Investigation of MoOx/Al2O3 under Cyclic Operation for Oxidative and Non-Oxidative Dehydrogenation of Propane


 , , and
, , and
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Activity Tests
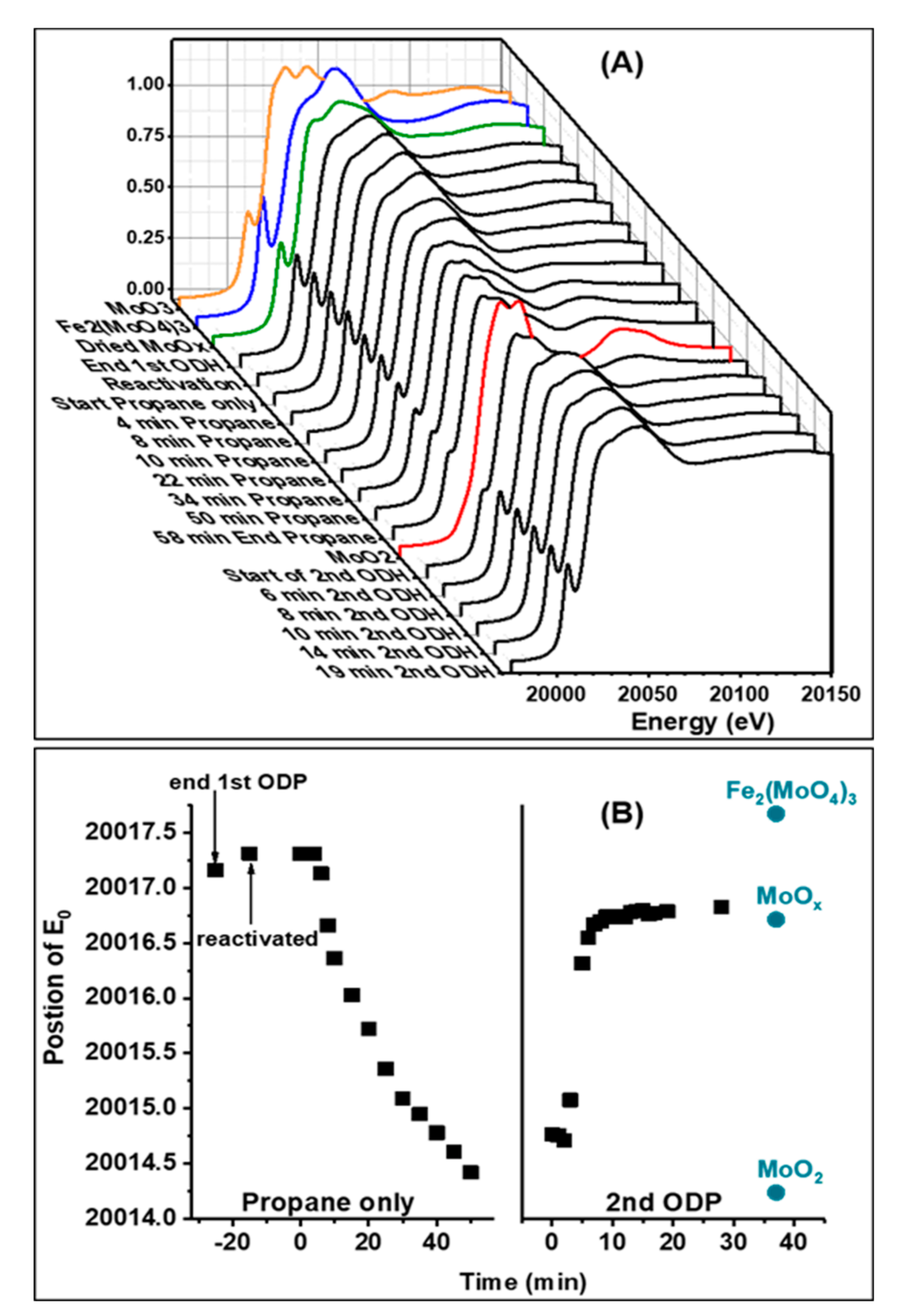
2.2. In-Situ XANES
2.3. Raman Spectra
2.4. Diffuse Reflectance Infrared Fourier Transform Spectroscopy (DRIFTS)
2.5. Inelastic Neutron Scattering (INS) Spectroscopy
3. Materials and Methods
3.1. Catalyst Synthesis
3.2. Activity Tests
3.3. In Situ X-ray Absorption Near Edge Structure (XANES)
3.4. Characterization
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Data Availability
References
- Kung, H.H. Oxidative dehydrogenation of light (C2 to C4) alkanes. Adv. Catal. 1995, 40, 1–38. [Google Scholar] [CrossRef]
- Cavani, F.; Trifirò, F. The oxidative dehydrogenation of ethane and propane as an alternative way for the production of light olefins. Catal. Today 1995, 24, 307–313. [Google Scholar] [CrossRef]
- Mamedov, E.A.; Cortés-Corberan, V. Oxidative dehydrogenation of lower alkanes on vanadium oxide-based catalysts. The present state of the art and outlooks. Appl. Catal. A 1995, 127, 1–40. [Google Scholar] [CrossRef]
- Wachs, I.E.; Weckhuysen, B.M. Structure and reactivity of surface vanadium oxide species on oxide supports. Appl. Catal. A 1997, 157, 67–90. [Google Scholar] [CrossRef] [Green Version]
- Blasco, T.; López-Nieto, J.M. Oxidative dyhydrogenation of short chain alkanes on supported vanadium oxide catalysts. Appl. Catal. A 1997, 157, 117–142. [Google Scholar] [CrossRef]
- Bañares, M.A. Supported metal oxide and other catalysts for ethane conversion: A review. Catal. Today 1999, 51, 319–348. [Google Scholar] [CrossRef]
- Wachs, I.E. Raman and IR studies of surface metal oxide species on oxide supports: Supported metal oxide catalysts. Catal. Today 1996, 27, 437–455. [Google Scholar] [CrossRef]
- Radhakrishnan, R.; Reed, C.; Oyama, S.T.; Seman, M.; Kondo, J.N.; Domen, K.; Ohminami, Y.; Asakura, K. Variability in the structure of supported MoO3 Catalysts: Studies using Raman and x-ray absorption spectroscopy with ab initio calculations. J. Phys. Chem. B 2001, 105, 8519–8530. [Google Scholar] [CrossRef]
- Bañares, M.A.; Wachs, I.E. Molecular structures of supported metal oxide catalysts under different environments. J. Raman Spectrosc. 2002, 33, 359–380. [Google Scholar] [CrossRef]
- Briand, L.E.; Tkachenko, O.P.; Guraya, M.; Wachs, I.E.; Grünert, W. Methodical aspects in the surface analysis of supported molybdena catalysts. Surf. Interface Anal. 2004, 36, 238–245. [Google Scholar] [CrossRef]
