chem. 1996, 271, 6159–6163. [Google Scholar] [CrossRef] [Green Version]
- Grossmann, K.S.; Rosário, M.; Birchmeier, C.; Birchmeier, W. The Tyrosine Phosphatase Shp2 in Development and Cancer. Adv. Cancer Res. 2010, 106, 53–89. [Google Scholar] [CrossRef]
- Bunda, S.; Heir, P.; Srikumar, T.; Cook, J.D.; Burrell, K.; Kano, Y.; Lee, J.E.; Zadeh, G.; Raught, B.; Ohh, M. Src promotes GTPase activity of Ras via tyrosine 32 phosphorylation. Proc. Natl. Acad. Sci. USA 2014, 111, E3785–E3794. [Google Scholar] [CrossRef] [Green Version]
- Rauen, K.A. The RASopathies. Annu. Rev. Genomics Hum. Genet. 2013, 14, 355. [Google Scholar] [CrossRef] [Green Version]
- Steinemann, D.; Arning, L.; Praulich, I.; Stuhrmann, M.; Hasle, H.; Starý, J.; Schlegelberger, B.; Niemeyer, C.M.; Flotho, C. Mitotic recombination and compound-heterozygous mutations are predominant NF1-inactivating mechanisms in children with juvenile myelomonocytic leukemia and neurofibromatosis type 1. Haematologica 2010, 95, 320–323. [Google Scholar] [CrossRef] [Green Version]
- Martin, G.A.; Viskoohil, D.; Bollag, G.; McCabe, P.C.; Crosier, W.J.; Haubruck, H.; Conroy, L.; Clark, R.; O’Connell, P.; Cawthon, R.M.; et al. The GAP-related domain of the neurofibromatosis type 1 gene product interacts with ras p21. Cell 1990, 63, 843–849. [Google Scholar] [CrossRef]
- Xu, G.; Lin, B.; Tanaka, K.; Dunn, D.; Wood, D.; Gesteland, R.; White, R.; Weiss, R.; Tamanoi, F.; GF, X.; et al. The catalytic domain of the neurofibromatosis type 1 gene product stimulates ras GTPase and complements ira mutants of S. cerevisiae. Cell 1990, 63, 835–841. [Google Scholar] [CrossRef]
- Scheffzek, K.; Ahmadian, M.R.; Wiesmüller, L.; Kabsch, W.; Stege, P.; Schmitz, F.; Wittinghofer, A. Structural analysis of the GAP-related domain from neurofibromin and its implications. EMBO J. 1998, 17, 4313. [Google Scholar] [CrossRef] [Green Version]
- Flotho, C.; Steinemann, D.; Mullighan, C.G.; Neale, G.; Mayer, K.; Kratz, C.P.; Schlegelberger, B.; Downing, J.R.; Niemeyer, C.M. Genome-wide single-nucleotide polymorphism analysis in juvenile myelomonocytic leukemia identifies uniparental disomy surrounding the NF1 locus in cases associated with neurofibromatosis but not in cases with mutant RAS or PTPN11. Oncogene 2007, 26, 5816–5821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Side, L.E.; Emanuel, P.D.; Taylor, B.; Franklin, J.; Thompson, P.; Castleberry, R.P.; Shannon, K.M. Mutations of the NF1 Gene in Children with Juvenile Myelomonocytic Leukemia Without Clinical Evidence of Neurofibromatosis, Type 1. Blood 1998, 92, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Cichowski, K.; Santiago, S.; Jardim, M.; Johnson, B.W.; Jacks, T. Dynamic regulation of the Ras pathway via proteolysis of the NF1 tumor suppressor. Genes Dev. 2003, 17, 449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stieglitz, E.; Taylor-Weiner, A.N.; Chang, T.Y.; Gelston, L.C.; Wang, Y.-D.D.; Mazor, T.; Esquivel, E.; Yu, A.; Seepo, S.; Olsen, S.R.; et al. The genomic landscape of juvenile myelomonocytic leukemia. Nat. Genet. 2015, 47, 1326–1333. [Google Scholar] [CrossRef] [PubMed]
- Morerio, C.; Acquila, M.; Rosanda, C.; Rapella, A.; Dufour, C.; Locatelli, F.; Maserati, E.; Pasquali, F.; Panarello, C. HCMOGT-1 Is a Novel Fusion Partner to PDGFRB in Juvenile Myelomonocytic Leukemia with t(5;17)(q33;p11.2). Cancer Res. 2004, 64, 2649–2651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byrgazov, K.; Kastner, R.; Dworzak, M.; Hoermann, G.; Haas, O.A.; Ulreich, R.; Urban, C.E.; Hantschel, O.D.; Valent, P.; Lion, T. A Novel Fusion Gene NDEL1-Pdgfrb in a Patient with JMML with a New Variant of TKI-Resistant Mutation in the Kinase Domain of PDGFRβ. Blood 2014, 124, 613. [Google Scholar] [CrossRef]
