
Differences in Tumor Growth and Differentiation in NSG and Humanized-BLT Mice; Analysis of Human vs. Humanized-BLT-Derived NK Expansion and Functions
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Tumor Cell Lines and Reagents
2.2. Tumor Implantation in NSG and hu-BLT Mice
2.3. Single-Cell Isolation of Tumors, Bone Marrow, Spleen, and Peripheral Blood of NSG and hu-BLT Mice
2.4. Human and hu-BLT NK Cells’ and Monocytes’ Purifications
2.5. Generation of Human and hu-BLT Osteoclasts
2.6. Expansion of Human and hu-BLT NK Cells
2.7. NK Cells’ Supernatant Induced Differentiation of OSCSCs and MP2 Tumors
2.8. Enzyme-Linked Immunosorbent Assays (ELISAs)
2.9. 4-Hour Standard 51Cr Release Cytotoxicity Assay
2.10. Surface Marker Analysis Assay
2.11. Statistical Analysis
3. Results
3.1. Tumor-Bearing NSG Mice Lost Higher Body Weight in Comparison to Tumor-Bearing hu-BLT Mice
3.2. Higher Tumor Growth in NSG Mice in Comparison to hu-BLT Mice
3.3. Tumor Resected from NSG Mice Exhibited Higher Growth Rate in Cultures in Comparison to Those Resected from hu-BLT Mice
3.4. Increased Level of IFN-γ Secretion Seen in Tumors Harvested from Hu-BLT Mice
3.5. Tumors Isolated from NSG Mice Exhibited More Stem-like Phenotype in Comparison to Those from Hu-BLT Mice
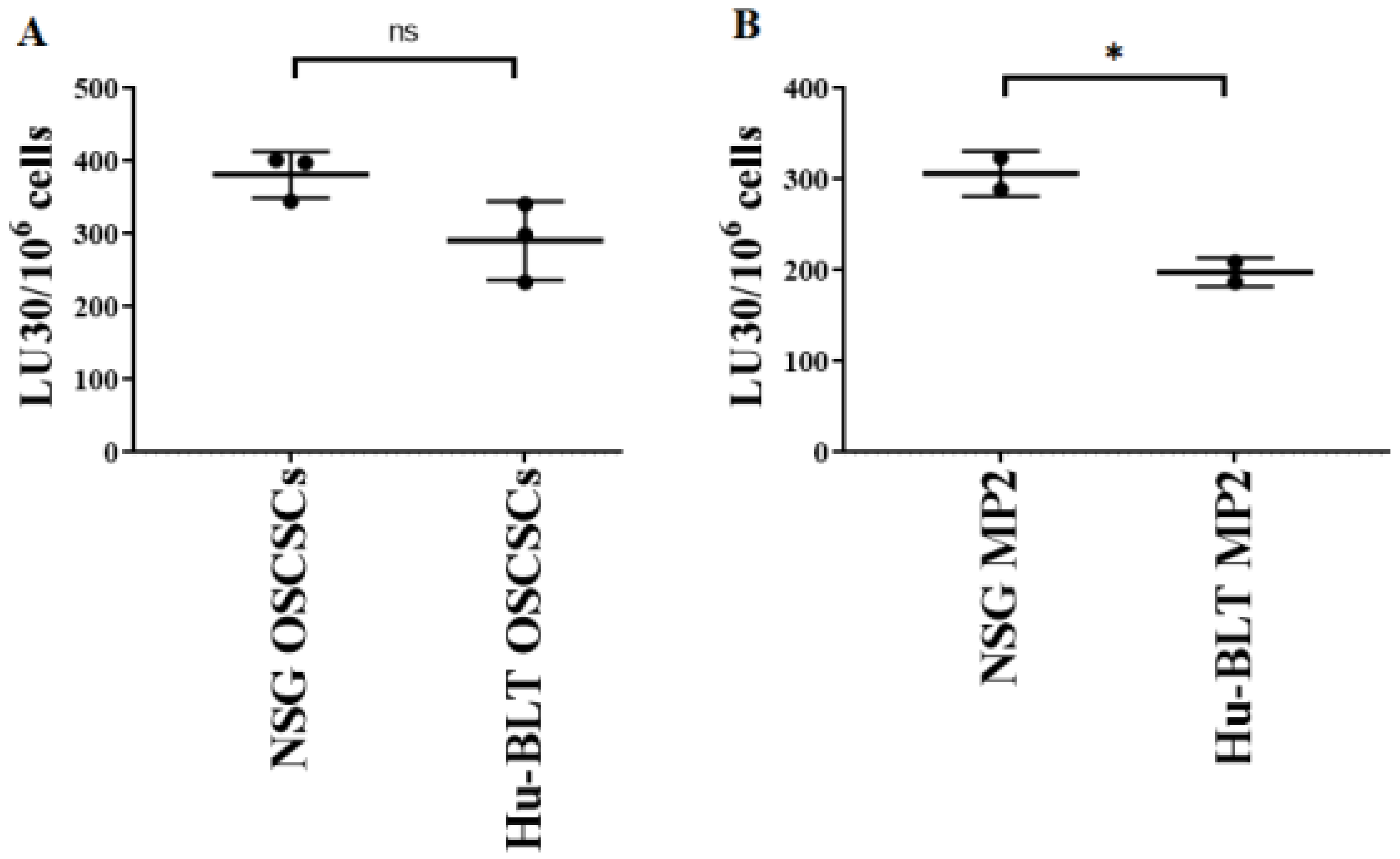
3.6. Tumors Expressing Higher MHC-Class I and CD54 on Their Surface Were Found Resistant to NK Cell-Mediated Cytotoxicity
3.7. hu-BLT NK Cells Are Capable of Expansion in the Presence of Osteoclasts Like Human NK Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shimizu, S.; Hong, P.; Arumugam, B.; Pokomo, L.; Boyer, J.; Koizumi, N.; Kittipongdaja, P.; Chen, A.; Bristol, G.; Galic, Z.; et al. A highly efficient short hairpin RNA potently down-regulates CCR5 expression in systemic lymphoid organs in the hu-BLT mouse model. Blood 2010, 115, 1534–1544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vatakis, D.N.; Bristol, G.C.; Kim, S.; Levin, B.; Liu, W.; Radu, C.G.; Kitchen, S.G.; Zack, J.A. Using the BLT Humanized Mouse as a Stem Cell based Gene Therapy Tumor Model. J. Vis. Exp. 2012, 70, e4181. [Google Scholar] [CrossRef] [PubMed]
- Kozlowska, A.K.; Kaur, K.; Topchyan, P.; Jewett, A. Adoptive transfer of osteoclast-expanded natural killer cells for immunotherapy targeting cancer stem-like cells in humanized mice. Cancer Immunol. Immunother. 2016, 65, 835–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shultz, L.D.; Brehm, M.A.; Garcia-Martinez, J.V.; Greiner, D.L. Humanized mice for immune system investigation: Progress, promise and challenges. Nat. Rev. Immunol. 2012, 12, 786–798. [Google Scholar] [CrossRef] [PubMed]
