Comparison of the Reactivity and Structures for the Neutral and Cationic Bis(imino)pyridyl Iron and Cobalt Species by DFT Calculations




Abstract
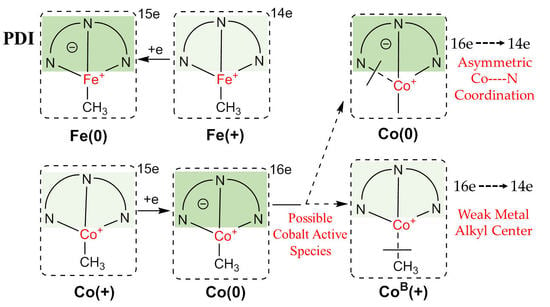
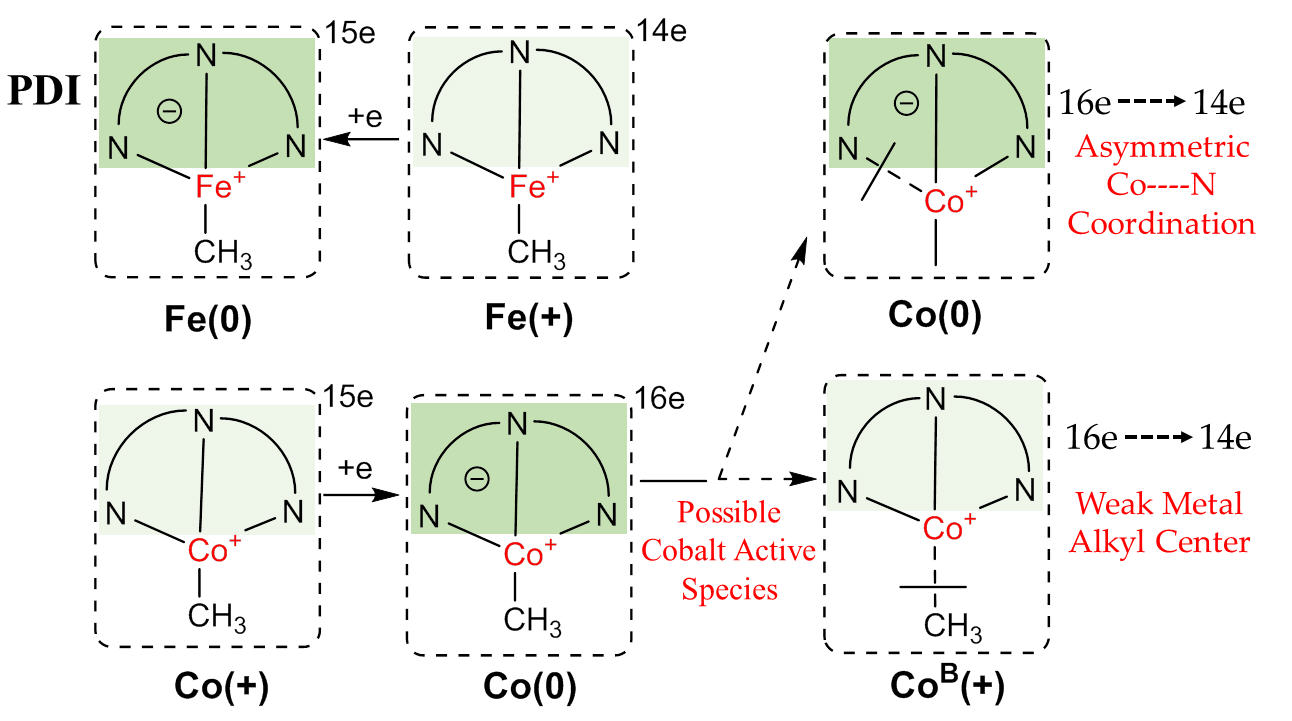
:
1. Introduction
2. Results and Discussion
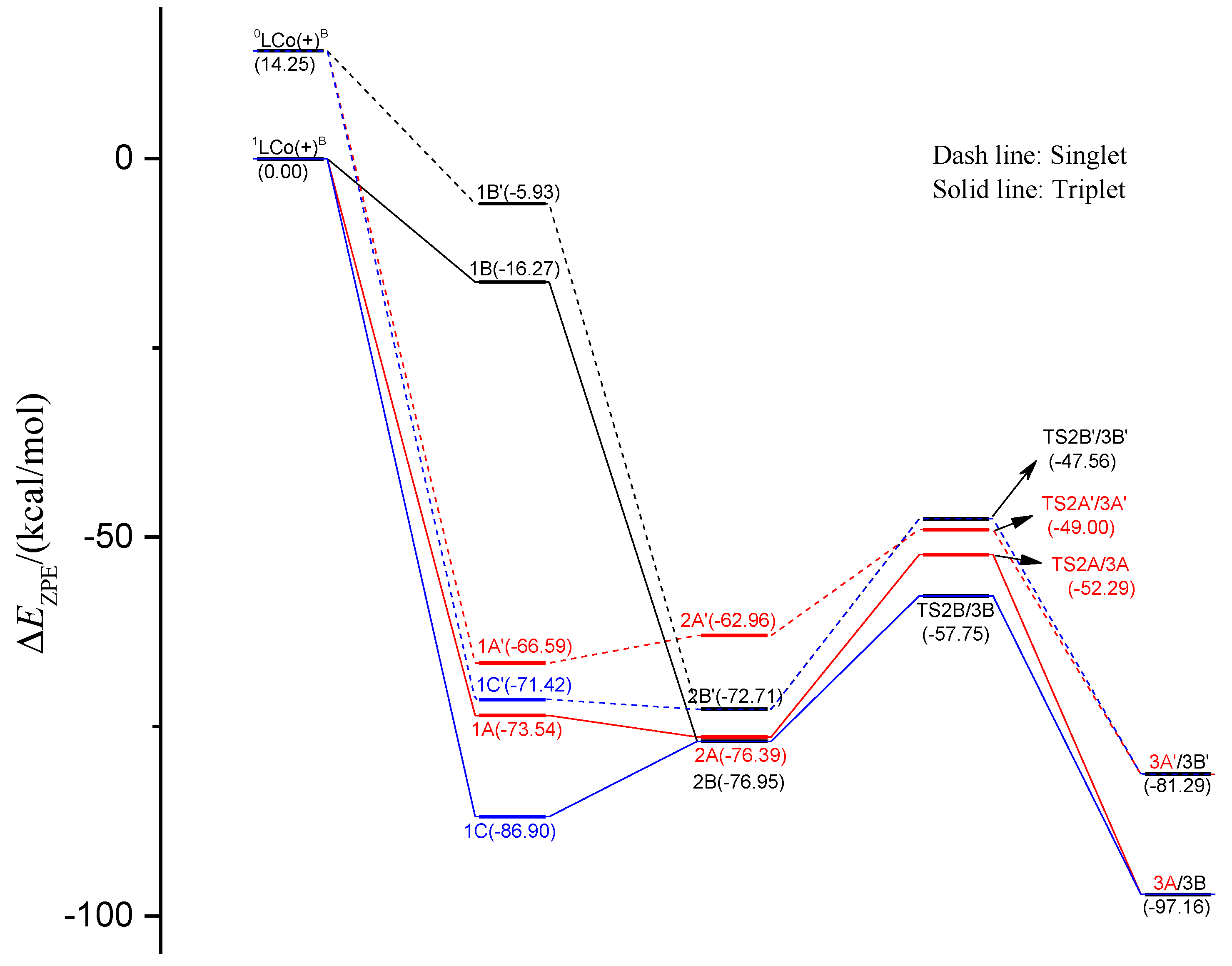
2.1. Energy Profiles and Structures
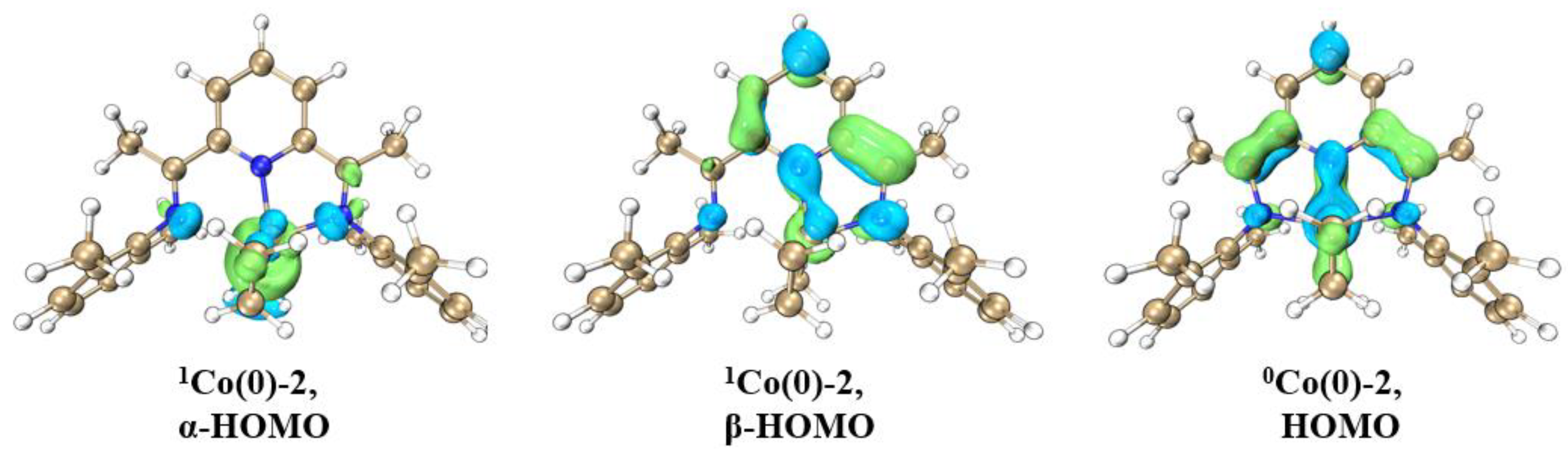
2.1.1. Energy Profiles and Structures of the Neutral LCoCH3 System
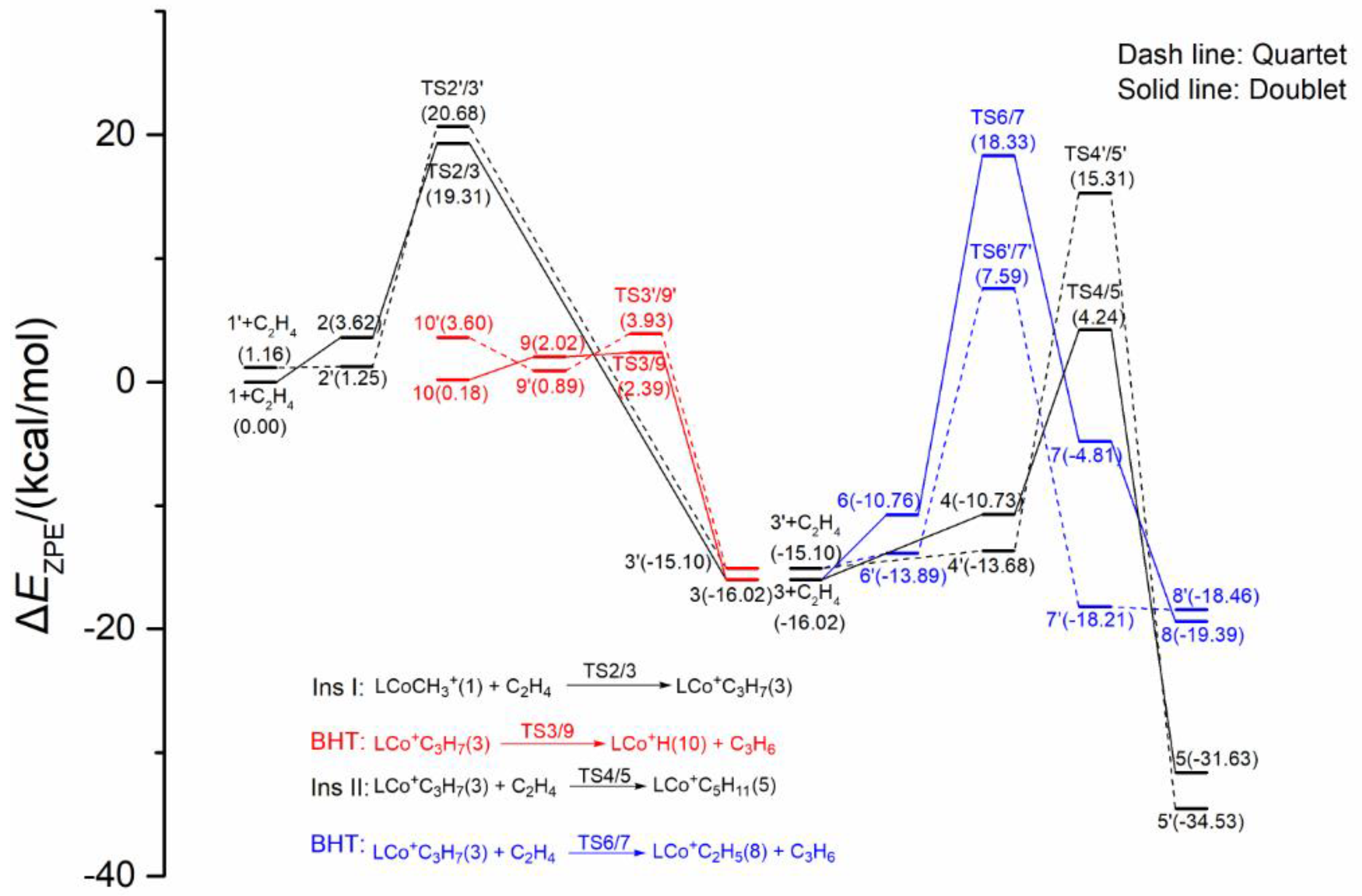
2.1.2. Energy Profiles and Structures of the Cationic LCo+CH3 System
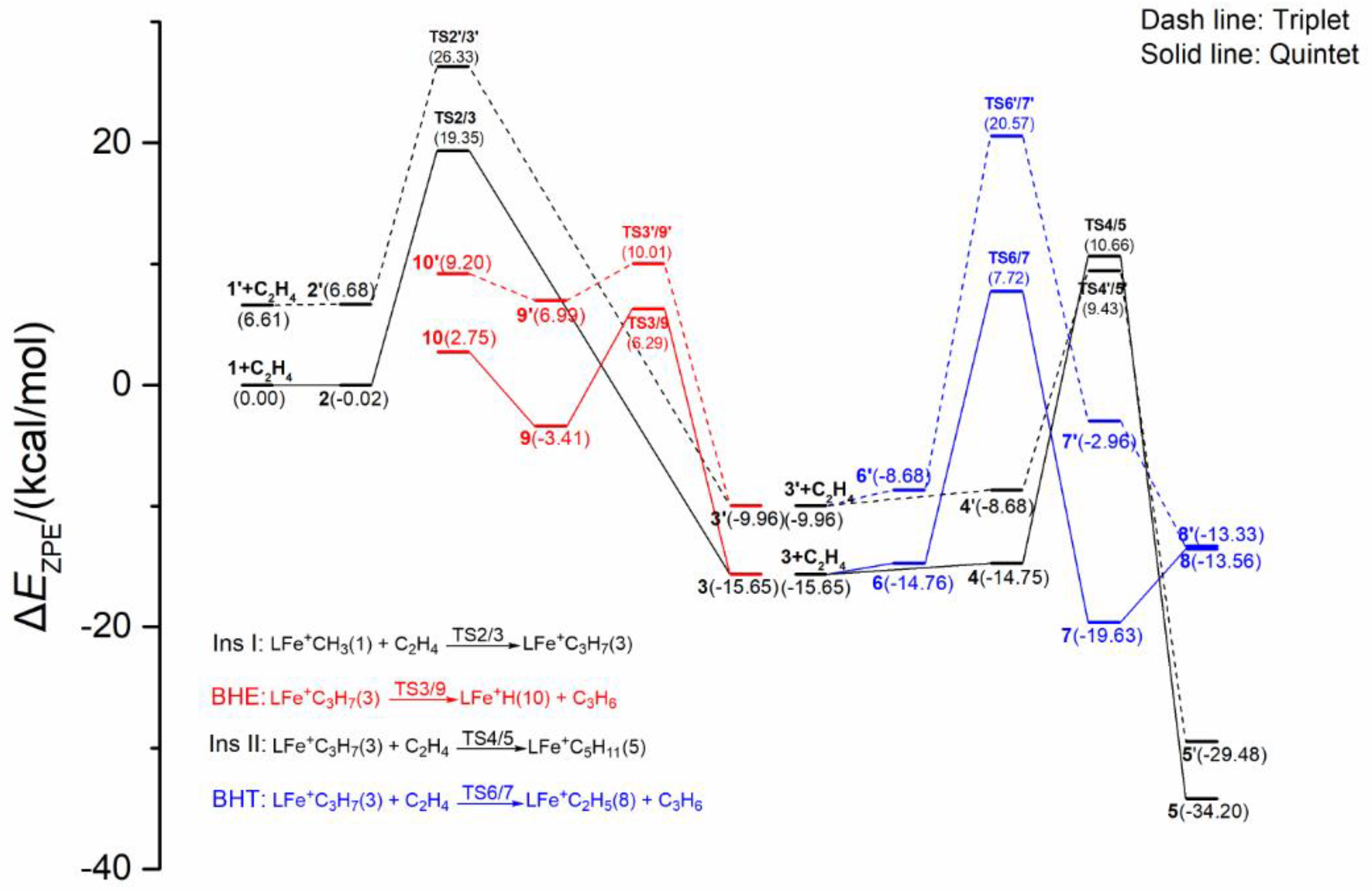
2.1.3. Energy Profiles and Structures of the Cationic LFe+CH3 System
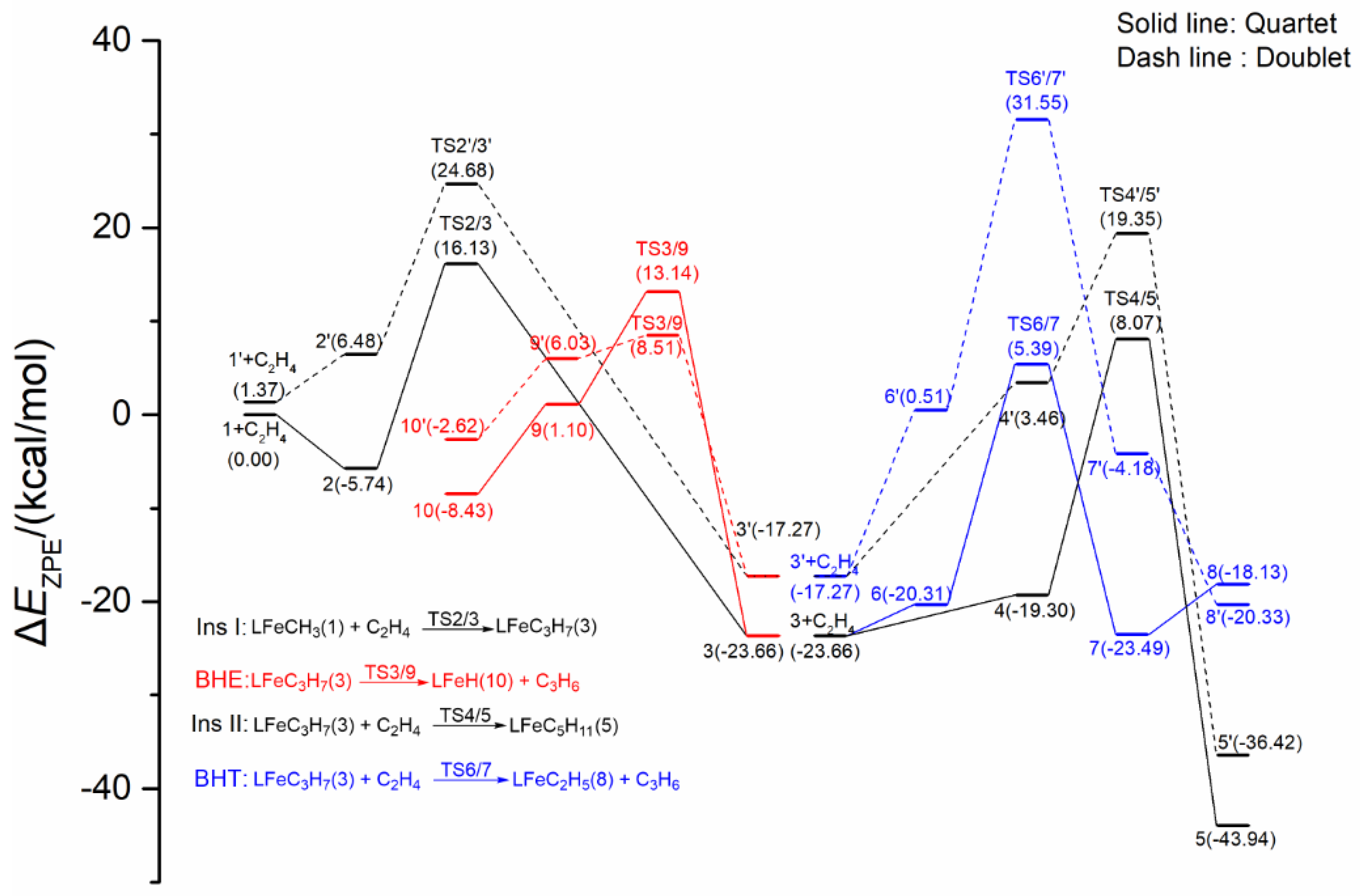
2.1.4. Energy Profiles and Structures of the Neutral LFeCH3 System
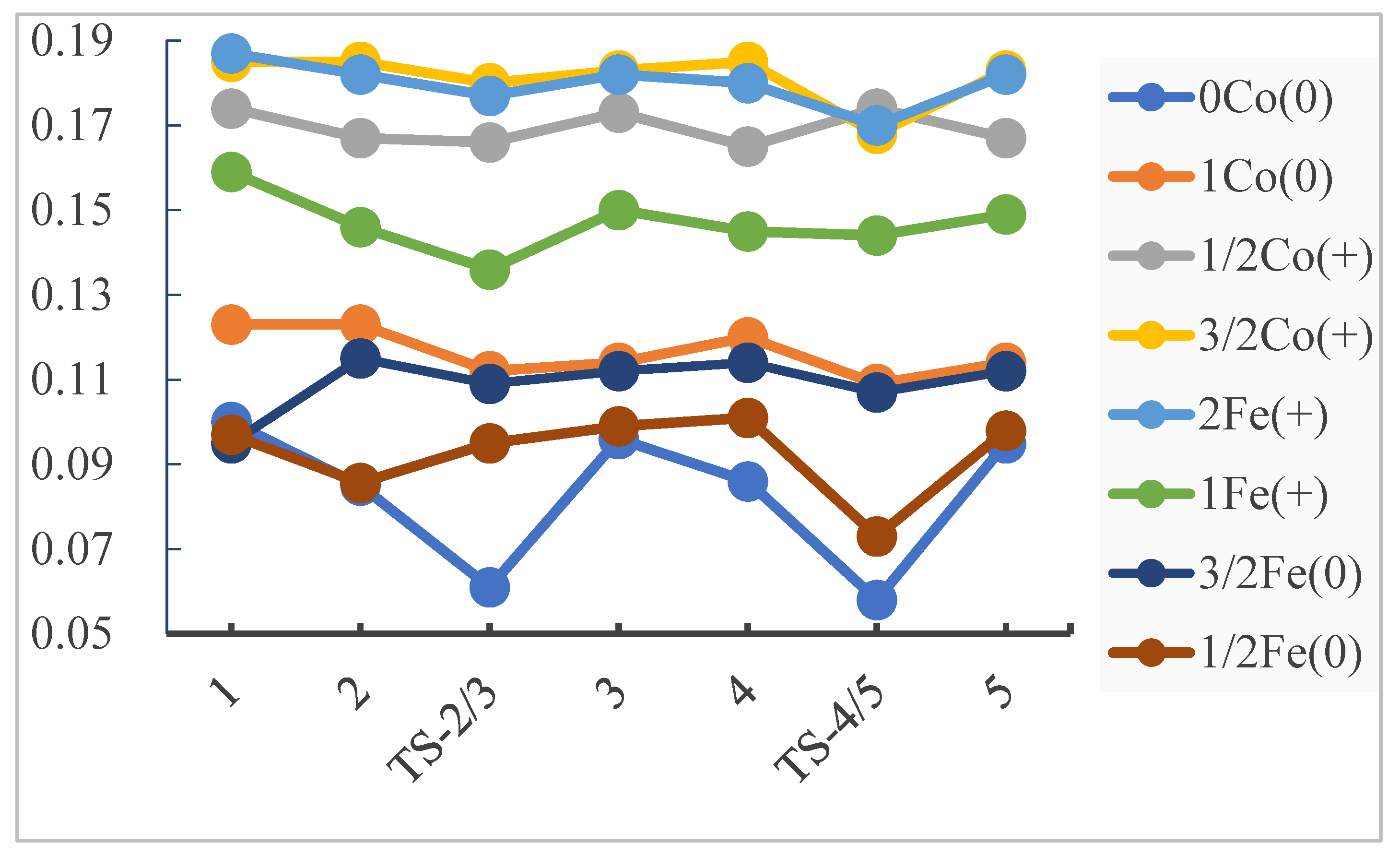
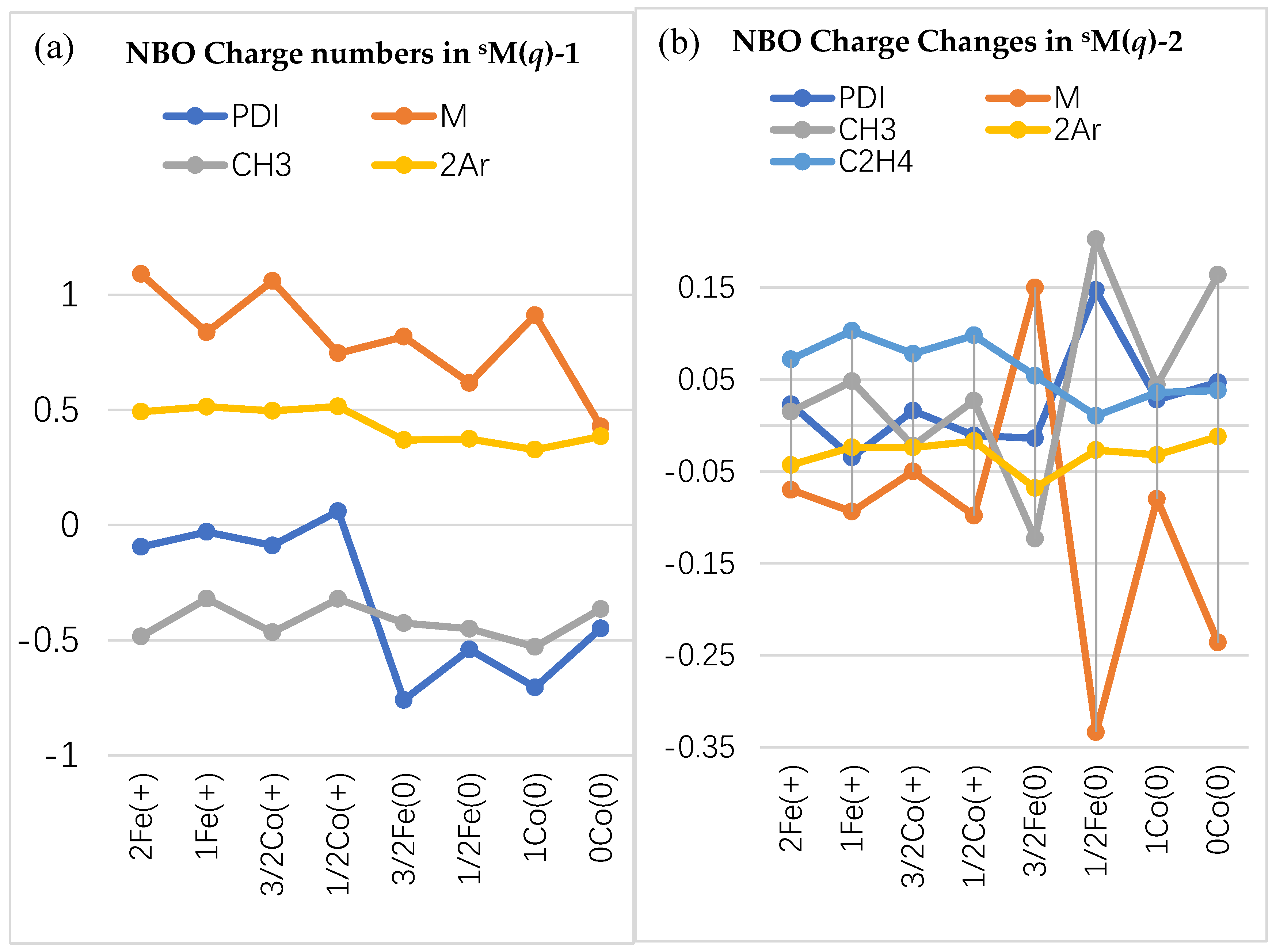
2.2. General Electronic Properties
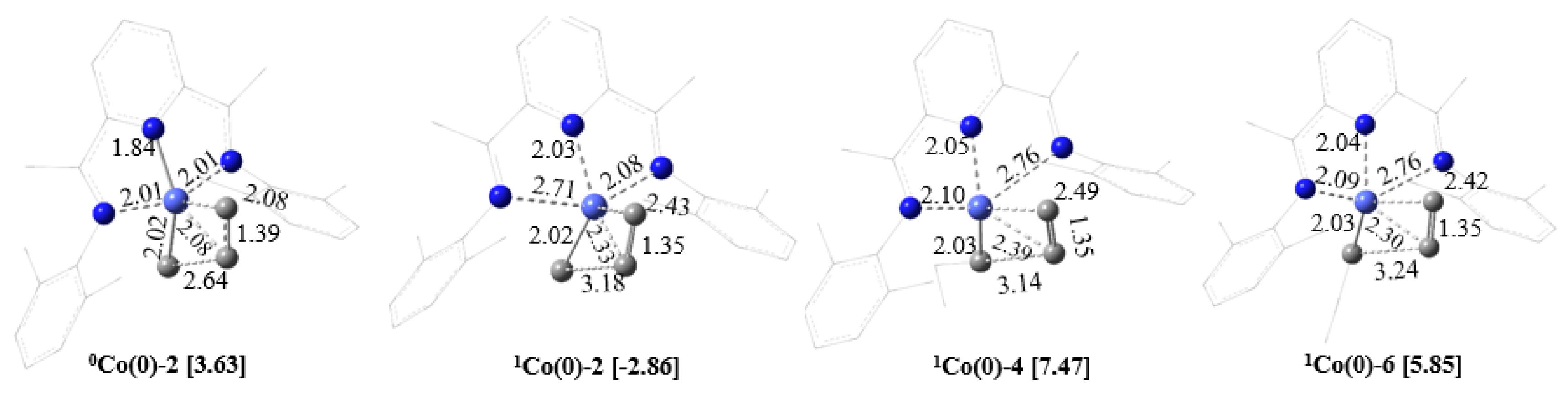
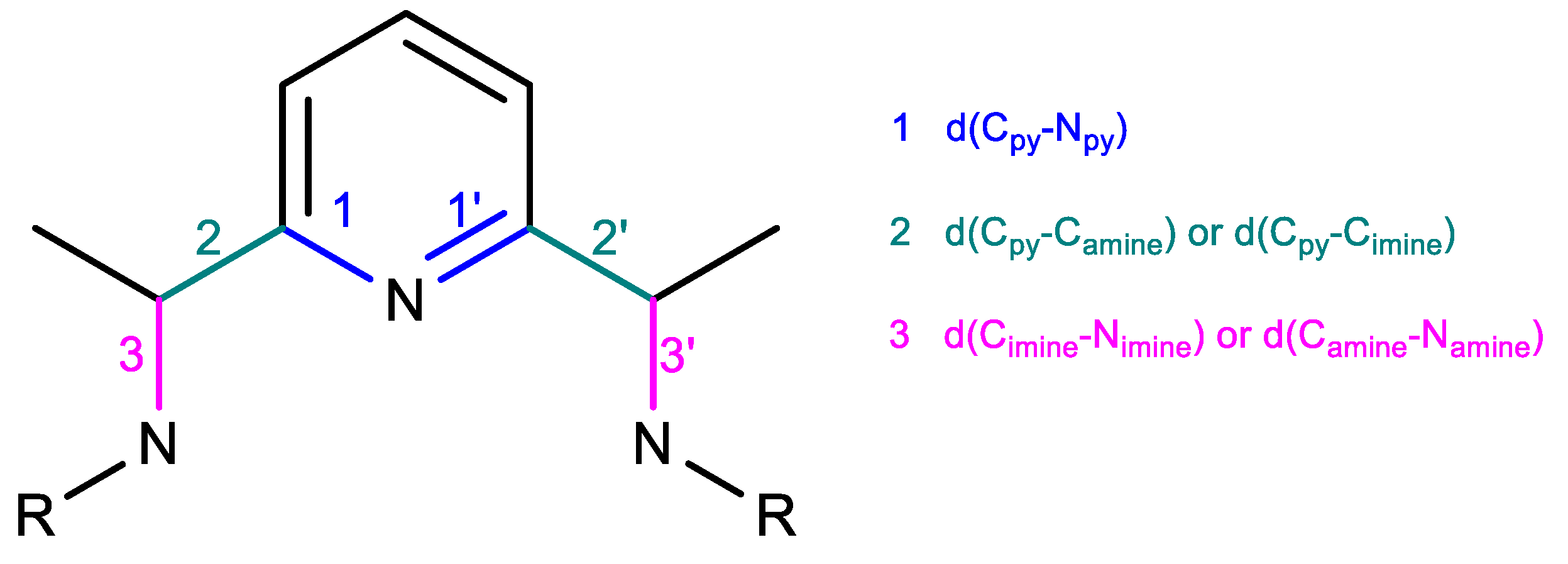
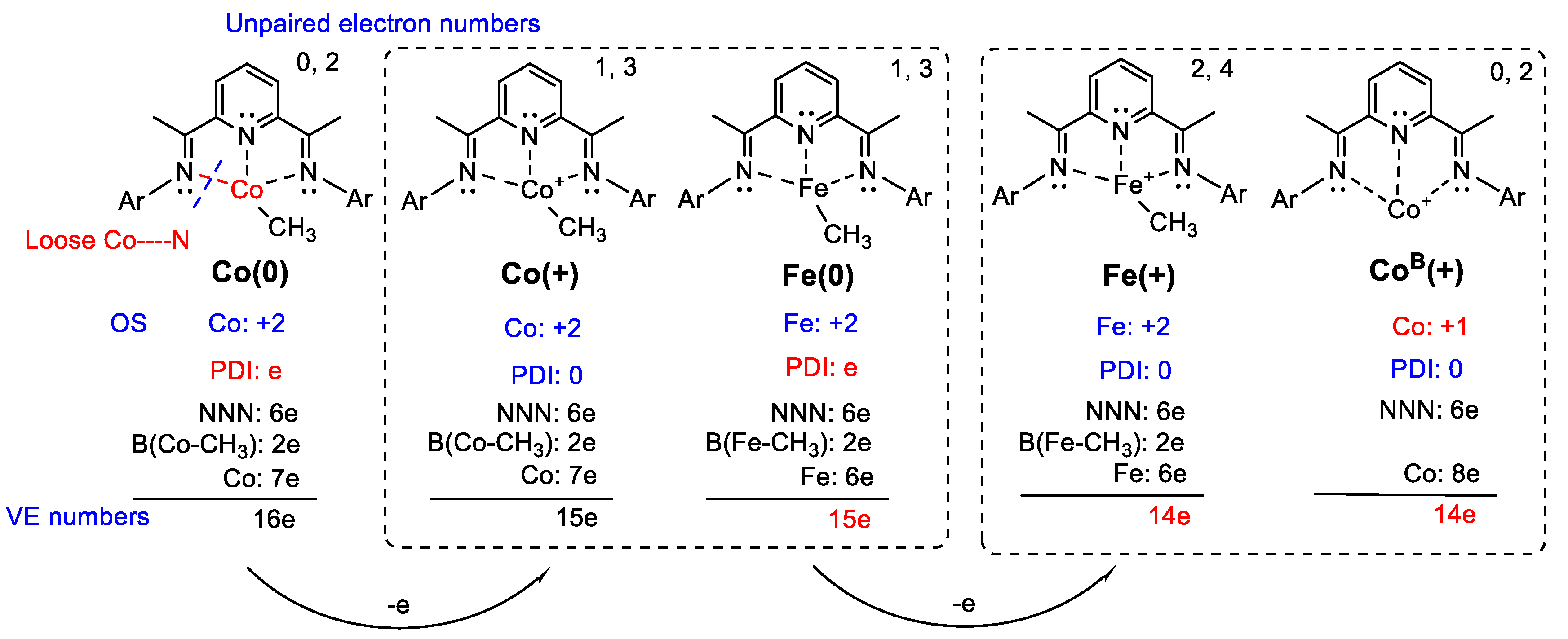
2.2.1. Oxidation Level of the PDI Ligand
2.2.2. Charge Distribution
2.2.3. General Comparison on Valence Electrons
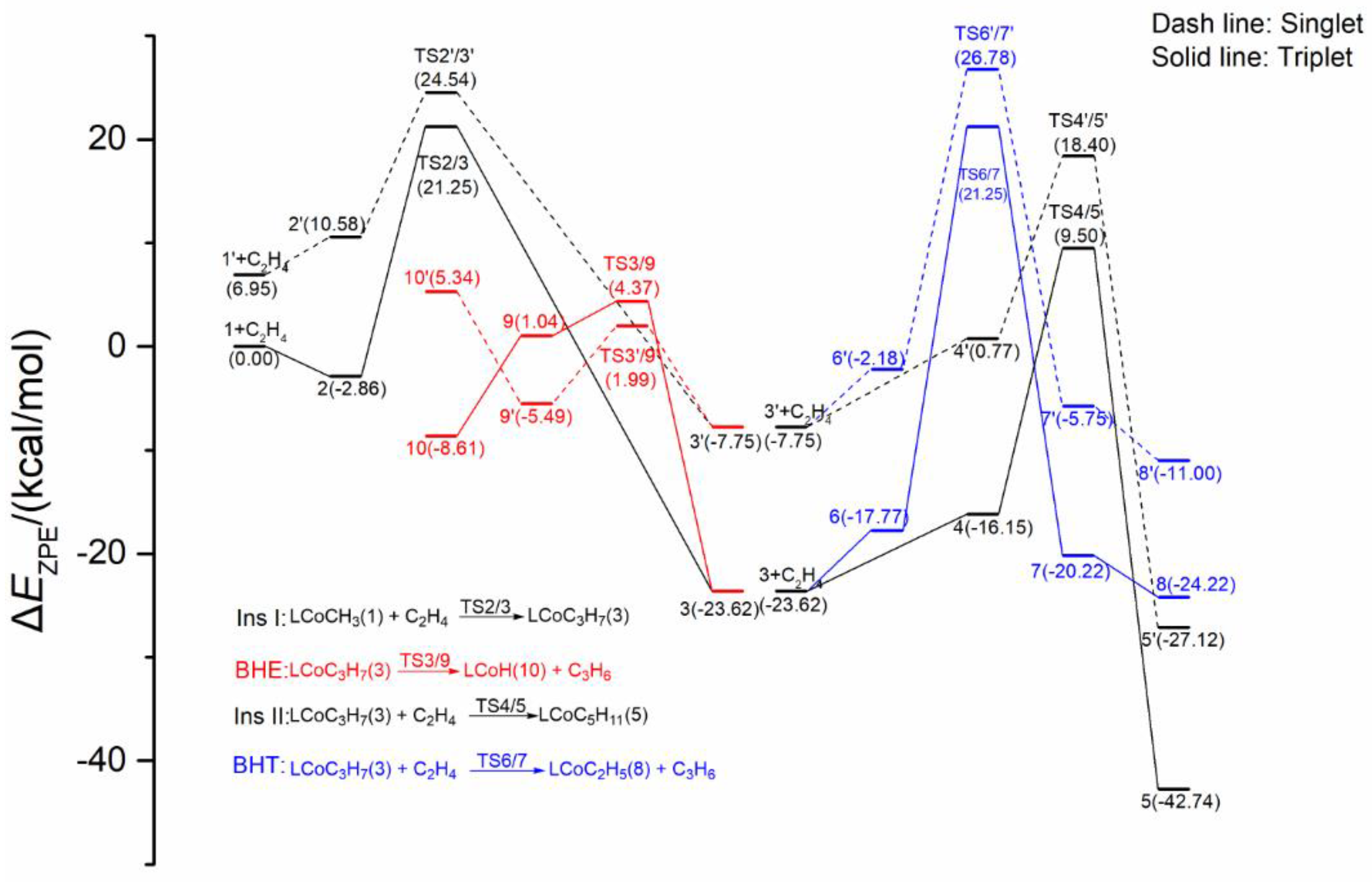
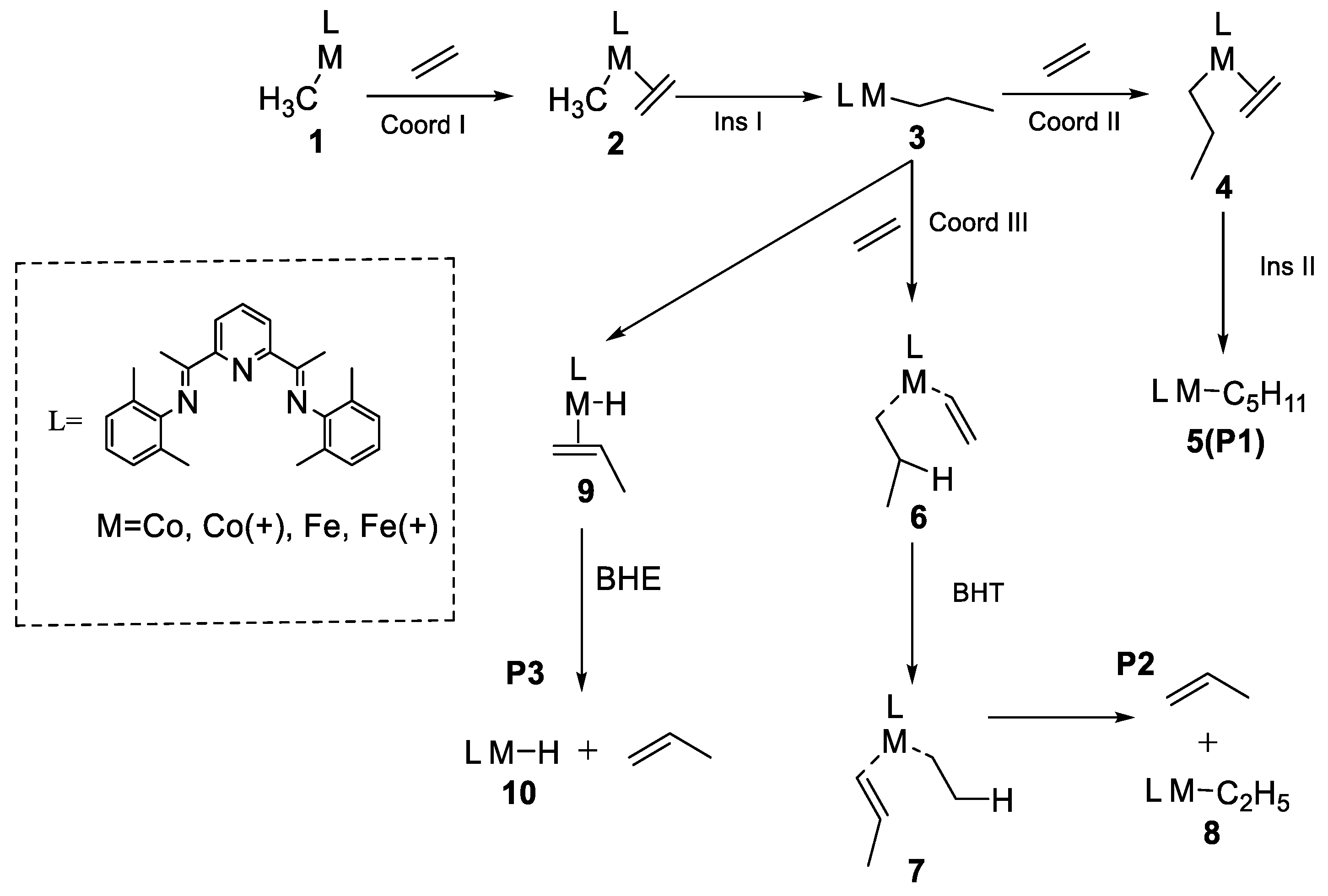
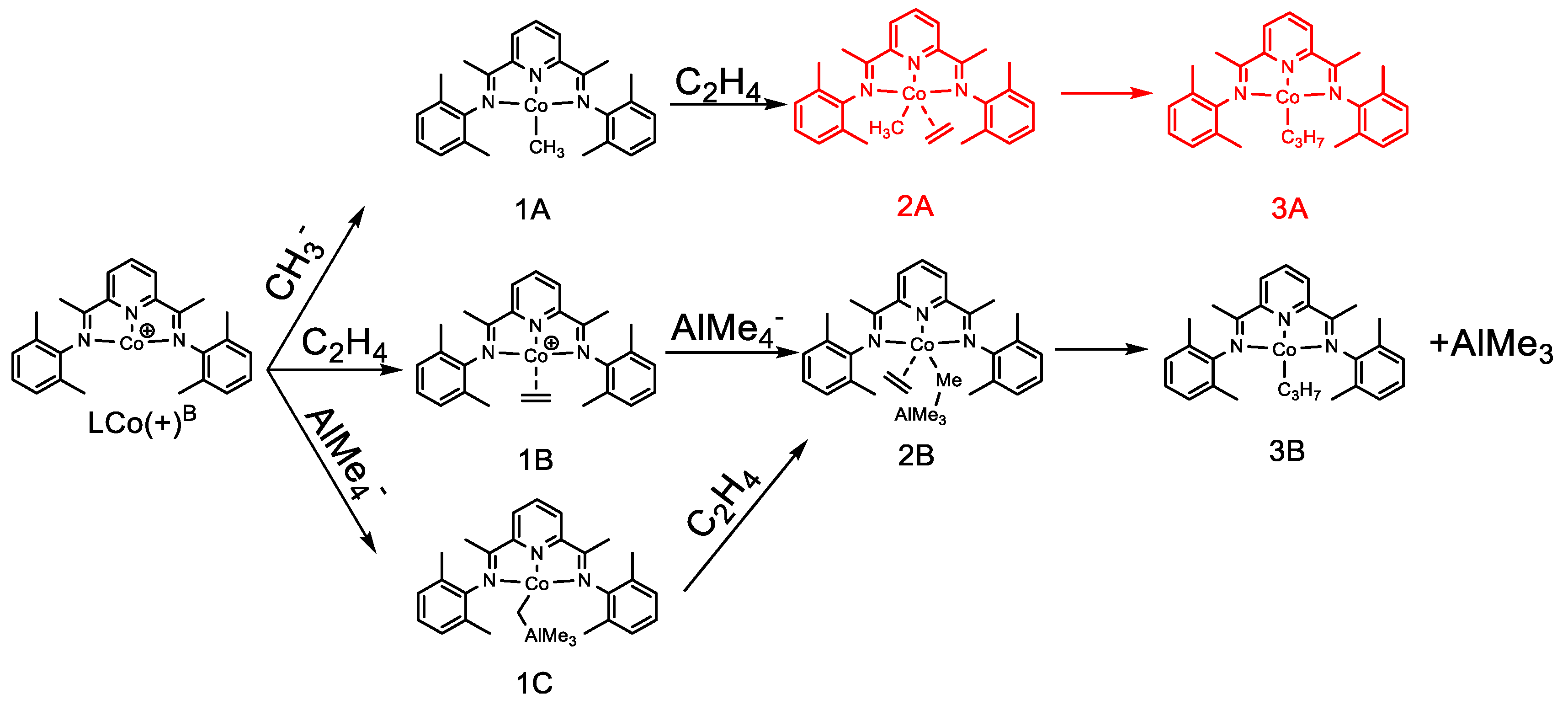
2.3. Possible Catalytic Process for the Cobalt Species
3. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Small, B.L.; Brookhart, M.; Bennett, A.M.A. Highly active iron and cobalt catalysts for the polymerization of ethylene. J. Am. Chem. Soc. 1998, 120, 4049–4050. [Google Scholar] [CrossRef]
- Britovsek, G.J.P.; Gibson, V.C.; Kimberley, B.S.; Maddox, P.J.; McTavish, S.J.; Solan, G.A.; White, A.J.P.; Williams, D.J. Novel olefin polymerization catalysts based on iron and cobalt. Chem. Commun. 1998, 849–850. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, S.; Du, S.; Guo, C.-Y.; Hao, X.; Sun, W.-H. 2-(1-(2,4-Bis((di(4-fluorophenyl)methyl)-6-methylphenylimino)ethyl)-6-(1-(arylimino)ethyl)pyridylmetal (iron or cobalt) Complexes: Synthesis, Characterization, and Ethylene Polymerization Behavior. Macromol. Chem. Phys. 2014, 215, 1797–1809. [Google Scholar] [CrossRef]
