
MEOX2 Regulates the Growth and Survival of Glioblastoma Stem Cells by Modulating Genes of the Glycolytic Pathway and Response to Hypoxia

 , , , ,
, , , ,  and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Lentiviral Vectors and Infections
- shRNA53: 5′-CCGGGCATTCATATTAGCTGATGAACTCGAGTTCATCAGCTAATATGAATGCTTTTT-3′
- shRNA18: 5′-CCGGCATCAGAGCTGTCGGGAATTGCTCGAGCAATTCCCGACAGCTCTGATGTTTTTTG-3′
2.3. RNA Extraction and qRT-PCR
- ACTIN Forward: 5′-GCACTCTTCCAGCCTTCC-3′
- ACTIN Reverse: 5′-TGTCCACGTCACACTTCATG-3′
- MEOX2 Forward: 5′-GCAAGAGGAAAAGCGACAG-3′
- MEOX2 Reverse: 5′-CTTTCCTGGGTTTGCTGTTG-3′
- PPP2CA Forward: 5′-AGGAGCTGGTTACACCTTTG-3′
- PPP2CA Reverse: 5′-GCACCAGTTATATCCCTCCATC-3′
2.4. Protein Extraction and Western Blot Analysis
2.5. Sphere Formation Analysis
2.6. Growth Assay on Geltrex® Coated Plates
2.7. Cytofluorimetric Analysis of Apoptosis
2.8. RNA-Seq Analysis of BT273 and BT379 GSCs
2.9. Transcription Factors (TF) Binding Sites Enrichment Analysis
3. Results
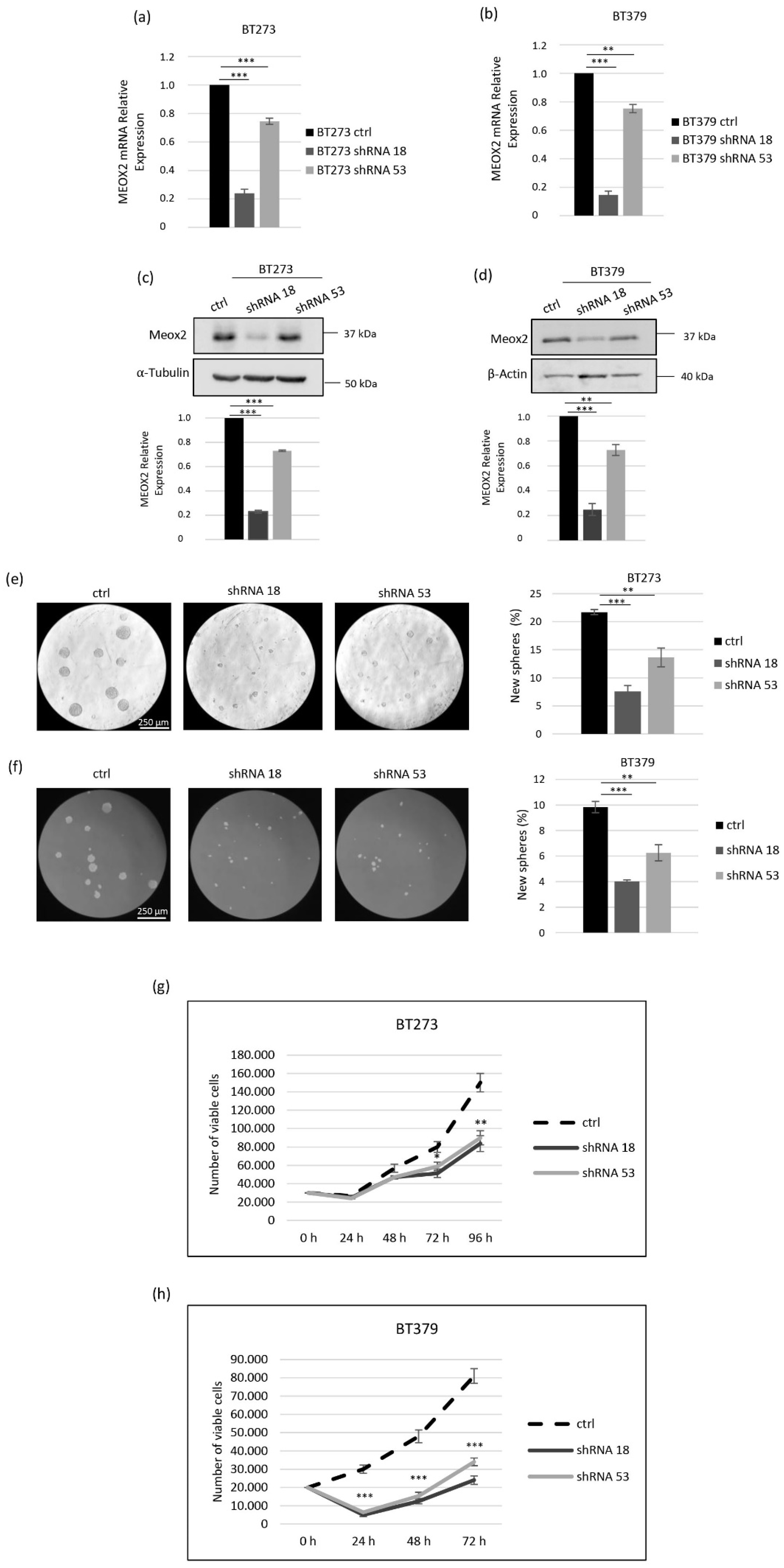
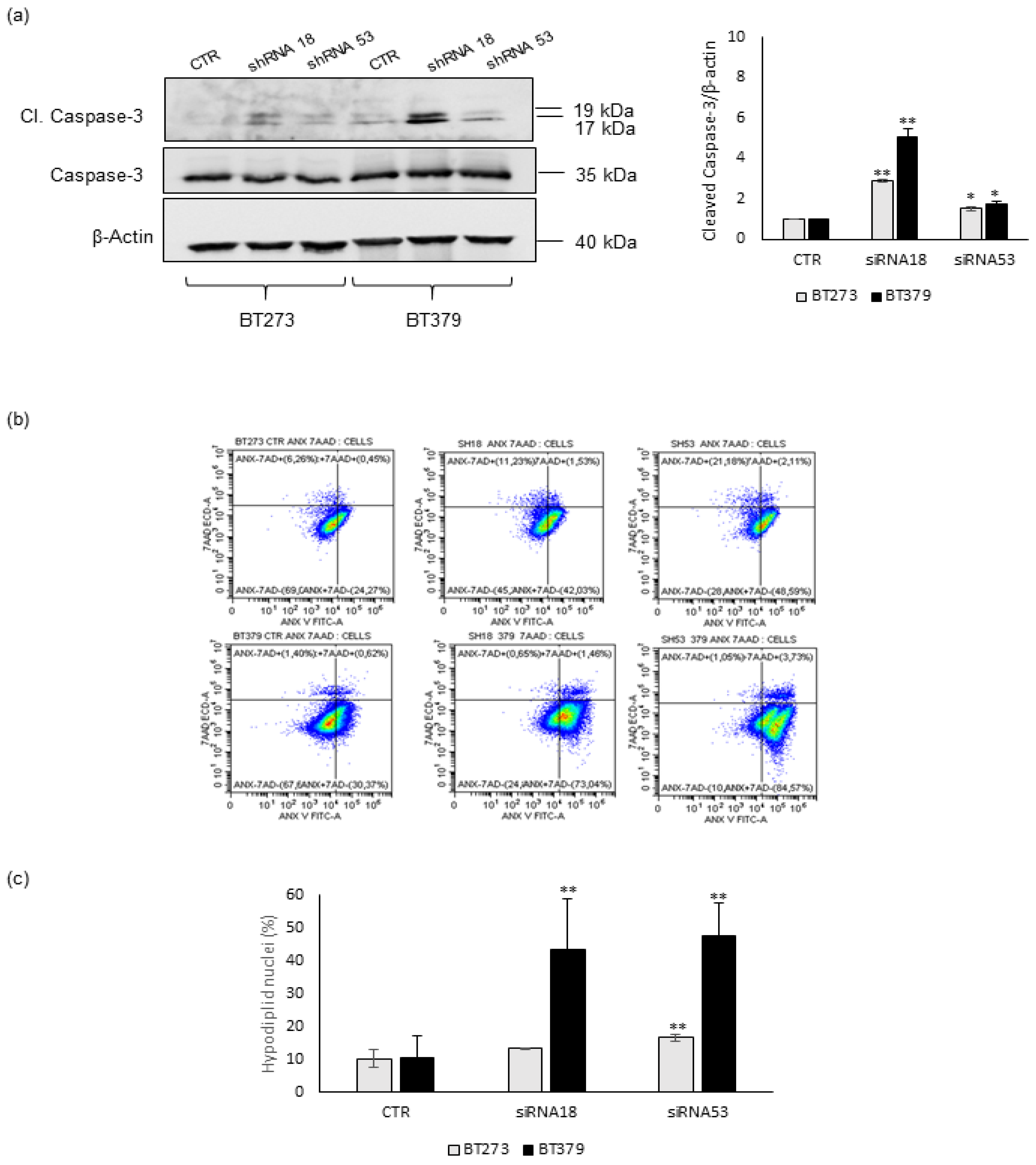
3.1. MEOX2 Depletion Inhibits the Sphere-Forming Ability and Induces Apoptosis in Glioblastoma Stem Cells
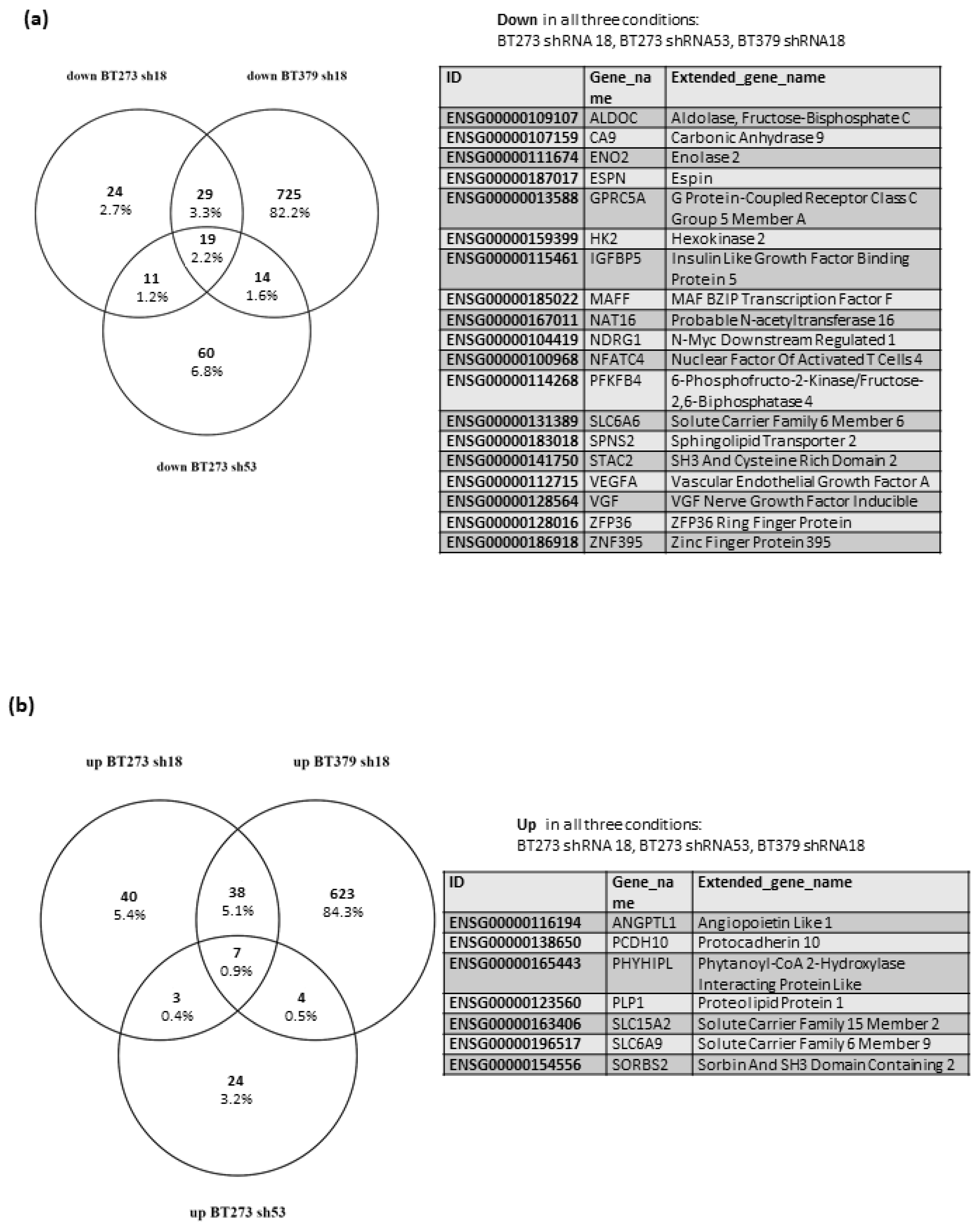
3.2. MEOX2 Knock down Variably Modulates Gene Expression in Different GSC Lines, but Consistently Affects the Glycolytic Pathway and the Response to Hypoxia
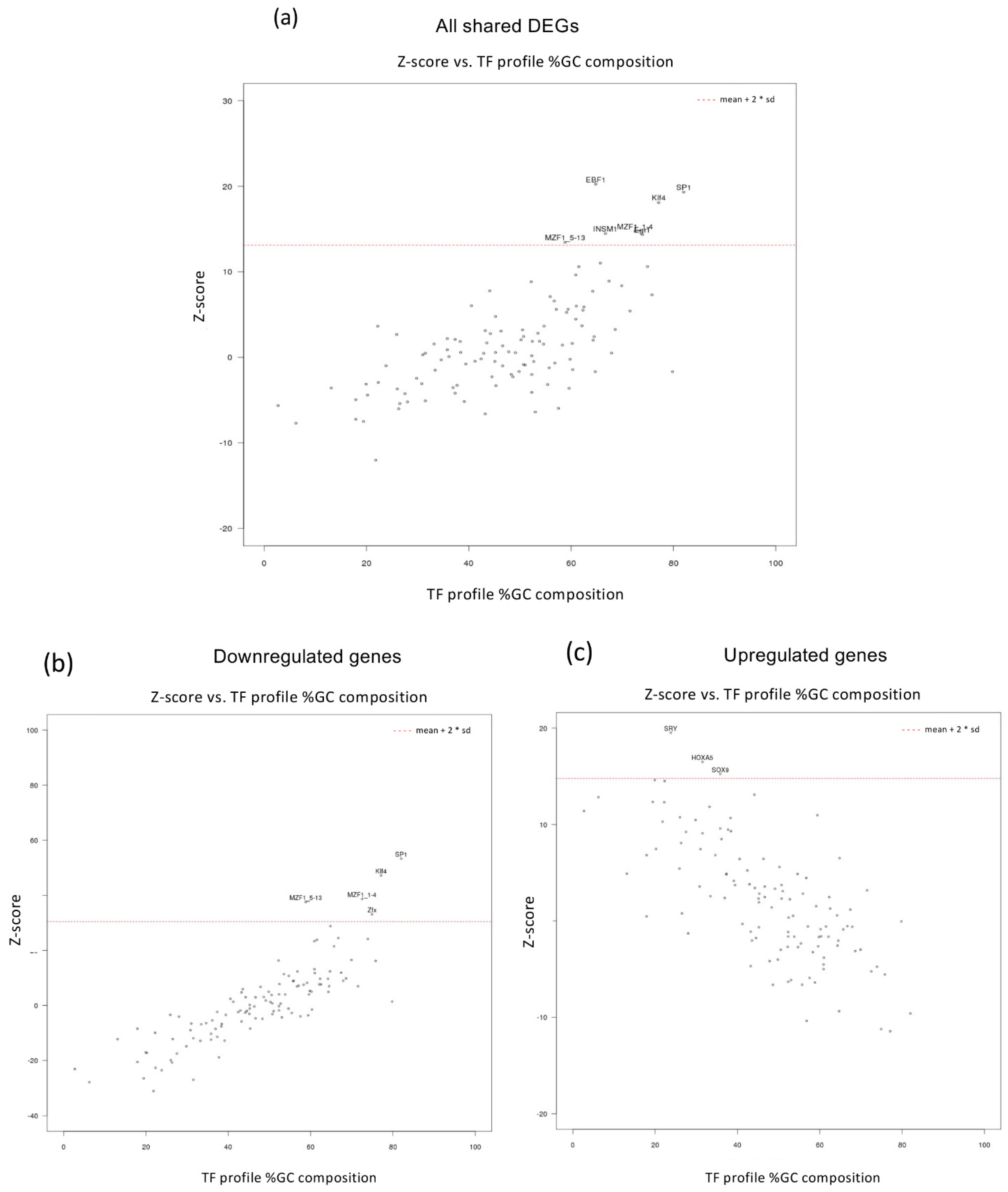
3.3. Up- and Down-Regulated Genes in MEOX2-Depleted GSCs Differ for the GC Content in Regulatory Regions
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, C.; Costa, A.; Osório, L.; Lago, R.C.; Linhares, P.; Carvalho, B.; Caeiro, C. Current Standards of Care in Glioblastoma Therapy. In Glioblastoma; De Vleeschouwer, S., Ed.; Codon Publications: Brisbane, Australia, 2017; Chapter 11. [Google Scholar]
- Stupp, R.; Taillibert, S.; Kanner, A.; Read, W.; Steinberg, D.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; Fink, K.; et al. Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs. Maintenance Temozolomide Alone on Survival in Patients With Glioblastoma: A Randomized Clinical Trial. JAMA 2017, 318, 2306–2316, Erratum in JAMA 2018, 319, 1824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Cancer Genome Atlas Research Network. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; de Carvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017, 32, 42–56.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Sun, D.; Chen, Y.J.; Xie, X.; Shi, Y.; Tabar, V.; Brennan, C.W.; Bale, T.A.; Jayewickreme, C.D.; Laks, D.R.; et al. Cell Lineage-Based Stratification for Glioblastoma. Cancer Cell 2020, 38, 366–379.e8. [Google Scholar] [CrossRef] [PubMed]
- Vieira de Castro, J.; Gonçalves, C.S.; Hormigo, A.; Costa, B.M. Exploiting the Complexities of Glioblastoma Stem Cells: Insights for Cancer Initiation and Therapeutic Targeting. Int. J. Mol. Sci. 2020, 21, 5278. [Google Scholar] [CrossRef]
- Singh, S.K.; Clarke, I.D.; Terasaki, M.; Bonn, V.E.; Hawkins, C.; Squire, J.; Dirks, P.B. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003, 63, 5821–5828. [Google Scholar]
- Galli, R.; Binda, E.; Orfanelli, U.; Cipelletti, B.; Gritti, A.; De Vitis, S.; Fiocco, R.; Foroni, C.; Dimeco, F.; Vescovi, A. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004, 64, 7011–7021. [Google Scholar] [CrossRef] [Green Version]
- Rheinbay, E.; Suvà, M.L.; Gillespie, S.M.; Wakimoto, H.; Patel, A.P.; Shahid, M.; Oksuz, O.; Rabkin, S.D.; Martuza, R.L.; Rivera, M.N.; et al. An aberrant transcription factor network essential for Wnt signaling and stem cell maintenance in glioblastoma. Cell Rep. 2013, 3, 1567–1579. [Google Scholar] [CrossRef] [Green Version]
- MacLeod, G.; Bozek, D.A.; Rajakulendran, N.; Monteiro, V.; Ahmadi, M.; Steinhart, Z.; Kushida, M.M.; Yu, H.; Coutinho, F.J.; Cavalli, F.M.G.; et al. Genome-Wide CRISPR-Cas9 Screens Expose Genetic Vulnerabilities and Mechanisms of Temozolomide Sensitivity in Glioblastoma Stem Cells. Cell Rep. 2019, 27, 971–986.e9. [Google Scholar] [CrossRef] [Green Version]
- Guardia, G.D.A.; Correa, B.R.; Araujo, P.R.; Qiao, M.; Burns, S.; Penalva, L.O.F.; Galante, P.A.F. Proneural and mesenchymal glioma stem cells display major differences in splicing and lncRNA profiles. NPJ Genom. Med. 2020, 5, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozuka-Hata, H.; Nasu-Nishimura, Y.; Koyama-Nasu, R.; Ao-Kondo, H.; Tsumoto, K.; Akiyama, T.; Oyama, M. Phosphoproteome of human glioblastoma initiating cells reveals novel signaling regulators encoded by the transcriptome. PLoS ONE 2012, 7, e43398. [Google Scholar] [CrossRef] [PubMed]
