
Elucidation of Focal Adhesion Kinase as a Modulator of Migration and Invasion and as a Potential Therapeutic Target in Chronic Lymphocytic Leukemia

 , , , and
, , , and 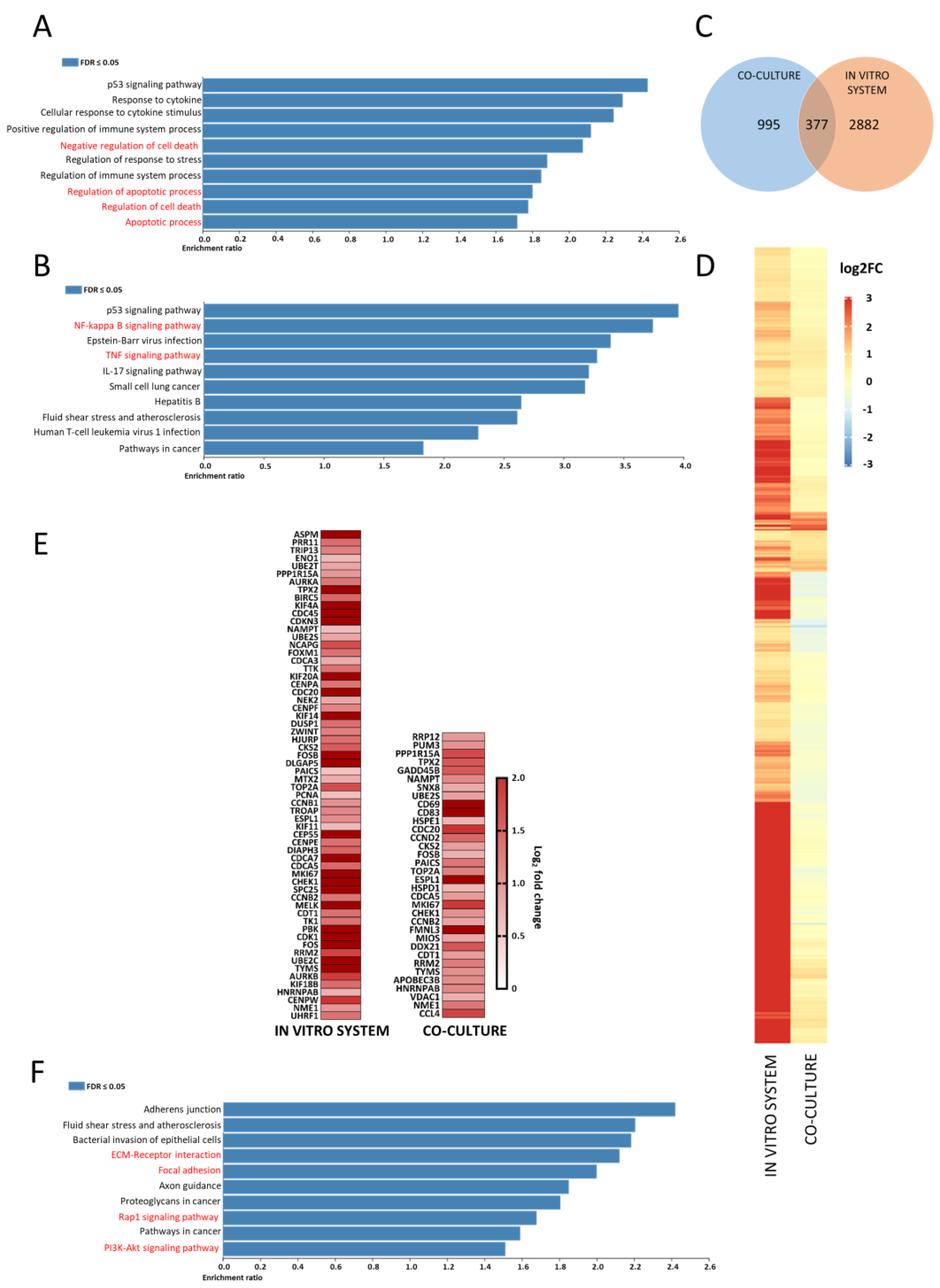{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and Processing
2.2. In Vitro Circulatory System
2.3. CD40L Fibroblast Co-Culture
2.4. RNA-Sequencing and Analysis
2.5. MiRNA-Seq and Analysis
2.6. Quantitative Real-Time PCR (qRT-PCR) of FAK (PTK2) RNA Expression
2.7. Apoptosis/p-FAK Immunostaining
2.8. Migration Assay
2.9. Invasion Assay
2.10. Synergy between Defactinib and Ibrutinib
2.11. Statistics
3. Results
3.1. CLL 2D Cell Culture with CD40L Fibroblasts Upregulates Pro-Survival and Anti-Apoptotic Gene Sets
3.2. Comparative RNA-Sequencing of CLL 2D Cell Culture with CD40L-Expressing Fibro Blasts Produced Distinct Differential Expression Profiles to Those of Migratory CLL Cells from the In Vitro System
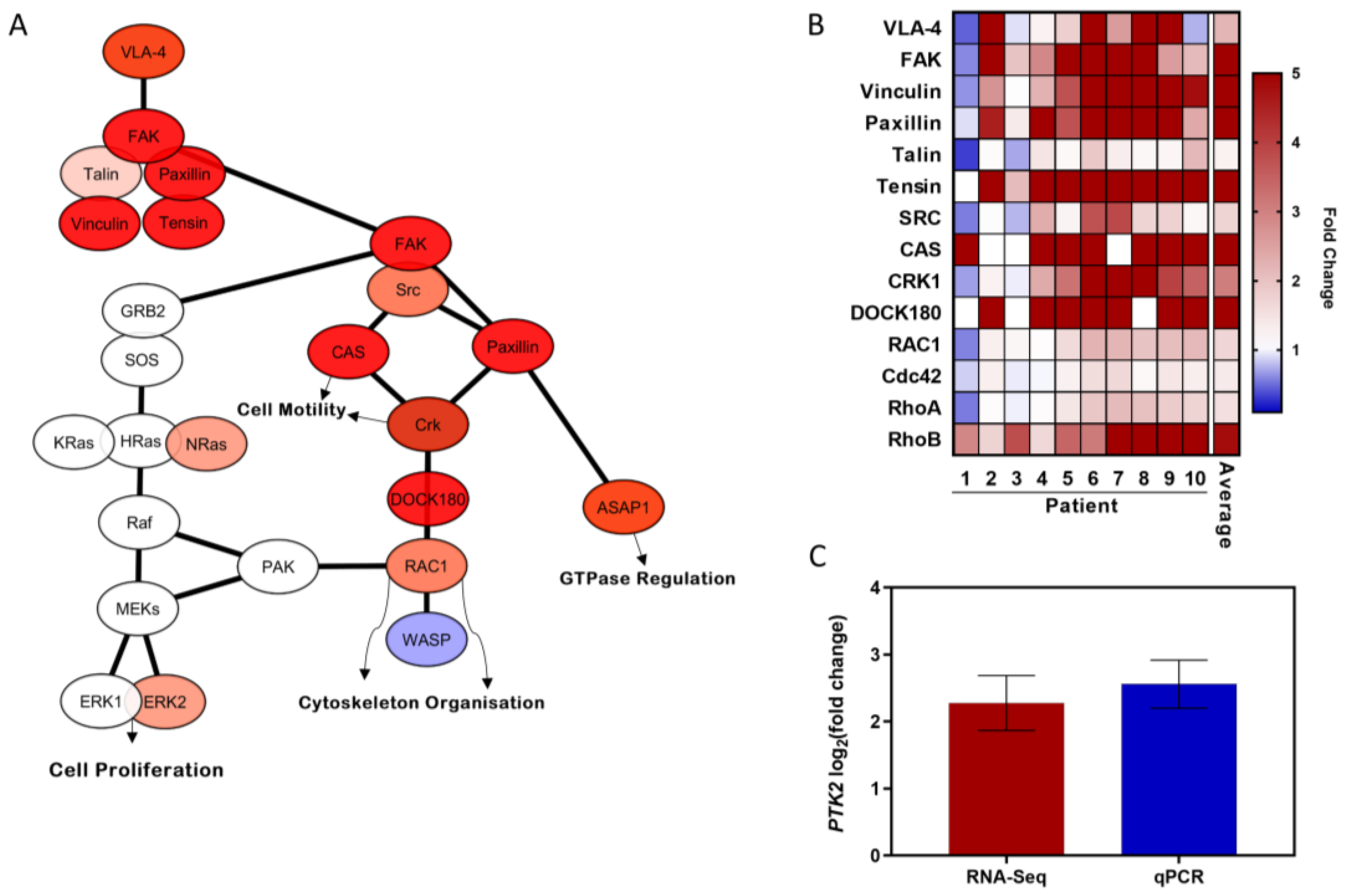
3.3. Migratory CLL Cells Have a Striking Gene Set Enrichment of Adhesion, RAP1 and PI3K-AKT Signalling Pathways
3.4. No Clear miRNomic Role in CLL Migration Identified
3.5. FAK Signalling Pathway Is Upregulated during Transendothelial CLL Migration
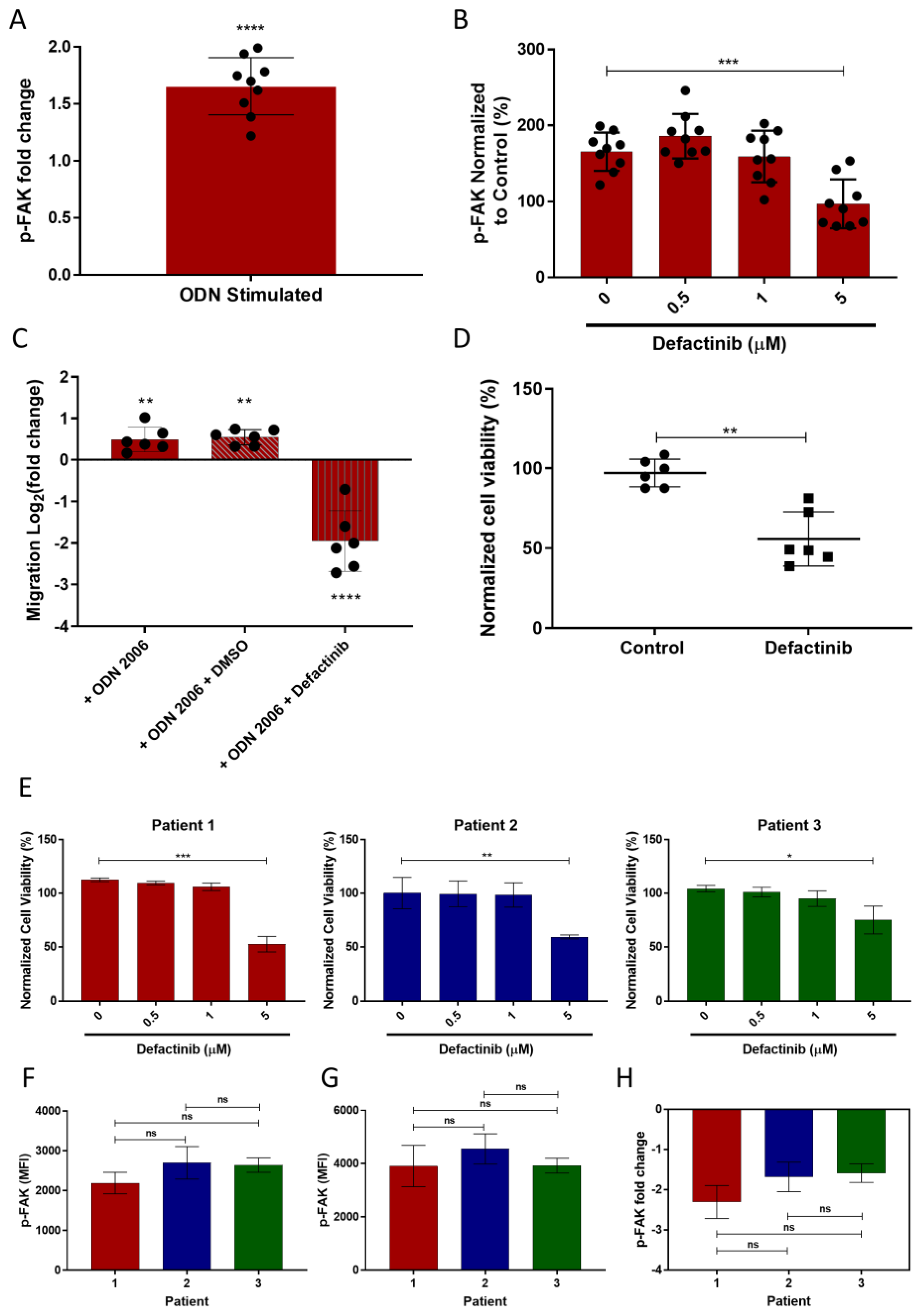
3.6. TLR9 Stimulation Induces CLL Migration through FAK Activation and This Is Inhibited by Defactinib
3.7. FAK Inhibition Results in a Heterogenic CLL Apoptotic Response
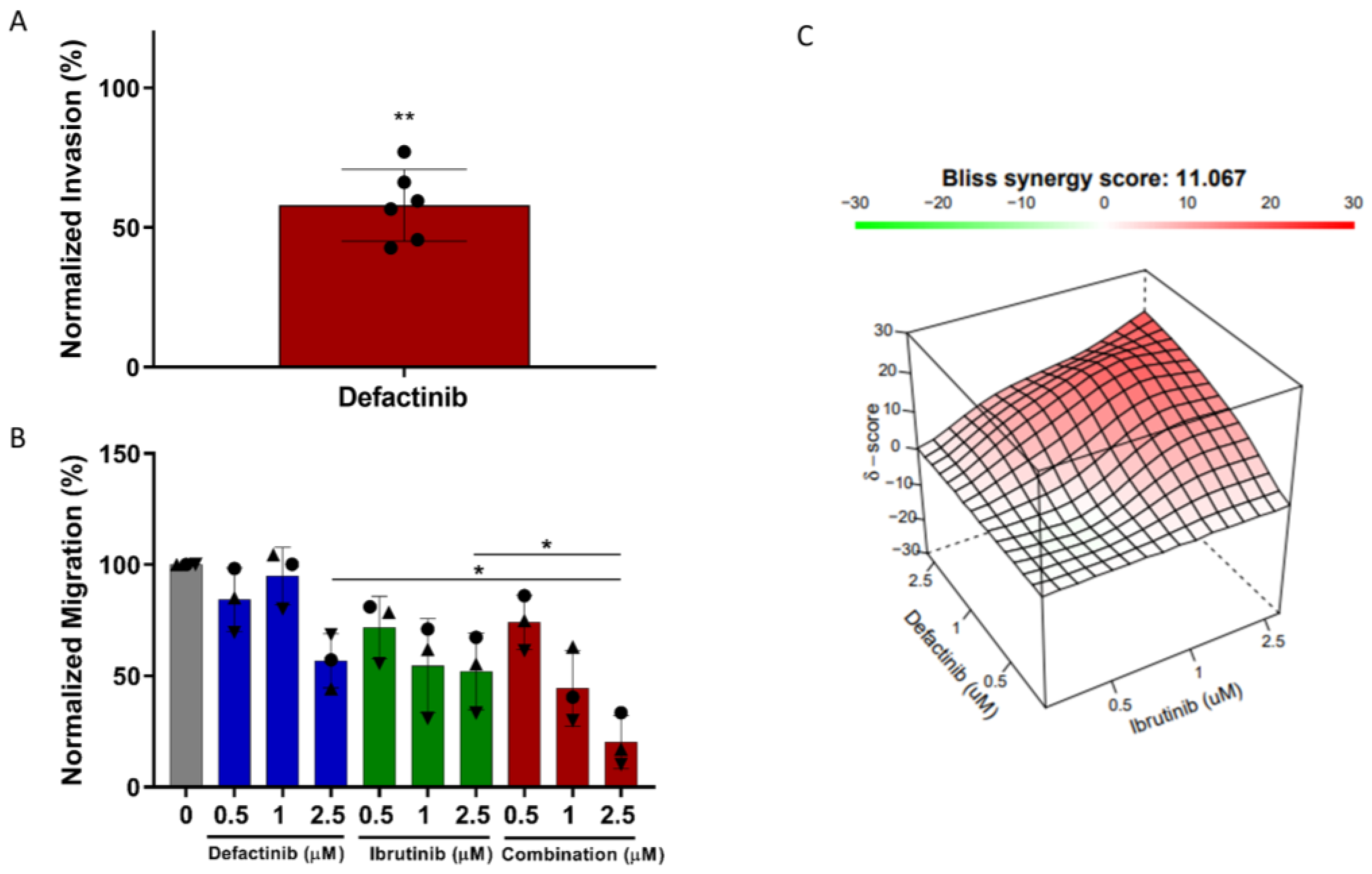
3.8. CLL Invasion Was Inhibited by Defactinib Treatment
3.9. Defactinib Synergises with Ibrutinib in CLL Migration Assays
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Delgado, J.; Nadeu, F.; Colomer, D.; Campo, E. Chronic lymphocytic leukemia: From molecular pathogenesis to novel therapeutic strategies. Haematologica 2020, 105, 2205–2217. [Google Scholar] [CrossRef] [PubMed]
- Shanafelt, T.D.; Wang, X.V.; Kay, N.E.; Hanson, C.A.; O’Brien, S.; Barrientos, J.; Jelinek, D.F.; Braggio, E.; Leis, J.F.; Zhang, C.C.; et al. Ibrutinib-Rituximab or Chemoimmunotherapy for Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2019, 381, 432–443. [Google Scholar] [CrossRef] [PubMed]
