Hydrogen Evolution Reaction-From Single Crystal to Single Atom Catalysts
by
 , , and
, , and
Sanjin J. Gutić
1 ,
,
Ana S. Dobrota
2,
Edvin Fako
3 ,
,
Natalia V. Skorodumova
4,5,
Núria López
3 and
Igor A. Pašti
2,4,*
1
Department of Chemistry, Faculty of Science, University of Sarajevo, Zmaja od Bosne 33-35, 71000 Sarajevo, Bosnia and Herzegovina
2
Faculty of Physical Chemistry, University of Belgrade, Studentski trg 12-16, 11158 Belgrade, Serbia
3
Institute of Chemical Research of Catalonia (ICIQ), The Barcelona Institute of Science and Technology, Països Catalans 16, 43007 Tarragona, Spain
4
Department of Materials Science and Engineering, School of Industrial Engineering and Management, KTH – Royal Institute of Technology, Brinellvägen 23, 100 44 Stockholm, Sweden
5
Department of Physics and Astronomy, Uppsala University, Box 516, 751 20 Uppsala, Sweden
*
Author to whom correspondence should be addressed.
Catalysts 2020, 10(3), 290; https://0-doi-org.brum.beds.ac.uk/10.3390/catal10030290
Submission received: 13 February 2020
/
Revised: 1 March 2020
/
Accepted: 2 March 2020
/
Published: 4 March 2020
(This article belongs to the Section Electrocatalysis)
Abstract
:Hydrogen evolution reaction (HER) is one of the most important reactions in electrochemistry. This is not only because it is the simplest way to produce high purity hydrogen and the fact that it is the side reaction in many other technologies. HER actually shaped current electrochemistry because it was in focus of active research for so many years (and it still is). The number of catalysts investigated for HER is immense, and it is not possible to overview them all. In fact, it seems that the complexity of the field overcomes the complexity of HER. The aim of this review is to point out some of the latest developments in HER catalysis, current directions and some of the missing links between a single crystal, nanosized supported catalysts and recently emerging, single-atom catalysts for HER.
1. Introduction
The hydrogen evolution reaction (HER) takes a special place in electrochemistry. While it has been investigated for more than a century, there are still some burning issues related to this reaction. HER is possibly the most direct and easiest way to produce high purity hydrogen, which is considered by many the fuel of the future. However, after so many years of research, a proper cheap and abundant electrocatalyst for HER, traditionally metal or a generally metallic compound, is missing. To understand this problem, we have to look into the mechanism. It depends on the pH of the solution [1,2] and in acidic media it can be summarized as [3]:
| (i.1) H+ + e− + *→ Hads | |
| (ii.1) Hads + Hads → H2 + 2* | |
| (iii.1) H+ + Hads + e− → H2 + * |
In the given mechanism, * represents an empty active site on the catalyst surface. The reaction starts with a Volmer step (i), and the intermediate Hads is removed from the surface either by Tafel reaction (ii) or Heyrovsky reaction (iii) [4,5]. Highly acidic media allows easy H+ transfer, but proton diffusion limitations are already visible at pH around 3. Therefore, HER shall be performed at a very low pH to have non-limited H2 production at the desired rate. Clearly, the rate of the reaction is greatly affected by the way Hads is interacting with the catalyst surface. Moreover, the number of available active sites (*) is crucial. At very low pH the reaction is fast on platinum and Pt-group metals, being practically reversible. However, these metals are expensive, and Pt-based catalysts cannot be used for industrial-scale production of H2. Cheaper metals like Ni perform poorly compared to Pt, but the problem is that these metals are much less noble and have stability issues as they dissolve in acids [6]. When it comes to the comparison of different catalysts, one has to quantify performance somehow. There are two main kinetic parameters that are important for the characterization of the catalyst. The first one is exchange current density, while the second one is the Tafel slope. The latter one is particularly (ab)used as different combinations of reactions (i)–(iii) give different Tafel slopes (between −30 and −120 mV dec−1), so usually, this is diagnostics tool for determining the mechanism of HER. However, without going into deeper kinetic analysis, we emphasize that the values of the Tafel slope can be the same for different mechanisms [7], so the value of the Tafel slope is not sufficient to determine the mechanism of HER. Moreover, the reported Tafel slopes often fall outside of the theoretical range (see Appendix), but this is still not enough to prevent “unambiguous” determination of the HER mechanism only from the Tafel slope.
