
iPSC-Derived Glioblastoma Cells Have Enhanced Stemness Wnt/β-Catenin Activity Which Is Negatively Regulated by Wnt Antagonist sFRP4

Abstract
:Simple Summary
Abstract
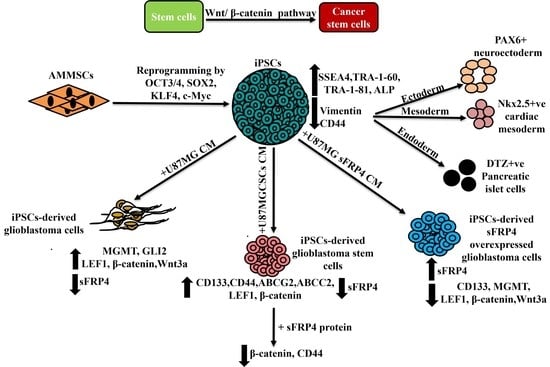
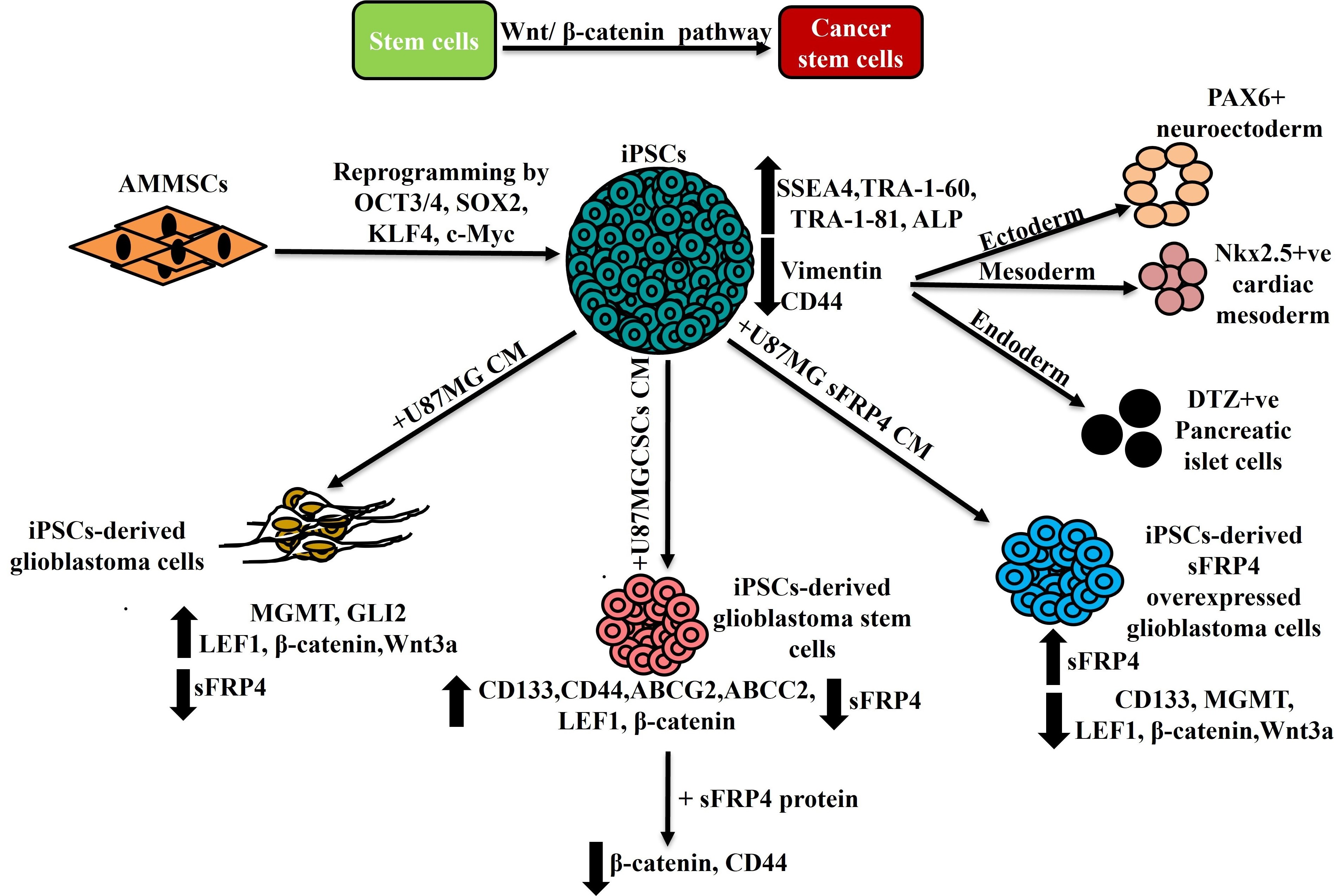
1. Introduction
2. Methods
2.1. Isolation, Cell Culture and Maintenance
2.2. Reprogramming of AMMSCs, Generation of Induced Pluripotent Stem Cells (iPSCs), Maintenance and Cryopreservation
2.3. Lipid Body-Associated Retinyl Ester Fluorescence Imaging
2.4. Alkaline Phosphatase Staining
2.5. RNA Extraction, cDNA Synthesis, Quantitative Real-Time PCR (qRT-PCR)
2.6. Immunocytochemistry
2.7. In Vitro Differentiation of iPSCs
2.7.1. Ectoderm Differentiation
2.7.2. Mesoderm Differentiation
2.7.3. Endoderm Differentiation
Dithizone (DTZ) Staining
2.8. Cell Culture
2.9. Enrichment and Characterization of Cancer Stem Cells (CSCs)
2.10. Flow Cytometry
2.11. Transient Transfections
2.12. Conditioned Media (CM)
2.13. Indirect Enzyme-Linked Immunosorbent Assay (ELISA)
2.14. Conversion of iPSCs to Glioblastoma Cells, Glioblastoma Stem Cells and sFRP4 Overexpressed Glioblastoma Cells
2.15. Co-Immunostaining
2.16. Statistical Analysis
3. Results
3.1. Reprogramming and Characterization of AMMSC-Derived iPSCs
3.2. In Vitro Differentiation of iPSCs
3.3. Conversion of iPSCs into Glioblastoma Cells and Glioblastoma Stem Cells and Their Characterization
3.4. sFRP4 Reverses the Conversion of iPSC into Glioblastoma Cells and Inhibits iPSC-Derived GSCs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Afify, S.M.; Chen, L.; Yan, T.; Sanchez Calle, A.; Nair, N.; Murakami, C.; Zahra, M.H.; Okada, N.; Iwasaki, Y.; Seno, A. Method to convert stem cells into cancer stem cells. Methods Protoc. 2019, 2, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Swingen, C.; Zhang, J. Induced pluripotent stem cells and their potential for basic and clinical sciences. Curr. Cardiol. Rev. 2013, 9, 63–72. [Google Scholar] [PubMed] [Green Version]
- Moradi, S.; Mahdizadeh, H.; Šarić, T.; Kim, J.; Harati, J.; Shahsavarani, H.; Greber, B.; Moore, J.B. Research and therapy with induced pluripotent stem cells (iPSCs): Social, legal, and ethical considerations. Stem Cell Res. Ther. 2019, 10, 341. [Google Scholar] [PubMed] [Green Version]
- Yamanaka, S. Induced pluripotent stem cells: Past, present, and future. Cell Stem Cell 2012, 10, 678–684. [Google Scholar] [CrossRef] [Green Version]
- Nicholson, M.W.; Ting, C.-Y.; Chan, D.Z.; Cheng, Y.-C.; Lee, Y.-C.; Hsu, C.-C.; Huang, C.-Y.; Hsieh, P.C. Utility of iPSC-Derived Cells for Disease Modeling, Drug Development, and Cell Therapy. Cells 2022, 11, 1853. [Google Scholar] [CrossRef]
- Prager, B.C.; Bhargava, S.; Mahadev, V.; Hubert, C.G.; Rich, J.N. Glioblastoma stem cells: Driving resilience through chaos. Trends Cancer 2020, 6, 223–235. [Google Scholar] [CrossRef]
- Tan, B.T.; Park, C.Y.; Ailles, L.E.; Weissman, I.L. The cancer stem cell hypothesis: A work in progress. Lab. Investig. 2006, 86, 1203–1207. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Shi, P.; Zhao, G.; Xu, J.; Peng, W.; Zhang, J.; Zhang, G.; Wang, X.; Dong, Z.; Chen, F. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct. Target. Ther. 2020, 5, 8. [Google Scholar]
- Du, Z.; Jia, D.; Liu, S.; Wang, F.; Li, G.; Zhang, Y.; Cao, X.; Ling, E.A.; Hao, A. Oct4 is expressed in human gliomas and promotes colony formation in glioma cells. Glia 2009, 57, 724–733. [Google Scholar] [CrossRef]