- Ahmed, S.; Yang, W.; Ma, Z.; Sun, W.-H. Catalytic Activities of Bis(pentamethylene)pyridyl(Fe/Co) Complex Analogues in Ethylene Polymerization by Modeling Method. J. Phys. Chem. A 2018, 122, 9637–9644. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Zakharov, V.A.; Semikolenova, N.V.; Matsko, M.A.; Solan, G.A.; Sun, W.-H. A comparative kinetic study of ethylene polymerization mediated by iron, cobalt and chromium catalysts bearing the same N,N,N-bis(imino)trihydroquinoline. J. Catal. 2019, 369, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Solan, G.A.; Zhang, W.; Sun, W.-H. Carbocyclic-fused N,N,N-pincer ligands as ring-strain adjustable supports for iron and cobalt catalysts in ethylene oligo-/polymerization. Coord. Chem. Rev. 2018, 363, 92–108. [Google Scholar] [CrossRef] [Green Version]
- Phillips, A.M.F.; Suo, H.; Guedes da Silva, M.d.F.C.; Pombeiro, A.J.L.; Sun, W.-H. Recent developments in vanadium-catalyzed olefin coordination polymerization. Coord. Chem. Rev. 2020, 416, 213332. [Google Scholar] [CrossRef]
- Bariashir, C.; Huang, C.; Solan, G.A.; Sun, W.-H. Recent advances in homogeneous chromium catalyst design for ethylene tri-, tetra-, oligo- and polymerization. Coord. Chem. Rev. 2019, 385, 208–229. [Google Scholar] [CrossRef] [Green Version]
- Suo, H.; Solan, G.A.; Ma, Y.; Sun, W.-H. Developments in compartmentalized bimetallic transition metal ethylene polymerization catalysts. Coord. Chem. Rev. 2018, 372, 101–116. [Google Scholar] [CrossRef] [Green Version]
- Chirik, P.J. Carbon-Carbon Bond Formation in a Weak Ligand Field: Leveraging Open-Shell First-Row Transition-Metal Catalysts. Angew. Chem. Int. Ed. 2017, 56, 5170–5181. [Google Scholar] [CrossRef]
- Zhang, W.; Chai, W.; Sun, W.-H.; Hu, X.; Redshaw, C.; Hao, X. 2-(1-(Arylimino)ethyl)-8-arylimino-5,6,7-trihydroquinoline Iron(II) Chloride Complexes: Synthesis, Characterization, and Ethylene Polymerization Behavior. Organometallics 2012, 31, 5039–5048. [Google Scholar] [CrossRef]
