
Targeting the Non-Canonical NF-κB Pathway in Chronic Lymphocytic Leukemia and Multiple Myeloma

 , , and
, , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Culture Conditions for Cell Lines, Primary CLL Cells and Normal Lymphocytes
2.2. Cell Separation and Counting
2.3. CD40L Co-Culture Conditions
2.4. Flow Cytometric Quantification of NF-κB p52
2.5. Reagents
2.6. Measurement of In Vitro Apoptosis
2.7. Measurement of Apoptosis in Normal B- and T-Lymphocytes
2.8. Proliferation and Cell Cycle Analysis
2.9. Enzyme Linked Immuno-Sorbent Assay (ELISA) for NF-κB Subunits
2.10. Transwell Migration Assay
2.11. Synergy between CW15337 and ABT-199 or Fludarabine
2.12. RNA Isolation
2.13. RNA-Sequencing and Analysis
2.14. Reverse Transcription of RNA
2.15. Quantitative Real-Time PCR (QPCR)
- β-Actin primers: Forward sequence, CACCATTGGCAATGAGCGGTTC; reverse sequence, AGGTCTTTGCGGATGTCCACGT.
- BCL2L1 primers: Forward sequence, GCCACTTACCTGAATGACCACC; reverse sequence, AACCAGCGGTTGAAGCGTTCCT.
- BCL2A1 primers: Forward sequence, GGATAAGGCAAAACGGAGGCTG; reverse sequence, CAGTATTGCTTCAGGAGAGATAGC.
- MCL1 primers: Forward sequence, CCAAGAAAGCTGCATCGAACCAT; reverse sequence, CAGCACATTCCTGATGCCACCT.
2.16. Statistical Analysis
3. Results
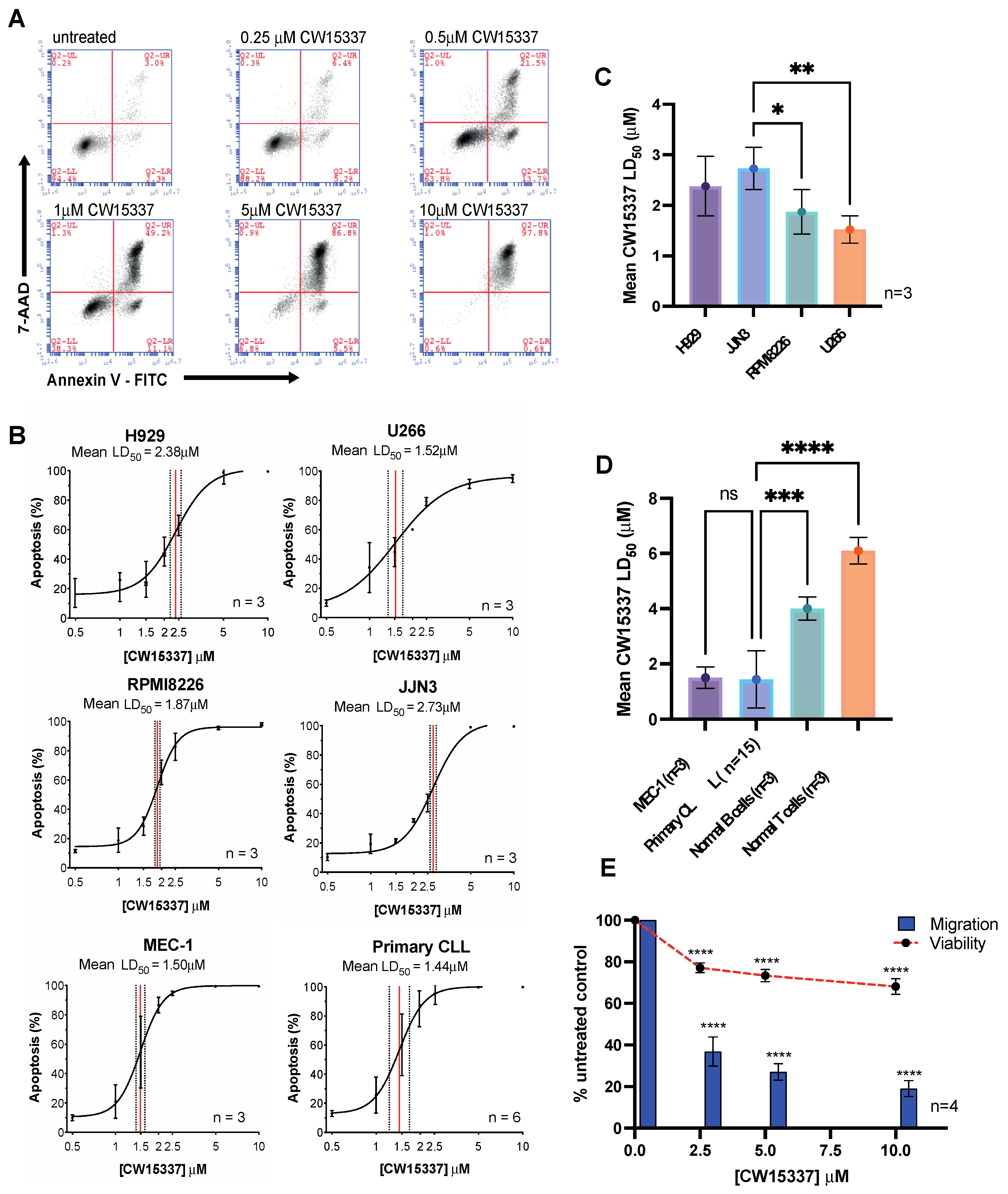
3.1. Evaluation of the Effects If the NIK Inhibitor, CW15337
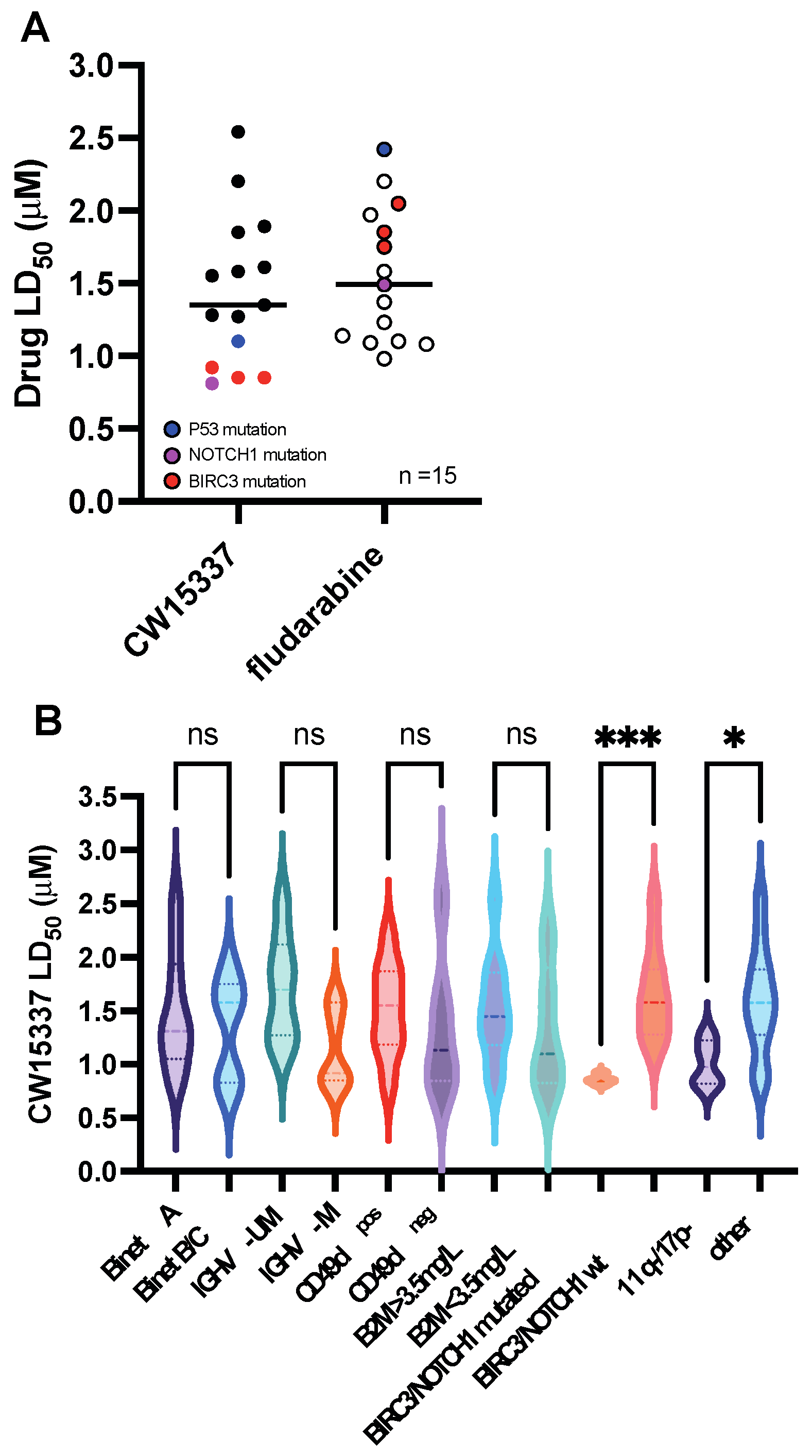
3.2. Comparative Sensitivity to CW15337 in CLL Prognostic Subsets
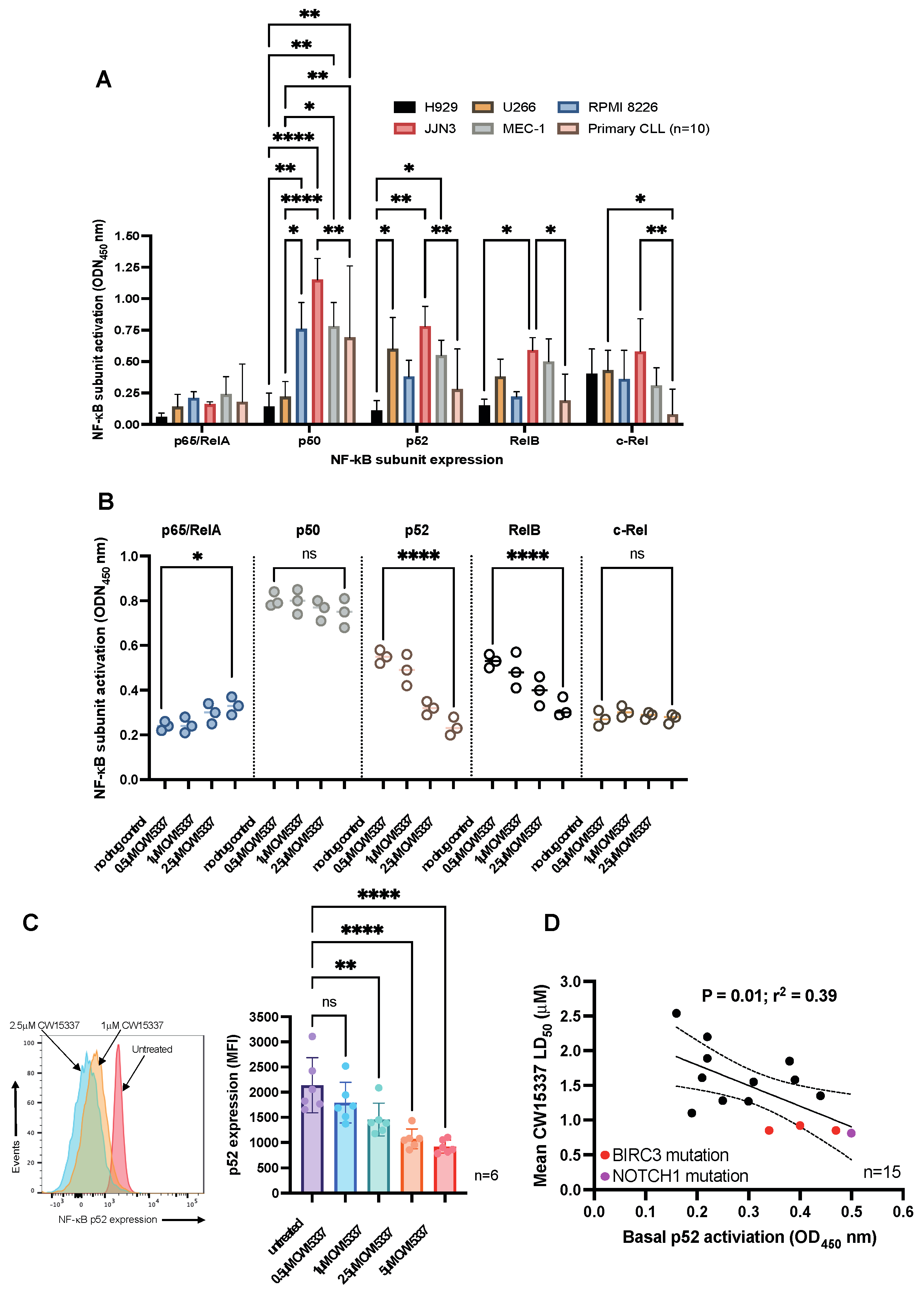
3.3. Basal Nuclear p52 Expression Predicts for an In Vitro Response to CW15337
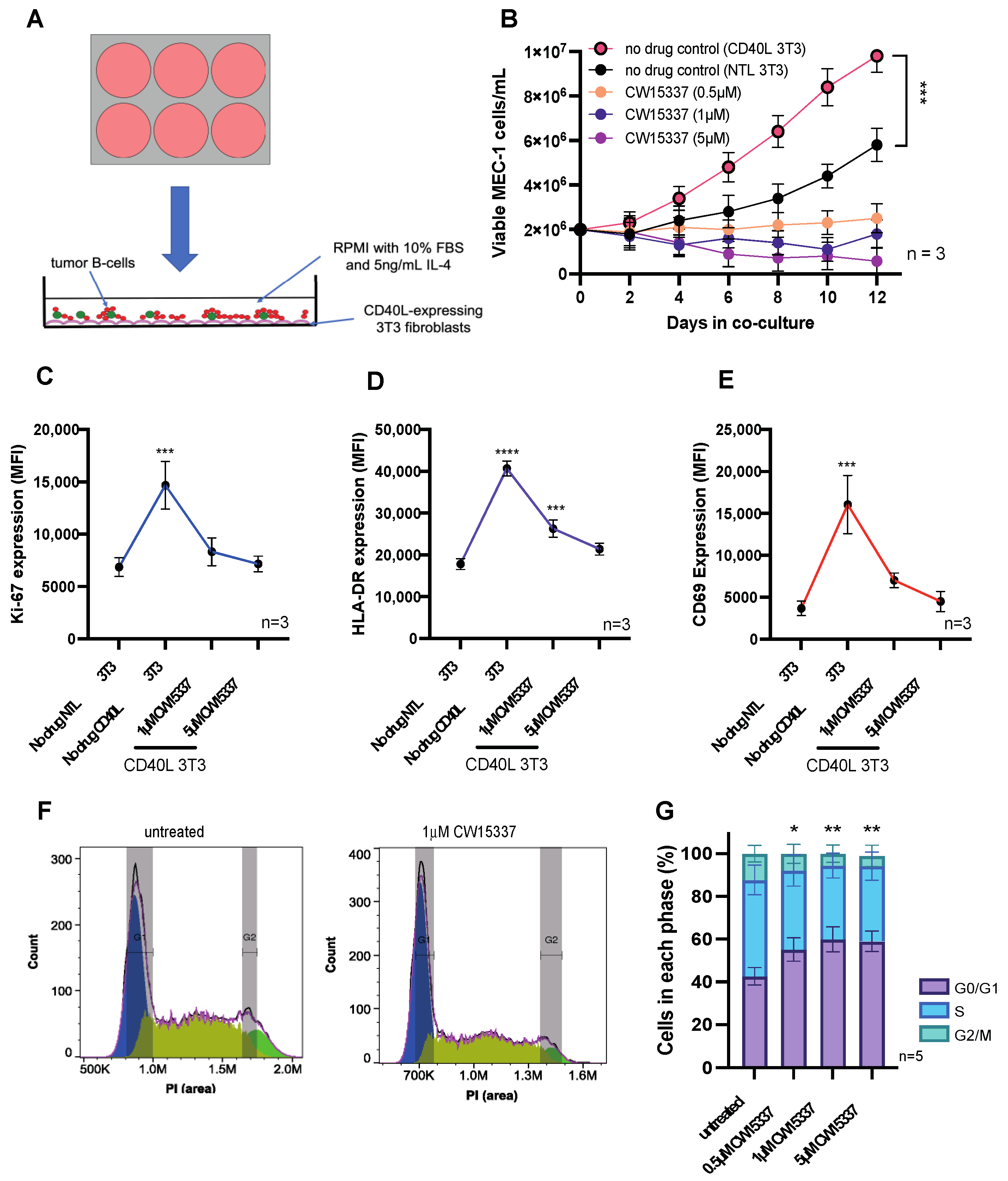
3.4. The Pro-Proliferative Effects of CD40L Co-Culture Are Reversed by NIK Inhibition
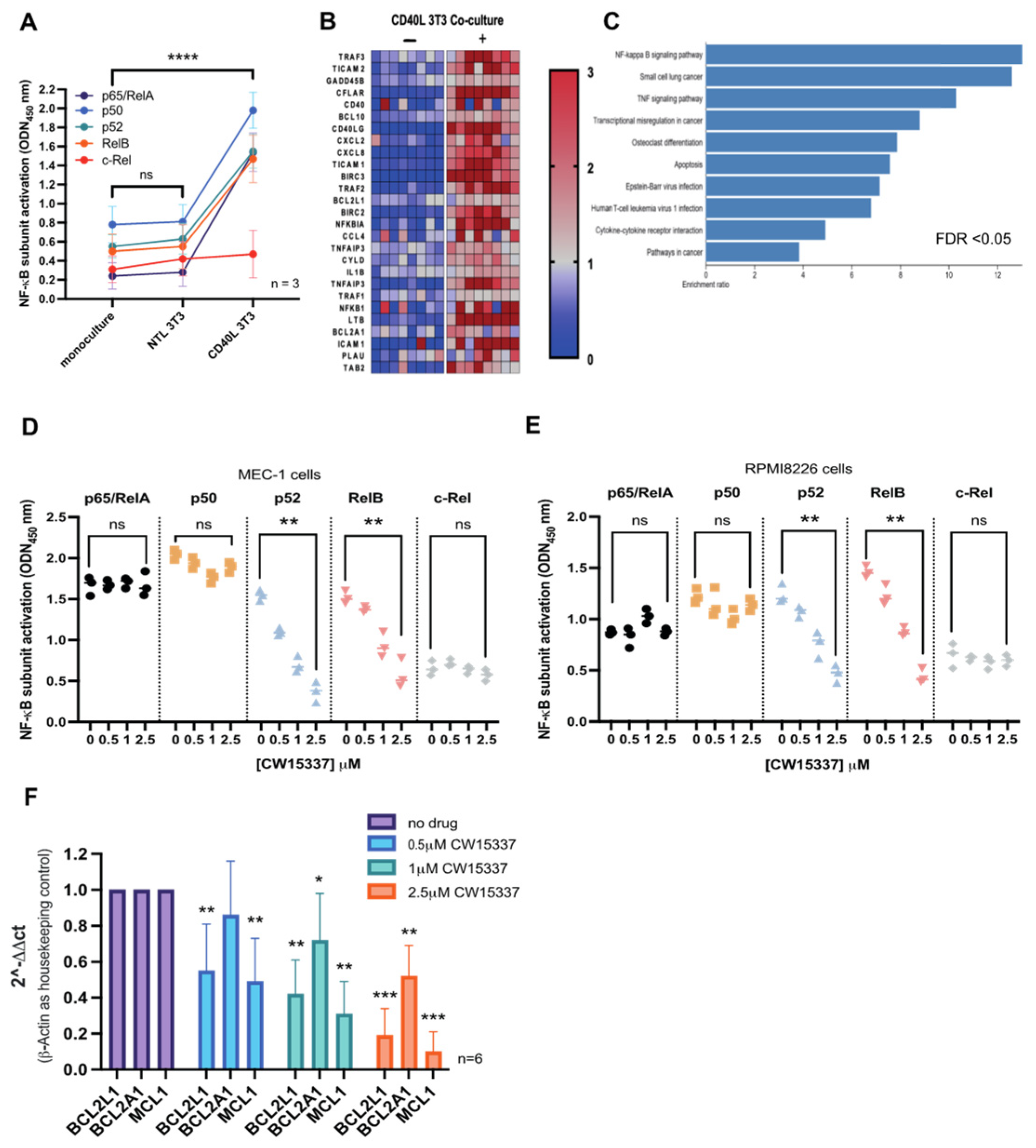
3.5. CD40L 3T3 Co-Culture Induced Both Canonical and Non-Canonical NF-κB Activation
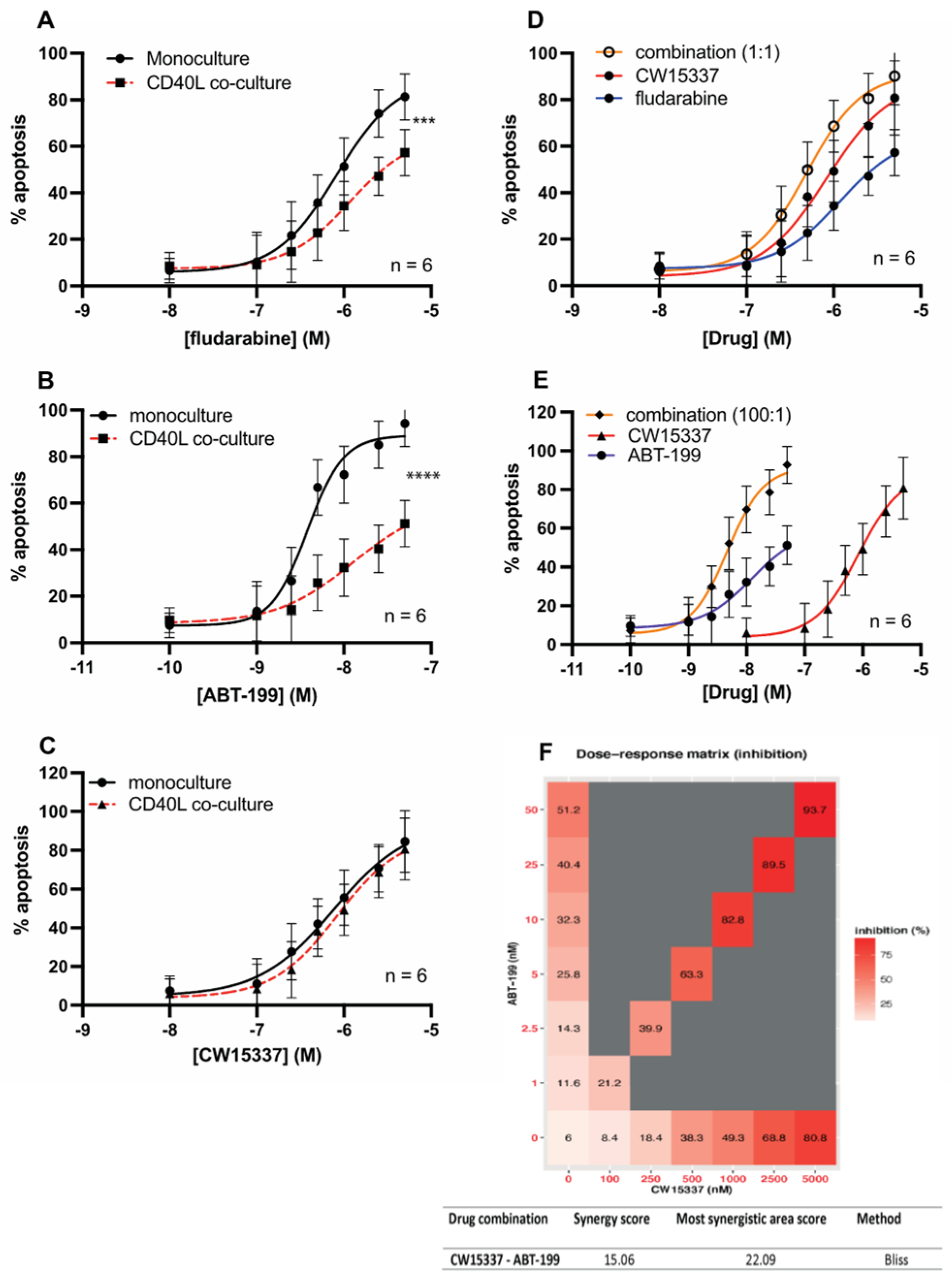
3.6. CW15337 Synergises with ABT-199 and Fludarabine under CD40L Co-Culture Conditions
