1. Introduction
The vast majority of cancer mortality is due to metastasis, the process by which cancerous cells spread to distant organs, and provides limited therapeutic options. In breast cancer specifically, greater than 95% of the mortality is associated with the spread of cancer to organs outside of the primary tissue [
1]. The process by which cancerous cells migrate from the primary tumor and establish distant metastases is complex and multiphasic. In order to improve outcomes for cancer patients, it is important to better understand the underlying cellular processes involved in metastasis to develop new therapeutic interventions. Research into drugs that target the metastatic cascade is especially needed since nearly all current breast cancer treatments aim to reduce the size of established tumors by slowing cell growth or inducing cell death [
2]. These therapeutics tend to take advantage of the fact that cancer cells divide more rapidly than many other tissues in the body. One common target of anti-neoplastic agents is the microtubule network to inhibit rapid proliferation by blocking mitosis [
3]. Microtubule-targeted drugs are frequently used in cancer treatment and fall into two main categories: taxanes/epothilones, which promote microtubule stabilization, and vinca alkaloids/halichondrins, which promote microtubule depolymerization and collapse [
3]. Microtubules are known to play an essential role in cellular processes such as replication, cell division, motility, cell polarity, and intracellular trafficking.
Microtubules are essential cellular structures formed by polymerizing stacks of α-tubulin/β-tubulin heterodimers that originate at the microtubule-organizing center (MTOC) in most mammalian cells. Microtubules are dynamic structures within the cell, and as they form and grow, post-translational modifications (PTMs) can be made to both α- and β-tubulin, modulating their stabilities and half-lives [
4]. These modifications are known to play specific roles in animal cells, often with tissue- or cell type-specific functions [
4]. Removal of the carboxy-terminal tyrosine of α-tubulin generates the PTM termed detyrosinated tubulin (deTyr-Tub), previously known as glu-tubulin. This tubulin PTM can then be further modified through the removal of revealed glutamic acid residues via a family of tubulin deglutamylases, CCP1-6, to generate Δ2 or Δ3 tubulin. Importantly, microtubules with increased stability often contain higher levels of deTyr-Tub, with those enriched in deTyr-Tub displaying an increased half-life of >1 h compared to 5–10 min for microtubules composed of the tyrosinated form of α-tubulin [
5].
The detyrosination of α-tubulin has been known for more than 40 years [
6], with two important enzymes composing the complete cycle of tyrosine removal and replacement, the tubulin tyrosine ligase (TTL) and the tubulin carboxypeptidase (TCP). The genetic identity of the TTL was first discovered in 1993 [
7], but the genetic identity of the TCP remained elusive for decades. Research into the mechanism of action of the TTL revealed the TTL enzyme has affinity for α- and β-tubulin heterodimers, the building blocks of microtubules, rather than polymerized microtubules [
8,
9,
10], whereas evidence has suggested that the TCP is acting on polymerized microtubules [
11,
12,
13]. Despite not knowing the identity of the TCP, the resulting deTyr-Tub modification was able to be studied through direct observation of the PTM. Given the previously unknown identity of the TCP, very few compounds were available to specifically inhibit the formation of deTyr-Tub. One such compound is parthenolide, a sesquiterpene lactone isolated from the feverfew plant. It has been previously shown that parthenolide decreases levels of deTyr-Tub; however, determining a specific mechanism by which this drug modulates tubulin is hampered by its broad-spectrum anti-inflammatory effect [
14]. One study revealed the ability of parthenolide to bind with
VASH1 via crystal structure [
15], while another has suggested that a specific enantiomer of parthenolide is responsible for its function in reducing deTyr-Tub [
16]. Recent work determined that parthenolide binds covalently to microtubules to non-specifically prevent the generation of deTyr-Tub [
17].
When examining the role of deTyr-Tub in cancer development and progression, research performed prior to the discovery of the identity of the TCP revealed a connection between detyrosinated microtubules, poor patient prognosis, and connection to migratory phenotypes. Early research revealed that microtubules composed of deTyr-Tub orient towards a wound edge in vitro using a scratch assay [
18]. Before the discovery of the TCP, levels of deTyr-Tub were studied in primary breast tumors. One such study examined the relationships between elevated deTyr-Tub and other clinical markers in patients with all types of breast cancer (ER+, PR+ and TNBC) revealing a correlation with a high pathologic mitotic score [
19]. In this somewhat limited dataset of breast cancers, patient outcomes were compared between those with high and low expression of deTyr-Tub and (
n = 134), 46 patients had adverse outcomes following initial treatment and elevated deTyr-Tub trended with worse prognosis, but the difference was not statistically significant (
p = 0.27). When deTyr-Tub status was combined with tumor grade (high deTyr-Tub score and high grade), patients were statistically more likely to have a poor prognosis (
p = 0.02). These findings paved the way for additional studies in the breast cancer field. More recently, expression of deTyr-Tub has been examined in circulating tumor cells (CTCs) isolated from patients with breast cancer [
20,
21]. In one study, levels of deTyr-Tub, along with other cytoskeletal markers, were compared in CTCs isolated from patients with early-stage breast cancer to those from patients with metastatic disease [
21]. This study identified that CTCs from patients with metastatic disease expressed higher levels of deTyr-Tub, total α-tubulin, and vimentin compared to those with early-stage disease. Similar patterns of cytoskeletal markers have been seen in breast cancer cells which have undergone epithelial-to-mesenchymal transition (EMT) [
22].
