
CMGC Kinases in Health and Cancer

1
Institute of Biotechnology, University of Helsinki, 00014 Helsinki, Finland
2
Helsinki Institute of Life Science, University of Helsinki, 00014 Helsinki, Finland
*
Author to whom correspondence should be addressed.
Cancers 2023, 15(15), 3838; https://0-doi-org.brum.beds.ac.uk/10.3390/cancers15153838
Submission received: 21 June 2023
/
Revised: 18 July 2023
/
Accepted: 26 July 2023
/
Published: 28 July 2023
(This article belongs to the Special Issue Kinase Signaling in Cancer)
Abstract
:Simple Summary
CMGC kinases, named after the initials of its main kinase families, CDKs, MAPKs, GSKs, and CLKs, play critical roles in many cellular processes. Dysregulation of these kinases has been associated with cancer development and progression, highlighting their significance in cancer biology. The success of certain kinase inhibitors demonstrated the therapeutic potential of targeting CMGC kinases. However, the clinical application of CMGC kinase inhibitors is not without its challenges. Here, we discuss the challenges including resistance; off-target effects; patient stratification, which can be addressed through the development of next-generation inhibitors; combination therapies; and exploiting protein–protein interactions involving CMGC kinases. Understanding these interactions could reveal novel targets and allow for the design of drugs that disrupt these specific interactions, offering another layer of precision in targeting these key players in cancer biology. Through continued research and development, next-generation inhibitors and novel therapeutic strategies could play a pivotal role in improving outcomes for cancer patients.
Abstract
CMGC kinases, encompassing cyclin-dependent kinases (CDKs), mitogen-activated protein kinases (MAPKs), glycogen synthase kinases (GSKs), and CDC-like kinases (CLKs), play pivotal roles in cellular signaling pathways, including cell cycle regulation, proliferation, differentiation, apoptosis, and gene expression regulation. The dysregulation and aberrant activation of these kinases have been implicated in cancer development and progression, making them attractive therapeutic targets. In recent years, kinase inhibitors targeting CMGC kinases, such as CDK4/6 inhibitors and BRAF/MEK inhibitors, have demonstrated clinical success in treating specific cancer types. However, challenges remain, including resistance to kinase inhibitors, off-target effects, and the need for better patient stratification. This review provides a comprehensive overview of the importance of CMGC kinases in cancer biology, their involvement in cellular signaling pathways, protein–protein interactions, and the current state of kinase inhibitors targeting these kinases. Furthermore, we discuss the challenges and future perspectives in targeting CMGC kinases for cancer therapy, including potential strategies to overcome resistance, the development of more selective inhibitors, and novel therapeutic approaches, such as targeting protein–protein interactions, exploiting synthetic lethality, and the evolution of omics in the study of the human kinome. As our understanding of the molecular mechanisms and protein–protein interactions involving CMGC kinases expands, so too will the opportunities for the development of more selective and effective therapeutic strategies for cancer treatment.
1. Introduction
Eukaryotic cells are characterized by the simultaneous activity of multiple molecular networks. Central to these networks are the reversible reactions of protein phosphorylation, catalyzed by protein kinases, and dephosphorylation, catalyzed by protein phosphatases. Protein kinases play a crucial role in signal transduction by phosphorylating specific protein substrates. Changes in the phosphorylation status of numerous proteins are closely associated with their activity, cellular location, and interactions with other proteins. Consequently, alterations in protein phosphorylation can influence a wide range of cellular processes, including metabolism, transcription, cell cycle progression, cytoskeletal rearrangement, cell motility, apoptosis, and differentiation. Dysregulation of protein phosphorylation has been implicated in various human diseases, including cancer. Phosphorylation involves the transfer of a phosphate group from an ATP molecule to a target protein’s serine (S), threonine (T), or tyrosine (Y) residues by protein kinases. Based on their target amino acid selectivity, protein kinases can be classified into serine/threonine (Ser-/Thr-specific), tyrosine (tyrosine-specific), and dual-specificity protein kinases, which primarily phosphorylate serine/threonine residues but can also target tyrosine residues. Serine/threonine protein kinases are the predominant regulators of most cellular processes, with phosphorylation event rates yielding ratios of 1800(Ser):200(Thr):1(Tyr) [1].
All eukaryotic protein kinases share a conserved catalytic core, comprising N- and C-terminal lobes with an ATP-binding cleft between them. The N-lobe contains five β-strands and an α-helix, while the C-lobe is primarily composed of helices [2]. The N-lobe β-strands serve as ATP-binding sites, and the α-helix acts as a regulatory unit crucial for kinase activation, connecting the N- and C-lobes. The C-lobe houses the substrate binding groove and the activation segment, which encompasses the catalytic loop, DFG motif, activation loop, P + 1 loop, and APE region [3]. The activation segment’s catalytic and regulatory functions facilitate the accurate transfer of the phosphate residue to the substrate. Mammalian protein kinases are tightly regulated, with the DFG-motif-regulated phosphorylation of the activation loop functioning as an activation/deactivation switch [2]. Activation loop phosphorylation represents the most common mechanism for regulating kinase activity [4]. Phosphorylated protein kinases are typically in an active ‘on’ state, while dephosphorylated kinases assume a basal ‘off’ state. The three-dimensional structure of the activation segment in different active protein kinases is highly conserved, whereas inactive conformations exhibit significant variation, allowing for kinase-specific activation mechanisms and high specificity [2]. Protein kinase activation can also be dependent on the priming phosphorylation of the substrate [3].
Manning and colleagues identified 518 protein kinases in the human genome in 2002, constituting one of the largest gene families in eukaryotes and accounting for approximately 2% of all human genes [5]. This collection of over 500 protein kinases, which forms the human ‘kinome’, was further categorized into seven major families based on structural similarities within their catalytic domains. Cyclin-dependent kinases (CDKs), mitogen-activated protein kinases (MAPKs), glycogen synthase kinases (GSK3s), CDC-like kinases (CLKs), and related kinase families were grouped together to form the CMGC kinase family based on similarities in their kinase domains [5]. The CMGC family is highly conserved across various species, from nematodes to humans, suggesting that their functions are essential for the survival of these organisms.
CMGC kinases possess a conserved kinase core, similar to other protein kinases. However, they also share a unique CMGC-insert segment absent in other protein kinases [3]. Located in the C-lobe, the CMGC-insert is involved in binding co-proteins that participate in kinase function [6]. This domain exhibits low homology among family members, thereby enabling substrate specificity. CMGC kinases are primarily regulated through tyrosine phosphorylation in the activation loop [7] or pre-phosphorylation of the substrate, which prepares it for sequential recognition and phosphorylation by these kinases [8]. Another shared functional feature is their preference for phosphorylating substrates with proline in the P + 1 position.
In total, the CMGC family comprises 62 members (uniprot.org (accessed on 29 May 2023) [9]), which can be further subdivided into eight subfamilies [5] (Figure 1). CDKs and MAPKs constitute the two largest and most extensively studied families, with 21 and 14 members, respectively [10]. Other families include DYRKs (dual-specificity tyrosine (Y)-phosphorylation-regulated kinases; 10), CLKs (4), RCKs (tyrosine kinase gene v-ros cross-hybridizing kinase; 3), CDKL (cyclin-dependent kinase-like; 5), GSK (2), and SRPK (SR-specific protein kinase; 3). Most CMGC kinases are associated with specific biological functions, such as controlling the cell cycle (CDKs), determining cell fate (MAPKs), regulating multiple signaling pathways (GSKs), and facilitating RNA splicing (CLKs). The individual subfamilies and their unique features will be described in detail below.
Given the crucial roles of CMGC kinases in regulating various cellular processes, their dysregulation can contribute to cancer development and progression. In this review, we will delve into the importance of CMGC kinases in cancer biology, their involvement in cellular signaling pathways, protein–protein interactions, and the current state of kinase inhibitors targeting these kinases. Furthermore, we will discuss the challenges and future perspectives in targeting CMGC kinases for cancer therapy, including potential strategies to overcome resistance, the development of more selective inhibitors, and novel therapeutic approaches.
CMGC kinases have gained considerable attention in the context of targeted cancer therapies, with several FDA-approved inhibitors already available and numerous others in various stages of clinical trials. While these inhibitors have demonstrated efficacy and safety to varying degrees, challenges persist, such as resistance development and off-target effects. The need for selective kinase inhibitors and novel therapeutic approaches, including combination therapies, underscores the importance of continued research in this area. Furthermore, the discovery and validation of specific biomarkers for patient stratification and personalized medicine approaches are crucial for selecting the most appropriate therapeutic strategies for individual patients, ultimately improving treatment outcomes and minimizing potential side effects.
In summary, this review highlights the importance of CMGC kinases in eukaryotic cells and their roles in regulating critical cellular processes. As such, understanding the dysregulation of these kinases and their contribution to cancer development provides a foundation for the development of targeted therapies. By delving into the structure, function, and regulation of CMGC kinases, this review aims to provide a comprehensive overview of their involvement in cancer biology and therapeutic potential. The subsequent sections will explore each subfamily in greater depth and discuss the current state of research on kinase inhibitors targeting CMGC kinases, as well as the challenges and future directions in this area of cancer therapy.
2. CMGC Kinase Subfamilies
2.1. Cyclin-Dependent Kinases (CDKs)
CDKs or cyclin-dependent kinases form a family of 21 constitutively expressed serine/threonine kinases that regulate the cell cycle, transcription, mRNA processing, and the differentiation of cells [11]. As their name suggests, CDKs depend on cyclins, proteins whose levels constantly fluctuate in a tightly coordinated manner during the cell cycle. In CDK/cyclin complexes, CDKs function as catalytic subunits, while cyclins serve as regulatory subunits required for CDKs to exhibit enzymatic activity. The primary mechanism for regulating CDK enzymatic activity is the production and degradation rate of cyclins. The CDK activation loop contains residues for both cyclin and subsequent ATP binding. Like most eukaryotic protein kinases, CDKs are also regulated by the phosphorylation of the activation segment. Phosphorylation can either inhibit by interfering with ATP binding at the catalytic cleft or activate when the phosphorylation site serves as a substrate for CDK-activating kinases, including other CDKs. Activating phosphorylation enhances substrate binding by promoting full kinase activity [12,13].
CDK activity is downregulated by two families of small proteins that function as CDK inhibitors during the cell cycle. The INK4 protein family acts during the G1 phase, specifically inhibiting CDK4/CDK6 binding with cyclin D [11,14]. The broader-spectrum inhibitors of the Cip/Kip family inhibit CDK/cyclin complex activity throughout the cell cycle [11,14]. Based on evolutionary clustering, CDKs form kinase subfamilies that regulate separate cellular functions. CDKs 1–4 and 6 primarily regulate cell cycle progression, whereas CDKs 7–9, 11–13, and 19 have established roles in transcription [11]. Other CDKs have varying roles that will be discussed later.
2.1.1. Cell Cycle Regulation by CDKs (CDK 1–4, and 6)
Most living organisms are unicellular, but others, like humans, consist of millions of cells. During the development of these multicellular organisms, multiple rounds of cell growth and division must occur, a process that continues throughout an individual’s life. This elegant, tightly regulated universal process of cell growth and division, where a cell duplicates its contents and divides into two, is known as the cell cycle. Under normal conditions, the cell cycle is tightly regulated at each stage through the activation or deactivation of various proteins. The mammalian cell cycle has five phases: G0 (resting state), G1 and G2 (RNA and protein synthesis), S (DNA replication), and M (mitosis and the completion of cell division). Multiple checkpoints ensure normal progression from one phase to another, with specific cyclin/CDK complexes controlling cell cycle progression. Briefly, the cyclin C/CDK3 complex assists cells in the G0–G1 transition ([15]; uniprot.org (accessed on 29 May 2023)). The cyclin D/CDK4 (CDK6) complex initiates G1 progression through the phosphorylation of the retinoblastoma protein, leading to the transcription of genes needed for DNA synthesis and further cell cycle progression (Figure 2A). The cyclin E/CDK2 complex serves as a gatekeeper in the G1/S transition and is responsible for the hyper-phosphorylation of the retinoblastoma protein. During the S phase, cyclin A accumulates, and the activity of the cyclin A/CDK2 complex is essential for S phase termination via phosphorylation of transcription factor E2F1. Cyclin A/CDK1 and cyclin B/CDK1 complexes are crucial for progression through the G2 and M phases [11,14]. CDK7 can also influence the cell cycle by acting as a CDK-activating kinase, phosphorylating, for example, CDK1 and CDK2 [16].
To study the functional roles of individual cell-cycle-related CDKs in vivo, multiple knockout mice have been generated. The analyses of knockout animals lacking one or more of the cell-cycle-related CDKs—CDK1 [17], CDK2 [18,19], CDK4 [20,21], and CDK6 [22]—have shown that only CDK1 is essential for embryonic cell division, leading to early embryonic lethality. CDK2, CDK4, and CDK6 are mainly needed for the development of distinct cell types, and the knockout animals develop normally into adulthood [23]. CDK4/CDK6 double-knockout mutant embryos die in utero [22].
2.1.2. Transcription Initiation, Elongation, and Termination Regulation by CDKs (‘Transcriptional CDKs’: CDK 7–9, 11–13, and 19)
Transcription, the tightly regulated process of copying genetic information from DNA to RNA, relies on RNA polymerase II (RNA pol II) for all protein-coding genes. RNA pol II has a C-terminal domain (CTD) composed of polypeptide repeats, which are phosphorylated in a gene-specific manner to regulate its activity. The first step in transcription involves recruiting RNA pol II to the promoter of the transcribed gene, a task performed by the transcription preinitiation complex (PIC) that comprises several general transcription factors (TFIIA, TFIIB, TFIID, TFIIE, IFIIF, TFIIH), the Mediator complex, and RNA pol II itself (Figure 2B). The TFIIH complex, which includes CDK7, cyclin H, and MAT1, is crucial at the transcription start site. It unwinds the DNA, positions it correctly in the RNA pol II active site, and phosphorylates a serine (primarily Ser-5, but it can also be Ser-7) at the CTD of RNA pol II to initiate RNA production. As the transcript reaches a certain length, RNA pol II sheds most of the initial transcription factors, and new factors are recruited. During elongation, one of these newly recruited factors is the CDK9/cyclin T-containing positive transcription elongation factor, pTEFb. The CDK9 subunit phosphorylates another serine (Ser-2) in the CTD of RNA pol II. CDK12 interacts with cyclin K and has been shown to phosphorylate the same serine-2 in the CTD. CDK12, CDK11, and CDK13 also play roles in alternative RNA splicing (uniprot.org (accessed on 29 May 2023); [24,25]). The last step of transcription is termination, which releases both RNA pol II from the template DNA and mRNA from the transcriptional complex. The presence of CDK8 and the CDK19-containing Mediator complex is also necessary for RNA pol II phosphorylation, although the primary function of the Mediator is to transfer gene-specific regulatory signals from multiple transcription factors to RNA pol II [26]. To date, knockout animal models of transcriptional CDKs are available only for CDK7 [27], CDK8 [28], CDK11 [29], and CDK12 [30]. All of these knockouts are embryonic-lethal during early development.
2.1.3. Other CDKs (CDK5–10, 14–18, and 20)
CDK5 is expressed in post-mitotic cells like neurons [31]. The presence of CDK5 is essential for the developing brain [32], and later, in the adult brain, it is involved in multiple neuronal processes, including learning and memory, survival, synaptic plasticity, and pain signaling [33]. In the brain, CDK5 is activated by interaction with the non-cyclins CDK5R1 (p35) or CDK5R2 (p39). Cyclins D1, D3, and E have been reported to form a complex with CDK5 [33], and in 2009 [34], cyclin I was identified as a new CDK5 activator. Recently, cyclin I-like (CCNI2), a homolog of cyclin I, was shown to activate CDK5, affect its cellular localization, and has a role in cell cycle progression and cell proliferation [35].
The functional role and activating cyclin of CDK10 were only recently detected [36]. Previously, the silencing of CDK10 was linked to ETS2-driven increased expression of c-RAF, leading to MAPK pathway activation, and a loss of estrogen responsiveness of breast cancer cells, causing tamoxifen resistance in certain breast tumors [37]. The study of Guen (2013) showed that cyclin M interacts with CDK10, and this complex phosphorylates the ETS2 transcription factor, positively controlling ETS2 degradation by the proteasome.
