
Non-Conventional Allogeneic Anti-BCMA Chimeric Antigen Receptor-Based Immune Cell Therapies for Multiple Myeloma Treatment

{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
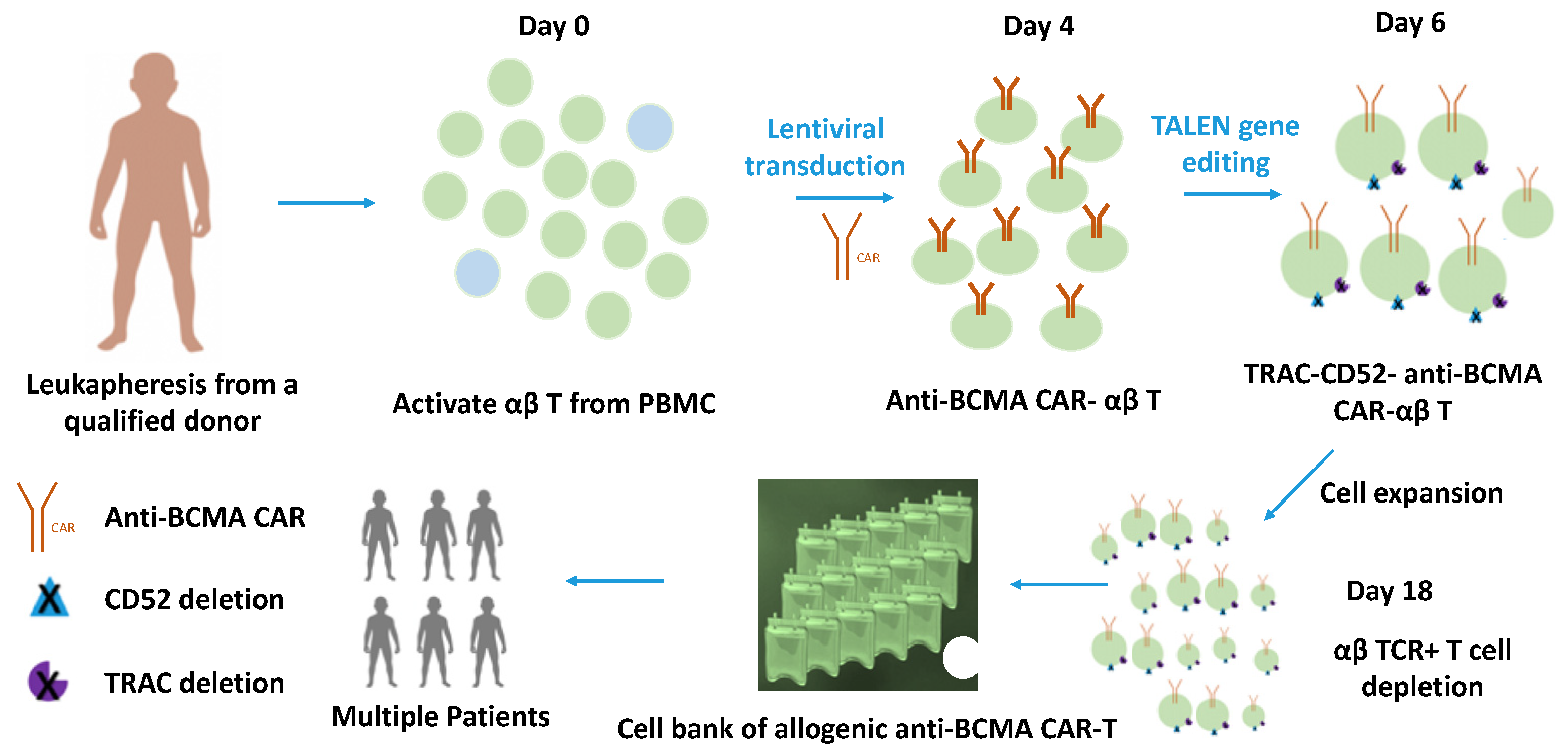
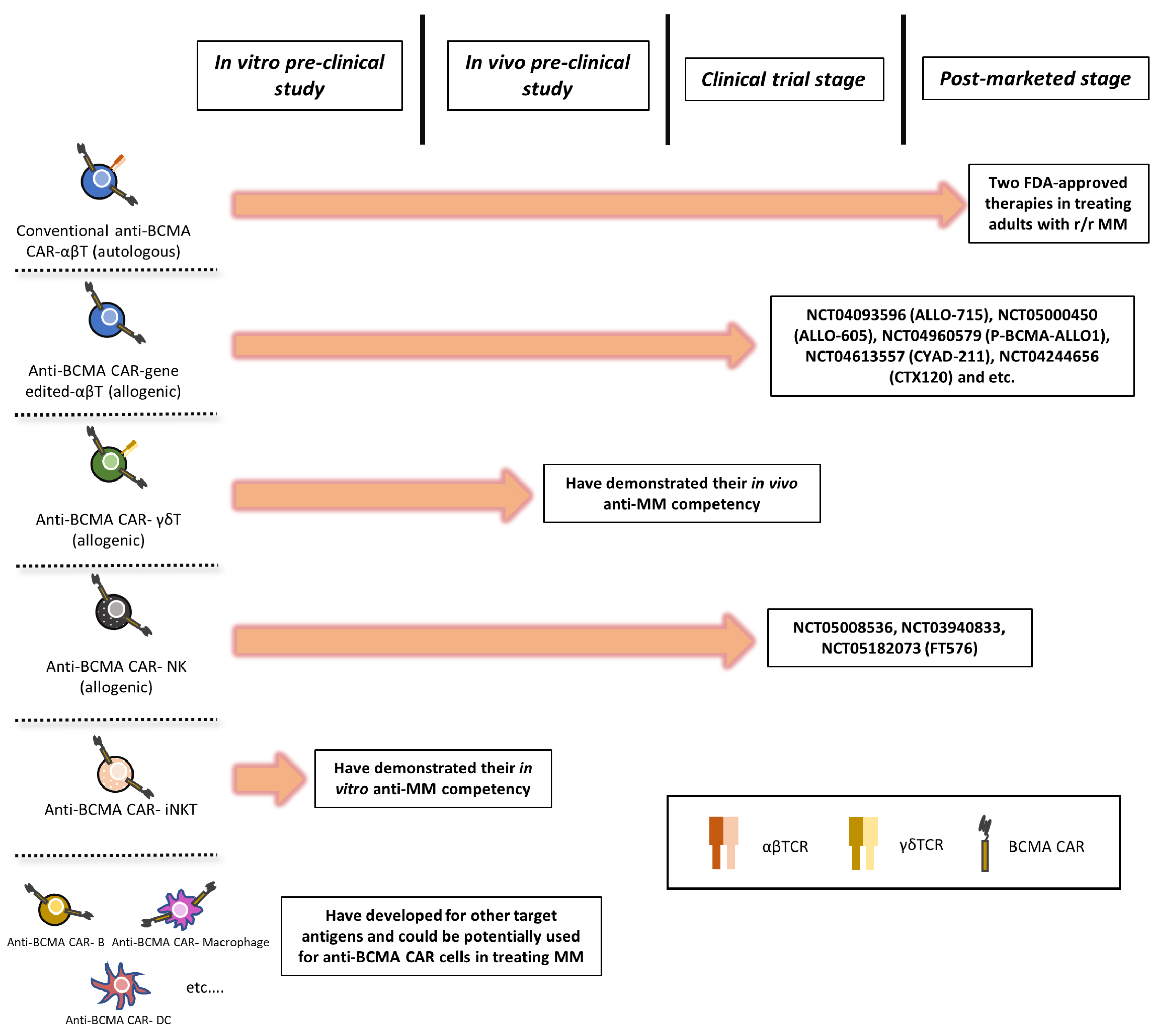
2. Gene-Edited-αβ T Cells
3. γδ T Cells
4. NK Cells
5. Other Potential Candidates (e.g., Macrophages, iNKT Cells, etc.)
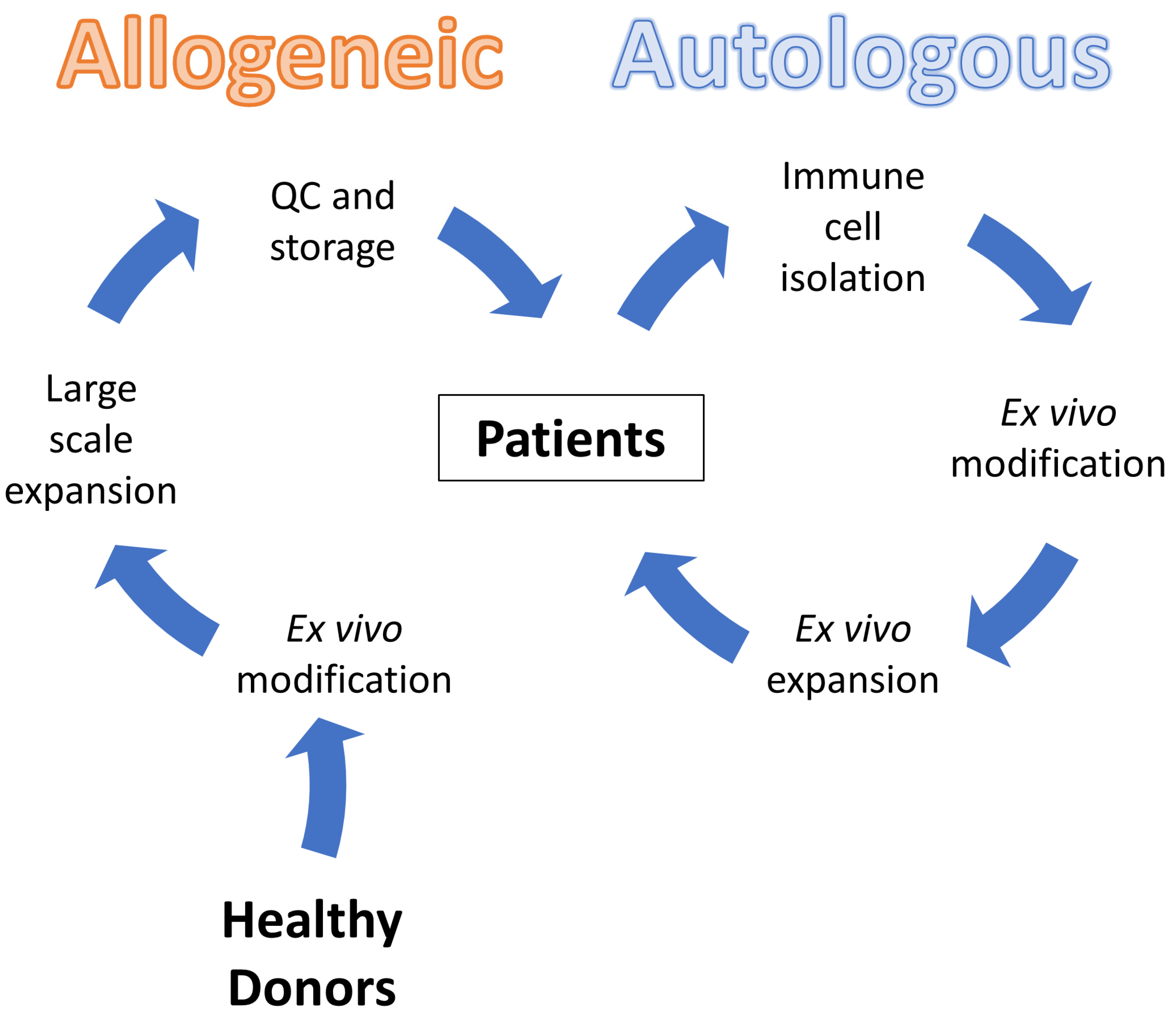
6. Current Landscape of Clinical Trial Regulations: Autologous vs. Allogeneic CAR Immune Cells
7. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Kazandjian, D. Multiple myeloma epidemiology and survival: A unique malignancy. Semin. Oncol. 2016, 43, 676–681. [Google Scholar] [CrossRef] [Green Version]
- Marino, S.; Roodman, G.D. Multiple Myeloma and Bone: The Fatal Interaction. Cold Spring Harb. Perspect. Med. 2018, 8, a031286. [Google Scholar] [CrossRef] [PubMed]
- Cowan, A.J.; Allen, C.; Barac, A.; Basaleem, H.; Bensenor, I.; Curado, M.P.; Foreman, K.; Gupta, R.; Harvey, J.; Hosgood, H.D.; et al. Global Burden of Multiple Myeloma: A Systematic Analysis for the Global Burden of Disease Study 2016. JAMA Oncol 2018, 4, 1221–1227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padala, S.A.; Barsouk, A.; Barsouk, A.; Rawla, P.; Vakiti, A.; Kolhe, R.; Kota, V.; Ajebo, G.H. Epidemiology, Staging, and Management of Multiple Myeloma. Med. Sci. 2021, 9, 3. [Google Scholar] [CrossRef]