- Huang, F.; Xing, Q.; Liang, T.; Flisak, Z.; Ye, B.; Hu, X.; Yang, W.; Sun, W.-H. 2-(1-Aryliminoethyl)-9-arylimino-5,6,7,8-tetrahydrocycloheptapyridyl iron(II) dichloride: Synthesis, characterization, and the highly active and tunable active species in ethylene polymerization. Dalton Trans. 2014, 43, 16818–16829. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.-H.; Kong, S.; Chai, W.; Shiono, T.; Redshaw, C.; Hu, X.; Guo, C.; Hao, X. 2-(1-(Arylimino)ethyl)-8-arylimino-5,6,7-trihydroquinolylcobalt dichloride: Synthesis and polyethylene wax formation. Appl. Catal. A 2012, 447–448, 67–73. [Google Scholar] [CrossRef]
- Huang, F.; Zhang, W.; Sun, Y.; Hu, X.; Solan, G.A.; Sun, W.-H. Thermally stable and highly active cobalt precatalysts for vinyl-polyethylenes with narrow polydispersities: Integrating fused-ring and imino-carbon protection into ligand design. New J. Chem. 2016, 40, 8012–8023. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Suo, H.; Huang, F.; Liang, T.; Hu, X.; Sun, W.-H. Thermo-stable 2-(arylimino)benzylidene-9-arylimino-5,6,7,8-tetrahydro cyclohepta[b]pyridyliron(II) precatalysts toward ethylene polymerization and highly linear polyethylenes. J. Polym. Sci. Part A Polym. Chem. 2017, 55, 830–842. [Google Scholar] [CrossRef]
- Appukuttan, V.K.; Liu, Y.; Son, B.C.; Ha, C.-S.; Suh, H.; Kim, I. Iron and Cobalt Complexes of 2,3,7,8-Tetrahydroacridine-4,5(1H,6H)-diimine Sterically Modulated by Substituted Aryl Rings for the Selective Oligomerization to Polymerization of Ethylene. Organometallics 2011, 30, 2285–2294. [Google Scholar] [CrossRef]
- Du, S.; Wang, X.; Zhang, W.; Flisak, Z.; Sun, Y.; Sun, W.-H. A practical ethylene polymerization for vinyl-polyethylenes: Synthesis, characterization and catalytic behavior of α,α′-bisimino-2,3:5,6-bis(pentamethylene)pyridyliron chlorides. Polym. Chem. 2016, 7, 4188–4197. [Google Scholar] [CrossRef]
- Zada, M.; Guo, L.; Ma, Y.; Zhang, W.; Flisak, Z.; Sun, Y.; Sun, W.-H. Activity and Thermal Stability of Cobalt(II)-Based Olefin Polymerization Catalysts Adorned with Sterically Hindered Dibenzocycloheptyl Groups. Molecules 2019, 24, 2007. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Ma, Y.; Guo, J.; Liu, Q.; Solan, G.A.; Liang, T.; Sun, W.-H. Bis(imino)pyridines fused with 6- and 7-membered carbocylic rings as N,N,N-scaffolds for cobalt ethylene polymerization catalysts. Dalton Trans. 2019, 48, 2582–2591. [Google Scholar] [CrossRef] [Green Version]