- Wierda, W.G.; Allan, J.N.; Siddiqi, T.; Kipps, T.J.; Opat, S.; Tedeschi, A.; Badoux, X.C.; Kuss, B.J.; Jackson, S.; Moreno, C.; et al. Ibrutinib Plus Venetoclax for First-Line Treatment of Chronic Lymphocytic Leukemia: Primary Analysis Results From the Minimal Residual Disease Cohort of the Randomized Phase II CAPTIVATE Study. J. Clin. Oncol. 2021, 39, 3853–3865. [Google Scholar] [CrossRef] [PubMed]
- Buggins, A.G.; Pepper, C.J. The role of Bcl-2 family proteins in chronic lymphocytic leukaemia. Leuk. Res. 2010, 34, 837–842. [Google Scholar] [CrossRef] [PubMed]
- Herishanu, Y.; Perez-Galan, P.; Liu, D.; Biancotto, A.; Pittaluga, S.; Vire, B.; Gibellini, F.; Njuguna, N.; Lee, E.; Stennett, L.; et al. The lymph node microenvironment promotes B-cell receptor signaling, NF-kappaB activation, and tumor proliferation in chronic lymphocytic leukemia. Blood 2011, 117, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Messmer, B.T.; Messmer, D.; Allen, S.L.; Kolitz, J.E.; Kudalkar, P.; Cesar, D.; Murphy, E.J.; Koduru, P.; Ferrarini, M.; Zupo, S.; et al. In vivo measurements document the dynamic cellular kinetics of chronic lymphocytic leukemia B cells. J. Clin. Investig. 2005, 115, 755–764. [Google Scholar] [CrossRef] [Green Version]
- Andreeff, M.; Darzynkiewicz, Z.; Sharpless, T.K.; Clarkson, B.D.; Melamed, M.R. Discrimination of human leukemia subtypes by flow cytometric analysis of cellular DNA and RNA. Blood 1980, 55, 282–293. [Google Scholar] [CrossRef] [Green Version]
- Buggins, A.G.; Pepper, C.; Patten, P.E.; Hewamana, S.; Gohil, S.; Moorhead, J.; Folarin, N.; Yallop, D.; Thomas, N.S.; Mufti, G.J.; et al. Interaction with vascular endothelium enhances survival in primary chronic lymphocytic leukemia cells via NF-kappaB activation and de novo gene transcription. Cancer Res. 2010, 70, 7523–7533. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, E.; Pearce, L.; Morgan, L.; Robinson, S.; Ware, V.; Brennan, P.; Thomas, N.S.; Yallop, D.; Devereux, S.; Fegan, C.; et al. Mimicking the tumour microenvironment: Three different co-culture systems induce a similar phenotype but distinct proliferative signals in primary chronic lymphocytic leukaemia cells. Br. J. Haematol. 2012, 158, 589–599. [Google Scholar] [CrossRef]