- Chao, A.K.; Meyer, J.A.; Lee, A.G.; Hecht, A.; Tarver, T.; Van Ziffle, J.; Koegel, A.K.; Golden, C.; Braun, B.S.; Sweet-Cordero, E.A.; et al. Fusion driven JMML: A novel CCDC88C–FLT3 fusion responsive to sorafenib identified by RNA sequencing. Leukemia 2020, 34, 662–666. [Google Scholar] [CrossRef] [PubMed]
- Morris, S.W.; Kirstein, M.N.; Valentine, M.B.; Dittmer, K.G.; Shapiro, D.N.; Saltman, D.L.; Look, A.T. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science 1994, 263, 1281–1284. [Google Scholar] [CrossRef] [PubMed]
- Maesako, Y.; Izumi, K.; Okamori, S.; Takeoka, K.; Kishimori, C.; Okumura, A.; Honjo, G.; Akasaka, T.; Ohno, H. inv(2)(p23q13)/RAN-binding protein 2 (RANBP2)-ALK fusion gene in myeloid leukemia that developed in an elderly woman. Int. J. Hematol. 2014, 99, 202–207. [Google Scholar] [CrossRef]
- Ma, Z.; Hill, D.A.; Collins, M.H.; Morris, S.W.; Sumegi, J.; Zhou, M.; Zuppan, C.; Bridge, J.A. Fusion of ALK to the Ran-binding protein 2 (RANBP2) gene in inflammatory myofibroblastic tumor. Genes. Chromosomes Cancer 2003, 37, 98–105. [Google Scholar] [CrossRef]
- Heilmann, A.M.; Schrock, A.B.; He, J.; Nahas, M.; Curran, K.; Shukla, N.; Cramer, S.; Draper, L.; Verma, A.; Erlich, R.; et al. Novel PDGFRB fusions in childhood B- and T-acute lymphoblastic leukemia. Leukemia 2017, 31, 1989–1992. [Google Scholar] [CrossRef]
- Zhang, H.; Paliga, A.; Hobbs, E.; Moore, S.; Olson, S.; Long, N.; Dao, K.H.T.; Tyner, J.W. Two myeloid leukemia cases with rare FLT3 fusions. Cold Spring Harb. Mol. Case Stud. 2018, 4, a003079. [Google Scholar] [CrossRef] [Green Version]
- Sakaguchi, H.; Okuno, Y.; Muramatsu, H.; Yoshida, K.; Shiraishi, Y.; Takahashi, M.; Kon, A.; Sanada, M.; Chiba, K.; Tanaka, H.; et al. Exome sequencing identifies secondary mutations of SETBP1 and JAK3 in juvenile myelomonocytic leukemia. Nat. Genet. 2013, 45, 937–941. [Google Scholar] [CrossRef]
- Bresolin, S.; De Filippi, P.; Vendemini, F.; D’Alia, M.; Zecca, M.; Meyer, L.H.; Danesino, C.; Locatelli, F.; Masetti, R.; Basso, G.; et al. Mutations of SETBP1 and JAK3 in juvenile myelomonocytic leukemia: A report from the Italian AIEOP study group. Oncotarget 2016, 7, 28914. [Google Scholar] [CrossRef]
- Cristóbal, I.; Blanco, F.J.; Garcia-Orti, L.; Marcotegui, N.; Vicente, C.; Rifon, J.; Novo, F.J.; Bandres, E.; Calasanz, M.J.; Bernabeu, C.; et al. SETBP1 overexpression is a novel leukemogenic mechanism that predicts adverse outcome in elderly patients with acute myeloid leukemia. Blood 2010, 115, 615–625. [Google Scholar] [CrossRef]
- Piazza, R.; Valletta, S.; Winkelmann, N.; Redaelli, S.; Spinelli, R.; Pirola, A.; Antolini, L.; Mologni, L.; Donadoni, C.; Papaemmanuil, E.; et al. Recurrent SETBP1 mutations in atypical chronic myeloid leukemia. Nat. Genet. 2013, 45, 18. [Google Scholar] [CrossRef]
- Bayarkhangai, B.; Noureldin, S.; Yu, L.; Zhao, N.; Gu, Y.; Xu, H.; Guo, C. A comprehensive and perspective view of oncoprotein SET in cancer. Cancer Med. 2018, 7, 3084–3094. [Google Scholar] [CrossRef]
- Acuna-Hidalgo, R.; Deriziotis, P.; Steehouwer, M.; Gilissen, C.; Graham, S.A.; van Dam, S.; Hoover-Fong, J.; Telegrafi, A.B.; Destree, A.; Smigiel, R.; et al. Overlapping SETBP1 gain-of-function mutations in Schinzel-Giedion syndrome and hematologic malignancies. PLoS Genet. 2017, 13, e1006683. [Google Scholar] [CrossRef]
- Oakley, K.; Han, Y.; Vishwakarma, B.A.; Chu, S.; Bhatia, R.; Gudmundsson, K.O.; Keller, J.; Chen, X.; Vasko, V.; Jenkins, N.A.; et al. Setbp1 promotes the self-renewal of murine myeloid progenitors via activation of Hoxa9 and Hoxa10. Blood 2012, 119, 6099–6108. [Google Scholar] [CrossRef] [Green Version]
