
T-Cells Expressing a Highly Potent PRAME-Specific T-Cell Receptor in Combination with a Chimeric PD1-41BB Co-Stimulatory Receptor Show a Favorable Preclinical Safety Profile and Strong Anti-Tumor Reactivity
 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Isolation of PRAME-Reactive T-Cell Clones
2.2. TCR Sequencing, Cloning, and Transduction
2.3. Effector Cell Preparation
2.4. Cell Culture
2.5. Cell Surface Staining for Flow Cytometry and FACS
2.6. Quantitative REAL-Time PCR (qPCR)
2.7. Cytokine Release Assay
2.8. Cytotoxicity Assay
2.9. Analysis of Homologous Peptides
2.10. Single-Cell Secretome Analysis
2.11. Xenograft Models
3. Results
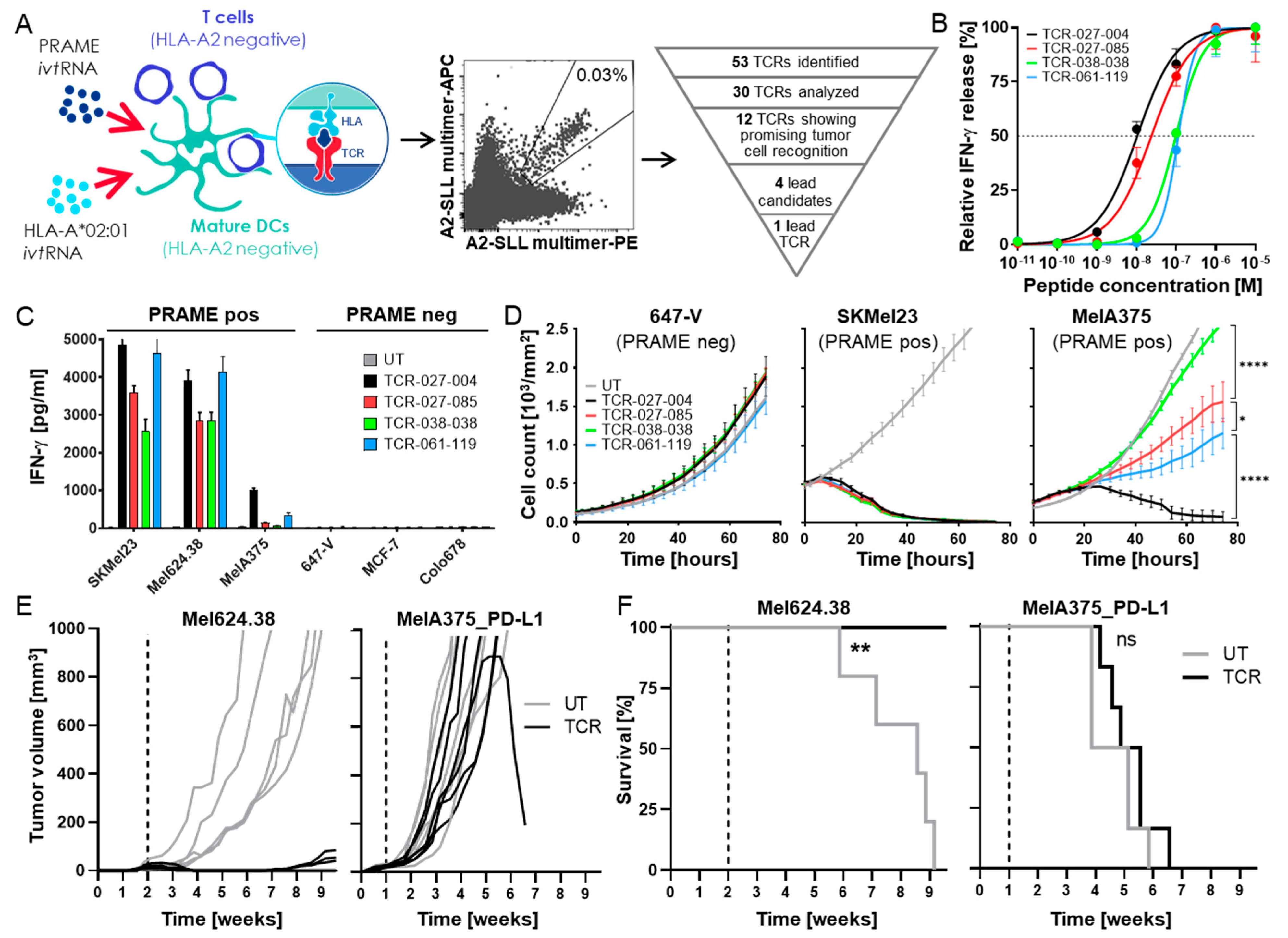
3.1. Isolation of a High-Affinity PRAME-Specific TCR from a Non-Tolerized T-Cell Repertoire
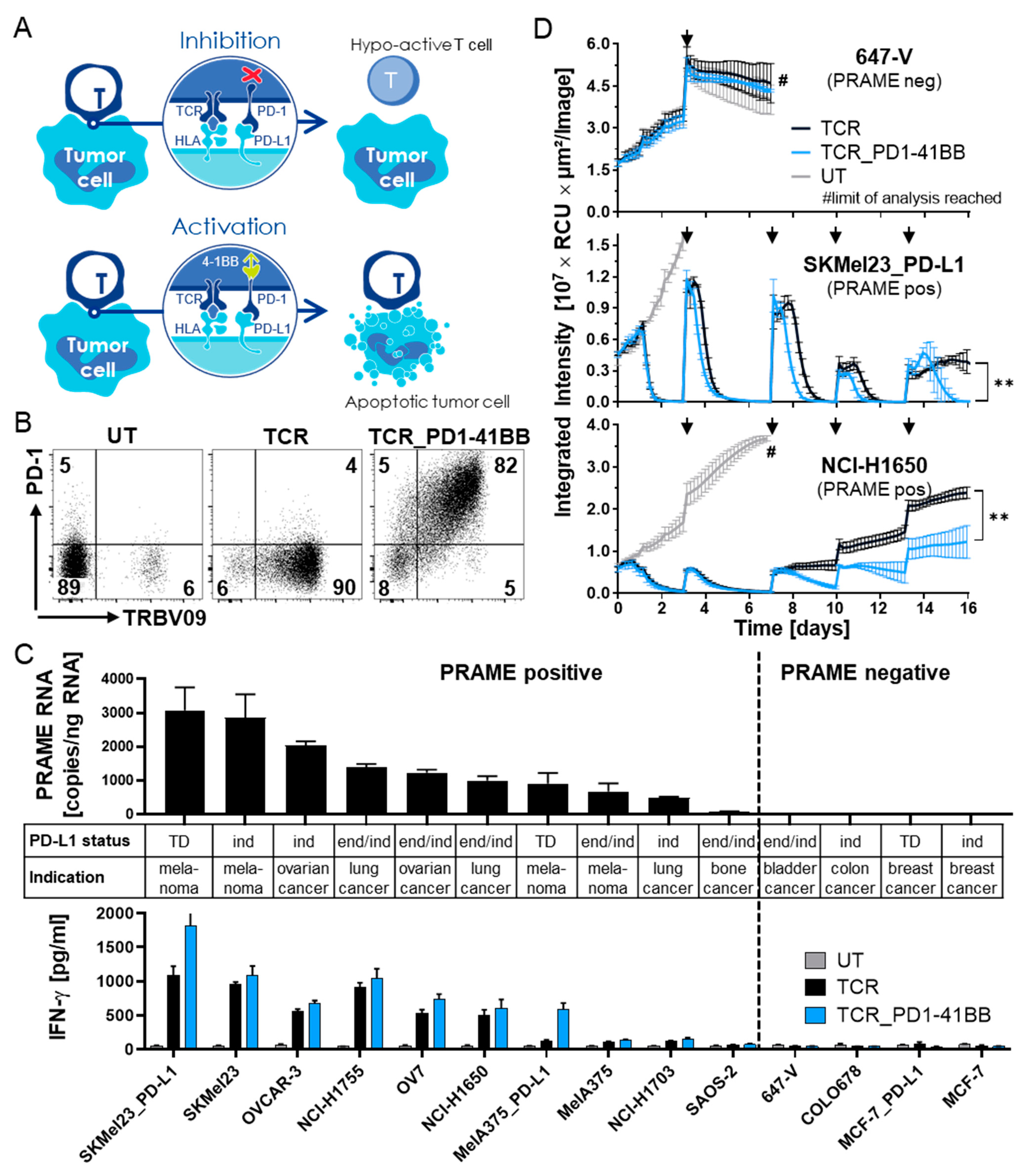
3.2. Chimeric PD1-41BB Co-Stimulatory Receptor Improves Effector Functions of TCR-Ts
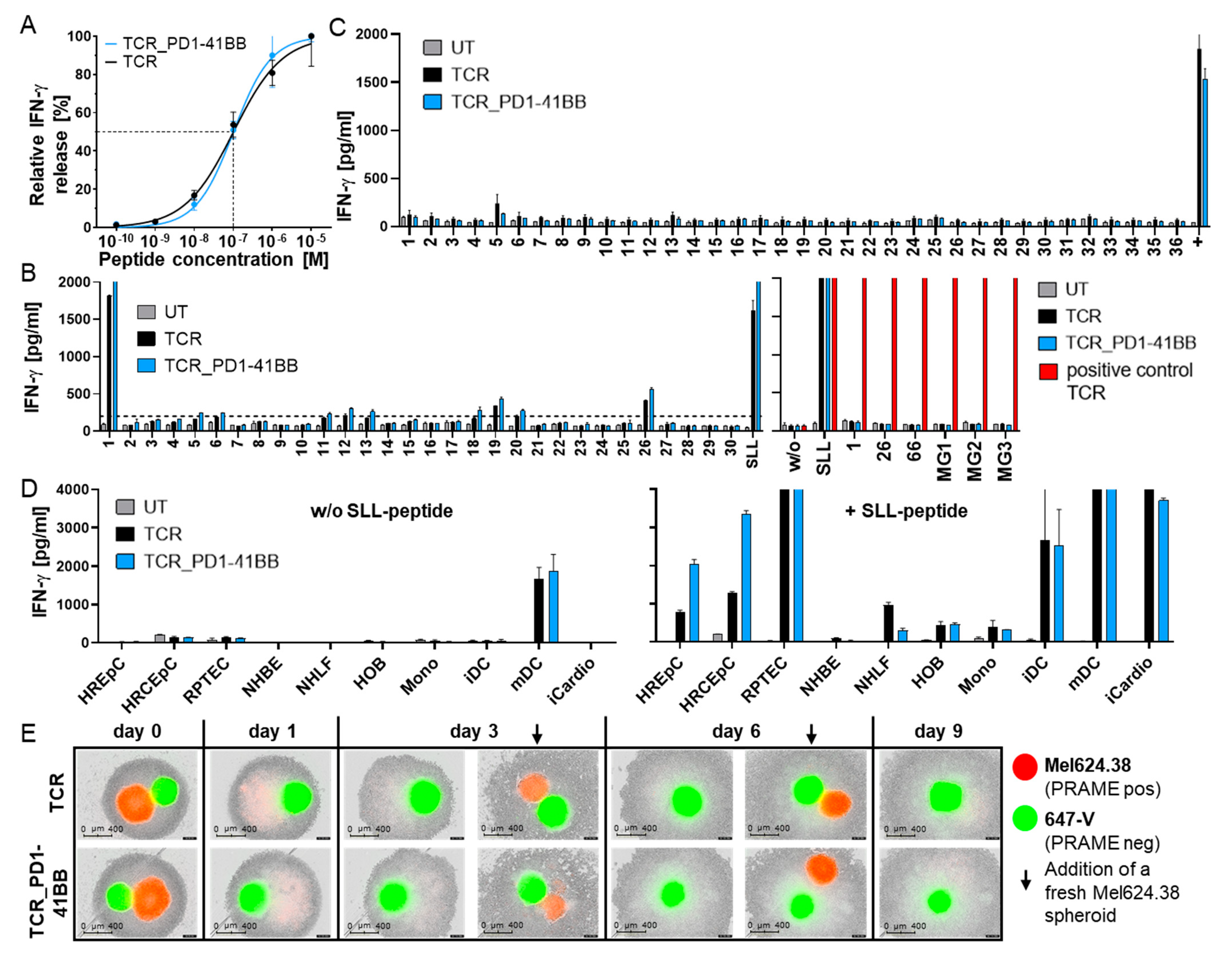
3.3. TCR-T In Vitro Safety Profile Is Not Changed by Co-Expression of PD1-41BB
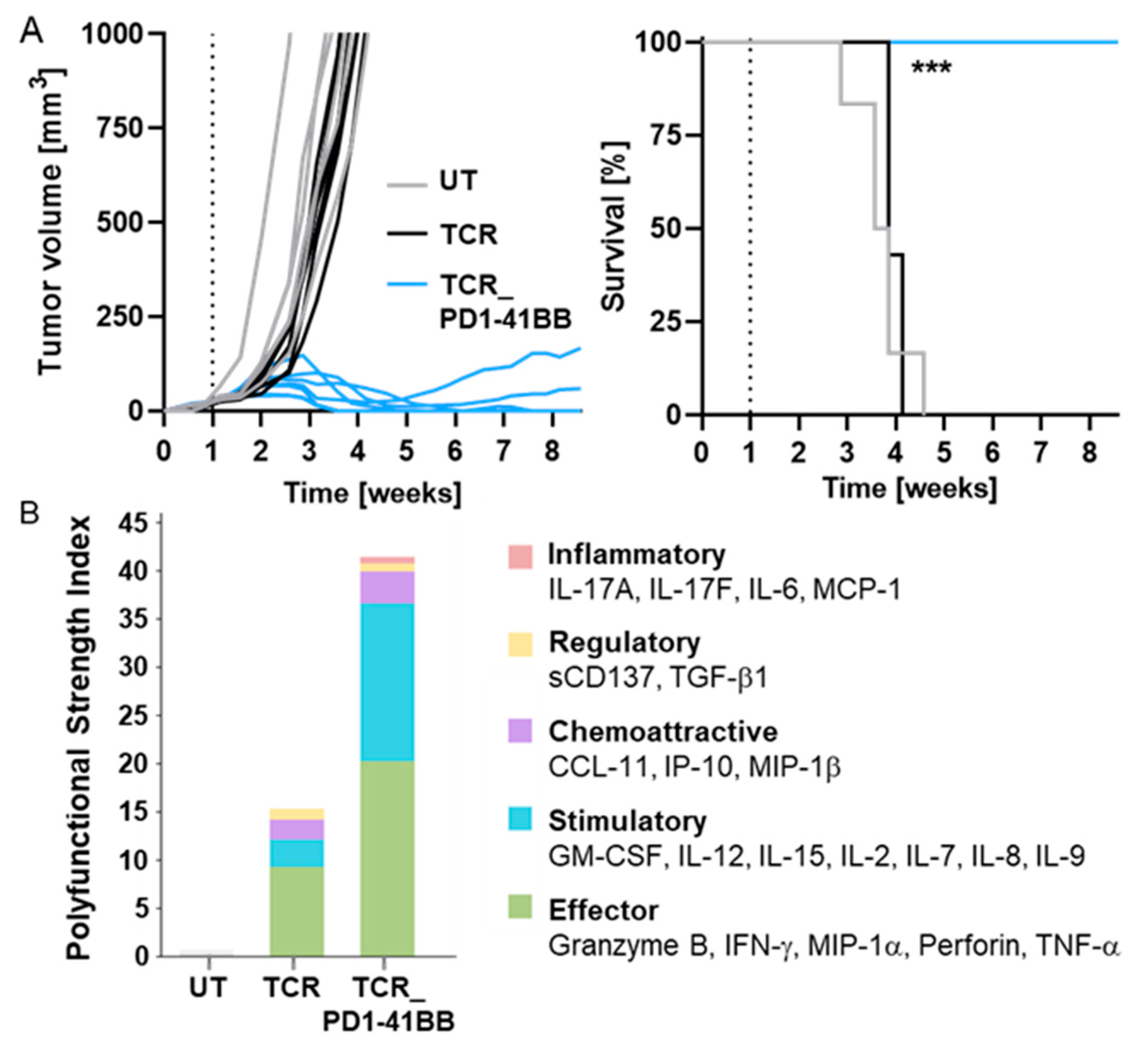
3.4. TCR-Ts Co-Expressing the Chimeric PD1-41BB Co-Stimulatory Receptor Reject Tumors In Vivo and Have a Poly-Cytokine Profile
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jiang, Y.; Li, Y.; Zhu, B. T-cell exhaustion in the tumor microenvironment. Cell Death Dis. 2015, 6, e1792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maimela, N.R.; Liu, S.; Zhang, Y. Fates of CD8+ T cells in Tumor Microenvironment. Comput. Struct. Biotechnol. J. 2018, 17, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, Activity, and Immune Correlates of Anti-PD-1 Antibody in Cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Allison, J.P. Immune Checkpoint Targeting in Cancer Therapy: Toward Combination Strategies with Curative Potential. Cell 2015, 161, 205–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callahan, M.K.; Postow, M.A.; Wolchok, J.D. CTLA-4 and PD-1 Pathway Blockade: Combinations in the Clinic. Front. Oncol. 