- Herreros, B.; Rodriguez-Pinilla, S.M.; Pajares, R.; Martinez-Gonzalez, M.A.; Ramos, R.; Munoz, I.; Montes-Moreno, S.; Lozano, M.; Sanchez-Verde, L.; Roncador, G.; et al. Proliferation centers in chronic lymphocytic leukemia: The niche where NF-kappaB activation takes place. Leukemia 2010, 24, 872–876. [Google Scholar] [CrossRef] [Green Version]
- Hayden, R.E.; Pratt, G.; Roberts, C.; Drayson, M.T.; Bunce, C.M. Treatment of chronic lymphocytic leukemia requires targeting of the protective lymph node environment with novel therapeutic approaches. Leuk. Lymphoma 2012, 53, 537–549. [Google Scholar] [CrossRef] [PubMed]
- Haselager, M.V.; Kater, A.P.; Eldering, E. Proliferative Signals in Chronic Lymphocytic Leukemia; What Are We Missing? Front. Oncol. 2020, 10, 2205. [Google Scholar] [CrossRef]
- Carrasco, Y.R.; Batista, F.D. B-cell activation by membrane-bound antigens is facilitated by the interaction of VLA-4 with VCAM-1. EMBO J. 2006, 25, 889–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiorcari, S.; Maffei, R.; Atene, C.G.; Potenza, L.; Luppi, M.; Marasca, R. Nurse-Like Cells and Chronic Lymphocytic Leukemia B Cells: A Mutualistic Crosstalk inside Tissue Microenvironments. Cells 2021, 10, 217. [Google Scholar] [CrossRef] [PubMed]
- Ahn, I.E.; Farooqui, M.Z.H.; Tian, X.; Valdez, J.; Sun, C.; Soto, S.; Lotter, J.; Housel, S.; Stetler-Stevenson, M.; Yuan, C.M.; et al. Depth and durability of response to ibrutinib in CLL: 5-year follow-up of a phase 2 study. Blood 2018, 131, 2357–2366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burger, J.A.; Barr, P.M.; Robak, T.; Owen, C.; Ghia, P.; Tedeschi, A.; Bairey, O.; Hillmen, P.; Coutre, S.E.; Devereux, S.; et al. Long-term efficacy and safety of first-line ibrutinib treatment for patients with CLL/SLL: 5 years of follow-up from the phase 3 RESONATE-2 study. Leukemia 2020, 34, 787–798. [Google Scholar] [CrossRef] [Green Version]
