
The Versatile Attributes of MGMT: Its Repair Mechanism, Crosstalk with Other DNA Repair Pathways, and Its Role in Cancer

Department of Biochemistry and Structural Biology, Greehey Children’s Cancer Research Institute, University of Texas Health San Antonio, San Antonio, TX 78229, USA
Cancers 2024, 16(2), 331; https://0-doi-org.brum.beds.ac.uk/10.3390/cancers16020331
Submission received: 20 December 2023
/
Revised: 9 January 2024
/
Accepted: 10 January 2024
/
Published: 11 January 2024
(This article belongs to the Special Issue Genome Maintenance in Cancer Biology and Therapy)
Abstract
:Simple Summary
This review offers a thorough examination of the potential role and structural properties of MGMT, the progression in targeting MGMT, and the interactions between MGMT and DNA repair pathways involved in processing DNA lesions.
Abstract
O6-methylguanine-DNA methyltransferase (MGMT or AGT) is a DNA repair protein with the capability to remove alkyl groups from O6-AlkylG adducts. Moreover, MGMT plays a crucial role in repairing DNA damage induced by methylating agents like temozolomide and chloroethylating agents such as carmustine, and thereby contributes to chemotherapeutic resistance when these agents are used. This review delves into the structural roles and repair mechanisms of MGMT, with emphasis on the potential structural and functional roles of the N-terminal domain of MGMT. It also explores the development of cancer therapeutic strategies that target MGMT. Finally, it discusses the intriguing crosstalk between MGMT and other DNA repair pathways.
1. Introduction
Chemical insults, both endogenous (e.g., S-adenosylmethionine) and exogenous (e.g., alkylating agents), constantly attack cellular DNA, resulting in DNA lesions [1,2,3,4]. Among these chemicals, alkylating agents play a significant role in causing DNA damage [5]. These agents are widely present in the environment, used as anticancer compounds in clinical settings [1,2,3,4,6,7,8,9,10], and can be endogenously generated during normal cellular processes. For example, S-adenosylmethionine, a methyl donor involved in various cellular reactions, has been shown to induce DNA methylation damage [3,11,12]. The attack of alkylating agents on DNA can lead to different types of lesions on the heterocyclic bases or backbone [3,5,13,14,15,16]. Most of these resulting adducts are mutagenic or toxic, prompting cells to develop various proteins for their detection and repair [13,17]. Interestingly, many of these alkylation lesions are repaired through direct removal of the adduct [5]. O6-alkylguanine-DNA alkyltransferase or O6-methylguanine-DNA methyltransferase, known as AGT or MGMT, is an enzyme responsible for the repair of alkylated DNA through a process known as “suicide” repair [3,4,13]. MGMT expression is primarily regulated through epigenetic modifications [18,19,20], and extensive research has indicated that reduced MGMT expression is attributed to the methylation of the CpG island that spans 762 base pairs and contains 98 CpG sites found in the MGMT promoter region [21,22,23,24]. Mechanistically, the primary function of MGMT involves the transfer of the methyl group located at the O6 site of guanine to cysteine residues located within the MGMT active site, thereby preventing mutations, cell death, and the development of tumors caused by alkylating agents [3,13]. Apart from being an avenue for cancer therapeutic strategies, MGMT has also been investigated as a valuable research tool for the specific labeling of proteins [5,13,25,26,27,28,29,30].
This review covers three main areas of focus. First, it explores the structural role of the N-terminal domain of MGMT. Second, it delves into the development of cancer therapeutic strategies that target MGMT. Lastly, it examines the crosstalk between MGMT and other DNA repair pathways. Readers are directed to several excellent reviews and articles that have been published [3,4,5,13,14,15,24,31,32,33,34,35,36,37,38,39].
2. Repair Mechanism
The initial fully characterized MGMT or AGT was the Ada gene product from E. coli [40,41]. It performs the repair of O6-methylguanine (m6G) in DNA through the direct transfer of the methyl group from the DNA to a cysteine residue within the protein itself (Figure 1) [40,41]. The inducibility of the Ada protein in response to alkylation damage facilitated its biochemical examination, enabling purification in substantial quantities after such treatment [42,43]. Subsequent investigations unveiled another MGMT in E. coli, Ogt [44,45]. Unlike the Ada protein, Ogt is not inducible by alkylating agents and has more limited expression levels. Both proteins are commonly referred to MGMT.
In the early 1990s, accumulating evidence indicated the existence of a similar protein in mammalian tissues, leading to the cloning of human MGMT (hMGMT) [46,47]. Studies have demonstrated that human MGMT is highly effective in repairing larger and bulky adducts, such as O6-[4-oxo-4-(3-pyridyl)butyl]guanine, which is formed by the tobacco-derived carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) [48], as well as seven- or four-carbon O6-2′-deoxyguanosine-alkyl-O6-2′-deoxyguanosine interstrand DNA cross-links [49,50]. However, it exhibits low efficiency in repairing O4-methylthymine [51,52]. For more information on MGMT substrates, please refer to reference [13].
MGMT is widely present in almost all organisms. It is easily identifiable due to the characteristic protein sequence surrounding the central Cys site that acts as the acceptor for alkyl groups: (I/V)PCHR(V/I)-(I/V) (Figure 1) [13]. Multiple structures of hMGMT have been elucidated, providing valuable insights into its functioning [53,54,55,56]. The structure of MGMT comprises two domains. The C-terminal domain encompasses the active site pocket and DNA binding region, interconnected by a hinge formed by the conserved residue Asn137 (amino acid numbers provided refer to hMGMT) (Figure 1) [53,54,55,56]. The N-terminal domain plays a vital role in maintaining structural stability, and can stabilize the complex formed when the two domains are expressed separately [57]. The initial published structure revealed a similar fold to the C-terminal domain of Ada (C-Ada: One of MGMTs in E. coli), even in the N-terminal domain, despite a lower sequence similarity between the two proteins [53](Figure 1). Another native structure, along with the benzylated form, uncovered an intriguing revelation—the N-terminus of hMGMT (N-hMGMT) contains a zinc(II) ion coordinated by Cys5, Cys24, His29, and His85, arranged tetrahedrally [54] (Figure 2). These four residues are conserved across mammalian sequences of the protein but are absent in yeast (S. cerevisiae) and bacterial homologues [58].
Further biochemical investigations have revealed that although the zinc(II) site in MGMT is located away from the reaction center, its binding to a zinc(II) ion effectively lowers the pKa of Cys145 compared to the apo protein, thereby enhancing the reactivity of the protein [59]. hMGMT utilizes a helix-turn-helix motif to interact with DNA, but exhibits an unusual behavior of specifically targeting the minor groove [55]. The recognition helix, characterized by hydrophobic residues, tightly fits into the minor groove with minimal sequence-specificity [55]. The structural findings have led to the proposal of a comprehensive mechanism for the activation and repair of Cys145. This mechanism involves a hydrogen bonding network that activates the Cys145 residue, with a crucial Tyr residue (Tyr114) playing a role in identifying the damaged guanine, and providing a proton to facilitate the removal of the alkyl adduct. Additionally, an arginine finger (Arg128) stabilizes the displacement of the base in the DNA helix (Figure 2) [55,60]. These residues are conserved across different MGMTs. The preference of hMGMT for larger aromatic substrates is evident in the benzylated structure, where the benzyl group, covalently attached to the active site cysteine, is positioned between Pro140, Ser159, and Tyr158 through hydrophobic interactions (Figure 2) [54]. In contrast, in C-Ada, the proline residue is replaced by alanine, and a glycine residue (Gly160) on the opposite side of the hMGMT pocket is substituted with tryptophan. This structural difference in C-Ada results in a narrower substrate binding pocket (Figure 1), which explains its inclination towards smaller lesions [58].
3. The Function of N-Ada and the Potential Role of N-hMGMT
The Ada protein consists of two domains: a 20 kDa N-terminal domain (N-Ada) and a 19 kDa C-terminal domain (C-Ada) containing 354 amino acids (Figure 1) [61,62]. In E. coli, the Ada protein serves multiple functions. N-Ada is responsible for repairing the Sp-configurated methylphosphotriester, while C-Ada repairs the highly mutagenic m6G and m4T [61,62,63]. Additionally, Ada acts as a transcriptional activator that induces the robust expression of the ada, alkA, alkB, and aidB genes, thereby regulating the adaptive response to methylation resistance [64]. This response occurs after transferring the methyl group from the Sp-methylphosphotriester to the Cys38 residue [63]. The methylated N-Ada transforms into a potent DNA binder, enabling it to recognize the promoter regions of the Ada regulon and recruit RNA polymerase to initiate the transcription of the aforementioned four genes [17,63,65,66,67]. This specific Cys38 residue demonstrates selective activation, for it is the only Cys ligand that is not involved in any hydrogen-bonding interactions, and exhibits higher nucleophilicity compared to other Cys residues (Figure 3) [63,68,69,70]. Additionally, this residue acts as a ligand to a zinc(II) ion present in the active site, which is also bound by three other Cys residues (Figure 3) [63,68,69,70]. A Zn cluster with four Cys acquires negative charges. Within this cluster, a Cys residue, potentially in a transiently dissociated state from the zinc(II) center, but remaining deprotonated, is capable of attacking a methylphosphotriester (Figure 3). This attack results in the transfer of the methyl group [63]. This zinc ion also plays an essential role to keep the structure of N-Ada [63].
Currently, no N-Ada homologues have been identified in humans, and there is no evidence to suggest that methylphosphotriesters are repaired in human cells. The adaptive response to the methylation challenge is not clearly observed in eukaryotes. All the residues identified as being associated with the activity and DNA binding of hMGMT are located in the C-terminal domain (Figure 2) [54,55,58]. Structural and biochemical studies of hMGMT indicate that N-hMGMT may primarily serve a passive structural role, rather than directly participating in the protein’s functional activity [53,54,55,56]. Previous biochemical studies indicated that hMGMT exhibits more similarity to Ogt than to Ada [71,72]. However, the published structures of hMGMT have revealed a similar fold to Ada [53,54,55,56] (Figure 1 and Figure 4). The role of N-hMGMT remains unclear. When the two domains are expressed separately, we found that N-hMGMT plays an important structural function in stabilizing the C-terminal domain of hMGMT (hMGMT-C) and forming the N-hMGMT/hMGMT-C complex [57]. Unexpectedly, we discovered that N-hMGMT exhibits Zn2+-dependent DNA repair activity in vitro, and can repair m6G DNA damage but not methylphosphotriester damage [57]. N-hMGMT shows very weak activity toward double-stranded DNA (dsDNA) containing m6G, while it is highly sensitive to MGMT inhibitor-O6-BenzylGuanine (O6-BG) [57], suggesting that N-hMGMT may prefer m6G in single-stranded DNA (ssDNA) or the dGTP pool rather than dsDNA. This conclusion is consistent with the crystal structures of hMGMT, which show that all the residues related to DNA binding are located in the hMGMT-C. Although the structural difference in C-Ada results in a smaller substrate binding pocket, this may explain why hMGMT is more sensitive to large DNA lesions such as O6-BG [58]. However, the active site of hMGMT-C is not located on the protein surface. How can O6-BG be recognized and located in the active site of hMGMT-C? Evidence suggests that the binding of zinc in N-hMGMT plays an essential role in recognizing O6-BG. N-hMGMT is more sensitive to O6-BG than hMGMT, and the loss of zinc results in the inactivation of N-hMGMT and the N-hMGMT/hMGMT-C complex (see Table 1 and Figure 2).
To explore the potential role of N-hMGMT, we employed protein structure prediction online server (Phyre2, Imperial College, London, UK). The results showed that the highest scoring template, with 99.6% confidence, indicates that the structure of N-hMGMT exhibits a ribonuclease H-like motif (Table 2). Genotoxic agents have the ability to cause damage to dsDNA, ssDNA, RNA, and the dGTP pool, leading to the formation of DNA and RNA adducts [14,74,75,76,77,78]. Interestingly, it has been observed that higher levels of methylation occur in RNA compared to DNA in human cells treated with N-methyl-N-nitrosourea (MNU) [79]. Mammalian cells have developed repair mechanisms to address alkylation damage in RNA [80,81]. The existence of RNA repair mechanisms serves as an important defense mechanism for cells, helping them mitigate dysfunctional RNA resulting from alkylated damage [76,77,78,82]. In hMGMT, the repair of RNA is impeded by a steric clash between the 2′-hydroxyl group and the Cα atom of Gly131. This clash hinders the ability of hMGMT-C to efficiently repair RNA lesions [5,56]. The structural characteristics of N-hMGMT suggest that it may have a propensity for repairing RNA lesions despite of the inefficiency of hMGMT-C, but additional research is required to establish its actual repair capabilities and preferences. It is unknown whether the alkylated N-hMGMT plays a similar role as alkylated N-Ada.
4. The Fate of Alkylated MGMT
Early reports suggested that the addition of an alkyl group to the Cys145 of hMGMT causes structural alterations, leading to its recognition by ubiquitin ligases and subsequent degradation by the proteasome [83,84,85]. This degradation process may be necessary for continuous repair, as the presence of alkylated MGMT or inactive mutants like C145A can interfere with the repair function of active MGMT molecules [86]. It has been proposed that the disruption of a salt bridge between Asn137 and the carbonyl oxygen of Met134, caused by a steric clash with the newly formed S-alkylcysteine, serves as the signal for this degradation [54]. These alterations destabilize the protein but have minimal impact on DNA binding [87,88].
Accumulating evidence suggests that alkylated MGMT functions as a transcriptional regulator, regulating the DNA damage response. hMGMT is found at active transcription sites, facilitated by interacting with RNA polymerase II, and it exhibits a preference for repairing m6G damage specifically in the transcription strands [89]. Upon alkylation, MGMT undergoes a conformational change that exposes VLWKLLKVV residues (codons 98 to 106), allowing it to bind to the estrogen receptor (ER), a critical transcription factor involved in regulating cell proliferation [90]. The binding of alkylated MGMT to ER prevents its interaction with steroid receptor coactivator-1, thereby inhibiting the activation of ER-regulated gene expression [90]. This suggests that alkylated MGMT functions as a transcription regulator, leading to cell cycle arrest. Additionally, alkylated MGMT suppresses the expression of deubiquitinating enzyme 3 (DUB3), thereby impacting chemoresistance in ovarian cancer [91]. It is currently unknown whether this effect is associated with alkylated N-hMGMT or alkylated hMGMT-C. Further research is necessary to elucidate this relationship. Previous studies have indicated that the expression of hMGMT can be modulated by glucocorticoids and partial hepatectomy [92,93]. The promoter region of hMGMT has been disclosed to contain estrogen-responsive elements and antioxidant-responsive elements recently, and the expression of hMGMT can be influenced by estrogen and nuclear factor-erythroid 2-related factor-2 (Nrf-2) [94]. The adaptive response triggered by alkylating agents is not clearly observed in mammalian cells [13]. However, clinical data has shown that chemotherapy utilizing alkylating agents may lead to an up-regulation of hMGMT expression in gliomas, although the underlying mechanisms remain unclear [24,95,96]. The factors that determine the different fates of alkylated MGMT remain unclear. One potential clue is that alkylated hMGMT has a half-life of several hours, suggesting the involvement of regulation mechanisms within this time period [13]. Further research is needed to uncover the conformational changes of alkylated hMGMT, its interactions with other proteins, and the detailed mechanism of ubiquitination. Another clue pertains to the post-translational modification (PTM) of hMGMT. Protein kinases can phosphorylate hMGMT [97], and PARP1 can cause the PARylation of MGMT in response to alkylating agent treatment [98,99]. While these modifications have an impact on its function [98,99,100], additional research is required to determine whether these modifications affect the stability and conformational alterations of hMGMT.
5. The Role of hMGMT in Cancer Prevention and Chemotherapy
The induction of O6-alkylGuanine (O6-alkylG) by alkylating agents can result in significant biological consequences such as mutagenicity, cytotoxicity, and carcinogenesis if not repaired by MGMT [13]. The expression of hMGMT in various cultured cell lines has been shown to effectively reduce GC to AT mutations and cytotoxicity caused by alkylating agents [36,101,102,103,104,105]. Several excellent reviews have focused on the role of hMGMT in cancer prevention and chemotherapy [13,24,37,106,107]. We briefly highlight the important findings from animal models and clinical trials here. N-nitroso compounds, which are present in many foods, play a crucial role in the development of colorectal cancer [107]. The study utilized a transgenic mouse model where mice had high expression of MGMT in the colon [108]. These mice demonstrated reduced rates of aberrant crypt foci (ACF) formation following intraperitoneal administrations of the alkylating agent azoxymethane (AOM). Additionally, the overexpression of MGMT provided protection against G:C to A:T mutations in the KRAS oncogene induced by AOM [108]. Consistent with these findings, depleting MGMT in rats using the potent MGMT inhibitor B6G resulted in an increased frequency of colonic tumors following AOM treatment [109]. MGMT knockout mice, in an inflammation-driven colon carcinogenesis model induced by AOM and dextran sodium sulfate, exhibit high susceptibility to colon tumorigenesis [110,111]. NNK, a carcinogenic N-nitrosamine found in tobacco and tobacco smoke, leads to the development of lung tumors [112,113]. However, A/J mice with high levels of MGMT display smaller lung tumors and a lower frequency of K-Ras mutation, indicating reduced susceptibility to NNK-induced carcinogenesis [114,115]. The protective role of MGMT has also been observed in animal models of thymic lymphoma [116,117], hepatocellular carcinoma [118,119,120], skin cancer [121,122,123], and brain cancer [124,125,126].
