
Dual Effect of Combined Metformin and 2-Deoxy-D-Glucose Treatment on Mitochondrial Biogenesis and PD-L1 Expression in Triple-Negative Breast Cancer Cells

 , , , and
, , , and
Abstract
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
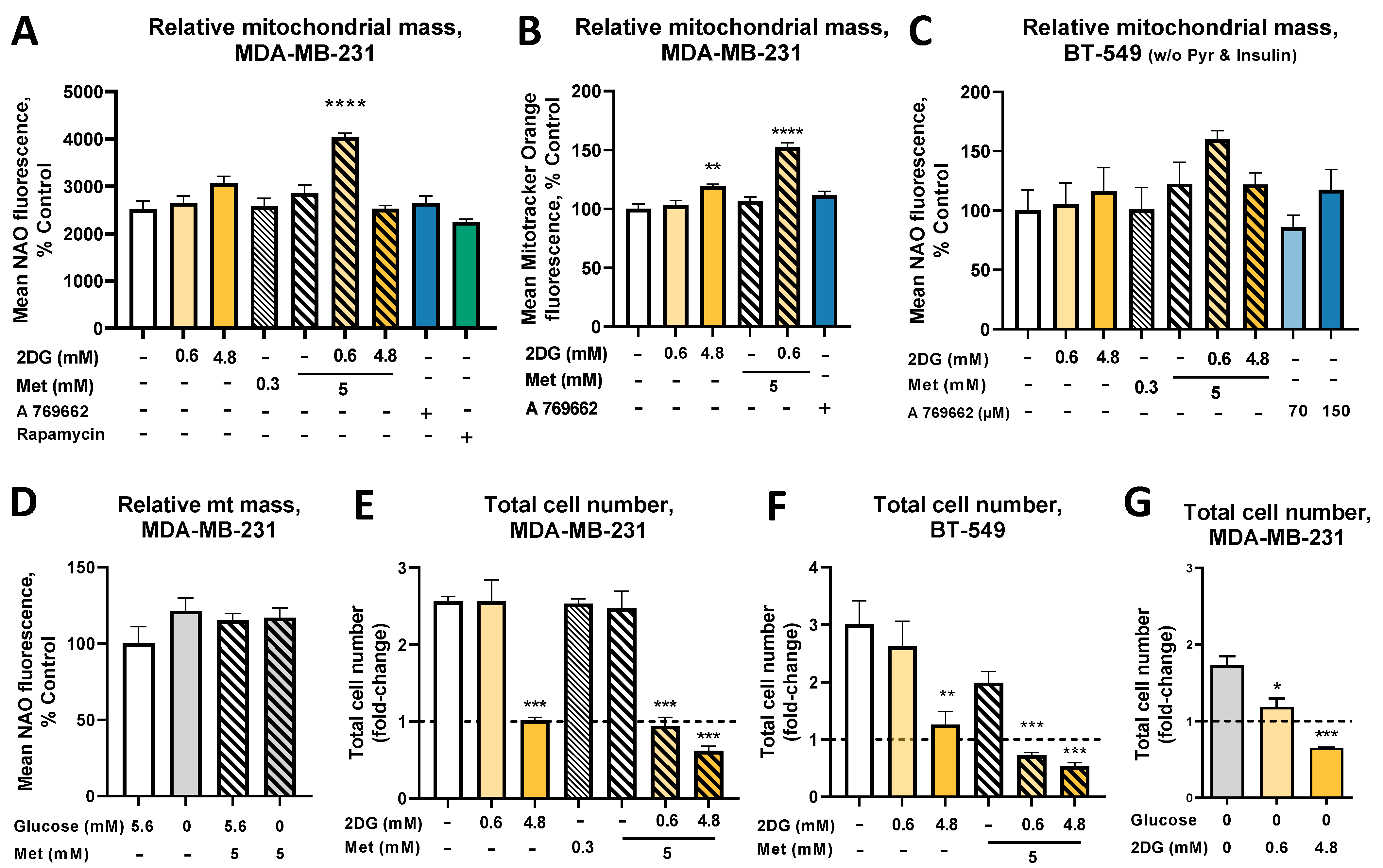
2.1. The Combined Treatment with Metformin and 2DG Increases Mitochondrial Mass in TNBC Cells
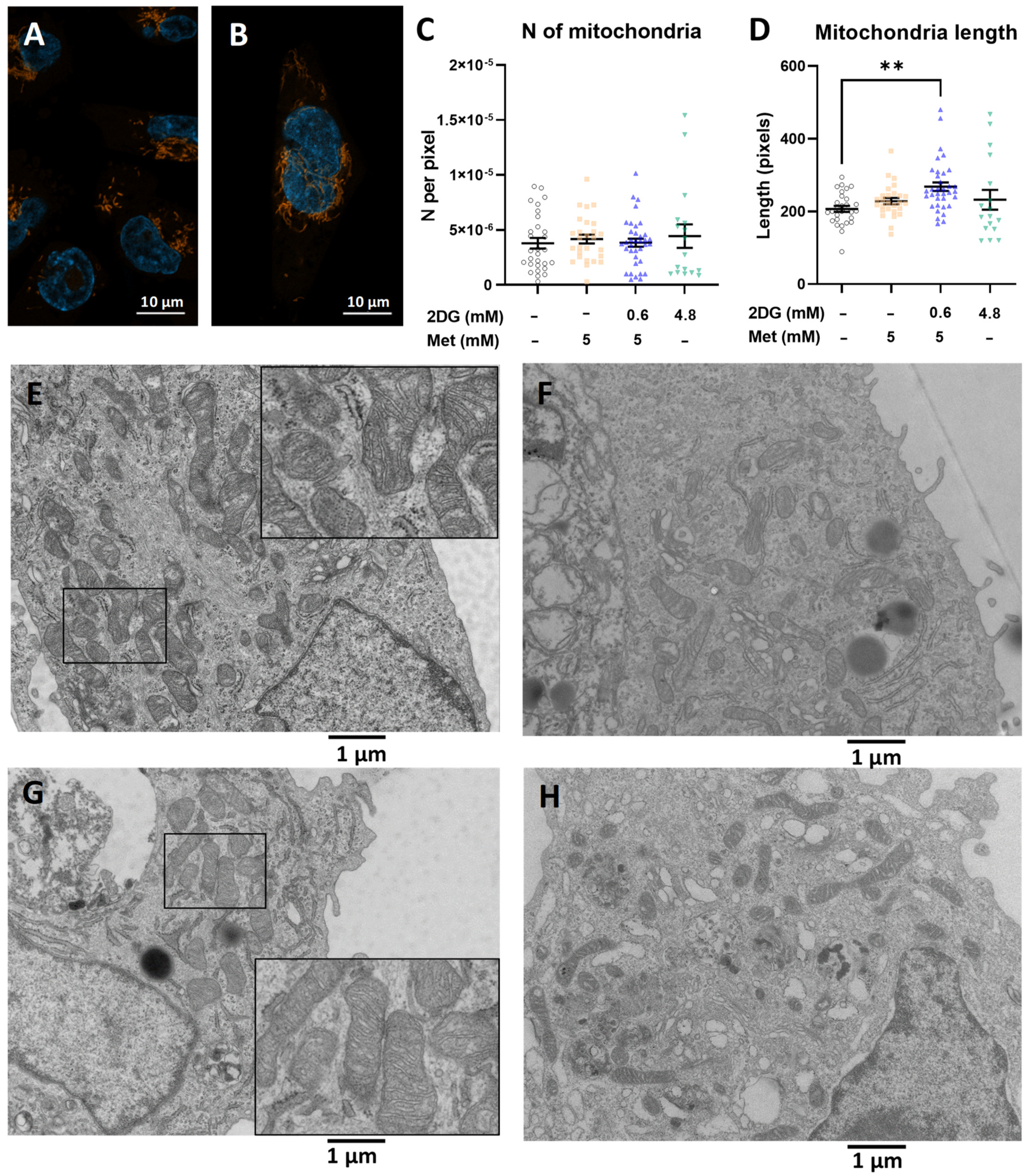
2.2. The Increased Mitochondrial Mass with Metformin and 0.6 mM 2DG Treatment Is Associated with Increased Mitochondrial Size in MDA-MB-231 Cells
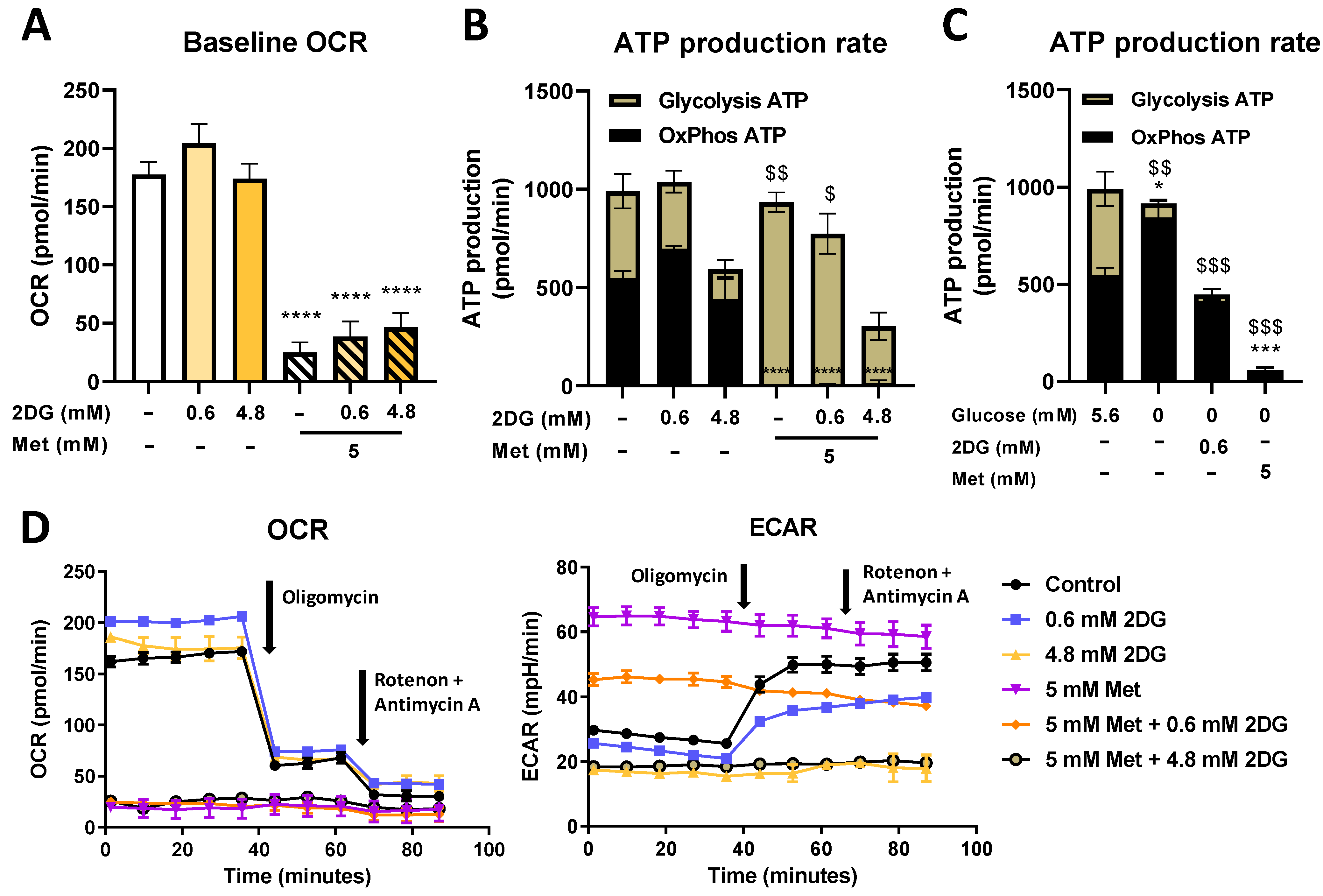
2.3. Seahorse Real Time ATP Production and Mito Stress Assay
2.4. Metformin and 2DG Treated MDA-MB-231 Cells Maintain Their Mitophagy
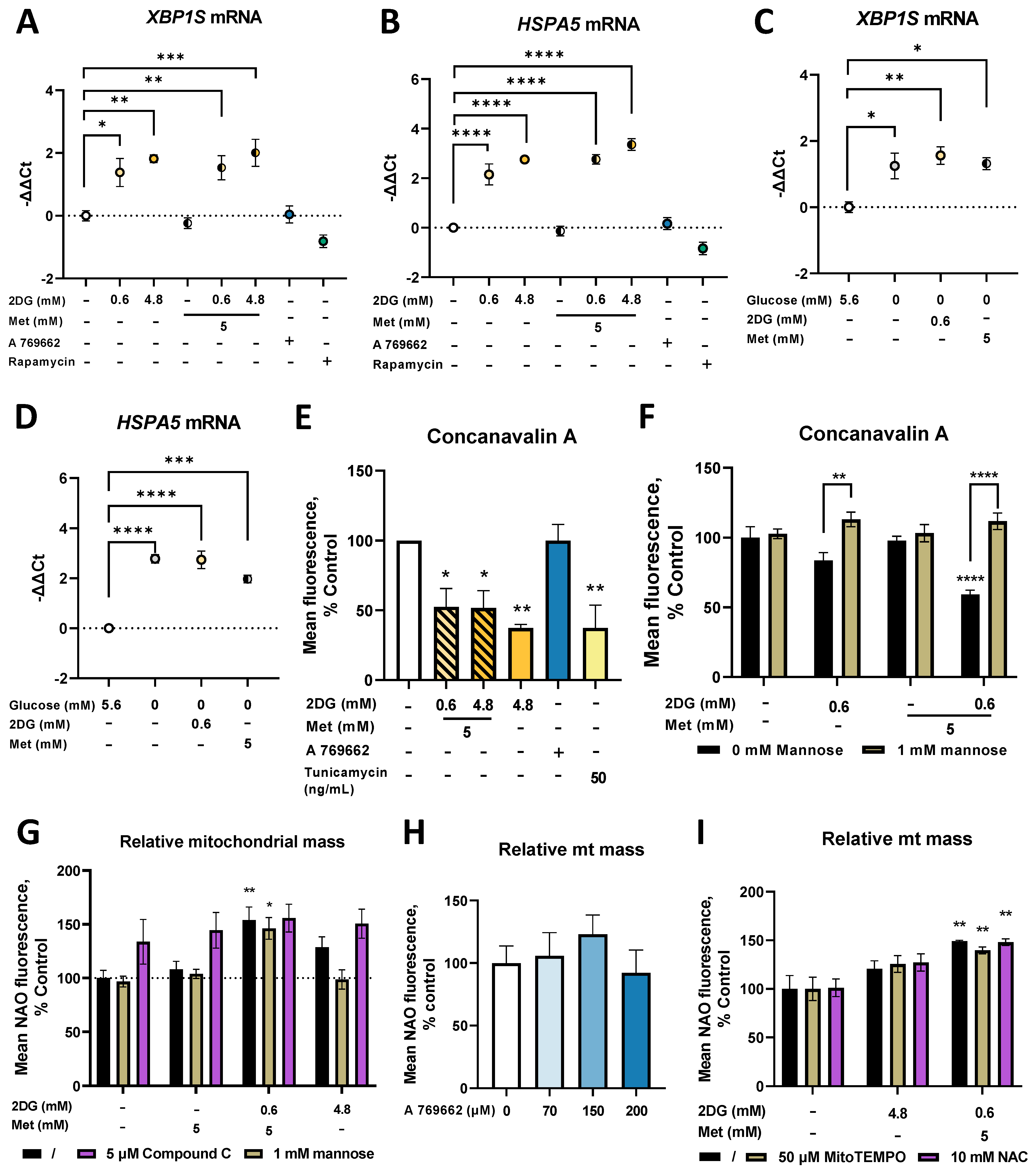
2.5. Combined Metformin + 2-Deoxyglucose Treatment Induces Mitochondrial Biogenesis in MDA-MB-231
2.6. Metformin and 2DG Induce ER Stress by Suppressing N-Glycosylation in MDA-MB-231 Cells
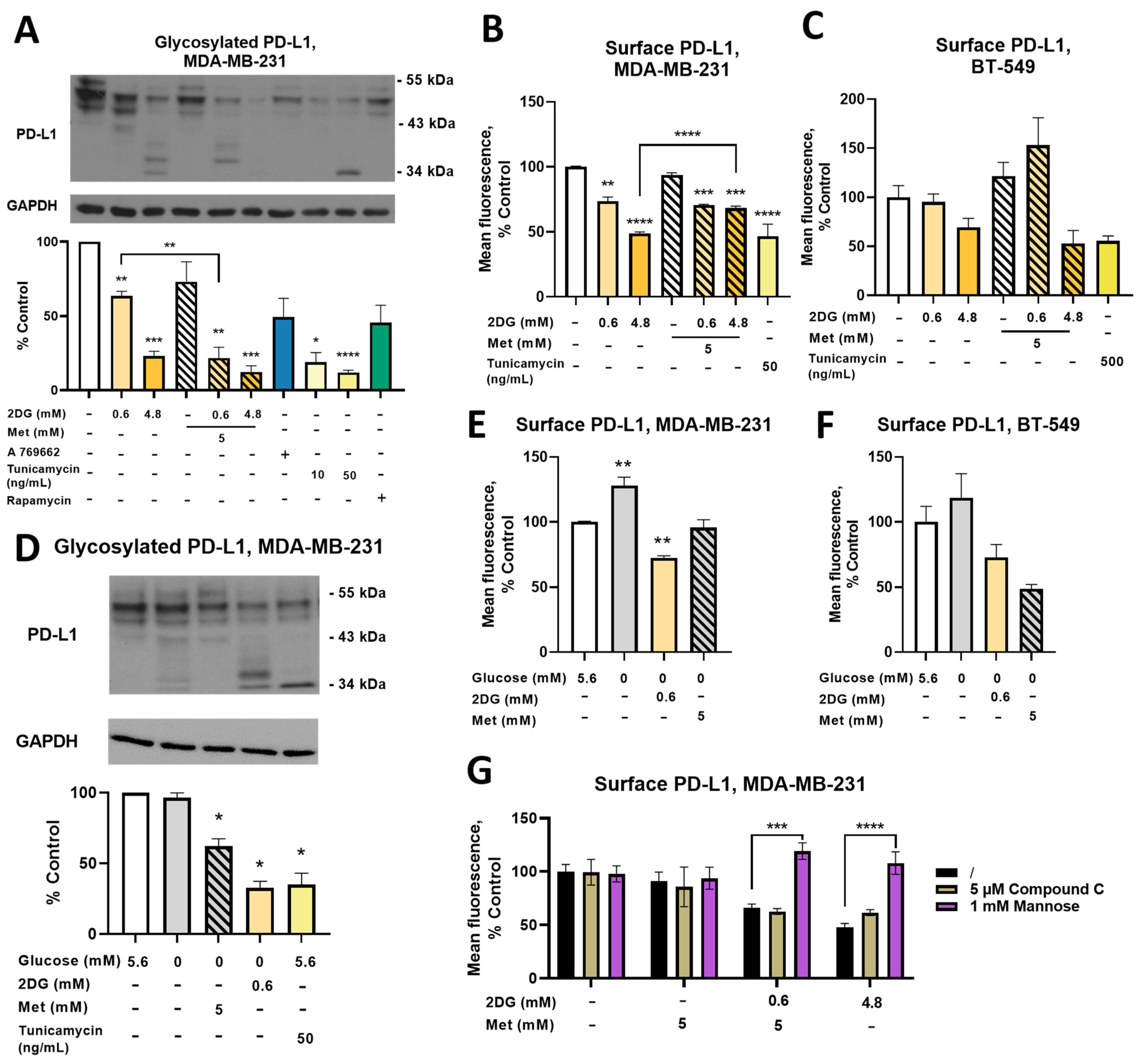
2.7. Combined Treatment with Metformin and 2DG Decreases PD-L1 Expression in MDA-MB-231 Cells
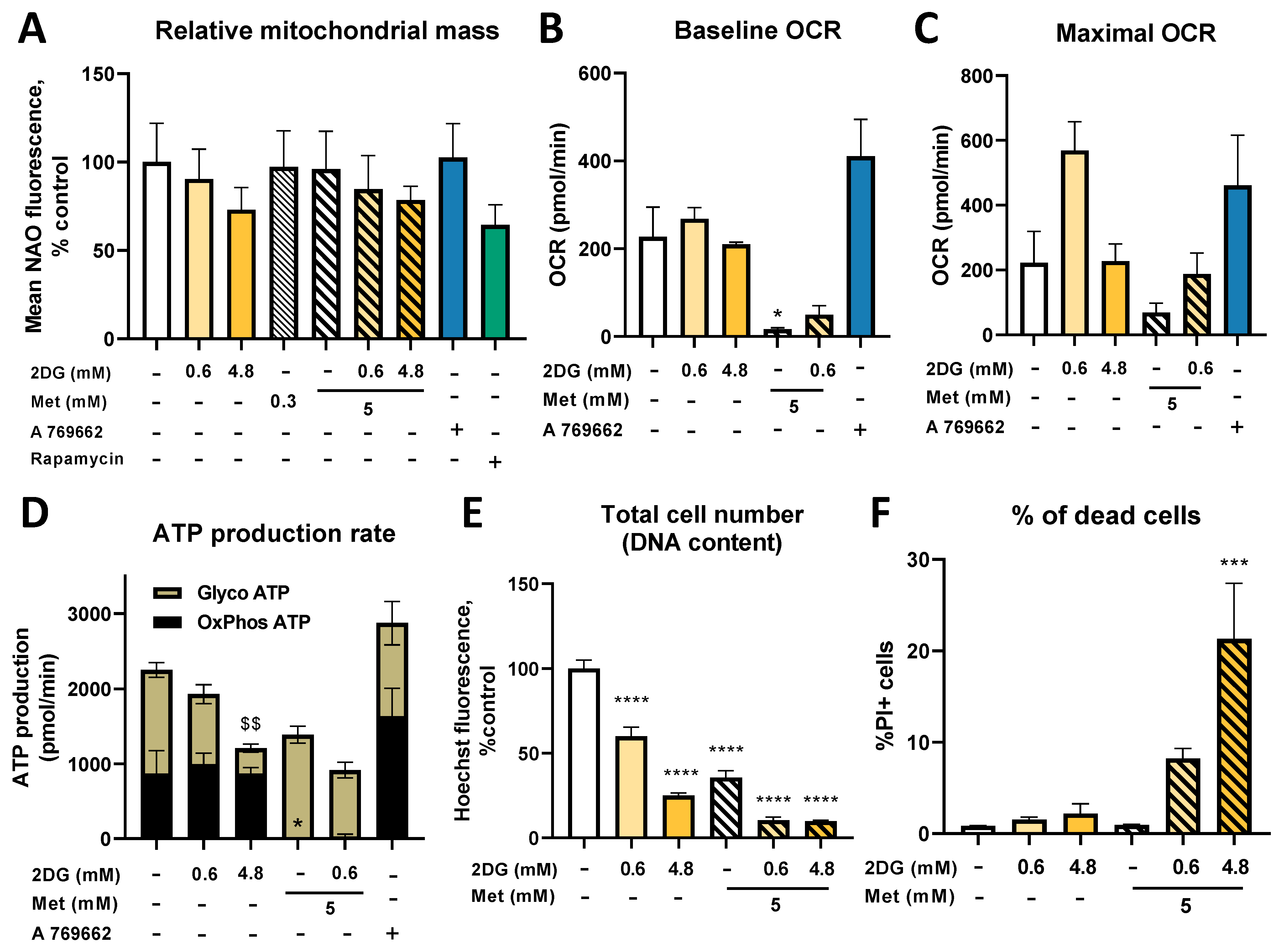
2.8. Metformin and 2DG Do Not Increase Mitochondrial Mass in Jurkat Cells
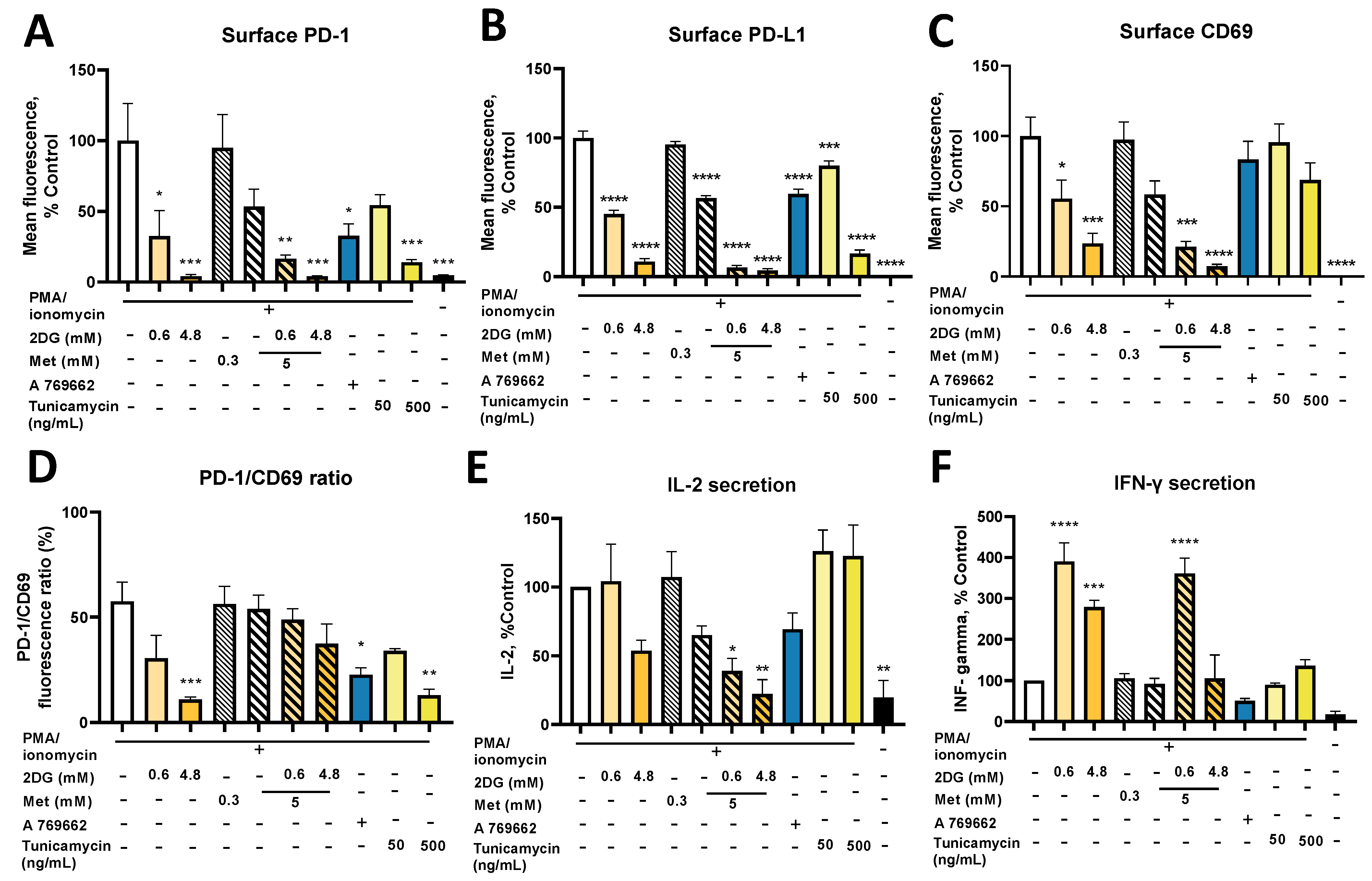
2.9. Combined Treatment with Metformin and 2DG Suppresses PD-L1 and PD-1 Expression in Jurkat Cells
3. Discussion
3.1. Metformin and 2DG Induce Mitochondria Enlargement in TNBC Cells
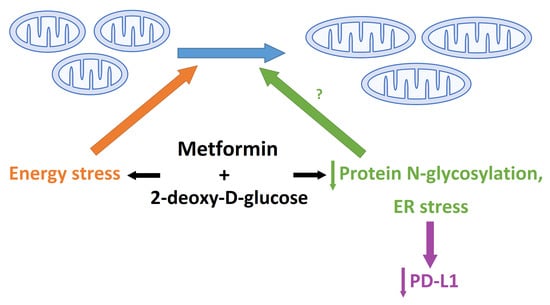
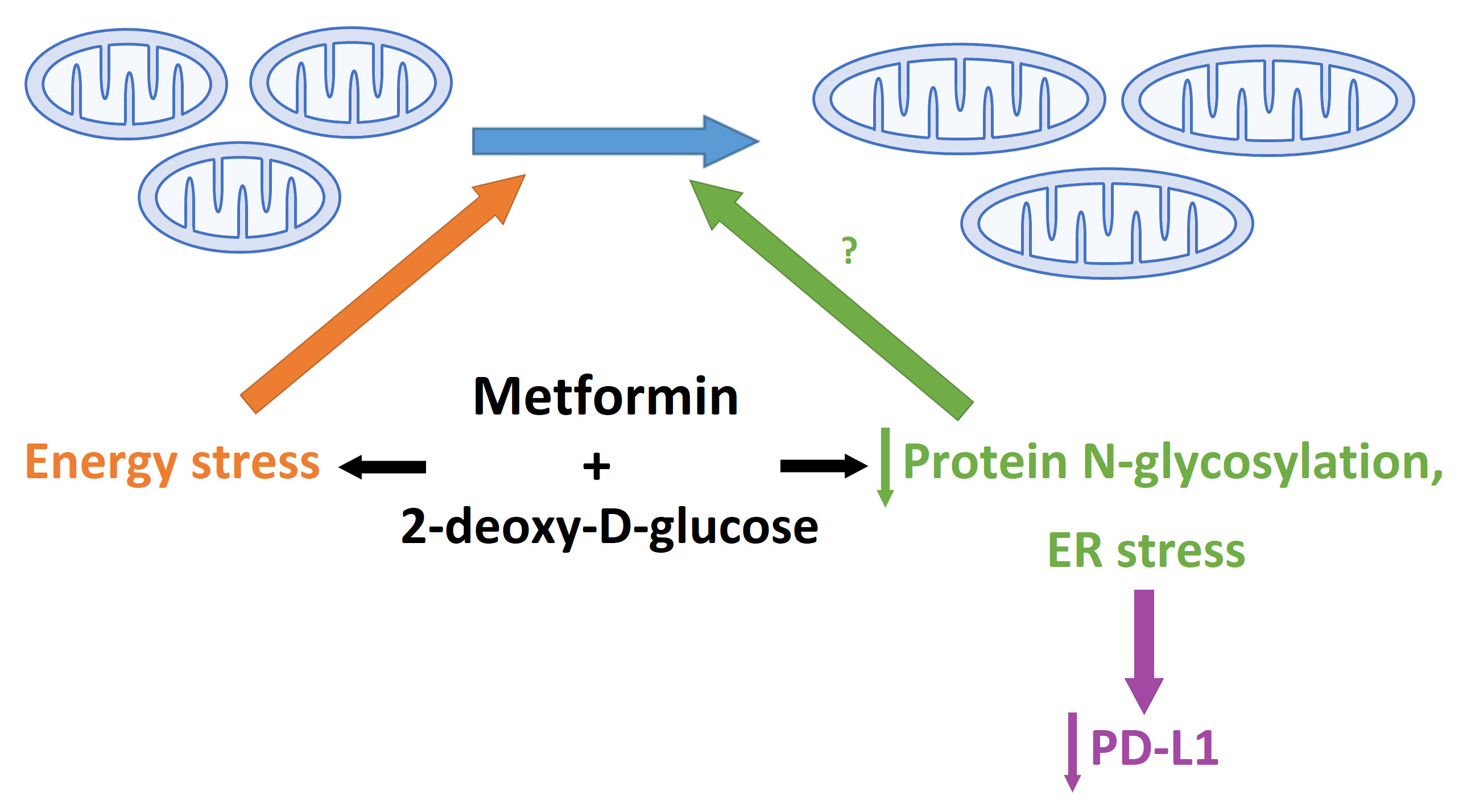
3.2. The Role of Energy Stress and Endoplasmic Reticulum Stress in Inducing Mitochondrial Biogenesis in MDA-MB-231 Cells
3.3. Metformin and 2DG Do Not Increase Mitochondrial Mass in Jurkat Cells
3.4. The Effect of Metformin and 2DG on PD-L1/PD-1 Axis through Suppressed Protein N-Glycosylation
4. Materials and Methods
4.1. Cell Culture and Treatments
4.2. Flow Cytometry
4.3. Determination of Total Cell Number
4.4. Transmission Electron Microscopy
4.5. RNA Quantification by Quantitative Real-Time PCR
4.6. Western Blotting
4.7. Seahorse Real-Time ATP Production Rate Assay and Mito Stress Assay
4.8. ELISA
4.9. Mitochondrial DNA Quantification
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Bost, F.; Decoux-Poullot, A.-G.; Tanti, J.F.; Clavel, S. Energy Disruptors: Rising Stars in Anticancer Therapy? Oncogenesis 2016, 5, e188. [Google Scholar] [CrossRef] [PubMed]
- Bodmer, M.; Meier, C.; Krähenbühl, S.; Jick, S.S.; Meier, C.R. Long-Term Metformin Use Is Associated With Decreased Risk of Breast Cancer. Diabetes Care 2010, 33, 1304–1308. [Google Scholar] [CrossRef]
- Chae, Y.K.; Arya, A.; Malecek, M.-K.; Shin, D.S.; Carneiro, B.; Chandra, S.; Kaplan, J.; Kalyan, A.; Altman, J.K.; Platanias, L.; et al. Repurposing Metformin for Cancer Treatment: Current Clinical Studies. Oncotarget 2016, 7, 40767–40780. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Luo, J.; Yu, T.; Zhou, L.; Lv, H.; Shang, P. Anticancer Mechanisms of Metformin: A Review of the Current Evidence. Life Sci. 2020, 254, 117717. [Google Scholar] [CrossRef] [PubMed]