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cuní, S.; Pérez-Aciego, P.; Pérez-Chacón, G.; Vargas, J.A.; Sánchez, A.; Martín-Saavedra, F.M.; Ballester, S.; García-Marco, J.; Jordá, J.; Durántez, A. A sustained activation of PI3K/NF-kappaB pathway is critical for the survival of chronic lymphocytic leukemia B cells. Leukemia 2004, 18, 1391–1400. [Google Scholar] [CrossRef] [PubMed]
- Hewamana, S.; Alghazal, S.; Lin, T.T.; Clement, M.; Jenkins, C.; Guzman, M.L.; Jordan, C.T.; Neelakantan, S.; Crooks, P.A.; Burnett, A.K.; et al. The NF-kappaB subunit Rel A is associated with in vitro survival and clinical disease progression in chronic lymphocytic leukemia and represents a promising therapeutic target. Blood 2008, 111, 4681–4689. [Google Scholar] [CrossRef] [PubMed]
- Prescott, J.A.; Cook, S.J. Targeting IKKβ in Cancer: Challenges and Opportunities for the Therapeutic Utilisation of IKKβ Inhibitors. Cells 2018, 7, 115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Annunziata, C.M.; Davis, R.E.; Demchenko, Y.; Bellamy, W.; Gabrea, A.; Zhan, F.; Lenz, G.; Hanamura, I.; Wright, G.; Xiao, W.; et al. Frequent engagement of the classical and alternative NF-kappaB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell 2007, 12, 115–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, P.; Sarkar, U.A.; Basak, S. The NF-κB Activating Pathways in Multiple Myeloma. Biomedicines 2018, 6, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puente, X.S.; Beà, S.; Valdés-Mas, R.; Villamor, N.; Gutiérrez-Abril, J.; Martín-Subero, J.I.; Munar, M.; Rubio-Pérez, C.; Jares, P.; Aymerich, M.; et al. Non-coding recurrent mutations in chronic lymphocytic leukaemia. Nature 2015, 526, 519–524. [Google Scholar] [CrossRef] [PubMed]
- Mansouri, L.; Sutton, L.-A.; Ljungström, V.; Bondza, S.; Arngården, L.; Bhoi, S.; Larsson, J.; Cortese, D.; Kalushkova, A.; Plevova, K.; et al. Functional loss of IκBε leads to NF-κB deregulation in aggressive chronic lymphocytic leukemia. J. Exp. Med. 2015, 212, 833–843. [Google Scholar] [CrossRef]