- Hägerstrand, D.; He, X.; Bradic Lindh, M.; Hoefs, S.; Hesselager, G.; Östman, A.; Nister, M. Identification of a SOX2-dependent subset of tumor-and sphere-forming glioblastoma cells with a distinct tyrosine kinase inhibitor sensitivity profile. Neuro-oncology 2011, 13, 1178–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galardi, S.; Savino, M.; Scagnoli, F.; Pellegatta, S.; Pisati, F.; Zambelli, F.; Illi, B.; Annibali, D.; Beji, S.; Orecchini, E. Resetting cancer stem cell regulatory nodes upon MYC inhibition. EMBO Rep. 2016, 17, 1872–1889. [Google Scholar] [CrossRef] [Green Version]
- Jin, X.; Jin, X.; Kim, H. Cancer stem cells and differentiation therapy. Tumor Biol. 2017, 39, 1010428317729933. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.; Zuo, C.; Fang, P.; Liu, G.; Qiu, Y.; Huang, Y.; Tang, R. Targeting glioblastoma stem cells: A review on biomarkers, signal pathways and targeted therapy. Front. Oncol. 2021, 11, 701291. [Google Scholar] [PubMed]
- El-Khayat, S.M.; Arafat, W.O. Therapeutic strategies of recurrent glioblastoma and its molecular pathways ‘Lock up the beast’. Ecancermedicalscience 2021, 15, 1176. [Google Scholar] [CrossRef]
- Shi, Y.; Inoue, H.; Wu, J.C.; Yamanaka, S. Induced pluripotent stem cell technology: A decade of progress. Nat. Rev. Drug Discov. 2017, 16, 115–130. [Google Scholar] [PubMed]
- Prieto-Vila, M.; Yan, T.; Calle, A.S.; Nair, N.; Hurley, L.; Kasai, T.; Kakuta, H.; Masuda, J.; Murakami, H.; Mizutani, A.; et al. iPSC-derived cancer stem cells provide a model of tumor vasculature. Am. J. Cancer Res. 2016, 6, 1906. [Google Scholar]
- Chen, L.; Kasai, T.; Li, Y.; Sugii, Y.; Jin, G.; Okada, M.; Vaidyanath, A.; Mizutani, A.; Satoh, A.; Kudoh, T.; et al. A model of cancer stem cells derived from mouse induced pluripotent stem cells. PLoS ONE 2012, 7, e33544. [Google Scholar]
- Afify, S.M.; Calle, A.S.; Hassan, G.; Kumon, K.; Nawara, H.M.; Zahra, M.H.; Mansour, H.M.; Khayrani, A.C.; Alam, J.; Du, J.; et al. A novel model of liver cancer stem cells developed from induced pluripotent stem cells. Br. J. Cancer 2020, 122, 1378–1390. [Google Scholar]
- Nair, N.; Calle, A.S.; Zahra, M.H.; Prieto-Vila, M.; Oo, A.K.K.; Hurley, L.; Vaidyanath, A.; Seno, A.; Masuda, J.; Iwasaki, Y.; et al. A cancer stem cell model as the point of origin of cancer-associated fibroblasts in tumor microenvironment. Sci. Rep. 2017, 7, 6838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, J.W.; Loisel-Duwattez, J.; Desterke, C.; Latsis, T.; Pagliaro, S.; Griscelli, F.; Bennaceur-Griscelli, A.; Turhan, A.G. A novel neuronal organoid model mimicking glioblastoma (GBM) features from induced pluripotent stem cells (iPSC). Biochim. Biophys. Acta (BBA)-Gen. Subj. 2020, 1864, 129540. [Google Scholar] [CrossRef]
- Azzarelli, R.; Ori, M.; Philpott, A.; Simons, B.D. Three-dimensional model of glioblastoma by co-culturing tumor stem cells with human brain organoids. Biol. Open 2021, 10, bio056416. [Google Scholar] [CrossRef]
- Schiefer, L.; Visweswaran, M.; Perumal, V.; Arfuso, F.; Groth, D.; Newsholme, P.; Warrier, S.; Dharmarajan, A. Epigenetic regulation of the secreted frizzled-related protein family in human glioblastoma multiforme. Cancer Gene Ther. 2014, 21, 297–303. [Google Scholar] [CrossRef]
- Yasmin, I.A.; Sundaram, S.M.; Banerjee, A.; Varier, L.; Dharmarajan, A.; Warrier, S. Netrin-like domain of sFRP4, a Wnt antagonist inhibits stemness, metastatic and invasive properties by specifically blocking MMP-2 in cancer stem cells from human glioma cell line U87MG. Exp. Cell Res. 2021, 409, 112912. [Google Scholar] [CrossRef]
- Warrier, S.; Haridas, N.; Bhonde, R. Inherent propensity of amnion-derived mesenchymal stem cells towards endothelial lineage: Vascularization from an avascular tissue. Placenta 2012, 33, 850–858. [Google Scholar] [CrossRef]