- Piazza, R.; Magistroni, V.; Redaelli, S.; Mauri, M.; Massimino, L.; Sessa, A.; Peronaci, M.; Lalowski, M.; Soliymani, R.; Mezzatesta, C.; et al. SETBP1 induces transcription of a network of development genes by acting as an epigenetic hub. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Carratt, S.A.; Braun, T.P.; Coblentz, C.; Schonrock, Z.; Callahan, R.; Smith, B.M.; Maloney, L.; Foley, A.C.; Maxson, J.E.; SA, C.; et al. Mutant SETBP1 enhances NRAS-driven MAPK pathway activation to promote aggressive leukemia. Leukemia 2021, 35, 3594–3599. [Google Scholar] [CrossRef] [PubMed]
- Niemeyer, C.M. JMML genomics and decisions. Hematol. Am. Soc. Hematol. Educ. Progr. 2018, 2018, 307–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flex, E.; Jaiswal, M.; Pantaleoni, F.; Martinelli, S.; Strullu, M.; Fansa, E.K.; Caye, A.; De Luca, A.; Lepri, F.; Dvorsky, R.; et al. Activating mutations in RRAS underlie a phenotype within the RASopathy spectrum and contribute to leukaemogenesis. Hum. Mol. Genet. 2014, 23, 4315–4327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caye, A.; Rouault-Pierre, K.; Strullu, M.; Lainey, E.; Abarrategi, A.; Fenneteau, O.; Arfeuille, C.; Osman, J.; Cassinat, B.; Pereira, S.; et al. Despite mutation acquisition in hematopoietic stem cells, JMML-propagating cells are not always restricted to this compartment. Leukemia 2020, 34, 1658–1668. [Google Scholar] [CrossRef]
- Stieglitz, E.; Troup, C.B.; Gelston, L.C.; Haliburton, J.; Chow, E.D.; Yu, K.B.; Akutagawa, J.; Taylor-Weiner, A.N.; Liu, Y.L.; Wang, Y.D.; et al. Subclonal mutations in SETBP1 confer a poor prognosis in juvenile myelomonocytic leukemia. Blood 2015, 125, 516–524. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Bauer, D.E.; Loh, M.L.; Bhagat, G.; Cantor, A.B.; Kung, A.L. Potential Role of RUNX1 In the Pathogenesis of Juvenile Myelomonocytic Leukemia (JMML). Blood 2013, 122, 45. [Google Scholar] [CrossRef]
- Buijs, A.; Bruin, M. Fusion of FIP1L1 and RARA as a result of a novel t(4;17)(q12;q21) in a case of juvenile myelomonocytic leukemia. Leukemia 2007, 21, 1104–1108. [Google Scholar] [CrossRef]
- Mizoguchi, Y.; Fujita, N.; Taki, T.; Hayashi, Y.; Hamamoto, K. Juvenile myelomonocytic leukemia with t(7;11)(p15;p15) and NUP98-HOXA11 fusion. Am. J. Hematol. 2009, 84, 295–297. [Google Scholar] [CrossRef]
- Coppe, A.; Nogara, L.; Pizzuto, M.S.; Cani, A.; Cesaro, S.; Masetti, R.; Locatelli, F.; Kronnie, G.; Basso, G.; Bortoluzzi, S.; et al. Somatic mutations activating Wiskott–Aldrich syndrome protein concomitant with RAS pathway mutations in juvenile myelomonocytic leukemia patients. Hum. Mutat. 2018, 39, 579–587. [Google Scholar] [CrossRef]
- Yang, L.; Rau, R.; Goodell, M.A. DNMT3A in haematological malignancies. Nat. Rev. Cancer 2015, 15, 152. [Google Scholar] [CrossRef]
- Laugesen, A.; Højfeldt, J.W.; Helin, K. Role of the Polycomb Repressive Complex 2 (PRC2) in Transcriptional Regulation and Cancer. Cold Spring Harb. Perspect. Med. 2016, 6, a026575. [Google Scholar] [CrossRef] [Green Version]
- Shen, H.; Laird, P.W. Interplay between the cancer genome and epigenome. Cell 2013, 153, 38–55. [Google Scholar] [CrossRef] [Green Version]
- Galm, O.; Herman, J.G.; Baylin, S.B. The fundamental role of epigenetics in hematopoietic malignancies. Blood Rev. 2006, 20, 1–13. [Google Scholar] [CrossRef]
- Olk-Batz, C.; Poetsch, A.R.; Nöllke, P.; Claus, R.; Zucknick, M.; Sandrock, I.; Witte, T.; Strahm, B.; Hasle, H.; Zecca, M.; et al. Aberrant DNA methylation characterizes juvenile myelomonocytic leukemia with poor outcome. Blood 2011, 117, 4871–4880. [Google Scholar] [CrossRef]
- Sakaguchi, H.; Muramatsu, H.; Okuno, Y.; Makishima, H.; Xu, Y.; Furukawa-Hibi, Y.; Wang, X.; Narita, A.; Yoshida, K.; Shiraishi, Y.; et al. Aberrant DNA Methylation Is Associated with a Poor Outcome in Juvenile Myelomonocytic Leukemia. PLoS ONE 2015, 10, e0145394. [Google Scholar] [CrossRef] [Green Version]