- Fabbri, G.; Rasi, S.; Rossi, D.; Trifonov, V.; Khiabanian, H.; Ma, J.; Grunn, A.; Fangazio, M.; Capello, D.; Monti, S.; et al. Analysis of the chronic lymphocytic leukemia coding genome: Role of NOTCH1 mutational activation. J. Exp. Med. 2011, 208, 1389–1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosati, E.; Sabatini, R.; Rampino, G.; Tabilio, A.; Di Ianni, M.; Fettucciari, K.; Bartoli, A.; Coaccioli, S.; Screpanti, I.; Marconi, P. Constitutively activated Notch signaling is involved in survival and apoptosis resistance of B-CLL cells. Blood 2009, 113, 856–865. [Google Scholar] [CrossRef] [PubMed]
- Baliakas, P.; Hadzidimitriou, A.; Sutton, L.A.; Rossi, D.; Minga, E.; Villamor, N.; Larrayoz, M.; Kminkova, J.; Agathangelidis, A.; Davis, Z.; et al. Recurrent mutations refine prognosis in chronic lymphocytic leukemia. Leukemia 2015, 29, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Chiaretti, S.; Marinelli, M.; Del Giudice, I.; Bonina, S.; Piciocchi, A.; Messina, M.; Vignetti, M.; Rossi, D.; Di Maio, V.; Mauro, F.R.; et al. NOTCH1, SF3B1, BIRC3 and TP53 mutations in patients with chronic lymphocytic leukemia undergoing first-line treatment: Correlation with biological parameters and response to treatment. Leuk. Lymphoma 2014, 55, 2785–2792. [Google Scholar] [CrossRef] [PubMed]
- Dejardin, E. The alternative NF-kappaB pathway from biochemistry to biology: Pitfalls and promises for future drug development. Biochem. Pharmacol. 2006, 72, 1161–1179. [Google Scholar] [CrossRef] [PubMed]