- Cai, J.; Li, W.; Su, H.; Qin, D.; Yang, J.; Zhu, F.; Xu, J.; He, W.; Guo, X.; Labuda, K. Generation of human induced pluripotent stem cells from umbilical cord matrix and amniotic membrane mesenchymal cells. J. Biol. Chem. 2010, 285, 11227–11234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muthusamy, T.; Mukherjee, O.; Menon, R.; Bangalore, M.P.; Panicker, M.M. A method to identify and isolate pluripotent human stem cells and mouse epiblast stem cells using lipid body-associated retinyl ester fluorescence. Stem Cell Rep. 2014, 3, 169–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, U.; Quintanilla, R.H.; Grecian, S.; Gee, K.R.; Rao, M.S.; Lakshmipathy, U. Novel live alkaline phosphatase substrate for identification of pluripotent stem cells. Stem Cell Rev. Rep. 2012, 8, 1021–1029. [Google Scholar] [CrossRef] [Green Version]
- Patani, R.; Compston, A.; Puddifoot, C.A.; Wyllie, D.J.; Hardingham, G.E.; Allen, N.D.; Chandran, S. Activin/Nodal inhibition alone accelerates highly efficient neural conversion from human embryonic stem cells and imposes a caudal positional identity. PLoS ONE 2009, 4, e7327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chambers, S.M.; Fasano, C.A.; Papapetrou, E.P.; Tomishima, M.; Sadelain, M.; Studer, L. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol. 2009, 27, 275–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, M.; Tang, Y.; Zhou, Y.; Zhang, J. Deciphering role of Wnt signalling in cardiac mesoderm and cardiomyocyte differentiation from human iPSCs: Four-dimensional control of Wnt pathway for hiPSC-CMs differentiation. Sci. Rep. 2019, 9, 19389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sung, T.-C.; Liu, C.-H.; Huang, W.-L.; Lee, Y.-C.; Kumar, S.S.; Chang, Y.; Ling, Q.-D.; Hsu, S.-T.; Higuchi, A. Efficient differentiation of human ES and iPS cells into cardiomyocytes on biomaterials under xeno-free conditions. Biomater. Sci. 2019, 7, 5467–5481. [Google Scholar] [CrossRef]
- Burridge, P.W.; Matsa, E.; Shukla, P.; Lin, Z.C.; Churko, J.M.; Ebert, A.D.; Lan, F.; Diecke, S.; Huber, B.; Mordwinkin, N.M. Chemically defined generation of human cardiomyocytes. Nat. Methods 2014, 11, 855–860. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Liu, H.; Klein, M.; Ostrominski, J.; Hong, S.G.; Yada, R.C.; Chen, G.; Navarengom, K.; Schwartzbeck, R.; San, H. Efficient differentiation of cardiomyocytes and generation of calcium-sensor reporter lines from nonhuman primate iPSCs. Sci. Rep. 2018, 8, 5907. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Qiu, H.; Dong, X.; Wang, C.; Na, J.; Zhou, J.; Wang, C. Transferrin improved the generation of cardiomyocyte from human pluripotent stem cells for myocardial infarction repair. J. Mol. Histol. 2021, 52, 87–99. [Google Scholar] [CrossRef]
- Ghorbani-Dalini, S.; Azarpira, N.; Sangtarash, M.H.; Soleimanpour-Lichaei, H.R.; Yaghobi, R.; Lorzadeh, S.; Sabet, A.; Sarshar, M.; Al-Abdullah, I.H. Optimization of activin-A: A breakthrough in differentiation of human induced pluripotent stem cell into definitive endoderm. 3 Biotech 2020, 10, 215. [Google Scholar] [CrossRef]
- Kunisada, Y.; Tsubooka-Yamazoe, N.; Shoji, M.; Hosoya, M. Small molecules induce efficient differentiation into insulin-producing cells from human induced pluripotent stem cells. Stem Cell Res. 2012, 8, 274–284. [Google Scholar] [CrossRef] [Green Version]
- Thakur, G.; Lee, H.-J.; Jeon, R.-H.; Lee, S.-L.; Rho, G.-J. Small molecule-induced pancreatic β-like cell development: Mechanistic approaches and available strategies. Int. J. Mol. Sci. 2020, 21, 2388. [Google Scholar] [CrossRef] [Green Version]