2015, 4, 385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berman, D.; Korman, A.; Peck, R.; Feltquate, D.; Lonberg, N.; Canetta, R. The development of immunomodulatory monoclonal antibodies as a new therapeutic modality for cancer: The Bristol-Myers Squibb experience. Pharmacol. Ther. 2014, 148, 132–153. [Google Scholar] [CrossRef] [Green Version]
- O’Kane, G.M.; Labbé, C.; Doherty, M.K.; Young, K.; Albaba, H.; Leighl, N.B. Monitoring and Management of Immune-Related Adverse Events Associated with Programmed Cell Death Protein-1 Axis Inhibitors in Lung Cancer. Oncologist 2017, 22, 70–80. [Google Scholar] [CrossRef] [Green Version]
- Cruz, E.; Kayser, V. Monoclonal antibody therapy of solid tumors: Clinical limitations and novel strategies to enhance treatment efficacy. Biol. Targets Ther. 2019, 13, 33–51. [Google Scholar] [CrossRef] [Green Version]
- Ankri, C.; Shamalov, K.; Horovitz-Fried, M.; Mauer, S.; Cohen, C.J. Human T Cells Engineered To Express a Programmed Death 1/28 Costimulatory Retargeting Molecule Display Enhanced Antitumor Activity. J. Immunol. 2013, 191, 4121–4129. [Google Scholar] [CrossRef] [Green Version]
- Kobold, S.; Grassmann, S.; Chaloupka, M.; Lampert, C.; Wenk, S.; Kraus, F.; Rapp, M.; Duewell, P.; Zeng, Y.; Schmollinger, J.C.; et al. Impact of a New Fusion Receptor on PD-1–Mediated Immunosuppression in Adoptive T Cell Therapy. J. Natl. Cancer Inst. 2015, 107, djv146. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Ranganathan, R.; Jiang, S.; Fang, C.; Sun, J.; Kim, S.; Newick, K.; Lo, A.; June, C.H.; Zhao, Y.; et al. A Chimeric Switch-Receptor Targeting PD1 Augments the Efficacy of Second-Generation CAR T Cells in Advanced Solid Tumors. Cancer Res. 2016, 76, 1578–1590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlenker, R.; Olguín-Contreras, L.F.; Leisegang, M.; Schnappinger, J.; Disovic, A.; Ruhland, S. Chimeric PD-1:28 Receptor Upgrades Low-Avidity T cells and Restores Effector Function of Tumor-Infiltrating Lymphocytes for Adoptive Cell Therapy. Cancer Res. 2017, 77, 3577–3590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hui, E.; Cheung, J.; Zhu, J.; Su, X.; Taylor, M.J.; Wallweber, H.A.; Sasmal, D.K.; Huang, J.; Kim, J.M.; Mellman, I.; et al. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science 2017, 355, 1428–1433. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, P.; Okeke, E.; Clay, M.; Haydar, D.; Justice, J.; O’Reilly, C.; Pruett-Miller, S.; Papizan, J.; Moore, J.; Zhou, S.; et al. Route of 41BB/41BBL Costimulation Determines Effector Function of B7-H3-CAR.CD28ζ T Cells. Mol. Ther. Oncolytics 2020, 18, 202–214. [Google Scholar] [CrossRef]
- Al-Khadairi, G.; Decock, J. Cancer testis antigens and immunotherapy: Where do we stand in the targeting of PRAME? Cancers 2019, 11, 984. [Google Scholar] [CrossRef] [Green Version]
- Almeida, L.G.; Sakabe, N.J.; Deoliveira, A.R.; Silva, M.C.C.; Mundstein, A.S.; Cohen, T.; Chen, Y.-T.; Chua, R.; Gurung, S.; Gnjatic, S.; et al. CTdatabase: A knowledge-base of high-throughput and curated data on cancer-testis antigens. Nucleic Acids Res. 2009, 37, D816–D819. [Google Scholar] [CrossRef] [Green Version]
- Epping, M.T.; Bernards, R. A Causal Role for the Human Tumor Antigen Preferentially Expressed Antigen of Melanoma in Cancer. Cancer Res. 2006, 66, 10639–10642. [Google Scholar] [CrossRef] [Green Version]
- Lezcano, C.; Jungbluth, A.A.; Nehal, K.S.; Hollmann, T.J.; Busam, K.J. PRAME Expression in Melanocytic Tumors. Am. J. Surg. Pathol. 2018, 42, 1456–1465. [Google Scholar] [CrossRef]