- Marziali, G.; Signore, M.; Buccarelli, M.; Grande, S.; Palma, A.; Biffoni, M.; Rosi, A.; D’Alessandris, Q.G.; Martini, M.; Larocca, L.M.; et al. Metabolic/Proteomic Signature Defines Two Glioblastoma Subtypes With Different Clinical Outcome. Sci. Rep. 2016, 6, 21557. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.C.; Lu, G.X.; Zhang, H.W.; Zhong, X.M.; Cong, X.L.M.; Xue, S.B.; Kong, R.; Li, D.; Chang, Z.Y.; Wang, X.F.; et al. Proteogenomic characterization and integrative analysis of glioblastoma multiforme. Oncotarget 2017, 8, 97304–97312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mostovenko, E.; Végvári, Á.; Rezeli, M.; Lichti, C.F.; Fenyö, D.M.; Wang, Q.; Lang, F.F.; Sulman, E.P.; Sahlin, K.B.; Marko-Varga, G.; et al. Large Scale Identification of Variant Proteins in Glioma Stem Cells. ACS Chem. Neurosci. 2018, 9, 73–79. [Google Scholar] [CrossRef]
- Asif, S.; Fatima, R.; Krc, R.; Bennett, J.; Raza, S. Comparative proteogenomic characterization of glioblastoma. CNS Oncol. 2019, 8, CNS37. [Google Scholar] [CrossRef]
- Le Boiteux, E.; Court, F.; Guichet, P.O.; Vaurs-Barrière, C.; Vaillant, I.; Chautard, E.; Verrelle, P.; Costa, B.M.; Karayan-Tapon, L.; Fogli, A.; et al. Widespread overexpression from the four DNA hypermethylated HOX clusters in aggressive (IDHwt) glioma is associated with H3K27me3 depletion and alternative promoter usage. Mol. Oncol. 2021, 15, 1995–2010. [Google Scholar] [CrossRef]
- Gonçalves, C.E.; Le Boiteux, E.; Arnaud, P.; Costa, B.M. HOX gene cluster (de)regulation in brain: From neurodevelopment to malignant glial tumours. Cell Mol. Life Sci. 2020, 77, 3797–3821. [Google Scholar] [CrossRef]
- Abdel-Fattah, R.; Xiao, A.; Bomgardner, D.; Pease, C.S.; Lopes, M.B.; Hussaini, I.M. Differential expression of HOX genes in neoplastic and non-neoplastic human astrocytes. J. Pathol. 2006, 209, 15–24. [Google Scholar] [CrossRef]
- Costa, B.M.; Smith, J.S.; Chen, Y. Reversing HOXA9 oncogene activation by PI3K inhibition: Epigenetic mechanism and prognostic significance in human glioblastoma. Cancer Res. 2010, 70, 453–462. [Google Scholar] [CrossRef] [Green Version]