- Onoe, T.; Kalscheuer, H.; Danzl, N.; Chittenden, M.; Zhao, G.; Yang, Y.-G.; Sykes, M. Human Natural Regulatory T Cell Development, Suppressive Function, and Postthymic Maturation in a Humanized Mouse Model. J. Immunol. 2011, 187, 3895–3903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, M.; Hiramatsu, H.; Kobayashi, K.; Suzue, K.; Kawahata, M.; Hioki, K.; Ueyama, Y.; Koyanagi, Y.; Sugamura, K.; Tsuji, K.; et al. NOD/SCID/gamma(c)(null) mouse: An excellent recipient mouse model for engraftment of human cells. Blood 2002, 100, 3175–3182. [Google Scholar] [CrossRef] [PubMed]
- Traggiai, E.; Chicha, L.; Mazzucchelli, L.; Bronz, L.; Piffaretti, J.-C.; Lanzavecchia, A.; Manz, M.G. Development of a Human Adaptive Immune System in Cord Blood Cell-Transplanted Mice. Science 2004, 304, 104–107. [Google Scholar] [CrossRef]
- Ishikawa, F.; Yasukawa, M.; Lyons, B.; Yoshida, S.; Miyamoto, T.; Yoshimoto, G.; Watanabe, T.; Akashi, K.; Shultz, L.D.; Harada, M. Development of functional human blood and immune systems in NOD/SCID/IL2 receptor {gamma} chain(null) mice. Blood 2005, 106, 1565–1573. [Google Scholar] [CrossRef] [Green Version]
- Strowig, T.; Chijioke, O.; Carrega, P.; Arrey, F.; Meixlsperger, S.; Rämer, P.C.; Ferlazzo, G.; Münz, C. Human NK cells of mice with reconstituted human immune system components require preactivation to acquire functional competence. Blood 2010, 116, 4158–4167. [Google Scholar] [CrossRef] [Green Version]
- Stoddart, C.A.; Maidji, E.; Galkina, S.A.; Kosikova, G.; Rivera, J.M.; Moreno, M.E.; Sloan, B.; Joshi, P.; Long, B.R. Superior human leukocyte reconstitution and susceptibility to vaginal HIV transmission in humanized NOD-scid IL-2Rgamma(-/-) (NSG) BLT mice. Virology 2011, 417, 154–160. [Google Scholar] [CrossRef]
- Kaur, K.; Kozlowska, A.K.; Topchyan, P.; Ko, M.-W.; Ohanian, N.; Chiang, J.; Cook, J.; Maung, P.O.; Park, S.-H.; Cacalano, N.; et al. Probiotic-Treated Super-Charged NK Cells Efficiently Clear Poorly Differentiated Pancreatic Tumors in Hu-BLT Mice. Cancers 2019, 12, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, K.; Topchyan, P.; Kozlowska, A.K.; Ohanian, N.; Chiang, J.; Maung, P.O.; Park, S.-H.; Ko, M.-W.; Fang, C.; Nishimura, I.; et al. Super-charged NK cells inhibit growth and progression of stem-like/poorly differentiated oral tumors in vivo in humanized BLT mice; effect on tumor differentiation and response to chemotherapeutic drugs. OncoImmunology 2018, 7, e1426518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, K.; Chang, H.