DNA alkylating agents, such as methylating and chloroethylating agents, have been used in cancer therapy for over four decades [31,37,39]. Among these agents, the m6G and O6-chloroethylG products are the main toxic lesions in most cases, particularly at lower doses [39]. However, at higher doses of these agents, N-alkyl purines also contribute to cellular cytotoxicity, and the significance of MGMT in terms of overall cytotoxicity diminishes [39]. The MGMT gene plays a critical role in repairing DNA damage caused by alkylating agents, thereby promoting resistance to chemotherapy. The methylation of the MGMT promoter leads to the reduced or absent expression of hMGMT by inhibiting transcription, thereby increasing sensitivity to alkylating agents [127]. The methylated MGMT promoter has garnered substantial support as a predictive marker for the effectiveness of temozolomide (TMZ), an alkylating agent used in treating glioblastoma and low-grade gliomas. Multiple studies have provided evidence supporting this notion [128,129,130,131,132,133,134,135]. Additionally, two distinct clinical trials have demonstrated that the methylation status of the MGMT promoter can serve as an indicator of the prognosis of glioma patients treated with TMZ, but not when treated with the chemotherapy regimen comprising procarbacine, lomustine (BCNU), and vincristine (PCV) [136,137]. Moreover, clinical data has revealed a significant difference in the overall median survival among patients with malignant astrocytoma treated with BCNU, depending on their MGMT levels [138]. This finding highlights that MGMT can also function as a predictive marker for the treatment of malignant astrocytoma with chloroethylating agents. In clinical trials involving anaplastic oligodendroglioma patients, it has been observed that the methylation of the MGMT promoter is associated with improved overall survival and progression-free survival, regardless of whether patients underwent radiotherapy alone or sequential radiotherapy and chemotherapy with PCV [139]. The combination of MGMT promoter methylation and an isocitrate dehydrogenase 1 (IDH1) mutation was found to reduce the risk of progression in anaplastic glioma patients [140].
Emerging evidence indicates that hMGMT plays a role in the chemoresistance of cisplatin. In ovarian cancer, MCL1 is stabilized by MGMT-activated DUB3, leading to resistance to platinum/paclitaxel-based chemotherapy in ovarian cancer cells [91]. Conversely, in gastric cancer, a study has shown that hMGMT inhibits the autophagy-related gene (ATG) 4B, resulting in the suppression of autophagy. Cisplatin, on the other hand, counteracts MGMT-mediated autophagy suppression, thereby reducing chemosensitivity in gastric cancer. High MGMT expression and low ATG4B expression are significantly associated with survival in gastric cancer [141]. It is noteworthy that the impact of MGMT levels appears to differ in ovarian cancer and gastric cancer. However, further investigations are warranted to validate these findings.
6. The Development of Strategies That Target MGMT
hMGMT plays a crucial role in repairing DNA damage induced by methylating agents like TMZ, as well as chloroethylating agents such as BCNU, ACNU, and MeCCNU, thereby promoting resistance to chemotherapy using these agents [3,4,13,31,39]. hMGMT activity has also been associated with the resistance to 6-thioguanine that a medication used to treat leukemia [142]. 6-thioguanine is a poor substrate for hMGMT but binds to hMGMT after it is methylated to form S6-methylthioguanine [143,144,145,146]. Therefore, it is plausible that the binding of 6-thioguanine to hMGMT contributes to cellular resistance to this compound. Based on these findings, inhibiting MGMT shows potential in overcoming resistance to alkylating agents [3,4,13,31,39]. Numerous compounds that inhibit MGMT activity have been synthesized and utilized as adjuvants to enhance the cytotoxic effects of alkylating agents.
6.1. Non Cancer-Selective MGMT Inhibitors
O6-BG and O6-(4-bromothenyl)guanine (O6-4-BTG) are analogs of m6G that serve as irreversible pseudosubstrates of MGMT [147,148,149,150]. These compounds have been utilized as inhibitors of MGMT in clinical trials [13,39]. O6-BG has gained significant use in sensitizing glioma cells to the alkylating agent TMZ [13,39,151,152]. It possesses the ability to penetrate the blood–brain barrier and deactivate MGMT [24]. Clinical trials combining O6-BG with TMZ have shown promise in delaying brain tumor recurrence and increasing survival time [153,154,155,156]. However, it is important to note that this approach carries an elevated risk of side effects such as hydrocephalus, cerebrospinal fluid leak, and brain infection [157]. Additionally, the positive effects of this treatment have been observed in other types of tumors, including melanoma, colon cancer, and lymphoma [24,158]. O6-4-BTG serves as a highly potent MGMT inhibitor, surpassing the effectiveness of O6-BG [149,150]. Data from in vivo and in vitro studies across various tumor types have demonstrated that O6-4-BTG efficiently deactivates MGMT, resulting in a notable enhancement of tumor sensitivity to TMZ [159,160,161,162]. Phase II clinical trials have further supported these findings, demonstrating that the combination of O6-4-BTG and TMZ more effectively inhibits hMGMT compared to TMZ alone in patients with melanoma, prostate cancer, primary CNS cancer, and colorectal cancer [163,164,165,166]. However, it is worth noting that both O6-BG and O6-4-BTG have been associated with increased myelosuppression, without significantly improving the response rate to TMZ [24,39,155,156,157,167].
Due to the limited water solubility of O6-BG, it is necessary to develop more soluble derivatives to enhance its bioavailability. One approach is the meta-substitution of the aminomethyl group on the benzyl moiety of O6-BG. This modification enhances the water solubility of the compound and results in a more potent inhibitory activity against MGMT [168]. Several other 6-(benzyloxy)pyrimidine derivatives have been synthesized as potential inhibitors of MGMT [13,169]. In addition to the previously mentioned MGMT inhibitors, other types of inhibitors have been discovered. Acrolein and chloromethyltriazoles are highly reactive molecules with nucleophilic sites that can react with cysteine residues, effectively inhibiting MGMT [170]. Another inhibitor, 6-carboxyfluorescein, acts on MGMT in a non-covalent manner [171]. Lipoic acid, a natural compound containing a disulfide structure, has demonstrated potent MGMT inhibition and can enhance the cytotoxicity of TMZ in colorectal tumor cells that are resistant to TMZ [172]. Nitric oxide and disulfiram function by inactivating hMGMT through their interaction with the active Cys145 residue of the protein [173,174]. These studies have provided clear evidence of the correlation between the inhibitory effects of these compounds on MGMT and the increased cytotoxicity of chemotherapeutic drugs [37].
The primary concern regarding the use of non-cancer selective MGMT inhibitors is the increased risk of myelosuppression in bone marrow cells and other normal cells, which can lead to severe hematological toxicity such as leukemia and myelodysplastic syndrome [13,24,169,175]. To address these concerns and for various other reasons, researchers are actively developing cancer-selective inhibitors.
6.2. Cancer-Selective MGMT Inhibitors
The primary strategy for developing a cancer-specific MGMT inhibitor involves modifying the inhibitor with tumor-targeting groups [13,39]. The objective of this approach is to prevent MGMT inhibition in normal tissues while sensitizing cancer cells to chemotherapy [13,39]. Aerobic glycolysis is a prevalent metabolic characteristic observed in numerous tumors. In light of this, the conjugation of a glucose group to the MGMT inhibitor represents a promising concept for the development of cancer-selective MGMT inhibitors [176]. Studies have reported the high efficacy of both O6-BG-Glucose (O6-BG-Glu) and O6-BTG-Glu in inhibiting MGMT in various cancer cell lines, including T98G glioblastoma [150,176,177]. Furthermore, these agents have been shown to enhance the cytotoxic effect of temozolomide [150,176,177]. However, it should be noted that glucose conjugates are susceptible to transport out of the cell through ATP-binding cassette (ABC) transporter-mediated efflux, which can affect the efficiency of MGMT inhibition [177]. Folate receptors have shown great potential as carriers for the targeted delivery of chemotherapeutic drugs, particularly in squamous cell carcinomas, ovarian cancers, and certain non-small cell lung carcinomas, where they are frequently overexpressed [178,179]. The conjugation of a folate group to the MGMT inhibitor is used to develop specific MGMT inhibitors. O4-benzylfolate has demonstrated an MGMT inhibitory potency approximately 30 times higher than O6-BG. It has also been found to be effective in deactivating the P140K mutant MGMT, which is resistant to O6-BG-mediated inhibition [180]. Additionally, the efficacy of O4-benzylfolate in enhancing BCNU-induced cell death is dependent on the expression of an α-folate receptor [179]. In another approach, the synthetic 3′-γ-folate ester of O6-benzyl-2′-deoxyguanosine not only increased MGMT inhibitory activity in tumor cells but also sensitized HT29 and A549 cells to BCNU cytotoxicity [179]. Furthermore, this modification improved the water solubility of the inhibitor [179]. However, none of these cancer-selective MGMT inhibitors enters clinical trial.
Prodrugs have the potential to enhance tumor specificity and improve pharmacokinetic profiles [13,37]. Many solid tumors exhibit a characteristic of hypoxia, and hypoxia-activated O6-BG prodrugs have already been developed and utilized [181,182]. In particular, the release of β-glucuronidase is commonly observed from necrotic tumor cells. Therefore, designing O6-BG prodrugs that are substrate-related to β-glucuronidase could be a promising strategy to exploit [183].
6.3. Local Drug Delivery
Drug delivery plays a critical role in improving targeted therapy by utilizing diverse delivery systems and strategies to enhance the effectiveness and specificity of therapeutic agents [184]. Local drug delivery can achieve targeted therapy. Gliadel (BCNU wafers) marked the initial clinical application of polymer drug delivery in the treatment of brain tumors. It involves the insertion of BCNU wafers into the resection cavity of patients following surgery [185,186]. These wafers gradually degrade, enabling localized delivery of BCNU to the target area [187]. TMZ was encapsulated within a biologically inert matrix for localized administration to patients with GBM. This encapsulation strategy demonstrated superior efficacy compared to standard therapy alone, resulting in a remarkable increase in overall survival of up to 33 weeks [188]. An injectable enzyme-responsive hydrogel was developed, capable of delivering TMZ and O6-BG. This hydrogel demonstrated effectiveness in reducing the recurrence of TMZ-resistant glioma after surgery, while also enhancing the inhibitory efficiency against tumors [189].
Nanoparticle-based delivery offers unique properties that enable targeted delivery to specific cells or tissues [190]. In a recent study, a combination of O6-BG formulation with a redox-responsive theranostic superparamagnetic iron oxide nanoparticle (SPION) platform was employed to enhance the intracellular delivery of O6-BG to glioblastoma multiforme (GBM) cells while minimizing drug accumulation in healthy tissues [191]. This improved formulation of O6-BG showed a significant decrease in MGMT activity and enhanced the cytotoxic effect of TMZ in vitro. Additionally, in an orthotopic primary human GBM xenograft mouse model, it resulted in a three-fold increase in survival compared to untreated controls [191]. Another formulation involved nanoparticles (NPs) coated with a pH-sensitive polymer and a modified analog of MGMT inhibitor, specifically dialdehyde-modified O6-benzylguanosine (DABGS). This formulation demonstrated a remarkable inhibition of MGMT activity and enhanced the cytotoxicity of TMZ in vitro [192]. Furthermore, a systematic nano delivery platform (SCL) was developed to encapsulate the p53 gene, facilitating the targeted delivery of p53 to brain tumor tissue. This delivery system successfully depleted MGMT levels and significantly enhanced the therapeutic efficacy of TMZ [193]. The SCL exhibited a notable improvement in the therapeutic effect of TMZ. The CRISPR/Cas9 system, in conjunction with lipid-polymer hybrid nanoparticles (LPHNs-cRGD), was employed to achieve an efficient delivery of pCas9/MGMT plasmids into glioblastoma cells. This delivery approach successfully downregulated MGMT expression levels, resulting in increased chemotherapy sensitivity of tumor cells [194].
In addition, gene delivery can be utilized to efficiently deliver genes to the desired cells. Retroviral and lentiviral vectors expressing inhibitor-resistant MGMT mutants have been utilized to protect against the myelosuppressive toxicity of chemotherapy drugs and prevent therapy-related secondary hematopoietic malignancies [195,196,197,198,199,200].
6.4. Targeting the Expression of hMGMT
Several strategies have been developed to target the expression of MGMT. Among them, the identification of microRNAs (miRNAs) that regulate MGMT expression by degrading MGMT mRNA shows promise as an innovative treatment approach to enhance TMZ sensitivity in patients with unmethylated MGMT [38]. Notably, miRNAs such as miR-142-3p, miR-181d, miR-370-3p, and others have been discovered to downregulate MGMT expression and enhance sensitivity to TMZ in GBM cell lines [38,201,202,203,204,205,206,207]. Another promising strategy is the use of siRNA to target MGMT. The combination of TMZ with the MGMT–siRNA/liposome complex has shown a strong synergistic antitumor effect [24,208]. A small-molecule compound, EPIC-0412, was discovered to enhance the chemotherapeutic effect of TMZ by epigenetically silencing the expression of MGMT. It achieved this by targeting two key pathways: the p21-E2F1 DNA damage repair axis and the ATF3-p-p65-MGMT axis [209]. These findings provide compelling evidence for the potential of combining epigenetic drugs to enhance sensitization to TMZ in GBM patients.
6.5. Others
Autoantibodies against MGMT were detected in patients’ serum [212]. The researchers screened the most responsive peptides using these autoantibodies and discovered that these peptides conferred resistance to TMZ in glioma cells both in vivo and in vitro [213]. This finding suggests that monoclonal antibodies targeting these peptides could serve as a novel strategy to overcome resistance in GBM cases with unmethylated MGMT promoters to alkylating agents. The new agent-KL-50 has been found to induce cell killing selectively in MGMT-silenced tumors, independent of mismatch repair (MMR). It creates a dynamic DNA lesion that can be repaired by MGMT. However, in MGMT-deficient conditions, this lesion slowly evolves into an interstrand cross-link, leading to MMR-independent cell death. Notably, this process exhibits low toxicity in both in vitro and in vivo settings [214]. This agent represents a novel approach in designing chemotherapeutics that exploit specific DNA repair defects. All of the mentioned strategies are summarized in Table 3.
7. The Crosstalk of MGMT with Other DNA Repair Pathways
The attack of alkylating agents on DNA can lead to various types of lesions on the heterocyclic bases or backbone. Repairing methylated DNA adducts involves several pathways, primarily the base excision repair (BER) pathway, the family of AlkB homolog proteins (ALKBH), and MGMT (Figure 5) [5,14,30]. The BER pathway plays a critical role in repairing the main N-alkylation DNA adducts, such as N3-methyladenine, N3-methylguanine, and N7-methylguanine, with the first step of this process involving the alkyladenine-DNA glycosylase (AAG, MPG) [5,14,30,215]. Members of the ALKBH family are responsible for the demethylation of N1-methyladenine and N3-methylcytosine. In cases where the removal of m6G fails, the resulting m6G:T mispair is recognized by the MMR system, leading to a variety of downstream effects [5,14,30]. However, it remains unclear how the different repair pathways collaborate and compete with each other.
7.1. MGMT and BER
MGMT and BER are the primary pathways to process DNA lesions induced by alkylating agents. As of now, no glycosylase capable of recognizing m6G has been discovered. AAG, on the other hand, specifically recognizes N-alkylated purines and does not compete with MGMT. Nevertheless, there are reports suggesting that MGMT activity is regulated by the BER protein-PARP, which plays a critical role in BER for processing N-alkylpurines [30]. The PARP-mediated PARylation of MGMT, induced by alkylating agents, enhances its binding to chromatin and its ability to facilitate the removal of m6G adducts from DNA (Figure 6) [98,99]. This indicates a significant interplay between PARP and MGMT in the repair process.
7.2. MGMT and Nucleotide Excision Repair (NER)
The NER pathway plays a crucial role in repairing bulky helix-distorting DNA adducts, like cyclobutene-pyrimidine dimers induced by UV light [216]. Experimental evidence suggests that cells expressing both MGMT and NER proteins efficiently repair O6-ethylG, indicating a collaborative effort between MGMT and NER in processing this type of DNA damage [217]. The proposed mechanism involves NER proteins opening up the tightly packed chromatin, thereby aiding MGMT in locating and addressing the DNA adducts [217]. In a recent report, it was discovered that MGMT also collaborates with NER in processing O6-carboxymethylG, which has implications in colorectal cancer development and is associated with meat consumption (Figure 6) [218]. Interestingly, the expression of hMGMT in E. coli has been observed to hinder the removal of m4T by NER, likely due to the significantly slower removal of m4T by hMGMT [52]. Notably, alkyltransferase-like proteins (ATLs) that share sequence similarities with the C-terminal domain of MGMT are highly conserved across all three domains of life. When ATLs bind to O6-alkylG in DNA, they form a complex that is readily recognized by NER to promote the repair of O6-alkylG by NER, effectively obstructing the repair process by MGMT [13,219,220].
7.3. MGMT and MMR
The expression of MGMT plays a critical role in preventing cell death induced by alkylating agents, since it directly converts m6G back to G. Additionally, MMR also has a significant impact on generating cytotoxic effects triggered by m6G lesions [221]. Deficiency in MMR results in resistance against these effects, both in vitro and in vivo [222,223]. Unrepaired m6G is highly stable and tends to pair with Thymine (T) instead of G. This m6G:T mispair is extremely mutagenic, and can be recognized by the MSH2/MSH6 heterodimer of the MMR pathway [224]. The persistence of m6G on the template strand leads to the formation of m6G:T mispairs during MMR-directed strand resynthesis, initiating repeated cycles of futile repair [30,221,225,226]. Consequently, unproductive replication cycles across m6Gs create unreplicated gaps that interfere with DNA replication in the subsequent S-phase, generating double-strand breaks (DSBs) that ultimately cause cell cycle arrest and cell death (Figure 5) [30,221,225,226]. The futile cycle model finds support through in vitro experiments, which demonstrate that the cytotoxicity of m6G takes place during the second cell cycle following treatment [221,225,227]. Circular DNA substrates containing m6G/T mismatches, rather than regular G/T mismatches, elicit a MMR-dependent preferential recruitment of ATR-ATRIP, leading to ATR activation (Figure 5) [228]. The observation that the MSH2G674A or MSH6T1217D missense mutants are unable to function in MMR but can still bind to mismatches and initiate apoptosis in response to m6G lesions supports the notion that MSH2-MSH6 complexes serve as DNA damage sensors [229,230]. This suggests that the excision of DNA lesions is not necessary for the DNA damage response function of MMR. These findings indicate that ATR activation does not require abortive excision and resynthesis cycles. This direct signaling model proposes that MMR proteins serve as a protein scaffold at m6G/T lesions, facilitating the direct recruitment and activation of a global damage response (Figure 5) [221]. The two models of m6G-induced cell death are consistent with the following observations: in the absence of MGMT, the MMR pathway is essential for recognizing m6G/T mismatches, leading to the induction of DNA strand breaks and ultimately triggering m6G-induced cell death. However, the direct signaling model fails to explain why an MMR protein-scaffold at m6G/T in the first S-phase does not lead to cell cycle arrest or cell death only after the second S-phase. One possible resolution could entail a combination of these two mechanisms, each contributing to a different outcome [221]. This proposition finds support in data obtained from human pluripotent stem cells (hPSCs). In hPSCs, the apoptosis response induced by an alkylating agent occurs within a few hours, whereas no observable downstream effectors activation is induced by the MMR-signaling complex [221,231,232].