- Gorski, D.H.; LePage, D.F.; Patel, C.V.; Copeland, N.G.; Jenkins, N.A.; Walsh, K. Molecular cloning of a diverged homeobox gene that is rapidly down-regulated during the G0/G1 transition in vascular smooth muscle cells. Mol. Cell Biol. 1993, 13, 3722–3733. [Google Scholar] [CrossRef] [PubMed]
- Ducray, F.; de Reyniès, A.; Chinot, O.; Idbaih, A.; Figarella-Branger, D.; Colin, C.; Karayan-Tapon, L.; Chneiweiss, H.; Wager, M.; Vallette, F.; et al. An ANOCEF genomic and transcriptomic microarray study of the response to radiotherapy or to alkylating first-line chemotherapy in glioblastoma patients. Mol. Cancer 2010, 9, 234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tachon, G.; Cortes, U.; Guichet, P.O.; Rivet, P.; Balbous, A.; Masliantsev, K.; Berger, A.; Boissonnade, O.; Wager, M.; Karayan-Tapon, L. Cell Cycle Changes after Glioblastoma Stem Cell Irradiation: The Major Role of RAD51. Int. J. Mol. Sci. 2018, 19, 3018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Q.; Huang, C.; Cao, H.; Lin, J.; Gong, X.M.; Li, J.; Chen, Y.; Tian, Z.; Fang, Z.; Huang, J. A Novel Prognostic Signature of Transcription Factors for the Prediction in Patients With GBM. Front. Genet. 2019, 10, 906. [Google Scholar] [CrossRef]
- Bao, Z.S.; Zhang, C.B.; Wang, H.J.; Yan, W.; Liu, Y.W.; Li, M.Y.; Zhang, W. Whole-genome mRNA expression profiling identifies functional and prognostic signatures in patients with mesenchymal glioblastoma multiforme. CNS Neurosci. Ther. 2013, 19, 714–720. [Google Scholar] [CrossRef] [PubMed]
- Mack, S.C.; Singh, I.; Wang, X.; Hirsch, R.; Wu, Q.; Villagomez, R.; Bernatchez, J.A.; Zhu, Z.; Gimple, R.C.; Kim, L.J.Y.; et al. Chromatin landscapes reveal developmentally encoded transcriptional states that define human glioblastoma. J. Exp. Med. 2019, 216, 1071–1090. [Google Scholar] [CrossRef] [Green Version]
- Tachon, G.; Masliantsev, K.; Rivet, P.; Desette, A.; Milin, S.; Gueret, E.; Wager, M.; Karayan-Tapon, L.; Guichet, P.O. MEOX2 Transcription Factor Is Involved in Survival and Adhesion of Glioma Stem-like Cells. Cancers 2021, 13, 5943. [Google Scholar] [CrossRef]
- De Bacco, F.; Casanova, E.; Medico, E.; Pellegatta, S.; Orzan, F.; Albano, R.; Luraghi, P.; Reato, G.; D’Ambrosio, A.; Porrati, P.; et al. The MET oncogene is a functional marker of a glioblastoma stem cell subtype. Cancer Res. 2012, 72, 4537–4550. [Google Scholar] [CrossRef] [Green Version]