CDK14 to CDK18 belong to the PFTAIRE and PCTAIRE kinase subfamilies (HUGO Gene Nomenclature Committee, genenames.org, accessed on 29 May 2023). CDK14 and CDK16 are activated at the plasma membrane by cyclin Y and participate in a multitude of signaling cascades, such as the Wnt pathway [38,39]. This pathway is known to control transcription, and its regulators are also important during mitosis. Notably, cyclin Y expression has been shown to peak during the G2-M phase of the cell cycle, suggesting these enzymes play a role in cell division [11]. CDK15, CDK17, and CDK18 are the least studied CDKs. CDK15 has been shown to phosphorylate survivin and lead to a reduction in tumor-necrosis-factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis [40]. The elevated phosphorylation of CDK17 and CDK18 was recently identified in a neuroblastoma cell line expressing amyloid precursor protein (APP) [41]. Cyclin-dependent kinase 20 (CDK20) plays a key role in the regulation of primary cilia, essential sensory organelles in cells. It controls ciliary length and contributes to cilia disassembly. The overexpression of CDK20 results in shorter cilia, whereas its knockdown leads to longer cilia and more ciliated cells, due to increased cilia stability in the absence of kinase activity [42]. Given the critical role of cilia in various cellular processes and diseases, understanding CDK20′s function provides important insights into these areas.
In sum, the cyclin-dependent kinases (CDKs) are a diverse family of proteins that play crucial roles in regulating cell cycle progression, transcription, and various signaling pathways. The functions of CDKs are tightly regulated through interactions with their activating cyclin partners and various phosphorylation events. Given their significant roles in cellular processes, the dysregulation of CDKs has been implicated in numerous diseases, including cancer and developmental abnormalities. Consequently, CDKs have emerged as potential therapeutic targets, and efforts are being made to develop specific inhibitors that can modulate their activity. In 2020, the FDA approved the CDK4/6 inhibitor abemaciclib in combination with endocrine therapy for the adjuvant treatment of hormone-receptor-positive, HER2-negative, high-risk early breast cancer [43].
Studies have revealed a critical role for CDK2 in renal carcinogenesis. The overexpression of CDK2 has been found in clear cell renal cell carcinoma (ccRCC), the most common type of kidney cancer, and it is associated with a higher grade and stage of the disease [44]. The study showed that higher CDK2 expression promotes cell proliferation and inhibits apoptosis in ccRCC. Furthermore, CDK2 overexpression has been associated with poor prognosis in patients with ccRCC, indicating its potential as a prognostic biomarker. The role of CDK1 and -2 activity as a biomarker in renal cell carcinoma has been studied [45] and downregulating CDK2 by miRNA in a mouse model suppressed tumor growth [44].
Future research will likely continue to uncover novel functions and regulatory mechanisms involving CDKs, providing new insights into their roles in both physiological and pathological processes. A deeper understanding of CDKs will aid in the development of more effective therapeutic strategies to target these kinases and combat cancer.
2.2. Mitogen-Activated Protein Kinases (MAPKs)
The mitogen-activated protein kinase (MAPK) family consists of 14 serine/threonine kinases that can be divided into atypical and conventional kinases.
The atypical mitogen-activated protein kinases (MAPKs) form a distinct, unique subgroup within the larger MAPK family, a major regulator of cellular processes. This group includes extracellular signal-regulated kinase 3 (ERK3), ERK4, ERK7, and Nemo-like kinase (NLK) [46]. Unlike their typical counterparts, atypical MAPKs feature a divergence from the canonical MAPK architecture in their kinase domain, deviating from the conserved TXY motif crucial for activation [47]. Moreover, they exhibit unique activation and regulatory mechanisms, such as autophosphorylation [48]. Their specific physiological roles and functions remain more undefined compared to classical MAPKs. However, emerging evidence links atypical MAPKs to several critical cellular processes, including cell differentiation, proliferation, motility, and apoptosis [49]. For instance, ERK3 and ERK4 are associated with cell migration and actin cytoskeleton reorganization, while ERK7/8 plays roles in cell cycle progression and response to stress [50]. In spite of this, the mechanisms by which atypical MAPKs exert their influence and their potential implications in human diseases, such as cancer, are areas of ongoing research and discovery.
The conventional kinases can be further subdivided into four subfamilies: (1) extracellular signal-regulated kinases 1/2 (ERK1/2), (2) c-Jun amino-terminal kinases 1, 2, and 3 (JNK1-3), (3) p38 MAPKs, and (4) extracellular signal-regulated kinase 5 (ERK5). These enzymes are characterized by a system in which three kinases act sequentially to phosphorylate the downstream kinase (Figure 2C). Activators for ERKs are MEK1 and MEK2; for JNKs-, MKK4 and MKK7; for p38-, MKK3 and MKK6; and for ERK5-, MEK5. MAPKs are highly conserved, respond to extracellular signaling cues, and regulate numerous essential cellular processes such as proliferation, differentiation, stress responses, and the transcriptional control of CDKs in organisms ranging from yeast to humans. Over the past few decades, extensive research has been conducted on MAPKs, from their substrates and functions to their roles in health and cancer.
Extracellular signal-regulated kinases 1/2 respond to various stimuli, including growth factors, insulin, cytokines, and carcinogens [50,51]. They are components of the cell surface receptor (receptor tyrosine kinase)/RasGTP/Raf/MEK/ERK signaling cascade. Components of this signaling cascade are essential for proper cell proliferation. ERKs function both in the cytoplasm to promote cell proliferation (G1 to S phase transition) and in the nucleus to phosphorylate and activate numerous transcription factors (TFs) [50,52]. Upon TF activation, c-Jun is expressed and stabilized by direct phosphorylation by ERK1/2 [53], allowing association with c-Fos and the subsequent formation of the AP-1 complex. AP-1 is essential for cyclin D1 expression, which interacts with CDKs to drive G1 to S transition.
- c-Jun N-terminal kinases are highly homologous (>85%) but have a distinct tissue distribution. JNK1 and 2 are ubiquitously expressed, while JNK3 expression is mainly limited to the brain. In contrast to ERKs, JNKs are primarily activated by stress signals such as oxidative stress, radiation, and DNA-damaging agents. JNKs are mainly localized in the cytoplasm, but their identified substrates are mostly TFs, including c-Jun, p53, STAT3, and c-Myc. The phosphorylation of c-Jun leads to AP-1 complex formation and thus the transcription of cyclin D1, promoting cell cycle progression, similar to ERK1/2 [54]. To date, only a few cytoplasmic interaction partners of JNKs have been identified [50,55].
- The p38 subfamily consists of four members (α, β, γ, δ), which respond to various environmental stress stimuli and cytokines such as interleukin-1 and tumor necrosis factor α(TNF). Interestingly, p38 both regulates the production of cytokines and responds to them. Other targets of p38 regulation are TFs and other protein kinases. Based on observations of p38 activation, it plays a role in inflammation, cell cycle regulation, and apoptosis [50,56].
- ERK5 (BMK1 or big MAP kinase 1) has a kinase domain similar to ERK1/2, sharing 51% similarity with ERK2. ERK5 is essential during normal embryogenesis [57]. An upstream activator of ERK5 is MEK5, whose expression is elevated in metastatic prostate cancer [58]. Similar to ERK1/2 and JNK, ERK5 also promotes cyclin D1 expression and cell cycle progression [59], as well as plays a crucial role in the maintenance of mitochondrial function and neuronal survival [60]. ERK5 is involved in various cellular processes, including cell survival, differentiation, and angiogenesis. Its activation has been linked to growth factors, oxidative stress, and other extracellular stimuli.
- The MAPK pathway has a critical role in cancer biology, extending beyond the extensively studied BRAF mutation in melanoma. The MAPK/ERK pathway, for instance, has been implicated in colorectal cancer, with mutations in KRAS and NRAS genes leading to its persistent activation, promoting uncontrolled cell proliferation and tumor growth [61]. These mutations, unfortunately, render the tumors resistant to EGFR-targeted therapies, highlighting the need for novel therapeutic strategies [62].
- Additionally, the JNK MAPK pathway, associated primarily with responses to stress signals and apoptosis, has shown links to cancer biology. Aberrations in JNK signaling can lead to an imbalance between cell proliferation and death, thereby contributing to oncogenesis. For instance, overactive JNK signaling has been found in several cancers, including breast and gastric cancer, often correlating with a worse prognosis [63]. Furthermore, the p38 MAPK pathway, typically associated with inflammation and cell differentiation, is also relevant in cancer research. Its complex, dual role in tumorigenesis is being unraveled; while its activation can suppress tumor growth by promoting cell cycle arrest and apoptosis, chronic activation can also enhance cancer cell survival, contributing to chemoresistance [64].
In summary, the MAPK family comprises a diverse group of kinases that play crucial roles in cellular signaling pathways. These kinases are involved in various cellular processes, including proliferation, differentiation, and stress responses, and are integral to the transcriptional control of CDKs. Alterations in the function of the components of the cellular signaling cascade can lead to cancer. Thus, understanding the mechanisms and functions of MAPKs is essential for advancing our knowledge of cellular processes and uncovering potential therapeutic targets in various diseases.
2.3. Glycogen Synthase Kinase-3 (GSK-3)
Glycogen synthase kinase-3 is a conserved, ubiquitously expressed serine/threonine kinase with unique characteristics, such as its high activity in resting cells. Humans have two forms of GSK-3s: GSK-3α and GSK-3β, which share an 85% overall sequence homology [65]. The kinase domain of these kinases is highly homologous (98%), while the N- and C-terminal domains differ [65]. GSK-3α and GSK-3β have distinct functions, as evidenced by the fact that homozygous GSK-3β knockout mice are embryonic-lethal, whereas GSK3α knockout mice are viable [66,67].
Unlike the CDK and MAPK families, GSK activity is regulated through multiple levels and mechanisms, ranging from phosphorylation by other kinases, autophosphorylation, and priming phosphorylation of the target substrate by another kinase [68,69,70]. In most cases, the phosphorylation of serine residues inhibits GSK’s kinase activity, while tyrosine phosphorylation increases its activity [71]. GSK-3 (mainly GSK-3α) has been extensively studied and is known to function in various signaling pathways, including Wnt, Notch, and Hedgehog (proliferation), as well as growth factors affecting differentiation/survival. It has been implicated in multiple roles in many human pathological conditions, ranging from neurodegenerative diseases to diabetes and several types of cancers [70,72].
GSK is a critical component of the canonical Wnt signaling pathway, which is essential during normal development and is often dysregulated in cancers. The main events during Wnt-β-catenin signaling involve a central component known as the destruction complex that regulates cytoplasmic β-catenin levels. This complex comprises tumor suppressors Axin and adenomatosis polyposis coli (APC), as well as casein kinase-1α (CK1α) and GSK-3 [73]. In the absence of the Wnt-ligand, β-catenin is assembled into this complex and phosphorylated by CK1 and GSK-3, marking it for proteasomal degradation and thus preventing the transcription of β-catenin-dependent genes. When Wnt binds to its receptor Frizzled, the destruction complex cannot phosphorylate and ubiquitinate β-catenin, allowing it to translocate to the nucleus, where it forms complexes with transcription factors and promotes transcription. Phosphorylation by GSK-3 is also essential for the targeting of multiple other proteins for proteasomal degradation [72,74]. In addition to its role in Wnt signaling, GSK-3 has been implicated in other critical signaling pathways. For example, GSK-3 plays a role in the insulin signaling pathway, which is essential for glucose homeostasis. Abnormalities in GSK-3 function have been linked to insulin resistance and the development of type 2 diabetes [75]. GSK-3 inhibitors have shown promise in preclinical studies as potential therapeutic agents for the treatment of type 2 diabetes [76].
GSK-3 has been found to be overactive in various types of cancers, including colon, breast, and prostate cancer. The overactivation of GSK-3 can lead to increased cell proliferation, migration, and invasion, as well as reduced apoptosis [70]. Targeting GSK-3 may represent a potential therapeutic strategy for cancer treatment. Preclinical studies have shown that GSK-3 inhibitors can suppress tumor growth and metastasis, as well as sensitize cancer cells to chemotherapy and radiation therapy [72].
In summary, GSK-3 is a multifunctional kinase that plays a pivotal role in numerous signaling pathways and cellular processes. The dysregulation of GSK-3 activity has been implicated in various pathological conditions. Consequently, GSK-3 has emerged as a potential therapeutic target, and the development of GSK-3 inhibitors may hold promise for the treatment of these diseases. However, due to the pleiotropic nature of GSK-3, further research is needed to understand the precise mechanisms of GSK-3 regulation and its involvement in different pathologies to develop safe and effective therapeutic strategies.
2.4. Dual-Specificity Tyrosine (Y)-Phosphorylation-Regulated Kinases (DYRKs)
DYRK is a relatively large, mammalian, dual-specificity protein kinase family that, based on homology analysis, consists of three subfamilies with a total of 10 members: DYRK (DYRK1A-B, 2–4), HIPKs (homeodomain-interacting protein kinase 1–4), and PRP4s (pre-mRNA processing protein 4 kinase).
2.4.1. DYRK1–4
The DYRK subfamily members are autophosphorylated on tyrosine within the activation loop during their translation, resulting in a constitutively active mature protein [77]. Autophosphorylation of this conserved tyrosine is essential for achieving full kinase activity [78]. As DYRKs are not subject to classical MAPK-like regulation by an upstream protein kinase, other regulatory mechanisms have been proposed. All of these subfamily members experience changes in their cytoplasmic versus nuclear localization, which are speculated to limit substrate accessibility and, thus, regulate DYRK1–4 [79]. Other regulatory mechanisms for DYRKs might include alterations in gene expression and protein abundance or interactions with regulatory proteins. Thus far, DYRKs have not been shown to undergo regulation by reversible phosphorylation or dephosphorylation [79].
The major function of mammalian DYRKs is the regulation of the cell cycle. Both DYRK1A and DYRK1B can be classified as survival- and differentiation-promoting factors. DYRK1A has been shown to control the length of the G1 phase by regulating cyclin D1 levels and promoting the transition between quiescence and differentiation [80]. DYRK1B is active in quiescent cells, preventing entry into G1 by stabilizing CDK inhibitors and destabilizing cyclin D [81,82]. In apoptosis, DYRK1A, DYRK3, and DYRK2 have opposite roles, as DYRK1A and DYRK3 phosphorylate either pro-apoptotic proteins or their inhibitors, leading to the inhibition of apoptotic activity [83,84], while the phosphorylation of p53 by DYRK2 (or HIPK2) promotes apoptosis [85].
Another common characteristic of the DYRKs is their action as priming serine/threonine kinases for subsequent phosphorylation by GSK-3, thus targeting proteins for proteasomal degradation [79]. To date, DYRK family members have been identified as priming kinases for oncoproteins c-Jun and c-Myc, in addition to transcription factor GLI2 [9,85]. Although the functions of the fourth member of this subfamily, DYRK4, are ambiguous, based on the molecular roles of other DYRKs, one can conclude that the members of this subfamily are involved in cell survival, cell differentiation, gene transcription, and translation [79].
2.4.2. Homeodomain-Interacting Protein Kinase (HIPK)
HIPKs, like DYRKs, are autophosphorylated to reach full kinase activity [86]. HIPK1–3 are highly homologous, whereas the later identified fourth member, HIPK4, is only related to others in its catalytic domain [87]. Most of the knowledge about HIPKs comes from HIPK2, which is known to act in the cell cycle, apoptosis, and responses to DNA damage, mostly via interactions with transcription factors. The activity of HIPK2 itself is orchestrated by multiple post-translational modifications and caspase-mediated cleavage [86]. Due to structural similarity, HIPK1 is thought to have a similar role in cells to HIPK2 [88]. A recent study identified HIPK2 as a facilitator of Disheveled phosphorylation, maintaining cells responsive to Wnt ligand stimulation [89]. In 2019, a study revealed the role of HIPK1 in the regulation of cellular stress response and its potential as a therapeutic target for the treatment of glioblastoma [90]. The molecular functions of both HIPK3 and, especially, HIPK4 are still rather uncharacterized.
2.4.3. Pre-mRNA Processing Protein 4 Kinase (PRP4)
PRP4 is a subfamily of ubiquitously expressed kinases that regulate pre-mRNA splicing and act as spindle assembly checkpoint proteins [91,92]. The kinase domain of PRP4 was identified as essential for pancreatic and colorectal tumor cell viability [93]. The PRP4 family members play a critical role in maintaining the proper function of spliceosomes, the cellular machinery responsible for splicing introns from pre-mRNA [94]. PRP4 kinase phosphorylates several spliceosome components, including the U4/U6-U5 tri-snRNP, to facilitate the assembly and disassembly of the spliceosome [95]. A 2016 study reported the role of PRP4 in regulating the pre-mRNA splicing of the POLD1 gene, which is involved in DNA replication and repair, suggesting a potential link between PRP4 and cancer development [96]. Due to the association between aberrant splicing and various diseases, PRP4 kinases are interesting targets for further investigation.
2.5. Cdc2-like Kinase (CLK) and Other Less-Studied Kinases
The Cdc2-like kinase (CLK) family consists of four dual-specificity kinases (CLK1–4) that autophosphorylate on tyrosine residues and specifically phosphorylate serine/threonine residues on their substrates [97]. CLKs are found in both the cytoplasm and the nucleus, where they phosphorylate the arginine- and serine-rich (RS) domains of serine/arginine (SR) splicing factor proteins, thereby controlling their nuclear distribution [98]. SR splicing factor proteins are extensively phosphorylated by multiple kinases, and the proper phosphorylation status has been shown to be essential for SR protein activity [99]. These proteins play a crucial role in alternative mRNA splicing, a fundamental process responsible for generating the complex human proteome during normal development.