- Pujol, J.-L.; De Pas, T.; Rittmeyer, A.; Vallières, E.; Kubisa, B.; Levchenko, E.; Wiesemann, S.; Masters, G.A.; Shen, R.; Tjulandin, S.A.; et al. Safety and Immunogenicity of the PRAME Cancer Immunotherapeutic in Patients with Resected Non-Small Cell Lung Cancer: A Phase I Dose Escalation Study. J. Thorac. Oncol. 2016, 11, 2208–2217. [Google Scholar] [CrossRef] [Green Version]
- Roszik, J.; Wang, W.-L.; Livingston, J.A.; Roland, C.L.; Ravi, V.; Yee, C.; Hwu, P.; Futreal, A.; Lazar, A.J.; Patel, D.R.; et al. Overexpressed PRAME is a potential immunotherapy target in sarcoma subtypes. Clin. Sarcoma Res. 2017, 7, 11. [Google Scholar] [CrossRef] [Green Version]
- Luk, S.J.; Van Der Steen, D.M.; Hagedoorn, R.S.; Jordanova, E.S.; Schilham, M.W.; Bovée, J.V.; Cleven, A.H.; Falkenburg, J.F.; Szuhai, K.; Heemskerk, M.H. PRAME and HLA Class I expression patterns make synovial sarcoma a suitable target for PRAME specific T-cell receptor gene therapy. Oncoimmunology 2018, 7, e1507600. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Barger, C.J.; Eng, K.H.; Klinkebiel, D.; Link, P.A.; Omilian, A.; Bshara, W.; Odunsi, K.; Karpf, A.R. PRAME expression and promoter hypomethylation in epithelial ovarian cancer. Oncotarget 2016, 7, 45352–45369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davari, K.; Holland, T.; Prassmayer, L.; Longinotti, G.; Ganley, K.P.; Pechilis, L.J.; Diaconu, I.; Nambiar, P.R.; Magee, M.S.; Schendel, D.J.; et al. Development of a CD8 co-receptor independent T-cell receptor specific for tumor-associated antigen MAGE-A4 for next generation T-cell-based immunotherapy. J. Immunother. Cancer 2021, 9, e002035. [Google Scholar] [CrossRef] [PubMed]