The complex mechanisms of metastatic dissemination are not fully known; however, the mechanism of EMT is thought to play an important role [
23]. EMT is a biologically conserved process which occurs during embryonic development, where cells lose their tight cell-to-cell contacts in order to migrate in the developing organism [
24]. Research has shown that when breast cancer cells are forced to undergo EMT via exogenous expression of the mesenchymal transcription factors, Twist or Snail, levels of deTyr-Tub increase. Conversely, this effect can be observed through the suppression of Twist in mesenchymal-like cells, leading to a decrease in the levels of deTyr-Tub. Increased levels of deTyr-Tub are observed in cells undergoing EMT at the invasive fronts of patient breast tumors, where invasion of adjacent tissues is starting to occur [
25]. These findings complement much earlier work which revealed cells at a wound edge generate polarized deTyr-Tub-rich microtubules oriented in the direction of the wound [
18,
26].
All of these earlier studies into the function and regulation of deTyr-Tub were hampered by the inability to target the unknown TCP enzyme and this has limited our understanding of the role of TCP in cancer. However, the major TCP enzymes were recently discovered to be vasohibin 1 (
VASH1) or vasohibin 2 (
VASH2) coupled with a small vasohibin-binding protein (
SVBP) in late 2017 [
27,
28]. Recent publications have shown the importance of SVBP as a core element essential for tubulin detyrosination [
29,
30]. The discovery of the TCP to be
VASH1 or
VASH2 coupled with
SVBP has revealed the mechanism by which α-tubulin is converted to deTyr-Tub. This detyrosinated α-tubulin can subsequently have the tyrosine replaced by the TTL. Given our current understand of the tubulin detyrosination cycle and the enzymes involved allows for the functional effects of this tubulin PTM to be directly tested for the first time.
The process by which deTyr-Tub and EMT are connected remains unknown; however, recent studies show the inhibition of
VASH2 expression through siRNA leads to a restoration of E-cadherin [
31]. This work was performed in breast cancer cell lines that are predominantly mesenchymal in phenotype and this manipulation forces these cells to undergo a mesenchymal-to-epithelial conversion. This observation, combined with the recent elucidation of the role of VASH/SVBP as the TCP, implies that microtubule modifications may play an essential role in the EMT process.
Existing knowledge of the role of deTyr-Tub in cells coupled with the recently established genetic identity of the TCP allows for us to directly increase deTyr-Tub through the systematic addition of the TCP components. Transfection of TCP constructs into immortalized breast epithelial cells induced apoptosis that was suppressed by activated KRas, Bcl-2 and Bcl-xL overexpression. Breast cancer cells could tolerate elevated TCP expression and demonstrated greater tumor cell invasion. Together, these results demonstrate that oncogenic signaling pathways can enable tolerance of an elevated level of deTyr-Tub and that TCP transfection promotes increased invasiveness.
2. Materials and Methods
2.1. Antibodies and Reagents
Bcl-xL antibody (1:1000, Cat#: ab32370) and VASH1 (1:1000, Cat#: ab199732) were purchased from Abcam. Bcl-2 antibody (1:1000, Cat #: 15071S), and Cleaved Caspase-3 (1:1000, Cat#: 9661S) were purchased from Cell Signaling Technologies. α-tubulin (1:1000, Cat#: T6199), FLAG (1:1000, Cat#: F3165), Tyr-Tub antibody (1:1000, Cat# MAB1864-I), and VASH2 (1:1000, Cat#: MABC536) were purchased from Sigma. GAPDH (1:5000, Cat#: sc-32233) was purchased from Santa Cruz. SVBP (CCDC23, 1:1000, Cat#: PA5-52569) was purchased from Invitrogen. The deTyr-Tub antibody was developed by Takashi Hotta and Ryoma Ohi [
17]. This antibody is now commercially available through RevMab under catalogue number RM444 (1:10000). Alexa Fluor 568 (1:1000, Cat#: A11011), Alexa Fluor 594 (1:1000, Cat#: A11012) and Hoechst 33258 (1:5000, Cat#: H3569) were purchased from Invitrogen (Waltham, MA, USA).
2.2. Cell Culture
MCF-10A cells were purchased from ATCC and cultured in DMEM/F-12 Media (Invitrogen, Cat#10565-018) supplemented with 5% Horse Serum (Invitrogen, Cat# 26050-088), 1% Pen/Strep (Gemini Bio-products, Cat# 400-109), Recombinant Human EGF (Invitrogen, 100 µg/500 mL), Hydrocortisone (Sigma H-0135, 50 µg/mL), Cholera Toxin (Sigma C-8052, 50 µg/500 mL), and Insulin (Sigma I-9278, 10 µg/mL). The parental MCF-10A cells were not passed beyond 35 passages in our laboratory, in accordance with protocols published from the Brugge Laboratory, to preserve their growth profile, and sensitivity to apoptosis and anoikis [
32].