In a recent study, CLKs were identified as upstream regulators of Aurora B, acting as novel components in the final steps of cytokinesis, the division of the cytoplasm into two daughter cells [100]. However, as Petsalaki and Zachos concluded, our current understanding of CLKs’ cellular functions remains limited [100]. A 2017 study reported that CLK inhibition could modulate the alternative splicing of Mcl-1, an anti-apoptotic protein, and promote cancer cell death, suggesting a potential therapeutic approach for cancer treatment [101]. Given the importance of alternative mRNA splicing in the regulation of gene expression and the emerging roles of CLKs in cell division, further investigation into the functions and regulation of CLKs could provide valuable insights into their role in various cellular processes and their potential as therapeutic targets in human diseases.
2.6. SR-Specific Protein Kinase (SRPK)
Serine–arginine protein kinases (SRPKs) specifically phosphorylate the serine residues in serine/arginine (SR) dipeptides. They are primarily localized in the cytoplasm, where these constitutively active kinases regiospecifically phosphorylate SR splicing factors on multiple serines, inducing the translocation of these factors into the nucleus. In the nucleus, SR splicing factors are further phosphorylated by CLKs [102,103]. The change in the cellular localization of SR proteins is essential for phosphorylation by CLKs [98]. As mentioned earlier, SR protein phosphorylation is necessary for correct mRNA splicing and maturation.
Over 100 SR-domain-containing proteins have been identified in the human genome [104], indicating that there are many open questions regarding the functional roles of SRPKs in mammalian cells. SRPK1 expression is elevated in breast, colon, and pancreatic cancers [105], as well as in acute T-cell leukemia [106]. A 2020 study identified SRPK1 as a potential therapeutic target in glioblastoma, with the SRPK1 inhibitor SPHINX31 showing promising results in preclinical models [107]. SRPK2 has a specific role in phosphorylating apoptosis-promoting protein ACIN1 and increasing cyclin A1 expression in leukemia cells [108]. Given the importance of SRPKs in the regulation of alternative splicing and their association with various types of cancer, further studies on the functional roles of these kinases and their potential as therapeutic targets in human cancer are warranted.
2.7. Tyrosine Kinase Gene v-Ros Cross-Hybridizing Kinase (RCK)
The RCK serine/threonine-protein kinase family consists of three kinases: MAK, ICK, and MOK. The functions of these kinases are poorly understood. Structurally, the RCK family resembles both MAPKs and CDKs [5]. MAK and ICK autophosphorylate on a tyrosine residue, but they need a second phosphorylation on their MAPK-like motif in their activation loop by an upstream kinase for full enzymatic activity [109,110,111]. Different from MAPK, the activating kinase for MAK and ICK is cell-cycle-related kinase (CCRK), but for MOK, it remains unknown [111,112].
- MAK (male germ cell-associated kinase) is mainly expressed in testicular germ cells during spermatogenesis and in the retina. In the retina, MAK localizes to connecting cilia in photoreceptor cells, negatively regulates the length of their cilia, and is essential for the survival of these cells [113]. Not surprisingly, MAK mutations are associated with retinitis pigmentosa, a photoreceptor degeneration disease in the retina [114,115].
- ICK (intestinal cell kinase) is highly conserved, constitutively, and widely expressed. Similarly to MAK, it negatively regulates ciliary length and is identified as an essential component of sonic hedgehog signaling [116]. These two factors seem to be the underlying cause of human ECO syndrome, a multi-organ illness affecting the endocrine, cerebral, and skeletal systems, caused by a missense mutation in the ICK gene [117]. In 2017, a study reported the role of ICK in colorectal cancer progression and its potential as a therapeutic target for the treatment of colorectal cancer [118].
The cellular function of the third RCK, MOK (MAPK/MAK/MRK-overlapping kinase), is largely unknown. To date, only the expression of MOK in mouse wild-type and cancerous intestinal cells has been analyzed by Western blotting, showing the downregulation of MOK in adenomas [80]. This observation suggests that MOK might have a role in intestinal cancer development, but further studies are needed.
2.8. Cyclin-Dependent Kinase-like (CDKL)
This family of five members of serine/threonine kinases with homology to MAPK and CDK families is relatively uncharacterized. Studies have focused on CDKL5, a ubiquitous protein mainly expressed in the brain, testes, and thymus [119]. In the context of cancer, CDKL proteins are now emerging as potential players. Studies suggest that the dysregulation of CDKL5 may contribute to tumorigenesis in certain cancers. For instance, the aberrant expression of CDKL5 has been suggested in glioma and some evidence exists on its role in breast cancer, indicating its possible role in disease progression and patient prognosis [120].
Similarly, another family member, CDKL1, has been associated with colorectal cancer, with its overexpression correlating with poor survival outcomes [121]. These preliminary findings suggest that CDKL proteins could potentially act as novel markers for cancer diagnosis or targets for therapeutic intervention.
3. Protein Kinase Therapeutics—Kinase Inhibitors
The protein kinase domain is among the most commonly encountered domain in known cancer genes [122]. As described earlier, most of the CMGC kinases affect various cellular signaling pathways that control critical processes like cell cycle progression, proliferation, differentiation, apoptosis, or survival. Often, abnormal phosphorylation is either the cause or consequence in cancer [123]. Therefore, protein kinases are among the most studied druggable targets in pharmacological research [124,125]. In human cancers, kinases are often found overexpressed or overactive, due to a vast array of genetic and epigenetic events, including point mutations, chromosomal gene amplifications (copy number alterations), and chromosomal translocations giving rise to gene fusions [126] (Table 1).
3.1. Oncogenic Relations of CMGC Family Members
Combined data from six recent studies identified a total of 1100 genes that drive cancer development. From this list, an enrichment of protein kinases was observed (91 kinases were present) [149]. Six of them (6/91; ~7%) belonged to the CMGC kinase family [149], involving family members of CDK, MAPK, and DYRK, namely CDK4, CDK6, CDK12, MAPK1 (Erk2), MAPK8 (JNK1)), and DYRK1A. Expectedly, the alterations observed in these genes were either point mutations or copy number changes [149]. Other studies have also identified amplified CDK12 expression in breast cancers and inactivated in ovarian cancers [150,151,152]. In addition, over-expression of CDK7 together with MAT1 and cyclin H was detected in >900 estrogen-receptor-positive breast cancer samples [153].
The deregulation of MAPK signaling pathways can result in the activation of oncogenic signaling, leading to increased cell proliferation, survival, and migration [154]. Mutations in components of the MAPK pathways, such as RAS and RAF, can lead to the constitutive activation of MAPK signaling, promoting cancer development and progression [155]. Aberrant activation of MAPK signaling has been implicated in various cancer types, including melanoma, colorectal, and lung cancers [154].
DYRK1B overexpression has been observed in pancreatic and ovarian cancer [156], and in osteosarcomas and rhabdomyosarcomas [141,142]. Interestingly, the depletion of DYRK1B drove pancreatic and ovarian cancer cells to apoptosis [157,158].
Dysregulation of GSK-3 can contribute to cancer development by affecting various cellular processes, such as cell cycle regulation, differentiation, and apoptosis [69]. GSK-3 can act as a tumor suppressor or an oncogene, depending on the cellular context and specific signaling pathways involved [159]. For example, GSK-3 promotes the degradation of β-catenin, a key mediator of the Wnt signaling pathway, thereby preventing the activation of oncogenic Wnt target genes [69]. However, GSK-3 can also promote cancer progression by stabilizing the oncoprotein c-Myc and enhancing its activity [69].
The aberrant activation of CLKs has been associated with cancer-associated splicing alterations, which can lead to the production of aberrant protein isoforms that contribute to tumor progression [160]. For example, CLKs have been implicated in the regulation of alternative splicing events that promote cell survival, angiogenesis, and metastasis in various cancer types [161,162]. Moreover, the overexpression of CLKs has been observed in several cancer types, such as breast and prostate cancers, and is associated with poor prognosis [160].
3.2. Therapeutic Targeting of CMGC Kinases
Based on the common structural features of protein kinases, most protein kinase inhibitors target the ATP-binding site in the activation segment, thus competing with ATP. The second-generation inhibitors target protein kinase in a specific conformation and are thought to be more specific. However, there are highly selective and broad range inhibitors within both generations of kinase inhibitors [163]. Unfortunately, a common shortcoming of kinase inhibitors is the relatively low percentage of patients presenting a satisfactory response [125]. The best clinical outcome is usually reached with a polypharmacological approach, using drug combinations either within a single kinase pathway or targeting parallel kinase pathways in preselected patient populations [125,126] (Table 2).
3.2.1. CDK Inhibitors
Several CDK inhibitors have been developed to target specific CDKs involved in cell cycle regulation and other cellular processes [164]. The first US Food and Drug Administration (FDA)-approved CMGC family kinase inhibitor was palbociclib, a selective CDK4/6 inhibitor [165]. Altogether, the FDA has approved three CDK4/6 inhibitors, palbociclib, ribociclib, and abemaciclib, for the treatment of hormone-receptor-positive, HER2-negative, advanced or metastatic breast cancer [165,166,167]. These inhibitors have shown promising results in clinical trials, improving progression-free survival and overall response rates. Further research is exploring their potential in other cancer types and combination therapies.
3.2.2. MAPK Inhibitors
The success of kinase inhibitors targeting the MAPK pathway components, such as BRAF and MEK inhibitors, has demonstrated the therapeutic potential of targeting MAPK signaling in cancer [132,133]. The FDA has approved BRAF inhibitors (vemurafenib, dabrafenib) and MEK inhibitors (trametinib, cobimetinib) for the treatment of metastatic melanoma harboring BRAF mutations. Additionally, combined BRAF and MEK inhibition has shown improved outcomes compared to single-agent therapy [133]. Efforts to develop MAPK inhibitors targeting other components of the MAPK pathways are ongoing.
3.2.3. DYRK Inhibitors
A polyphenolic green tea flavonol, epigallocatechin-3-gallate (EGCG), is a DYRK1A inhibitor that was shown to normalize DYRK1A activity in Down syndrome patients in a pilot study reversing cognitive deficits [168]. The inhibition of DYRK1A is an enchanting therapeutic approach for the treatment of cognitive impairment in Down syndrome together with inhibiting tau hyperphosphorylation in Alzheimer’s disease [124]. DYRK-family inhibitors have also been shown effective in vitro in reducing proliferation and inducing apoptosis either as single agents or as dual-inhibitors targeting CKL1 [124].
3.2.4. GSK Inhibitors
A plethora of GSK-3 inhibitors have been published and studied in clinical trials for the treatment of cancer or Alzheimer’s disease [169]. However, the results of the studies have been controversial, and none of the inhibitors have made it into clinical use [169]. One of the studied inhibitors, tideglusib, showed promising preclinical results in different cancer types, such as glioblastoma and pancreatic cancer [69,170]. Further development and clinical evaluation of GSK-3 inhibitors are needed to determine their therapeutic potential in cancer treatment.
3.2.5. CLK Inhibitors
Several small molecule inhibitors targeting CLKs are being investigated for their ability to modulate alternative splicing and their potential as anticancer agents [171]. One such CLK inhibitor, TG-003, has shown promising effects in preclinical studies, modulating alternative splicing and reducing tumor cell viability [172]. Still, no CLK inhibitors have been approved for clinical use.
All in all, the development of kinase inhibitors targeting CMGC kinases has shown promise in cancer treatment (Table 2). The approval of CDK4/6 and MAPK pathway inhibitors demonstrates the potential of targeting these kinases for therapeutic intervention. Ongoing research efforts aim to develop novel inhibitors targeting GSKs and CLKs and explore their therapeutic potential in cancer and other diseases. Future research should focus on overcoming resistance to kinase inhibitors, developing more selective inhibitors with fewer off-target effects, improving patient stratification, and exploring novel therapeutic strategies, such as targeting protein–protein interactions and exploiting synthetic lethality. Addressing these challenges will pave the way for more effective and personalized cancer treatments based on CMGC kinase inhibition.
3.3. Challenges in Targeting CMGC Kinases
3.3.1. Resistance to Kinase Inhibitors
One major challenge in targeting CMGC kinases is the development of resistance to kinase inhibitors. Resistance can occur through various mechanisms, such as the acquisition of secondary mutations in the targeted kinase, the activation of compensatory signaling pathways, or alterations in drug metabolism [173,174]. Strategies to overcome resistance include the development of next-generation inhibitors with improved target selectivity, combination therapies targeting multiple signaling pathways, or sequential treatment strategies [175,176].
3.3.2. Off-Target Effects and Toxicity
Kinase inhibitors may have off-target effects, leading to unwanted toxicity and limiting their therapeutic window. To address this issue, researchers are focusing on developing more selective inhibitors with higher specificity for their target kinases [177]. Additionally, understanding the molecular basis of these off-target effects can guide the development of inhibitors with fewer side effects.
3.4. Approaches to Overcome the Challenges
3.4.1. Patient Stratification and Biomarkers
The identification of predictive biomarkers and proper patient stratification is essential for the successful implementation of targeted therapies. Efforts to identify biomarkers that predict response to CMGC kinase inhibitors can help select patients who are more likely to benefit from these treatments [178]. Additionally, the development of non-invasive techniques, such as liquid biopsies, can facilitate the real-time monitoring of treatment response and early detection of resistance [179].
3.4.2. Exploiting Protein–Protein Interactions
Another approach to overcome the challenges associated with kinase inhibitor resistance is to target protein–protein interactions involving CMGC kinases. This strategy may provide a more selective way to modulate CMGC kinase activity and minimize off-target effects [180]. Further research is needed to identify and validate novel protein–protein interaction targets in CMGC kinase signaling pathways.
3.4.3. Combination Therapies and Synthetic Lethality
Combination therapies that target multiple signaling pathways or exploit synthetic lethality can help overcome resistance and enhance the efficacy of CMGC kinase inhibitors [181] (https://clinicaltrials.gov/, accessed on 8 May 2023) (Table 3). Synthetic lethality arises when the simultaneous inhibition of two genes or pathways leads to cell death, while the inhibition of either one alone is insufficient [182]. Identifying synthetic lethal interactions involving CMGC kinases may reveal new therapeutic opportunities in cancer treatment.
As our understanding of the molecular mechanisms and protein–protein interactions involving CMGC kinases expands, so too will the opportunities for the development of more selective and effective therapeutic strategies.
4. Protein–Protein Interactions of the CMGC Kinases
CMGC kinases participate in various protein–protein interactions that are crucial for their activity, functions, and signaling pathways. Understanding these interactions is essential to elucidate the molecular mechanisms governing CMGC-kinase-mediated cellular processes. In this section, we will discuss some of the known interactions involving CMGC kinases and their importance (Figure 3).
CMGC kinases contribute to the development and progression of various diseases. Therefore, understanding the intricate network of protein–protein interactions involving CMGC kinases can provide valuable insights into the molecular mechanisms underlying their functions and offer potential therapeutic targets for the treatment of diseases associated with their dysregulation. For instance, the development of small molecule inhibitors that specifically target the protein–protein interactions of CMGC kinases could represent a promising approach to modulate their activity and restore normal cellular functions in disease settings [180].
The development of small molecule inhibitors that target the protein–protein interactions (PPIs) of these kinases has emerged as a promising strategy in cancer therapeutics. Traditional kinase inhibitors target the ATP-binding site of kinases. However, this site’s high conservation across different kinases challenges the development of inhibitors with high specificity. In contrast, the PPI interfaces are often less conserved, presenting an opportunity for creating more specific inhibitors [183,184]. Small molecule inhibitors act by obstructing the critical protein interactions necessary for the activation or functioning of these kinases. For instance, inhibitors may preclude a kinase from interacting with its activator or substrate, consequently inhibiting the kinase’s activity and downstream signaling [185]. In cancer, these inhibitors can restore normal cellular functions by blocking the aberrant signaling engendered by dysregulated kinases. For example, if a kinase is overactive in a cancer cell, promoting uncontrolled cell proliferation, an inhibitor that obstructs this kinase’s interactions could impede or slow down the proliferation [186]. Considerable progress has been made in the development of such inhibitors. High-throughput screening techniques have enabled researchers to identify small molecules that can bind to the PPI interfaces of CMGC kinases [185]. Furthermore, advances in structural biology have provided intricate details of these kinases’ 3D structures and their complexes with interacting proteins, aiding in the design of more effective inhibitors [187]. Nonetheless, challenges persist in developing PPI inhibitors. The interfaces involved in PPIs are often expansive and flat, complicating the ability of small molecules to bind with high affinity. Also, these inhibitors must navigate cellular barriers to reach their intracellular targets [186].