- Foretz, M.; Guigas, B.; Bertrand, L.; Pollak, M.; Viollet, B. Metformin: From Mechanisms of Action to Therapies. Cell Metab. 2014, 20, 953–966. [Google Scholar] [CrossRef] [PubMed]
- Leclerc, G.M.; Leclerc, G.J.; Kuznetsov, J.N.; DeSalvo, J.; Barredo, J.C. Metformin Induces Apoptosis through AMPK-Dependent Inhibition of UPR Signaling in ALL Lymphoblasts. PLoS ONE 2013, 8, e74420. [Google Scholar] [CrossRef]
- Sahra, I.B.; Regazzetti, C.; Robert, G.; Laurent, K.; Marchand-Brustel, Y.L.; Auberger, P.; Tanti, J.-F.; Giorgetti-Peraldi, S.; Bost, F. Metformin, Independent of AMPK, Induces MTOR Inhibition and Cell-Cycle Arrest through REDD1. Cancer Res. 2011, 71, 4366–4372. [Google Scholar] [CrossRef]
- Yan, M.; Qi, H.; Xia, T.; Zhao, X.; Wang, W.; Wang, Z.; Lu, C.; Ning, Z.; Chen, H.; Li, T.; et al. Metabolomics Profiling of Metformin-Mediated Metabolic Reprogramming Bypassing AMPKα. Metabolism 2019, 91, 18–29. [Google Scholar] [CrossRef]
- Sullivan, L.B.; Gui, D.Y.; Hosios, A.M.; Bush, L.N.; Freinkman, E.; Vander Heiden, M.G. Supporting Aspartate Biosynthesis Is an Essential Function of Respiration in Proliferating Cells. Cell 2015, 162, 552–563. [Google Scholar] [CrossRef]
- Corominas-Faja, B.; Quirantes-Piné, R.; Oliveras-Ferraros, C.; Vazquez-Martin, A.; Cufí, S.; Martin-Castillo, B.; Micol, V.; Joven, J.; Segura-Carretero, A.; Menendez, J.A. Metabolomic Fingerprint Reveals That Metformin Impairs One-Carbon Metabolism in a Manner Similar to the Antifolate Class of Chemotherapy Drugs. Aging 2012, 4, 480–498. [Google Scholar] [CrossRef]
- Kang, S.W.S.; Haydar, G.; Taniane, C.; Farrell, G.; Arias, I.M.; Lippincott-Schwartz, J.; Fu, D. AMPK Activation Prevents and Reverses Drug-Induced Mitochondrial and Hepatocyte Injury by Promoting Mitochondrial Fusion and Function. PLoS ONE 2016, 11, e0165638. [Google Scholar] [CrossRef] [PubMed]
- Marin, T.L.; Gongol, B.; Zhang, F.; Martin, M.; Johnson, D.A.; Xiao, H.; Wang, Y.; Subramaniam, S.; Chien, S.; Shyy, J.Y.-J. AMPK Promotes Mitochondrial Biogenesis and Function by Phosphorylating the Epigenetic Factors DNMT1, RBBP7, and HAT1. Sci. Signal 2017, 10, eaaf7478. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; An, H.; Liu, T.; Qin, C.; Sesaki, H.; Guo, S.; Radovick, S.; Hussain, M.; Maheshwari, A.; Wondisford, F.E.; et al. Metformin Improves Mitochondrial Respiratory Activity through Activation of AMPK. Cell Rep. 2019, 29, 1511–1523.e5. [Google Scholar] [CrossRef] [PubMed]
- Andrzejewski, S.; Klimcakova, E.; Johnson, R.M.; Tabariès, S.; Annis, M.G.; McGuirk, S.; Northey, J.J.; Chénard, V.; Sriram, U.; Papadopoli, D.J.; et al. PGC-1α Promotes Breast Cancer Metastasis and Confers Bioenergetic Flexibility against Metabolic Drugs. Cell Metab. 2017, 26, 778–787.e5. [Google Scholar] [CrossRef]
- Mori, S.; Chang, J.T.; Andrechek, E.R.; Matsumura, N.; Baba, T.; Yao, G.; Kim, J.W.; Gatza, M.; Murphy, S.; Nevins, J.R. Anchorage-Independent Cell Growth Signature Identifies Tumors with Metastatic Potential. Oncogene 2009, 28, 2796–2805. [Google Scholar] [CrossRef] [PubMed]