- De Bacco, F.; Orzan, F.; Erriquez, J.; Casanova, E.; Barault, L.; Albano, R.; D’Ambrosio, A.; Bigatto, V.; Reato, G.; Patanè, M.; et al. ERBB3 overexpression due to miR-205 inactivation confers sensitivity to FGF, metabolic activation, and liability to ERBB3 targeting in glioblastoma. Cell Rep. 2021, 36, 109455. [Google Scholar] [CrossRef]
- Matteucci, C.; Minutolo, A.; Balestrieri, E.; Marino-Merlo, F.; Bramanti, P.; Garaci, E.; Macchi, B.; Mastino, A. Inhibition of NF-κB activation sensitizes U937 cells to 3’-azido-3’-deoxythymidine induced apoptosis. Cell Death Dis. 2010, 1, e81. [Google Scholar] [CrossRef] [Green Version]
- Kwon, A.T.; Arenillas, D.J.; Worsley Hunt, R.; Wasserman, W.W. oPOSSUM-3: Advanced analysis of regulatory motif over-representation across genes or ChIP-Seq datasets. G3 Bethesda 2012, 2, 987–1002. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.C.; Branellec, D.; Gorski, D.H.; Guo, K.; Perlman, H.; Dedieu, J.F.; Pastore, C.; Mahfoudi, A.; Denèfle, P.; Isner, J.M.; et al. p21CIP1-mediated inhibition of cell proliferation by overexpression of the gax homeodomain gene. Genes Dev. 1997, 11, 1674–1689. [Google Scholar] [CrossRef] [Green Version]
- Irelan, J.T.; Gutierrez Del Arroyo, A.; Gutierrez, A.; Peters, G.; Quon, K.C.; Miraglia, L.; Chanda, S.K. A functional screen for regulators of CKDN2A reveals MEOX2 as a transcriptional activator of INK4a. PLoS ONE 2009, 4, e5067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, S.; Leal, A.D.; Gorski, D.H. The homeobox gene Gax inhibits angiogenesis through inhibition of nuclear factor-kappaB-dependent endothelial cell gene expression. Cancer Res. 2005, 65, 1414–1424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Rabson, A.B.; Gorski, D.H. MEOX2 regulates nuclear factor-kappaB activity in vascular endothelial cells through interactions with p65 and IkappaBbeta. Cardiovasc. Res. 2010, 87, 723–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Shi, X.; Wei, S.; Ma, D.; Oyinlade, O.; Lv, S.Q.; Ying, M.; Zhang, Y.A.; Claypool, S.M.; Watkins, P.; et al. Krüppel-like factor 4 (KLF4) induces mitochondrial fusion and increases spare respiratory capacity of human glioblastoma cells. J. Biol. Chem. 2018, 293, 6544–6555. [Google Scholar] [CrossRef] [Green Version]