The mechanism can be presented in an analogous way for alkaline media [8]:
| (i.2) H2O + e− + *→ Hads + OH− | |
| (ii.2) Hads + Hads → H2 + 2* | |
| (iii.2) H2O + Hads + e− → H2 + OH− + * |
As H2 is being formed from H2O, there are no diffusion limitations in alkaline solutions. In addition, most of the metals are stable, but HER is much slower in alkalis. This is because the Volmer step requires H2O dissociation which is slow, so even on Pt reaction is much slower than in acids. In practice, large scale production of H2 via water electrolysis is done in concentrated alkalis using Ni-based catalyst—materials are stable and the price compensates for lower activity compared to Pt [9].
Following these few lines, one can easily devise strategies for making HER catalyst. First, one has to tackle the problem of the catalyst stability by choosing the catalyst which is stable under HER conditions. This should not be very difficult as stability can be devised from Pourbaix diagrams for most of the known materials. It can also be tested relatively easily by measuring the corrosion rate under given conditions, or simply HER performance over a long time. Second, one has to deal with the energetics of Hads formation. Current state-of-the-art understanding says that thermoneutral Hads formation (versus H2) is the target, and this point is elaborated and discussed further in Section 2. Third, one has to provide a sufficient number of active sites. For bulk materials, the surface-to-volume ratio is low; therefore, the number of active sites is low compared to a possible number of sites. As a consequence, the catalyst can be dispersed and reduced to nanosizes, but in this case, it should be supported on some substrate (catalyst support). This support can actually help H2 production due to interfacial effects, so it is not necessarily inactive. HER at supported catalysts is elaborated in Section 3. Still, one must not forget the first and the second points, as the Hads energetics and stability requirements still have to be satisfied. However, this can be problematic as at nanoscale materials can behave differently than bulk materials. There is a large number of nonequivalent sites on the surface of nanoparticles, while low coordination can change thermodynamic tendencies towards dissolution. Moreover, some metals are so highly oxophilic, like W, Mo, and Ti, that they are partially oxidized even under cathodic potentials corresponding to HER (see further). This also reduces the number of active sites for HER. The ultimate limit of catalyst utilization is a single atom catalyst (SAC). Metal is dispersed in such a way that every atom bears catalytic function. It is clear that in this case thermodynamics of metal atom (which is always incorporated into the suitably chosen matrix) is different from the thermodynamics of bulk parental metal and that is ultimately coupled to the properties of the support (matrix). Such catalysts are presented in Section 4.
Providing a complete overview of the work done on the development of HER catalysts is an impossible task. However, its consideration must start from single crystal and bulk electrodes where fundamental knowledge is accumulated. This knowledge is further passed to the development of supported and SACs, where the situation is complicated due to the presence of the second phase in addition to the catalysts. In this contribution, we follow this line. We present a brief overview of some classical works related to single crystal and bulk electrodes and then present new results in the field considering supported HER catalyst and SACs. For the supported catalysts, we especially focus on the interaction between the catalyst and the support in terms of the role of the support on the catalytic activity.
2. HER at Single Crystal Surfaces and Bulk Surfaces
A crystalline solid can be considered as a series of mutually parallel lattice planes, arranged in a periodic way. Close to the surface, the spatial arrangement of the lattice planes, their crystallographic structure, as well as the dynamics of the constituent atoms may differ from the corresponding features in the bulk [10]. Most clean metals show the tendency to minimize their surface energy. One of the mechanisms is relaxation, the shift of one or more lattice planes located near to the surface, usually perpendicularly to the surface, so that the interlayer distance changes compared to the bulk lattice. The other one is reconstruction-change in equilibrium positions and bonding of surface atoms so that the projection of bulk unit cell to the given surface does not correspond to the surface unit cell. Different crystallographic planes (with different Miller indices) of the same metal exhibit unequal surface densities (number of atoms per surface area) and therefore undergo surface relaxation/reconstruction to different extents [11]. For example, Pt(100) has lower surface density compared to densely packed Pt(111) and therefore has a stronger tendency to relax/reconstruct. Since the main driving force for the mentioned surface modifications is the low coordination number (non-saturation) of surface atoms, it is obvious that atom/molecule adsorption on clean metal surfaces can have a significant effect on its surface structure, inducing relaxation/reconstruction changes. This is of huge importance for HER, as it involves adsorbed atomic hydrogen (Hads) as an intermediate. Additionally, in an electrochemical system, the electrode will be modified by adsorption of “spectator” species (ions/molecules from the electrolyte), so HER will not be taking place on clean metal surfaces, but rather on modified ones. Obviously, there is a complex dynamic interplay between the catalyst surface, reactants, intermediates, and electrolyte.