MCF7 and MDA-MB-231 cells were purchased from ATCC and cultured in DMEM Media supplemented with 10% FBS (Atlanta Biologicals, R&D Systems, Minneapolis, MI, USA) and 1% Pen/Strep (Gemini Bio-products, Cat# 400-109).
MCF10A cells with homozygous loss of
PTEN or stable expression of activated KRas have been described previously, and characterized for effects on primary tumor formation and metastasis. The MCF10As with activated KRas pathway are able to form tumor in vivo and show a strong upregulation of p-ERK [
33,
34,
35]. MCF10A cells with stable expression of Bcl-2 and Bcl-xL were generated using lentiviral transduction of Bcl-2 or Bcl-xL plasmids containing a puromycin selection vector obtained from addgene (see ‘Lentivirus production and transduction’ below).
2.3. Lentivirus Production and Transduction
Overexpression of Bcl-2 and Bcl-xL in MCF10A cells was performed by lentiviral transduction using the Lenti-X Packaging System (Clontech, Mountain View, CA, USA) according to the manufacturer’s instructions. The pCDH-puro-Bcl2 plasmid (Addgene plasmid #46971) and pCDH-puro-Bcl-xL (Addgene plasmid #46972) were added into supplied nanoparticle complexes for 10 min and applied to Lenti-X 293T cells to produce the virus. The medium was changed after 24 h and viral supernatant was harvested after 48 h, filtered, and used to infect cells at an approximate MOI of 10 along with 1 µg/mL Polybrene. The plate was then immediately centrifuged for 1.5 h at 1000 rpm. Cells were selected with 1 µg/mL puromycin at 2 days after infections and maintained in puromycin-containing media. Puromycin was removed from the medium for experimental conditions. pCDH-puro-Bcl2 and pCDH-puro-Bcl-xL were gifts from Jialiang Wang [
36].
2.4. Transient Transfections
TCP and Individual Component Plasmids
FLAG-sfGFP-His
FLAG-VASH1-sfGFP-His
FLAG-VASH2-sfGFP-His
FLAG-SVBP-myc
FLAG-VASH1-sfGFP-His + IRES + SVBP-myc
FLAG-VASH2-sfGFP-His + IRES + SVBP-myc
These plasmids for expression of the TCP components were generously provided through our collaboration with the Moutin group [
27].
Transfections were performed 24 h after plating cells at the indicated density for each given experiment after being harvested from a ~80–90% confluent tissue culture dish. The plasmid of interest was prepared at a concentration of 2 µgDNA/100 µL at a ratio of 1 µg to 3 µL of FuGene HD transfection reagent (Promega, Cat#: E2311).
2.5. Live Imaging
Epi-fluorescence images of transfected cells were captured with a Nikon Ti2E microscope with High Content Analysis (HCA). Images were collected with equal exposure times between conditions. A 10× phase-contrast objective was utilized to capture images of the central 5 mm square of each well on the IBIDI 8-well chamber slide with IBITreat (Fitchburg, WI, USA; Cat.No:80826) and images were stitched together by the Nikon Elements software AR 5.21.03 (Nikon). Using the GFP channel of these images, GFP+ objects were detected using the Nikon GA3 analysis suite. Objects were determined based on having an intensity greater than 12,000 RFUs and a size greater than 10 µm but less than 150 µm to identify individual cells rather than cellular debris or aggregates, respectively. A 2× clean and separate filter was employed to remove additional debris and separate adjacent cells into unique objects. With individual GFP+ objects identified, the number of objects was enumerated using the Nikon General Analysis software, as well as the cell length and circularity for each object.
2.6. Single Cell Imaging and Tracking
MDA-MB-231 or Hs578t cells were transfected with sfGFP or TCP constructs and moved to IBIDI 8-well chamber slide with IBITreat (Fitchburg, WI, USA; Cat.No:80826) 12 h after transfection and allowed to attach for 12 h before imaging. A total of 1 to 2 fields of cells were imaged in each condition using a 10× phase-contrast objective in GFP and brightfield channels every 15 min for 19 h. Images were processed using the ImageJ plugin TrackMate [
37] to identify GFP+ cells and track them across the time series. Tracks were filtered based on a minimum duration of 5 h with no maximum duration in length. These tracks were exported and analyzed using the MotilityLab platform [
38]. Plots were generated for mean squared displacement (MSD) and mean track speed in MotilityLab using the aggregate data across biological and technical replicates.