- Key Statistics about Multiple Myeloma. Available online: https://www.cancer.org/cancer/multiple-myeloma/about/key-statistics.html (accessed on 28 September 2022).
- Xia, C.; Dong, X.; Li, H.; Cao, M.; Sun, D.; He, S.; Yang, F.; Yan, X.; Zhang, S.; Li, N.; et al. Cancer statistics in China and United States, 2022: Profiles, trends, and determinants. Chin. Med. J. 2022, 135, 584–590. [Google Scholar] [CrossRef] [PubMed]
- Kyle, R.A.; Rajkumar, S.V. Treatment of multiple myeloma: A comprehensive review. Clin. Lymphoma Myeloma 2009, 9, 278–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharya, K.; Bentley, J.P.; Ramachandran, S.; Chang, Y.; Banahan, B.F., 3rd; Shah, R.; Bhakta, N.; Yang, Y. Phase-Specific and Lifetime Costs of Multiple Myeloma Among Older Adults in the US. JAMA Netw. Open 2021, 4, e2116357. [Google Scholar] [CrossRef] [PubMed]
- Pimentel, M.; Espinal, O.; Godinez, F.; Jimenez, F.; Martinez, D.; Mendoza, N.; Quintana, A.; Richmond, J.E.; Romero, E. Consensus Statement: Importance of Timely Access to Multiple Myeloma Diagnosis and Treatment in Central America and the Caribbean. J. Hematol. 2022, 11, 1–7. [Google Scholar] [CrossRef]