- Coutre, S.E.; Furman, R.R.; Flinn, I.W.; Burger, J.A.; Blum, K.; Sharman, J.; Jones, J.; Wierda, W.; Zhao, W.; Heerema, N.A.; et al. Extended Treatment with Single-Agent Ibrutinib at the 420 mg Dose Leads to Durable Responses in Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma. Clin. Cancer Res. 2017, 23, 1149–1155. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, S.; Jones, J.A.; Coutre, S.E.; Mato, A.R.; Hillmen, P.; Tam, C.; Osterborg, A.; Siddiqi, T.; Thirman, M.J.; Furman, R.R.; et al. Ibrutinib for patients with relapsed or refractory chronic lymphocytic leukaemia with 17p deletion (RESONATE-17): A phase 2, open-label, multicentre study. Lancet Oncol. 2016, 17, 1409–1418. [Google Scholar] [CrossRef]
- Byrd, J.C.; Hillmen, P.; O’Brien, S.; Barrientos, J.C.; Reddy, N.M.; Coutre, S.; Tam, C.S.; Mulligan, S.P.; Jaeger, U.; Barr, P.M.; et al. Long-term follow-up of the RESONATE phase 3 trial of ibrutinib vs. ofatumumab. Blood 2019, 133, 2031–2042. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, S.; Furman, R.R.; Coutre, S.; Flinn, I.W.; Burger, J.A.; Blum, K.; Sharman, J.; Wierda, W.; Jones, J.; Zhao, W.; et al. Single-agent ibrutinib in treatment-naive and relapsed/refractory chronic lymphocytic leukemia: A 5-year experience. Blood 2018, 131, 1910–1919. [Google Scholar] [CrossRef]
- Herman, S.E.; Niemann, C.U.; Farooqui, M.; Jones, J.; Mustafa, R.Z.; Lipsky, A.; Saba, N.; Martyr, S.; Soto, S.; Valdez, J.; et al. Ibrutinib-induced lymphocytosis in patients with chronic lymphocytic leukemia: Correlative analyses from a phase II study. Leukemia 2014, 28, 2188–2196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davids, M.S.; Burger, J.A. Cell Trafficking in Chronic Lymphocytic Leukemia. Open J. Hematol. 2012, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tissino, E.; Benedetti, D.; Herman, S.E.M.; Ten Hacken, E.; Ahn, I.E.; Chaffee, K.G.; Rossi, F.M.; Dal Bo, M.; Bulian, P.; Bomben, R.; et al. Functional and clinical relevance of VLA-4 (CD49d/CD29) in ibrutinib-treated chronic lymphocytic leukemia. J. Exp. Med. 2018, 215, 