2.7. Immunofluorescence
Cells were fixed using freshly prepared 3.7% formaldehyde diluted in 1x phosphate-buffered saline (PBS) for 10 min. The fixation reagent was removed and 2 washes of 1× PBS were performed 5 min apart. Fixed cells were permeabilized using a 0.1% v/v ratio of Triton-X100 in PBS. The permeabilization solution was left on the cells for 10 min followed by 2 washes of PBS 5 min apart. Cells were blocked in a solution containing 5% bovine serum albumin (BSA) w/v, a 0.5% v/v ratio of NP-40 substitute in PBS for one hour at room temperature while being protected from light. Primary antibodies were added at the outlined ratios in primary antibody solution consisting of a 2.5% BSA w/v ratio, with 0.5% v/v ratio of NP-40 substitute in 1× PBS. Primary antibodies were allowed to bind overnight at 4 degrees Celsius with gentle rocking. Removal of unbound and non-specific antibody was accomplished with 3 consecutive washes with PBS 10 min apart with gentle rocking during the incubation steps. Secondary antibodies conjugated to indicated Alexa fluorophores were diluted in fresh primary antibody solution and diluted at 1:1000, for nuclear co-staining a 1:5000 dilution of Hoescht 33258 is added to the secondary antibody solution. Cells were stained for 2 h at room temperature. Removal of unbound and non-specific antibodies was accomplished with 3 consecutive washes with PBS 10 min apart with gentle rocking during the incubation steps. A final quick wash with double-distilled water was performed before adding Fluoromount-G (Invitrogen, Cat# 00-4958-02, Waltham, MA, USA).
2.8. Active Caspase-3 Immunofluorescence Assay
Cells were seeded at 15,000 cell/well onto IbiTreat 8-well chambers and allowed to adhere for 24 h. They were subsequently transfected with sfGFP, TCP-VASH1, or TCP-VASH2 plasmids for 24 h. The cells were then fixed, permeabilized and stained as described above with a Cleaved Caspase-3 antibody and an Alexa Fluor 594 secondary antibody. Images were acquired on a Nikon Ti2-E inverted microscope at 10× magnification. To analyze these images, first, the boundary of a successfully transfected cell was identified using the GFP channel in Nikon HCA to create an object for each cell. The same metrics to identify GFP+ objects were utilized as from the imaging of transfected live cells. With each object identified, the boundary of the object was slightly eroded to ensure that the AF549 intensity associated with Caspase-3 is only a result of that cell, and not any adjacent cells or debris. The average AF594 intensity of the cell is then calculated for each eroded object. Cells were determined to be positive for active Caspase-3 when the average 594 intensity of the GFP+ object was calculated to be >10,000 RFUs.
2.9. Western Blotting
Cells were lysed at the indicated time point using a chilled solution of 1× RIPA containing phenylmethylsulfonyl fluoride, protease inhibitor cocktail, and phosphatase inhibitor cocktail II. Dishes or plates containing the cells of interest were placed on a bed of ice for 15 min following the addition of the RIPA solution. Cells were scraped from the dish using a cell scraper and lysates collected in a 1.5 mL microcentrifuge tubes. These cells were vortexed every 5 min for the next 15 min, and then placed in the −30 °C freezer until ice crystals formed. The tubes were then thawed on wet ice and centrifuged at 15,000 rcf for 15 min. The supernatants were removed and placed in a new microcentrifuge tube, while the resulting pellets were discarded. The resulting total protein was quantified using the BioRad DC Protein Assay Kit according to the manufacturer’s recommended protocol. Samples and standards were read on a BioTek Synergy HT plate reader after a 15 min incubation period. Samples were diluted to a final concentration of 1 µg/µL, boiled at 95 °C for 10 min, and loaded in gels at equal volumes and total protein quantity.
An amount of 20 µg of total protein was added to each lane of a 1.5 mm × 10 well NuPAGE 4–12% Bis-Tris Gel (Invitrogen, Cat# NP0335BPX). Loaded gels were run using 1× NuPAGE MES SDS Running Buffer using 2 constant voltage phases, the first phase is 90 volts for 30 min, followed by 120 volts for 90 min. Gels are trimmed and transferred to a PVDF membrane using the eBlot™ L1 Fast Wet Transfer System (GenScript, Piscataway, NJ, USA). Membranes were blocked in either 5% BSA in 1× TBST or 5% non-fat dry milk in 1× TBST rocking for 1 h at room temperature. Primary antibodies were added to 2.5% BSA or non-fat dry milk in 1× TBST shaking overnight at 4 °C. Removal of unbound and non-specific antibody was accomplished with 3 consecutive washes of 1× TBST shaking for 10 min each. HRP-conjugated secondary antibody was diluted at 1:5000 in 2.5% BSA or non-fat dry milk in 1× TBST and incubated for 2 h at room temperature with gentle rocking. Removal of unbound and non-specific antibody was accomplished with 3 consecutive washes of 1× TBST rocking for 10 min each. Electrochemiluminescence reagent (Amersham) was added to blots for 2 min before capturing images on the iBright imager (ThermoFisher, Waltham, MA, USA).
2.10. Chemiluminescence and Molecular Weights
Using the iBright Software (ThermoFisher, Waltham, MA, USA), the molecular weights of bands of interest can be determined. For each blot, the SeeBlue™ Plus2 Pre-stained Protein Standard (LC5925, Invitrogen, Waltham, MA, USA) was used. The software allows for each band of the marker to be identified. Using these as the standard, the molecular weight of a band of interest is calculated by the software.