- Sun, Z.; Zheng, F.; Wu, S.; Liu, Y.; Guo, H.; Liu, Y. Triplet versus doublet combination regimens for the treatment of relapsed or refractory multiple myeloma: A meta-analysis of phase III randomized controlled trials. Crit. Rev. Oncol. Hematol. 2017, 113, 249–255. [Google Scholar] [CrossRef]
- Chinese Medical Doctor Association Hematology Physicians Branch. Chinese guidelines for the diagnosis and treatment of multiple myeloma (2022 revision). Chin. J. Intern. Med. 2022, 61, 480–487. [Google Scholar] [CrossRef]
- National Comprehensive Cancer Network, Inc. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for Multiple Myeloma Version 1.2023. Available online: https://www.nccn.org/guidelines/guidelines-detail?category=1&id=1445. (accessed on 28 September 2022).
- Moreau, P.; de Wit, E. Recent progress in relapsed multiple myeloma therapy: Implications for treatment decisions. Br. J. Haematol. 2017, 179, 198–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, N.; Chari, A.; Scott, E.; Mezzi, K.; Usmani, S.Z. B-cell maturation antigen (BCMA) in multiple myeloma: Rationale for targeting and current therapeutic approaches. Leukemia 2020, 34, 985–1005. [Google Scholar] [CrossRef] [Green Version]
- DARZALEX® (Daratumumab) Injection Package Insert. Available online: https://www.janssenlabels.com/package-insert/product-monograph/prescribing-information/DARZALEX-pi.pdf (accessed on 28 September 2022).