- Tian, H.; Roberts, C.A.; Wachs, I.E. Molecular structural determination of molybdena in different environments: Aqueous solutions, bulk mixed oxides, and supported MoO3 catalysts. J. Phys. Chem. C 2010, 114, 14110–14120. [Google Scholar] [CrossRef]
- Bergwerff, J.A.; Visser, T.; Leliveld, B.R.G.; Rossenaar, B.D.; de Jong, K.P.; Weckhuysen, B.M. Envisaging the physicochemical processes during the preparation of supported catalysts: Raman microscopy on the impregnation of Mo onto Al2O3 extrudates. J. Am. Chem. Soc. 2004, 126, 14548–14556. [Google Scholar] [CrossRef] [Green Version]
- Bentrup, U.; Radnik, J.; Armbruster, U.; Martin, A.; Leiterer, J.; Emmerling, F.; Brückner, A. Linking simultaneous in situ WAXS/SAXS/Raman with Raman/ATR/UV–vis Spectroscopy: Comprehensive insight into the synthesis of molybdate catalyst precursors. Top. Catal. 2009, 52, 1350–1359. [Google Scholar] [CrossRef]
- Gibson, E.K.; Zandbergen, M.W.; Jacques, S.D.M.; Biao, C.; Cernik, R.J.; O’Brien, M.G.; Di Michiel, M.; Weckhuysen, B.M.; Beale, A.M. Noninvasive spatiotemporal profiling of the processes of impregnation and drying within Mo/Al2O3 catalyst bodies by a combination of X-ray absorption tomography and diagonal offset Raman spectroscopy. ACS Catal. 2013, 3, 339–347. [Google Scholar] [CrossRef]
- Chakrabarti, A.; Wachs, I.E. Molecular structure–reactivity relationships for olefin metathesis by Al2O3-supported surface MoOx sites. ACS Catal. 2018, 8, 949–959. [Google Scholar] [CrossRef]
- Chen, K.; Xie, S.; Iglesia, E.; Bell, A.T. Structure and Properties of zirconia-supported molybdenum oxide catalysts for oxidative dehydrogenation of propane. J. Catal. 2000, 189, 421–430. [Google Scholar] [CrossRef] [Green Version]
- Christodoulakis, A.; Heracleous, E.; Lemonidou, A.A.; Boghosin, S. An operando Raman study of structure and reactivity of alumina-supported molybdenum oxide catalysts for the oxidative dehydrogenation of ethane. J. Catal. 2006, 242, 16–25. [Google Scholar] [CrossRef]
- Grünert, W.; Stakheev, A.Y.; Feldhaus, R.; Anders, K.; Shpiro, E.S.; Minachev, K.M. Reduction and aromatization activity of MoO3/Al2O3 Catalysts: The identification of the active Mo oxidation state on the basis of reinterpreted Mo 3d XPS spectra. Studies. Surf. Sci. Catal. 1993, 75, 1053–1064. [Google Scholar] [CrossRef]
- Banks, R.L. Olefin metathesis: Technology and applications. In Applied Industrial Catalysis; Leach, B.E., Ed.; Academic Press: Orlando, FL, USA, 1984; pp. 215–239. [Google Scholar]
- Brandhorst, M.; Cristol, S.; Capron, M.; Dujadin, C.; Vezin, H.; Le bourdon, G.; Payen, E. Catalytic oxidation of methanol on Mo/Al2O3 catalyst: An EPR and Raman/infrared operando spectroscopies study. Catal. Today 2006, 113, 34–39. [Google Scholar] [CrossRef]