- Wilde, S.; Sommermeyer, D.; Frankenberger, B.; Schiemann, M.; Milosevic, S.; Spranger, S.; Pohla, H.; Uckert, W.; Busch, D.H.; Schendel, D.J. Dendritic cells pulsed with RNA encoding allogeneic MHC and antigen induce T cells with superior anti-tumor activity and higher TCR functional avidity. Blood 2009, 114, 2131–2139. [Google Scholar] [CrossRef]
- Wilde, S.; Geiger, C.; Milosevic, S.; Mosetter, B.; Eichenlaub, S.; Schendel, D.J. Generation of allo-restricted peptide-specific T cells using RNA-pulsed dendritic cells: A three phase experimental procedure. Oncoimmunology 2012, 1, 129–140. [Google Scholar] [CrossRef] [Green Version]
- Javorovic, M.; Wilde, S.; Zobywalski, A.; Noessner, E.; Lennerz, V.; Wölfel, T.; Schendel, D.J. Inhibitory Effect of RNA Pool Complexity on Stimulatory Capacity of RNA-pulsed Dendritic Cells. J. Immunother. 2008, 31, 52–62. [Google Scholar] [CrossRef]
- Sommermeyer, D.; Uckert, W. Minimal Amino Acid Exchange in Human TCR Constant Regions Fosters Improved Function of TCR Gene-Modified T Cells. J. Immunol. 2010, 184, 6223–6231. [Google Scholar] [CrossRef] [Green Version]
- Leisegang, M.; Engels, B.; Meyerhuber, P.; Kieback, E.; Sommermeyer, D.; Xue, S.-A.; Reuß, S.; Stauss, H.; Uckert, W. Enhanced functionality of T cell receptor-redirected T cells is defined by the transgene cassette. J. Mol. Med. 2008, 86, 573–583. [Google Scholar] [CrossRef]
- Scholten, K.B.; Kramer, D.; Kueter, E.W.; Graf, M.; Schoedl, T.; Meijer, C.J.; Schreurs, M.W.; Hooijberg, E. Codon modification of T cell receptors allows enhanced functional expression in transgenic human T cells. Clin. Immunol. 2006, 119, 135–145. [Google Scholar] [CrossRef]
- Riddell, S.R.; Greenberg, P.D. The use of anti-CD3 and anti-CD28 monoclonal antibodies to clone and expand human antigen-specific T cells. J. Immunol. Methods 1990, 128, 189–201. [Google Scholar] [CrossRef]