- Asslaber, D.; Wacht, N.; Leisch, M.; Qi, Y.; Maeding, N.; Hufnagl, C.; Jansko, B.; Zaborsky, N.; Villunger, A.; Hartmann, T.N.; et al. BIRC3 Expression Predicts CLL Progression and Defines Treatment Sensitivity via Enhanced NF-κB Nuclear Translocation. Clin. Cancer Res. 2019, 25, 1901–1912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puente, X.S.; Pinyol, M.; Quesada, V.; Conde, L.; Ordóñez, G.R.; Villamor, N.; Escaramis, G.; Jares, P.; Beà, S.; González-Díaz, M.; et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature 2011, 475, 101–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pascutti, M.F.; Jak, M.; Tromp, J.M.; Derks, I.A.M.; Remmerswaal, E.B.M.; Thijssen, R.; Van Attekum, M.H.A.; Van Bochove, G.G.; Luijks, D.M.; Pals, S.T.; et al. IL-21 and CD40L signals from autologous T cells can induce antigen-independent proliferation of CLL cells. Blood 2013, 122, 3010–3019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keats, J.; Fonseca, R.; Chesi, M.; Schop, R.; Baker, A.; Chng, W.-J.; Van Wier, S.; Tiedemann, R.; Shi, C.-X.; Sebag, M.; et al. Promiscuous mutations activate the noncanonical NF-kappaB pathway in multiple myeloma. Cancer Cell 2007, 12, 131–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herishanu, Y.; Pérez-Galán, P.; Liu, D.; Biancotto, A.; Pittaluga, S.; Vire, B.; Gibellini, F.; Njuguna, N.; Lee, E.; Stennett, L.; et al. The lymph node microenvironment promotes B-cell receptor signaling, NF-kappaB activation, and tumor proliferation in chronic lymphocytic leukemia. Blood 2011, 117, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Rosén, A.; Murray, F.; Evaldsson, C.; Rosenquist, R. Antigens in chronic lymphocytic leukemia--implications for cell origin and leukemogenesis. Semin. Cancer Biol. 2010, 20, 400–409. [Google Scholar] [CrossRef] [PubMed]
- Jayappa, K.D.; Portell, C.A.; Gordon, V.L.; Capaldo, B.J.; Bekiranov, S.; Axelrod, M.J.; Brett, L.K.; Wulfkuhle, J.D.; Gallagher, R.I.; Petricoin, E.F.; et al. Microenvironmental agonists generate de novo phenotypic resistance to combined ibrutinib plus venetoclax in CLL and MCL. Blood Adv. 2017, 1, 933–946. [Google Scholar] [CrossRef] [Green Version]