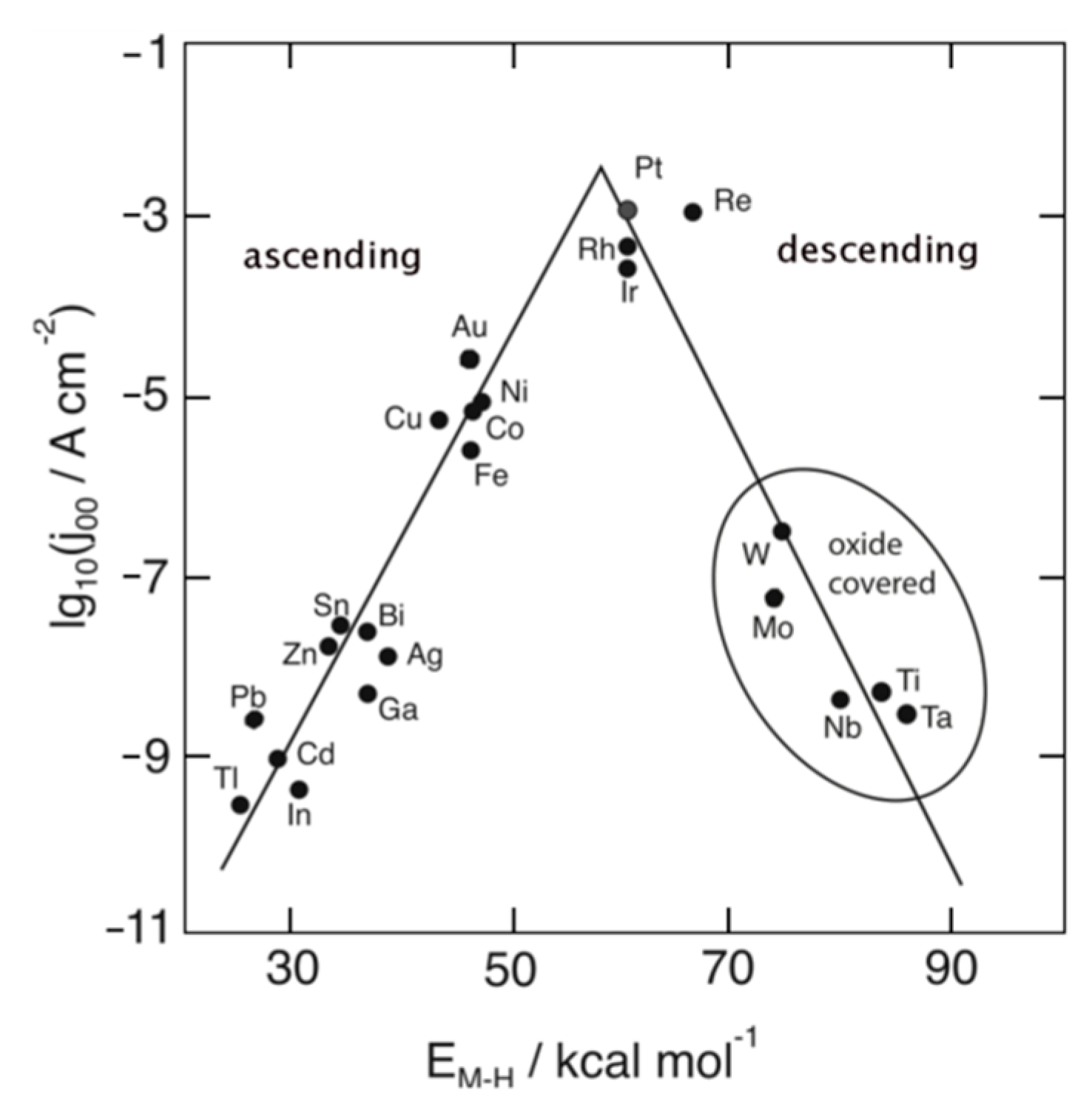
According to the Sabatier principle, the interaction of the reaction reactants/intermediates with the catalyst has to be optimal in strength. If it is too weak, the reactants will not bind to the catalyst and the reaction will not be able to take place. In other words, reactants do not get activated by such catalysts. On the other hand, if the interaction is too strong, the catalyst active sites will be blocked by reactant, intermediate or product molecules, leaving no active sites available for new reactant molecules that would continue the reaction. In other words, the catalyst surface will be (irreversibly) poisoned. Obviously, the intermediate’s adsorption energy is of crucial importance for catalyst efficiency. In the case of HER, this relates to the adsorption energy of atomic hydrogen (Eads(H)). The relationship between the logarithm of the exchange current density (log|j00|) and the energy of hydride formation (EM–H) was found by Trasatti [12] in the form of a “volcano” curve (Figure 1). EM–H was used instead of Eads(H) since that data was not available at the time [13]. HER exchange current density varies by 5–6 orders of magnitude depending on the electrode material (Figure 1), with the highest rates on electrode materials with an optimal interaction of hydrogen intermediate with the catalyst surface-Pt-group metals.