- Schafer, Z.T.; Grassian, A.R.; Song, L.; Jiang, Z.; Gerhart-Hines, Z.; Irie, H.Y.; Gao, S.; Puigserver, P.; Brugge, J.S. Antioxidant and Oncogene Rescue of Metabolic Defects Caused by Loss of Matrix Attachment. Nature 2009, 461, 109–113. [Google Scholar] [CrossRef]
- Porporato, P.E.; Payen, V.L.; Pérez-Escuredo, J.; De Saedeleer, C.J.; Danhier, P.; Copetti, T.; Dhup, S.; Tardy, M.; Vazeille, T.; Bouzin, C.; et al. A Mitochondrial Switch Promotes Tumor Metastasis. Cell Rep. 2014, 8, 754–766. [Google Scholar] [CrossRef]
- LeBleu, V.S.; O’Connell, J.T.; Gonzalez Herrera, K.N.; Wikman, H.; Pantel, K.; Haigis, M.C.; de Carvalho, F.M.; Damascena, A.; Domingos Chinen, L.T.; Rocha, R.M.; et al. PGC-1α Mediates Mitochondrial Biogenesis and Oxidative Phosphorylation in Cancer Cells to Promote Metastasis. Nat. Cell Biol. 2014, 16, 992–1003. [Google Scholar] [CrossRef]
- Ben Sahra, I.; Tanti, J.-F.; Bost, F. The Combination of Metformin and 2-Deoxyglucose Inhibits Autophagy and Induces AMPK-Dependent Apoptosis in Prostate Cancer Cells. Autophagy 2010, 6, 670–671. [Google Scholar] [CrossRef]
- Cheong, J.-H.; Park, E.S.; Liang, J.; Dennison, J.B.; Tsavachidou, D.; Nguyen-Charles, C.; Wa Cheng, K.; Hall, H.; Zhang, D.; Lu, Y.; et al. Dual Inhibition of Tumor Energy Pathway by 2-Deoxyglucose and Metformin Is Effective against a Broad Spectrum of Preclinical Cancer Models. Mol. Cancer Ther. 2011, 10, 2350–2362. [Google Scholar] [CrossRef]
- Nile, D.L.; Rae, C.; Walker, D.J.; Waddington, J.C.; Vincent, I.; Burgess, K.; Gaze, M.N.; Mairs, R.J.; Chalmers, A.J. Inhibition of Glycolysis and Mitochondrial Respiration Promotes Radiosensitisation of Neuroblastoma and Glioma Cells. Cancer Metab. 2021, 9, 24. [Google Scholar] [CrossRef] [PubMed]
- Mathews Samuel, S.; Satheesh, N.J.; Ghosh, S.; Büsselberg, D.; Majeed, Y.; Ding, H.; Triggle, C.R. Treatment with a Combination of Metformin and 2-Deoxyglucose Upregulates Thrombospondin-1 in Microvascular Endothelial Cells: Implications in Anti-Angiogenic Cancer Therapy. Cancers 2019, 11, 1737. [Google Scholar] [CrossRef] [PubMed]
- Nakada, H.I.; Wick, A.N. The Effect of 2-Deoxyglucose on the Metabolism of Glucose, Fructose, and Galactose by Rat Diaphragm. J. Biol. Chem. 1956, 222, 671–676. [Google Scholar] [CrossRef]
- Ahadova, A.; Gebert, J.; von Knebel Doeberitz, M.; Kopitz, J.; Kloor, M. Dose-Dependent Effect of 2-Deoxy-D-Glucose on Glycoprotein Mannosylation in Cancer Cells. IUBMB Life 2015, 67, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Urakami, K.; Zangiacomi, V.; Yamaguchi, K.; Kusuhara, M. Impact of 2-Deoxy-D-Glucose on the Target Metabolome Profile of a Human Endometrial Cancer Cell Line. Biomed. Res. 2013, 34, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Berthe, A.; Zaffino, M.; Muller, C.; Foulquier, F.; Houdou, M.; Schulz, C.; Bost, F.; De Fay, E.; Mazerbourg, S.; Flament, S. Protein N-Glycosylation Alteration and Glycolysis Inhibition Both Contribute to the Antiproliferative Action of 2-Deoxyglucose in Breast Cancer Cells. Breast Cancer Res. Treat. 2018, 171, 581–591. [Google Scholar] [CrossRef]