- Tsubaki, M.; Seki, S.; Takeda, T.; Chihara, A.; Arai, Y.; Morii, Y.; Imano, M.; Satou, T.; Shimomura, K.; Nishida, S. The HGF/Met/NF-κB Pathway Regulates RANKL Expression in Osteoblasts and Bone Marrow Stromal Cells. Int. J. Mol. Sci. 2020, 21, 7905. [Google Scholar] [CrossRef]
- Markovina, S.; Callander, N.S.; O’Connor, S.L.; Xu, G.; Shi, Y.; Leith, C.P.; Kim, K.; Trivedi, P.; Kim, J.; Hematti, P.; et al. Bone marrow stromal cells from multiple myeloma patients uniquely induce bortezomib resistant NF-kappaB activity in myeloma cells. Mol. Cancer 2010, 9, 176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roux, S.; Mariette, X. The high rate of bone resorption in multiple myeloma is due to RANK (receptor activator of nuclear factor-kappaB) and RANK Ligand expression. Leuk. Lymphoma 2004, 45, 1111–1118. [Google Scholar] [CrossRef] [PubMed]
- Kawano, Y.; Moschetta, M.; Manier, S.; Glavey, S.; Görgün, G.T.; Roccaro, A.M.; Anderson, K.C.; Ghobrial, I.M. Targeting the bone marrow microenvironment in multiple myeloma. Immunol. Rev. 2015, 263, 160–172. [Google Scholar] [CrossRef] [PubMed]
- Podar, K.; Chauhan, D.; Anderson, K.C. Bone marrow microenvironment and the identification of new targets for myeloma therapy. Leukemia 2009, 23, 10–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haselager, M.; Thijssen, R.; West, C.; Young, L.; Van Kampen, R.; Willmore, E.; Mackay, S.; Kater, A.; Eldering, E. Regulation of Bcl-XL by non-canonical NF-κB in the context of CD40-induced drug resistance in CLL. Cell Death Differ. 2021, 28, 1658–1668. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, E.; Pearce, L.; Morgan, L.; Robinson, S.; Ware, V.L.; Brennan, P.; Thomas, N.S.B.; Yallop, D.; Devereux, S.; Fegan, C.; et al. Mimicking the tumour microenvironment: Three different co-culture systems induce a similar phenotype but distinct proliferative signals in primary chronic lymphocytic leukaemia cells. Br. J. Haematol. 2012, 158, 589–599. [Google Scholar] [CrossRef] [PubMed]