- Mullard, A. FDA approves second BCMA-targeted CAR-T cell therapy. Nat. Rev. Drug Discov. 2022, 21, 249. [Google Scholar] [CrossRef] [PubMed]
- Kleber, M.; Ntanasis-Stathopoulos, I.; Terpos, E. BCMA in Multiple Myeloma-A Promising Key to Therapy. J. Clin. Med. 2021, 10, 4088. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, R.O.; Evbuomwan, M.O.; Pittaluga, S.; Rose, J.J.; Raffeld, M.; Yang, S.; Gress, R.E.; Hakim, F.T.; Kochenderfer, J.N. B-cell maturation antigen is a promising target for adoptive T-cell therapy of multiple myeloma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 2048–2060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tai, Y.T.; Anderson, K.C. Targeting B-cell maturation antigen in multiple myeloma. Immunotherapy 2015, 7, 1187–1199. [Google Scholar] [CrossRef] [Green Version]
- Sadelain, M.; Brentjens, R.; Riviere, I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013, 3, 388–398. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Zhou, G.; Zhang, L.; Zhao, Q. Building Potent Chimeric Antigen Receptor T Cells With CRISPR Genome Editing. Front. Immunol. 2019, 10, 456. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, J.; Zhong, J.F.; Zhang, X. Engineering CAR-T cells. Biomark. Res. 2017, 5, 22. [Google Scholar] [CrossRef] [Green Version]
- Tay, J.C.K.; Wang, J.; Du, Z.; Ng, Y.Y.; Li, Z.; Ren, Y.; Zhang, C.; Zhu, J.; Xu, X.H.; Wang, S. Manufacturing NKG2D CAR-T cells with piggyBac transposon vectors and K562 artificial antigen-presenting cells. Mol. Methods Clin. Dev. 2021, 21, 107–120. [Google Scholar] [CrossRef]
- Rafiq, S.; Hackett, C.S.; Brentjens, R.J. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat. Rev. Clin. Oncol. 2020, 17, 147–167. [Google Scholar] [CrossRef] [PubMed]
- Brocker, T.; Karjalainen, K. Signals through T cell receptor-zeta chain alone are insufficient to prime resting T lymphocytes. J. Exp. Med. 1995, 181, 1653–1659. [Google Scholar] [CrossRef] [PubMed]
- Cappell, K.M.; Kochenderfer, J.N. A comparison of chimeric antigen receptors containing CD28 versus 4-1BB costimulatory domains. Nat. Rev. Clin. Oncol. 2021, 18, 715–727. [Google Scholar] [CrossRef] [PubMed]
- Hombach, A.A.; Rappl, G.; Abken, H. Arming Cytokine-induced Killer Cells With Chimeric Antigen Receptors: CD28 Outperforms Combined CD28–OX40 “Super-stimulation”. Mol. Ther. 2013, 21, 2268–2277. [Google Scholar] [CrossRef] [Green Version]
- Package Insert—ABECMA. Available online: https://www.fda.gov/media/147055/download (accessed on 28 September 2022).
- Package Insert—CARVYKTI. Available online: https://www.fda.gov/media/156560/download (accessed on 28 September 2022).
- Depil, S.; Duchateau, P.; Grupp, S.A.; Mufti, G.; Poirot, L. ‘Off-the-shelf’ allogeneic CAR T cells: Development and challenges. Nat. Rev. Drug Discov. 2020, 19, 185–199. [Google Scholar] [CrossRef]
- Levine, B.L.; Miskin, J.; Wonnacott, K.; Keir, C. Global Manufacturing of CAR T Cell Therapy. Mol. Methods Clin. Dev. 2017, 4, 92–101. [Google Scholar] [CrossRef] [Green Version]
- Available online: https://www.drugs.com/price-guide/ (accessed on 28 September 2022).
- REVLIMID (Lenalidomide) Capsules Package Insert. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/021880s057lbl.pdf (accessed on 28 September 2022).
- VELCADE (Bortezomib) Package Insert. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/021602s040lbl.pdf (accessed on 28 September 2022).
- Vucinic, V.; Quaiser, A.; Luckemeier, P.; Fricke, S.; Platzbecker, U.; Koehl, U. Production and Application of CAR T Cells: Current and Future Role of Europe. Front. Med. 2021, 8, 713401. [Google Scholar] [CrossRef]
- Caldwell, K.J.; Gottschalk, S.; Talleur, A.C. Allogeneic CAR Cell Therapy-More Than a Pipe Dream. Front. Immunol. 2020, 11, 618427. [Google Scholar] [CrossRef]
- Rådestad, E.; Sundin, M.; Törlén, J.; Thunberg, S.; Önfelt, B.; Ljungman, P.; Watz, E.; Mattsson, J.; Uhlin, M. Individualization of Hematopoietic Stem Cell Transplantation Using Alpha/Beta T-Cell Depletion. Front. Immunol. 2019, 10, 189. [Google Scholar] [CrossRef] [Green Version]
- Felix, N.J.; Allen, P.M. Specificity of T-cell alloreactivity. Nat. Rev. Immunol. 2007, 7, 942–953. [Google Scholar] [CrossRef]
- Mailankody, S.; Liedtke, M.; Sidana, S.; Matous, J.V.; Chhabra, S.; Oluwole, O.O.; Malik, S.A.; Kumar, S.; Nath, R.; Anwer, F.; et al. Universal Updated Phase 1 Data Validates the Feasibility of Allogeneic Anti-BCMA ALLO-715 Therapy for Relapsed/Refractory Multiple Myeloma. Blood 2021, 138, 651. [Google Scholar] [CrossRef]
- Allogene Therapeutics Reports Positive Results from Phase 1 UNIVERSAL Study of Single Dose ALLO-715 AlloCAR T™ Cell Therapy in Relapsed/Refractory Multiple Myeloma at the 63rd American Society of Hematology Annual Meeting. Available online: https://allogene.gcs-web.com/node/8501/pdf (accessed on 28 September 2022).