- Kurtoglu, M.; Gao, N.; Shang, J.; Maher, J.C.; Lehrman, M.A.; Wangpaichitr, M.; Savaraj, N.; Lane, A.N.; Lampidis, T.J. Under Normoxia, 2-Deoxy-d-Glucose Elicits Cell Death in Select Tumor Types Not by Inhibition of Glycolysis but by Interfering with N-Linked Glycosylation. Mol. Cancer Ther. 2007, 6, 3049–3058. [Google Scholar] [CrossRef]
- Ishino, K.; Kudo, M.; Peng, W.-X.; Kure, S.; Kawahara, K.; Teduka, K.; Kawamoto, Y.; Kitamura, T.; Fujii, T.; Yamamoto, T.; et al. 2-Deoxy-d-Glucose Increases GFAT1 Phosphorylation Resulting in Endoplasmic Reticulum-Related Apoptosis via Disruption of Protein N-Glycosylation in Pancreatic Cancer Cells. Biochem. Biophys. Res. Commun. 2018, 501, 668–673. [Google Scholar] [CrossRef]
- Bizjak, M.; Malavašič, P.; Dolinar, K.; Pohar, J.; Pirkmajer, S.; Pavlin, M. Combined Treatment with Metformin and 2-Deoxy Glucose Induces Detachment of Viable MDA-MB-231 Breast Cancer Cells in Vitro. Sci. Rep. 2017, 7, 1761. [Google Scholar] [CrossRef]
- Jiang, L.; Shestov, A.A.; Swain, P.; Yang, C.; Parker, S.J.; Wang, Q.A.; Terada, L.S.; Adams, N.D.; McCabe, M.T.; Pietrak, B.; et al. Reductive Carboxylation Supports Redox Homeostasis during Anchorage-Independent Growth. Nature 2016, 532, 255–258. [Google Scholar] [CrossRef] [PubMed]
- Luca, A.D.; Fiorillo, M.; Peiris-Pagès, M.; Ozsvari, B.; Smith, D.L.; Sanchez-Alvarez, R.; Martinez-Outschoorn, U.E.; Cappello, A.R.; Pezzi, V.; Lisanti, M.P.; et al. Mitochondrial Biogenesis Is Required for the Anchorage-Independent Survival and Propagation of Stem-like Cancer Cells. Oncotarget 2015, 6, 14777–14795. [Google Scholar] [CrossRef] [PubMed]
- Palikaras, K.; Daskalaki, I.; Markaki, M.; Tavernarakis, N. Mitophagy and Age-Related Pathologies: Development of New Therapeutics by Targeting Mitochondrial Turnover. Pharmacol. Ther. 2017, 178, 157–174. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Alvarez, R.; Martinez-Outschoorn, U.E.; Lamb, R.; Hulit, J.; Howell, A.; Gandara, R.; Sartini, M.; Rubin, E.; Lisanti, M.P.; Sotgia, F. Mitochondrial Dysfunction in Breast Cancer Cells Prevents Tumor Growth: Understanding Chemoprevention with Metformin. Cell Cycle Georget. Tex. 2013, 12, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Xi, H.; Kurtoglu, M.; Liu, H.; Wangpaichitr, M.; You, M.; Liu, X.; Savaraj, N.; Lampidis, T.J. 2-Deoxy-d-Glucose Activates Autophagy via Endoplasmic Reticulum Stress Rather than ATP Depletion. Cancer Chemother. Pharmacol. 2011, 67, 899–910. [Google Scholar] [CrossRef] [PubMed]
- Xi, H.; Barredo, J.C.; Merchan, J.R.; Lampidis, T.J. Endoplasmic Reticulum Stress Induced by 2-Deoxyglucose but Not Glucose Starvation Activates AMPK through CaMKKβ Leading to Autophagy. Biochem. Pharmacol. 2013, 85, 1463–1477. [Google Scholar] [CrossRef]
- Bénéteau, M.; Zunino, B.; Jacquin, M.A.; Meynet, O.; Chiche, J.; Pradelli, L.A.; Marchetti, S.; Cornille, A.; Carles, M.; Ricci, J.-E. Combination of Glycolysis Inhibition with Chemotherapy Results in an Antitumor Immune Response. Proc. Natl. Acad. Sci. USA 2012, 109, 20071–20076. [Google Scholar] [CrossRef]