- Guo, L.; Zada, M.; Zhang, W.; Vignesh, A.; Zhu, D.; Ma, Y.; Liang, T.; Sun, W.-H. Highly linear polyethylenes tailored with 2,6-bis[1-(p-dibenzo-cycloheptylarylimino)ethyl]pyridylcobalt dichlorides. Dalton Trans. 2019, 48, 5604–5613. [Google Scholar] [CrossRef]
- Chen, Q.; Zhang, W.; Solan, G.A.; Zhang, R.; Guo, L.; Hao, X.; Sun, W.-H. CH(phenol)-Bridged Bis(imino)pyridines as Compartmental Supports for Diiron Precatalysts for Ethylene Polymerization: Exploring Cooperative Effects on Performance. Organometallics 2018, 37, 4002–4014. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Zhang, W.; Solan, G.A.; Liang, T.; Sun, W.-H. Methylene-bridged bimetallic bis(imino)pyridine-cobaltous chlorides as precatalysts for vinyl-terminated polyethylene waxes. Dalton Trans. 2018, 47, 6124–6133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khoshsefat, M.; Dechal, A.; Ahmadjo, S.; Mortazavi, S.M.M.; Zohuri, G.; Soares, J.B.P. Amorphous to high crystalline PE made by mono and dinuclear Fe-based catalysts. Eur. Polym. J. 2019, 119, 229–238. [Google Scholar] [CrossRef]
- Huang, F.; Zhang, W.; Yue, E.; Liang, T.; Hu, X.; Sun, W.-H. Controlling the molecular weights of polyethylene waxes using the highly active precatalysts of 2-(1-aryliminoethyl)-9-arylimino-5,6,7,8-tetrahydrocycloheptapyridylcobalt chlorides: Synthesis, characterization, and catalytic behavior. Dalton Trans. 2016, 45, 657–666. [Google Scholar] [CrossRef] [PubMed]
- Du, S.; Zhang, W.; Yue, E.; Huang, F.; Liang, T.; Sun, W.-H. α,α′-Bis(arylimino)-2,3:5,6-bis(pentamethylene)pyridylcobalt Chlorides: Synthesis, Characterization, and Ethylene Polymerization Behavior. Eur. J. Inorg. Chem. 2016, 2016, 1748–1755. [Google Scholar] [CrossRef]
- Britovsek, G.J.P.; Clentsmith, G.K.B.; Gibson, V.C.; Goodgame, D.M.L.; McTavish, S.J.; Pankhurst, Q.A. The nature of the active site in bis(imino)pyridine iron ethylene polymerisation catalysts. Catal. Commun. 2002, 3, 207–211. [Google Scholar] [CrossRef]
- Humphries, M.J.; Tellmann, K.P.; Gibson, V.C.; White, A.J.P.; Williams, D.J. Investigations into the mechanism of activation and initiation of ethylene polymerization by bis(imino)pyridine cobalt catalysts: Synthesis, structures, and deuterium labeling studies. Organometallics 2005, 24, 2039–2050. [Google Scholar] [CrossRef]