Most of the studies regarding kinase interactions have focused on a single kinase and its function or dysfunction in a disease. Additionally, the names of the CDK kinases were unified as late as in 2009 [10], and that might have contributed to the slowness of studies of some these kinases. It is, after all, rather difficult to assign a kinase to this interesting family when the name is very different, such as PFTK1 or CRKRS.
4.1. Affinity Purification and BioID Proximity Labeling
The first high-throughput screenings of protein–protein interactions were performed using the yeast two-hybrid technique [188]. This method allows the identification of binary interactions, thus assigning proteins to a biological context. Also, human protein–protein interactions were first studied in the yeast two-hybrid system as an important intermediate step towards a more systematic and comprehensive analysis of human interactome [189]. The next development in interactomics was the detailed characterization of yeast protein complexes utilizing affinity purification coupled to mass spectrometry (AP-MS) [190,191]. In 2009, Glatter and co-workers [192] presented a sensitive, reproducible, high-throughput AP-MS workflow for human cells. The general workflow of an AP-MS experiment has three steps: (1) expression of the epitope-tagged bait protein, (2) single- or double-step affinity purification of the protein complexes, and (3) analysis of the complex components with liquid chromatography coupled to a mass spectrometer.
This system offers several significant advantages. Some key benefits include the utilization of Gateway compatible human Orfeome [193] collections, the establishment of isogenic cell lines, the capability of inducible expression of bait proteins at nearly physiological levels, resulting in high yield, and reproducibility of affinity purification. Additionally, this system facilitates direct, “gel-free” analysis with LC-MS, enhancing its potential for precise and comprehensive proteomic studies [192]. Later, this approach was also shown to be robust and highly reproducible between laboratories [194].
Proteins are not static components of cells forming only binary interactions, but can also form indirect interactions within a protein complex or with nearby proteins [195]. The BioID method for the proximity labeling of interacting proteins is based on the ability of a modified biotin ligase (BirA*) to covalently attach a biotin on the neighboring proteins’ lysine residues upon biotin supplementation in the growth media [195]. A workflow for BioID sample analysis resembles AP-MS: the protein-of-interest (bait) is tagged with the BirA*, affinity-purified with streptavidin beads, and analyzed with LC-MS. Endogenous biotinylation in mammalian cells is mainly limited to carboxylases, thus providing a rather specific way to label close proximity interactors in living cells and to detect transient and weak interactions with a reasonable background [195,196]. It is noteworthy that the labeling radius is about 10 nm [197], indicating that spatiotemporally similar proteins will be labeled. Biotin will remain attached to the protein even though the BirA*-containing protein has moved further away, providing a molecular picture of the proteomics landscape in the cell.
4.2. Interactions of CDKs
CDKs interact with cyclins, a family of regulatory proteins, to form active CDK–cyclin complexes that regulate cell cycle progression [198]. Cyclins modulate CDK activity by promoting their catalytic activity and determining their substrate specificity [199]. CDK–cyclin complexes are regulated by CDK inhibitors (CKIs), which bind to CDKs and inhibit their activity, thus ensuring proper cell cycle control [200]. CDKs can also interact with various other proteins, such as transcription factors and chromatin remodeling enzymes, to regulate gene expression and cellular processes beyond cell cycle control [201].
4.3. Interactions of MAPKs
MAPKs are involved in highly conserved signaling cascades known as the MAPK pathways [202]. These pathways consist of a series of protein–protein interactions and phosphorylation events involving three classes of kinases: MAP kinase kinase kinases (MAP3Ks), MAP kinase kinases (MAP2Ks), and MAPKs [203]. MAP3Ks are activated in response to extracellular stimuli and phosphorylate MAP2Ks, which in turn phosphorylate and activate MAPKs. Activated MAPKs can then phosphorylate and regulate various downstream substrates, such as transcription factors, to modulate gene expression and cellular responses [202].
4.4. Interactions of GSKs
GSK-3 is known to interact with numerous proteins and is involved in various signaling pathways, such as the Wnt, insulin, and PI3K/Akt pathways [69,170]. GSK-3 can phosphorylate and modulate the activity of key proteins in these pathways, such as β-catenin in the Wnt pathway, glycogen synthase in the insulin pathway, and Mdm2 in the PI3K/Akt pathway [69]. GSK-3 also interacts with proteins involved in cell cycle regulation, apoptosis, and cellular differentiation, such as cyclin D1, c-Myc, and Bcl-2 family members [69].
4.5. Interactions of CLKs
CLKs interact with and phosphorylate serine/arginine-rich (SR) proteins, which are essential components of the spliceosome and play a critical role in the regulation of pre-mRNA splicing [204]. The CLK-mediated phosphorylation of SR proteins affects their localization, stability, and interaction with other spliceosome components, thereby modulating alternative splicing events [161,162]. The dysregulation of CLK-mediated alternative splicing has been implicated in various diseases, including cancer [160].
Further research is needed to identify additional interaction partners of CMGC kinases and elucidate their roles in the regulation of cellular signaling pathways. This knowledge will not only enhance our understanding of the molecular basis of CMGC kinase-mediated cellular processes but also facilitate the development of targeted therapeutic strategies for diseases associated with CMGC kinase dysfunction.
5. Assessment of Kinase Activity
As the function of protein kinases is to transfer a phosphate group, Western blotting with antibodies against a substrate’s phosphoserine, -threonine or -tyrosine is the most common and widely used method to indirectly identify kinase activity. This approach is very labor-intensive, highly dependent on the specificity and sensitivity of the available antibodies, and not directly comparable with high-throughput approaches.
A more sensitive, direct approach to quantitatively measure kinase activity in vitro is to measure the incorporation of radiolabeled phosphate from γ-33P ATP into the substrate [205]. A prerequisite to the solution-based in vitro kinase assays is a purified kinase, which in most cases is compared to the kinase dead mutant that abolishes ATP binding. An extensive study of 84 pairs of wild-type kinases and their kinase-dead counterparts [9] confirmed, using in vitro kinase assays, that indeed the mutated “VAIK” or “HRD” motif is essential for kinase activity. The lysine in the “VAIK” motif is among the most conserved amino acids in all kinases as it is a central catalyzer of the phosphate transfer [5].
Functional protein microarrays are an efficient ‘omics’-scale tool to detect kinase-substrate interactions [206,207]. The commercially available protein microarrays have thousands of purified proteins spotted onto a glass slide, providing an efficient means to identify the phosphorylation of candidate substrates for the kinases of interest when incubated together with radiolabeled ATP [208]. To date, the only large-scale study utilizing protein microarrays in kinase substrate identification has been performed with yeast protein kinases [209], although human protein microarrays are nowadays commercially available.
6. Conclusions
CMGC kinases play crucial roles in cellular signaling pathways, such as cell cycle regulation, proliferation, differentiation, apoptosis, and gene expression regulation [69,198,202,204]. Aberrant activation and dysregulation of these kinases have been implicated in cancer development and progression, highlighting their importance in cancer biology [162,201,203].
The therapeutic potential of targeting CMGC kinases has been demonstrated by the development and clinical success of kinase inhibitors, such as CDK4/6 inhibitors (palbociclib, ribociclib, and abemaciclib) for hormone-receptor-positive, HER2-negative, advanced or metastatic breast cancer [165,166,167], and BRAF and MEK inhibitors (vemurafenib, dabrafenib, trametinib, and cobimetinib) for metastatic melanoma-harboring BRAF mutations [132,133]. Furthermore, ongoing research efforts aim to develop novel inhibitors targeting GSKs and CLKs and explore their therapeutic potential in cancer and other diseases [171,210,211].
However, several challenges remain in targeting CMGC kinases for cancer therapy, including resistance to kinase inhibitors, off-target effects, and the need for better patient stratification [173,174,177,178]. To overcome these challenges, future research should focus on developing next-generation inhibitors with improved target selectivity, combination therapies targeting multiple signaling pathways, sequential treatment strategies, and exploiting protein–protein interactions involving CMGC kinases [175,176,180]. Additionally, identifying synthetic lethal interactions involving CMGC kinases and using non-invasive techniques, such as liquid biopsies, can facilitate the real-time monitoring of treatment response and early detection of resistance [179,182].
In summary, CMGC kinases are important in cancer biology, and targeting these kinases holds great therapeutic potential. Despite the challenges associated with their inhibition, ongoing research efforts and the development of novel therapeutic strategies promise to improve the efficacy and specificity of CMGC-kinase-targeted therapies. As our understanding of the molecular mechanisms and protein–protein interactions involving CMGC kinases expands, so too will the opportunities for the development of more selective and effective therapeutic strategies for cancer treatment.
Author Contributions
Conceptualization, S.K.; writing and editing, I.C. and S.K.; visualization, I.C. and G.D. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Academy of Finland.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The publicly available interaction dataset from https://www.ebi.ac.uk/intact (accessed on 8 May 2023) was used to generate the protein–protein interaction landscape of CMGC kinases which is visualized in Figure 3.
Acknowledgments
Open access funding provided by University of Helsinki.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Mann, M.; Ong, S.-E.; Grønborg, M.; Steen, H.; Jensen, O.N.; Pandey, A. Analysis of protein phosphorylation using mass spectrometry: Deciphering the phosphoproteome. Trends Biotechnol. 2002, 20, 261–268. [Google Scholar] [CrossRef]
- Taylor, S.S.; Kornev, A.P. Protein kinases: Evolution of dynamic regulatory proteins. Trends Biochem. Sci. 2011, 36, 65–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kannan, N.; Neuwald, A.F. Evolutionary constraints associated with functional specificity of the CMGC protein kinases MAPK, CDK, GSK, SRPK, DYRK, and CK2α. Protein Sci. 2004, 13, 2059–2077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nolen, B.; Taylor, S.; Ghosh, G. Regulation of Protein Kinases: Controlling Activity through Activation Segment Conformation. Mol. Cell 2004, 15, 661–675. [Google Scholar] [CrossRef]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The Protein Kinase Complement of the Human Genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [Green Version]
- Pawson, T.; Nash, P. Assembly of Cell Regulatory Systems Through Protein Interaction Domains. Science 2003, 300, 445–452. [Google Scholar] [CrossRef] [Green Version]
- Johnson, L.N.; Noble, M.E.; Owen, D.J. Active and Inactive Protein Kinases: Structural Basis for Regulation. Cell 1996, 85, 149–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haar, E. Structure of GSK3beta reveals a primed phosphorylation mechanism. Nat. Struct. Biol. 2001, 8, 593. [Google Scholar] [CrossRef] [PubMed]
- Varjosalo, M.; Björklund, M.; Cheng, F.; Syvänen, H.; Kivioja, T.; Kilpinen, S.; Sun, Z.; Kallioniemi, O.; Stunnenberg, H.G.; He, W.-W.; et al. Application of Active and Kinase-Deficient Kinome Collection for Identification of Kinases Regulating Hedgehog Signaling. Cell 2008, 133, 537–548. [Google Scholar] [CrossRef] [Green Version]
- Malumbres, M.; Harlow, E.; Hunt, T.; Hunter, T.; Lahti, J.M.; Manning, G.; Morgan, D.O.; Tsai, L.-H.; Wolgemuth, D.J. Cyclin-dependent kinases: A family portrait. Nat. Cell Biol. 2009, 11, 1275–1276. [Google Scholar] [CrossRef] [Green Version]
- Malumbres, M. Cyclin-dependent kinases. Genome Biol. 2014, 15, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeffrey, P.D. Mechanism of CDK Activation Revealed by the Structure of a CyclinA-CDK2 Complex. Nature 1995, 376, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Russo, A.A.; Jeffrey, P.D.; Pavletich, N.P. Structural basis of cyclin-dependent kinase activation by phosphorylation. Nat. Struct. Biol. 1996, 3, 696–700. [Google Scholar] [CrossRef] [PubMed]
- Harper, J.; Brooks, G. The Mammalian Cell Cycle—An Overwiew. From Methods in Molecular Biology. In Cell Cycle Control: Mechanisms and Protocols; Brooks, T.H., Ed.; Humana Press Inc.: Totowa, NJ, USA, 2005; Volume 296. [Google Scholar]
- Ren, S.; Rollins, B.J. Cyclin C/Cdk3 Promotes Rb-Dependent G0 Exit. Cell 2004, 117, 239–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, R.P. The CDK Network: Linking Cycles of Cell Division and Gene Expression. Genes Cancer 2012, 3, 731–738. [Google Scholar] [CrossRef] [Green Version]
- Santamaría, D.; Barrière, C.; Cerqueira, A.; Hunt, S.; Tardy, C.; Newton, K.; Cáceres, J.F.; Dubus, P.; Malumbres, M.; Barbacid, M. Cdk1 is sufficient to drive the mammalian cell cycle. Nature 2007, 448, 811–815. [Google Scholar] [CrossRef] [PubMed]
- Berthet, C.; Aleem, E.; Coppola, V.; Tessarollo, L.; Kaldis, P. Cdk2 Knockout Mice Are Viable. Curr. Biol. 2003, 13, 1775–1785. [Google Scholar] [CrossRef] [Green Version]
- Ortega, S.; Prieto, I.; Odajima, J.; Martín, A.; Dubus, P.; Sotillo, R.; Barbero, J.L.; Malumbres, M.; Barbacid, M. Cyclin-dependent kinase 2 is essential for meiosis but not for mitotic cell division in mice. Nat. Genet. 2003, 35, 25–31. [Google Scholar] [CrossRef]
- Rane, S.G. Loss of Cdk4 Expression Causes Insulin-Deficient Diabetes and Cdk4 Activation Results in Beta-Islet Cell Hyperplasia. Nat. Genet. 1999, 22, 44–52. [Google Scholar] [CrossRef]
- Tsutsui, T.; Hesabi, B.; Moons, D.S.; Pandolfi, P.P.; Hansel, K.S.; Koff, A.; Kiyokawa, H. Targeted Disruption of CDK4 Delays Cell Cycle Entry with Enhanced p27Kip1 Activity. Mol. Cell. Biol. 1999, 19, 7011–7019. [Google Scholar] [CrossRef] [Green Version]
- Malumbres, M.; Sotillo, R.; Santamaría, D.; Galán, J.; Cerezo, A.; Ortega, S.; Dubus, P.; Barbacid, M. Mammalian Cells Cycle without the D-Type Cyclin-Dependent Kinases Cdk4 and Cdk6. Cell 2004, 118, 493–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrière, C.; Santamaría, D.; Cerqueira, A.; Galán, J.; Martín, A.; Ortega, S.; Malumbres, M.; Dubus, P.; Barbacid, M. Mice thrive without Cdk4 and Cdk2. Mol. Oncol. 2007, 1, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Even, Y.; Durieux, S.; Escande, M.-L.; Lozano, J.C.; Peaucellier, G.; Weil, D.; Genevière, A.-M. CDC2L5, a Cdk-like kinase with RS domain, interacts with the ASF/SF2-associated protein p32 and affects splicing in vivo. J. Cell. Biochem. 2006, 99, 890–904. [Google Scholar] [CrossRef] [PubMed]
- Loyer, P. Characterization of Cyclin L1 and L2 Interactions with CDK11 and Splicing Factors: Influence of Cyclin L Isoforms on Splice Site Selection. J. Biol. Chem. 2008, 283, 7721. [Google Scholar] [CrossRef] [Green Version]
- Allen, B.L.; Taatjes, D.J. The Mediator complex: A central integrator of transcription. Nat. Rev. Mol. Cell Biol. 2015, 16, 155–166. [Google Scholar] [CrossRef]
- Ganuza, M.; Sáiz-Ladera, C.; Cañamero, M.; Gómez, G.; Schneider, R.; Blasco, M.A.; Pisano, D.; Paramio, J.M.; Santamaría, D.; Barbacid, M. Genetic inactivation of Cdk7 leads to cell cycle arrest and induces premature aging due to adult stem cell exhaustion. EMBO J. 2012, 31, 2498–2510. [Google Scholar] [CrossRef] [Green Version]
- Westerling, T.; Kuuluvainen, E.; Maäkelaä, T.P. Cdk8 Is Essential for Preimplantation Mouse Development. Mol. Cell. Biol. 2007, 27, 6177–6182. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Inoue, A.; Lahti, J.M.; Kidd, V.J. Failure to proliferate and mitotic arrest of CDK11(p110/p58)-null mutant mice at the blastocyst stage of embryonic cell development. Mol. Cell. Biol. 2004, 24, 3188–3197. [Google Scholar] [CrossRef] [Green Version]
- Juan, H.-C.; Lin, Y.; Chen, H.-R.; Fann, M.-J. Cdk12 is essential for embryonic development and the maintenance of genomic stability. Cell Death Differ. 2015, 23, 1038–1048. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Li, J.; Song, Y.-S.; Li, Y.; Jia, Y.-H.; Zhao, H.-D. Cdk5 links with DNA damage response and cancer. Mol. Cancer 2017, 16, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Ohshima, T.; Ward, J.M.; Huh, C.G.; Longenecker, G.; Veeranna; Pant, H.C.; Brady, R.O.; Martin, L.J.; Kulkarni, A.B. Targeted disruption of the cyclin-dependent kinase 5 gene results in abnormal corticogenesis, neuronal pathology and perinatal death. Proc. Natl. Acad. Sci. USA 1996, 93, 11173–11178. [Google Scholar] [CrossRef] [PubMed]
- Shah, K.; Lahiri, D.K. Cdk5 activity in the brain—Multiple paths of regulation. J. Cell Sci. 2014, 127, 2391–2400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brinkkoetter, P.T.; Olivier, P.; Wu, J.S.; Henderson, S.; Krofft, R.D.; Pippin, J.W.; Hockenbery, D.; Roberts, J.M.; Shankland, S.J. Cyclin I activates Cdk5 and regulates expression of Bcl-2 and Bcl-XL in postmitotic mouse cells. J. Clin. Investig. 2009, 119, 3089–3101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Zhai, X.; Zhao, B.; Wang, Y.; Xu, Z. Cyclin I-like (CCNI2) is a cyclin-dependent kinase 5 (CDK5) activator and is involved in cell cycle regulation. Sci. Rep. 2017, 7, 40979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guen, V.J. CDK10/cyclin M is a protein kinase that controls ETS2 degradation and is deficient in STAR syndrome. Proc. Natl. Acad. Sci. USA 2013, 110, 19525–19530. [Google Scholar] [CrossRef] [PubMed]
- Iorns, E.; Turner, N.C.; Elliott, R.; Syed, N.; Garrone, O.; Gasco, M.; Tutt, A.N.; Crook, T.; Lord, C.J.; Ashworth, A. Identification of CDK10 as an Important Determinant of Resistance to Endocrine Therapy for Breast Cancer. Cancer Cell 2008, 13, 91–104. [Google Scholar] [CrossRef] [Green Version]
- Jiang, M.; Gao, Y.; Yang, T.; Zhu, X.; Chen, J. Cyclin Y, a novel membrane-associated cyclin, interacts with PFTK1. FEBS Lett. 2009, 583, 2171–2178. [Google Scholar] [CrossRef] [Green Version]
- Shehata, S.N.; Hunter, R.W.; Ohta, E.; Peggie, M.W.; Lou, H.J.; Sicheri, F.; Zeqiraj, E.; Turk, B.E.; Sakamoto, K. Analysis of substrate specificity and cyclin Y binding of PCTAIRE-1 kinase. Cell. Signal. 2012, 24, 2085–2094. [Google Scholar] [CrossRef] [Green Version]
- Park, M.H.; Kim, S.Y.; Kim, Y.J.; Chung, Y.-H. ALS2CR7 (CDK15) attenuates TRAIL induced apoptosis by inducing phosphorylation of survivin Thr34. Biochem. Biophys. Res. Commun. 2014, 450, 129–134. [Google Scholar] [CrossRef]
- Chaput, D.; Kirouac, L.; Stevens, S.M., Jr.; Padmanabhan, J. Potential role of PCTAIRE-2, PCTAIRE-3 and P-Histone H4 in amyloid precursor protein-dependent Alzheimer pathology. Oncotarget 2016, 7, 8481–8497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Roine, N.; Mäkelä, T.P. CCRK depletion inhibits glioblastoma cell proliferation in a cilium-dependent manner. EMBO Rep. 2013, 14, 741–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- FDA Approves Abemaciclib with Endocrine Therapy for Early Breast Cancer. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-abemaciclib-endocrine-therapy-early-breast-cancer (accessed on 26 July 2023).