-H.; Cook, J.; Eibl, G.; Jewett, A. Suppression of Gingival NK Cells in Precancerous and Cancerous Stages of Pancreatic Cancer in KC and BLT-Humanized Mice. Front. Immunol. 2017, 8, 1606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olesen, R.; Wahl, A.; Denton, P.W.; Garcia, J.V. Immune reconstitution of the female reproductive tract of humanized BLT mice and their susceptibility to human immunodeficiency virus infection. J. Reprod. Immunol. 2011, 88, 195–203. [Google Scholar] [CrossRef] [Green Version]
- Denton, P.W.; Olesen, R.; Choudhary, S.K.; Archin, N.M.; Wahl, A.; Swanson, M.D.; Chateau, M.; Nochi, T.; Krisko, J.F.; Spagnuolo, R.A.; et al. Generation of HIV Latency in Humanized BLT Mice. J. Virol. 2012, 86, 630–634. [Google Scholar] [CrossRef] [Green Version]
- Shultz, L.D.; Goodwin, N.; Ishikawa, F.; Hosur, V.; Lyons, B.L.; Greiner, D.L. Human Cancer Growth and Therapy in Immunodeficient Mouse Models. Cold Spring Harb. Protoc. 2014, 2014, 694–708. [Google Scholar] [CrossRef] [Green Version]
- Tseng, H.-C.; Bui, V.; Man, Y.-G.; Cacalano, N.; Jewett, A. Induction of Split Anergy Conditions Natural Killer Cells to Promote Differentiation of Stem Cells through Cell–Cell Contact and Secreted Factors. Front. Immunol. 2014, 5, 269. [Google Scholar] [CrossRef] [Green Version]
- Tseng, H.-C.; Arasteh, A.; Paranjpe, A.; Teruel, A.; Yang, W.; Behel, A.; Alva, J.A.; Walter, G.; Head, C.; Ishikawa, T.-O.; et al. Increased Lysis of Stem Cells but Not Their Differentiated Cells by Natural Killer Cells; De-Differentiation or Reprogramming Activates NK Cells. PLoS ONE 2010, 5, e11590. [Google Scholar] [CrossRef] [Green Version]
- Bui, V.T.; Tseng, H.-C.; Kozlowska, A.; Maung, P.O.; Kaur, K.; Topchyan, P.; Jewett, A. Augmented IFN-γ and TNF-α Induced by Probiotic Bacteria in NK Cells Mediate Differentiation of Stem-Like Tumors Leading to Inhibition of Tumor Growth and Reduction in Inflammatory Cytokine Release; Regulation by IL-10. Front. Immunol. 2015, 6, 576. [Google Scholar] [CrossRef] [Green Version]
- Tseng, H.-C.; Inagaki, A.; Bui, V.T.; Cacalano, N.; Kasahara, N.; Man, Y.-G.; Jewett, A. Differential Targeting of Stem Cells and Differentiated Glioblastomas by NK Cells. J. Cancer 2015, 6, 866–876. [Google Scholar] [CrossRef]
- Vatakis, D.N.; Koya, R.C.; Nixon, C.C.; Wei, L.; Kim, S.G.; Avancena, P.; Bristol, G.; Baltimore, D.; Kohn, D.B.; Ribas, A.; et al. Antitumor activity from antigen-specific CD8 T cells generated in vivo from genetically engineered human hematopoietic stem cells. Proc. Natl. Acad. Sci. USA 2011, 108, E1408–E1416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tseng, H.C.; Kanayama, K.; Kaur, K.; Park, S.H.; Park, S.; Kozlowska, A.; Sun, S.; McKenna, C.E.; Nishimura, I.; Jewett, A. Bisphospho-nate-induced differential modulation of immune cell function in gingiva and bone marrow in vivo: Role in osteo-clast-mediated NK cell activation. Oncotarget 2015, 6, 20002–20025. [Google Scholar] [CrossRef] [PubMed]