- Jonuleit, H.; Kühn, U.; Müller, G.; Steinbrink, K.; Paragnik, L.; Schmitt, E.; Knop, J.; Enk, A.H. Pro-inflammatory cytokines and prostaglandins induce maturation of potent immunostimulatory dendritic cells under fetal calf serum-free conditions. Eur. J. Immunol. 1997, 27, 3135–3142. [Google Scholar] [CrossRef] [PubMed]
- Jaravine, V.; Mösch, A.; Raffegerst, S.; Schendel, D.J.; Frishman, D. Expitope 2.0: A tool to assess immunotherapeutic antigens for their potential cross-reactivity against naturally expressed proteins in human tissues. BMC Cancer 2017, 17, 892. [Google Scholar] [CrossRef] [PubMed]
- Wilde, S.; Sommermeyer, D.; Leisegang, M.; Frankenberger, B.; Mosetter, B.; Uckert, W.; Schendel, D.J. Human Antitumor CD8+ T Cells Producing Th1 Polycytokines Show Superior Antigen Sensitivity and Tumor Recognition. J. Immunol. 2012, 189, 598–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amir, A.L.; van der Steen, D.M.; van Loenen, M.M.; Hagedoorn, R.S.; de Boer, R.; Kester, M.D.G.; de Ru, A.H.; Lugthart, G.-J.; van Kooten, C.; Hiemstra, P.S.; et al. PRAME-Specific Allo-HLA-Restricted T cells with Potent Antitumor Reactivity Useful for Therapeutic T-Cell Receptor Gene Transfer. Clin. Cancer Res. 2011, 17, 5615–5625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenberg, S.A.; Restifo, N.P. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015, 348, 62–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mardiana, S.; Solomon, B.J.; Darcy, P.K.; Beavis, P.A. Supercharging adoptive T cell therapy to overcome solid tumor-induced immunosuppression. Sci. Transl. Med. 2019, 11, eaaw2293. [Google Scholar] [CrossRef]
- Janelle, V.; Delisle, J.-S. T-Cell Dysfunction as a Limitation of Adoptive Immunotherapy: Current Concepts and Mitigation Strategies. Cancers 2021, 13, 598. [Google Scholar] [CrossRef]
- Ravetch, J.V.; Lanier, L.L. Immune Inhibitory Receptors. Science 2000, 290, 84–89. [Google Scholar] [CrossRef]