The data obtained from Xenopus laevis eggs, where plasmid DNA was exposed to alkylating agents under non-replicating conditions, suggest that concurrent BER and MMR processes on the same DNA molecule may inadvertently lead to the formation of DSBs [233]. This occurrence arises when the repair intermediates of BER and MMR pathways encounter each other, and it may represent an additional mechanism for TMZ-induced cytotoxicity in non-dividing or quiescent cells [233]. This phenomenon is referred to as the “Repair Accident” model [234].
7.4. MGMT and DSB Repair
Concomitant with DNA replication, MMR gives rise to DSBs induced by m6G lesions, which are responsible for provoking apoptosis signaling [30,39,127]. Considering the crucial role of DSBs in m6G induced cytotoxicity, DSB repair is expected to significantly impact m6G-induced chromosomal changes. This is supported by the finding that m6G lesions caused higher aberration frequencies in cells with ATM deficiency [235]. Thus, MGMT serves as a key defense against clastogenicity by O6-methylating agents, acting in concert with MMR, BER, and DSB repair (Figure 5) [30]. After DSBs are formed, they undergo repair through homologous recombination (HR) or non-homologous end joining (NHEJ). BRCA2 plays a crucial role in HR and interacts with hMGMT to facilitate the degradation of methylated or benzylated hMGMT [236].
8. Conclusions
Biochemical studies and crystal structures of MGMT reveal the repair mechanism of hMGMT. Additionally, cell-based assays and animal studies unveil the cancer prevention function of MGMT. Interestingly, when the two domains of hMGMT are expressed separately, N-hMGMT demonstrates Zn2+-dependent DNA repair activity in vitro, effectively repairing m6G DNA damage but not methylphosphotriester damage [57]. This raises questions about the physiological significance of N-hMGMT and how it repairs m6G. Since all the residues responsible for DNA binding and repair are located in hMGMT-C [54,55,58], the mechanism by which m6G is repaired by N-hMGMT remains a fascinating aspect to explore. Notably, the active site of hMGMT-C is buried inside the protein [54,55], which prompts the question of how O6-BG can be recognized and rapidly located in the active site of hMGMT-C. The fate of alkylated MGMT is still ambiguous. Evidence suggests that alkylated MGMT will be ubiquitinated and degraded [83,84,85], other accumulating evidence shows that alkylated MGMT becomes a transcription regulator to regulate the expression of genes [90,91]. The mechanisms governing the diverse outcomes of alkylated MGMT remain unclear.
MGMT inhibitors, despite promising preclinical results, have not exhibited significant benefits in clinical settings for tumor patients treated with alkylating agents. The main challenge is the increased risk of severe hematological toxicity [13,24,169,175], limiting widespread clinical application. Researchers explore alternative strategies like cancer-selective inhibitors, MGMT expression downregulation, autoantibodies, and localized drug delivery to attenuate MGMT activity and minimize normal cell exposure [13,24,37,39]. While showing promise in cells and animal models, further research and clinical trials are needed to refine these strategies for enhanced efficacy in alkylating agent treatments with minimal harm to healthy tissues.
Alkylating agents exert their effects by inducing various types of DNA lesions, which are repaired by multiple pathways, including the BER pathway, ALKBH, and MGMT [5,14,30]. However, our increasing understanding of the mechanisms of cell killing by alkylating agents reveals that certain cancers, such as gliomas and malignant melanomas, may inherently exhibit resistance to a wide range of these and other anticancer drugs. To address this challenge, an integrated strategy involving MGMT inhibition in combination with cancer chemotherapy is likely necessary. This approach combines the inactivation of MGMT with the inhibition of other repair pathways that protect against the toxicity induced by methylating and chloroethylating agents, along with targeting downstream drug resistance factors [39]. The discovery of crosstalk between MGMT and other DNA repair pathways opens new avenues for improving the clinical response to treatment. For example, the PARylation of MGMT by PARP can regulate the activity of MGMT, potentially enhancing the sensitivity of cancer patients when combining alkylating agents with PARP inhibitors [98,99]. Additionally, the identification of alkylated MGMT and MGMT inhibitors promoting the degradation of BRCA2 presents a promising strategy for targeting homologous recombination proficient cancers with MGMT inactivators in conjunction with DNA crosslinking agents [236]. These novel insights hold significant potential for developing more effective and tailored approaches to cancer treatment, with the aim of overcoming drug resistance and improving therapeutic outcomes for patients. However, further research and clinical studies are essential to validate and refine these strategies before their implementation in clinical practice.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available in this article.
Acknowledgments
We are grateful to Patrick Sung at UT health science center for providing financial support and instructions, Cody M. Rogers and Youngho Kwon at UT health science center for reviewing and editing the manuscript, and Richard Fang at the University of Texas in Dallas for the manuscript editing. Y.K. is supported by R50 CA265315.
Conflicts of Interest
The author declares no conflict of interest.
References
- Lindahl, T. Instability and decay of the primary structure of DNA. Nature 1993, 362, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Friedberg, E.C. Out of the shadows and into the light: The emergence of DNA repair. Trends Biochem. Sci. 1995, 20, 381. [Google Scholar] [CrossRef] [PubMed]
- Pegg, A.E. Repair of O(6)-alkylguanine by alkyltransferases. Mutat. Res. 2000, 462, 83–100. [Google Scholar] [CrossRef]
- Pegg, A.E.; Dolan, M.E.; Moschel, R.C. Structure, function, and inhibition of O6-alkylguanine-DNA alkyltransferase. Prog. Nucleic Acid Res. Mol. Biol. 1995, 51, 167–223. [Google Scholar] [CrossRef]
- Mishina, Y.; Duguid, E.M.; He, C. Direct reversal of DNA alkylation damage. Chem. Rev. 2006, 106, 215–232. [Google Scholar] [CrossRef]
- Hecht, S.S. DNA adduct formation from tobacco-specific N-nitrosamines. Mutat. Res. 1999, 424, 127–142. [Google Scholar] [CrossRef]
- Hurley, L.H. DNA and its associated processes as targets for cancer therapy. Nat. Rev. Cancer 2002, 2, 188–200. [Google Scholar] [CrossRef]
- Rajski, S.R.; Williams, R.M. DNA Cross-Linking Agents as Antitumor Drugs. Chem. Rev. 1998, 98, 2723–2796. [Google Scholar] [CrossRef] [PubMed]
- Sedgwick, B. Repairing DNA-methylation damage. Nat. Rev. Mol. Cell Biol. 2004, 5, 148–157. [Google Scholar] [CrossRef]
- Vaughan, P.; Lindahl, T.; Sedgwick, B. Induction of the adaptive response of Escherichia coli to alkylation damage by the environmental mutagen, methyl chloride. Mutat. Res. 1993, 293, 249–257. [Google Scholar] [CrossRef]
- Barrows, L.R.; Magee, P.N. Nonenzymatic methylation of DNA by S-adenosylmethionine in vitro. Carcinogenesis 1982, 3, 349–351. [Google Scholar] [CrossRef]
- Rydberg, B.; Lindahl, T. Nonenzymatic methylation of DNA by the intracellular methyl group donor S-adenosyl-L-methionine is a potentially mutagenic reaction. EMBO J. 1982, 1, 211–216. [Google Scholar] [CrossRef]
- Pegg, A.E. Multifaceted roles of alkyltransferase and related proteins in DNA repair, DNA damage, resistance to chemotherapy, and research tools. Chem. Res. Toxicol. 2011, 24, 618–639. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, M.D.; Pittman, D.L. Methylating agents and DNA repair responses: Methylated bases and sources of strand breaks. Chem. Res. Toxicol. 2006, 19, 1580–1594. [Google Scholar] [CrossRef] [PubMed]
- Margison, G.P.; Povey, A.C.; Kaina, B.; Santibanez Koref, M.F. Variability and regulation of O6-alkylguanine-DNA alkyltransferase. Carcinogenesis 2003, 24, 625–635. [Google Scholar] [CrossRef]
- Beranek, D.T. Distribution of methyl and ethyl adducts following alkylation with monofunctional alkylating agents. Mutat. Res. 1990, 231, 11–30. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, T.; Sedgwick, B.; Sekiguchi, M.; Nakabeppu, Y. Regulation and expression of the adaptive response to alkylating agents. Annu. Rev. Biochem. 1988, 57, 133–157. [Google Scholar] [CrossRef]
- Fornace, A.J., Jr.; Papathanasiou, M.A.; Hollander, M.C.; Yarosh, D.B. Expression of the O6-methylguanine-DNA methyltransferase gene MGMT in MER+ and MER- human tumor cells. Cancer Res. 1990, 50, 7908–7911. [Google Scholar] [PubMed]
- Bouras, E.; Karakioulaki, M.; Bougioukas, K.I.; Aivaliotis, M.; Tzimagiorgis, G.; Chourdakis, M. Gene promoter methylation and cancer: An umbrella review. Gene 2019, 710, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Kroes, R.A.; Erickson, L.C. The role of mRNA stability and transcription in O6-methylguanine DNA methyltransferase (MGMT) expression in Mer+ human tumor cells. Carcinogenesis 1995, 16, 2255–2257. [Google Scholar] [CrossRef]
- Gardiner-Garden, M.; Frommer, M. CpG islands in vertebrate genomes. J. Mol. Biol. 1987, 196, 261–282. [Google Scholar] [CrossRef]
- Costello, J.F.; Futscher, B.W.; Tano, K.; Graunke, D.M.; Pieper, R.O. Graded methylation in the promoter and body of the O6-methylguanine DNA methyltransferase (MGMT) gene correlates with MGMT expression in human glioma cells. J. Biol. Chem. 1994, 269, 17228–17237. [Google Scholar] [CrossRef]
- Watts, G.S.; Pieper, R.O.; Costello, J.F.; Peng, Y.M.; Dalton, W.S.; Futscher, B.W. Methylation of discrete regions of the O6-methylguanine DNA methyltransferase (MGMT) CpG island is associated with heterochromatinization of the MGMT transcription start site and silencing of the gene. Mol. Cell. Biol. 1997, 17, 5612–5619. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Zhang, L.; Wei, Q.; Shao, A. O(6)-Methylguanine-DNA Methyltransferase (MGMT): Challenges and New Opportunities in Glioma Chemotherapy. Front. Oncol. 2019, 9, 1547. [Google Scholar] [CrossRef]
- Juillerat, A.; Gronemeyer, T.; Keppler, A.; Gendreizig, S.; Pick, H.; Vogel, H.; Johnsson, K. Directed evolution of O6-alkylguanine-DNA alkyltransferase for efficient labeling of fusion proteins with small molecules in vivo. Chem. Biol. 2003, 10, 313–317. [Google Scholar] [CrossRef]
- Damoiseaux, R.; Keppler, A.; Johnsson, K. Synthesis and applications of chemical probes for human O6-alkylguanine-DNA alkyltransferase. Chembiochem 2001, 2, 285–287. [Google Scholar] [CrossRef]
- Keppler, A.; Gendreizig, S.; Gronemeyer, T.; Pick, H.; Vogel, H.; Johnsson, K. A general method for the covalent labeling of fusion proteins with small molecules in vivo. Nat. Biotechnol. 2003, 21, 86–89. [Google Scholar] [CrossRef] [PubMed]
- Keppler, A.; Kindermann, M.; Gendreizig, S.; Pick, H.; Vogel, H.; Johnsson, K. Labeling of fusion proteins of O6-alkylguanine-DNA alkyltransferase with small molecules in vivo and in vitro. Methods 2004, 32, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Keppler, A.; Pick, H.; Arrivoli, C.; Vogel, H.; Johnsson, K. Labeling of fusion proteins with synthetic fluorophores in live cells. Proc. Natl. Acad. Sci. USA 2004, 101, 9955–9959. [Google Scholar] [CrossRef]
- Kaina, B.; Christmann, M.; Naumann, S.; Roos, W.P. MGMT: Key node in the battle against genotoxicity, carcinogenicity and apoptosis induced by alkylating agents. DNA Repair 2007, 6, 1079–1099. [Google Scholar] [CrossRef]
- Gerson, S.L. MGMT: Its role in cancer aetiology and cancer therapeutics. Nat. Rev. Cancer 2004, 4, 296–307. [Google Scholar] [CrossRef]
- Tubbs, J.L.; Pegg, A.E.; Tainer, J.A. DNA binding, nucleotide flipping, and the helix-turn-helix motif in base repair by O6-alkylguanine-DNA alkyltransferase and its implications for cancer chemotherapy. DNA Repair 2007, 6, 1100–1115. [Google Scholar] [CrossRef]
- Eker, A.P.; Quayle, C.; Chaves, I.; van der Horst, G.T. DNA repair in mammalian cells: Direct DNA damage reversal: Elegant solutions for nasty problems. Cell. Mol. Life Sci. 2009, 66, 968–980. [Google Scholar] [CrossRef] [PubMed]
- Dalhus, B.; Laerdahl, J.K.; Backe, P.H.; Bjoras, M. DNA base repair--recognition and initiation of catalysis. FEMS Microbiol. Rev. 2009, 33, 1044–1078. [Google Scholar] [CrossRef]
- Sharma, S.; Salehi, F.; Scheithauer, B.W.; Rotondo, F.; Syro, L.V.; Kovacs, K. Role of MGMT in tumor development, progression, diagnosis, treatment and prognosis. Anticancer Res. 2009, 29, 3759–3768. [Google Scholar]
- Shrivastav, N.; Li, D.; Essigmann, J.M. Chemical biology of mutagenesis and DNA repair: Cellular responses to DNA alkylation. Carcinogenesis 2010, 31, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Bai, P.; Fan, T.; Sun, G.; Wang, X.; Zhao, L.; Zhong, R. The dual role of DNA repair protein MGMT in cancer prevention and treatment. DNA Repair 2023, 123, 103449. [Google Scholar] [CrossRef]
- Kirstein, A.; Schmid, T.E.; Combs, S.E. The Role of miRNA for the Treatment of MGMT Unmethylated Glioblastoma Multiforme. Cancers 2020, 12, 1099. [Google Scholar] [CrossRef]
- Kaina, B.; Margison, G.P.; Christmann, M. Targeting O(6)-methylguanine-DNA methyltransferase with specific inhibitors as a strategy in cancer therapy. Cell. Mol. Life Sci. 2010, 67, 3663–3681. [Google Scholar] [CrossRef]
- Lindahl, T.; Demple, B.; Robins, P. Suicide inactivation of the E. coli O6-methylguanine-DNA methyltransferase. EMBO J. 1982, 1, 1359–1363. [Google Scholar] [CrossRef] [PubMed]
- Olsson, M.; Lindahl, T. Repair of alkylated DNA in Escherichia coli. Methyl group transfer from O6-methylguanine to a protein cysteine residue. J. Biol. Chem. 1980, 255, 10569–10571. [Google Scholar] [CrossRef] [PubMed]
- Samson, L. The suicidal DNA repair methyltransferases of microbes. Mol. Microbiol. 1992, 6, 825–831. [Google Scholar] [CrossRef]
- Samson, L.; Cairns, J. A new pathway for DNA repair in Escherichia coli. Nature 1977, 267, 281–283. [Google Scholar] [CrossRef]