681–697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lafouresse, F.; Bellard, E.; Laurent, C.; Moussion, C.; Fournie, J.J.; Ysebaert, L.; Girard, J.P. L-selectin controls trafficking of chronic lymphocytic leukemia cells in lymph node high endothelial venules in vivo. Blood 2015, 126, 1336–1345. [Google Scholar] [CrossRef] [Green Version]
- Gooden, C.E.; Jones, P.; Bates, R.; Shallenberger, W.M.; Surti, U.; Swerdlow, S.H.; Roth, C.G. CD49d shows superior performance characteristics for flow cytometric prognostic testing in chronic lymphocytic leukemia/small lymphocytic lymphoma. Cytometry B Clin. Cytom. 2018, 94, 129–135. [Google Scholar] [CrossRef]
- Rossi, D.; Zucchetto, A.; Rossi, F.M.; Capello, D.; Cerri, M.; Deambrogi, C.; Cresta, S.; Rasi, S.; De Paoli, L.; Bodoni, C.L.; et al. CD49d expression is an independent risk factor of progressive disease in early stage chronic lymphocytic leukemia. Haematologica 2008, 93, 1575–1579. [Google Scholar] [CrossRef] [Green Version]
- Walsby, E.; Buggins, A.; Devereux, S.; Jones, C.; Pratt, G.; Brennan, P.; Fegan, C.; Pepper, C. Development and characterization of a physiologically relevant model of lymphocyte migration in chronic lymphocytic leukemia. Blood 2014, 123, 3607–3617. [Google Scholar] [CrossRef] [Green Version]
- Pasikowska, M.; Walsby, E.; Apollonio, B.; Cuthill, K.; Phillips, E.; Coulter, E.; Longhi, M.S.; Ma, Y.; Yallop, D.; Barber, L.D.; et al. Phenotype and immune function of lymph node and peripheral blood CLL cells are linked to transendothelial migration. Blood 2016, 128, 563–573. [Google Scholar] [CrossRef] [Green Version]
- Lezina, L.; Spriggs, R.V.; Beck, D.; Jones, C.; Dudek, K.M.; Bzura, A.; Jones, G.D.D.; Packham, G.; Willis, A.E.; Wagner, S.D. CD40L/IL-4-stimulated CLL demonstrates variation in translational regulation of DNA damage response genes including ATM. Blood Adv. 2018, 2, 1869–1881. [Google Scholar] [CrossRef]