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992, 11, 3887–3895. [Google Scholar] [CrossRef]
- Simon, S.; Labarriere, N. PD-1 expression on tumor-specific T cells: Friend or foe for immunotherapy? Oncoimmunology 2018, 7, e1364828. [Google Scholar] [CrossRef]
- Baumeister, S.H.; Freeman, G.J.; Dranoff, G.; Sharpe, A.H. Coinhibitory Pathways in Immunotherapy for Cancer. Annu. Rev. Immunol. 2016, 34, 539–573. [Google Scholar] [CrossRef] [PubMed]
- Boussiotis, V.A. Molecular and Biochemical Aspects of the PD-1 Checkpoint Pathway. N. Engl. J. Med. 2016, 375, 1767–1778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blank, C.; Brown, I.; Peterson, A.C.; Spiotto, M.; Iwai, Y.; Honjo, T.; Gajewski, T.F. PD-L1/B7H-1 Inhibits the Effector Phase of Tumor Rejection by T Cell Receptor (TCR) Transgenic CD8+ T Cells. Cancer Res. 2004, 64, 1140–1145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.S.; Irving, B.A.; Hodi, F.S. Molecular pathways: Next-generation immunotherapy-inhibiting programmed death-ligand 1 and programmed death-1. Clin. Cancer Res. 2012, 18, 6580–6587. [Google Scholar] [CrossRef] [Green Version]
- Costa, R.; Carneiro, B.A.; Agulnik, M.; Rademaker, A.W.; Pai, S.G.; Villaflor, V.M.; Cristofanilli, M.; Sosman, J.A.; Giles, F.J. Toxicity profile of approved anti-PD-1 monoclonal antibodies in solid tumors: A systematic review and meta-analysis of randomized clinical trials. Oncotarget 2017, 8, 8910–8920. [Google Scholar] [CrossRef] [Green Version]
- Schoenfeld, A.J.; Hellmann, M.D. Acquired Resistance to Immune Checkpoint Inhibitors. Cancer Cell. 2020, 37, 443–455. [Google Scholar] [CrossRef]
- Su, S.; Hu, B.; Shao, J.; Shen, B.; Du, J.; Du, Y.; Zhou, J.; Yu, L.; Zhang, L.; Chen, F.; et al. CRISPR-Cas9 mediated efficient PD-1 disruption on human primary T cells from cancer patients. Sci. Rep. 2016, 6, 20070. [Google Scholar] [CrossRef] [Green Version]