2.11. Gelatin Degradation Assay
The Cy-3-labeled gelatin degradation assay kit (Sigma, Cat#: ECM671) was purchased from EMD Millipore and the manufacturer’s protocol was followed to generate gelatin coated 8 chamber IBIDI well slides (Cat: 80826, poly-L-lysine coated). To generate a more consistent layer of fluorescent gelatin, 100 µL of prepared gelatin was added to each well and constantly rotated during the 5 min solidification time in the protocol. MDA-MB-231 cells were transfected 24 h prior to plating on prepared gelatin and cells were fixed using 3.7% formaldehyde diluted in 1× PBS after 24 h on the gelation. Hs578t cells were transfected 24 h prior to plating on prepared gelatin, and cells were fixed 48 h after plating using 3.7% formaldehyde diluted in 1× PBS. A total of 10 GFP+ cells were imaged at random on an Olympus FV1000 point-scanning confocal microscope at 60× magnification from each well to generate 20 or 30 images per condition per biological replicate. Four biological replicates were performed. A total of 110 cells were imaged per condition per cell line.
For each image, degradation percentages were calculated in MATLAB based on a maximum intensity projection (MIP) of the original images. GFP MIP images were filtered with multiple sizes of Laplacian of Gaussian filters ranging from a sigma of 2 pixels to 20 pixels, increasing in 1 pixel increments. A maximum intensity projection across the filtered images resulted in an image highlighting the cell body; a threshold for this image was set at values greater than the 75th percentile of the filtered maximum projection. Objects less than 10 pixels were removed and values in a 20 pixel border around the edge of the image were set to 0 to remove edge effects. This image was further process by closing the binary image with a disk of radius 4 pixels, filling holes in the image, and then removing objects less than 50,000 pixels. The MIP of the Cy-3 images were contrast adjusted using the MATLAB function imadjust (default of 1% saturation) and then the image was binarized to keep values less than 10,000. A further constraint that the original intensity be less than 1200 was then added and objects smaller than 10 pixels were removed. Percent degradation was calculated as the number of degraded pixels in the Cy-3 image that were inside the cell boundary, divided by the number of pixels inside the cell boundary.
After cells were fixed, one slide per cell line was permeabilized for 10 min with 0.25% TritonX100 in PBS. These cells were then stained with phalloidin conjugated to AF647 at a 5× stock concentration in PBS for 30 min. Cells were washed 3 times with PBS and preserved using Fluoromount-G. Cells were imaged on an Olympus FV1000 point-scanning confocal microscope at 60× magnification and representative overlay of image channels were generated using ImageJ.
2.12. Statistical Analysis
GraphPad Prism (version 9) was used to determine all statistical comparisons. One-way and two-way ANOVA tests were performed with a Tukey multiple comparisons post-test as indicated. A p-value of 0.05 or less was considered statistically significant. Multiple technical replicates were averaged for each experiment, and the means were compared across biological replicates.
4. Discussion
The establishment of
VASH1 and
VASH2 coupled with
SVBP to be the major TCPs has provided the opportunity to better understand the function of deTyr-Tub in various cell types. The necessity of both the vasohibin and
SVBP to effectively remove the C-terminal tyrosine from α-tubulin likely contributed to the delay in the discovery of the TCP. Leveraging this discovery, we aimed to understand for the first time the effect of specifically enhancing deTyr-Tub in breast epithelial cells. Through this study, it was shown that addition of the TCP-containing constructs to both breast epithelial and tumor cell lines resulted in a dramatic increase in levels of filamentous deTyr-Tub (
Figure 1). The conversion of Tyr-Tub to deTyr-Tub following transfection with the TCP constructs can be observed through immunofluorescence staining in
Figure S1. By Western blot, the conversion of Tyr-Tub to deTyr-Tub can be observed, but the decrease is minimal given the heterogeneity of the transfection (
Figure S2). Importantly, the increases in deTyr-Tub occur to the greatest extent with constructs expressing the vasohibin (
VASH1/
2) and its binding partner (
SVBP), while expression of the vasohibin alone provides a smaller increase in deTyr-Tub (
Figure 2). This increase in deTyr-Tub with vasohibin alone is likely occurring through the interaction with endogenous
SVBP already expressed by the cells. Expression of
SVBP alone does not lead to an increase in deTyr-Tub, indicating that the level of SVBP is not a limiting factor.