- Allogene Therapeutics Receives FDA Orphan-Drug Designation for ALLO-605, Its First TurboCAR™ T Cell Product Candidate, for the Treatment of Multiple Myeloma. Available online: https://ir.allogene.com/news-releases/news-release-details/allogene-therapeutics-receives-fda-orphan-drug-designation-allo/ (accessed on 28 September 2022).
- Sommer, C.; Lin, R.; Sutton, J.; Bentley, T.; Nguyen, D.; Yoon, H.; Au, M.; Vargas-Inchaustegui, D.; Cheng, H.-Y.; Van Blarcom, T.; et al. Preclinical Evaluation of ALLO-605, an Allogeneic BCMA Turbocar TTM Cell Therapy for the Treatment of Multiple Myeloma. Blood 2020, 136, 8. [Google Scholar] [CrossRef]
- Safety and Efficacy of ALLO-605 an Anti-BCMA Allogeneic CAR T Cell Therapy in Patients With Relapsed/Refractory Multiple Myeloma. Available online: https://clinicaltrials.gov/ct2/show/NCT05000450 (accessed on 27 December 2022).
- Dar, H.; Henderson, D.; Padalia, Z.; Porras, A.; Mu, D.; Kyungah, M.; Police, S.; Kalaitzidis, D.; Terrett, J.; Sagert, J. Preclinical Development of CTX120, an Allogeneic CAR-T Cell Targeting Bcma. Blood 2018, 132, 1921. [Google Scholar] [CrossRef]
- CRISPR Therapeutics Provides Business Update and Reports Third Quarter 2021 Financial Results. Available online: https://crisprtx.com/about-us/press-releases-and-presentations/crispr-therapeutics-provides-business-update-and-reports-third-quarter-2021-financial-results (accessed on 30 December 2022).
- Ran, F.A.; Hsu, P.D.; Lin, C.Y.; Gootenberg, J.S.; Konermann, S.; Trevino, A.E.; Scott, D.A.; Inoue, A.; Matoba, S.; Zhang, Y.; et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 2013, 154, 1380–1389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madison, B.B.; Patil, D.; Richter, M.; Li, X.; Tong, M.; Cranert, S.; Wang, X.; Martin, R.; Xi, H.; Tan, Y. Cas-CLOVER is a novel high-fidelity nuclease for safe and robust generation of TSCM-enriched allogeneic CAR-T cells. Mol. Ther. -Nucleic Acids 2022, 29, 979–995. [Google Scholar] [CrossRef]
- Al-Homsi, A.-S.; Anguille, S.; Deeren, D.; Nishihori, T.; Meuleman, N.; Abdul-Hay, M.; Morgan, G.J.; Brayer, J.; Braun, N.; Lonez, C.; et al. Immunicy-1: Targeting BCMA with Cyad-211 to Establish Proof of Concept of an shRNA-Based Allogeneic CAR T Cell Therapy Platform. Blood 2021, 138, 2817. [Google Scholar] [CrossRef]
- Pistoia, V.; Tumino, N.; Vacca, P.; Veneziani, I.; Moretta, A.; Locatelli, F.; Moretta, L. Human γδ T-Cells: From Surface Receptors to the Therapy of High-Risk Leukemias. Front. Immunol. 2018, 9, 984. [Google Scholar] [CrossRef] [Green Version]
- Karunakaran, M.M.; Willcox, C.R.; Salim, M.; Paletta, D.; Fichtner, A.S.; Noll, A.; Starick, L.; Nöhren, A.; Begley, C.R.; Berwick, K.A.; et al. Butyrophilin-2A1 Directly Binds Germline-Encoded Regions of the Vγ9Vδ2 TCR and Is Essential for Phosphoantigen Sensing. Immunity 2020, 52, 487–498.e486. [Google Scholar] [CrossRef]
- Kakimi, K.; Matsushita, H.; Masuzawa, K.; Karasaki, T.; Kobayashi, Y.; Nagaoka, K.; Hosoi, A.; Ikemura, S.; Kitano, K.; Kawada, I.; et al. Adoptive transfer of zoledronate-expanded autologous Vγ9Vδ2 T-cells in patients with treatment-refractory non-small-cell lung cancer: A multicenter, open-label, single-arm, phase 2 study. J. Immunother. Cancer 2020, 8, e001185. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, M.; Smetak, M.; Schaefer-Eckart, K.; Kimmel, B.; Birkmann, J.; Einsele, H.; Kunzmann, V. Successful adoptive transfer and in vivo expansion of haploidentical γδ T cells. J. Transl. Med. 2014, 12, 45. [Google Scholar] [CrossRef]
- Bennouna, J.; Bompas, E.; Neidhardt, E.M.; Rolland, F.; Philip, I.; Galéa, C.; Salot, S.; Saiagh, S.; Audrain, M.; Rimbert, M.; et al. Phase-I study of Innacell gammadelta, an autologous cell-therapy product highly enriched in gamma9delta2 T lymphocytes, in combination with IL-2, in patients with metastatic renal cell carcinoma. Cancer Immunol. Immunother. CII 2008, 57, 1599–1609. [Google Scholar] [CrossRef] [PubMed]
- Aoki, T.; Matsushita, H.; Hoshikawa, M.; Hasegawa, K.; Kokudo, N.; Kakimi, K. Adjuvant combination therapy with gemcitabine and autologous γδ T-cell transfer in patients with curatively resected pancreatic cancer. Cytotherapy 2017, 19, 473–485. [Google Scholar] [CrossRef] [PubMed]
- Abe, Y.; Muto, M.; Nieda, M.; Nakagawa, Y.; Nicol, A.; Kaneko, T.; Goto, S.; Yokokawa, K.; Suzuki, K. Clinical and immunological evaluation of zoledronate-activated Vgamma9gammadelta T-cell-based immunotherapy for patients with multiple myeloma. Exp. Hematol. 2009, 37, 956–968. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.; Zhang, X.; Liang, S.; Luo, H.; Alnaggar, M.; Liu, A.; Yin, Z.; Chen, J.; Niu, L.; Jiang, Y. Irreversible electroporation plus allogenic Vγ9Vδ2 T cells enhances antitumor effect for locally advanced pancreatic cancer patients. Signal Transduct. Target. Ther. 2020, 5, 215. [Google Scholar] [CrossRef] [PubMed]
- IN8bio Observes Durable Morphologic Complete Responses in Ongoing Phase 1 Clinical Trial of INB-100, an Allogeneic Gamma-Delta T Cell Therapy in High-Risk Leukemia Patients. Available online: https://investors.in8bio.com/node/7501/pdf (accessed on 28 September 2022).