- Bridgewater, A.J.; Burch, R.; Mitchell, P.C.H. Molybdenum/carbon catalysts for reforming reactions. J. Chem. Soc. Faraday I 1980, 76, 1811–1820. [Google Scholar] [CrossRef]
- Meunier, F.C.; Yasmeen, A.; Ross, J.R.H. Oxidative dehydrogenation of propane over molybdenum-containing catalysts. Catal. Today 1997, 37, 33–42. [Google Scholar] [CrossRef]
- Abello, M.C.; Gomez, M.F.; Ferretti, O. Mo/γ-Al2O3 catalysts for the oxidative dehydrogenation of propane.: Effect of Mo loading. Appl. Catal. A 2001, 207, 421–431. [Google Scholar] [CrossRef]
- Heracleous, E.; Machli, M.; Lemonidou, A.A.; Vasalos, I.A. Oxidative dehydrogenation of ethane and propane over vanadia and molybdena supported catalysts. J. Mol. Catal. A Chem. 2005, 232, 29–39. [Google Scholar] [CrossRef]
- Abello, M.C.; Gomez, M.F.; Casella, M.; Ferretti, O.A.; Bañares, M.A.; Fierro, J.L.G. Characterization and performance for propane oxidative dehydrogenation of Li-modified MoO3/Al2O3 catalysts. Appl. Catal. A Gen. 2003, 251, 435–447. [Google Scholar] [CrossRef]
- Rostom, S.; de Lasa, H.I. Propane oxidative dehydrogenation using consecutive feed injections and fluidizable VOx/γAl2O3 and VOx/ZrO2–γAl2O3 catalysts. Ind. Eng. Chem. Res. 2017, 56, 13109–13124. [Google Scholar] [CrossRef]
- Chen, S.; Zeng, L.; Mu, R.; Xiong, C.; Zhao, Z.-J.; Zhao, C.; Pei, C.; Peng, L.; Luo, J.; Fan, L.-S.; et al. Modulating lattice oxygen in dual-functional Mo–V–O mixed oxides for chemical looping oxidative dehydrogenation. J. Am. Chem. Soc. 2019, 141, 18653–18657. [Google Scholar] [CrossRef] [PubMed]
- Beale, A.M.; van der Eerden, A.M.J.; Kervinen, K.; Newton, M.A.; Weckhuysen, B.M. Adding a third dimension to operando spectroscopy: A combined UV-Vis, Raman and XAFS setup to study heterogeneous catalysts under working conditions. Chem Commun. 2005, 3015–3017. [Google Scholar] [CrossRef]
- Brookes, C.; Wells, P.P.; Dimitratos, N.; Jones, W.; Gibson, E.K.; Morgan, D.J.; Cibin, G.; Nicklin, C.; Mora-Fonz, D.; Scanlon, D.O.; et al. The nature of the molybdenum surface in iron molybdate. The active phase in selective methanol oxidation. J. Phys. Chem. C 2014, 118, 26155–26161. [Google Scholar] [CrossRef]
- Chen, K.; Xie, S.; Bell, A.T.; Iglesia, E. Alkali effects on molybdenum oxide catalysts for the oxidative dehydrogenation of propane. J. Catal. 2000, 195, 244–252. [Google Scholar] [CrossRef] [Green Version]