- Afzal, M.Z.; Mercado, R.R.; Shirai, K. Efficacy of Metformin in Combination with Immune Checkpoint Inhibitors (Anti-PD-1/Anti-CTLA-4) in Metastatic Malignant Melanoma. J. Immunother. Cancer 2018, 6, 64. [Google Scholar] [CrossRef]
- Bahrambeigi, S.; Shafiei-Irannejad, V. Immune-Mediated Anti-Tumor Effects of Metformin; Targeting Metabolic Reprogramming of T Cells as a New Possible Mechanism for Anti-Cancer Effects of Metformin. Biochem. Pharmacol. 2020, 174, 113787. [Google Scholar] [CrossRef]
- Eikawa, S.; Nishida, M.; Mizukami, S.; Yamazaki, C.; Nakayama, E.; Udono, H. Immune-Mediated Antitumor Effect by Type 2 Diabetes Drug, Metformin. Proc. Natl. Acad. Sci. USA 2015, 112, 1809–1814. [Google Scholar] [CrossRef]
- Scharping, N.E.; Menk, A.V.; Whetstone, R.D.; Zeng, X.; Delgoffe, G.M. Efficacy of PD-1 Blockade Is Potentiated by Metformin-Induced Reduction of Tumor Hypoxia. Cancer Immunol. Res. 2017, 5, 9–16. [Google Scholar] [CrossRef]
- Cha, J.-H.; Yang, W.-H.; Xia, W.; Wei, Y.; Chan, L.-C.; Lim, S.-O.; Li, C.-W.; Kim, T.; Chang, S.-S.; Lee, H.-H.; et al. Metformin Promotes Antitumor Immunity via Endoplasmic-Reticulum-Associated Degradation of PD-L1. Mol. Cell 2018, 71, 606–620.e7. [Google Scholar] [CrossRef] [PubMed]
- Shao, B.; Li, C.-W.; Lim, S.-O.; Sun, L.; Lai, Y.-J.; Hou, J.; Liu, C.; Chang, C.-W.; Qiu, Y.; Hsu, J.-M.; et al. Deglycosylation of PD-L1 by 2-Deoxyglucose Reverses PARP Inhibitor-Induced Immunosuppression in Triple-Negative Breast Cancer. Am. J. Cancer Res. 2018, 8, 1837–1846. [Google Scholar] [PubMed]
- Kim, B.; Sun, R.; Oh, W.; Kim, A.M.J.; Schwarz, J.R.; Lim, S.-O. Saccharide Analog, 2-Deoxy-d-Glucose Enhances 4-1BB-Mediated Antitumor Immunity via PD-L1 Deglycosylation. Mol. Carcinog. 2020, 59, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Rajh, M.; Dolinar, K.; Miš, K.; Pavlin, M.; Pirkmajer, S. Medium Renewal Blocks Anti-Proliferative Effects of Metformin in Cultured MDA-MB-231 Breast Cancer Cells. PLoS ONE 2016, 11, e0154747. [Google Scholar] [CrossRef] [PubMed]
- Birsoy, K.; Possemato, R.; Lorbeer, F.K.; Bayraktar, E.C.; Thiru, P.; Yucel, B.; Wang, T.; Chen, W.W.; Clish, C.B.; Sabatini, D.M. Metabolic Determinants of Cancer Cell Sensitivity to Glucose Limitation and Biguanides. Nature 2014, 508, 108–112. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J. Tumor Microenvironment, Metabolism, and Immunotherapy. N. Engl. J. Med. 2020, 382, 869–891. [Google Scholar] [CrossRef]
- Chang, C.-H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; van der Windt, G.J.W.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef]
- Duksin, D.; Bornstein, P. Changes in Surface Properties of Normal and Transformed Cells Caused by Tunicamycin, an Inhibitor of Protein Glycosylation. Proc. Natl. Acad. Sci. USA 1977, 74, 3433–3437. [Google Scholar] [CrossRef]
- Leung, H.J.; Duran, E.M.; Kurtoglu, M.; Andreansky, S.; Lampidis, T.J.; Mesri, E.A. Activation of the Unfolded Protein Response by 2-Deoxy-D-Glucose Inhibits Kaposi’s Sarcoma-Associated Herpesvirus Replication and Gene Expression. Antimicrob. Agents Chemother. 2012, 56, 5794–5803. [Google Scholar] [CrossRef]