- Neelapu, S.S.; Hamadani, M.; Miklos, D.B.; Holmes, H.; Hinkle, J.; Kennedy-Wilde, J.; Maller, O.; Weinstein, M.; Galimi, F.; Lai, R.; et al. A phase 1 study of ADI-001: Anti-CD20 CAR-engineered allogeneic gamma delta (γδ) T cells in adults with B-cell malignancies. J. Clin. Oncol. 2022, 40, 7509. [Google Scholar] [CrossRef]
- Zhang, X.; Ng, Y.Y.; Du, Z.; Li, Z.; Chen, C.; Xiao, L.; Chng, W.J.; Wang, S. Vγ9Vδ2 T cells expressing a BCMA-Specific chimeric antigen receptor inhibit multiple myeloma xenograft growth. PloS ONE 2022, 17, e0267475. [Google Scholar] [CrossRef]
- Wallet, M.A.; Nishimura, T.; Del Casale, C.; Lebid, A.; Salantes, B.; Santostefano, K.E.; Bucher, S.; Mendonca, M.; Beqiri, M.; Thompson, L.J.; et al. Induced Pluripotent Stem Cell-Derived Gamma Delta CAR-T Cells for Cancer Immunotherapy. Blood 2021, 138, 2711. [Google Scholar] [CrossRef]
- Makkouk, A.; Yang, X.C.; Barca, T.; Lucas, A.; Turkoz, M.; Wong, J.T.S.; Nishimoto, K.P.; Brodey, M.M.; Tabrizizad, M.; Gundurao, S.R.Y.; et al. Off-the-shelf Vδ1 gamma delta T cells engineered with glypican-3 (GPC-3)-specific chimeric antigen receptor (CAR) and soluble IL-15 display robust antitumor efficacy against hepatocellular carcinoma. J. Immunother. Cancer 2021, 9, e003441. [Google Scholar] [CrossRef]
- Almeida, A.R.; Correia, D.V.; Fernandes-Platzgummer, A.; da Silva, C.L.; da Silva, M.G.; Anjos, D.R.; Silva-Santos, B. Delta One T Cells for Immunotherapy of Chronic Lymphocytic Leukemia: Clinical-Grade Expansion/Differentiation and Preclinical Proof of Concept. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 5795–5804. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.; Wu, P.; Wu, X.; Ye, J.; Wang, Z.; Zhao, S.; Ni, C.; Hu, G.; Xu, J.; Han, Y.; et al. Ex vivo expanded human circulating Vδ1 γδT cells exhibit favorable therapeutic potential for colon cancer. Oncoimmunology 2015, 4, e992749. [Google Scholar] [CrossRef]
- Deniger, D.C.; Maiti, S.N.; Mi, T.; Switzer, K.C.; Ramachandran, V.; Hurton, L.V.; Ang, S.; Olivares, S.; Rabinovich, B.A.; Huls, M.H.; et al. Activating and propagating polyclonal gamma delta T cells with broad specificity for malignancies. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 5708–5719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.N.; Wen, Q.; He, W.T.; Yang, J.H.; Zhou, C.Y.; Xiong, W.J.; Ma, L. Optimized protocols for γδ T cell expansion and lentiviral transduction. Mol. Med. Rep. 2019, 19, 1471–1480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoeres, T.; Smetak, M.; Pretscher, D.; Wilhelm, M. Improving the Efficiency of Vγ9Vδ2 T-Cell Immunotherapy in Cancer. Front. Immunol. 2018, 9, 800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, J.M.; Kaikobad, M.R.; Wallace, M.; Staab, M.J.; Horvath, D.L.; Wilding, G.; Liu, G.; Eickhoff, J.C.; McNeel, D.G.; Malkovsky, M. Pilot trial of interleukin-2 and zoledronic acid to augment γδ T cells as treatment for patients with refractory renal cell carcinoma. Cancer Immunol. Immunother. CII 2011, 60, 1447–1460. [Google Scholar] [CrossRef] [Green Version]
- Martinet, L.; Smyth, M.J. Balancing natural killer cell activation through paired receptors. Nat. Rev. Immunol. 2015, 15, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Clara, J.A.; Childs, R.W. Harnessing natural killer cells for the treatment of multiple myeloma. Semin. Oncol. 2022, 49, 69–85. [Google Scholar] [CrossRef]
- Leivas, A.; Risueno, R.M.; Guzman, A.; Sanchez-Vega, L.; Perez, M.; Megias, D.; Fernandez, L.; Alonso, R.; Perez-Martinez, A.; Rapado, I.; et al. Natural killer cells efficiently target multiple myeloma clonogenic tumor cells. Cancer Immunol. Immunother. CII 2021, 70, 2911–2924. [Google Scholar] [CrossRef]
- Myers, J.A.; Miller, J.S. Exploring the NK cell platform for cancer immunotherapy. Nat. Rev. Clin. Oncol. 2021, 18, 85–100. [Google Scholar] [CrossRef]
- Zhao, X.; Cai, L.; Hu, Y.; Wang, H. Cord-Blood Natural Killer Cell-Based Immunotherapy for Cancer. Front. Immunol. 2020, 11, 584099. [Google Scholar] [CrossRef]
- Zhu, H.; Kaufman, D.S. An Improved Method to Produce Clinical-Scale Natural Killer Cells from Human Pluripotent Stem Cells. Methods Mol. Biol. 2019, 2048, 107–119. [Google Scholar] [CrossRef]