- Potter, P.M.; Wilkinson, M.C.; Fitton, J.; Carr, F.J.; Brennand, J.; Cooper, D.P.; Margison, G.P. Characterisation and nucleotide sequence of ogt, the O6-alkylguanine-DNA-alkyltransferase gene of E. coli. Nucleic Acids Res. 1987, 15, 9177–9193. [Google Scholar] [CrossRef]
- Rebeck, G.W.; Smith, C.M.; Goad, D.L.; Samson, L. Characterization of the major DNA repair methyltransferase activity in unadapted Escherichia coli and identification of a similar activity in Salmonella typhimurium. J. Bacteriol. 1989, 171, 4563–4568. [Google Scholar] [CrossRef] [PubMed]
- Koike, G.; Maki, H.; Takeya, H.; Hayakawa, H.; Sekiguchi, M. Purification, structure, and biochemical properties of human O6-methylguanine-DNA methyltransferase. J. Biol. Chem. 1990, 265, 14754–14762. [Google Scholar] [CrossRef]
- Tano, K.; Shiota, S.; Collier, J.; Foote, R.S.; Mitra, S. Isolation and structural characterization of a cDNA clone encoding the human DNA repair protein for O6-alkylguanine. Proc. Natl. Acad. Sci. USA 1990, 87, 686–690. [Google Scholar] [CrossRef] [PubMed]
- Mijal, R.S.; Thomson, N.M.; Fleischer, N.L.; Pauly, G.T.; Moschel, R.C.; Kanugula, S.; Fang, Q.; Pegg, A.E.; Peterson, L.A. The repair of the tobacco specific nitrosamine derived adduct O6-[4-Oxo-4-(3-pyridyl)butyl]guanine by O6-alkylguanine-DNA alkyltransferase variants. Chem. Res. Toxicol. 2004, 17, 424–434. [Google Scholar] [CrossRef]
- Fang, Q.; Noronha, A.M.; Murphy, S.P.; Wilds, C.J.; Tubbs, J.L.; Tainer, J.A.; Chowdhury, G.; Guengerich, F.P.; Pegg, A.E. Repair of O6-G-alkyl-O6-G interstrand cross-links by human O6-alkylguanine-DNA alkyltransferase. Biochemistry 2008, 47, 10892–10903. [Google Scholar] [CrossRef]
- McManus, F.P.; Fang, Q.; Booth, J.D.; Noronha, A.M.; Pegg, A.E.; Wilds, C.J. Synthesis and characterization of an O(6)-2′-deoxyguanosine-alkyl-O(6)-2′-deoxyguanosine interstrand cross-link in a 5′-GNC motif and repair by human O(6)-alkylguanine-DNA alkyltransferase. Org. Biomol. Chem. 2010, 8, 4414–4426. [Google Scholar] [CrossRef]
- Samson, L.; Han, S.; Marquis, J.C.; Rasmussen, L.J. Mammalian DNA repair methyltransferases shield O4MeT from nucleotide excision repair. Carcinogenesis 1997, 18, 919–924. [Google Scholar] [CrossRef]
- Fang, Q.; Kanugula, S.; Tubbs, J.L.; Tainer, J.A.; Pegg, A.E. Repair of O4-alkylthymine by O6-alkylguanine-DNA alkyltransferases. J. Biol. Chem. 2010, 285, 8185–8195. [Google Scholar] [CrossRef] [PubMed]
- Wibley, J.E.; Pegg, A.E.; Moody, P.C. Crystal structure of the human O(6)-alkylguanine-DNA alkyltransferase. Nucleic Acids Res. 2000, 28, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Daniels, D.S.; Mol, C.D.; Arvai, A.S.; Kanugula, S.; Pegg, A.E.; Tainer, J.A. Active and alkylated human AGT structures: A novel zinc site, inhibitor and extrahelical base binding. EMBO J. 2000, 19, 1719–1730. [Google Scholar] [CrossRef] [PubMed]
- Daniels, D.S.; Woo, T.T.; Luu, K.X.; Noll, D.M.; Clarke, N.D.; Pegg, A.E.; Tainer, J.A. DNA binding and nucleotide flipping by the human DNA repair protein AGT. Nat. Struct. Mol. Biol. 2004, 11, 714–720. [Google Scholar] [CrossRef] [PubMed]
- Duguid, E.M.; Rice, P.A.; He, C. The structure of the human AGT protein bound to DNA and its implications for damage detection. J. Mol. Biol. 2005, 350, 657–666. [Google Scholar] [CrossRef] [PubMed]
- Fang, Q.; Kanugula, S.; Pegg, A.E. Function of domains of human O6-alkylguanine-DNA alkyltransferase. Biochemistry 2005, 44, 15396–15405. [Google Scholar] [CrossRef] [PubMed]
- Daniels, D.S.; Tainer, J.A. Conserved structural motifs governing the stoichiometric repair of alkylated DNA by O(6)-alkylguanine-DNA alkyltransferase. Mutat. Res. 2000, 460, 151–163. [Google Scholar] [CrossRef]
- Guengerich, F.P.; Fang, Q.; Liu, L.; Hachey, D.L.; Pegg, A.E. O6-alkylguanine-DNA alkyltransferase: Low pKa and high reactivity of cysteine 145. Biochemistry 2003, 42, 10965–10970. [Google Scholar] [CrossRef]
- Hu, J.; Ma, A.; Dinner, A.R. A two-step nucleotide-flipping mechanism enables kinetic discrimination of DNA lesions by AGT. Proc. Natl. Acad. Sci. USA 2008, 105, 4615–4620. [Google Scholar] [CrossRef]
- Demple, B.; Sedgwick, B.; Robins, P.; Totty, N.; Waterfield, M.D.; Lindahl, T. Active site and complete sequence of the suicidal methyltransferase that counters alkylation mutagenesis. Proc. Natl. Acad. Sci. USA 1985, 82, 2688–2692. [Google Scholar] [CrossRef] [PubMed]
- Sedgwick, B.; Robins, P.; Totty, N.; Lindahl, T. Functional domains and methyl acceptor sites of the Escherichia coli ada protein. J. Biol. Chem. 1988, 263, 4430–4433. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Hus, J.C.; Sun, L.J.; Zhou, P.; Norman, D.P.; Dotsch, V.; Wei, H.; Gross, J.D.; Lane, W.S.; Wagner, G.; et al. A methylation-dependent electrostatic switch controls DNA repair and transcriptional activation by E. coli ada. Mol. Cell 2005, 20, 117–129. [Google Scholar] [CrossRef]
- Teo, I.; Sedgwick, B.; Kilpatrick, M.W.; McCarthy, T.V.; Lindahl, T. The intracellular signal for induction of resistance to alkylating agents in E. coli. Cell 1986, 45, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Akimaru, H.; Sakumi, K.; Yoshikai, T.; Anai, M.; Sekiguchi, M. Positive and negative regulation of transcription by a cleavage product of Ada protein. J. Mol. Biol. 1990, 216, 261–273. [Google Scholar] [CrossRef]
- Landini, P.; Busby, S.J. The Escherichia coli Ada protein can interact with two distinct determinants in the sigma70 subunit of RNA polymerase according to promoter architecture: Identification of the target of Ada activation at the alkA promoter. J. Bacteriol. 1999, 181, 1524–1529. [Google Scholar] [CrossRef] [PubMed]
- Landini, P.; Volkert, M.R. Transcriptional activation of the Escherichia coli adaptive response gene aidB is mediated by binding of methylated Ada protein. Evidence for a new consensus sequence for Ada-binding sites. J. Biol. Chem. 1995, 270, 8285–8289. [Google Scholar] [CrossRef] [PubMed]
- Myers, L.C.; Terranova, M.P.; Ferentz, A.E.; Wagner, G.; Verdine, G.L. Repair of DNA methylphosphotriesters through a metalloactivated cysteine nucleophile. Science 1993, 261, 1164–1167. [Google Scholar] [CrossRef]
- Myers, L.C.; Jackow, F.; Verdine, G.L. Metal dependence of transcriptional switching in Escherichia coli Ada. J. Biol. Chem. 1995, 270, 6664–6670. [Google Scholar] [CrossRef]
- Myers, L.C.; Terranova, M.P.; Nash, H.M.; Markus, M.A.; Verdine, G.L. Zinc binding by the methylation signaling domain of the Escherichia coli Ada protein. Biochemistry 1992, 31, 4541–4547. [Google Scholar] [CrossRef]
- Moore, M.H.; Gulbis, J.M.; Dodson, E.J.; Demple, B.; Moody, P.C. Crystal structure of a suicidal DNA repair protein: The Ada O6-methylguanine-DNA methyltransferase from E. coli. EMBO J. 1994, 13, 1495–1501. [Google Scholar] [CrossRef] [PubMed]
- Paalman, S.R.; Sung, C.; Clarke, N.D. Specificity of DNA repair methyltransferases determined by competitive inactivation with oligonucleotide substrates: Evidence that Escherichia coli Ada repairs O6-methylguanine and O4-methylthymine with similar efficiency. Biochemistry 1997, 36, 11118–11124. [Google Scholar] [CrossRef] [PubMed]
- Pegg, A.E.; Boosalis, M.; Samson, L.; Moschel, R.C.; Byers, T.L.; Swenn, K.; Dolan, M.E. Mechanism of inactivation of human O6-alkylguanine-DNA alkyltransferase by O6-benzylguanine. Biochemistry 1993, 32, 11998–12006. [Google Scholar] [CrossRef] [PubMed]
- Pullman, A.; Pullman, B. Molecular electrostatic potential of the nucleic acids. Q. Rev. Biophys. 1981, 14, 289–380. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Laurent, B.; Hsu, C.H.; Nachtergaele, S.; Lu, Z.; Sheng, W.; Xu, C.; Chen, H.; Ouyang, J.; Wang, S.; et al. RNA m(6)A methylation regulates the ultraviolet-induced DNA damage response. Nature 2017, 543, 573–576. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Yang, C.G.; Yang, S.; Jian, X.; Yi, C.; Zhou, Z.; He, C. Oxidative demethylation of 3-methylthymine and 3-methyluracil in single-stranded DNA and RNA by mouse and human FTO. FEBS Lett. 2008, 582, 3313–3319. [Google Scholar] [CrossRef] [PubMed]
- Aas, P.A.; Otterlei, M.; Falnes, P.O.; Vagbo, C.B.; Skorpen, F.; Akbari, M.; Sundheim, O.; Bjoras, M.; Slupphaug, G.; Seeberg, E.; et al. Human and bacterial oxidative demethylases repair alkylation damage in both RNA and DNA. Nature 2003, 421, 859–863. [Google Scholar] [CrossRef]
- Ougland, R.; Zhang, C.M.; Liiv, A.; Johansen, R.F.; Seeberg, E.; Hou, Y.M.; Remme, J.; Falnes, P.O. AlkB restores the biological function of mRNA and tRNA inactivated by chemical methylation. Mol. Cell 2004, 16, 107–116. [Google Scholar] [CrossRef]
- Friedman, S.; Parsa, I. DNA adduct formation in rat, human and hamster pancreas treated with methylnitrosourea. Cancer Lett. 1985, 26, 269–276. [Google Scholar] [CrossRef]
- Falnes, P.O. RNA repair--the latest addition to the toolbox for macromolecular maintenance. RNA Biol. 2005, 2, 14–16. [Google Scholar] [CrossRef]
- Wang, T.; Pickard, A.J.; Gallo, J.M. Histone Methylation by Temozolomide; A Classic DNA Methylating Anticancer Drug. Anticancer Res. 2016, 36, 3289–3299. [Google Scholar] [PubMed]
- Yan, L.L.; Zaher, H.S. How do cells cope with RNA damage and its consequences? J. Biol. Chem. 2019, 294, 15158–15171. [Google Scholar] [CrossRef] [PubMed]
- Pegg, A.E.; Wiest, L.; Mummert, C.; Stine, L.; Moschel, R.C.; Dolan, M.E. Use of antibodies to human O6-alkylguanine-DNA alkyltransferase to study the content of this protein in cells treated with O6-benzylguanine or N-methyl-N′-nitro-N-nitrosoguanidine. Carcinogenesis 1991, 12, 1679–1683. [Google Scholar] [CrossRef] [PubMed]
- Srivenugopal, K.S.; Yuan, X.H.; Friedman, H.S.; Ali-Osman, F. Ubiquitination-dependent proteolysis of O6-methylguanine-DNA methyltransferase in human and murine tumor cells following inactivation with O6-benzylguanine or 1,3-bis(2-chloroethyl)-1-nitrosourea. Biochemistry 1996, 35, 1328–1334. [Google Scholar] [CrossRef] [PubMed]
- Xu-Welliver, M.; Pegg, A.E. Degradation of the alkylated form of the DNA repair protein, O(6)-alkylguanine-DNA alkyltransferase. Carcinogenesis 2002, 23, 823–830. [Google Scholar] [CrossRef]
- Edara, S.; Kanugula, S.; Pegg, A.E. Expression of the inactive C145A mutant human O6-alkylguanine-DNA alkyltransferase in E. coli increases cell killing and mutations by N-methyl-N′-nitro-N-nitrosoguanidine. Carcinogenesis 1999, 20, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Rasimas, J.J.; Dalessio, P.A.; Ropson, I.J.; Pegg, A.E.; Fried, M.G. Active-site alkylation destabilizes human O6-alkylguanine DNA alkyltransferase. Protein Sci. Publ. Protein Soc. 2004, 13, 301–305. [Google Scholar] [CrossRef]
- Rasimas, J.J.; Pegg, A.E.; Fried, M.G. DNA-binding mechanism of O6-alkylguanine-DNA alkyltransferase. Effects of protein and DNA alkylation on complex stability. J. Biol. Chem. 2003, 278, 7973–7980. [Google Scholar] [CrossRef]
- Ali, R.B.; Teo, A.K.; Oh, H.K.; Chuang, L.S.; Ayi, T.C.; Li, B.F. Implication of localization of human DNA repair enzyme O6-methylguanine-DNA methyltransferase at active transcription sites in transcription-repair coupling of the mutagenic O6-methylguanine lesion. Mol. Cell. Biol. 1998, 18, 1660–1669. [Google Scholar] [CrossRef]
- Teo, A.K.; Oh, H.K.; Ali, R.B.; Li, B.F. The modified human DNA repair enzyme O(6)-methylguanine-DNA methyltransferase is a negative regulator of estrogen receptor-mediated transcription upon alkylation DNA damage. Mol. Cell. Biol. 2001, 21, 7105–7114. [Google Scholar] [CrossRef]
- Wu, X.; Luo, Q.; Zhao, P.; Chang, W.; Wang, Y.; Shu, T.; Ding, F.; Li, B.; Liu, Z. MGMT-activated DUB3 stabilizes MCL1 and drives chemoresistance in ovarian cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 2961–2966. [Google Scholar] [CrossRef] [PubMed]
- Horiguchi, M.; Kim, J.; Matsunaga, N.; Kaji, H.; Egawa, T.; Makino, K.; Koyanagi, S.; Ohdo, S. Glucocorticoid-dependent expression of O(6)-methylguanine-DNA methyltransferase gene modulates dacarbazine-induced hepatotoxicity in mice. J. Pharmacol. Exp. Ther. 2010, 333, 782–787. [Google Scholar] [CrossRef] [PubMed]
- Pegg, A.E.; Perry, W.; Bennett, R.A. Effect of partial hepatectomy on removal of O6-methylguanine from alkylated DNA by rat liver extracts. Biochem. J. 1981, 197, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Paranjpe, A.; Bailey, N.I.; Konduri, S.; Bobustuc, G.C.; Ali-Osman, F.; Yusuf, M.A.; Punganuru, S.R.; Madala, H.R.; Basak, D.; Mostofa, A.; et al. New insights into estrogenic regulation of O(6)-methylguanine DNA-methyltransferase (MGMT) in human breast cancer cells: Co-degradation of ER-alpha and MGMT proteins by fulvestrant or O(6)-benzylguanine indicates fresh avenues for therapy. J. Biomed. Res. 2016, 30, 393–410. [Google Scholar] [CrossRef] [PubMed]
- Brandes, A.A.; Franceschi, E.; Tosoni, A.; Bartolini, S.; Bacci, A.; Agati, R.; Ghimenton, C.; Turazzi, S.; Talacchi, A.; Skrap, M.; et al. O(6)-methylguanine DNA-methyltransferase methylation status can change between first surgery for newly diagnosed glioblastoma and second surgery for recurrence: Clinical implications. Neuro Oncol. 2010, 12, 283–288. [Google Scholar] [CrossRef]
- Wiewrodt, D.; Nagel, G.; Dreimuller, N.; Hundsberger, T.; Perneczky, A.; Kaina, B. MGMT in primary and recurrent human glioblastomas after radiation and chemotherapy and comparison with p53 status and clinical outcome. Int. J. Cancer 2008, 122, 1391–1399. [Google Scholar] [CrossRef] [PubMed]
- Mullapudi, S.R.; Ali-Osman, F.; Shou, J.; Srivenugopal, K.S. DNA repair protein O6-alkylguanine-DNA alkyltransferase is phosphorylated by two distinct and novel protein kinases in human brain tumour cells. Biochem. J. 2000, 351 Pt 2, 393–402. [Google Scholar] [CrossRef]
- Cropper, J.D.; Alimbetov, D.S.; Brown, K.T.G.; Likhotvorik, R.I.; Robles, A.J.; Guerra, J.T.; He, B.; Chen, Y.; Kwon, Y.; Kurmasheva, R.T. PARP1-MGMT complex underpins pathway crosstalk in O(6)-methylguanine repair. J. Hematol. Oncol. 2022, 15, 146. [Google Scholar] [CrossRef]
- Wu, S.; Li, X.; Gao, F.; de Groot, J.F.; Koul, D.; Yung, W.K.A. PARP-mediated PARylation of MGMT is critical to promote repair of temozolomide-induced O6-methylguanine DNA damage in glioblastoma. Neuro Oncol. 2021, 23, 920–931. [Google Scholar] [CrossRef]