- Lee, K.; Tirasophon, W.; Shen, X.; Michalak, M.; Prywes, R.; Okada, T.; Yoshida, H.; Mori, K.; Kaufman, R.J. IRE1-Mediated Unconventional MRNA Splicing and S2P-Mediated ATF6 Cleavage Merge to Regulate XBP1 in Signaling the Unfolded Protein Response. Genes Dev. 2002, 16, 452–466. [Google Scholar] [CrossRef] [PubMed]
- Scharping, N.E.; Menk, A.V.; Moreci, R.S.; Whetstone, R.D.; Dadey, R.E.; Watkins, S.C.; Ferris, R.L.; Delgoffe, G.M. The Tumor Microenvironment Represses T Cell Mitochondrial Biogenesis to Drive Intratumoral T Cell Metabolic Insufficiency and Dysfunction. Immunity 2016, 45, 374–388. [Google Scholar] [CrossRef] [PubMed]
- Chi, H. Regulation and Function of MTOR Signalling in T Cell Fate Decisions. Nat. Rev. Immunol. 2012, 12, 325–338. [Google Scholar] [CrossRef]
- Repas, J.; Zügner, E.; Gole, B.; Bizjak, M.; Potočnik, U.; Magnes, C.; Pavlin, M. Metabolic Profiling of Attached and Detached Metformin and 2-Deoxy-D-Glucose Treated Breast Cancer Cells Reveals Adaptive Changes in Metabolome of Detached Cells. Sci. Rep. 2021, 11, 21354. [Google Scholar] [CrossRef] [PubMed]
- Knupp, J.; Arvan, P.; Chang, A. Increased Mitochondrial Respiration Promotes Survival from Endoplasmic Reticulum Stress. Cell Death Differ. 2019, 26, 487–501. [Google Scholar] [CrossRef] [PubMed]
- Neill, T.; Torres, A.; Buraschi, S.; Owens, R.T.; Hoek, J.B.; Baffa, R.; Iozzo, R.V. Decorin Induces Mitophagy in Breast Carcinoma Cells via Peroxisome Proliferator-Activated Receptor γ Coactivator-1α (PGC-1α) and Mitostatin*. J. Biol. Chem. 2014, 289, 4952–4968. [Google Scholar] [CrossRef] [PubMed]
- Scarpulla, R.C. Transcriptional Paradigms in Mammalian Mitochondrial Biogenesis and Function. Physiol. Rev. 2008, 88, 611–638. [Google Scholar] [CrossRef]
- Kozhukhar, N.; Alexeyev, M.F. Limited Predictive Value of TFAM in Mitochondrial Biogenesis. Mitochondrion 2019, 49, 156–165. [Google Scholar] [CrossRef]
- Twig, G.; Shirihai, O.S. The Interplay Between Mitochondrial Dynamics and Mitophagy. Antioxid. Redox Signal. 2011, 14, 1939–1951. [Google Scholar] [CrossRef]
- Antico Arciuch, V.G.; Elguero, M.E.; Poderoso, J.J.; Carreras, M.C. Mitochondrial Regulation of Cell Cycle and Proliferation. Antioxid. Redox Signal. 2012, 16, 1150–1180. [Google Scholar] [CrossRef]
- Gomes, L.C.; Benedetto, G.D.; Scorrano, L. During Autophagy Mitochondria Elongate, Are Spared from Degradation and Sustain Cell Viability. Nat. Cell Biol. 2011, 13, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, R.; Gilkerson, R.; Aggeler, R.; Yamagata, K.; Remington, S.J.; Capaldi, R.A. Energy Substrate Modulates Mitochondrial Structure and Oxidative Capacity in Cancer Cells. Cancer Res. 2004, 64, 985–993. [Google Scholar] [CrossRef] [PubMed]
- Mitra, K.; Wunder, C.; Roysam, B.; Lin, G.; Lippincott-Schwartz, J. A Hyperfused Mitochondrial State Achieved at G1–S Regulates Cyclin E Buildup and Entry into S Phase. Proc. Natl. Acad. Sci. USA 2009, 106, 11960–11965. [Google Scholar] [CrossRef] [PubMed]