- Poetsch, A.R.; Lipka, D.B.; Witte, T.; Claus, R.; Nöllke, P.; Zucknick, M.; Olk-Batz, C.; Fluhr, S.; Dworzak, M.; De Moerloose, B.; et al. RASA4 undergoes DNA hypermethylation in resistant juvenile myelomonocytic leukemia. Epigenetics 2014, 9, 1252–1260. [Google Scholar] [CrossRef] [Green Version]
- Fluhr, S.; Boerries, M.; Busch, H.; Symeonidi, A.; Witte, T.; Lipka, D.B.; Mücke, O.; Nöllke, P.; Krombholz, C.F.; Niemeyer, C.M.; et al. CREBBP is a target of epigenetic, but not genetic, modification in juvenile myelomonocytic leukemia. Clin. Epigenet. 2016, 8, 50. [Google Scholar] [CrossRef] [Green Version]
- Wilhelm, T.; Lipka, D.B.; Witte, T.; Wierzbinska, J.A.; Fluhr, S.; Helf, M.; Mücke, O.; Claus, R.; Konermann, C.; Nöllke, P.; et al. Epigenetic silencing of AKAP12 in juvenile myelomonocytic leukemia. Epigenetics 2016, 11, 110–119. [Google Scholar] [CrossRef] [Green Version]
- Stieglitz, E.; Mazor, T.; Olshen, A.B.; Geng, H.; Gelston, L.C.; Akutagawa, J.; Lipka, D.B.; Plass, C.; Flotho, C.; Chehab, F.F.; et al. Genome-wide DNA methylation is predictive of outcome in juvenile myelomonocytic leukemia. Nat. Commun. 2017, 8, 2127. [Google Scholar] [CrossRef] [Green Version]
- Schönung, M.; Meyer, J.; Nöllke, P.; Olshen, A.; Hartmann, M.; Murakami, N.; Wakamatsu, M.; Okuno, Y.; Plass, C.; Loh, M.L.; et al. International consensus definition of DNA methylation subgroups in juvenile myelomonocytic leukemia. Clin. Cancer Res. 2021, 27, 158. [Google Scholar] [CrossRef]
- Kitazawa, H.; Okuno, Y.; Muramatsu, H.; Aoki, K.; Murakami, N.; Wakamatsu, M.; Suzuki, K.; Narita, K.; Kataoka, S.; Ichikawa, D.; et al. A simple and robust methylation test for risk stratification of patients with juvenile myelomonocytic leukemia. Blood Adv. 2021, 5, 5507–5518. [Google Scholar] [CrossRef]
- Jelinek, J.; Lee, J.T.; Cesaroni, M.; Madzo, J.; Liang, S.; Lu, Y.; Issa, J.P.J. Digital Restriction Enzyme Analysis of Methylation (DREAM). Methods Mol. Biol. 2018, 1708, 247–265. [Google Scholar] [CrossRef] [PubMed]
- Niemeyer, C.M.; Flotho, C. Juvenile myelomonocytic leukemia: Who’s the driver at the wheel? Blood 2019, 133, 1060–1070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, N.; Yagasaki, H.; Xu, Y.; Matsuda, K.; Yoshimi, A.; Takahashi, Y.; Hama, A.; Nishio, N.; Muramatsu, H.; Watanabe, N.; et al. Correlation of clinical features with the mutational status of GM-CSF signaling pathway-related genes in juvenile myelomonocytic leukemia. Pediatr. Res. 2009, 65, 334–340. [Google Scholar] [CrossRef] [Green Version]
- Stephen, A.G.; Esposito, D.; Bagni, R.G.; McCormick, F. Dragging ras back in the ring. Cancer Cell 2014, 25, 272–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, A.F.; Braun, B.S.; Shannon, K.M. Targeting oncogenic Ras signaling in hematologic malignancies. Blood 2012, 120, 3397–3406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stieglitz, E.; Ward, A.F.; Gerbing, R.B.; Alonzo, T.A.; Arceci, R.J.; Liu, Y.L.; Emanuel, P.D.; Widemann, B.C.; Cheng, J.W.; Jayaprakash, N.; et al. Phase II/III trial of a pre-transplant farnesyl transferase inhibitor in juvenile myelomonocytic leukemia: A report from the Children’s Oncology Group. Pediatr. Blood Cancer 2015, 62, 629. [Google Scholar] [CrossRef] [Green Version]
- Kazandjian, D.; Blumenthal, G.M.; Chen, H.-Y.; He, K.; Patel, M.; Justice, R.; Keegan, P.; Pazdur, R. FDA approval summary: Crizotinib for the treatment of metastatic non-small cell lung cancer with anaplastic lymphoma kinase rearrangements. Oncologist 2014, 19, e5–e11. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, V.E.; Smith, C.C. FLT3 Mutations in Acute Myeloid Leukemia: Key Concepts and Emerging Controversies. Front. Oncol. 2020, 10, 612880. [Google Scholar] [CrossRef]
- Jenkins, C.; Luty, S.B.; Maxson, J.E.; Eide, C.A.; Abel, M.L.; Togiai, C.; Nemecek, E.R.; Bottomly, D.; McWeeney, S.K.; Wilmot, B.; et al. Synthetic lethality of TNK2 inhibition in PTPN11-mutant leukemia. Sci. Signal. 2018, 11, eaao5617. [Google Scholar] [CrossRef] [Green Version]