- D’Amour, K.A.; Bang, A.G.; Eliazer, S.; Kelly, O.G.; Agulnick, A.D.; Smart, N.G.; Moorman, M.A.; Kroon, E.; Carpenter, M.K.; Baetge, E.E. Production of pancreatic hormone–expressing endocrine cells from human embryonic stem cells. Nat. Biotechnol. 2006, 24, 1392–1401. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Au, M.; Lu, K.; Eshpeter, A.; Korbutt, G.; Fisk, G.; Majumdar, A.S. Generation of insulin-producing islet-like clusters from human embryonic stem cells. Stem Cells 2007, 25, 1940–1953. [Google Scholar] [CrossRef] [PubMed]
- Kroon, E.; Martinson, L.A.; Kadoya, K.; Bang, A.G.; Kelly, O.G.; Eliazer, S.; Young, H.; Richardson, M.; Smart, N.G.; Cunningham, J. Pancreatic endoderm derived from human embryonic stem cells generates glucose-responsive insulin-secreting cells in vivo. Nat. Biotechnol. 2008, 26, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Borowiak, M.; Fox, J.L.; Maehr, R.; Osafune, K.; Davidow, L.; Lam, K.; Peng, L.F.; Schreiber, S.L.; Rubin, L.L. A small molecule that directs differentiation of human ESCs into the pancreatic lineage. Nat. Chem. Biol. 2009, 5, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Thatava, T.; Nelson, T.J.; Edukulla, R.; Sakuma, T.; Ohmine, S.; Tonne, J.M.; Yamada, S.; Kudva, Y.; Terzic, A.; Ikeda, Y. Indolactam V/GLP-1-mediated differentiation of human iPS cells into glucose-responsive insulin-secreting progeny. Gene Ther. 2011, 18, 283–293. [Google Scholar] [CrossRef] [Green Version]
- Walczak, M.P.; Drozd, A.M.; Stoczynska-Fidelus, E.; Rieske, P.; Grzela, D.P. Directed differentiation of human iPSC into insulin producing cells is improved by induced expression of PDX1 and NKX6. 1 factors in IPC progenitors. J. Transl. Med. 2016, 14, 341. [Google Scholar] [CrossRef] [Green Version]
- Shiroi, A.; Yoshikawa, M.; Yokota, H.; Fukui, H.; Ishizaka, S.; Tatsumi, K.; Takahashi, Y. Identification of insulin-producing cells derived from embryonic stem cells by zinc-chelating dithizone. Stem Cells 2002, 20, 284–292. [Google Scholar] [CrossRef]
- Deshmukh, A.; Kumar, S.; Arfuso, F.; Newsholme, P.; Dharmarajan, A. Secreted Frizzled-related protein 4 (sFRP4) chemo-sensitizes cancer stem cells derived from human breast, prostate, and ovary tumor cell lines. Sci. Rep. 2017, 7, 2256. [Google Scholar] [CrossRef] [Green Version]
- Han, K.-H.; Kim, A.-K.; Kim, D.-I. Enhanced Anti-Cancer Effects of Conditioned Medium from Hypoxic Human Adult Dermal Fibroblasts on Cervical Cancer Cells. Int. J. Mol. Sci. 2022, 23, 5134. [Google Scholar] [CrossRef]
- Plotnikov, A.; Kozer, N.; Krupalnik, V.; Peles, S.; Mor, N.; Rais, Y.; Hanna, J.H.; Barr, H.M. A multiplexed screening method for pluripotency. Stem Cell Res. 2017, 23, 158–162. [Google Scholar] [CrossRef]
- Khiatah, B.; Qi, M.; Wu, Y.; Chen, K.-T.; Perez, R.; Valiente, L.; Omori, K.; Isenberg, J.S.; Kandeel, F.; Yee, J.-K. Pancreatic human islets and insulin-producing cells derived from embryonic stem cells are rapidly identified by a newly developed Dithizone. Sci. Rep. 2019, 9, 9295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, V.K.; Kalsan, M.; Kumar, N.; Saini, A.; Chandra, R. Induced pluripotent stem cells: Applications in regenerative medicine, disease modeling, and drug discovery. Front. Cell Dev. Biol. 2015, 3, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pipino, C.; Shangaris, P.; Resca, E.; Zia, S.; Deprest, J.; Sebire, N.J.; David, A.L.; Guillot, P.V.; De Coppi, P. Placenta as a reservoir of stem cells: An underutilized resource? Br. Med. Bull. 2013, 105. [Google Scholar] [CrossRef] [Green Version]