- Gibson, V.C.; Humphries, M.J.; Tellmann, K.P.; Wass, D.F.; White, A.J.; Williams, D.J. The nature of the active species in bis(imino)pyridyl cobalt ethylene polymerisation catalysts. Chem. Commun. 2001, 2252–2253. [Google Scholar] [CrossRef]
- Roemelt, C.; Weyhermueller, T.; Wieghardt, K. Structural characteristics of redox-active pyridine-1,6-diimine complexes: Electronic structures and ligand oxidation levels. Coord. Chem. Rev. 2019, 380, 287–317. [Google Scholar] [CrossRef]
- Bryliakov, K.P.; Semikolenova, N.V.; Zakharov, V.A.; Talsi, E.P. Active intermediates of ethylene polymerization over 2,6-bis(imino)pyridyl iron complex activated with aluminum trialkyls and methylaluminoxane. Organometallics 2004, 23, 5375–5378. [Google Scholar] [CrossRef]
- Bryliakov, K.P.; Talsi, E.P.; Semikolenova, N.V.; Zakharov, V.A. Formation and Nature of the Active Sites in Bis(imino)pyridine Iron-Based Polymerization Catalysts. Organometallics 2009, 28, 3225–3232. [Google Scholar] [CrossRef]
- Bryliakov, K.P.; Talsi, E.P. Frontiers of mechanistic studies of coordination polymerization and oligomerization of α-olefins. Coord. Chem. Rev. 2012, 256, 2994–3007. [Google Scholar] [CrossRef]
- Kleigrewe, N.; Steffen, W.; Blomker, T.; Kehr, G.; Frohlich, R.; Wibbeling, B.; Erker, G.; Wasilke, J.C.; Wu, G.; Bazan, G.C. Chelate bis(imino)pyridine cobalt complexes: Synthesis, reduction, and evidence for the generation of ethene polymerization catalysts by Li+ cation activation. J. Am. Chem. Soc. 2005, 127, 13955–13968. [Google Scholar] [CrossRef] [PubMed]
- Soshnikov, I.E.; Semikolenova, N.V.; Bushmelev, A.N.; Bryliakov, K.P.; Lyakin, O.V.; Redshaw, C.; Zakharov, V.A.; Talsi, E.P. Investigating the Nature of the Active Species in Bis(imino)pyridine Cobalt Ethylene Polymerization Catalysts. Organometallics 2009, 28, 6003–6013. [Google Scholar] [CrossRef]
- Deng, L.Q.; Margl, P.; Ziegler, T. Mechanistic aspects of ethylene polymerization by iron(II)-bisimine pyridine catalysts: A combined density functional theory and molecular mechanics study. J. Am. Chem. Soc. 1999, 121, 6479–6487. [Google Scholar] [CrossRef]