- Wright, C.J.M.; McCormack, P.L. Trametinib: First global approval. Drugs 2013, 73, 1245–1254. [Google Scholar] [CrossRef]
- Tasian, S.K.; Casas, J.A.; Posocco, D.; Gandre-Babbe, S.; Gagne, A.L.; Liang, G.; Loh, M.L.; Weiss, M.J.; French, D.L.; Chou, S.T.; et al. Mutation-specific signaling profiles and kinase inhibitor sensitivities of juvenile myelomonocytic leukemia revealed by induced pluripotent stem cells. Leukemia 2019, 33, 181–190. [Google Scholar] [CrossRef]
- Goodwin, C.B.; Li, X.J.; Mali, R.S.; Chan, G.; Kang, M.; Liu, Z.; Vanhaesebroeck, B.; Neel, B.G.; Loh, M.L.; Lannutti, B.J.; et al. PI3K p110δ uniquely promotes gain-of-function Shp2-induced GM-CSF hypersensitivity in a model of JMML. Blood 2014, 123, 2838–2842. [Google Scholar] [CrossRef] [Green Version]
- Deng, L.; Virts, E.L.; Kapur, R.; Chan, R.J. Pharmacologic inhibition of PI3K p110δ in mutant Shp2E76K-expressing mice. Oncotarget 2017, 8, 84776–84781. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Yu, W.-M.M.; Zhang, J.; Chan, R.J.; Loh, M.L.; Zhang, Z.; Bunting, K.D. Inhibition of the Gab2/PI3K/mTOR signaling ameliorates myeloid malignancy caused by Ptpn11 (Shp2) gain-of-function mutations. Leukemia 2017, 31, 1415–1422. [Google Scholar] [CrossRef] [Green Version]
- Niemeyer, C.M.; Flotho, C.; Lipka, D.B.; Stary, J.; Rössig, C.; Baruchel, A.; Klingebiel, T.; Micalizzi, C.; Michel, G.; Nysom, K.; et al. Response to upfront azacitidine in juvenile myelomonocytic leukemia in the AZA-JMML-001 trial. Blood Adv. 2021, 5, 2901–2908. [Google Scholar] [CrossRef]
- Furlan, I.; Niemeyer, C.M.; Batz, C.; Flotho, C.; Mohr, B.; Lübbert, M.; Suttorp, M. Intriguing response to azacitidine in a patient with juvenile myelomonocytic leukemia and monosomy 7. Blood 2009, 113, 2867–2868. [Google Scholar] [CrossRef]
- Cseh, A.; Niemeyer, C.M.; Yoshimi, A.; Dworzak, M.; Hasle, H.; Van Den Heuvel-Eibrink, M.M.; Locatelli, F.; Masetti, R.; Schmugge, M.; Groß-Wieltsch, U.; et al. Bridging to transplant with azacitidine in juvenile myelomonocytic leukemia: A retrospective analysis of the EWOG-MDS study group. Blood 2015, 125, 2311–2313. [Google Scholar] [CrossRef] [Green Version]
- Chang, T.; Krisman, K.; Theobald, E.H.; Xu, J.; Akutagawa, J.; Lauchle, J.O.; Kogan, S.; Braun, B.S.; Shannon, K. Sustained MEK inhibition abrogates myeloproliferative disease in Nf1 mutant mice. J. Clin. Invest. 2013, 123, 335–339. [Google Scholar] [CrossRef]
- Lyubynska, N.; Gorman, M.F.; Lauchle, J.O.; Hong, W.X.; Akutagawa, J.K.; Shannon, K.; Braun, B.S. A MEK Inhibitor Abrogates Myeloproliferative Disease in Kras Mutant Mice. Sci. Transl. Med. 2011, 3, 76ra27. [Google Scholar] [CrossRef] [Green Version]
- Fabri, O.; Horakova, J.; Bodova, I.; Svec, P.; Striezencova, Z.L.; Bubanska, E.; Cermak, M.; Galisova, V.; Skalicka, K.; Vaska, A.; et al. Diagnosis and treatment of juvenile myelomonocytic leukemia in Slovak Republic: Novel approaches. Neoplasma 2019, 66, 818–824. [Google Scholar] [CrossRef]
- Hashmi, S.K.; Punia, J.N.; Marcogliese, A.N.; Gaikwad, A.S.; Fisher, K.E.; Roy, A.; Rao, P.; Lopez-Terrada, D.H.; Ringrose, J.; Loh, M.L.; et al. Sustained remission with azacitidine monotherapy and an aberrantprecursor B-lymphoblast population in juvenile myelomonocyticleukemia. Pediatr. Blood Cancer 2019, 66, e27905. [Google Scholar] [CrossRef]
- Chan, R.J.; Cooper, T.; Kratz, C.P.; Weiss, B.; Loh, M.L. Juvenile Myelomonocytic Leukemia: A Report from the 2nd International JMML Symposium. Leuk. Res. 2009, 33, 355. [Google Scholar] [CrossRef] [Green Version]
- Wehbe, Z.; Ghanjati, F.; Flotho, C. Induced Pluripotent Stem Cells to Model Juvenile Myelomonocytic Leukemia: New Perspectives for Preclinical Research. Cells 2021, 10, 2335. [Google Scholar] [CrossRef]