- Wang, W.; Jiang, J.; Wu, C. CAR-NK for tumor immunotherapy: Clinical transformation and future prospects. Cancer Lett. 2020, 472, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Klein Wolterink, R.G.J.; Wang, J.; Bos, G.M.J.; Germeraad, W.T.V. Chimeric antigen receptor natural killer (CAR-NK) cell design and engineering for cancer therapy. J. Hematol. Oncol. 2021, 14, 73. [Google Scholar] [CrossRef] [PubMed]
- Abel, A.M.; Yang, C.; Thakar, M.S.; Malarkannan, S. Natural Killer Cells: Development, Maturation, and Clinical Utilization. Front. Immunol. 2018, 9, 1869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bachanova, V.; Cooley, S.; Defor, T.E.; Verneris, M.R.; Zhang, B.; McKenna, D.H.; Curtsinger, J.; Panoskaltsis-Mortari, A.; Lewis, D.; Hippen, K.; et al. Clearance of acute myeloid leukemia by haploidentical natural killer cells is improved using IL-2 diphtheria toxin fusion protein. Blood 2014, 123, 3855–3863. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.S.; Soignier, Y.; Panoskaltsis-Mortari, A.; McNearney, S.A.; Yun, G.H.; Fautsch, S.K.; McKenna, D.; Le, C.; Defor, T.E.; Burns, L.J.; et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood 2005, 105, 3051–3057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, L.; Cen, D.; Gan, H.; Sun, Y.; Huang, N.; Xiong, H.; Jin, Q.; Su, L.; Liu, X.; Wang, K.; et al. Adoptive Transfer of NKG2D CAR mRNA-Engineered Natural Killer Cells in Colorectal Cancer Patients. Mol. Ther. J. Am. Soc. Gene Ther. 2019, 27, 1114–1125. [Google Scholar] [CrossRef] [Green Version]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Nassif Kerbauy, L.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef]
- Ng, Y.Y.; Du, Z.; Zhang, X.; Chng, W.J.; Wang, S. CXCR4 and anti-BCMA CAR co-modified natural killer cells suppress multiple myeloma progression in a xenograft mouse model. Cancer Gene 2022, 29, 475–483. [Google Scholar] [CrossRef]
- Maroto-Martín, E.; Encinas, J.; García-Ortiz, A.; Alonso, R.; Leivas, A.; Paciello, M.L.; Garrido, V.; Cedena, T.; Ugalde, L.; Powell, D.J.J.; et al. PS1209 NKG2D AND BCMA-CAR NK CELLS EFFICIENTLY ELIMINATE MULTIPLE MYELOMA CELLS. A COMPREHENSIVE COMPARISON BETWEEN TWO CLINICALLY RELEVANT CARS. HemaSphere 2019, 3, 550–551. [Google Scholar] [CrossRef]
- Mantesso, S.; Geerts, D.; Spanholtz, J.; Kucerova, L. Genetic Engineering of Natural Killer Cells for Enhanced Antitumor Function. Front. Immunol. 2020, 11, 607131. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, P.; Raftery, M.J.; Pecher, G. Engineering NK Cells for CAR Therapy-Recent Advances in Gene Transfer Methodology. Front. Immunol. 2020, 11, 611163. [Google Scholar] [CrossRef] [PubMed]
- Sutlu, T.; Nystrom, S.; Gilljam, M.; Stellan, B.; Applequist, S.E.; Alici, E. Inhibition of intracellular antiviral defense mechanisms augments lentiviral transduction of human natural killer cells: Implications for gene therapy. Hum. Gene 2012, 23, 1090–1100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, Z.; Ng, Y.Y.; Zha, S.; Wang, S. piggyBac system to co-express NKG2D CAR and IL-15 to augment the in vivo persistence and anti-AML activity of human peripheral blood NK cells. Mol. Methods Clin. Dev. 2021, 23, 582–596. [Google Scholar] [CrossRef] [PubMed]
- Carlsten, M.; Childs, R.W. Genetic Manipulation of NK Cells for Cancer Immunotherapy: Techniques and Clinical Implications. Front. Immunol. 2015, 6, 266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petty, A.J.; Heyman, B.; Yang, Y. Chimeric Antigen Receptor Cell Therapy: Overcoming Obstacles to Battle Cancer. Cancers 2020, 12, 842. [Google Scholar] [CrossRef] [Green Version]
- Beider, K.; Nagler, A.; Wald, O.; Franitza, S.; Dagan-Berger, M.; Wald, H.; Giladi, H.; Brocke, S.; Hanna, J.; Mandelboim, O.; et al. Involvement of CXCR4 and IL-2 in the homing and retention of human NK and NK T cells to the bone marrow and spleen of NOD/SCID mice. Blood 2003, 102, 1951–1958. [Google Scholar] [CrossRef]
- Bonanni, V.; Antonangeli, F.; Santoni, A.; Bernardini, G. Targeting of CXCR3 improves anti-myeloma efficacy of adoptively transferred activated natural killer cells. J. Immunother. Cancer 2019, 7, 290. [Google Scholar] [CrossRef] [Green Version]
- Kremer, V.; Ligtenberg, M.A.; Zendehdel, R.; Seitz, C.; Duivenvoorden, A.; Wennerberg, E.; Colon, E.; Scherman-Plogell, A.H.; Lundqvist, A. Genetic engineering of human NK cells to express CXCR2 improves migration to renal cell carcinoma. J. Immunother. Cancer 2017, 5, 73. [Google Scholar] [CrossRef]