- Girbl, T.; Hinterseer, E.; Grossinger, E.M.; Asslaber, D.; Oberascher, K.; Weiss, L.; Hauser-Kronberger, C.; Neureiter, D.; Kerschbaum, H.; Naor, D.; et al. CD40-mediated activation of chronic lymphocytic leukemia cells promotes their CD44-dependent adhesion to hyaluronan and restricts CCL21-induced motility. Cancer Res. 2013, 73, 561–570. [Google Scholar] [CrossRef] [Green Version]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kluin, R.J.C.; Kemper, K.; Kuilman, T.; de Ruiter, J.R.; Iyer, V.; Forment, J.V.; Cornelissen-Steijger, P.; de Rink, I.; Ter Brugge, P.; Song, J.Y.; et al. XenofilteR: Computational deconvolution of mouse and human reads in tumor xenograft sequence data. BMC Bioinform. 2018, 19, 366. [Google Scholar] [CrossRef] [Green Version]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Friedlander, M.R.; Mackowiak, S.D.; Li, N.; Chen, W.; Rajewsky, N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012, 40, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Kaucka, M.; Plevova, K.; Pavlova, S.; Janovska, P.; Mishra, A.; Verner, J.; Prochazkova, J.; Krejci, P.; Kotaskova, J.; Ovesna, P.; et al. The planar cell polarity pathway drives pathogenesis of chronic lymphocytic leukemia by the regulation of B-lymphocyte migration. Cancer Res. 2013, 73, 1491–1501. [Google Scholar] [CrossRef] [Green Version]
- Murphy, J.M.; Rodriguez, Y.A.R.; Jeong, K.; Ahn, E.E.; Lim, S.S. Targeting focal adhesion kinase in cancer cells and the tumor microenvironment. Exp. Mol. Med. 2020, 52, 877–886. [Google Scholar] [CrossRef]
- Kennedy, E.; Coulter, E.; Halliwell, E.; Profitos-Peleja, N.; Walsby, E.; Clark, B.; Phillips, E.H.; Burley, T.A.; Mitchell, S.; Devereux, S.; et al. TLR9 expression in chronic lymphocytic leukemia identifies a promigratory subpopulation and novel therapeutic target. Blood 2021, 137, 3064–3078. [Google Scholar] [CrossRef]
- Maa, M.C.; Chang, M.Y.; Li, J.; Li, Y.Y.; Hsieh, M.Y.; Yang, C.J.; Chen, Y.J.; Li, Y.; Chen, H.C.; Cheng, W.E.; et al. The iNOS/Src/FAK axis is critical in Toll-like receptor-mediated cell motility in macrophages. Biochim. Biophys. Acta 2011, 1813, 136–147. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.; Rai, K.R. Chronic lymphocytic leukemia (CLL) treatment: So many choices, such great options. Cancer 2019, 125, 1432–1440. [Google Scholar] [CrossRef]