- Yoshimi, A.; Balasis, M.E.; Vedder, A.; Feldman, K.; Ma, Y.; Zhang, H.; Lee, S.C.W.; Letson, C.; Niyongere, S.; Lu, S.X.; et al. Robust patient-derived xenografts of MDS/MPN overlap syndromes capture the unique characteristics of CMML and JMML. Blood 2017, 130, 397–407. [Google Scholar] [CrossRef] [Green Version]
- Gandre-Babbe, S.; Paluru, P.; Aribeana, C.; Chou, S.T.; Bresolin, S.; Lu, L.; Sullivan, S.K.; Tasian, S.K.; Weng, J.; Favre, H.; et al. Patient-derived induced pluripotent stem cells recapitulate hematopoietic abnormalities of juvenile myelomonocytic leukemia. Blood 2013, 121, 4925–4929. [Google Scholar] [CrossRef]
- Mulero-Navarro, S.; Sevilla, A.; Roman, A.C.; Lee, D.F.; D’Souza, S.L.; Pardo, S.; Riess, I.; Su, J.; Cohen, N.; Schaniel, C.; et al. Myeloid dysregulation in a human induced pluripotent stem cell model of PTPN11-associated juvenile myelomonocytic leukemia. Cell Rep. 2015, 13, 504. [Google Scholar] [CrossRef] [Green Version]
- Shigemura, T.; Matsuda, K.; Kurata, T.; Sakashita, K.; Okuno, Y.; Muramatsu, H.; Yue, F.; Ebihara, Y.; Tsuji, K.; Sasaki, K.; et al. Essential role of PTPN11 mutation in enhanced haematopoietic differentiation potential of induced pluripotent stem cells of juvenile myelomonocytic leukaemia. Br. J. Haematol. 2019, 187, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Sbrana, F.V.; Pinos, R.; Barbaglio, F.; Ribezzi, D.; Scagnoli, F.; Scarfò, L.; Redwan, I.N.; Martinez, H.; Farè, S.; Ghia, P.; et al. 3D Bioprinting Allows the Establishment of Long-Term 3D Culture Model for Chronic Lymphocytic Leukemia Cells. Front. Immunol. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Janagama, D.; Hui, S.K. 3-D Cell Culture Systems in Bone Marrow Tissue and Organoid Engineering, and BM Phantoms as In Vitro Models of Hematological Cancer Therapeutics-A Review. Materials 2020, 13, 5609. [Google Scholar] [CrossRef] [PubMed]
- Wolf, D.M.C.; Langhans, S.A. Moving Myeloid Leukemia Drug Discovery into the Third Dimension. Front. Pediatr. 2019, 7, 314. [Google Scholar] [CrossRef] [Green Version]
- Lau, R.C.; Squire, J.; Brisson, L.; Kamel-Reid, S.; Grunberger, T.; Dubé, I.; Letarte, M.; Shannon, K.; Freedman, M.H. Lymphoid blast crisis of B-lineage phenotype with monosomy 7 in a patient with juvenile chronic myelogenous leukemia (JCML). Leukemia 1994, 8, 903–908. [Google Scholar]
- Roy, A.; Wang, G.; Iskander, D.; O’Byrne, S.; Elliott, N.; O’Sullivan, J.; Buck, G.; Heuston, E.F.; Wen, W.X.; Meira, A.R.; et al. Transitions in lineage specification and gene regulatory networks in hematopoietic stem/progenitor cells over human development. Cell Rep. 2021, 36, 109698. [Google Scholar] [CrossRef]
- Louka, E.; Povinelli, B.; Rodriguez-Meira, A.; Buck, G.; Wen, W.X.; Wang, G.; Sousos, N.; Ashley, N.; Hamblin, A.; Booth, C.A.G.; et al. Heterogeneous disease-propagating stem cells in juvenile myelomonocytic leukemia. J. Exp. Med. 2021, 218, e20180853. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Gao, R.; Sei, E.; Brandt, R.; Hartman, J.; Hatschek, T.; Crosetto, N.; Foukakis, T.; Navin, N.E. Chemoresistance Evolution in Triple-Negative Breast Cancer Delineated by Single Cell Sequencing. Cell 2018, 173, 879. [Google Scholar] [CrossRef] [Green Version]
- Andor, N.; Lau, B.T.; Catalanotti, C.; Sathe, A.; Kubit, M.; Chen, J.; Blaj, C.; Cherry, A.; Bangs, C.D.; Grimes, S.M.; et al. Joint single cell DNA-seq and RNA-seq of gastric cancer cell lines reveals rules of in vitro evolution. NAR Genom. Bioinform. 2020, 2. [Google Scholar] [CrossRef] [Green Version]