- Ianevski, A.; Giri, A.K.; Aittokallio, T. SynergyFinder 2.0: Visual analytics of multi-drug combination synergies. Nucleic Acids Res. 2020, 48, W488–W493. [Google Scholar] [CrossRef] [PubMed]
- Qing, G.; Qu, Z.; Xiao, G. Stabilization of basally translated NF-kappaB-inducing kinase (NIK) protein functions as a molecular switch of processing of NF-kappaB2 p100. J. Biol. Chem. 2005, 280, 40578–40582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furman, R.R.; Asgary, Z.; Mascarenhas, J.O.; Liou, H.C.; Schattner, E.J. Modulation of NF-kappa B activity and apoptosis in chronic lymphocytic leukemia B cells. J. Immunol. 2000, 164, 2200–2206. [Google Scholar] [CrossRef] [PubMed]
- Diop, F.; Moia, R.; Favini, C.; Spaccarotella, E.; De Paoli, L.; Bruscaggin, A.; Spina, V.; Terzi-Di-Bergamo, L.; Arruga, F.; Tarantelli, C.; et al. Biological and clinical implications of BIRC3 mutations in chronic lymphocytic leukemia. Haematologica 2020, 105, 448–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogler, M.; Butterworth, M.; Majid, A.; Walewska, R.J.; Sun, X.M.; Dyer, M.J.; Cohen, G.M. Concurrent up-regulation of BCL-XL and BCL2A1 induces approximately 1000-fold resistance to ABT-737 in chronic lymphocytic leukemia. Blood 2009, 113, 4403–4413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tromp, J.M.; Geest, C.R.; Breij, E.C.; Elias, J.A.; van Laar, J.; Luijks, D.M.; Kater, A.P.; Beaumont, T.; van Oers, M.H.; Eldering, E. Tipping the Noxa/Mcl-1 balance overcomes ABT-737 resistance in chronic lymphocytic leukemia. Clin. Cancer Res. 2012, 18, 487–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, T.; Corcoran, D.B.; Thurston, D.E.; Giles, P.J.; Ashelford, K.; Walsby, E.J.; Fegan, C.D.; Pepper, A.G.S.; Miraz Rahman, K.; Pepper, C. Novel pyrrolobenzodiazepine benzofused hybrid molecules inhibit NF-κB activity and synergise with bortezomib and ibrutinib in hematological cancers. Haematologica 2021, 106, 958–967. [Google Scholar] [CrossRef] [PubMed]