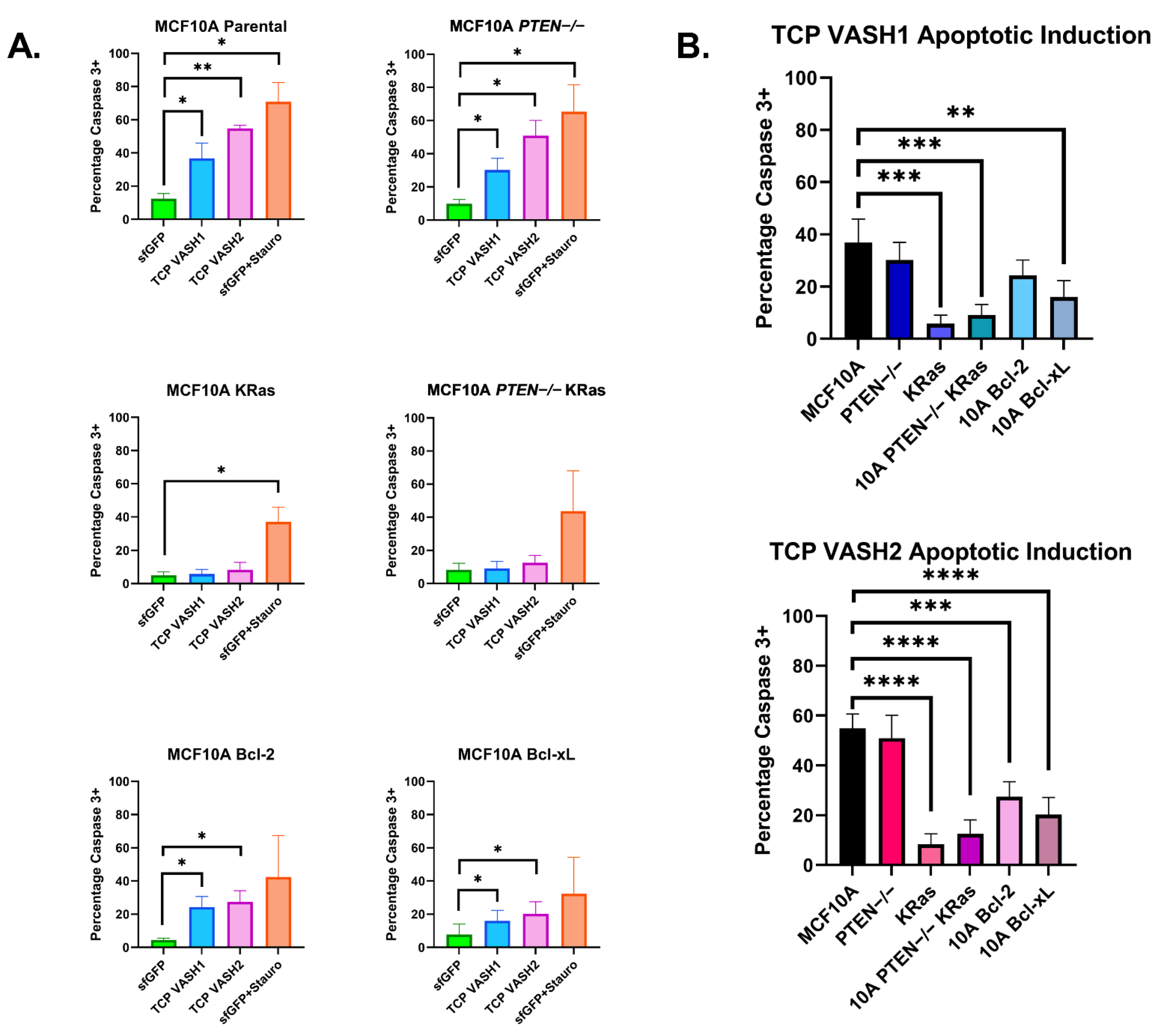
Our studies revealed that nontumorigenic breast epithelial cells and the modestly tumorigenic MCF7 cells did not tolerate the increase in deTyr-Tub and resulted in morphological changes consistent with increased apoptosis (
Figure 3 and
Figure 4). Given the knowledge that deTyr-Tub is increased at the invasive margin of breast epithelial carcinomas and elevated deTyr-Tub is associated with poor prognosis when combined with tumor grade, we sought to identify a molecular basis for tolerating enhanced deTyr-Tub through the induction of specific mutations in the background of breast epithelial cells.
To determine mechanisms of resistance, we first began by confirming that TCP-induced cell death was occurring through Caspase-3 activation in the immortalized nontumorigenic breast epithelial cell line MCF-10A (
Figure 4). With this Caspase-3 activation confirmed following TCP overexpression, we utilized the High Content Analysis (HCA) feature of our Nikon Ti2e to identify GFP(+) cells, indicating a successful transfection of the constructs of interest. Using these identified cells, the HCA software then quantified the intensity of active Caspase-3, as labeled with a AF594 secondary antibody. The quantification of active Caspase-3 intensity was used to determine the percentage of transfected cells undergoing apoptosis across the different constructs and cell lines.