- Van den Berge, K.; Roux de Bézieux, H.; Street, K.; Saelens, W.; Cannoodt, R.; Saeys, Y.; Dudoit, S.; Clement, L. Trajectory-based differential expression analysis for single-cell sequencing data. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Qiu, X.; Mao, Q.; Tang, Y.; Wang, L.; Chawla, R.; Pliner, H.A.; Trapnell, C. Reversed graph embedding resolves complex single-cell trajectories. Nat. Methods. 2017, 14, 979. [Google Scholar] [CrossRef] [Green Version]
- Lönnberg, T.; Svensson, V.; James, K.R.; Fernandez-Ruiz, D.; Sebina, I.; Montandon, R.; Soon, M.S.F.; Fogg, L.G.; Nair, A.S.; Liligeto, U.N.; et al. Single-cell RNA-seq and computational analysis using temporal mixture modelling resolves Th1/Tfh fate bifurcation in malaria. Sci. Immunol. 2017, 2, eaal2192. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Huang, K.; An, Q.; Du, G.; Hu, G.; Xue, J.; Zhu, X.; Wang, C.Y.; Xue, Z.; Fan, G. Simultaneous profiling of transcriptome and DNA methylome from a single cell. Genome Biol. 2016, 17, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Wierzbinska, J.A.; Toth, R.; Ishaque, N.; Rippe, K.; Mallm, J.P.; Klett, L.C.; Mertens, D.; Zenz, T.; Hielscher, T.; Seifert, M.; et al. Methylome-based cell-of-origin modeling (Methyl-COOM) identifies aberrant expression of immune regulatory molecules in CLL. Genome Med. 2020, 12, 1–19. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The RAS signaling pathway. NRAS and KRAS are small GTPase switch proteins that act downstream receptor and non-receptor tyrosine kinases (RTKs and TKs). RAS activation status is regulated by a two-stage molecular system directed by phosphorylation and dephosphorylation changes in RAS, which are regulated by the opposing activities of guanine nucleotide-exchange factors (GEFs) and GTPase activating proteins (GAPs). RTK/TK stimulation promotes the recruitment of adaptor proteins (such as GAB2, GRB2, and SHP2) and GEFs to mediate RAS-GDP phosphorylation to a RAS-GTP active status. Active RAS then triggers a signaling cascade that sequentially activates RAF, and phosphorylates MEK and ERK proteins, which ultimately signal to the nucleus to control specific cell functions such as proliferation, survival, and differentiation, among others. RAS pathway is inactivated by the activity of GAPs, such as NF1, which promote RAS-GTP dephosphorylation to a RAS-GDP inactive form. In addition, the ubiquitin ligase CBL can also act as a negative regulator of the RAS pathway by targeting active RTKs for proteasomal degradation. Figure was created with BioRender.com.
Figure 1.
The RAS signaling pathway. NRAS and KRAS are small GTPase switch proteins that act downstream receptor and non-receptor tyrosine kinases (RTKs and TKs). RAS activation status is regulated by a two-stage molecular system directed by phosphorylation and dephosphorylation changes in RAS, which are regulated by the opposing activities of guanine nucleotide-exchange factors (GEFs) and GTPase activating proteins (GAPs). RTK/TK stimulation promotes the recruitment of adaptor proteins (such as GAB2, GRB2, and SHP2) and GEFs to mediate RAS-GDP phosphorylation to a RAS-GTP active status. Active RAS then triggers a signaling cascade that sequentially activates RAF, and phosphorylates MEK and ERK proteins, which ultimately signal to the nucleus to control specific cell functions such as proliferation, survival, and differentiation, among others. RAS pathway is inactivated by the activity of GAPs, such as NF1, which promote RAS-GTP dephosphorylation to a RAS-GDP inactive form. In addition, the ubiquitin ligase CBL can also act as a negative regulator of the RAS pathway by targeting active RTKs for proteasomal degradation. Figure was created with BioRender.com.

Figure 2.
Distribution of RAS pathway mutations in children with JMML. Data reported by Lipka et al. [25] (A), Murakami et al. [37] (B), and Caye et al. [29] (C). Panel (D) summarizes the three studies. NS cases were excluded from the analysis.