- Kwon, D.H.; Small, B.L.; Sydora, O.L.; Bischof, S.M.; Ess, D.H. Challenge of Using Practical DFT to Model Fe Pendant Donor Diimine Catalyzed Ethylene Oligomerization. J. Phys. Chem. C 2019, 123, 3727–3739. [Google Scholar] [CrossRef]
- Cruz, V.L.; Ramos, J.; Martinez-Salazar, J.; Gutierrez-Oliva, S.; Toro-Labbe, A. Theoretical Study on a Multicenter Model Based on Different Metal Oxidation States for the Bis(imino)pyridine Iron Catalysts in Ethylene Polymerization. Organometallics 2009, 28, 5889–5895. [Google Scholar] [CrossRef]
- Griffiths, E.A.H.; Britovsek, G.J.P.; Gibson, V.C.; Gould, I.R. Highly active ethylene polymerisation catalysts based on iron: An ab initio study. Chem. Commun. 1999, 1333–1334. [Google Scholar] [CrossRef]
- Tondreau, A.M.; Milsmann, C.; Patrick, A.D.; Hoyt, H.M.; Lobkovsky, E.; Wieghardt, K.; Chirik, P.J. Synthesis and Electronic Structure of Cationic, Neutral, and Anionic Bis(imino)pyridine Iron Alkyl Complexes: Evaluation of Redox Activity in Single-Component Ethylene Polymerization Catalysts. J. Am. Chem. Soc. 2010, 132, 15046–15059. [Google Scholar] [CrossRef]
- Khoroshun, D.V.; Musaev, D.G.; Vreven, T.; Morokuma, K. Theoretical study on bis(imino)pyridyl-Fe(II) olefin poly- and oligomerization catalysts. Dominance of different spin states in propagation and beta-hydride transfer pathways. Organometallics 2001, 20, 2007–2026. [Google Scholar] [CrossRef]
- Boudene, Z.; Boudier, A.; Breuil, P.-A.R.; Olivier-Bourbigou, H.; Raybaud, P.; Toulhoat, H.; de Bruin, T. Understanding the role of aluminum-based activators in single site iron catalysts for ethylene oligomerization. J. Catal. 2014, 317, 153–157. [Google Scholar] [CrossRef] [Green Version]
- Bouwkamp, M.W.; Bart, S.C.; Hawrelak, E.J.; Trovitch, R.J.; Lobkovsky, E.; Chirik, P.J. Square planar bis(imino)pyridine iron halide and alkyl complexes. Chem. Commun. 2005, 3406–3408. [Google Scholar] [CrossRef] [PubMed]
- Bellows, S.M.; Cundari, T.R.; Holland, P.L. Spin Crossover during β-Hydride Elimination in High-Spin Iron(II)– and Cobalt(II)–Alkyl Complexes. Organometallics 2013, 32, 4741–4751. [Google Scholar] [CrossRef]