- Jiang, G.; Di Bernardo, J.; DeLong, C.J.; Monteiro da Rocha, A.; O’Shea, K.S.; Kunisaki, S.M. Induced pluripotent stem cells from human placental chorion for perinatal tissue engineering applications. Tissue Eng. Part C Methods 2014, 20, 731–740. [Google Scholar] [CrossRef] [Green Version]
- Thomson, J.A.; Itskovitz-Eldor, J.; Shapiro, S.S.; Waknitz, M.A.; Swiergiel, J.J.; Marshall, V.S.; Jones, J.M. Embryonic stem cell lines derived from human blastocysts. Science 1998, 282, 1145–1147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polo, J.M.; Anderssen, E.; Walsh, R.M.; Schwarz, B.A.; Nefzger, C.M.; Lim, S.M.; Borkent, M.; Apostolou, E.; Alaei, S.; Cloutier, J. A molecular roadmap of reprogramming somatic cells into iPS cells. Cell 2012, 151, 1617–1632. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Xu, Y.; He, M.; Zhang, M.; Cui, F.; Lu, L.; Yao, M.; Tian, W.; Benda, C.; Zhuang, Q. Transcriptional pause release is a rate-limiting step for somatic cell reprogramming. Cell Stem Cell 2014, 15, 574–588. [Google Scholar] [CrossRef] [Green Version]
- Quintanilla, R.H., Jr.; Asprer, J.S.; Vaz, C.; Tanavde, V.; Lakshmipathy, U. CD44 is a negative cell surface marker for pluripotent stem cell identification during human fibroblast reprogramming. PLoS ONE 2014, 9, e85419. [Google Scholar] [CrossRef] [Green Version]
- Secunda, R.; Vennila, R.; Mohanashankar, A.; Rajasundari, M.; Jeswanth, S.; Surendran, R. Isolation, expansion and characterisation of mesenchymal stem cells from human bone marrow, adipose tissue, umbilical cord blood and matrix: A comparative study. Cytotechnology 2015, 67, 793–807. [Google Scholar] [CrossRef]
- Samavarchi-Tehrani, P.; Golipour, A.; David, L.; Sung, H.-k.; Beyer, T.A.; Datti, A.; Woltjen, K.; Nagy, A.; Wrana, J.L. Functional genomics reveals a BMP-driven mesenchymal-to-epithelial transition in the initiation of somatic cell reprogramming. Cell Stem Cell 2010, 7, 64–77. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.-Y.; Lin, H.-H.; Tang, M.-J.; Wang, Y.-K. Vimentin contributes to epithelial-mesenchymal transition cancer cell mechanics by mediating cytoskeletal organization and focal adhesion maturation. Oncotarget 2015, 6, 15966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, R.; Liang, J.; Ni, S.; Zhou, T.; Qing, X.; Li, H.; He, W.; Chen, J.; Li, F.; Zhuang, Q. A mesenchymal-to-epithelial transition initiates and is required for the nuclear reprogramming of mouse fibroblasts. Cell Stem Cell 2010, 7, 51–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adewumi, O.; Aflatoonian, B.; Ahrlund-Richter, L.; Amit, M.; Andrews, P.W.; Beighton, G.; Bello, P.A.; Bennvenisty, N.; Berry, L.S.; Bevan, S.; et al. Characterization of human embryonic stem cell lines by the International Stem Cell Initiative. Nat. Biotechnol. 2007, 25, 803–816. [Google Scholar]
- Madhu, V.; Dighe, A.S.; Cui, Q.; Deal, D.N. Dual inhibition of activin/nodal/TGF-β and BMP signaling pathways by SB431542 and dorsomorphin induces neuronal differentiation of human adipose derived stem cells. Stem Cells Int. 2016, 2016, 1035374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Huang, C.T.; Chen, J.; Pankratz, M.T.; Xi, J.; Li, J.; Yang, Y.; LaVaute, T.M.; Li, X.-J.; Ayala, M. Pax6 is a human neuroectoderm cell fate determinant. Cell Stem Cell 2010, 7, 90–100. [Google Scholar] [CrossRef] [Green Version]