- Wang, X.; Chen, X.; Han, W.; Ruan, A.; Chen, L.; Wang, R.; Xu, Z.; Xiao, P.; Lu, X.; Zhao, Y.; et al. miR-200c Targets CDK2 and Suppresses Tumorigenesis in Renal Cell Carcinoma. Mol. Cancer Res. 2015, 13, 1567–1577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hongo, F.; Takaha, N.; Oishi, M.; Ueda, T.; Nakamura, T.; Naitoh, Y.; Naya, Y.; Kamoi, K.; Okihara, K.; Matsushima, T.; et al. CDK1 and CDK2 activity is a strong predictor of renal cell carcinoma recurrence. Urol. Oncol. Semin. Orig. Investig. 2014, 32, 1240–1246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coulombe, P.; Meloche, S. Atypical mitogen-activated protein kinases: Structure, regulation and functions. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2007, 1773, 1376–1387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deleris, A.; Stroud, H.; Bernatavichute, Y.; Johnson, E.; Klein, G.; Schubert, D.; Jacobsen, S.E. Loss of the DNA Methyltransferase MET1 Induces H3K9 Hypermethylation at PcG Target Genes and Redistribution of H3K27 Trimethylation to Transposons in Arabidopsis thaliana. PLoS Genet. 2012, 8, e1003062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, W.; Foulds, C.E.; Qin, J.; Liu, J.; Ding, C.; Lonard, D.M.; Solis, L.M.; Wistuba, I.I.; Qin, J.; Tsai, S.Y.; et al. ERK3 signals through SRC-3 coactivator to promote human lung cancer cell invasion. J. Clin. Investig. 2012, 122, 1869–1880. [Google Scholar] [CrossRef]
- Kostenko, S.; Dumitriu, G.; Lægreid, K.J.; Moens, U. Physiological roles of mitogen-activated-protein-kinase-activated p38-regulated/activated protein kinase. World J. Biol. Chem. 2011, 2, 73–89. [Google Scholar] [CrossRef]
- Cargnello, M.; Roux, P.P. Activation and Function of the MAPKs and Their Substrates, the MAPK-Activated Protein Kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef] [Green Version]
- Johnson, G.L.; Lapadat, R. Mitogen-Activated Protein Kinase Pathways Mediated by ERK, JNK, and p38 Protein Kinases. Science 2002, 298, 1911–1912. [Google Scholar] [CrossRef] [Green Version]
- Burotto, M.; Chiou, V.L.; Lee, J.-M.; Kohn, E.C. The MAPK pathway across different malignancies: A new perspective. Cancer 2014, 120, 3446–3456. [Google Scholar] [CrossRef] [Green Version]
- Murphy, L.O.; Smith, S.; Chen, R.-H.; Fingar, D.C.; Blenis, J. Molecular interpretation of ERK signal duration by immediate early gene products. Nat. Cell Biol. 2002, 4, 556–564. [Google Scholar] [CrossRef] [PubMed]
- Sabapathy, K.; Hochedlinger, K.; Nam, S.Y.; Bauer, A.; Karin, M.; Wagner, E.F. Distinct Roles for JNK1 and JNK2 in Regulating JNK Activity and c-Jun-Dependent Cell Proliferation. Mol. Cell 2004, 15, 713–725. [Google Scholar] [CrossRef]
- Bogoyevitch, M.A.; Ngoei, K.R.; Zhao, T.T.; Yeap, Y.Y.; Ng, D.C. c-Jun N-terminal kinase (JNK) signaling: Recent advances and challenges. Biochim. Biophys. Acta (BBA)—Proteins Proteom. 2010, 1804, 463–475. [Google Scholar] [CrossRef]
- Cuadrado, A.; Nebreda, A.R. Mechanisms and functions of p38 MAPK signalling. Biochem. J. 2010, 429, 403–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Regan, C.P.; Li, W.; Boucher, D.M.; Spatz, S.; Su, M.S.; Kuida, K. Erk5 null mice display multiple extraembryonic vascular and embryonic cardiovascular defects. Proc. Natl. Acad. Sci. USA 2002, 99, 9248–9253. [Google Scholar] [CrossRef] [PubMed]
- Mehta, P.B.; Jenkins, B.L.; McCarthy, L.; Thilak, L.; Robson, C.N.; Neal, D.E.; Leung, H.Y. MEK5 overexpression is associated with metastatic prostate cancer, and stimulates proliferation, MMP-9 expression and invasion. Oncogene 2003, 22, 1381–1389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulloy, R.; Salinas, S.; Philips, A.; Hipskind, R.A. Activation of cyclin D1 expression by the ERK5 cascade. Oncogene 2003, 22, 5387–5398. [Google Scholar] [CrossRef] [Green Version]
- Jo, M.; Lee, S.; Kim, K.; Lee, S.; Kim, S.R.; Kim, H.-J. Inhibition of MEK5 suppresses TDP-43 toxicity via the mTOR-independent activation of the autophagy-lysosome pathway. Biochem. Biophys. Res. Commun. 2019, 513, 925–932. [Google Scholar] [CrossRef]
- Markowitz, S.D.; Bertagnolli, M.M. Molecular Origins of Cancer: Molecular Basis of Colorectal Cancer. N. Engl. J. Med. 2009, 361, 2449–2460. [Google Scholar] [CrossRef] [Green Version]
- Van Cutsem, E.; Köhne, C.-H.; Hitre, E.; Zaluski, J.; Chien, C.-R.C.; Makhson, A.; D’Haens, G.; Pintér, T.; Lim, R.; Bodoky, G.; et al. Cetuximab and Chemotherapy as Initial Treatment for Metastatic Colorectal Cancer. N. Engl. J. Med. 2009, 360, 1408–1417. [Google Scholar] [CrossRef] [Green Version]
- Min, L.; He, B.; Hui, L. Mitogen-activated protein kinases in hepatocellular carcinoma development. Semin. Cancer Biol. 2011, 21, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Wagner, E.F.; Nebreda, Á.R. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat. Rev. Cancer 2009, 9, 537–549. [Google Scholar] [CrossRef] [PubMed]
- Woodgett, J.R. Molecular Cloning and Expression of Glycogen Synthase Kinase-3/Factor A. EMBO J. 1990, 9, 2431. [Google Scholar] [CrossRef] [PubMed]
- Hoeflich, K.P. Requirement for Glycogen Synthase Kinase-3beta in Cell Survival and NF-KappaB Activation. Nature 2000, 406, 86–90. [Google Scholar] [CrossRef]
- MacAulay, K.; Doble, B.W.; Patel, S.; Hansotia, T.; Sinclair, E.M.; Drucker, D.J.; Nagy, A.; Woodgett, J.R. Glycogen Synthase Kinase 3α-Specific Regulation of Murine Hepatic Glycogen Metabolism. Cell Metab. 2007, 6, 329–337. [Google Scholar] [CrossRef] [Green Version]
- Cole, A.R.; Knebel, A.; Morrice, N.A.; Robertson, L.A.; Irving, A.J.; Connolly, C.N.; Sutherland, C. GSK-3 Phosphorylation of the Alzheimer Epitope within Collapsin Response Mediator Proteins Regulates Axon Elongation in Primary Neurons. J. Biol. Chem. 2004, 279, 50176–50180. [Google Scholar] [CrossRef] [Green Version]
- Frame, S.; Cohen, P. GSK3 Takes Centre Stage More than 20 Years after Its Discovery. Biochem. J. 2001, 359, 1–16. [Google Scholar] [CrossRef]
- McCubrey, J.A. Effects of Mutations in Wnt/Beta-Catenin, Hedgehog, Notch and PI3K Pathways on GSK-3 Activity-Diverse Effects on Cell Growth, Metabolism and Cancer. Biochim. Biophys. Acta 2016, 1863, 2942. [Google Scholar] [CrossRef]
- Medina, M.; Wandosell, F. Deconstructing GSK-3: The Fine Regulation of Its Activity. Int. J. Alzheimer’s Dis. 2011, 2011, 479249. [Google Scholar] [CrossRef]
- Cormier, K.W.; Woodgett, J.R. Recent advances in understanding the cellular roles of GSK-3. F1000Research 2017, 6, 167. [Google Scholar] [CrossRef] [Green Version]
- Niehrs, C. The complex world of WNT receptor signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 767–779. [Google Scholar] [CrossRef]
- Kaidanovich-Beilin, O.; Woodgett, J.R. GSK-3: Functional Insights from Cell Biology and Animal Models. Front. Mol. Neurosci. 2011, 4, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eldar-Finkelman, H.; Krebs, E.G. Phosphorylation of insulin receptor substrate 1 by glycogen synthase kinase 3 impairs insulin action. Proc. Natl. Acad. Sci. USA 1997, 94, 9660–9664. [Google Scholar] [CrossRef] [PubMed]
- Ring, D.B.; Johnson, K.W.; Henriksen, E.J.; Nuss, J.M.; Goff, D.; Kinnick, T.R.; Ma, S.T.; Reeder, J.W.; Samuels, I.; Slabiak, T.; et al. Selective Glycogen Synthase Kinase 3 Inhibitors Potentiate Insulin Activation of Glucose Transport and Utilization In Vitro and In Vivo. Diabetes 2003, 52, 588–595. [Google Scholar] [CrossRef] [Green Version]
- Lochhead, P.A.; Sibbet, G.; Morrice, N.; Cleghon, V. Activation-Loop Autophosphorylation Is Mediated by a Novel Transitional Intermediate Form of DYRKs. Cell 2005, 121, 925–936. [Google Scholar] [CrossRef] [Green Version]
- Himpel, S. Identification of the Autophosphorylation Sites and Characterization of Their Effects in the Protein Kinase DYRK1A. Biochem. J. 2001, 359, 497. [Google Scholar] [CrossRef]
- Aranda, S.; Laguna, A.; de la Luna, S. DYRK family of protein kinases: Evolutionary relationships, biochemical properties, and functional roles. FASEB J. 2010, 25, 449–462. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-Y.; Lin, J.-R.; Tsai, F.-C.; Meyer, T. Dosage of Dyrk1a Shifts Cells within a p21-Cyclin D1 Signaling Map to Control the Decision to Enter the Cell Cycle. Mol. Cell 2013, 52, 87–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, X.; Mercer, S.E.; Shah, S.; Ewton, D.Z.; Friedman, E. The Cyclin-dependent Kinase Inhibitor p27Kip1 Is Stabilized in G0 by Mirk/dyrk1B Kinase. J. Biol. Chem. 2004, 279, 22498–22504. [Google Scholar] [CrossRef] [Green Version]
- Zou, Y.; Ewton, D.Z.; Deng, X.; Mercer, S.E.; Friedman, E. Mirk/dyrk1B Kinase Destabilizes Cyclin D1 by Phosphorylation at Threonine 288. J. Biol. Chem. 2004, 279, 27790–27798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.; Williams, J.G.; Schug, T.T.; Li, X. DYRK1A and DYRK3 Promote Cell Survival through Phosphorylation and Activation of SIRT1. J. Biol. Chem. 2010, 285, 13223–13232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seifert, A.; Allan, L.A.; Clarke, P.R. DYRK1A phosphorylates caspase 9 at an inhibitory site and is potently inhibited in human cells by harmine. FEBS J. 2008, 275, 6268–6280. [Google Scholar] [CrossRef] [PubMed]
- Taira, N.; Nihira, K.; Yamaguchi, T.; Miki, Y.; Yoshida, K. DYRK2 Is Targeted to the Nucleus and Controls p53 via Ser46 Phosphorylation in the Apoptotic Response to DNA Damage. Mol. Cell 2007, 25, 725–738. [Google Scholar] [CrossRef] [PubMed]
- van der Laden, J.; Soppa, U.; Becker, W. Effect of tyrosine autophosphorylation on catalytic activity and subcellular localisation of homeodomain-interacting protein kinases (HIPK). Cell Commun. Signal. 2015, 13, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arai, S.; Matsushita, A.; Du, K.; Yagi, K.; Okazaki, Y.; Kurokawa, R. Novel homeodomain-interacting protein kinase family member, HIPK4, phosphorylates human p53 at serine 9. FEBS Lett. 2007, 581, 5649–5657. [Google Scholar] [CrossRef] [Green Version]
- Aikawa, Y.; Nguyen, L.A.; Isono, K.; Takakura, N.; Tagata, Y.; Schmitz, M.L.; Koseki, H.; Kitabayashi, I. Roles of HIPK1 and HIPK2 in AML1- and p300-dependent transcription, hematopoiesis and blood vessel formation. EMBO J. 2006, 25, 3955–3965. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, N.; Ishitani, S.; Sato, A.; Shibuya, H.; Ishitani, T. Hipk2 and PP1c Cooperate to Maintain Dvl Protein Levels Required for Wnt Signal Transduction. Cell Rep. 2014, 8, 1391–1404. [Google Scholar] [CrossRef] [Green Version]
- Lv, C.; Fu, S.; Dong, Q.; Yu, Z.; Zhang, G.; Kong, C.; Fu, C.; Zeng, Y. PAGE4 promotes prostate cancer cells survive under oxidative stress through modulating MAPK/JNK/ERK pathway. J. Exp. Clin. Cancer Res. 2019, 38, 24. [Google Scholar] [CrossRef] [Green Version]
- Kojima, T.; Zama, T.; Wada, K.; Onogi, H.; Hagiwara, M. Cloning of Human PRP4 Reveals Interaction with Clk1. J. Biol. Chem. 2001, 276, 32247–32256. [Google Scholar] [CrossRef] [Green Version]
- Montembault, E.; Dutertre, S.; Prigent, C.; Giet, R. PRP4 is a spindle assembly checkpoint protein required for MPS1, MAD1, and MAD2 localization to the kinetochores. J. Cell Biol. 2007, 179, 601–609. [Google Scholar] [CrossRef]
- Gao, Q.; Mechin, I.; Kothari, N.; Guo, Z.; Deng, G.; Haas, K.; McManus, J.; Hoffmann, D.; Wang, A.; Wiederschain, D.; et al. Evaluation of Cancer Dependence and Druggability of PRP4 Kinase Using Cellular, Biochemical, and Structural Approaches. J. Biol. Chem. 2013, 288, 30125–30138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gross, T.; Lutzelberger, M.; Wiegmann, H.; Klingenhoff, A.; Shenoy, S.; Kaufer, N.F. Functional Analysis of the Fission Yeast Prp4 Protein Kinase Involved in Pre-Mrna Splicing and Isolation of a Putative Mammalian Homologue. Nucleic Acids Res. 1997, 25, 1028–1035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, M.; Will, C.L.; Anokhina, M.; Tazi, J.; Urlaub, H.; Lührmann, R. Exon Definition Complexes Contain the Tri-snRNP and Can Be Directly Converted into B-like Precatalytic Splicing Complexes. Mol. Cell 2010, 38, 223–235. [Google Scholar] [CrossRef]
- Bertram, K.; Agafonov, D.E.; Dybkov, O.; Haselbach, D.; Leelaram, M.N.; Will, C.L.; Urlaub, H.; Kastner, B.; Lührmann, R.; Stark, H. Cryo-EM Structure of a Pre-catalytic Human Spliceosome Primed for Activation. Cell 2017, 170, 701–713.e11. [Google Scholar] [CrossRef] [PubMed]
- Nayler, O.; Stamm, S.; Ullrich, A. Characterization and comparison of four serine- and arginine-rich (SR) protein kinases. Biochem. J. 1997, 326, 693–700. [Google Scholar] [CrossRef] [Green Version]
- Bullock, A.N.; Das, S.; Debreczeni, J.; Rellos, P.; Fedorov, O.; Niesen, F.H.; Guo, K.; Papagrigoriou, E.; Amos, A.L.; Cho, S.; et al. Kinase Domain Insertions Define Distinct Roles of CLK Kinases in SR Protein Phosphorylation. Structure 2009, 17, 352–362. [Google Scholar] [CrossRef] [Green Version]
- Prasad, J.; Colwill, K.; Pawson, T.; Manley, J.L. The Protein Kinase Clk/Sty Directly Modulates SR Protein Activity: Both Hyper- and Hypophosphorylation Inhibit Splicing. Mol. Cell. Biol. 1999, 19, 6991–7000. [Google Scholar] [CrossRef] [Green Version]
- Petsalaki, E.; Zachos, G. Clks 1, 2 and 4 prevent chromatin breakage by regulating the Aurora B-dependent abscission checkpoint. Nat. Commun. 2016, 7, 11451. [Google Scholar] [CrossRef]