- Shimasaki, N.; Jain, A.; Campana, D. NK cells for cancer immunotherapy. Nat Rev Drug Discov 2020, 19, 200–218. [Google Scholar] [CrossRef]
- Miller, J.S.; Rooney, C.M.; Curtsinger, J.; McElmurry, R.; McCullar, V.; Verneris, M.R.; Lapteva, N.; McKenna, D.; Wagner, J.E.; Blazar, B.R.; et al. Expansion and homing of adoptively transferred human natural killer cells in immunodeficient mice varies with product preparation and in vivo cytokine administration: Implications for clinical therapy. Biol. Blood Marrow Transpl. 2014, 20, 1252–1257. [Google Scholar] [CrossRef]
- Tarannum, M.; Romee, R.; Shapiro, R.M. Innovative Strategies to Improve the Clinical Application of NK Cell-Based Immunotherapy. Front. Immunol. 2022, 13, 859177. [Google Scholar] [CrossRef] [PubMed]
- Ben-Shmuel, A.; Biber, G.; Barda-Saad, M. Unleashing Natural Killer Cells in the Tumor Microenvironment-The Next Generation of Immunotherapy? Front. Immunol. 2020, 11, 275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, E.; Tong, Y.; Dotti, G.; Shaim, H.; Savoldo, B.; Mukherjee, M.; Orange, J.; Wan, X.; Lu, X.; Reynolds, A.; et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia 2018, 32, 520–531. [Google Scholar] [CrossRef] [PubMed]
- Qin, V.M.; D’Souza, C.; Neeson, P.J.; Zhu, J.J. Chimeric Antigen Receptor beyond CAR-T Cells. Cancers 2021, 13, 404. [Google Scholar] [CrossRef] [PubMed]
- Poels, R.; Drent, E.; Lameris, R.; Katsarou, A.; Themeli, M.; van der Vliet, H.J.; de Gruijl, T.D.; van de Donk, N.; Mutis, T. Preclinical Evaluation of Invariant Natural Killer T Cells Modified with CD38 or BCMA Chimeric Antigen Receptors for Multiple Myeloma. Int. J. Mol. Sci. 2021, 22, 1096. [Google Scholar] [CrossRef]
- Krovi, S.H.; Gapin, L. Invariant Natural Killer T Cell Subsets-More Than Just Developmental Intermediates. Front. Immunol. 2018, 9, 1393. [Google Scholar] [CrossRef] [Green Version]
- Exley, M.A.; Wilson, B.; Balk, S.P. Isolation and functional use of human NKT cells. Curr. Protoc. Immunol. 2010, 119, 11–14. [Google Scholar] [CrossRef]
- Guidance on Pharmacological Research and Evaluation of Immune Cell Therapy Products (Trial Version). Available online: https://www.cde.org.cn/main/news/viewInfoCommon/0584963a84e01bb4d83022f559d22144 (accessed on 28 September 2022).
- Chemistry, Manufacturing, and Control (CMC) Information for Human Gene Therapy Investigational New Drug Applications (INDs). Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/chemistry-manufacturing-and-control-cmc-information-human-gene-therapy-investigational-new-drug (accessed on 28 September 2022).
- Considerations for the Development of Chimeric Antigen Receptor (CAR) T Cell Products. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/considerations-development-chimeric-antigen-receptor-car-t-cell-products (accessed on 28 September 2022).
- Guideline on Quality, Non-Clinical and Clinical Aspects of Medicinal Products Containing Genetically Modified Cells. Available online: https://www.ema.europa.eu/en/quality-non-clinical-clinical-aspects-medicinal-products-containing-genetically-modified-cells (accessed on 28 September 2022).
- Guideline On Human Cell-Based Medicinal Products. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-human-cell-based-medicinal-products_en.pdf (accessed on 28 September 2022).



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Du, Z.; Zhu, S.; Zhang, X.; Gong, Z.; Wang, S. Non-Conventional Allogeneic Anti-BCMA Chimeric Antigen Receptor-Based Immune Cell Therapies for Multiple Myeloma Treatment. Cancers 2023, 15, 567. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers15030567
Du Z, Zhu S, Zhang X, Gong Z, Wang S. Non-Conventional Allogeneic Anti-BCMA Chimeric Antigen Receptor-Based Immune Cell Therapies for Multiple Myeloma Treatment. Cancers. 2023; 15(3):567. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers15030567
Chicago/Turabian StyleDu, Zhicheng, Sumin Zhu, Xi Zhang, Zhiyuan Gong, and Shu Wang. 2023. "Non-Conventional Allogeneic Anti-BCMA Chimeric Antigen Receptor-Based Immune Cell Therapies for Multiple Myeloma Treatment" Cancers 15, no. 3: 567. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers15030567