- Huang, C.; Huang, Y.; Ma, Y.; Solan, G.A.; Sun, Y.; Hu, X.; Sun, W.-H. Cycloheptyl-fused N,N,N′-chromium catalysts with selectivity for vinyl-terminated polyethylene waxes: Thermal optimization and polymer functionalization. Dalton Trans. 2018, 47, 13487–13497. [Google Scholar] [CrossRef] [Green Version]
- Bouwkamp, M.W.; Lobkovsky, E.; Chirik, P.J. Bis(imino)pyridine iron(II) alkyl cations for olefin polymerization. J. Am. Chem. Soc. 2005, 127, 9660–9661. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Chen, F.W. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Raucoules, R.; de Bruin, T.; Raybaud, P.; Adamo, C. Theoretical Unraveling of Selective 1-Butene Oligomerization Catalyzed by Iron−Bis(arylimino)pyridine. Organometallics 2009, 28, 5358–5367. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinstock, R.B.; Weinhold, F. Natural population analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar] [CrossRef]
- Ba, J.; Du, S.; Yue, E.; Hu, X.; Flisak, Z.; Sun, W.-H. Constrained formation of 2-(1-(arylimino)ethyl)-7-arylimino-6,6-dimethylcyclopentapyridines and their cobalt(ii) chloride complexes: Synthesis, characterization and ethylene polymerization. RSC Adv. 2015, 5, 32720–32729. [Google Scholar] [CrossRef]
- Pasha, F.A.; Basset, J.-M.; Toulhoat, H.; de Bruin, T. DFT Study on the Impact of the Methylaluminoxane Cocatalyst in Ethylene Oligomerization Using a Titanium-Based Catalyst. Organometallics 2015, 34, 426–431. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision E.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Minaev, B.; Baryshnikova, A.; Sun, W.-H. Spin-dependent effects in ethylene polymerization with bis(imino)pyridine iron(II) complexes. J. Organomet. Chem. 2016, 811, 48–65. [Google Scholar] [CrossRef]
- Ehm, C.; Budzelaar, P.H.M.; Busico, V. Calculating accurate barriers for olefin insertion and related reactions. J. Organomet. Chem. 2015, 775, 39–49. [Google Scholar] [CrossRef]