- Kitada, S.; Zapata, J.M.; Andreeff, M.; Reed, J.C. Bryostatin and CD40-ligand enhance apoptosis resistance and induce expression of cell survival genes in B-cell chronic lymphocytic leukaemia. Br. J. Haematol. 1999, 106, 995–1004. [Google Scholar] [CrossRef] [PubMed]
- Godwin, P.; Baird, A.M.; Heavey, S.; Barr, M.P.; O’Byrne, K.J.; Gately, K. Targeting nuclear factor-kappa B to overcome resistance to chemotherapy. Front. Oncol. 2013, 3, 120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hewamana, S.; Lin, T.T.; Rowntree, C.; Karunanithi, K.; Pratt, G.; Hills, R.; Fegan, C.; Brennan, P.; Pepper, C. Rel A is an independent biomarker of clinical outcome in chronic lymphocytic leukemia. J. Clin. Oncol. 2009, 27, 763–769. [Google Scholar] [CrossRef] [PubMed]
- Baumann, P.; Mandl-Weber, S.; Oduncu, F.; Schmidmaier, R. Alkylating agents induce activation of NFkappaB in multiple myeloma cells. Leuk. Res. 2008, 32, 1144–1147. [Google Scholar] [CrossRef] [PubMed]
- Murray, M.Y.; Auger, M.J.; Bowles, K.M. Overcoming bortezomib resistance in multiple myeloma. Biochem. Soc. Trans. 2014, 42, 804–808. [Google Scholar] [CrossRef] [PubMed]
- Haselager, M.V.; Kielbassa, K.; Ter Burg, J.; Bax, D.J.C.; Fernandes, S.M.; Borst, J.; Tam, C.; Forconi, F.; Chiodin, G.; Brown, J.R.; et al. Changes in Bcl-2 members after ibrutinib or venetoclax uncover functional hierarchy in determining resistance to venetoclax in CLL. Blood 2020, 136, 2918–2926. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Wan, J.; Zhang, W.; Hao, S. MCL-1 or BCL-xL-dependent resistance to the BCL-2 antagonist (ABT-199) can be overcome by specific inhibitor as single agents and in combination with ABT-199 in acute myeloid leukemia cells. Leuk. Lymphoma 2019, 60, 2170–2180. [Google Scholar] [CrossRef]