- Murai, A.; Ebara, S.; Sasaki, S.; Ohashi, T.; Miyazaki, T.; Nomura, T.; Araki, S. Synergistic apoptotic effects in cancer cells by the combination of CLK and Bcl-2 family inhibitors. PLoS ONE 2020, 15, e0240718. [Google Scholar] [CrossRef]
- Ghosh, G.; Adams, J.A. Phosphorylation mechanism and structure of serine-arginine protein kinases. FEBS J. 2010, 278, 587–597. [Google Scholar] [CrossRef] [Green Version]
- Giannakouros, T.; Nikolakaki, E.; Mylonis, I.; Georgatsou, E. Serine-arginine protein kinases: A small protein kinase family with a large cellular presence. FEBS J. 2011, 278, 570–586. [Google Scholar] [CrossRef] [PubMed]
- Calarco, J.A.; Superina, S.; O’Hanlon, D.; Gabut, M.; Raj, B.; Pan, Q.; Skalska, U.; Clarke, L.; Gelinas, D.; van der Kooy, D.; et al. Regulation of Vertebrate Nervous System Alternative Splicing and Development by an SR-Related Protein. Cell 2009, 138, 898–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayes, G.M.; Carrigan, P.E.; Miller, L.J. Serine-Arginine Protein Kinase 1 Overexpression Is Associated with Tumorigenic Imbalance in Mitogen-Activated Protein Kinase Pathways in Breast, Colonic, and Pancreatic Carcinomas. Cancer Res. 2007, 67, 2072–2080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hishizawa, M.; Imada, K.; Sakai, T.; Ueda, M.; Hori, T.; Uchiyama, T. Serological identification of adult T-cell leukaemia-associated antigens. Br. J. Haematol. 2005, 130, 382–390. [Google Scholar] [CrossRef]
- Chandra, A.; Ananda, H.; Singh, N.; Qamar, I. Identification of a novel and potent small molecule inhibitor of SRPK1: Mechanism of dual inhibition of SRPK1 for the inhibition of cancer progression. Aging 2020, 13, 163–180. [Google Scholar] [CrossRef]
- Jang, S.-W.; Yang, S.-J.; Ehlén, A.; Dong, S.; Khoury, H.; Chen, J.; Persson, J.L.; Ye, K. Serine/Arginine Protein–Specific Kinase 2 Promotes Leukemia Cell Proliferation by Phosphorylating Acinus and Regulating Cyclin A1. Cancer Res. 2008, 68, 4559–4570. [Google Scholar] [CrossRef] [Green Version]
- Fu, Z.; Schroeder, M.J.; Shabanowitz, J.; Kaldis, P.; Togawa, K.; Rustgi, A.K.; Hunt, D.F.; Sturgill, T.W. Activation of a Nuclear Cdc2-Related Kinase within a Mitogen-Activated Protein Kinase-Like TDY Motif by Autophosphorylation and Cyclin-Dependent Protein Kinase-Activating Kinase. Mol. Cell. Biol. 2005, 25, 6047–6064. [Google Scholar] [CrossRef] [Green Version]
- Miyata, Y.; Akashi, M.; Nishida, E. Molecular cloning and characterization of a novel member of the MAP kinase superfamily. Genes Cells 1999, 4, 299–309. [Google Scholar] [CrossRef]
- Wang, L.-Y.; Kung, H.-J. Male germ cell-associated kinase is overexpressed in prostate cancer cells and causes mitotic defects via deregulation of APC/CCDH1. Oncogene 2011, 31, 2907–2918. [Google Scholar] [CrossRef] [Green Version]
- Fu, Z.; Larson, K.A.; Chitta, R.K.; Parker, S.A.; Turk, B.E.; Lawrence, M.W.; Kaldis, P.; Galaktionov, K.; Cohn, S.M.; Shabanowitz, J.; et al. Identification of Yin-Yang Regulators and a Phosphorylation Consensus for Male Germ Cell-Associated Kinase (MAK)-Related Kinase. Mol. Cell. Biol. 2006, 26, 8639–8654. [Google Scholar] [CrossRef] [Green Version]
- Omori, Y.; Chaya, T.; Katoh, K.; Kajimura, N.; Sato, S.; Muraoka, K.; Ueno, S.; Koyasu, T.; Kondo, M.; Furukawa, T. Negative regulation of ciliary length by ciliary male germ cell-associated kinase (Mak) is required for retinal photoreceptor survival. Proc. Natl. Acad. Sci. USA 2010, 107, 22671–22676. [Google Scholar] [CrossRef] [PubMed]
- Özgül, R.K.; Siemiatkowska, A.M.; Yücel, D.; Myers, C.A.; Collin, R.W.; Zonneveld, M.N.; Beryozkin, A.; Banin, E.; Hoyng, C.B.; Born, L.I.v.D.; et al. Exome Sequencing and cis-Regulatory Mapping Identify Mutations in MAK, a Gene Encoding a Regulator of Ciliary Length, as a Cause of Retinitis Pigmentosa. Am. J. Hum. Genet. 2011, 89, 253–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tucker, B.A.; Scheetz, T.E.; Mullins, R.F.; DeLuca, A.P.; Hoffmann, J.M.; Johnston, R.M.; Jacobson, S.G.; Sheffield, V.C.; Stone, E.M. Exome sequencing and analysis of induced pluripotent stem cells identify the cilia-related gene male germ cell-associated kinase (MAK) as a cause of retinitis pigmentosa. Proc. Natl. Acad. Sci. USA 2011, 108, E569–E576. [Google Scholar] [CrossRef]
- Moon, H.; Song, J.; Shin, J.-O.; Lee, H.; Kim, H.-K.; Eggenschwiller, J.T.; Bok, J.; Ko, H.W. Intestinal cell kinase, a protein associated with endocrine-cerebro-osteodysplasia syndrome, is a key regulator of cilia length and Hedgehog signaling. Proc. Natl. Acad. Sci. USA 2014, 111, 8541–8546. [Google Scholar] [CrossRef] [PubMed]
- Lahiry, P. A multiplex human syndrome implicates a key role for intestinal cell kinase in development of central nervous, skeletal, and endocrine systems. Am. J. Hum. Genet. 2009, 84, 134. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.-M.; Huang, Y.-T.; Wang, G.-C. Outcome of colon cancer initially presenting as colon perforation and obstruction. World J. Surg. Oncol. 2017, 15, 164. [Google Scholar] [CrossRef]
- Lin, C.; Franco, B.; Rosner, M.R. CDKL5/Stk9 Kinase Inactivation Is Associated with Neuronal Developmental Disorders. Hum. Mol. Genet. 2005, 14, 3775–3786. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Z.; Gong, T.; Wei, H. CDKL5 promotes proliferation, migration, and chemotherapeutic drug resistance of glioma cells via activation of the PI3K/AKT signaling pathway. FEBS Open Bio 2020, 10, 268–277. [Google Scholar] [CrossRef] [Green Version]
- Qin, C.; Ren, L.; Ji, M.; Lv, S.; Wei, Y.; Zhu, D.; Lin, Q.; Xu, P.; Chang, W.; Xu, J. CDKL1 promotes tumor proliferation and invasion in colorectal cancer. OncoTargets Ther. 2017, 10, 1613–1624. [Google Scholar] [CrossRef] [Green Version]
- Futreal, P.A.; Coin, L.; Marshall, M.; Down, T.; Hubbard, T.; Wooster, R.; Rahman, N.; Stratton, M.R. A census of human cancer genes. Nat. Rev. Cancer 2004, 4, 177–183. [Google Scholar] [CrossRef]
- Cohen, P. Protein kinases–the major drug targets of the twenty-first century? Nat. Rev. Drug Discov. 2002, 1, 309. [Google Scholar] [CrossRef] [PubMed]
- Abbassi, R.; Johns, T.G.; Kassiou, M.; Munoz, L. DYRK1A in neurodegeneration and cancer: Molecular basis and clinical implications. Pharmacol. Ther. 2015, 151, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Knight, Z.A.; Lin, H.; Shokat, K.M. Targeting the cancer kinome through polypharmacology. Nat. Rev. Cancer 2010, 10, 130–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gross, S.; Rahal, R.; Stransky, N.; Lengauer, C.; Hoeflich, K.P. Targeting cancer with kinase inhibitors. J. Clin. Investig. 2015, 125, 1780–1789. [Google Scholar] [CrossRef]
- Qian, J.-Y.; Gao, J.; Sun, X.; Cao, M.-D.; Shi, L.; Xia, T.-S.; Zhou, W.-B.; Wang, S.; Ding, Q.; Wei, J.-F. KIAA1429 acts as an oncogenic factor in breast cancer by regulating CDK1 in an N6-methyladenosine-independent manner. Oncogene 2019, 38, 6123–6141. [Google Scholar] [CrossRef]
- Xing, Z.; Wang, X.; Liu, J.; Zhang, M.; Feng, K.; Wang, X. Expression and prognostic value of CDK1, CCNA2, and CCNB1 gene clusters in human breast cancer. J. Int. Med. Res. 2021, 49, 0300060520980647. [Google Scholar]
- Wang, N.; Zhang, H.; Li, D.; Jiang, C.; Zhao, H.; Teng, Y. Identification of novel biomarkers in breast cancer via integrated bioinformatics analysis and experimental validation. Bioengineered 2021, 12, 12431–12446. [Google Scholar] [CrossRef]
- Tong, W.; Han, T.-C.; Wang, W.; Zhao, J. LncRNA CASC11 Promotes the Development of Lung Cancer through Targeting MicroRNA-302/CDK1 Axis. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 6539–6547. [Google Scholar] [CrossRef]
- Piao, J.; Zhu, L.; Sun, J.; Li, N.; Dong, B.; Yang, Y.; Chen, L. High expression of CDK1 and BUB1 predicts poor prognosis of pancreatic ductal adenocarcinoma. Gene 2019, 701, 15–22. [Google Scholar] [CrossRef]
- Flaherty, K.T.; Infante, J.R.; Daud, A.; Gonzalez, R.; Kefford, R.F.; Sosman, J.; Hamid, O.; Schuchter, L.; Cebon, J.; Ibrahim, N.; et al. Combined BRAF and MEK Inhibition in Melanoma with BRAF V600 Mutations. N. Engl. J. Med. 2012, 367, 1694–1703. [Google Scholar] [CrossRef] [Green Version]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.J.; et al. Combined BRAF and MEK Inhibition versus BRAF Inhibition Alone in Melanoma. N. Engl. J. Med. 2014, 371, 1877–1888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ascierto, P.A.; Kirkwood, J.M.; Grob, J.-J.; Simeone, E.; Grimaldi, A.M.; Maio, M.; Palmieri, G.; Testori, A.; Marincola, F.M.; Mozzillo, N. The role of BRAF V600 mutation in melanoma. J. Transl. Med. 2012, 10, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murugan, A.K.; Dong, J.; Xie, J.; Xing, M. MEK1 mutations, but not ERK2 mutations, occur in melanomas and colon carcinomas, but none in thyroid carcinomas. Cell Cycle 2009, 8, 2122–2124. [Google Scholar] [CrossRef] [PubMed]
- Turke, A.B.; Zejnullahu, K.; Wu, Y.-L.; Song, Y.; Dias-Santagata, D.; Lifshits, E.; Toschi, L.; Rogers, A.; Mok, T.; Sequist, L.; et al. Preexistence and Clonal Selection of MET Amplification in EGFR Mutant NSCLC. Cancer Cell 2010, 17, 77–88. [Google Scholar] [CrossRef] [Green Version]
- Luna, J.; Boni, J.; Cuatrecasas, M.; Bofill-De Ros, X.; Núñez-Manchón, E.; Gironella, M.; Vaquero, E.C.; Arbones, M.; de la Luna, S.; Fillat, C. DYRK1A modulates c-MET in pancreatic ductal adenocarcinoma to drive tumour growth. Gut 2018, 68, 1465–1476. [Google Scholar] [CrossRef]
- Singh, R.; Dhanyamraju, P.K.; Lauth, M. DYRK1B blocks canonical and promotes non-canonical Hedgehog signaling through activation of the mTOR/AKT pathway. Oncotarget 2016, 8, 833–845. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Nakhla, H.; Friedman, E. Transient arrest in a quiescent state allows ovarian cancer cells to survive suboptimal growth conditions and is mediated by both Mirk/dyrk1b and p130/Rb2. Int. J. Cancer 2010, 129, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Thompson, F.H.; Nelson, M.A.; Trent, J.M.; Guan, X.-Y.; Liu, Y.; Yang, J.-M.; Emerson, J.; Adair, L.; Wymer, J.; Balfour, C.; et al. Amplification of 19q13.1–q13.2 sequences in ovarian cancer: G-band, FISH, and molecular studies. Cancer Genet. Cytogenet. 1996, 87, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Mercer, S.E.; Ewton, D.Z.; Shah, S.; Naqvi, A.; Friedman, E. Mirk/Dyrk1b Mediates Cell Survival in Rhabdomyosarcomas. Cancer Res. 2006, 66, 5143–5150. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Ji, D.; Weinstein, E.J.; Choy, E.; Hornicek, F.J.; Wood, K.B.; Liu, X.; Mankin, H.; Duan, Z. The kinase Mirk is a potential therapeutic target in osteosarcoma. Carcinogenesis 2009, 31, 552–558. [Google Scholar] [CrossRef]
- Ugolkov, A.; Qiang, W.; Bondarenko, G.; Procissi, D.; Gaisina, I.; James, C.D.; Chandler, J.; Kozikowski, A.; Gunosewoyo, H.; O’Halloran, T.; et al. Combination Treatment with the GSK-3 Inhibitor 9-ING-41 and CCNU Cures Orthotopic Chemoresistant Glioblastoma in Patient-Derived Xenograft Models. Transl. Oncol. 2017, 10, 669–678. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Madamsetty, V.S.; Kiers, S.; Alekhina, O.; Ugolkov, A.; Dube, J.; Zhang, Y.; Zhang, J.-S.; Wang, E.; Dutta, S.K.; et al. Glycogen Synthase Kinase-3 Inhibition Sensitizes Pancreatic Cancer Cells to Chemotherapy by Abrogating the TopBP1/ATR-Mediated DNA Damage Response. Clin. Cancer Res. 2019, 25, 6452–6462. [Google Scholar] [CrossRef] [PubMed]
- Kuroki, H.; Anraku, T.; Kazama, A.; Bilim, V.; Tasaki, M.; Schmitt, D.; Mazar, A.P.; Giles, F.J.; Ugolkov, A.; Tomita, Y. 9-ING-41, a small molecule inhibitor of GSK-3beta, potentiates the effects of anticancer therapeutics in bladder cancer. Sci. Rep. 2019, 9, 19977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, T.; Kim, J.H.; Carver, K.; Su, Y.; Weremowicz, S.; Mulvey, L.; Yamamoto, S.; Brennan, C.; Mei, S.; Long, H.; et al. CLK2 Is an Oncogenic Kinase and Splicing Regulator in Breast Cancer. Cancer Res. 2015, 75, 1516–1526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muraki, M.; Ohkawara, B.; Hosoya, T.; Onogi, H.; Koizumi, J.; Koizumi, T.; Sumi, K.; Yomoda, J.-I.; Murray, M.V.; Kimura, H.; et al. Manipulation of Alternative Splicing by a Newly Developed Inhibitor of Clks. J. Biol. Chem. 2004, 279, 24246–24254. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Conaway, L.; Bethard, J.R.; Al-Ayoubi, A.M.; Bradley, A.T.; Zheng, H.; Weed, S.A.; Eblen, S.T. Phosphorylation of the alternative mRNA splicing factor 45 (SPF45) by Clk1 regulates its splice site utilization, cell migration and invasion. Nucleic Acids Res. 2013, 41, 4949–4962. [Google Scholar] [CrossRef] [Green Version]
- Fleuren, E.D.; Zhang, L.; Wu, J.; Daly, R.J. The Kinome “at Large” in Cancer. Nat. Rev. Cancer 2016, 16, 83–98. [Google Scholar] [CrossRef]
- Joshi, P.M.; Sutor, S.L.; Huntoon, C.J.; Karnitz, L.M. Ovarian Cancer-associated Mutations Disable Catalytic Activity of CDK12, a Kinase That Promotes Homologous Recombination Repair and Resistance to Cisplatin and Poly(ADP-ribose) Polymerase Inhibitors. J. Biol. Chem. 2014, 289, 9247–9253. [Google Scholar] [CrossRef] [Green Version]