- Jewett, A.; Bonavida, B. Target-induced inactivation and cell death by apoptosis in a subset of human NK cells. J. Immunol. 1996, 156, 907–915. [Google Scholar] [PubMed]
- Jewett, A.; Wang, M.Y.; Teruel, A.; Poupak, Z.; Bostanian, Z.; Park, N.H. Cytokine dependent inverse regulation of CD54 (ICAM1) and major histocompatibility complex class I antigens by nuclear factor kappaB in HEp2 tumor cell line: Effect on the function of natural killer cells. Hum. Immunol. 2003, 64, 505–520, PubMed. [Google Scholar] [CrossRef] [PubMed]
- Jewett, A.; Man, Y.-G.; Tseng, H.-C. Dual Functions of Natural Killer Cells in Selection and Differentiation of Stem Cells; Role in Regulation of Inflammation and Regeneration of Tissues. J. Cancer 2013, 4, 12–24. [Google Scholar] [CrossRef]
- Kozlowska, A.K.; Topchyan, P.; Kaur, K.; Tseng, H.-C.; Teruel, A.; Hiraga, T.; Jewett, A. Differentiation by NK cells is a prerequisite for effective targeting of cancer stem cells/poorly differentiated tumors by chemopreventive and chemotherapeutic drugs. J. Cancer 2017, 8, 537–554. [Google Scholar] [CrossRef]
- Kaur, K.; Cook, J.; Park, S.-H.; Topchyan, P.; Kozlowska, A.K.; Ohanian, N.; Fang, C.; Nishimura, I.; Jewett, A. Novel Strategy to Expand Super-Charged NK Cells with Significant Potential to Lyse and Differentiate Cancer Stem Cells: Differences in NK Expansion and Function between Healthy and Cancer Patients. Front. Immunol. 2017, 8, 297. [Google Scholar] [CrossRef] [Green Version]
- Kaur, K.; Ko, M.-W.; Chen, F.; Jewett, A. Defective NK cell expansion, cytotoxicity, and lack of ability to differentiate tumors from a pancreatic cancer patient in a long term follow-up: Implication in the progression of cancer. Cancer Immunol. Immunother. 2022, 71, 1033–1047. [Google Scholar] [CrossRef]
- Pek, E.A.; Chan, T.; Reid, S.; Ashkar, A.A. Characterization and IL-15 dependence of NK cells in humanized mice. Immunobiology 2010, 216, 218–224. [Google Scholar] [CrossRef]
- Liu, S.; Galat, V.; Galat, Y.; Lee, Y.K.A.; Wainwright, D.; Wu, J. NK cell-based cancer immunotherapy: From basic biology to clinical development. J. Hematol. Oncol. 2021, 14, 7. [Google Scholar] [CrossRef]