- Herman, S.E.; Mustafa, R.Z.; Jones, J.; Wong, D.H.; Farooqui, M.; Wiestner, A. Treatment with Ibrutinib Inhibits BTK- and VLA-4-Dependent Adhesion of Chronic Lymphocytic Leukemia Cells In Vivo. Clin. Cancer Res. 2015, 21, 4642–4651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Rooij, M.F.; Kuil, A.; Geest, C.R.; Eldering, E.; Chang, B.Y.; Buggy, J.J.; Pals, S.T.; Spaargaren, M. The clinically active BTK inhibitor PCI-32765 targets B-cell receptor- and chemokine-controlled adhesion and migration in chronic lymphocytic leukemia. Blood 2012, 119, 2590–2594. [Google Scholar] [CrossRef] [PubMed]
- Ponader, S.; Chen, S.S.; Buggy, J.J.; Balakrishnan, K.; Gandhi, V.; Wierda, W.G.; Keating, M.J.; O’Brien, S.; Chiorazzi, N.; Burger, J.A. The Bruton tyrosine kinase inhibitor PCI-32765 thwarts chronic lymphocytic leukemia cell survival and tissue homing in vitro and in vivo. Blood 2012, 119, 1182–1189. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.; Dehart, J.P.; Murphy, J.M.; Lim, S.T. Understanding the roles of FAK in cancer: Inhibitors, genetic models, and new insights. J. Histochem. Cytochem. 2015, 63, 114–128. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Guan, J.L. Focal adhesion kinase and its signaling pathways in cell migration and angiogenesis. Adv. Drug Deliv. Rev. 2011, 63, 610–615. [Google Scholar] [CrossRef] [Green Version]
- Rudelius, M.; Rosenfeldt, M.T.; Leich, E.; Rauert-Wunderlich, H.; Solimando, A.G.; Beilhack, A.; Ott, G.; Rosenwald, A. Inhibition of focal adhesion kinase overcomes resistance of mantle cell lymphoma to ibrutinib in the bone marrow microenvironment. Haematologica 2018, 103, 116–125. [Google Scholar] [CrossRef] [Green Version]
- Glodek, A.M.; Le, Y.; Dykxhoorn, D.M.; Park, S.Y.; Mostoslavsky, G.; Mulligan, R.; Lieberman, J.; Beggs, H.E.; Honczarenko, M.; Silberstein, L.E. Focal adhesion kinase is required for CXCL12-induced chemotactic and pro-adhesive responses in hematopoietic precursor cells. Leukemia 2007, 21, 1723–1732. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Mak, P.Y.; Mu, H.; Tao, W.; Rao, A.; Visweswaran, R.; Ruvolo, V.; Pachter, J.A.; Weaver, D.T.; Andreeff, M.; et al. Combinatorial Inhibition of Focal Adhesion Kinase and BCL-2 Enhances Antileukemia Activity of Venetoclax in Acute Myeloid Leukemia. Mol. Cancer Ther. 2020, 19, 1636–1648. [Google Scholar] [CrossRef]
- Kong, D.; Chen, F.; Sima, N.I. Inhibition of focal adhesion kinase induces apoptosis in bladder cancer cells via Src and the phosphatidylinositol 3-kinase/Akt pathway. Exp. Ther. Med. 2015, 10, 1725–1731. [Google Scholar] [CrossRef] [Green Version]
- Jayappa, K.D.; Portell, C.A.; Gordon, V.L.; Capaldo, B.J.; Bekiranov, S.; Axelrod, M.J.; Brett, L.K.; Wulfkuhle, J.D.; Gallagher, R.I.; Petricoin, E.F.; et al. Microenvironmental agonists generate de novo phenotypic resistance to combined ibrutinib plus venetoclax in CLL and MCL. Blood Adv. 2017, 1, 933–946. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burley, T.A.; Hesketh, A.; Bucca, G.; Kennedy, E.; Ladikou, E.E.; Towler, B.P.; Mitchell, S.; Smith, C.P.; Fegan, C.; Johnston, R.; et al. Elucidation of Focal Adhesion Kinase as a Modulator of Migration and Invasion and as a Potential Therapeutic Target in Chronic Lymphocytic Leukemia. Cancers 2022, 14, 1600. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14071600
Burley TA, Hesketh A, Bucca G, Kennedy E, Ladikou EE, Towler BP, Mitchell S, Smith CP, Fegan C, Johnston R, et al. Elucidation of Focal Adhesion Kinase as a Modulator of Migration and Invasion and as a Potential Therapeutic Target in Chronic Lymphocytic Leukemia. Cancers. 2022; 14(7):1600. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14071600
Chicago/Turabian StyleBurley, Thomas A., Andrew Hesketh, Giselda Bucca, Emma Kennedy, Eleni E. Ladikou, Benjamin P. Towler, Simon Mitchell, Colin P. Smith, Christopher Fegan, Rosalynd Johnston, and et al. 2022. "Elucidation of Focal Adhesion Kinase as a Modulator of Migration and Invasion and as a Potential Therapeutic Target in Chronic Lymphocytic Leukemia" Cancers 14, no. 7: 1600. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14071600