- Rambold, A.S.; Kostelecky, B.; Elia, N.; Lippincott-Schwartz, J. Tubular Network Formation Protects Mitochondria from Autophagosomal Degradation during Nutrient Starvation. Proc. Natl. Acad. Sci. USA 2011, 108, 10190–10195. [Google Scholar] [CrossRef]
- SLP-2 Is Required for Stress-Induced Mitochondrial Hyperfusion. EMBO J. 2009, 28, 1589–1600. [CrossRef]
- Ricciardiello, F.; Votta, G.; Palorini, R.; Raccagni, I.; Brunelli, L.; Paiotta, A.; Tinelli, F.; D’Orazio, G.; Valtorta, S.; De Gioia, L.; et al. Inhibition of the Hexosamine Biosynthetic Pathway by Targeting PGM3 Causes Breast Cancer Growth Arrest and Apoptosis. Cell Death Dis. 2018, 9, 377. [Google Scholar] [CrossRef]
- Wu, J.; Ruas, J.L.; Estall, J.L.; Rasbach, K.A.; Choi, J.H.; Ye, L.; Boström, P.; Tyra, H.M.; Crawford, R.W.; Campbell, K.P.; et al. The Unfolded Protein Response Mediates Adaptation to Exercise in Skeletal Muscle through a PGC-1α/ATF6α Complex. Cell Metab. 2011, 13, 160–169. [Google Scholar] [CrossRef]
- Wu, J.; Chen, S.; Liu, H.; Zhang, Z.; Ni, Z.; Chen, J.; Yang, Z.; Nie, Y.; Fan, D. Tunicamycin Specifically Aggravates ER Stress and Overcomes Chemoresistance in Multidrug-Resistant Gastric Cancer Cells by Inhibiting N-Glycosylation. J. Exp. Clin. Cancer Res. CR 2018, 37, 272. [Google Scholar] [CrossRef]
- Cullinan, S.B.; Zhang, D.; Hannink, M.; Arvisais, E.; Kaufman, R.J.; Diehl, J.A. Nrf2 Is a Direct PERK Substrate and Effector of PERK-Dependent Cell Survival. Mol. Cell. Biol. 2003, 23, 7198–7209. [Google Scholar] [CrossRef]
- Han, C.; Jin, L.; Mei, Y.; Wu, M. Endoplasmic Reticulum Stress Inhibits Cell Cycle Progression via Induction of P27 in Melanoma Cells. Cell. Signal. 2013, 25, 144–149. [Google Scholar] [CrossRef]
- Shiota, T.; Traven, A.; Lithgow, T. Mitochondrial Biogenesis: Cell-Cycle-Dependent Investment in Making Mitochondria. Curr. Biol. 2015, 25, R78–R80. [Google Scholar] [CrossRef] [PubMed]
- Harbauer, A.B.; Opalińska, M.; Gerbeth, C.; Herman, J.S.; Rao, S.; Schönfisch, B.; Guiard, B.; Schmidt, O.; Pfanner, N.; Meisinger, C. Mitochondria. Cell Cycle-Dependent Regulation of Mitochondrial Preprotein Translocase. Science 2014, 346, 1109–1113. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Li, Z.; Lu, Y.; Du, R.; Katiyar, S.; Yang, J.; Fu, M.; Leader, J.E.; Quong, A.; Novikoff, P.M.; et al. Cyclin D1 Repression of Nuclear Respiratory Factor 1 Integrates Nuclear DNA Synthesis and Mitochondrial Function. Proc. Natl. Acad. Sci. USA 2006, 103, 11567–11572. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Y.; Miskimins, W.K. Cell Cycle Arrest in Metformin Treated Breast Cancer Cells Involves Activation of AMPK, Downregulation of Cyclin D1, and Requires P27Kip1 or P21Cip1. J. Mol. Signal. 2008, 3, 18. [Google Scholar] [CrossRef]
- Liu, B.; Fan, Z.; Edgerton, S.M.; Deng, X.-S.; Alimova, I.N.; Lind, S.E.; Thor, A.D. Metformin Induces Unique Biological and Molecular Responses in Triple Negative Breast Cancer Cells. Cell Cycle Georget. Tex. 2009, 8, 2031–2040. [Google Scholar] [CrossRef]