- Huntington, N.D.; Alves, N.L.; Legrand, N.; Lim, A.; Strick-Marchand, H.; Mention, J.-J.; Plet, A.; Weijer, K.; Jacques, Y.; Becker, P.D.; et al. IL-15 transpresentation promotes both human T-cell reconstitution and T-cell–dependent antibody responses in vivo. Proc. Natl. Acad. Sci. USA 2011, 108, 6217–6222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szczepanski, M.J.; Szajnik, M.; Welsh, A.; Foon, K.A.; Whiteside, T.L.; Boyiadzis, M. Interleukin-15 enhances natural killer cell cytotoxicity in patients with acute myeloid leukemia by upregulating the activating NK cell receptors. Cancer Immunol. Immunother. 2010, 59, 73–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Felices, M.; Lenvik, T.R.; Kodal, B.; Lenvik, A.J.; Hinderlie, P.; Bendzick, L.E.; Schirm, D.K.; Kaminski, M.F.; McElmurry, R.T.; Geller, M.A.; et al. Potent Cytolytic Activity and Specific IL15 Delivery in a Second-Generation Trispecific Killer Engager. Cancer Immunol. Res. 2020, 8, 1139–1149. [Google Scholar] [CrossRef] [PubMed]
- Tseng, H.-C.; Cacalano, N.; Jewett, A. Split anergized natural killer cells halt inflammation by inducing stem cell differentiation, resistance to NK cell cytotoxicity and prevention of cytokine and chemokine secretion. Oncotarget 2015, 6, 8947–8959. [Google Scholar] [CrossRef] [Green Version]
- Poli, A.; Michel, T.; Thérésine, M.; Andrès, E.; Hentges, F.; Zimmer, J. CD56brightnatural killer (NK) cells: An important NK cell subset. Immunology 2009, 126, 458–465. [Google Scholar] [CrossRef]
- Melsen, J.E.; Lugthart, G.; Lankester, A.C.; Schilham, M.W. Human Circulating and Tissue-Resident CD56bright Natural Killer Cell Populations. Front. Immunol. 2016, 7, 262. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| A | ||
|---|---|---|
| PBMCs | IFN-γ | LU 30/106 |
| Hu-BLT (n = 6) | 1423 +/− 237 | 53 +/− 9 |
| Human (n = 6) | 546 +/− 79 | 185 +/− 81 |
| B | ||
| PBMCs | IFN-γ/1% NK | LU 30/106/1% NK |
| Hu-BLT (n = 6) | 189.7 +/− 142 | 7.1 +/− 4.3 |
| Human (n = 6) | 45.5 +/− 38 | 15.4 +/− 2.7 |
| A: Human (PBMC-Derived NK) | |
|---|---|
| Primary NK Cells | |
| IFN-γ secretion (pg/mL) | 297 +/− 53 |
| NK cell-mediated cytotoxicity (LU 30/106 cells) LU 30/106 cells/1% NK cell | 228 +/− 72 2.49 +/− 1.7 |
| Supercharged NK Cells | |
| Fold expansion (cells recovered/cells cultured) | Day 6: 1.4 +/− 0.6 Day 10: 3.1 +/− 0.3 Day 14: 3.8 +/− 1.1 Day 18: 4.6 +/− 1.2 Day 22: 3.01 +/− 0.7 Day 25: 1.94 +/− 0.2 Day 30:1.4 +/− 0.4 Day 34: 0.8 +/− 0.6 |
| CD16 + cells % | Day 6: 93 +/− 3 Day 10: 90 +/− 7 Day 14: 89 +/− 4 Day 18: 92 +/− 2 Day 22: 91 +/− 6 |
| CD3+ cells % | Day 6: 4 +/− 3 Day 10: 8 +/− 7 Day 14: 12 +/− 4 Day 18: 8 +/− 8 Day 22: 6 +/− 4 |
| IFN-γ secretion (pg/mL) | Day 6: 3562 +/− 134 Day 10: 10,561 +/− 435 Day 14: 8972 +/− 276 Day 22: 4512 +/− 89 |