- Cao, N.; Liu, Z.; Chen, Z.; Wang, J.; Chen, T.; Zhao, X.; Ma, Y.; Qin, L.; Kang, J.; Wei, B. Ascorbic acid enhances the cardiac differentiation of induced pluripotent stem cells through promoting the proliferation of cardiac progenitor cells. Cell Res. 2012, 22, 219–236. [Google Scholar] [CrossRef] [Green Version]
- Drowley, L.; Koonce, C.; Peel, S.; Jonebring, A.; Plowright, A.T.; Kattman, S.J.; Andersson, H.; Anson, B.; Swanson, B.J.; Wang, Q.-D. Human induced pluripotent stem cell-derived cardiac progenitor cells in phenotypic screening: A transforming growth factor-β type 1 receptor kinase inhibitor induces efficient cardiac differentiation. Stem Cells Transl. Med. 2016, 5, 164–174. [Google Scholar] [CrossRef] [Green Version]
- Kreuser, U.; Buchert, J.; Haase, A.; Richter, W.; Diederichs, S. Initial WNT/β-catenin activation enhanced mesoderm commitment, extracellular matrix expression, cell aggregation and cartilage tissue yield from induced pluripotent stem cells. Front. Cell Dev. Biol. 2020, 8, 581331. [Google Scholar] [CrossRef]
- Wang, H.; Hao, J.; Hong, C.C. Cardiac induction of embryonic stem cells by a small molecule inhibitor of Wnt/β-catenin signaling. ACS Chem. Biol. 2011, 6, 192–197. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Nomura-Kitabayashi, A.; Sultana, N.; Cai, W.; Cai, X.; Moon, A.M.; Cai, C.-L. Mesodermal Nkx2. 5 is necessary and sufficient for early second heart field development. Dev. Biol. 2014, 390, 68–79. [Google Scholar] [CrossRef] [Green Version]
- Nostro, M.C.; Sarangi, F.; Ogawa, S.; Holtzinger, A.; Corneo, B.; Li, X.; Micallef, S.J.; Park, I.-H.; Basford, C.; Wheeler, M.B. Stage-specific signaling through TGFβ family members and WNT regulates patterning and pancreatic specification of human pluripotent stem cells. Development 2011, 138, 861–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rezania, A.; Bruin, J.E.; Riedel, M.J.; Mojibian, M.; Asadi, A.; Xu, J.; Gauvin, R.; Narayan, K.; Karanu, F.; O’Neil, J.J. Maturation of human embryonic stem cell–derived pancreatic progenitors into functional islets capable of treating pre-existing diabetes in mice. Diabetes 2012, 61, 2016–2029. [Google Scholar] [CrossRef] [Green Version]
- Stafford, D.; White, R.J.; Kinkel, M.D.; Linville, A.; Schilling, T.F.; Prince, V.E. Retinoids signal directly to zebrafish endoderm to specify insulin-expressing β-cells. Development 2006, 133, 949–956. [Google Scholar] [CrossRef] [Green Version]
- Naujok, O.; Diekmann, U.; Lenzen, S. The generation of definitive endoderm from human embryonic stem cells is initially independent from activin A but requires canonical Wnt-signaling. Stem Cell Rev. Rep. 2014, 10, 480–493. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Jiang, W.; Liu, M.; Sui, X.; Yin, X.; Chen, S.; Shi, Y.; Deng, H. Highly efficient differentiation of human ES cells and iPS cells into mature pancreatic insulin-producing cells. Cell Res. 2009, 19, 429–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Xu, J.; Ren, Z.; Meng, Y.; Liu, W.; Lu, L.; Zhou, Z.; Chen, G. Nicotinamide promotes pancreatic differentiation through the dual inhibition of CK1 and ROCK kinases in human embryonic stem cells. Stem Cell Res. Ther. 2021, 12, 362. [Google Scholar] [CrossRef]
- Walcher, L.; Kistenmacher, A.-K.; Suo, H.; Kitte, R.; Dluczek, S.; Strauß, A.; Blaudszun, A.-R.; Yevsa, T.; Fricke, S.; Kossatz-Boehlert, U. Cancer stem cells—Origins and biomarkers: Perspectives for targeted personalized therapies. Front. Immunol. 2020, 11, 1280. [Google Scholar] [CrossRef]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef]