Figure 3.
Association of secondary mutations with specific genetic drivers in JMML. Data reported by Murakami et al. [37], Caye et al. [29], and Stieglitz et al. [64,85]. Colored circles represent the relative frequency of a secondary mutation in patients with each of the five JMML canonical drivers (green > 2%; light blue 5–10%; dark blue > 10%).
Figure 3.
Association of secondary mutations with specific genetic drivers in JMML. Data reported by Murakami et al. [37], Caye et al. [29], and Stieglitz et al. [64,85]. Colored circles represent the relative frequency of a secondary mutation in patients with each of the five JMML canonical drivers (green > 2%; light blue 5–10%; dark blue > 10%).

Figure 4.
Association of epigenetic alterations with specific genetic drivers in JMML. Data reported by Olk-Batz et al. [94] and Poetsch et al. [96]. Colored circles represent the relative frequency of a specific epigenetic alteration in patients with each of the five JMML canonical drivers (yellow > 2%; orange 20–50%; red > 50%).
Figure 4.
Association of epigenetic alterations with specific genetic drivers in JMML. Data reported by Olk-Batz et al. [94] and Poetsch et al. [96]. Colored circles represent the relative frequency of a specific epigenetic alteration in patients with each of the five JMML canonical drivers (yellow > 2%; orange 20–50%; red > 50%).

Table 1.
Recurrent genetic alterations in JMML.
| Pathway | Affected Gene | Alteration | % | References | |
|---|---|---|---|---|---|
| RAS pathway | Tyrosine phosphatases | PTPN11 | GoF-M | 39% | [12,25,37,82] |
| Tyrosine kinases | ALK | GoF-F | <3% | [37] | |
| PDGFRB | GoF-F | <3% | [65,66] | ||
| RAS signaling components | KRAS | GoF-M | 16% | [37,82] | |
| NRAS | GoF-M | 15% | [37,82] | ||
| RRAS | GoF-M | <3% | [29,64,83,84] | ||
| RRAS2 | GoF-M | <3% | [37,64] | ||
| RAS regulators | NF1 | LoF-M | 11% | [37,57,82] | |
| CBL | LoF-M | 11% | [38] | ||
| SOS1 | MS | <3% | [37] | ||
| JAK/STAT pathway | SETBP1 | GoF-M | 30% | [37,73,81,84,85] | |
| JAK3 | GoF-M | 8% | [29,73,74] | ||
| SH2B3 | LoF-M | 7% | [64] | ||
| Hematopoietic commitment transcription factors | RUNX1 | LoF-M | <3% | [64,84] | |
| GATA2 | MS | <3% | [64] | ||
| Spliceosome components | ZRSR2 | LoF-M | <3% | [29,64] | |
| Epigenetic machinery | Histone modifiers | DNMT3A | LoF-M | 3% | [64] |
| PRC2 complex components and associated factors | ASXL1 | LoF-M | 8% | [64,84] | |
| EZH2 | LoF-M | 4% | [64] | ||
| SUZ12 | LOH | <3% | [29] | ||
| CDYL | LOH | <3% | [29] | ||
Abbreviations: GoF-M, gain-of-function mutations; GoF-F, gain-of-function gene fusions; LoF-M, loss-of-function mutations; MS, missense mutations (undetermined effect); LOH, loss of heterozygosity.
Table 2.
Recurrent epigenetic alterations in JMML.
| Affected Gene | Alteration | % | References | |
|---|---|---|---|---|
| Polypeptide from TGF-β superfamily of proteins | BMP4 | Hypermethylation | 36% | [94] |
| Family of G-protein-coupled receptors | CALCA | Hypermethylation | 54% | [94] |
| Cyclin-dependent kinase inhibitor | CDKN2B | Hypermethylation | 22% | [94] |
| Retinoic acid receptor | RARB | Hypermethylation | 13% | [94] |
| RAS Regulator | RASA4 | Hypermethylation | 51% | [96] |
| Histone acetylation | CREBBP | Hypermethylation | 77% | [97] |
| Scaffold protein in signal transduction | AKAP12 | Hypermethylation | 42% | [98] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Fiñana, C.; Gómez-Molina, N.; Alonso-Moreno, S.; Belver, L. Genomic and Epigenomic Landscape of Juvenile Myelomonocytic Leukemia. Cancers 2022, 14, 1335. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14051335
AMA Style
Fiñana C, Gómez-Molina N, Alonso-Moreno S, Belver L. Genomic and Epigenomic Landscape of Juvenile Myelomonocytic Leukemia. Cancers. 2022; 14(5):1335. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14051335
Chicago/Turabian StyleFiñana, Claudia, Noel Gómez-Molina, Sandra Alonso-Moreno, and Laura Belver. 2022. "Genomic and Epigenomic Landscape of Juvenile Myelomonocytic Leukemia" Cancers 14, no. 5: 1335. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14051335
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.