- Srivenugopal, K.S.; Mullapudi, S.R.; Shou, J.; Hazra, T.K.; Ali-Osman, F. Protein phosphorylation is a regulatory mechanism for O6-alkylguanine-DNA alkyltransferase in human brain tumor cells. Cancer Res. 2000, 60, 282–287. [Google Scholar]
- Choi, J.Y.; Chowdhury, G.; Zang, H.; Angel, K.C.; Vu, C.C.; Peterson, L.A.; Guengerich, F.P. Translesion synthesis across O6-alkylguanine DNA adducts by recombinant human DNA polymerases. J. Biol. Chem. 2006, 281, 38244–38256. [Google Scholar] [CrossRef]
- Loechler, E.L.; Green, C.L.; Essigmann, J.M. In vivo mutagenesis by O6-methylguanine built into a unique site in a viral genome. Proc. Natl. Acad. Sci. USA 1984, 81, 6271–6275. [Google Scholar] [CrossRef]
- Pence, M.G.; Choi, J.Y.; Egli, M.; Guengerich, F.P. Structural basis for proficient incorporation of dTTP opposite O6-methylguanine by human DNA polymerase iota. J. Biol. Chem. 2010, 285, 40666–40672. [Google Scholar] [CrossRef] [PubMed]
- Warren, J.J.; Forsberg, L.J.; Beese, L.S. The structural basis for the mutagenicity of O(6)-methyl-guanine lesions. Proc. Natl. Acad. Sci. USA 2006, 103, 19701–19706. [Google Scholar] [CrossRef]
- Woodside, A.M.; Guengerich, F.P. Effect of the O6 substituent on misincorporation kinetics catalyzed by DNA polymerases at O(6)-methylguanine and O(6)-benzylguanine. Biochemistry 2002, 41, 1027–1038. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.; Pongor, L.; Su, Y.T.; Xi, L.; Raffeld, M.; Quezado, M.; Trepel, J.; Aldape, K.; Pommier, Y.; Wu, J. MGMT Status as a Clinical Biomarker in Glioblastoma. Trends Cancer 2020, 6, 380–391. [Google Scholar] [CrossRef] [PubMed]
- Fahrer, J.; Kaina, B. O6-methylguanine-DNA methyltransferase in the defense against N-nitroso compounds and colorectal cancer. Carcinogenesis 2013, 34, 2435–2442. [Google Scholar] [CrossRef]
- Zaidi, N.H.; Pretlow, T.P.; O’Riordan, M.A.; Dumenco, L.L.; Allay, E.; Gerson, S.L. Transgenic expression of human MGMT protects against azoxymethane-induced aberrant crypt foci and G to A mutations in the K-ras oncogene of mouse colon. Carcinogenesis 1995, 16, 451–456. [Google Scholar] [CrossRef]
- Wali, R.K.; Skarosi, S.; Hart, J.; Zhang, Y.; Dolan, M.E.; Moschel, R.C.; Nguyen, L.; Mustafi, R.; Brasitus, T.A.; Bissonnette, M. Inhibition of O(6)-methylguanine-DNA methyltransferase increases azoxymethane-induced colonic tumors in rats. Carcinogenesis 1999, 20, 2355–2360. [Google Scholar] [CrossRef]
- Wirtz, S.; Nagel, G.; Eshkind, L.; Neurath, M.F.; Samson, L.D.; Kaina, B. Both base excision repair and O6-methylguanine-DNA methyltransferase protect against methylation-induced colon carcinogenesis. Carcinogenesis 2010, 31, 2111–2117. [Google Scholar] [CrossRef]
- Bugni, J.M.; Meira, L.B.; Samson, L.D. Alkylation-induced colon tumorigenesis in mice deficient in the Mgmt and Msh6 proteins. Oncogene 2009, 28, 734–741. [Google Scholar] [CrossRef]
- Hecht, S.S.; Hatsukami, D.K. Smokeless tobacco and cigarette smoking: Chemical mechanisms and cancer prevention. Nat. Rev. Cancer 2022, 22, 143–155. [Google Scholar] [CrossRef]
- Schrader, E.; Hirsch-Ernst, K.I.; Richter, E.; Foth, H. Metabolism of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) in isolated rat lung and liver. Naunyn-Schmiedebergs Arch. Pharmacol. 1998, 357, 336–343. [Google Scholar] [CrossRef]
- Liu, L.; Qin, X.; Gerson, S.L. Reduced lung tumorigenesis in human methylguanine DNA--methyltransferase transgenic mice achieved by expression of transgene within the target cell. Carcinogenesis 1999, 20, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Staretz, M.E.; Foiles, P.G.; Miglietta, L.M.; Hecht, S.S. Evidence for an important role of DNA pyridyloxobutylation in rat lung carcinogenesis by 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone: Effects of dose and phenethyl isothiocyanate. Cancer Res. 1997, 57, 259–266. [Google Scholar] [PubMed]
- Dumenco, L.L.; Allay, E.; Norton, K.; Gerson, S.L. The prevention of thymic lymphomas in transgenic mice by human O6-alkylguanine-DNA alkyltransferase. Science 1993, 259, 219–222. [Google Scholar] [CrossRef] [PubMed]
- Sakumi, K.; Shiraishi, A.; Shimizu, S.; Tsuzuki, T.; Ishikawa, T.; Sekiguchi, M. Methylnitrosourea-induced tumorigenesis in MGMT gene knockout mice. Cancer Res. 1997, 57, 2415–2418. [Google Scholar]
- Hanigan, M.H.; Kemp, C.J.; Ginsler, J.J.; Drinkwater, N.R. Rapid growth of preneoplastic lesions in hepatocarcinogen-sensitive C3H/HeJ male mice relative to C57BL/6J male mice. Carcinogenesis 1988, 9, 885–890. [Google Scholar] [CrossRef] [PubMed]
- Nakatsuru, Y.; Matsukuma, S.; Nemoto, N.; Sugano, H.; Sekiguchi, M.; Ishikawa, T. O6-methylguanine-DNA methyltransferase protects against nitrosamine-induced hepatocarcinogenesis. Proc. Natl. Acad. Sci. USA 1993, 90, 6468–6472. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Zhang, S.; Matsukuma, S.; Zarkovic, M.; Shimizu, S.; Ishikawa, T.; Nakatsuru, Y. Protection against malignant progression of spontaneously developing liver tumors in transgenic mice expressing O(6)-methylguanine-DNA methyltransferase. Jpn. J. Cancer Res. 2000, 91, 1085–1089. [Google Scholar] [CrossRef]
- Becker, K.; Gregel, C.M.; Kaina, B. The DNA repair protein O6-methylguanine-DNA methyltransferase protects against skin tumor formation induced by antineoplastic chloroethylnitrosourea. Cancer Res. 1997, 57, 3335–3338. [Google Scholar]
- Becker, K.; Dosch, J.; Gregel, C.M.; Martin, B.A.; Kaina, B. Targeted expression of human O(6)-methylguanine-DNA methyltransferase (MGMT) in transgenic mice protects against tumor initiation in two-stage skin carcinogenesis. Cancer Res. 1996, 56, 3244–3249. [Google Scholar] [PubMed]
- Becker, K.; Gregel, C.; Fricke, C.; Komitowski, D.; Dosch, J.; Kaina, B. DNA repair protein MGMT protects against N-methyl-N-nitrosourea-induced conversion of benign into malignant tumors. Carcinogenesis 2003, 24, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Bobola, M.S.; Blank, A.; Berger, M.S.; Silber, J.R. O6-methylguanine-DNA methyltransferase deficiency in developing brain: Implications for brain tumorigenesis. DNA Repair 2007, 6, 1127–1133. [Google Scholar] [CrossRef]
- Kisby, G.E.; Olivas, A.; Park, T.; Churchwell, M.; Doerge, D.; Samson, L.D.; Gerson, S.L.; Turker, M.S. DNA repair modulates the vulnerability of the developing brain to alkylating agents. DNA Repair 2009, 8, 400–412. [Google Scholar] [CrossRef]
- Silber, J.R.; Blank, A.; Bobola, M.S.; Mueller, B.A.; Kolstoe, D.D.; Ojemann, G.A.; Berger, M.S. Lack of the DNA repair protein O6-methylguanine-DNA methyltransferase in histologically normal brain adjacent to primary human brain tumors. Proc. Natl. Acad. Sci. USA 1996, 93, 6941–6946. [Google Scholar] [CrossRef] [PubMed]
- Kaina, B.; Christmann, M. DNA repair in personalized brain cancer therapy with temozolomide and nitrosoureas. DNA Repair 2019, 78, 128–141. [Google Scholar] [CrossRef] [PubMed]
- Hegi, M.E.; Diserens, A.C.; Godard, S.; Dietrich, P.Y.; Regli, L.; Ostermann, S.; Otten, P.; Van Melle, G.; de Tribolet, N.; Stupp, R. Clinical trial substantiates the predictive value of O-6-methylguanine-DNA methyltransferase promoter methylation in glioblastoma patients treated with temozolomide. Clin. Cancer Res. 2004, 10, 1871–1874. [Google Scholar] [CrossRef]
- Hegi, M.E.; Diserens, A.C.; Gorlia, T.; Hamou, M.F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef]
- Esteller, M.; Garcia-Foncillas, J.; Andion, E.; Goodman, S.N.; Hidalgo, O.F.; Vanaclocha, V.; Baylin, S.B.; Herman, J.G. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N. Engl. J. Med. 2000, 343, 1350–1354. [Google Scholar] [CrossRef]
- Aoki, K.; Natsume, A. Overview of DNA methylation in adult diffuse gliomas. Brain Tumor Pathol. 2019, 36, 84–91. [Google Scholar] [CrossRef]
- Reifenberger, G.; Hentschel, B.; Felsberg, J.; Schackert, G.; Simon, M.; Schnell, O.; Westphal, M.; Wick, W.; Pietsch, T.; Loeffler, M.; et al. Predictive impact of MGMT promoter methylation in glioblastoma of the elderly. Int. J. Cancer 2012, 131, 1342–1350. [Google Scholar] [CrossRef] [PubMed]
- Qi, F.; Yin, Z.; Wang, G.; Zeng, S. Clinical and Prognostic Significance of O(6)-Methylguanine-DNA Methyltransferase Promoter Methylation in Patients with Melanoma: A Systematic Meta-Analysis. Ann. Dermatol. 2018, 30, 129–135. [Google Scholar] [CrossRef]
- Pandith, A.A.; Qasim, I.; Zahoor, W.; Shah, P.; Bhat, A.R.; Sanadhya, D.; Shah, Z.A.; Naikoo, N.A. Concordant association validates MGMT methylation and protein expression as favorable prognostic factors in glioma patients on alkylating chemotherapy (Temozolomide). Sci. Rep. 2018, 8, 6704. [Google Scholar] [CrossRef] [PubMed]
- Dahlrot, R.H.; Larsen, P.; Boldt, H.B.; Kreutzfeldt, M.S.; Hansen, S.; Hjelmborg, J.B.; Kristensen, B.W. Posttreatment Effect of MGMT Methylation Level on Glioblastoma Survival. J. Neuropathol. Exp. Neurol. 2019, 78, 633–640. [Google Scholar] [CrossRef]
- Malmstrom, A.; Gronberg, B.H.; Marosi, C.; Stupp, R.; Frappaz, D.; Schultz, H.; Abacioglu, U.; Tavelin, B.; Lhermitte, B.; Hegi, M.E.; et al. Temozolomide versus standard 6-week radiotherapy versus hypofractionated radiotherapy in patients older than 60 years with glioblastoma: The Nordic randomised, phase 3 trial. Lancet Oncol. 2012, 13, 916–926. [Google Scholar] [CrossRef] [PubMed]
- Wick, W.; Platten, M.; Meisner, C.; Felsberg, J.; Tabatabai, G.; Simon, M.; Nikkhah, G.; Papsdorf, K.; Steinbach, J.P.; Sabel, M.; et al. Temozolomide chemotherapy alone versus radiotherapy alone for malignant astrocytoma in the elderly: The NOA-08 randomised, phase 3 trial. Lancet Oncol. 2012, 13, 707–715. [Google Scholar] [CrossRef] [PubMed]
- Jaeckle, K.A.; Eyre, H.J.; Townsend, J.J.; Schulman, S.; Knudson, H.M.; Belanich, M.; Yarosh, D.B.; Bearman, S.I.; Giroux, D.J.; Schold, S.C. Correlation of tumor O6 methylguanine-DNA methyltransferase levels with survival of malignant astrocytoma patients treated with bis-chloroethylnitrosourea: A Southwest Oncology Group study. J. Clin. Oncol. 1998, 16, 3310–3315. [Google Scholar] [CrossRef]
- Van den Bent, M.J.; Dubbink, H.J.; Sanson, M.; van der Lee-Haarloo, C.R.; Hegi, M.; Jeuken, J.W.; Ibdaih, A.; Brandes, A.A.; Taphoorn, M.J.; Frenay, M.; et al. MGMT promoter methylation is prognostic but not predictive for outcome to adjuvant PCV chemotherapy in anaplastic oligodendroglial tumors: A report from EORTC Brain Tumor Group Study 26951. J. Clin. Oncol. 2009, 27, 5881–5886. [Google Scholar] [CrossRef]
- Wick, W.; Hartmann, C.; Engel, C.; Stoffels, M.; Felsberg, J.; Stockhammer, F.; Sabel, M.C.; Koeppen, S.; Ketter, R.; Meyermann, R.; et al. NOA-04 randomized phase III trial of sequential radiochemotherapy of anaplastic glioma with procarbazine, lomustine, and vincristine or temozolomide. J. Clin. Oncol. 2009, 27, 5874–5880. [Google Scholar] [CrossRef]
- Lei, Y.; Tang, L.; Hu, J.; Wang, S.; Liu, Y.; Yang, M.; Zhang, J.; Tang, B. Inhibition of MGMT-mediated autophagy suppression decreases cisplatin chemosensitivity in gastric cancer. Biomed. Pharmacother. 2020, 125, 109896. [Google Scholar] [CrossRef]
- Gefen, N.; Brkic, G.; Galron, D.; Priel, E.; Ozer, J.; Benharroch, D.; Gopas, J. Acquired resistance to 6-thioguanine in melanoma cells involves the repair enzyme O6-methylguanine-DNA methyltransferase (MGMT). Cancer Biol. Ther. 2010, 9, 49–55. [Google Scholar] [CrossRef]
- Spratt, T.E.; Campbell, C.R. Synthesis of oligodeoxynucleotides containing analogs of O6-methylguanine and reaction with O6-alkylguanine-DNA alkyltransferase. Biochemistry 1994, 33, 11364–11371. [Google Scholar] [CrossRef] [PubMed]
- Spratt, T.E.; de los Santos, H. Reaction of O6-alkylguanine-DNA alkyltransferase with O6-methylguanine analogues: Evidence that the oxygen of O6-methylguanine is protonated by the protein to effect methyl transfer. Biochemistry 1992, 31, 3688–3694. [Google Scholar] [CrossRef] [PubMed]
- Swann, P.F.; Waters, T.R.; Moulton, D.C.; Xu, Y.Z.; Zheng, Q.; Edwards, M.; Mace, R. Role of postreplicative DNA mismatch repair in the cytotoxic action of thioguanine. Science 1996, 273, 1109–1111. [Google Scholar] [CrossRef]
- Waters, T.R.; Swann, P.F. Cytotoxic mechanism of 6-thioguanine: HMutSalpha, the human mismatch binding heterodimer, binds to DNA containing S6-methylthioguanine. Biochemistry 1997, 36, 2501–2506. [Google Scholar] [CrossRef] [PubMed]
- Dolan, M.E.; Pegg, A.E. O6-benzylguanine and its role in chemotherapy. Clin. Cancer Res. 1997, 3, 837–847. [Google Scholar] [PubMed]
- Dolan, M.E.; Moschel, R.C.; Pegg, A.E. Depletion of mammalian O6-alkylguanine-DNA alkyltransferase activity by O6-benzylguanine provides a means to evaluate the role of this protein in protection against carcinogenic and therapeutic alkylating agents. Proc. Natl. Acad. Sci. USA 1990, 87, 5368–5372. [Google Scholar] [CrossRef]
- Shibata, T.; Glynn, N.; McMurry, T.B.; McElhinney, R.S.; Margison, G.P.; Williams, D.M. Novel synthesis of O6-alkylguanine containing oligodeoxyribonucleotides as substrates for the human DNA repair protein, O6-methylguanine DNA methyltransferase (MGMT). Nucleic Acids Res. 2006, 34, 1884–1891. [Google Scholar] [CrossRef]
- Kaina, B.; Muhlhausen, U.; Piee-Staffa, A.; Christmann, M.; Garcia Boy, R.; Rosch, F.; Schirrmacher, R. Inhibition of O6-methylguanine-DNA methyltransferase by glucose-conjugated inhibitors: Comparison with nonconjugated inhibitors and effect on fotemustine and temozolomide-induced cell death. J. Pharmacol. Exp. Ther. 2004, 311, 585–593. [Google Scholar] [CrossRef]
- Middleton, M.R.; Margison, G.P. Improvement of chemotherapy efficacy by inactivation of a DNA-repair pathway. Lancet Oncol. 2003, 4, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Bobola, M.S.; Silber, J.R.; Ellenbogen, R.G.; Geyer, J.R.; Blank, A.; Goff, R.D. O6-methylguanine-DNA methyltransferase, O6-benzylguanine, and resistance to clinical alkylators in pediatric primary brain tumor cell lines. Clin. Cancer Res. 2005, 11, 2747–2755. [Google Scholar] [CrossRef] [PubMed]