- Brookes, C.; Bowker, M.; Gibson, E.K.; Gianolio, D.; Mohammed, K.M.H.; Parry, S.; Rogers, S.M.; Silverwood, I.P.; Wells, P.P. In situ spectroscopic investigations of MoOx/Fe2O3 catalysts for the selective oxidation of methanol. Catal. Sci. Technol. 2016, 6, 722–730. [Google Scholar] [CrossRef] [Green Version]
- Hu, H.; Wachs, I.E.; Bare, S.R. Surface Structures of supported molybdenum oxide catalysts: Characterization by Raman and Mo L3-edge XANES. J. Phys. Chem. 1995, 99, 10897–10910. [Google Scholar] [CrossRef]
- Payen, E.; Grimblot, J.; Kasztelan, S. Study of Oxidic and Reduced Alumina-Supported Molybdate and Heptamolybdate Species by in Situ Laser Raman Spectroscopy. J. Phys. Chem. 1987, 91, 6642–6648. [Google Scholar] [CrossRef]
- Jawhari, T.; Roid, A.; Casado, J. Raman spectroscopic characterization of some commercially available carbon black materials. Carbon 1995, 33, 1561–1565. [Google Scholar] [CrossRef]
- Coccato, A.; Jehlicka, J.; Moens, L.; Vandenabeele, P. Raman spectroscopy for the investigation of carbon-based black pigments. J. Raman Spectrosc. 2015, 46, 1003–1015. [Google Scholar] [CrossRef] [Green Version]
- Eberly, P.E.; Kimberlin, C.N.; Miller, W.H.; Drushel, H.V. Coke formation on silica-alumina cracking catalysts. Ind. Eng. Chem. Process. Des. Dev. 1966, 5, 193–198. [Google Scholar] [CrossRef]
- Al-Abadleh, H.A.; Grassian, V.H. FT-IR study of water adsorption on aluminum oxide surfaces. Langmuir 2003, 19, 341–347. [Google Scholar] [CrossRef]
- Matam, S.K.; Newton, M.A.; Weidenkaff, A.; Ferri, D. Time resolved operando spectroscopic study of the origin of phosphorus induced chemical aging of model three-way catalysts Pd/Al2O3. Catal. Today 2013, 205, 3–9. [Google Scholar] [CrossRef]
- Wijnja, H.; Schulthess, C.P. ATR–FTIR and DRIFT spectroscopy of carbonate species at the aged γ-Al2O3/water interface. Spectrochimica Acta Part. A 1999, 55, 861–872. [Google Scholar] [CrossRef]
- Davydov, A. Molecular Spectroscopy of Oxide Catalyst Surfaces; John Wiley & Sons Ltd.: Chichester, UK, 2003; p. 32. [Google Scholar]
- Magg, N.; Immaraporn, B.; Giorgi, J.B.; Schroeder, T.; Baumer, M.; Dobler, J.; Wu, Z.; Kondratenko, E.; Cherian, M.; Baerns, M.; et al. Vibrational spectra of alumina- and silica-supported vanadia revisited: An experimental and theoretical model catalyst study. J. Catal. 2004, 226, 88–100. [Google Scholar] [CrossRef]
- Pazè, C.; Sazak, B.; Zecchina, A.; Dwyer, J. FTIR and UV−vis spectroscopic study of interaction of 1-butene on H−ferrierite zeolite. J. Phys. Chem. B 1999, 103, 9978–9986. [Google Scholar] [CrossRef]
- Lin-Vien, D.; Colthup, N.B.; Fateley, W.G.; Grasselli, J.G. The Handbook of Infrared and Raman Characteristic Frequencies of Organic Molecules; Academic Press: Boston, MA, USA, 1991. [Google Scholar]