| NK cell-mediated cytotoxicity (LU 30/106 cells) | Day 10: 127 +/− 53 Day 14: 314 +/− 90 Day 18: 450 +/− 42 |
| LU 30/106 cells/1% NK cell | Day 10: 1.52 +/− 0.9 Day 14: 3.8 +/− 1.3 Day 18: 5.1 +/− 0.7 |
| B: Hu-BLT (Spleen-Derived NK) | |
| Primary NK Cells | |
| IFN-γ secretion (pg/mL) | 112 +/− 27 |
| NK cell-mediated cytotoxicity (LU 30/106 cells) LU 30/106 cells/1% NK cell | 113 +/− 31 2.28 +/− 0.56 |
| Supercharged NK Cells | |
| Fold expansion (cells recovered/cells cultured) | Day 6: 2.2 +/− 0.1 Day 10: 3.4 +/− 1.2 Day 14: 2.8 +/− 0.7 Day 18: 1.9 +/− 0.3 Day 22: 0.78 +/− 0.2 Note: Hu-BLT NK cells expanded for day 22 only |
| CD16 + cells % | Day 6: 56 +/− 12 Day 10: 76 +/− 9 Day 14: 87 +/− 6 Day 18: 89 +/− 8 Day 22: 91 +/− 2 |
| CD3+ cells % | Day 6: 47 +/− 11 Day 10: 23 +/− 17 Day 14: 21 +/− 13 Day 18: 14 +/− 9 Day 22: 5 +/− 4 |
| IFN-γ secretion (pg/mL) | Day 6: 2344 +/− 38 Day 10: 3781 +/− 155 Day 14: 2678 +/− 209 Day 22: 1459 +/− 63 |
| NK cell-mediated cytotoxicity (LU 30/106 cells) | Day 10: 89 +/− 24 Day 14: 201 +/− 65 Day 18: 159 +/− 49 |
| LU 30/106 cells/1% NK cell | Day 10: 1.19 +/− 0.2 Day 14: 2.41 +/− 1.09 Day 18: 1.6 +/− 0.7 |
| A | |
|---|---|
| sNK/Primary NK IFN-γ (n = 5) | Human (PBMC-Derived NK) |
| Day 6 | 10 +/− 3 |
| Day 10 | 29 +/− 2.3 |
| Day 14 | 25 +/− 1.7 |
| Day 22 | 15 +/− 1.1 |
| B | |
| sNK/Primary NK IFN-γ (n = 5) | Hu-BLT (Spleen-Derived NK) |
| Day 6 | 23 +/− 4 |
| Day 10 | 32 +/− 2.5 |
| Day 14 | 24 +/− 7 |
| Day 22 | 15 +/− 2.1 |
| Hu-BLT | IFN-γ | LU 30/106 |
|---|---|---|
| BM (n = 6) | 43 +/− 6 | 28 +/− 6.3 |
| Spleen (n = 6) | 288 +/− 24 | 154 +/− 23 |
| PBMCs (n = 6) | 1423 +/− 237 | 53 +/− 9 |
| Gingiva (n = 2) | 12 +/− 4 | 9 +/− 4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaur, K.; Jewett, A. Differences in Tumor Growth and Differentiation in NSG and Humanized-BLT Mice; Analysis of Human vs. Humanized-BLT-Derived NK Expansion and Functions. Cancers 2023, 15, 112. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers15010112
Kaur K, Jewett A. Differences in Tumor Growth and Differentiation in NSG and Humanized-BLT Mice; Analysis of Human vs. Humanized-BLT-Derived NK Expansion and Functions. Cancers. 2023; 15(1):112. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers15010112
Chicago/Turabian StyleKaur, Kawaljit, and Anahid Jewett. 2023. "Differences in Tumor Growth and Differentiation in NSG and Humanized-BLT Mice; Analysis of Human vs. Humanized-BLT-Derived NK Expansion and Functions" Cancers 15, no. 1: 112. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers15010112