- Choudhary, G.S.; Al-Harbi, S.; Mazumder, S.; Hill, B.T.; Smith, M.R.; Bodo, J.; Hsi, E.D.; Almasan, A. MCL-1 and BCL-xL-dependent resistance to the BCL-2 inhibitor ABT-199 can be overcome by preventing PI3K/AKT/mTOR activation in lymphoid malignancies. Cell Death Dis. 2015, 6, e1593. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Number |
|---|---|
| Number of CLL cases | 15 |
| Median age at sample collection (range) | 64 |
| (48–80) | |
| Gender | |
| Male | 9 |
| Female | 6 |
| Binet stage at diagnosis | |
| A | 5 |
| B | 4 |
| C | 6 |
| IGHV-mutated | 7 |
| IGHV-unmutated | 8 |
| CD49neg (<30%) | 6 |
| CD49pos (330%) | 9 |
| CD38neg (<20%) | 7 |
| CD38pos (320%) | 8 |
| B2M (<3.5mg/L) | 5 |
| B2M (33.5 mg/L) | 10 |
| Chromosomal aberrations | 3 |
| 11q- | 1 |
| 17p- | 9 |
| 13q-Trisomy 12 | 1 |
| Genetic mutations | |
| TP53 mutation | 1 |
| BIRC3 mutation | 3 |
| NOTCH1 mutation | 1 |
| SF3B1 mutation | 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burley, T.A.; Kennedy, E.; Broad, G.; Boyd, M.; Li, D.; Woo, T.; West, C.; Ladikou, E.E.; Ashworth, I.; Fegan, C.; et al. Targeting the Non-Canonical NF-κB Pathway in Chronic Lymphocytic Leukemia and Multiple Myeloma. Cancers 2022, 14, 1489. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14061489
Burley TA, Kennedy E, Broad G, Boyd M, Li D, Woo T, West C, Ladikou EE, Ashworth I, Fegan C, et al. Targeting the Non-Canonical NF-κB Pathway in Chronic Lymphocytic Leukemia and Multiple Myeloma. Cancers. 2022; 14(6):1489. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14061489
Chicago/Turabian StyleBurley, Thomas A., Emma Kennedy, Georgia Broad, Melanie Boyd, David Li, Timothy Woo, Christopher West, Eleni E. Ladikou, Iona Ashworth, Christopher Fegan, and et al. 2022. "Targeting the Non-Canonical NF-κB Pathway in Chronic Lymphocytic Leukemia and Multiple Myeloma" Cancers 14, no. 6: 1489. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14061489