- Rozwadowski, M.; Lezanska, M.; Wloch, J.; Erdmann, K.; Golembiewski, R.; Kornatowski, J. Investigation of coke deposits on Al-MCM-41. Chem. Mater. 2001, 13, 1609–1616. [Google Scholar] [CrossRef]
- Lercher, J.A.; Colombier, C.; Noller, H. Acid–base properties of alumina–magnesia mixed oxides. Part 4.—Infrared study of adsorption of carbon dioxide. J. Chem. Soc. Faraday Trans. I 1984, 80, 949–959. [Google Scholar] [CrossRef]
- Parker, S.F.; Lennon, D.; Albers, P.W. Vibrational spectroscopy with neutrons—A review of new directions. Appl. Spectrosc. 2011, 65, 1325–1341. [Google Scholar] [CrossRef]
- Albers, P.W.; Bösing, S.; Rotgerink, H.L.; Ross, D.K.; Parker, S.F. Inelastic neutron scattering study on the influence of after-treatments on different technical cokes of varying impurity level and sp2/sp3 character. Carbon 2002, 40, 1549–1558. [Google Scholar] [CrossRef]
- Albers, P.W.; Weber, W.; Möbus, K.; Wieland, S.D.; Parker, S.F. Neutron scattering study of the terminating protons in the basic structural units of non-graphitising and graphitising carbons. Carbon 2016, 109, 239–245. [Google Scholar] [CrossRef] [Green Version]
- Jackson, S.D.; Grenfell, J.; Matheson, I.M.; Munro, S.; Raval, R.; Webb, G. Deactivation and regeneration of alkane dehydrogenation catalysts. In Proceedings of the 7th International Symposium on Catalyst Deactivation, Cancun, Mexico, 5–8 October 1997; Bartholomew, C.H., Fuentes, G.A., Eds.; Studies in Surface Science and Catalysis. Elsevier: Amsterdam, The Netherlands, 1997; Volume 111, pp. 167–174. [Google Scholar] [CrossRef]
- Gibson, E.K.; Beale, A.M.; Catlow, C.R.A.; Chutia, A.; Gianolio, D.; Gould, A.; Kroner, A.; Mohammed, K.M.H.; Perdjon, M.; Rogers, S.M.; et al. Restructuring of AuPd nanoparticles studied by a combined XAFS/DRIFTS approach. Chem. Mater. 2015, 27, 3714–3720. [Google Scholar] [CrossRef] [Green Version]
- Newville, M. IFEFFIT: Interactive XAFS analysis and FEFF fitting. J. Synchrotron Rad. 2001, 8, 322–324. [Google Scholar] [CrossRef]
- Ravel, B.; Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: Data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Rad. 2005, 12, 537–541. [Google Scholar] [CrossRef] [Green Version]
- Gibson, E.; Craswell, J.; Hellier, P.; Catlow, C.R.A.; Parker, S.F.; Matam, S.K. Investigating the Speciation of Hydrocarbonaceous Deposits Formed during Oxidative Dehydrogenation of Propane, STFC ISIS Neutron and Muon Source. 2017. Available online: https://data.isis.stfc.ac.uk/doi/STUDY/103200776/ (accessed on 23 October 2020).