- Li, C.-W.; Lim, S.-O.; Chung, E.M.; Kim, Y.-S.; Park, A.H.; Yao, J.; Cha, J.-H.; Xia, W.; Chan, L.-C.; Kim, T.; et al. Eradication of Triple-Negative Breast Cancer Cells by Targeting Glycosylated PD-L1. Cancer Cell 2018, 33, 187–201.e10. [Google Scholar] [CrossRef]
- Wang, Y.-N.; Lee, H.-H.; Hsu, J.L.; Yu, D.; Hung, M.-C. The Impact of PD-L1 N-Linked Glycosylation on Cancer Therapy and Clinical Diagnosis. J. Biomed. Sci. 2020, 27, 77. [Google Scholar] [CrossRef]
- Yu, Y.; Liang, Y.; Li, D.; Wang, L.; Liang, Z.; Chen, Y.; Ma, G.; Wu, H.; Jiao, W.; Niu, H. Glucose Metabolism Involved in PD-L1-Mediated Immune Escape in the Malignant Kidney Tumour Microenvironment. Cell Death Discov. 2021, 7, 15. [Google Scholar] [CrossRef]
- Li, C.-W.; Lim, S.-O.; Xia, W.; Lee, H.-H.; Chan, L.-C.; Kuo, C.-W.; Khoo, K.-H.; Chang, S.-S.; Cha, J.-H.; Kim, T.; et al. Glycosylation and Stabilization of Programmed Death Ligand-1 Suppresses T-Cell Activity. Nat. Commun. 2016, 7, 12632. [Google Scholar] [CrossRef]
- Schmitz, M.L.; Shaban, M.S.; Albert, B.V.; Gökçen, A.; Kracht, M. The Crosstalk of Endoplasmic Reticulum (ER) Stress Pathways with NF-ΚB: Complex Mechanisms Relevant for Cancer, Inflammation and Infection. Biomedicines 2018, 6, 58. [Google Scholar] [CrossRef]
- Bregar, V.B.; Lojk, J.; Šuštar, V.; Veranič, P.; Pavlin, M. Visualization of Internalization of Functionalized Cobalt Ferrite Nanoparticles and Their Intracellular Fate. Int. J. Nanomed. 2013, 8, 919–931. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.; Kim, K.C.; Lee, D.C.; Lee, H.R.; Shim, J.Y. Association between Salivary Mitochondrial DNA Copy Number and Chronic Fatigue According to Combined Symptoms in Korean Adults. Korean J. Fam. Med. 2017, 38, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-C.; Lee, W.-C.; Liao, S.-C.; Lee, L.-C.; Su, Y.-J.; Lee, C.-T.; Chen, J.-B. Mitochondrial DNA Copy Number Correlates with Oxidative Stress and Predicts Mortality in Nondiabetic Hemodialysis Patients. J. Nephrol. 2011, 24, 351–358. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Repas, J.; Zupin, M.; Vodlan, M.; Veranič, P.; Gole, B.; Potočnik, U.; Pavlin, M. Dual Effect of Combined Metformin and 2-Deoxy-D-Glucose Treatment on Mitochondrial Biogenesis and PD-L1 Expression in Triple-Negative Breast Cancer Cells. Cancers 2022, 14, 1343. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14051343
Repas J, Zupin M, Vodlan M, Veranič P, Gole B, Potočnik U, Pavlin M. Dual Effect of Combined Metformin and 2-Deoxy-D-Glucose Treatment on Mitochondrial Biogenesis and PD-L1 Expression in Triple-Negative Breast Cancer Cells. Cancers. 2022; 14(5):1343. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14051343
Chicago/Turabian StyleRepas, Jernej, Mateja Zupin, Maja Vodlan, Peter Veranič, Boris Gole, Uroš Potočnik, and Mojca Pavlin. 2022. "Dual Effect of Combined Metformin and 2-Deoxy-D-Glucose Treatment on Mitochondrial Biogenesis and PD-L1 Expression in Triple-Negative Breast Cancer Cells" Cancers 14, no. 5: 1343. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14051343