- Dolg, M.; Wedig, U.; Stoll, H.; Preuss, H. Energy-adjusted abinitio pseudopotentials for the 1st-row transition-elements. J. Chem. Phys. 1987, 86, 866–872. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez, C.; Schlegel, H.B. Reaction-path following in mass-weighted internal coordinates. J. Phys. Chem. 1990, 94, 5523–5527. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinhold, F. Natural localized molecular-orbitals. J. Chem. Phys. 1985, 83, 1736–1740. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Reed, A.E.; Schleyer, P.V. chemical bonding in hypervalent molecules—The dominance of ionic bonding and negative hyperconjugation over d-orbital participation. J. Am. Chem. Soc. 1990, 112, 1434–1445. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
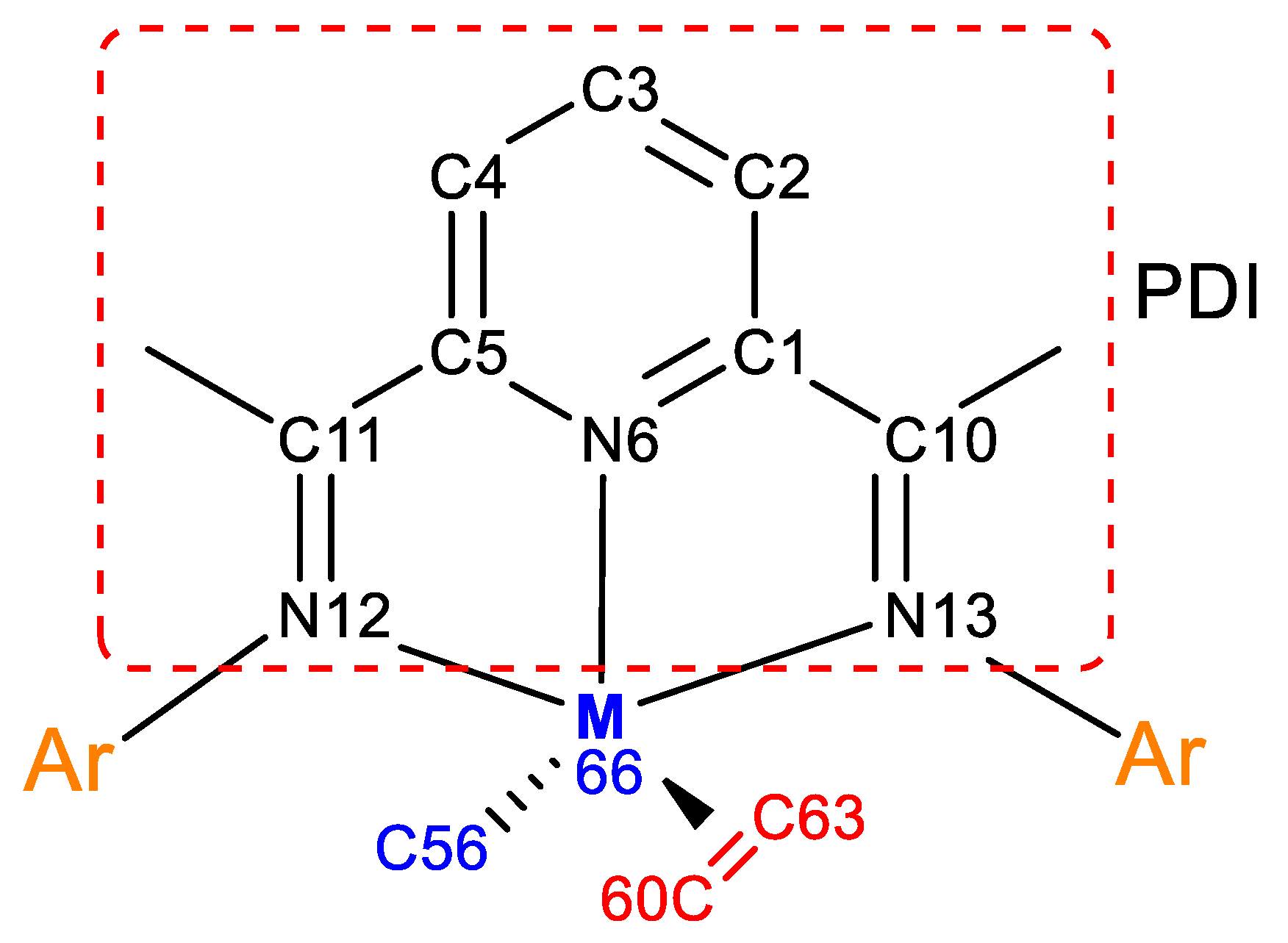
| Bonds | Bond Order | Bonds | Bond Order | ||
|---|---|---|---|---|---|
| 1Co(0)-2 | 0Co(0)-2 | 1Co(0)-2 | 0Co(0)-2 | ||
| C(1)-–C(2) | 1.36 | 1.42 | C(10)–N(13) | 1.44 | 1.47 |
| C(2)–C(3) | 1.49 | 1.41 | C(11)–N(12) | 1.82 | 1.48 |
| C(3)–C(4) | 1.33 | 1.41 | N(12)---Co(66) | 0.11 | 0.47 |
| C(4)–C(5) | 1.47 | 1.42 | N(13)---Co(66) | 0.39 | 0.48 |
| C(1)–N(6) | 1.13 | 1.15 | C(56)–Co(66) | 0.73 | 0.79 |
| C(5)–N(6) | 1.25 | 1.15 | C(60)–C(63) | 1.63 | 1.29 |
| C(1)–C(10) | 1.23 | 1.24 | C(60)---Co(66) | 0.24 | 0.54 |
| C(5)–C(11) | 1.08 | 1.24 | C(63)---Co(66) | 0.18 | 0.49 |
| N(6)---Co(66) | 0.42 | 0.61 | |||
| 0Co(+)B | 1A | 1B | 1C | 2A | 2B | TS2A/3A | TS2B/3B | 3A/3B | |
|---|---|---|---|---|---|---|---|---|---|
| Singlet | 0.15 | 0.1 | 0.158 | 0.114 | 0.085 | 0.084 | 0.061 | 0.077 | 0.095 |
| Triplet | 0.181 | 0.123 | 0.18 | 0.119 | 0.123 | 0.168 | 0.112 | 0.116 | 0.114 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Z.; Ma, Y.; Sun, W.-H. Comparison of the Reactivity and Structures for the Neutral and Cationic Bis(imino)pyridyl Iron and Cobalt Species by DFT Calculations. Catalysts 2020, 10, 1396. https://0-doi-org.brum.beds.ac.uk/10.3390/catal10121396
Li Z, Ma Y, Sun W-H. Comparison of the Reactivity and Structures for the Neutral and Cationic Bis(imino)pyridyl Iron and Cobalt Species by DFT Calculations. Catalysts. 2020; 10(12):1396. https://0-doi-org.brum.beds.ac.uk/10.3390/catal10121396
Chicago/Turabian StyleLi, Zilong, Yanping Ma, and Wen-Hua Sun. 2020. "Comparison of the Reactivity and Structures for the Neutral and Cationic Bis(imino)pyridyl Iron and Cobalt Species by DFT Calculations" Catalysts 10, no. 12: 1396. https://0-doi-org.brum.beds.ac.uk/10.3390/catal10121396