Previously, our lab has generated a number of specific genetic changes in the stable background of the MCF10A cell line [
33,
34,
35]. These altered cell lines allow us to determine how individual genetic alterations contribute to cancer phenotypes such as migration, dormancy, and metastasis [
33,
34,
35]. Several of these changes are anti-apoptotic in function such as
PTEN−/−, Bcl-2 and Bcl-xL overexpression, while others such as the activating KRas mutation alter migration and in vivo tumor formation. While exploring the effects of these mutations on deTyr-Tub, we discovered that the MCF10A KRas mutation has a modest effect on increasing this tubulin PTM alone or in combination with
PTEN−/−. When transfecting the TCP constructs into the altered MCF10A cells, we anticipated that anti-apoptotic genes would provide the maximum protective effect against TCP-induced apoptosis. Previously, it has been reported that MCF10As with
PTEN loss and Bcl-2 overexpression were more resistant to intrinsic apoptosis, as determined by placing cells in suspension, treatment with latrunculin A to induce cell rounding, and deprivation of essential growth factors [
35]. Unexpectedly,
PTEN−/− provided no protection against TCP-induced apoptosis and Bcl-2 and Bcl-xL overexpression cells only provided moderate protection (
Figure 4). Meanwhile, KRas and
PTEN−/− KRas cells were highly protected from TCP-induced apoptosis. Since the KRas and
PTEN−/− KRas cells were developed from independent parental clones, this reinforces the conclusion that the protective effect from apoptosis induced by deTyr-Tub results from KRas activation, rather than clonal variation. While we cannot completely rule out that this apoptotic effect may be through the action of the TCP on another cellular process, we have increased confidence that it is due to its action on microtubules given the occurrence in both VASH1 and VASH2 TCP constructs. Of importance, VASH1 and VASH2 share only ~52.5% homology and have been shown to display differential functions in their roles with angiogenesis [
39]. Therefore, we have increased confidence that their common function of generating deTyr-Tub is the cause of apoptotic induction.
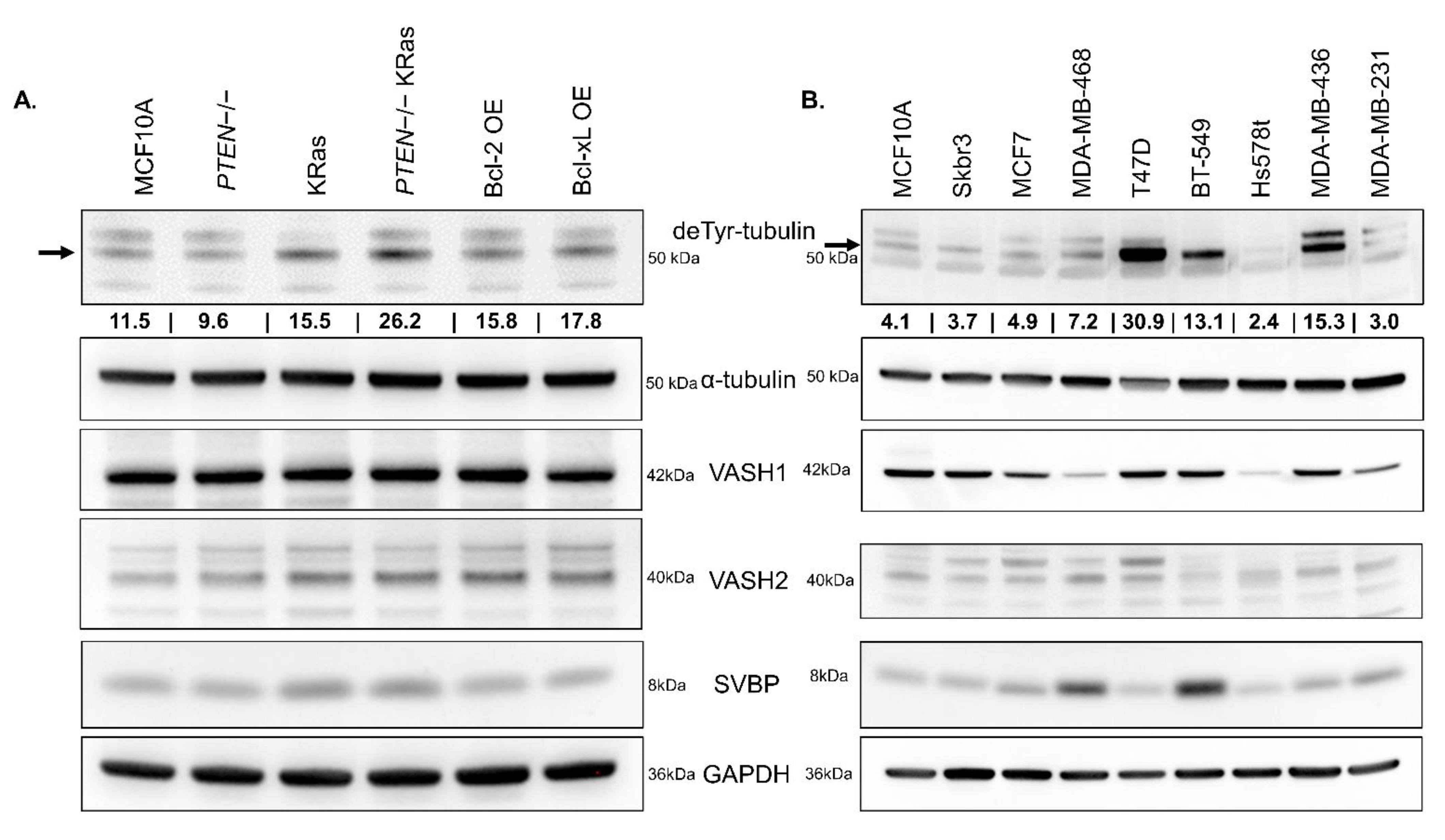
When comparing components of the TCP across these cell lines, we discovered that
VASH1,
VASH2, or
SVBP do not vary in the MCF10As and variants despite moderate changes in basal deTyr-Tub presence (
Figure 5A). When comparing the abundance of deTyr-Tub across commonly utilized breast cancer cell lines, the level of the TCP components,
VASH1,
VASH2, and
SVBP, did not show a clear correlation with the amount of deTyr-Tub (
Figure 5B). Given the ability for the
VASH2 antibody to detect both
VASH1 and
VASH2 (
Figure S5), we have increased confidence that the relative amount of vasohibin does not correspond to level of deTyr-Tub. The full, uncropped images for all Western blots can be found in
Figure S8. This led us to conclude that the generation of deTyr-Tub may be controlled at an enzymatic regulation step rather than at the level of TCP protein abundance.