- Koch, D.; Hundsberger, T.; Boor, S.; Kaina, B. Local intracerebral administration of O(6)-benzylguanine combined with systemic chemotherapy with temozolomide of a patient suffering from a recurrent glioblastoma. J. Neurooncol. 2007, 82, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Broniscer, A.; Gururangan, S.; MacDonald, T.J.; Goldman, S.; Packer, R.J.; Stewart, C.F.; Wallace, D.; Danks, M.K.; Friedman, H.S.; Poussaint, T.Y.; et al. Phase I trial of single-dose temozolomide and continuous administration of O6-benzylguanine in children with brain tumors: A pediatric brain tumor consortium report. Clin. Cancer Res. 2007, 13, 6712–6718. [Google Scholar] [CrossRef] [PubMed]
- Quinn, J.A.; Desjardins, A.; Weingart, J.; Brem, H.; Dolan, M.E.; Delaney, S.M.; Vredenburgh, J.; Rich, J.; Friedman, A.H.; Reardon, D.A.; et al. Phase I trial of temozolomide plus O6-benzylguanine for patients with recurrent or progressive malignant glioma. J. Clin. Oncol. 2005, 23, 7178–7187. [Google Scholar] [CrossRef] [PubMed]
- Quinn, J.A.; Jiang, S.X.; Reardon, D.A.; Desjardins, A.; Vredenburgh, J.J.; Rich, J.N.; Gururangan, S.; Friedman, A.H.; Bigner, D.D.; Sampson, J.H.; et al. Phase I trial of temozolomide plus O6-benzylguanine 5-day regimen with recurrent malignant glioma. Neuro-Oncology 2009, 11, 556–561. [Google Scholar] [CrossRef] [PubMed]
- Quinn, J.A.; Jiang, S.X.; Reardon, D.A.; Desjardins, A.; Vredenburgh, J.J.; Rich, J.N.; Gururangan, S.; Friedman, A.H.; Bigner, D.D.; Sampson, J.H.; et al. Phase II trial of temozolomide plus O6-benzylguanine in adults with recurrent, temozolomide-resistant malignant glioma. J. Clin. Oncol. 2009, 27, 1262–1267. [Google Scholar] [CrossRef]
- Verbeek, B.; Southgate, T.D.; Gilham, D.E.; Margison, G.P. O6-Methylguanine-DNA methyltransferase inactivation and chemotherapy. Br. Med. Bull. 2008, 85, 17–33. [Google Scholar] [CrossRef]
- Barvaux, V.A.; Lorigan, P.; Ranson, M.; Gillum, A.M.; McElhinney, R.S.; McMurry, T.B.; Margison, G.P. Sensitization of a human ovarian cancer cell line to temozolomide by simultaneous attenuation of the Bcl-2 antiapoptotic protein and DNA repair by O6-alkylguanine-DNA alkyltransferase. Mol. Cancer Ther. 2004, 3, 1215–1220. [Google Scholar] [CrossRef]
- Clemons, M.; Kelly, J.; Watson, A.J.; Howell, A.; McElhinney, R.S.; McMurry, T.B.; Margison, G.P. O6-(4-bromothenyl)guanine reverses temozolomide resistance in human breast tumour MCF-7 cells and xenografts. Br. J. Cancer 2005, 93, 1152–1156. [Google Scholar] [CrossRef]
- Middleton, M.R.; Kelly, J.; Thatcher, N.; Donnelly, D.J.; McElhinney, R.S.; McMurry, T.B.; McCormick, J.E.; Margison, G.P. O(6)-(4-bromothenyl)guanine improves the therapeutic index of temozolomide against A375M melanoma xenografts. Int. J. Cancer 2000, 85, 248–252. [Google Scholar] [CrossRef]
- Turriziani, M.; Caporaso, P.; Bonmassar, L.; Buccisano, F.; Amadori, S.; Venditti, A.; Cantonetti, M.; D’Atri, S.; Bonmassar, E. O6-(4-bromothenyl)guanine (PaTrin-2), a novel inhibitor of O6-alkylguanine DNA alkyl-transferase, increases the inhibitory activity of temozolomide against human acute leukaemia cells in vitro. Pharmacol. Res. 2006, 53, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Watson, A.J.; Middleton, M.R.; McGown, G.; Thorncroft, M.; Ranson, M.; Hersey, P.; McArthur, G.; Davis, I.D.; Thomson, D.; Beith, J.; et al. O(6)-methylguanine-DNA methyltransferase depletion and DNA damage in patients with melanoma treated with temozolomide alone or with lomeguatrib. Br. J. Cancer 2009, 100, 1250–1256. [Google Scholar] [CrossRef] [PubMed]
- Watson, A.J.; Sabharwal, A.; Thorncroft, M.; McGown, G.; Kerr, R.; Bojanic, S.; Soonawalla, Z.; King, A.; Miller, A.; Waller, S.; et al. Tumor O(6)-methylguanine-DNA methyltransferase inactivation by oral lomeguatrib. Clin. Cancer Res. 2010, 16, 743–749. [Google Scholar] [CrossRef]
- Ranson, M.; Hersey, P.; Thompson, D.; Beith, J.; McArthur, G.A.; Haydon, A.; Davis, I.D.; Kefford, R.F.; Mortimer, P.; Harris, P.A.; et al. Randomized trial of the combination of lomeguatrib and temozolomide compared with temozolomide alone in chemotherapy naive patients with metastatic cutaneous melanoma. J. Clin. Oncol. 2007, 25, 2540–2545. [Google Scholar] [CrossRef] [PubMed]
- Khan, O.A.; Ranson, M.; Michael, M.; Olver, I.; Levitt, N.C.; Mortimer, P.; Watson, A.J.; Margison, G.P.; Midgley, R.; Middleton, M.R. A phase II trial of lomeguatrib and temozolomide in metastatic colorectal cancer. Br. J. Cancer 2008, 98, 1614–1618. [Google Scholar] [CrossRef] [PubMed]
- Tawbi, H.A.; Villaruz, L.; Tarhini, A.; Moschos, S.; Sulecki, M.; Viverette, F.; Shipe-Spotloe, J.; Radkowski, R.; Kirkwood, J.M. Inhibition of DNA repair with MGMT pseudosubstrates: Phase I study of lomeguatrib in combination with dacarbazine in patients with advanced melanoma and other solid tumours. Br. J. Cancer 2011, 105, 773–777. [Google Scholar] [CrossRef]
- Pauly, G.T.; Loktionova, N.A.; Fang, Q.; Vankayala, S.L.; Guida, W.C.; Pegg, A.E. Substitution of aminomethyl at the meta-position enhances the inactivation of O6-alkylguanine-DNA alkyltransferase by O6-benzylguanine. J. Med. Chem. 2008, 51, 7144–7153. [Google Scholar] [CrossRef]
- Sun, G.; Zhao, L.; Zhong, R.; Peng, Y. The specific role of O(6)-methylguanine-DNA methyltransferase inhibitors in cancer chemotherapy. Future Med. Chem. 2018, 10, 1971–1996. [Google Scholar] [CrossRef]
- Wang, C.; Abegg, D.; Hoch, D.G.; Adibekian, A. Chemoproteomics-Enabled Discovery of a Potent and Selective Inhibitor of the DNA Repair Protein MGMT. Angew. Chem. Int. Ed. 2016, 55, 2911–2915. [Google Scholar] [CrossRef]
- Melikishvili, M.; Rodgers, D.W.; Fried, M.G. 6-Carboxyfluorescein and structurally similar molecules inhibit DNA binding and repair by O(6)-alkylguanine DNA alkyltransferase. DNA Repair 2011, 10, 1193–1202. [Google Scholar] [CrossRef]
- Goder, A.; Nagel, G.; Kraus, A.; Dorsam, B.; Seiwert, N.; Kaina, B.; Fahrer, J. Lipoic acid inhibits the DNA repair protein O 6-methylguanine-DNA methyltransferase (MGMT) and triggers its depletion in colorectal cancer cells with concomitant autophagy induction. Carcinogenesis 2015, 36, 817–831. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Xu-Welliver, M.; Kanugula, S.; Pegg, A.E. Inactivation and degradation of O(6)-alkylguanine-DNA alkyltransferase after reaction with nitric oxide. Cancer Res. 2002, 62, 3037–3043. [Google Scholar] [PubMed]
- Paranjpe, A.; Zhang, R.; Ali-Osman, F.; Bobustuc, G.C.; Srivenugopal, K.S. Disulfiram is a direct and potent inhibitor of human O6-methylguanine-DNA methyltransferase (MGMT) in brain tumor cells and mouse brain and markedly increases the alkylating DNA damage. Carcinogenesis 2014, 35, 692–702. [Google Scholar] [CrossRef]
- Geiger, H.; Schleimer, D.; Nattamai, K.J.; Dannenmann, S.R.; Davies, S.M.; Weiss, B.D. Mutagenic potential of temozolomide in bone marrow cells in vivo. Blood 2006, 107, 3010–3011. [Google Scholar] [CrossRef] [PubMed]
- Reinhard, J.; Eichhorn, U.; Wiessler, M.; Kaina, B. Inactivation of O(6)-methylguanine-DNA methyltransferase by glucose-conjugated inhibitors. Int. J. Cancer 2001, 93, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Tomaszowski, K.H.; Schirrmacher, R.; Kaina, B. Multidrug Efflux Pumps Attenuate the Effect of MGMT Inhibitors. Mol. Pharm. 2015, 12, 3924–3934. [Google Scholar] [CrossRef]
- Ladino, C.A.; Chari, R.V.; Bourret, L.A.; Kedersha, N.L.; Goldmacher, V.S. Folate-maytansinoids: Target-selective drugs of low molecular weight. Int. J. Cancer 1997, 73, 859–864. [Google Scholar] [CrossRef]
- Javanmard, S.; Loktionova, N.A.; Fang, Q.; Pauly, G.T.; Pegg, A.E.; Moschel, R.C. Inactivation of O6-alkylguanine-DNA alkyltransferase by folate esters of O6-benzyl-2′-deoxyguanosine and of O6-[4-(hydroxymethyl)benzyl]guanine. J. Med. Chem. 2007, 50, 5193–5201. [Google Scholar] [CrossRef]
- Nelson, M.E.; Loktionova, N.A.; Pegg, A.E.; Moschel, R.C. 2-amino-O4-benzylpteridine derivatives: Potent inactivators of O6-alkylguanine-DNA alkyltransferase. J. Med. Chem. 2004, 47, 3887–3891. [Google Scholar] [CrossRef]
- Zhu, R.; Baumann, R.P.; Penketh, P.G.; Shyam, K.; Sartorelli, A.C. Hypoxia-selective O6-alkylguanine-DNA alkyltransferase inhibitors: Design, synthesis, and evaluation of 6-(benzyloxy)-2-(aryldiazenyl)-9H-purines as prodrugs of O6-benzylguanine. J. Med. Chem. 2013, 56, 1355–1359. [Google Scholar] [CrossRef]
- Zhu, R.; Liu, M.C.; Luo, M.Z.; Penketh, P.G.; Baumann, R.P.; Shyam, K.; Sartorelli, A.C. 4-nitrobenzyloxycarbonyl derivatives of O(6)-benzylguanine as hypoxia-activated prodrug inhibitors of O(6)-alkylguanine-DNA alkyltransferase (AGT), which produces resistance to agents targeting the O-6 position of DNA guanine. J. Med. Chem. 2011, 54, 7720–7728. [Google Scholar] [CrossRef] [PubMed]
- Wei, G.; Loktionova, N.A.; Pegg, A.E.; Moschel, R.C. Beta-glucuronidase-cleavable prodrugs of O6-benzylguanine and O6-benzyl-2′-deoxyguanosine. J. Med. Chem. 2005, 48, 256–261. [Google Scholar] [CrossRef] [PubMed]
- Tseng, Y.Y.; Kau, Y.C.; Liu, S.J. Advanced interstitial chemotherapy for treating malignant glioma. Expert Opin. Drug Deliv. 2016, 13, 1533–1544. [Google Scholar] [CrossRef] [PubMed]
- Guerin, C.; Olivi, A.; Weingart, J.D.; Lawson, H.C.; Brem, H. Recent advances in brain tumor therapy: Local intracerebral drug delivery by polymers. Investig. New Drugs 2004, 22, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, B.D.; Blanco, E.; Gao, J. Polymer implants for intratumoral drug delivery and cancer therapy. J. Pharm. Sci. 2008, 97, 1681–1702. [Google Scholar] [CrossRef] [PubMed]
- Weingart, J.; Grossman, S.A.; Carson, K.A.; Fisher, J.D.; Delaney, S.M.; Rosenblum, M.L.; Olivi, A.; Judy, K.; Tatter, S.B.; Dolan, M.E. Phase I trial of polifeprosan 20 with carmustine implant plus continuous infusion of intravenous O6-benzylguanine in adults with recurrent malignant glioma: New approaches to brain tumor therapy CNS consortium trial. J. Clin. Oncol. 2007, 25, 399–404. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, I.; Veevnik, D.; Fedulov, A.; Yurkshtovich, N.; Yurkshtovich, T.; Pejler, G.; Lokot, I. Local delivery of temozolomide via a biologically inert carrier (Temodex) prolongs survival in glioma patients, irrespectively of the methylation status of MGMT. Neoplasma 2019, 66, 288–293. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Shen, J.; Zhang, L.; Wang, L.; Xu, H.; Han, Y.; Jia, J.; Lu, Y.; Yu, R.; Liu, H. Injectable postoperative enzyme-responsive hydrogels for reversing temozolomide resistance and reducing local recurrence after glioma operation. Biomater. Sci. 2020, 8, 5306–5316. [Google Scholar] [CrossRef]
- Kievit, F.M.; Wang, F.Y.; Fang, C.; Mok, H.; Wang, K.; Silber, J.R.; Ellenbogen, R.G.; Zhang, M. Doxorubicin loaded iron oxide nanoparticles overcome multidrug resistance in cancer in vitro. J. Control. Release 2011, 152, 76–83. [Google Scholar] [CrossRef]
- Stephen, Z.R.; Kievit, F.M.; Veiseh, O.; Chiarelli, P.A.; Fang, C.; Wang, K.; Hatzinger, S.J.; Ellenbogen, R.G.; Silber, J.R.; Zhang, M. Redox-responsive magnetic nanoparticle for targeted convection-enhanced delivery of O6-benzylguanine to brain tumors. ACS Nano 2014, 8, 10383–10395. [Google Scholar] [CrossRef] [PubMed]
- Stephen, Z.R.; Gebhart, R.N.; Jeon, M.; Blair, A.A.; Ellenbogen, R.G.; Silber, J.R.; Zhang, M. pH-Sensitive O6-Benzylguanosine Polymer Modified Magnetic Nanoparticles for Treatment of Glioblastomas. Bioconjug. Chem. 2017, 28, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.S.; Rait, A.; Kim, E.; Pirollo, K.F.; Nishida, M.; Farkas, N.; Dagata, J.A.; Chang, E.H. A nanoparticle carrying the p53 gene targets tumors including cancer stem cells, sensitizes glioblastoma to chemotherapy and improves survival. ACS Nano 2014, 8, 5494–5514. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Zhou, Y.; Chen, J.; Huang, N.; Wang, Z.; Cheng, Y. Gene Therapy for Drug-Resistant Glioblastoma via Lipid-Polymer Hybrid Nanoparticles Combined with Focused Ultrasound. Int. J. Nanomed. 2021, 16, 185–199. [Google Scholar] [CrossRef] [PubMed]
- Allay, J.A.; Dumenco, L.L.; Koc, O.N.; Liu, L.; Gerson, S.L. Retroviral transduction and expression of the human alkyltransferase cDNA provides nitrosourea resistance to hematopoietic cells. Blood 1995, 85, 3342–3351. [Google Scholar] [CrossRef] [PubMed]
- Maze, R.; Carney, J.P.; Kelley, M.R.; Glassner, B.J.; Williams, D.A.; Samson, L. Increasing DNA repair methyltransferase levels via bone marrow stem cell transduction rescues mice from the toxic effects of 1,3-bis(2-chloroethyl)-1-nitrosourea, a chemotherapeutic alkylating agent. Proc. Natl. Acad. Sci. USA 1996, 93, 206–210. [Google Scholar] [CrossRef] [PubMed]
- Moritz, T.; Mackay, W.; Glassner, B.J.; Williams, D.A.; Samson, L. Retrovirus-mediated expression of a DNA repair protein in bone marrow protects hematopoietic cells from nitrosourea-induced toxicity in vitro and in vivo. Cancer Res. 1995, 55, 2608–2614. [Google Scholar]
- Davis, B.M.; Roth, J.C.; Liu, L.; Xu-Welliver, M.; Pegg, A.E.; Gerson, S.L. Characterization of the P140K, PVP(138-140)MLK, and G156A O6-methylguanine-DNA methyltransferase mutants: Implications for drug resistance gene therapy. Hum. Gene Ther. 1999, 10, 2769–2778. [Google Scholar] [CrossRef]
- Davis, B.M.; Reese, J.S.; Koc, O.N.; Lee, K.; Schupp, J.E.; Gerson, S.L. Selection for G156A O6-methylguanine DNA methyltransferase gene-transduced hematopoietic progenitors and protection from lethality in mice treated with O6-benzylguanine and 1,3-bis(2-chloroethyl)-1-nitrosourea. Cancer Res. 1997, 57, 5093–5099. [Google Scholar]
- Koc, O.N.; Reese, J.S.; Davis, B.M.; Liu, L.; Majczenko, K.J.; Gerson, S.L. DeltaMGMT-transduced bone marrow infusion increases tolerance to O6-benzylguanine and 1,3-bis(2-chloroethyl)-1-nitrosourea and allows intensive therapy of 1,3-bis(2-chloroethyl)-1-nitrosourea-resistant human colon cancer xenografts. Hum. Gene Ther. 1999, 10, 1021–1030. [Google Scholar] [CrossRef]
- Cabrini, G.; Fabbri, E.; Lo Nigro, C.; Dechecchi, M.C.; Gambari, R. Regulation of expression of O6-methylguanine-DNA methyltransferase and the treatment of glioblastoma (Review). Int. J. Oncol. 2015, 47, 417–428. [Google Scholar] [CrossRef]