- Kauraniemi, P.; Bärlund, M.; Monni, O.; Kallioniemi, A. New amplified and highly expressed genes discovered in the ERBB2 amplicon in breast cancer by cDNA microarrays. Cancer Res. 2001, 61, 8235. [Google Scholar] [PubMed]
- Kauraniemi, P.; Kuukasjärvi, T.; Sauter, G.; Kallioniemi, A. Amplification of a 280-Kilobase Core Region at the ERBB2 Locus Leads to Activation of Two Hypothetical Proteins in Breast Cancer. Am. J. Pathol. 2003, 163, 1979–1984. [Google Scholar] [CrossRef]
- Patel, H. Expression of CDK7, Cyclin H, and MAT1 Is Elevated in Breast Cancer and Is Prognostic in Estrogen Receptor-Positive Breast Cancer. Clin. Cancer Res. 2016, 22, 5929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP kinase signalling pathways in cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef] [Green Version]
- Roberts, P.J.; Der, C.J. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 2007, 26, 3291–3310. [Google Scholar] [CrossRef] [Green Version]
- Friedman, E. Mirk/Dyrk1B in cancer. J. Cell. Biochem. 2007, 102, 274–279. [Google Scholar] [CrossRef]
- Deng, X.; Ewton, D.Z.; Li, S.; Naqvi, A.; Mercer, S.E.; Landas, S.; Friedman, E. The Kinase Mirk/Dyrk1B Mediates Cell Survival in Pancreatic Ductal Adenocarcinoma. Cancer Res. 2006, 66, 4149–4158. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Deng, H.; Friedman, E.A. Ovarian cancer cells, not normal cells, are damaged by Mirk/Dyrk1B kinase inhibition. Int. J. Cancer 2012, 132, 2258–2269. [Google Scholar] [CrossRef]
- McCubrey, J.A.; Steelman, L.S.; Bertrand, F.E.; Davis, N.M.; Abrams, S.L.; Montalto, G.; D’Assoro, A.B.; Libra, M.; Nicoletti, F.; Maestro, R.; et al. Multifaceted roles of GSK-3 and Wnt/β-catenin in hematopoiesis and leukemogenesis: Opportunities for therapeutic intervention. Leukemia 2013, 28, 15–33. [Google Scholar] [CrossRef] [Green Version]
- Uzor, S.; Porazinski, S.R.; Li, L.; Clark, B.; Ajiro, M.; Iida, K.; Hagiwara, M.; Alqasem, A.A.; Perks, C.M.; Wilson, I.D.; et al. CDC2-like (CLK) protein kinase inhibition as a novel targeted therapeutic strategy in prostate cancer. Sci. Rep. 2021, 11, 7963. [Google Scholar] [CrossRef]
- Dominski, Z.; Marzluff, W.F. Formation of the 3′ end of histone mRNA: Getting closer to the end. Gene 2007, 396, 373–390. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.C.W.; Abdel-Wahab, O. Therapeutic targeting of splicing in cancer. Nat. Med. 2016, 22, 976–986. [Google Scholar] [CrossRef]
- Treiber, D.K.; Shah, N.P. Ins and Outs of Kinase DFG Motifs. Chem. Biol. 2013, 20, 745–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asghar, U.; Witkiewicz, A.K.; Turner, N.C.; Knudsen, E.S. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat. Rev. Drug Discov. 2015, 14, 130–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finn, R.S.; Crown, J.P.; Lang, I.; Boer, K.; Bondarenko, I.M.; Kulyk, S.O.; Ettl, J.; Patel, R.; Pinter, T.; Schmidt, M.; et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): A randomised phase 2 study. Lancet Oncol. 2015, 16, 25–35. [Google Scholar] [CrossRef]
- Hortobagyi, G.N.; Stemmer, S.M.; Burris, H.A.; Yap, Y.-S.; Sonke, G.S.; Paluch-Shimon, S.; Campone, M.; Blackwell, K.L.; André, F.; Winer, E.P.; et al. Ribociclib as First-Line Therapy for HR-Positive, Advanced Breast Cancer. N. Engl. J. Med. 2016, 375, 1738–1748. [Google Scholar] [CrossRef]
- Sledge, G.W., Jr.; Toi, M.; Neven, P.; Sohn, J.; Inoue, K.; Pivot, X.; Burdaeva, O.; Okera, M.; Masuda, N.; Kaufman, P.A.; et al. MONARCH 2: Abemaciclib in Combination with Fulvestrant in Women With HR+/HER2− Advanced Breast Cancer Who Had Progressed While Receiving Endocrine Therapy. J. Clin. Oncol. 2017, 35, 2875–2884. [Google Scholar] [CrossRef]
- Torre, R. Epigallocatechin-3-Gallate, a DYRK1A Inhibitor, Rescues Cognitive Deficits in Down Syndrome Mouse Models and in Humans. Mol. Nutr. Food Res. 2014, 58, 278. [Google Scholar] [CrossRef] [PubMed]
- Pandey, M.K.; DeGrado, T.R. Glycogen Synthase Kinase-3 (GSK-3)-Targeted Therapy and Imaging. Theranostics 2016, 6, 571–593. [Google Scholar] [CrossRef]
- McCubrey, J.A.; Steelman, L.S.; Bertrand, F.E.; Davis, N.M.; Sokolosky, M.; Abrams, S.L.; Montalto, G.; D’assoro, A.B.; Libra, M.; Nicoletti, F.; et al. GSK-3 as potential target for therapeutic intervention in cancer. Oncotarget 2014, 5, 2881–2911. [Google Scholar] [CrossRef] [Green Version]
- Fedoriw, A.; Rajapurkar, S.R.; O’Brien, S.; Gerhart, S.V.; Mitchell, L.H.; Adams, N.D.; Rioux, N.; Lingaraj, T.; Ribich, S.A.; Pappalardi, M.B.; et al. Anti-tumor Activity of the Type I PRMT Inhibitor, GSK3368715, Synergizes with PRMT5 Inhibition through MTAP Loss. Cancer Cell 2019, 36, 100–114.e25. [Google Scholar] [CrossRef]
- Yomoda, J.-I.; Muraki, M.; Kataoka, N.; Hosoya, T.; Suzuki, M.; Hagiwara, M.; Kimura, H. Combination of Clk family kinase and SRp75 modulates alternative splicing of Adenovirus E1A. Genes Cells 2008, 13, 233–244. [Google Scholar] [CrossRef] [Green Version]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef] [PubMed]
- Montero, J.; Letai, A. Why do BCL-2 inhibitors work and where should we use them in the clinic? Cell Death Differ. 2018, 25, 56–64. [Google Scholar] [CrossRef]
- Ackerley, S.J.; Byblow, W.D.; Barber, P.A.; MacDonald, H.; McIntyre-Robinson, A.; Stinear, C.M. Primed Physical Therapy Enhances Recovery of Upper Limb Function in Chronic Stroke Patients. Neurorehabilit. Neural Repair 2015, 30, 339–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glickman, M.S.; Sawyers, C.L. Converting Cancer Therapies into Cures: Lessons from Infectious Diseases. Cell 2012, 148, 1089–1098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.; Qu, X. Cancer biomarker detection: Recent achievements and challenges. Chem. Soc. Rev. 2015, 44, 2963–2997. [Google Scholar] [CrossRef] [PubMed]
- Arnedos, M.; Vicier, C.; Loi, S.; Lefebvre, C.; Michiels, S.; Bonnefoi, H.; Andre, F. Precision medicine for metastatic breast cancer—Limitations and solutions. Nat. Rev. Clin. Oncol. 2015, 12, 693–704. [Google Scholar] [CrossRef]
- Siravegna, G.; Marsoni, S.; Siena, S.; Bardelli, A. Integrating liquid biopsies into the management of cancer. Nat. Rev. Clin. Oncol. 2017, 14, 531–548. [Google Scholar] [CrossRef]
- Bogoyevitch, M.; Fairlie, D.P. A new paradigm for protein kinase inhibition: Blocking phosphorylation without directly targeting ATP binding. Drug Discov. Today 2007, 12, 622–633. [Google Scholar] [CrossRef]
- O’Neil, N.J.; Bailey, M.L.; Hieter, P. Synthetic lethality and cancer. Nat. Rev. Genet. 2017, 18, 613–623. [Google Scholar] [CrossRef]
- Kaelin, W.G., Jr. The Concept of Synthetic Lethality in the Context of Anticancer Therapy. Nat. Rev. Cancer 2005, 5, 689–698. [Google Scholar] [CrossRef]
- Bosc, N.; Meyer, C.; Bonnet, P. The use of novel selectivity metrics in kinase research. BMC Bioinform. 2017, 18, 17. [Google Scholar] [CrossRef] [Green Version]
- Ran, X.; Gestwicki, J.E. Inhibitors of protein–protein interactions (PPIs): An analysis of scaffold choices and buried surface area. Curr. Opin. Chem. Biol. 2018, 44, 75–86. [Google Scholar] [CrossRef]
- Klaeger, S.; Heinzlmeir, S.; Wilhelm, M.; Polzer, H.; Vick, B.; Koenig, P.-A.; Reinecke, M.; Ruprecht, B.; Petzoldt, S.; Meng, C.; et al. The target landscape of clinical kinase drugs. Science 2017, 358, eaan4368. [Google Scholar] [CrossRef] [Green Version]
- Arkin, M.R.; Tang, Y.; Wells, J.A. Small-Molecule Inhibitors of Protein-Protein Interactions: Progressing toward the Reality. Chem. Biol. 2014, 21, 1102–1114. [Google Scholar] [CrossRef] [Green Version]
- Hu, L.; Yang, Y.; Zheng, S.; Xu, J.; Ran, T.; Chen, H. Kinase Inhibitor Scaffold Hopping with Deep Learning Approaches. J. Chem. Inf. Model. 2021, 61, 4900–4912. [Google Scholar] [CrossRef]
- Uetz, P.; Giot, L.; Cagney, G.; Mansfield, T.A.; Judson, R.S.; Knight, J.R.; Lockshon, D.; Narayan, V.; Srinivasan, M.; Pochart, P.; et al. A comprehensive analysis of protein–protein interactions in Saccharomyces cerevisiae. Nature 2000, 403, 623–627. [Google Scholar] [CrossRef]
- Rual, J.-F.; Venkatesan, K.; Hao, T.; Hirozane-Kishikawa, T.; Dricot, A.; Li, N.; Berriz, G.F.; Gibbons, F.D.; Dreze, M.; Ayivi-Guedehoussou, N.; et al. Towards a proteome-scale map of the human protein–protein interaction network. Nature 2005, 437, 1173–1178. [Google Scholar] [CrossRef] [PubMed]
- Gavin, A.-C.; Aloy, P.; Grandi, P.; Krause, R.; Boesche, M.; Marzioch, M.; Rau, C.; Jensen, L.J.; Bastuck, S.; Dümpelfeld, B.; et al. Proteome survey reveals modularity of the yeast cell machinery. Nature 2006, 440, 631–636. [Google Scholar] [CrossRef]
- Krogan, N.J.; Cagney, G.; Yu, H.; Zhong, G.; Guo, X.; Ignatchenko, A.; Li, J.; Pu, S.; Datta, N.; Tikuisis, A.P.; et al. Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature 2006, 440, 637–643. [Google Scholar] [CrossRef]
- Glatter, T.; Wepf, A.; Aebersold, R.; Gstaiger, M. An integrated workflow for charting the human interaction proteome: Insights into the PP2A system. Mol. Syst. Biol. 2009, 5, 237. [Google Scholar] [CrossRef]
- Rual, J.-F.; Hirozane-Kishikawa, T.; Hao, T.; Bertin, N.; Li, S.; Dricot, A.; Li, N.; Rosenberg, J.; Lamesch, P.; Vidalain, P.-O.; et al. Human ORFeome Version 1.1: A Platform for Reverse Proteomics. Genome Res. 2004, 14, 2128–2135. [Google Scholar] [CrossRef] [Green Version]
- Varjosalo, M.; Sacco, R.; Stukalov, A.; van Drogen, A.; Planyavsky, M.; Hauri, S.; Aebersold, R.; Bennett, K.L.; Colinge, J.; Gstaiger, M.; et al. Interlaboratory reproducibility of large-scale human protein-complex analysis by standardized AP-MS. Nat. Methods 2013, 10, 307–314. [Google Scholar] [CrossRef]
- Roux, K.J.; Kim, D.I.; Raida, M.; Burke, B. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell Biol. 2012, 196, 801–810. [Google Scholar] [CrossRef] [Green Version]
- Varnaite, R.; MacNeill, S.A. Meet the Neighbors: Mapping Local Protein Interactomes by Proximity-Dependent Labeling with BioID. Proteomics 2016, 16, 2503. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.I.; Kc, B.; Zhu, W.; Motamedchaboki, K.; Doye, V.; Roux, K.J. Probing nuclear pore complex architecture with proximity-dependent biotinylation. Proc. Natl. Acad. Sci. USA 2014, 111, E2453–E2461. [Google Scholar] [CrossRef]
- Morgan, D.O. Cyclin-Dependent Kinases: Engines, Clocks, and Microprocessors. Annu. Rev. Cell Dev. Biol. 1997, 13, 261–291. [Google Scholar] [CrossRef]
- Nurse, P. Ordering S phase and M phase in the cell cycle. Cell 1994, 79, 547–550. [Google Scholar] [CrossRef]
- Sherr, C.J.; Roberts, J.M. CDK inhibitors: Positive and negative regulators of G1-phase progression. Genes Dev. 1999, 13, 1501–1512. [Google Scholar] [CrossRef] [Green Version]
- Malumbres, M.; Barbacid, M. Cell cycle, CDKs and cancer: A changing paradigm. Nat. Rev. Cancer 2009, 9, 153–166. [Google Scholar] [CrossRef]
- Chang, L.; Karin, M. Mammalian MAP kinase signalling cascades. Nature 2001, 410, 37–40. [Google Scholar] [CrossRef]
- Kyriakis, J.M.; Avruch, J. Mammalian MAPK Signal Transduction Pathways Activated by Stress and Inflammation: A 10-Year Update. Physiol. Rev. 2012, 92, 689–737. [Google Scholar] [CrossRef] [Green Version]
- Colwill, K.; Pawson, T.; Andrews, B.; Prasad, J.; Manley, J.; Bell, J.C.; Duncan, P.I. The Clk/Sty protein kinase phosphorylates SR splicing factors and regulates their intranuclear distribution. EMBO J. 1996, 15, 265–275. [Google Scholar] [CrossRef]
- Hastie, C.J.; McLauchlan, H.J.; Cohen, P. Assay of protein kinases using radiolabeled ATP: A protocol. Nat. Protoc. 2006, 1, 968–971. [Google Scholar] [CrossRef]
- Mok, J.; Im, H.; Snyder, M. Global identification of protein kinase substrates by protein microarray analysis. Nat. Protoc. 2009, 4, 1820–1827. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Bilgin, M.; Bangham, R.; Hall, D.; Casamayor, A.; Bertone, P.; Lan, N.; Jansen, R.; Bidlingmaier, S.; Houfek, T.; et al. Global Analysis of Protein Activities Using Proteome Chips. Science 2001, 293, 2101–2105. [Google Scholar] [CrossRef]
- Meng, L.; Michaud, G.A.; Merkel, J.S.; Zhou, F.; Huang, J.; Mattoon, D.R.; Schweitzer, B. Protein kinase substrate identification on functional protein arrays. BMC Biotechnol. 2008, 8, 22. [Google Scholar] [CrossRef] [Green Version]
- Fasolo, J.; Sboner, A.; Sun, M.G.; Yu, H.; Chen, R.; Sharon, D.; Kim, P.M.; Gerstein, M.; Snyder, M. Diverse protein kinase interactions identified by protein microarrays reveal novel connections between cellular processes. Genes Dev. 2011, 25, 767–778. [Google Scholar] [CrossRef] [Green Version]
- Hao, Q.; Gao, L.; Niu, W.; Chen, L.; Zhang, P.; Chen, Z. POTEE stimulates the proliferation of pancreatic cancer by activating the PI3K/Akt/GSK-3β/β-catenin signaling. Biofactors 2020, 46, 685–692. [Google Scholar] [CrossRef]
- Wagman, A.S.; Johnson, K.W.; Bussiere, D.E. Discovery and Development of GSK3 Inhibitors for the Treatment of Type 2 Diabetes. Curr. Pharm. Des. 2004, 10, 1105–1137. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
(A) A schematic diagram illustrating the taxonomy of the eight subfamilies of CMGC kinases based on their kinase domain sequence similarity. (B) CMGC kinase subfamily dysregulation in most common cancer types with the prevalent deregulated mechanisms involved in cancer progression. Images created with BioRender.com (accessed on 11 July 2023).