Prior research in our laboratory has revealed an increase in deTyr-Tub, “glu-tubulin” at that time, which occurs in combination with the EMT transcription factor, Twist, at the invasive margin of patient ductal carcinoma in situ samples [
25]. These data suggested a functional relationship between deTyr-Tub and invasion. Additionally, we have observed that treating many different cell types with the microtubule stabilizing chemotherapeutic paclitaxel leads to a dramatic increase in the abundance of detyrosinated microtubules [
40,
41]. To better understand the connection between the enhanced deTyr-Tub observed at the invasive margin of patient samples, we began looking in the literature for a connection between established methods of increasing deTyr-Tub and mechanisms of invasion. A study by the Courtneidge group determined the effects of a wide variety of compounds on invadopodia formation [
42]. Invadopodia are thought to be some of the earliest structures necessary for cells to escape the confines of the basement membrane [
43]. This step is a requirement for breast cancers to be categorized as invasive [
43]. The Courtneidge study revealed that breast cancer cells treated with paclitaxel lead to increased invadopodia formation [
42]. Previously, we have observed that paclitaxel treatment greatly elevates the amount of deTyr-Tub [
40,
41], and therefore we sought to determine if directly increasing deTyr-Tub through the addition of TCP constructs will alter the invasive capability of breast cancer cells.
MDA-MB-231 cells harbor a KRas mutation as do the genetically engineered MCF10A KRas cells and both cell lines tolerate the transfection with TCP constructs. The Hs578t cell line contains an HRas mutation which may contribute to these cells tolerating elevated levels of deTyr-Tub. Interestingly, both the MDA-MB-231 and Hs578t cell lines are classified as Basal-B triple-negative breast cancer (TNBC) [
44]. While there are many differences between the MCF10A KRas, MDA-MB-231, and Hs578t cell lines the activation of the Ras-pathway and survival following TCP transfection appear to be connected in a manner currently unknown to us. To better understand the impact of increasing deTyr-Tub in these cell lines, transfected cells were imaged every 15 min for 19 h, starting at 24 h post-transfection. The mean squared displacement (MSD) and mean speed were quantified for both MDA-MB-231s and Hs578t (
Figure S6). The TCP constructs had differential effects on MSD and speed based on the cell line. These data suggest that deTyr-Tub may have an impact on migration, but more research must be done to understand its role and effect in this phenotype.
When plated on a gelatin matrix, MDA-MB-231 and Hs578t cells transfected with TCP constructs degraded the gelatin more effectively than cells transfected with the
sfGFP control vector (
Figure 6). The pattern of matrix degradation is consistent with the focal, punctate degradation that characterizes invadopodia, but we cannot conclusively determine that the increase in gelatin degradation is solely due to an increase in invadopodia. Cells at the end of the gelatin degradation assay were stained with phalloidin to look for co-localization of f-actin foci and gelatin voids (
Supplementary Figure S7). It is not yet known how deTyr-Tub increases gelatin degradation, but it is possible that deTyr-Tub could increase outward cell extension [
45] or promote the recruitment of components to interface between the cell and extracellular matrix and elongation of invadopodia [
46,
47]. Recently, invadopodia have been shown to be a component in early cellular invasion and metastasis [
48,
49]. The interaction between microtubules, invadopodia, and invasion has been examined with findings suggesting that specific trafficking along microtubules may aid in the degradation of the extracellular matrix [
50,
51]. Alternatively, enhanced deTyr-Tub may stimulate programmed cellular mechanisms to enhance invasive capacity, or microtubules with enhanced deTyr-Tub may increase the outward force at established points of cell attachment. It is also possible that the increase in TCP activity in transfected cells may be altering their regulation of proteases necessary to degrade gelatin. Utilizing the published structure of the components of the TCP, especially the vasohibins harboring the site of enzymatic activity, there is currently great interest in developing new compounds to reduce the formation of deTyr-Tub. The evidence presented here indicates that TCP could be a potential therapeutic target to reduce metastatic phenotypes.


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