- Chiou, G.Y.; Chien, C.S.; Wang, M.L.; Chen, M.T.; Yang, Y.P.; Yu, Y.L.; Chien, Y.; Chang, Y.C.; Shen, C.C.; Chio, C.C.; et al. Epigenetic regulation of the miR142-3p/interleukin-6 circuit in glioblastoma. Mol. Cell 2013, 52, 693–706. [Google Scholar] [CrossRef]
- Lee, Y.Y.; Yarmishyn, A.A.; Wang, M.L.; Chen, H.Y.; Chiou, S.H.; Yang, Y.P.; Lin, C.F.; Huang, P.I.; Chen, Y.W.; Ma, H.I.; et al. MicroRNA-142-3p is involved in regulation of MGMT expression in glioblastoma cells. Cancer Manag. Res. 2018, 10, 775–785. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.T.; Chen, X.B.; Liu, H.L. Up-regulation of miR-370-3p restores glioblastoma multiforme sensitivity to temozolomide by influencing MGMT expression. Sci. Rep. 2016, 6, 32972. [Google Scholar] [CrossRef] [PubMed]
- Nadaradjane, A.; Briand, J.; Bougras-Cartron, G.; Disdero, V.; Vallette, F.M.; Frenel, J.S.; Cartron, P.F. miR-370-3p Is a Therapeutic Tool in Anti-glioblastoma Therapy but Is Not an Intratumoral or Cell-free Circulating Biomarker. Mol. Ther. Nucleic Acids 2018, 13, 642–650. [Google Scholar] [CrossRef] [PubMed]
- Quintavalle, C.; Mangani, D.; Roscigno, G.; Romano, G.; Diaz-Lagares, A.; Iaboni, M.; Donnarumma, E.; Fiore, D.; De Marinis, P.; Soini, Y.; et al. MiR-221/222 target the DNA methyltransferase MGMT in glioma cells. PLoS ONE 2013, 8, e74466. [Google Scholar] [CrossRef]
- Zhang, W.; Zhang, J.; Hoadley, K.; Kushwaha, D.; Ramakrishnan, V.; Li, S.; Kang, C.; You, Y.; Jiang, C.; Song, S.W.; et al. miR-181d: A predictive glioblastoma biomarker that downregulates MGMT expression. Neuro-Oncology 2012, 14, 712–719. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Natsume, A.; Toda, H.; Iwamizu, H.; Sugita, T.; Hachisu, R.; Watanabe, R.; Yuki, K.; Motomura, K.; Bankiewicz, K.; et al. Efficient delivery of liposome-mediated MGMT-siRNA reinforces the cytotoxity of temozolomide in GBM-initiating cells. Gene Ther. 2010, 17, 1363–1371. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Yang, S.; Cui, X.; Wang, Q.; Yang, E.; Tong, F.; Hong, B.; Xiao, M.; Xin, L.; Xu, C.; et al. A novel compound EPIC-0412 reverses temozolomide resistance via inhibiting DNA repair/MGMT in glioblastoma. Neuro-Oncology 2023, 25, 857–870. [Google Scholar] [CrossRef]
- Bhakat, K.K.; Mitra, S. Regulation of the human O(6)-methylguanine-DNA methyltransferase gene by transcriptional coactivators cAMP response element-binding protein-binding protein and p300. J. Biol. Chem. 2000, 275, 34197–34204. [Google Scholar] [CrossRef]
- Alonso, M.M.; Gomez-Manzano, C.; Bekele, B.N.; Yung, W.K.; Fueyo, J. Adenovirus-based strategies overcome temozolomide resistance by silencing the O6-methylguanine-DNA methyltransferase promoter. Cancer Res. 2007, 67, 11499–11504. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Deng, Z.; Wang, H.; Li, X.; Sun, T.; Tao, Z.; Yao, L.; Jin, Y.; Wang, X.; Yang, L.; et al. MGMT autoantibodies as a potential prediction of recurrence and treatment response biomarker for glioma patients. Cancer Med. 2019, 8, 4359–4369. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhang, K.; Wang, H.; Chen, G.; Liu, Y.; Li, W.; Zhou, Y. Experimental study of selective MGMT peptides mimicking TMZ drug resistance in glioma. Biochem. Biophys. Rep. 2022, 32, 101386. [Google Scholar] [CrossRef]
- Lin, K.; Gueble, S.E.; Sundaram, R.K.; Huseman, E.D.; Bindra, R.S.; Herzon, S.B. Mechanism-based design of agents that selectively target drug-resistant glioma. Science 2022, 377, 502–511. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Heras, G.; Castro-Robles, B.; Romero-Sanchez, C.M.; Carrion, B.; Barbella-Aponte, R.; Sandoval, H.; Segura, T. Involvement of N-methylpurine DNA glycosylase in resistance to temozolomide in patient-derived glioma cells. Sci. Rep. 2020, 10, 22185. [Google Scholar] [CrossRef] [PubMed]
- Marteijn, J.A.; Lans, H.; Vermeulen, W.; Hoeijmakers, J.H. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat. Rev. Mol. Cell Biol. 2014, 15, 465–481. [Google Scholar] [CrossRef]
- Bronstein, S.M.; Skopek, T.R.; Swenberg, J.A. Efficient repair of O6-ethylguanine, but not O4-ethylthymine or O2-ethylthymine, is dependent upon O6-alkylguanine-DNA alkyltransferase and nucleotide excision repair activities in human cells. Cancer Res. 1992, 52, 2008–2011. [Google Scholar]
- Aloisi, C.M.N.; Escher, N.A.; Kim, H.S.; Geisen, S.M.; Fontana, G.A.; Yeo, J.E.; Scharer, O.D.; Sturla, S.J. A combination of direct reversion and nucleotide excision repair counters the mutagenic effects of DNA carboxymethylation. DNA Repair 2022, 110, 103262. [Google Scholar] [CrossRef]
- Tubbs, J.L.; Latypov, V.; Kanugula, S.; Butt, A.; Melikishvili, M.; Kraehenbuehl, R.; Fleck, O.; Marriott, A.; Watson, A.J.; Verbeek, B.; et al. Flipping of alkylated DNA damage bridges base and nucleotide excision repair. Nature 2009, 459, 808–813. [Google Scholar] [CrossRef]
- Pearson, S.J.; Ferguson, J.; Santibanez-Koref, M.; Margison, G.P. Inhibition of O6-methylguanine-DNA methyltransferase by an alkyltransferase-like protein from Escherichia coli. Nucleic Acids Res. 2005, 33, 3837–3844. [Google Scholar] [CrossRef]
- Gupta, D.; Heinen, C.D. The mismatch repair-dependent DNA damage response: Mechanisms and implications. DNA Repair 2019, 78, 60–69. [Google Scholar] [CrossRef]
- Fink, D.; Aebi, S.; Howell, S.B. The role of DNA mismatch repair in drug resistance. Clin. Cancer Res. 1998, 4, 1–6. [Google Scholar] [PubMed]
- Yip, S.; Miao, J.; Cahill, D.P.; Iafrate, A.J.; Aldape, K.; Nutt, C.L.; Louis, D.N. MSH6 mutations arise in glioblastomas during temozolomide therapy and mediate temozolomide resistance. Clin. Cancer Res. 2009, 15, 4622–4629. [Google Scholar] [CrossRef] [PubMed]
- Duckett, D.R.; Drummond, J.T.; Murchie, A.I.; Reardon, J.T.; Sancar, A.; Lilley, D.M.; Modrich, P. Human MutSalpha recognizes damaged DNA base pairs containing O6-methylguanine, O4-methylthymine, or the cisplatin-d(GpG) adduct. Proc. Natl. Acad. Sci. USA 1996, 93, 6443–6447. [Google Scholar] [CrossRef] [PubMed]
- Mojas, N.; Lopes, M.; Jiricny, J. Mismatch repair-dependent processing of methylation damage gives rise to persistent single-stranded gaps in newly replicated DNA. Genes Dev. 2007, 21, 3342–3355. [Google Scholar] [CrossRef]
- Kaina, B.; Ziouta, A.; Ochs, K.; Coquerelle, T. Chromosomal instability, reproductive cell death and apoptosis induced by O6-methylguanine in Mex-, Mex+ and methylation-tolerant mismatch repair compromised cells: Facts and models. Mutat. Res. 1997, 381, 227–241. [Google Scholar] [CrossRef] [PubMed]
- Karran, P.; Marinus, M.G. Mismatch correction at O6-methylguanine residues in E. coli DNA. Nature 1982, 296, 868–869. [Google Scholar] [CrossRef]
- Yoshioka, K.; Yoshioka, Y.; Hsieh, P. ATR kinase activation mediated by MutSalpha and MutLalpha in response to cytotoxic O6-methylguanine adducts. Mol. Cell 2006, 22, 501–510. [Google Scholar] [CrossRef]
- Lin, D.P.; Wang, Y.; Scherer, S.J.; Clark, A.B.; Yang, K.; Avdievich, E.; Jin, B.; Werling, U.; Parris, T.; Kurihara, N.; et al. An Msh2 point mutation uncouples DNA mismatch repair and apoptosis. Cancer Res. 2004, 64, 517–522. [Google Scholar] [CrossRef]
- Yang, G.; Scherer, S.J.; Shell, S.S.; Yang, K.; Kim, M.; Lipkin, M.; Kucherlapati, R.; Kolodner, R.D.; Edelmann, W. Dominant effects of an Msh6 missense mutation on DNA repair and cancer susceptibility. Cancer Cell 2004, 6, 139–150. [Google Scholar] [CrossRef]
- Gupta, D.; Lin, B.; Cowan, A.; Heinen, C.D. ATR-Chk1 activation mitigates replication stress caused by mismatch repair-dependent processing of DNA damage. Proc. Natl. Acad. Sci. USA 2018, 115, 1523–1528. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.; Gupta, D.; Heinen, C.D. Human pluripotent stem cells have a novel mismatch repair-dependent damage response. J. Biol. Chem. 2014, 289, 24314–24324. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, R.P.; Isogawa, A.; Paulo, J.A.; Onizuka, K.; Takahashi, T.; Amunugama, R.; Duxin, J.P.; Fujii, S. Crosstalk between repair pathways elicits double-strand breaks in alkylated DNA and implications for the action of temozolomide. Elife 2021, 10, e69544. [Google Scholar] [CrossRef]
- Fujii, S.; Sobol, R.W.; Fuchs, R.P. Double-strand breaks: When DNA repair events accidentally meet. DNA Repair 2022, 112, 103303. [Google Scholar] [CrossRef] [PubMed]
- Debiak, M.; Nikolova, T.; Kaina, B. Loss of ATM sensitizes against O6-methylguanine triggered apoptosis, SCEs and chromosomal aberrations. DNA Repair 2004, 3, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Philip, S.; Swaminathan, S.; Kuznetsov, S.G.; Kanugula, S.; Biswas, K.; Chang, S.; Loktionova, N.A.; Haines, D.C.; Kaldis, P.; Pegg, A.E.; et al. Degradation of BRCA2 in alkyltransferase-mediated DNA repair and its clinical implications. Cancer Res. 2008, 68, 9973–9981. [Google Scholar] [CrossRef]
Figure 1.
The reaction of MGMT (top panel) and the structure overlay of Ada and hMGMT (bottom panel). Ada: red; hMGMT: green; the side chains of PCHRV are shown. The overlay is to show the structural similarity of both proteins.
Figure 1.
The reaction of MGMT (top panel) and the structure overlay of Ada and hMGMT (bottom panel). Ada: red; hMGMT: green; the side chains of PCHRV are shown. The overlay is to show the structural similarity of both proteins.

Figure 2.
The structure of human MGMT repairing (modified from PDB ID: 1EH7). Y114: blue; R128: yellow; PCHRV: green; hMGMT: red; dsDNA: green and yellow. Nucleotides: blue. The key residues involved in the repair are shown.
Figure 2.
The structure of human MGMT repairing (modified from PDB ID: 1EH7). Y114: blue; R128: yellow; PCHRV: green; hMGMT: red; dsDNA: green and yellow. Nucleotides: blue. The key residues involved in the repair are shown.

Figure 3.
The structure of N-Ada and its interaction with DNA chain (modified from PDB ID: 1U8B). N-Ada: green; zinc cluster with side chains of C38, C42, C69, and C72: orange; phosphodiesterase chains: red and orange. Nucleotides: blue and grey.
Figure 3.
The structure of N-Ada and its interaction with DNA chain (modified from PDB ID: 1U8B). N-Ada: green; zinc cluster with side chains of C38, C42, C69, and C72: orange; phosphodiesterase chains: red and orange. Nucleotides: blue and grey.

Figure 4.
The overlay of N-Ada and N-hMGMT. N-Ada: orange; N-hMGMT: light sea green; Zn2+: small ball. Both N-Ada and N-hMGMT contain zinc binding sites and these sites locate in the surface of protein.
Figure 4.
The overlay of N-Ada and N-hMGMT. N-Ada: orange; N-hMGMT: light sea green; Zn2+: small ball. Both N-Ada and N-hMGMT contain zinc binding sites and these sites locate in the surface of protein.

Figure 5.
Model of DNA repair pathways involved in DNA alkylation damage. DNA lesions caused by alkylating agents are processed by BER, ALKBH, and MGMT. If m6G is unaffected by MGMT during DNA replication, m6G forms mismatches with T, generating m6G–T pairs. MMR rectifies these mismatches by removing T from m6G–T pairs. This futile cycling replication process results in unreplicated gaps opposite the m6G lesions, which remain tolerated until the subsequent S-phase (middle panel). In this phase, they impede DNA replication, causing DSBs that trigger cell cycle arrest or eventual cell death. Alternatively, mismatch repair proteins recognizing m6G/T lesions might function as a scaffold, directly recruiting DNA damage signaling molecules (middle panel), leading to the activation of cell cycle checkpoints and/or apoptosis. The circles with different color mean different types of DNA lesions.
Figure 5.
Model of DNA repair pathways involved in DNA alkylation damage. DNA lesions caused by alkylating agents are processed by BER, ALKBH, and MGMT. If m6G is unaffected by MGMT during DNA replication, m6G forms mismatches with T, generating m6G–T pairs. MMR rectifies these mismatches by removing T from m6G–T pairs. This futile cycling replication process results in unreplicated gaps opposite the m6G lesions, which remain tolerated until the subsequent S-phase (middle panel). In this phase, they impede DNA replication, causing DSBs that trigger cell cycle arrest or eventual cell death. Alternatively, mismatch repair proteins recognizing m6G/T lesions might function as a scaffold, directly recruiting DNA damage signaling molecules (middle panel), leading to the activation of cell cycle checkpoints and/or apoptosis. The circles with different color mean different types of DNA lesions.

Figure 6.
The crosstalk of MGMT with other DNA repair pathways. The interplay of MGMT with other DNA repair mechanisms involves several notable connections. PARP1, a protein in the BER pathway, can perform poly-ADP-ribosylation on hMGMT, resulting in the formation of poly-ADP-ribosylated hMGMT. This modification facilitates the localization of hMGMT within chromatin, enabling efficient repair of DNA lesions. The effectiveness of the NER process can assist MGMT in managing DNA adducts. Through a collaborative effort, MGMT and NER collectively process O6-carboxymethylguanine (O6-CMG) lesions, thereby thwarting potential carcinogenic outcomes. Intriguingly, the alkylation of hMGMT and the existence of alkylating damage can accelerate the degradation of BRCA2, potentially heightening the sensitivity of cancer cells to the treatment of crosslinking agents. ↑: increase.
Figure 6.
The crosstalk of MGMT with other DNA repair pathways. The interplay of MGMT with other DNA repair mechanisms involves several notable connections. PARP1, a protein in the BER pathway, can perform poly-ADP-ribosylation on hMGMT, resulting in the formation of poly-ADP-ribosylated hMGMT. This modification facilitates the localization of hMGMT within chromatin, enabling efficient repair of DNA lesions. The effectiveness of the NER process can assist MGMT in managing DNA adducts. Through a collaborative effort, MGMT and NER collectively process O6-carboxymethylguanine (O6-CMG) lesions, thereby thwarting potential carcinogenic outcomes. Intriguingly, the alkylation of hMGMT and the existence of alkylating damage can accelerate the degradation of BRCA2, potentially heightening the sensitivity of cancer cells to the treatment of crosslinking agents. ↑: increase.