Figure 1.
(A) A schematic diagram illustrating the taxonomy of the eight subfamilies of CMGC kinases based on their kinase domain sequence similarity. (B) CMGC kinase subfamily dysregulation in most common cancer types with the prevalent deregulated mechanisms involved in cancer progression. Images created with BioRender.com (accessed on 11 July 2023).

Figure 2.
CMGC kinase signaling pathways in health and disease. A graphical representation of the major signaling pathways involving the two most studied families, (A,B) the CDK and (C) MAPK families, and their roles in promoting cancer hallmarks (e.g., cell proliferation, survival, migration, angiogenesis). Images created with BioRender.com (accessed on 16 July 2023).
Figure 2.
CMGC kinase signaling pathways in health and disease. A graphical representation of the major signaling pathways involving the two most studied families, (A,B) the CDK and (C) MAPK families, and their roles in promoting cancer hallmarks (e.g., cell proliferation, survival, migration, angiogenesis). Images created with BioRender.com (accessed on 16 July 2023).

Figure 3.
(A) A network of known high-confidence protein–protein interactions of CMGC kinases obtained from the IntAct Molecular Interaction Database are grouped according to their simplified GO process annotation. (B) Distribution of the number of known interactors of the CMGC kinases based on the affinity purification and proximity labeling detection method. The visualization of the protein–protein interaction (PPI) network was generated by importing the publicly available interaction dataset from https://www.ebi.ac.uk/intact (accessed on 8 May 2023) protein interaction data into Cytoscape v3.9.
Figure 3.
(A) A network of known high-confidence protein–protein interactions of CMGC kinases obtained from the IntAct Molecular Interaction Database are grouped according to their simplified GO process annotation. (B) Distribution of the number of known interactors of the CMGC kinases based on the affinity purification and proximity labeling detection method. The visualization of the protein–protein interaction (PPI) network was generated by importing the publicly available interaction dataset from https://www.ebi.ac.uk/intact (accessed on 8 May 2023) protein interaction data into Cytoscape v3.9.


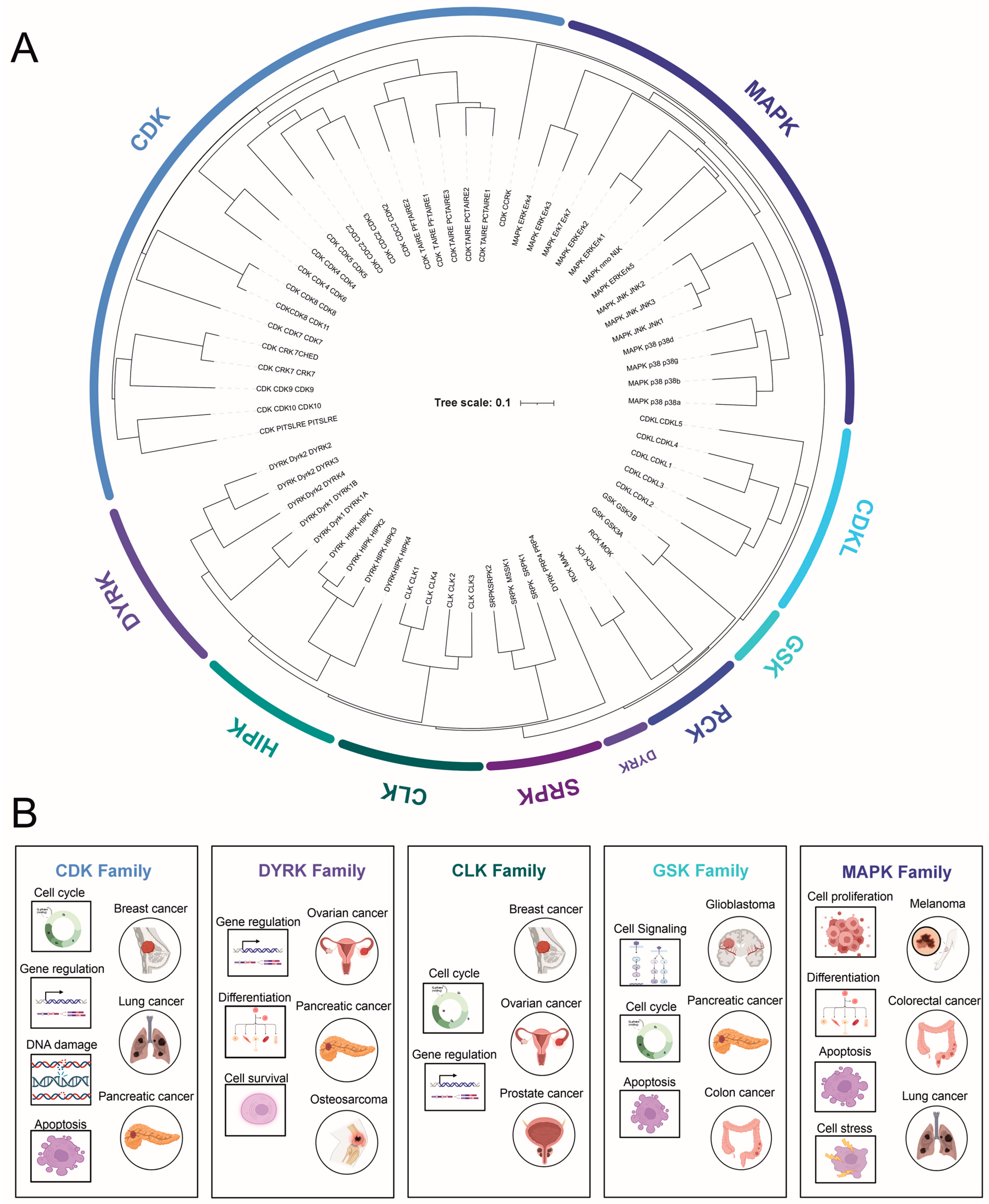{kind=link}
{kind=link}
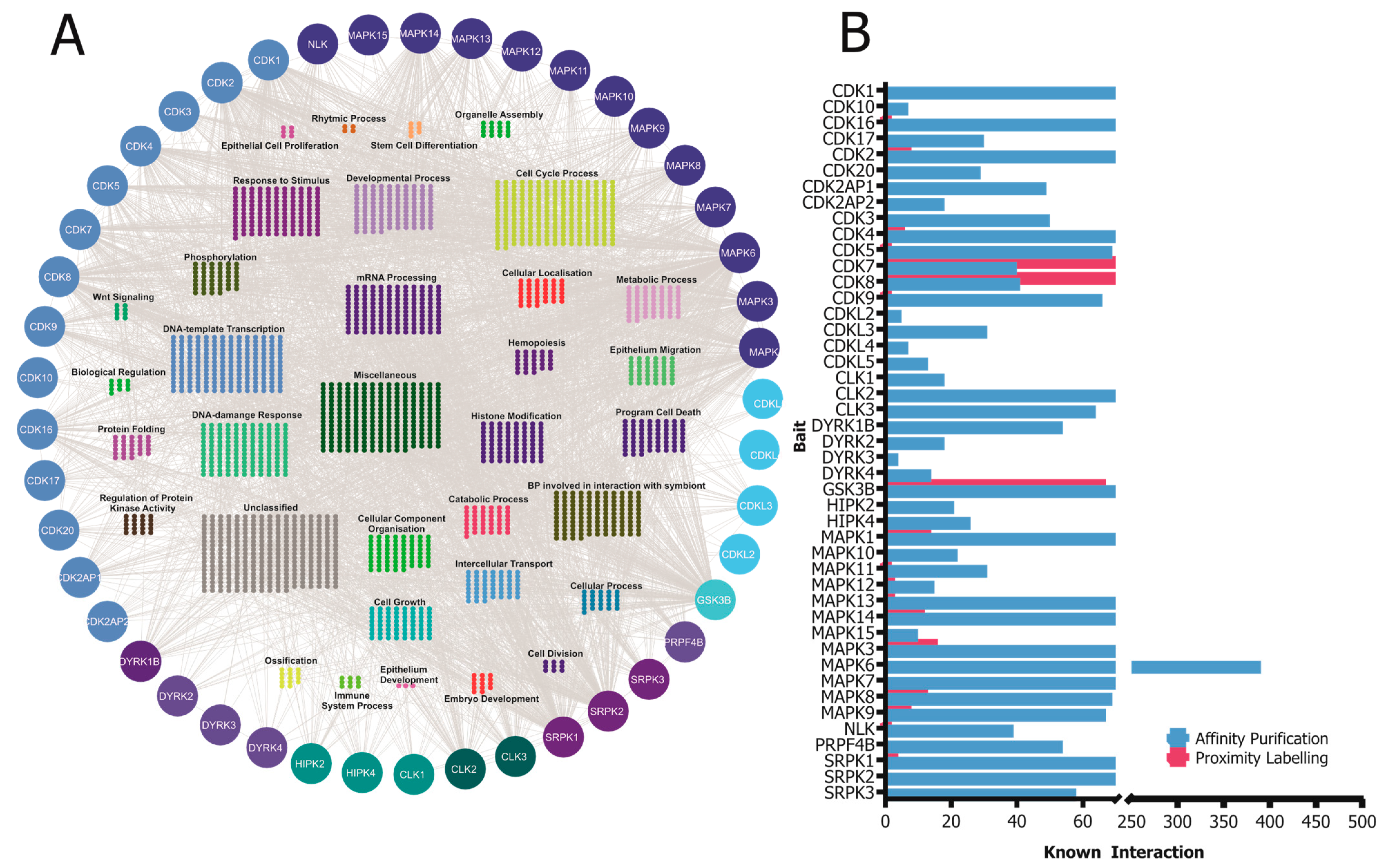{kind=link}
Table 1.
Prevalence and pattern of CMGC kinase subfamily dysregulation in cancer.
| Kinase Family | Deregulation Mechanism | Examples of Cancer Types |
|---|---|---|
| CDKs | Mutations, amplifications, deletions, and altered expression levels | Breast cancer [127,128,129], lung cancer [130], pancreatic cancer [131] |
| MAPKs | Mutations in pathway components (e.g., RAS, RAF) | Melanoma [132,133,134], colorectal cancer [135], lung cancer [48,136] |
| DYRK | Overexpression | Pancreatic [137,138], ovarian cancer [139,140], osteosarcoma [141], rhabdomyosarcoma [142] |
| GSKs | Altered expression levels, post-translational modifications | Glioblastoma [143], pancreatic cancer [144], colon cancer [145] |
| CLKs | Overexpression, cancer-associated splicing alterations | Breast cancer [146], prostate cancer [147], ovarian cancer [148] |
Table 2.
Aberrant activation of CMGC kinases in cancer. N/A: information not available.
| Kinase Class | Inhibitor Name | FDA Approval Status | Approved Indications | Key Clinical Benefits | Ongoing Research Areas |
|---|---|---|---|---|---|
| CDK | Palbociclib | Approved (2015) | HR-positive, HER2-negative advanced/metastatic breast cancer | Improved progression-free survival and overall response rates | Potential in other cancer types, combination therapies |
| CDK | Ribociclib | Approved (2017) | HR-positive, HER2-negative advanced/metastatic breast cancer | Improved progression-free survival and overall response rates | Potential in other cancer types, combination therapies |
| CDK | Abemaciclib | Approved (2017) | HR-positive, HER2-negative advanced/metastatic breast cancer | Improved progression-free survival and overall response rates | Potential in other cancer types, combination therapies |
| MAPK (BRAF) | Vemurafenib | Approved (2011) | Metastatic melanoma with BRAF mutations | Effective in BRAF-mutated melanoma, improved response rates | Targeting other components of MAPK pathways |
| MAPK (BRAF) | Dabrafenib | Approved (2013) | Metastatic melanoma with BRAF mutations | Effective in BRAF-mutated melanoma, improved response rates | Targeting other components of MAPK pathways |
| MAPK (MEK) | Trametinib | Approved (2013) | Metastatic melanoma with BRAF mutations | Improved outcomes when combined with BRAF inhibitors | Targeting other components of MAPK pathways |
| MAPK (MEK) | Cobimetinib | Approved (2015) | Metastatic melanoma with BRAF mutations | Improved outcomes when combined with BRAF inhibitors | Targeting other components of MAPK pathways |
| GSK | Tideglusib | Not Approved | N/A | Promising preclinical results in glioblastoma, pancreatic cancer | Further development, clinical evaluation in cancer treatment |
| CLK | TG-003 | Not Approved | N/A | Modulates alternative splicing, reduces tumor cell viability (preclinical) | Therapeutic potential in cancer, diseases with aberrant splicing |
Table 3.
Potential combination therapies involving CMGC kinase inhibitors (https://clinicaltrials.gov/, accessed on 8 May 2023).
Table 3.
Potential combination therapies involving CMGC kinase inhibitors (https://clinicaltrials.gov/, accessed on 8 May 2023).
| CMGC Kinase Inhibitor | Combination Agent | Rationale | Synergistic Effects | Clinical Trial Status |
|---|---|---|---|---|
| Palbociclib | Immune checkpoint inhibitors | Enhance antitumor immune response | Improved response rates | Ongoing clinical trials (NCT00141297) |
| Ribociclib | Angiogenesis inhibitors | Block tumor vascularization and growth | Enhanced tumor growth inhibition | Preclinical studies (NCT03285412) |
| Abemaciclib | Other kinase inhibitors | Target multiple signaling pathways simultaneously | Increased cell death, decreased proliferation | Ongoing clinical trials (NCT02057133) |
| Vemurafenib + Trametinib | Immune checkpoint inhibitors | Enhance antitumor immune response in combination with MAPK pathway inhibition | Improved response rates, prolonged survival | Ongoing clinical trials (NCT01597908) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Chowdhury, I.; Dashi, G.; Keskitalo, S. CMGC Kinases in Health and Cancer. Cancers 2023, 15, 3838. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers15153838
AMA Style
Chowdhury I, Dashi G, Keskitalo S. CMGC Kinases in Health and Cancer. Cancers. 2023; 15(15):3838. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers15153838
Chicago/Turabian StyleChowdhury, Iftekhar, Giovanna Dashi, and Salla Keskitalo. 2023. "CMGC Kinases in Health and Cancer" Cancers 15, no. 15: 3838. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers15153838
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.