- Yin, J.; Shi, Z.; Wei, W.; Lu, C.; Wei, Y.; Yan, W.; Li, R.; Zhang, J.; You, Y.; Wang, X. MiR-181b suppress glioblastoma multiforme growth through inhibition of SP1-mediated glucose metabolism. Cancer Cell Int. 2020, 20, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fazi, B.; Felsani, A.; Grassi, L.; Moles, A.; D’Andrea, D.; Toschi, N.; Sicari, D.; De Bonis, P.; Anile, C.; Guerrisi, M.G.; et al. The transcriptome and miRNome profiling of glioblastoma tissues and peritumoral regions highlights molecular pathways shared by tumors and surrounding areas and reveals differences between short-term and long-term survivors. Oncotarget 2015, 6, 22526–22552. [Google Scholar] [CrossRef] [Green Version]
- Markmann, A.; Rauterberg, J.; Vischer, P.; Robenek, H.; Echtermeyer, F.; Will, H.; Seidler, D.G.; Young, M.F.; Kresse, H. Expression of transcription factors and matrix genes in response to serum stimulus in vascular smooth muscle cells. Eur. J. Cell Biol. 2003, 82, 119–129. [Google Scholar] [CrossRef]
- Mahase, S.; Rattenni, R.N.; Wesseling, P.; Leenders, W.; Baldotto, C.; Jain, R.; Zagzag, D. Hypoxia-Mediated Mechanisms Associated with Antiangiogenic Treatment Resistance in Glioblastomas. Am. J. Pathol. 2017, 187, 940–953. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Forbes, R.A.; Verma, A. Hypoxia-inducible factor 1 activation by aerobic glycolysis implicates the Warburg effect in carcinogenesis. J. Biol. Chem. 2002, 277, 23111–23115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jelluma, N.; Yang, X.; Stokoe, D.; Evan, G.I.; Dansen, T.B.; Haas-Kogan, D.A. Glucose withdrawal induces oxidative stress followed by apoptosis in glioblastoma cells but not in normal human astrocytes. Mol. Cancer Res. 2006, 4, 319–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuen, C.A.; Asuthkar, S.; Guda, M.R.; Tsung, A.J.; Velpula, K.K. Cancer stem cell molecular reprogramming of the Warburg effect in glioblastomas: A new target gleaned from an old concept. CNS Oncol. 