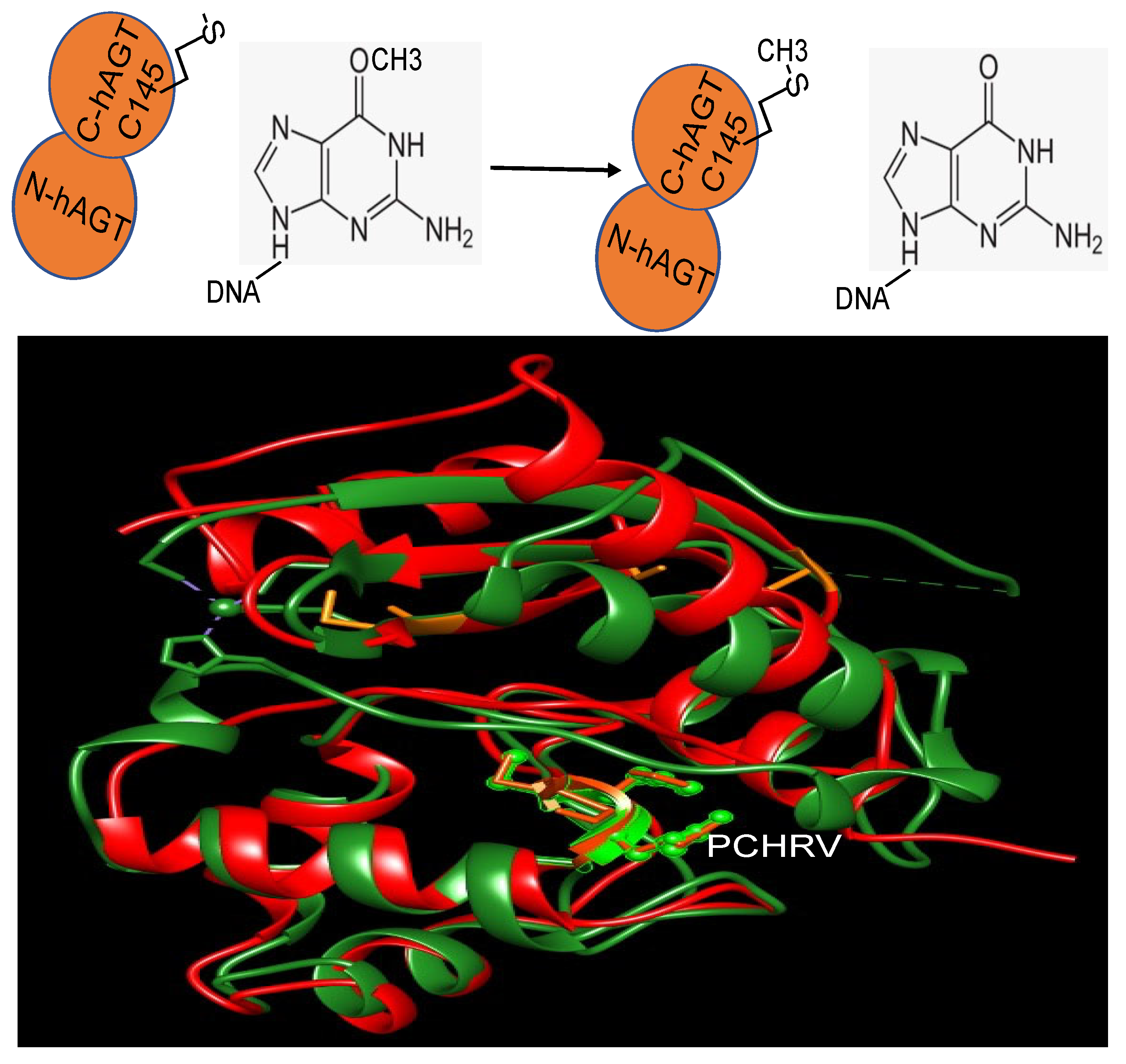{kind=link}
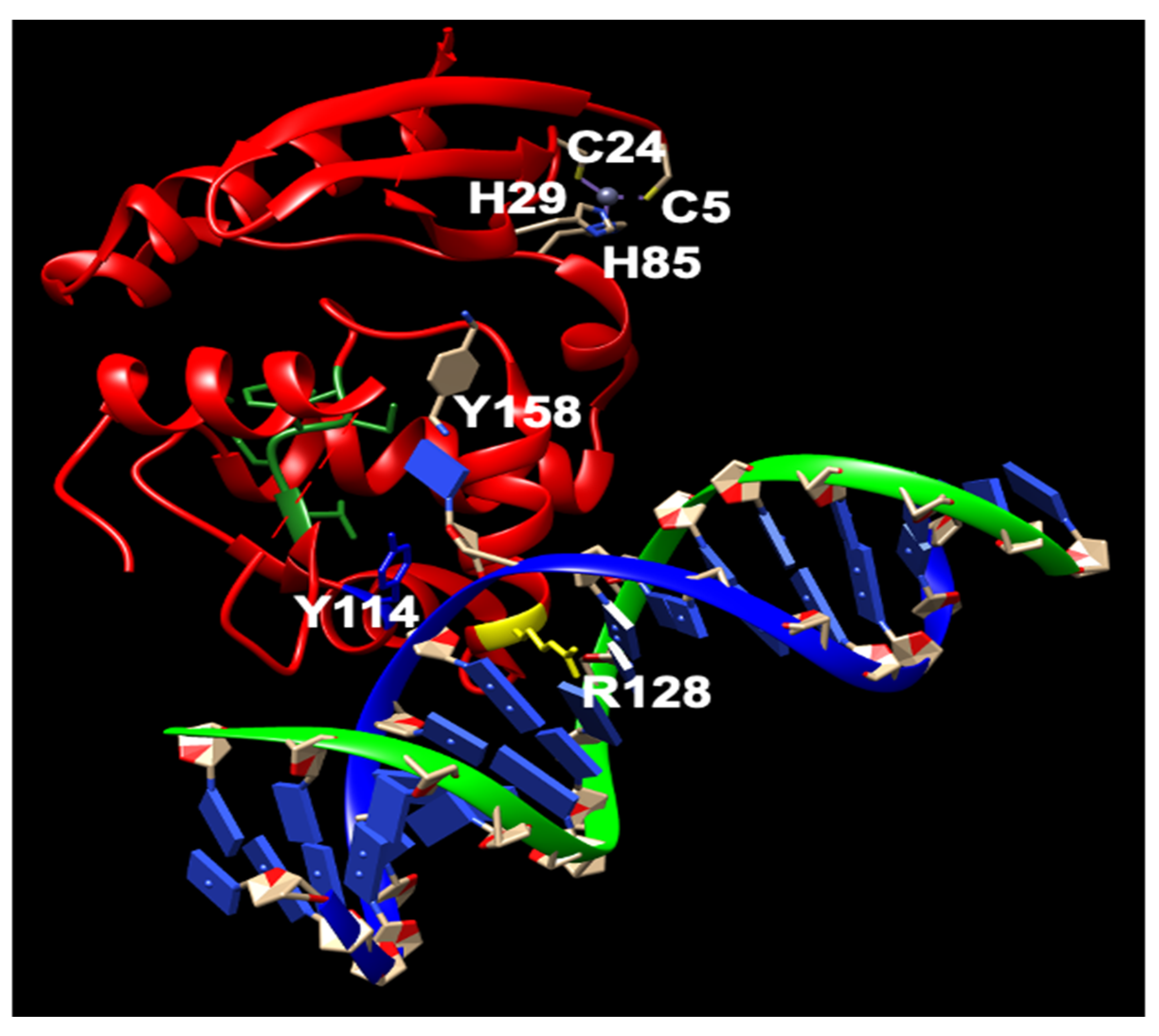{kind=link}
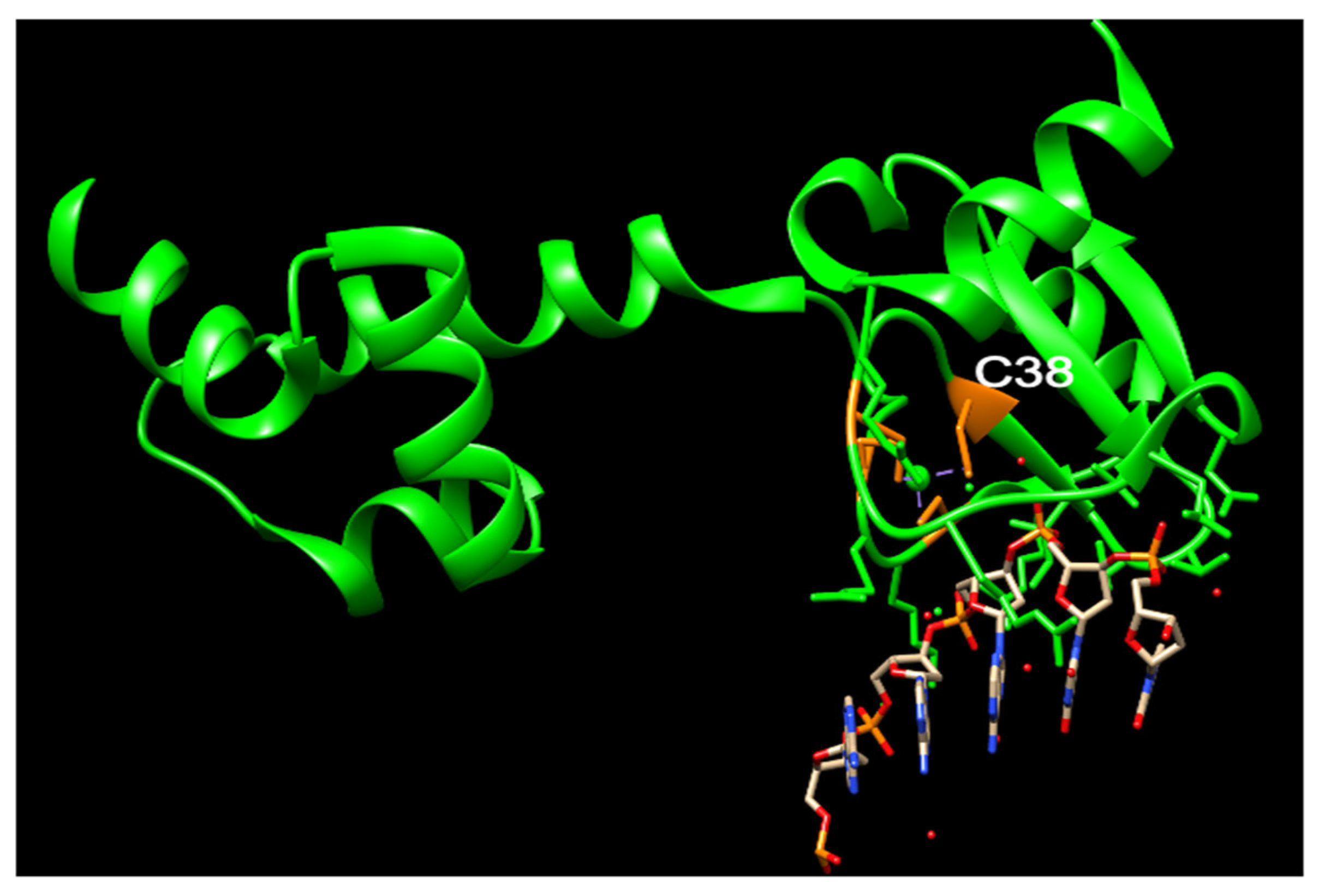{kind=link}
{kind=link}
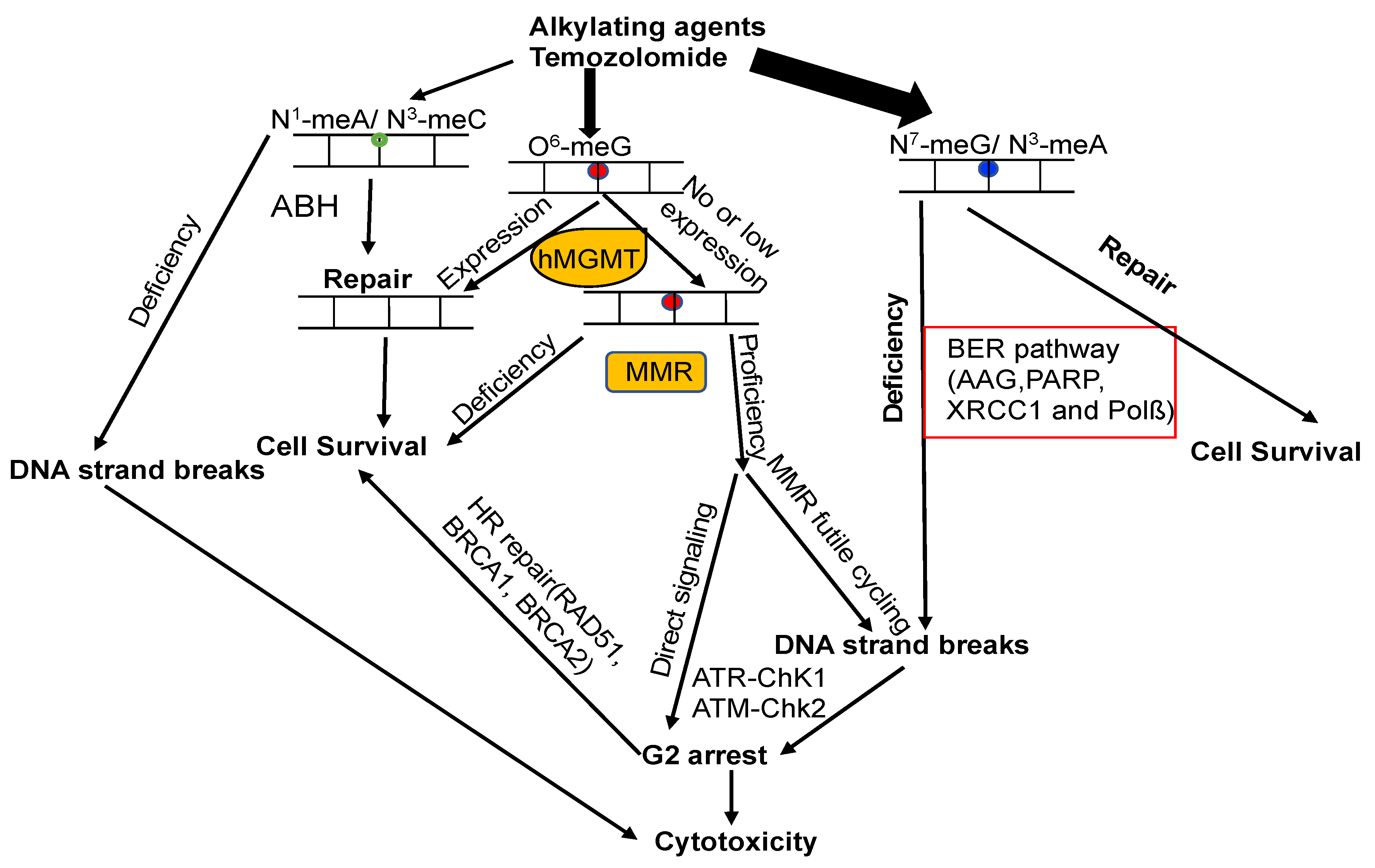{kind=link}
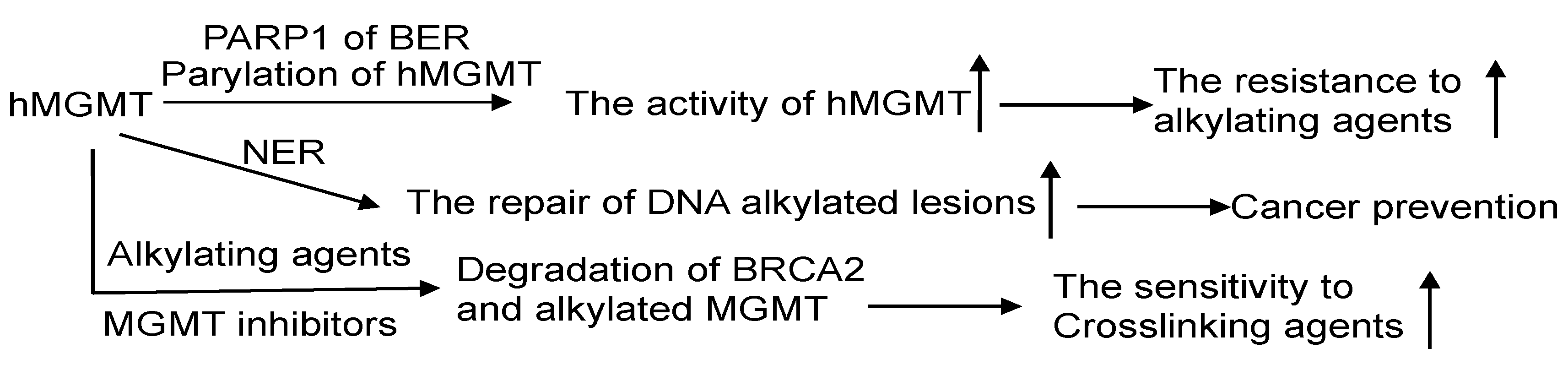{kind=link}
Table 1.
ED50 of MGMT to O6-BG.
| MGMT | ED50 (µM) |
|---|---|
| N-hMGMT | 0.15 ± 0.01 [57] |
| N-hMGMT/C-hMGMT | 0.20 ± 0.02 [57] |
| hMGMT | 0.24 ± 0.02 [57] |
| Ogt | 600 [73] |
| Ada | >1 mM (no repair) [73] |
| YMGMT * | >1 mM (no repair) [73] |
* YMGMT: MGMT of Yeast.
Table 2.
The structure match of N-hMGMT obtained with Phyre2 online server.
| # | Template | 3D Model | Confidence | Template Information |
|---|---|---|---|---|
| 1 | d1qnta2 |  | 100 | Fold: Ribonuclease H-like motif Family: Methylated DNA-protein cysteine methyltransferase domain |
| 2 | C1t39A |  | 98.7 | PDB header: transferase/DNA PDB title: human O6-alkylguanine-DNA alkyltransferase covalently crosslinked to DNA |
| 3 | C4bhcA |  | 97.7 | PDB header: transferase PDB title: crystal structure of the m.tuberculosis O6-methylguanine DNA methyltransferase r371 variant |
Table 3.
The list of strategies targeting MGMT.
| The Category of Strategies | Compounds or Molecules | Effects and Advantage |
|---|---|---|
| Non caner-selective inhibitors | O6-BG and O6-4-BTG | Efficiently inhibit the activity of MGMT in mammalian cells, animal models and patients [147,148,149,150]. |
| Meta-substitution of O6-BG | Improve the water solubility and more potent inhibitors of MGMT in mammalian cells [168]. | |
| 6-(benzyloxy)pyrimidine derivatives | Inhibit the activity of MGMT [13,169]. | |
| Acrolein and chloromethyltriazoles | React with cysteine residues to inhibit the activity of MGMT in cells efficiently [170]. | |
| 6-carboxyflfluorescein | Inhibit the activity of MGMT with a non-covalent manner [171]. | |
| Lipoic acid | A natural compound that efficiently inhibits the activity of MGMT and enhances the cytotoxic effects of TMZ in cancer cells that are resistant to TMZ [172]. | |
| Nitric oxide and disulfiram | Inactivate MGMT by interacting with Cys145 [173,174]. | |
| Cancer-selective inhibitors | O6-BG-Glu and O6-BTG-Glu | Conjugate a glucose group with an MGMT inhibitor. They inhibit the activity of MGMT in various cancer cell lines [150,176,177]. |
| O4-benzylfolate | Conjugate a folate group to an MGMT inhibitor. It is a more potent inhibitor than O6-BG and enhances BCNU-induced cell death dependent on the expression of the α-folate receptor [179,180]. | |
| O6-benzyl-2′-deoxyguanosine | Increase MGMT inhibitory activity in tumor cells and improve the water solubility [179]. | |
| Glucuronic acid linked prodrugs of O6-BG and O6-benzyl-2′-deoxyguanosine | These prodrugs are stable and less active. O6-BG and O6-benzyl-2′-deoxyguanosine can be released after the removal of beta-glucuronidase [181,182]. They may be useful in cancer cells that liberate beta-glucuronidase. | |
| Local drug delivery | Gliadel (BCNU wafers) | The first clinical application of polymer drug delivery to treat brain tumors [185,186]. |
| Encapsulated TMZ with inert matrix | Demonstrated superior efficacy compared to standard therapy alone, resulting in a remarkable increase in overall survival for GBM patients [188]. | |
| Injectable hydrogel capable of delivering TMZ and O6-BG | Demonstrated effectiveness in reducing the recurrence of TMZ-resistant glioma after surgery and enhancing the inhibitory efficiency against tumors [189]. | |
| Nanoparticle-based delivery of TMZ and O6-BG | These delivery systems successfully deplete MGMT and significantly enhance the therapeutic effect of TMZ in cancer cells and animal models [191,192,193,194]. | |
| Targeting the expression of MGMT | miRNAs including miR-142-3p, miR-181d, miR-370-3p | Downregulate the expression of MGMT and enhance the sensitivity to TMZ in GBM cell lines [38,201,202,203,204,205,206,207]. |
| EPIC-0412 | Enhances the chemotherapeutic effect of TMZ by epigenetically silencing the expression of MGMT [209]. | |
| Oncolytic viruses | Downregulate the expression of MGMT [210,211]. | |
| Others | Autoantibodies | May overcome the resistance to TMZ in glioma cells [213]. |
| New agent-KL-50 | Induce cell killing selectively in MGMT-silenced tumors, independent of MMR [214]. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Fang, Q. The Versatile Attributes of MGMT: Its Repair Mechanism, Crosstalk with Other DNA Repair Pathways, and Its Role in Cancer. Cancers 2024, 16, 331. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers16020331
AMA Style
Fang Q. The Versatile Attributes of MGMT: Its Repair Mechanism, Crosstalk with Other DNA Repair Pathways, and Its Role in Cancer. Cancers. 2024; 16(2):331. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers16020331
Chicago/Turabian StyleFang, Qingming. 2024. "The Versatile Attributes of MGMT: Its Repair Mechanism, Crosstalk with Other DNA Repair Pathways, and Its Role in Cancer" Cancers 16, no. 2: 331. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers16020331
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.