- Singh, S.K.; Clarke, I.D.; Terasaki, M.; Bonn, V.E.; Hawkins, C.; Squire, J.; Dirks, P.B. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003, 63, 5821–5828. [Google Scholar]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef]
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug resistance in cancer: Role of ATP–dependent transporters. Nat. Rev. Cancer 2002, 2, 48–58. [Google Scholar] [CrossRef] [Green Version]
- Ginestier, C.; Hur, M.H.; Charafe-Jauffret, E.; Monville, F.; Dutcher, J.; Brown, M.; Jacquemier, J.; Viens, P.; Kleer, C.G.; Liu, S. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 2007, 1, 555–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nasrolahi, A.; Azizidoost, S.; Radoszkiewicz, K.; Najafi, S.; Ghaedrahmati, F.; Anbiyaee, O.; Khoshnam, S.E.; Farzaneh, M.; Uddin, S. Signaling pathways governing glioma cancer stem cells behavior. Cell. Signal. 2022, 101, 110493. [Google Scholar] [CrossRef]
- Chen, J.-F.; Luo, X.; Xiang, L.-S.; Li, H.-T.; Zha, L.; Li, N.; He, J.-M.; Xie, G.-F.; Xie, X.; Liang, H.-J. EZH2 promotes colorectal cancer stem-like cell expansion by activating p21cip1-Wnt/β-catenin signaling. Oncotarget 2016, 7, 41540. [Google Scholar] [CrossRef]
- Giancotti, F.G. Mechanisms governing metastatic dormancy and reactivation. Cell 2013, 155, 750–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.; Dai, Y. Tumor microenvironment and therapeutic response. Cancer Lett. 2017, 387, 61–68. [Google Scholar] [CrossRef]
- López de Andrés, J.; Griñán-Lisón, C.; Jiménez, G.; Marchal, J.A. Cancer stem cell secretome in the tumor microenvironment: A key point for an effective personalized cancer treatment. J. Hematol. Oncol. 2020, 13, 136. [Google Scholar] [CrossRef]
- Peñuelas, S.; Anido, J.; Prieto-Sánchez, R.M.; Folch, G.; Barba, I.; Cuartas, I.; García-Dorado, D.; Poca, M.A.; Sahuquillo, J.; Baselga, J. TGF-β increases glioma-initiating cell self-renewal through the induction of LIF in human glioblastoma. Cancer Cell 2009, 15, 315–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, T.; Mizutani, A.; Chen, L.; Takaki, M.; Hiramoto, Y.; Matsuda, S.; Shigehiro, T.; Kasai, T.; Kudoh, T.; Murakami, H. Characterization of cancer stem-like cells derived from mouse induced pluripotent stem cells transformed by tumor-derived extracellular vesicles. J. Cancer 2014, 5, 572. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Zamarron, B.F.; Chen, W. Dual roles of immune cells and their factors in cancer development and progression. Int. J. Biol. Sci. 2011, 7, 651. [Google Scholar] [CrossRef]
- Bayarsaihan, D. Epigenetic mechanisms in inflammation. J. Dent. Res. 2011, 90, 9–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearson, J.R.; Regad, T. Targeting cellular pathways in glioblastoma multiforme. Signal Transduct. Target. Ther. 2017, 2, 17040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pantazi, E.; Gemenetzidis, E.; Teh, M.-T.; Reddy, S.V.; Warnes, G.; Evagora, C.; Trigiante, G.; Philpott, M.P. GLI2 is a regulator of β-catenin and is associated with loss of E-cadherin, cell invasiveness, and long-term epidermal regeneration. J. Investig. Dermatol. 2017, 137, 1719–1730. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Xu, Y.; Sasada, S.; Oo, A.K.K.; Hassan, G.; Mahmud, H.; Khayrani, A.C.; Alam, M.J.; Kumon, K.; Uesaki, R. Signaling inhibitors accelerate the conversion of mouse iPS cells into cancer stem cells in the tumor microenvironment. Sci. Rep. 2020, 10, 9955. [Google Scholar] [CrossRef] [PubMed]