- Sharpe, A.H.; Pauken, K.E. The diverse functions of the PD1 inhibitory pathway. Nat. Rev. Immunol. 2018, 18, 153–167. [Google Scholar] [CrossRef]
- Philipson, B.I.; O’Connor, R.S.; May, M.J.; June, C.H.; Albelda, S.M.; Milone, M.C. 4-1BB costimulation promotes CAR T cell survival through noncanonical NF-κB signaling. Sci. Signal. 2020, 13, eaay8248. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sailer, N.; Fetzer, I.; Salvermoser, M.; Braun, M.; Brechtefeld, D.; Krendl, C.; Geiger, C.; Mutze, K.; Noessner, E.; Schendel, D.J.; et al. T-Cells Expressing a Highly Potent PRAME-Specific T-Cell Receptor in Combination with a Chimeric PD1-41BB Co-Stimulatory Receptor Show a Favorable Preclinical Safety Profile and Strong Anti-Tumor Reactivity. Cancers 2022, 14, 1998. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14081998
Sailer N, Fetzer I, Salvermoser M, Braun M, Brechtefeld D, Krendl C, Geiger C, Mutze K, Noessner E, Schendel DJ, et al. T-Cells Expressing a Highly Potent PRAME-Specific T-Cell Receptor in Combination with a Chimeric PD1-41BB Co-Stimulatory Receptor Show a Favorable Preclinical Safety Profile and Strong Anti-Tumor Reactivity. Cancers. 2022; 14(8):1998. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14081998
Chicago/Turabian StyleSailer, Nadja, Ina Fetzer, Melanie Salvermoser, Monika Braun, Doris Brechtefeld, Christian Krendl, Christiane Geiger, Kathrin Mutze, Elfriede Noessner, Dolores J. Schendel, and et al. 2022. "T-Cells Expressing a Highly Potent PRAME-Specific T-Cell Receptor in Combination with a Chimeric PD1-41BB Co-Stimulatory Receptor Show a Favorable Preclinical Safety Profile and Strong Anti-Tumor Reactivity" Cancers 14, no. 8: 1998. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14081998