- Gibson, E.; Craswell, J.; Hellier, P.; Catlow, C.R.A.; Parker, S.F.; Matam, S.K. Investigating the speciation of hydrocarbonaceous deposits formed during oxidative dehydrogenation of propane, STFC ISIS Neutron and Muon Source. 2018. Available online: https://data.isis.stfc.ac.uk/doi/STUDY/103195237/ (accessed on 23 October 2020). [CrossRef]
- The University of Glasgow Data Repository. Available online: http://0-dx-doi-org.brum.beds.ac.uk/10.5525/gla.researchdata.1092 (accessed on 23 November 2020).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Abs-Scatterer | E0 (eV) | Coordination Number | R (Å) | σ2 | Rfactor |
|---|---|---|---|---|---|---|
| End of 1st ODP | Mo-O | −8.0 ± 0.3 | 4 ± 1 | 1.74 ± 0.01 | 0.006 ± 0.004 | 0.05 |
| End of DP | Mo-O | 5 ± 8 | 1.0 ± 0.7 | 1.76 ± 0.03 | 0.003 | 0.1 |
| Mo-O | 2.8 ± 0.8 | 2.06 ± 0.04 | 0.003 | |||
| End of 2nd ODP | Mo-O | −9 ± 7 | 4.2 ± 0.6 | 1.74 ± 0.04 | 0.005 | 0.04 |
| Mo-O | 1.3 ± 0.9 | 1.96 ± 0.07 | 0.003 |
| Sample | Wavenumber (cm−1) | Mode 2 | Mo Species |
|---|---|---|---|
| Fresh | 1025 | ν(Mo=O) | Al2(MoO4)3 |
| 1000 | ν(Mo=O) | Al2(MoO4)3 or MoO3 | |
| 993 | ν(Mo–O) | Al2(MoO4)3 or MoO3 | |
| Used Type 1 | 965 | ν(Mo–O) | Octahedral Mo species |
| 930 | νs(Mo–O) | Dimeric | |
| Fresh + Used Type 1 | 884 | νas(Mo–O) | Dimeric |
| Used Type 2 | 840 | ν(Mo–O–Mo) or ν(Mo–O–Al) | Mo5+ |
| Fresh + Used Type 1 | 814 | νas(Mo–O–Mo) | MoO3 |
| Fresh + Used Type 1 + 2 | 374 | δ(Mo=O) | Al2(MoO4)3 |
| Used Type 2 | 267 | δ(Mo–O–Mo) | Polymerized species |
| Catalyst | Vtotal (cm3/g) | SBET a (m2/g) | Pore Size b (nm) | Surface Mo Density (Mo/nm2) c | Oxidation State of Mo Oxide d |
|---|---|---|---|---|---|
| Fresh | 0.612 | 81 | 16 | 10.8 | VI |
| Used | 0.610 | 79.0 | 16 | 11.1 | IV < δ < VI |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matam, S.K.; Moffat, C.; Hellier, P.; Bowker, M.; Silverwood, I.P.; Catlow, C.R.A.; Jackson, S.D.; Craswell, J.; Wells, P.P.; Parker, S.F.; et al. Investigation of MoOx/Al2O3 under Cyclic Operation for Oxidative and Non-Oxidative Dehydrogenation of Propane. Catalysts 2020, 10, 1370. https://0-doi-org.brum.beds.ac.uk/10.3390/catal10121370
Matam SK, Moffat C, Hellier P, Bowker M, Silverwood IP, Catlow CRA, Jackson SD, Craswell J, Wells PP, Parker SF, et al. Investigation of MoOx/Al2O3 under Cyclic Operation for Oxidative and Non-Oxidative Dehydrogenation of Propane. Catalysts. 2020; 10(12):1370. https://0-doi-org.brum.beds.ac.uk/10.3390/catal10121370
Chicago/Turabian StyleMatam, Santhosh K., Caitlin Moffat, Pip Hellier, Michael Bowker, Ian P. Silverwood, C. Richard A. Catlow, S. David Jackson, James Craswell, Peter P. Wells, Stewart F. Parker, and et al. 2020. "Investigation of MoOx/Al2O3 under Cyclic Operation for Oxidative and Non-Oxidative Dehydrogenation of Propane" Catalysts 10, no. 12: 1370. https://0-doi-org.brum.beds.ac.uk/10.3390/catal10121370