- Hassan, G.; Ohara, T.; Afify, S.M.; Kumon, K.; Zahra, M.H.; Fu, X.; Al Kadi, M.; Seno, A.; Salomon, D.S.; Seno, M. Different pancreatic cancer microenvironments convert iPSCs into cancer stem cells exhibiting distinct plasticity with altered gene expression of metabolic pathways. J. Exp. Clin. Cancer Res. 2022, 41, 29. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhu, J.; Shi, B.; Wang, X.; Lu, Q.; Li, C.; Chen, H. The transcription factor LEF1 promotes tumorigenicity and activates the TGF-β signaling pathway in esophageal squamous cell carcinoma. J. Exp. Clin. Cancer Res. 2019, 38, 304. [Google Scholar] [CrossRef] [PubMed]
- Santiago, L.; Daniels, G.; Wang, D.; Deng, F.-M.; Lee, P. Wnt signaling pathway protein LEF1 in cancer, as a biomarker for prognosis and a target for treatment. Am. J. Cancer Res. 2017, 7, 1389. [Google Scholar]
- Gao, X.; Mi, Y.; Ma, Y.; Jin, W. LEF1 regulates glioblastoma cell proliferation, migration, invasion, and cancer stem-like cell self-renewal. Tumor Biol. 2014, 35, 11505–11511. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Huang, K.; Shi, Z.; Zou, J.; Wang, Y.; Jia, Z.; Zhang, A.; Han, L.; Yue, X.; Liu, N. High β-catenin/Tcf-4 activity confers glioma progression via direct regulation of AKT2 gene expression. Neuro-Oncol. 2011, 13, 600–609. [Google Scholar] [CrossRef] [PubMed]
- Kaur, N.; Chettiar, S.; Rathod, S.; Rath, P.; Muzumdar, D.; Shaikh, M.; Shiras, A. Wnt3a mediated activation of Wnt/β-catenin signaling promotes tumor progression in glioblastoma. Mol. Cell. Neurosci. 2013, 54, 44–57. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Sequences 5′-3′ | Product Length (bp) | Annealing Temperature (°C) |
|---|---|---|---|
| GAPDH | FP: CGACCACTTGTCAAGCTCA RP: AGGGGAGATTCAGTGTGGT | 202 | 60 °C |
| OCT3/4 | FP: GAAGGATGTGGTCCGAGTGT RP: TTGTGTTCCCAATTCCTTCC | 400 | 60 °C |
| KLF4 | FP: GGCGAGAAACCTTACCACTGT RP: TACTGAACTCTCTCTGCTGGCA | 370, 454, 622, 706, 1042, 538, 874, 958 | 60 °C |
| c-Myc | FP: GCGTCCTGGGAAGGGAGATCCGGAGC RP: TTGAGGGGCATCGTCGCGGGAGGCTG | 328,325 | 60 °C |
| NANOG | FP: TTTGTGGGCCTGAAGAAAACT RP: AGGGCTGTCCTGAATAAGCAG | 116 | 60 °C |
| CD44 | FP: CATCTACCCCAGCAACCCTA RP: GGTTGTGTTTGCTCCACCTT | 271 | 60 °C |
| CD133 (PROM1) | FP: TCAGTGAGAAAGTGGCATCG RP: TGTTGTGATGGGCTTGTCAT | 313, 244 | 60 °C |
| ABCG2 | FP: GTGGCATTAAACAGAGAAGAAGACT RP: CACCCCCGGAAAGTTGATGT | 148 | 60 °C |
| ABCC2 | FP: AGAGCTGGCCCTTGTACTCA RP: TGCGTTTCAAACTTGCTCAC | 492 | 60 °C |
| MGMT | FP: CACCGTTTGCGACTTGGTACTT RP: AGACCCTGCTCACAACCAGACA | 111 | 58 °C |
| GLI2 | FP: CATGGAGCACTACCTCCGTTC RP: CGACGGTCATCTGGTGGTAAT | 173 | 60 °C |
| LEF1 | FP: AAATGGGTCCCTTTCTCCAC RP: TCGTCGCTGTAGGTGATGAG | 108 | 59 °C |
| β-catenin | FP: CGTCCACAACACTCTGGCTA RP: GCCAGCACTTCACTGCAATA | 159 | 55 °C |
| Wnt3a | FP: ACTACGTGGAGATCATGCCC RP: ATGAGCGTGTCACTGCAAAG | 210 | 54 °C |
| sFRP4 | FP: CGATCGGTGCAAGTGTAAA RP: GACTTGAGTTCGAGGGATGG | 181 | 49 °C |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yasmin, I.A.; Dharmarajan, A.; Warrier, S. iPSC-Derived Glioblastoma Cells Have Enhanced Stemness Wnt/β-Catenin Activity Which Is Negatively Regulated by Wnt Antagonist sFRP4. Cancers 2023, 15, 3622. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers15143622
Yasmin IA, Dharmarajan A, Warrier S. iPSC-Derived Glioblastoma Cells Have Enhanced Stemness Wnt/β-Catenin Activity Which Is Negatively Regulated by Wnt Antagonist sFRP4. Cancers. 2023; 15(14):3622. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers15143622
Chicago/Turabian StyleYasmin, Ishmat Ara, Arun Dharmarajan, and Sudha Warrier. 2023. "iPSC-Derived Glioblastoma Cells Have Enhanced Stemness Wnt/β-Catenin Activity Which Is Negatively Regulated by Wnt Antagonist sFRP4" Cancers 15, no. 14: 3622. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers15143622