2016, 5, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Goidts, V.; Bageritz, J.; Puccio, L.; Nakata, S.; Zapatka, M.; Barbus, S.; Toedt, G.; Campos, B.; Korshunov, A.; Momma, S.; et al. RNAi screening in glioma stem-like cells identifies PFKFB4 as a key molecule important for cancer cell survival. Oncogene 2012, 31, 3235–3243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agnihotri, S.; Mansouri, S.; Burrell, K.; Li, M.; Mamatjan, Y.; Liu, J.; Nejad, R.; Kumar, S.; Jalali, S.; Singh, S.K.; et al. Ketoconazole and Posaconazole Selectively Target HK2-expressing Glioblastoma Cells. Clin. Cancer Res. 2019, 25, 844–855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanzey, M.; Abdul Rahim, S.A.; Oudin, A.; Dirkse, A.; Kaoma, T.; Vallar, L.; Herold-Mende, C.; Bjerkvig, R.; Golebiewska, A.; Niclou, S.P. Comprehensive analysis of glycolytic enzymes as therapeutic targets in the treatment of glioblastoma. PLoS ONE 2015, 10, e0123544. [Google Scholar] [CrossRef]
- Wang, W.; Li, J.; Lin, F.; Guo, J.; Zhao, J. Expression and prognostic value of mRNAs in lower grade glioma with MGMT promoter methylated. J. Clin. Neurosci. 2020, 75, 45–51. [Google Scholar] [CrossRef]
- Murat, A.; Migliavacca, E.; Hussain, S.F.; Heimberger, A.B.; Desbaillets, I.; Hamou, M.F.; Rüegg, C.; Stupp, R.; Delorenzi, M.; Hegi, M.E. Modulation of angiogenic and inflammatory response in glioblastoma by hypoxia. PLoS ONE 2009, 4, e5947. [Google Scholar] [CrossRef]
- Herwartz, C.; Castillo-Juárez, P.; Schröder, L.; Barron, B.L.; Steger, G. The Transcription Factor ZNF395 Is Required for the Maximal Hypoxic Induction of Proinflammatory Cytokines in U87-MG Cells. Mediat. Inflamm. 2015, 2015, 804264. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, K.; Shouguchi-Miyata, J.; Miyamoto, N.; Ikeda, J.E. Novel nuclear shuttle proteins, HDBP1 and HDBP2, bind to neuronal cell-specific cis-regulatory element in the promoter for the human Huntington’s disease gene. J. Biol. Chem. 2004, 279, 7275–7286. [Google Scholar] [CrossRef] [Green Version]
- Yao, X.; Tan, J.; Lim, K.J.; Koh, J.; Ooi, W.F.; Li, Z.; Huang, D.; Xing, M.; Chan, Y.S.; Qu, J.Z.; et al. VHL Deficiency Drives Enhancer Activation of Oncogenes in Clear Cell Renal Cell Carcinoma. Cancer Discov. 2017, 7, 1284–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.T.T.; Zhang, Y.; Shang, E.; Shu, C.; Torrini, C.; Zhao, J.; Bianchetti, E.; Mela, A.; Humala, N.; Mahajan, A.; et al. HDAC inhibitors elicit metabolic reprogramming by targeting super-enhancers in glioblastoma models. J. Clin. Investig. 2020, 130, 3699–3716. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Bergström, T.; Jiang, Y.; Johansson, P.; Marinescu, V.D.; Lindberg, N.; Segerman, A.; Wicher, G.; Niklasson, M.; Baskaran, S.; et al. The Human Glioblastoma Cell Culture Resource: Validated Cell Models Representing All Molecular Subtypes. EBioMedicine 2015, 2, 1351–1363. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cells | Percentage (%) | ANX+7AAD- | ANX+7AAD+ | ANX-7AAD+ | Hypodiploid Nuclei |
|---|---|---|---|---|---|
| BT273 | CTR | 41.92 ± 12,71 | 7.62 ± 10.58 | 2.67 ± 2.07 | 10.19 ± 2.76 |
| shRNA18 | * 62.19 ± 15.65 | 4.29 ± 4.14 | 3.40 ± 4.52 | 13.18 ± 0.05 | |
| shRNA53 | +** 71.79 ± 16.40 | 3.26 ± 1.01 | 7.79 ± 9.46 | * 16.55 ± 1.04 | |
| BT379 | CTR | 29.37 ± 2.25 | 5.35 ± 3.34 | 7.05 ± 4.26 | 10.52 ± 6.61 |
| shRNA18 | * 67.54 ± 5.25 | 6.46 ± 5.18 | 0.90 ± 0.37 | ** 43.52 ± 15.28 | |
| shRNA53 | ++** 79.82 ± 3.38 | 12.99 ± 6.55 | 4.51 ± 4.58 | ** 47.46 ± 10.19 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Proserpio, C.; Galardi, S.; Desimio, M.G.; Michienzi, A.; Doria, M.; Minutolo, A.; Matteucci, C.; Ciafrè, S.A. MEOX2 Regulates the Growth and Survival of Glioblastoma Stem Cells by Modulating Genes of the Glycolytic Pathway and Response to Hypoxia. Cancers 2022, 14, 2304. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14092304
Proserpio C, Galardi S, Desimio MG, Michienzi A, Doria M, Minutolo A, Matteucci C, Ciafrè SA. MEOX2 Regulates the Growth and Survival of Glioblastoma Stem Cells by Modulating Genes of the Glycolytic Pathway and Response to Hypoxia. Cancers. 2022; 14(9):2304. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14092304
Chicago/Turabian StyleProserpio, Carla, Silvia Galardi, Maria Giovanna Desimio, Alessandro Michienzi, Margherita Doria, Antonella Minutolo, Claudia Matteucci, and Silvia Anna Ciafrè. 2022. "MEOX2 Regulates the Growth and Survival of Glioblastoma Stem Cells by Modulating Genes of the Glycolytic Pathway and Response to Hypoxia" Cancers 14, no. 9: 2304. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14092304