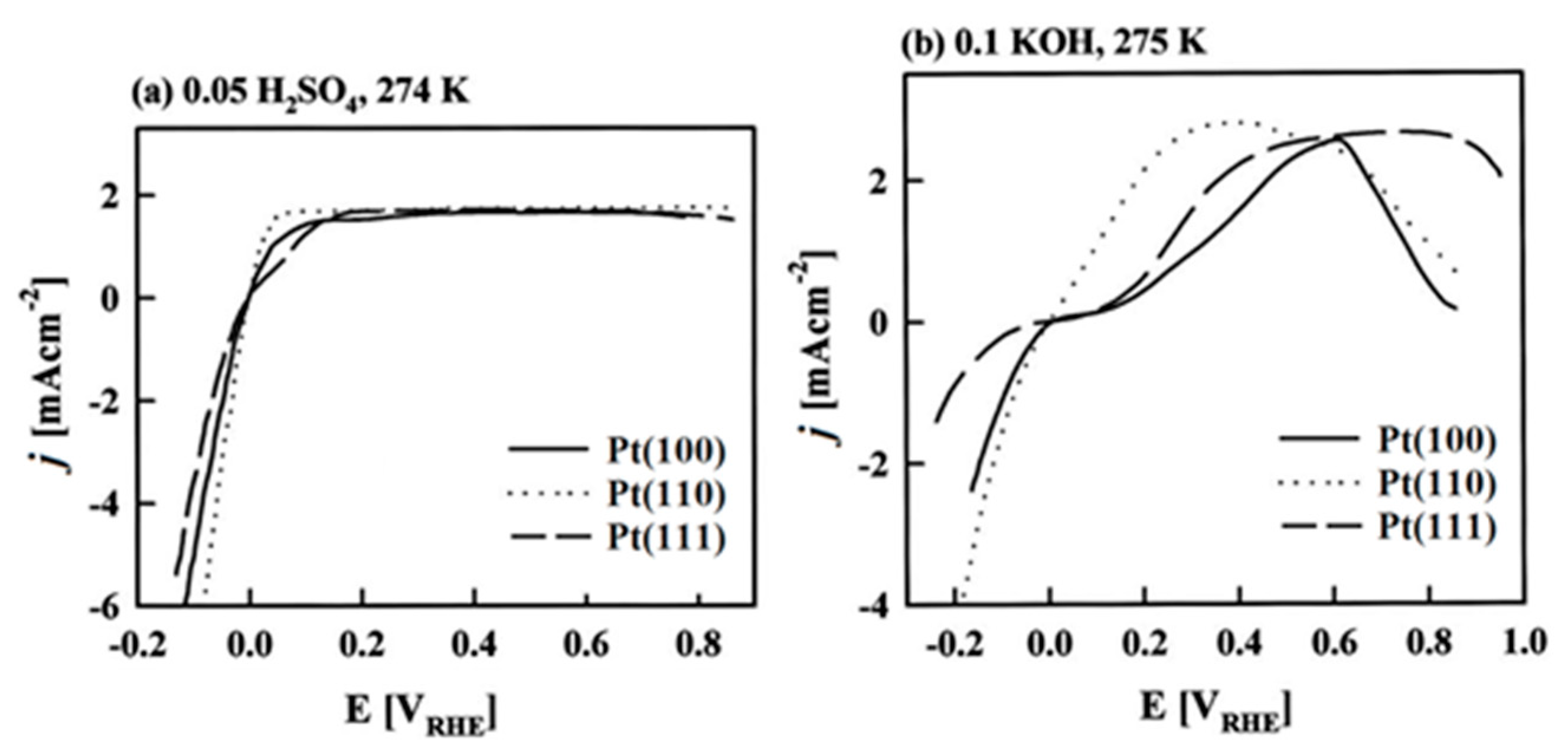
Since the highest |j00| is exhibited by Pt, atomic-scale studies of HER rate dependence on the Pt single crystal surfaces’ atomic-scale morphology have been in the research focus for years. Marković et al. [14] have first demonstrated that HER in acid solutions is a surface-sensitive process, i.e., that its rate depends on the Pt crystal orientation, as can be clearly seen in Figure 2, left. This might seem intuitively obvious today when Eads(H) is commonly used in HER activity studies and can be relatively easily obtained from Density Functional Theory (DFT) calculations. This energy depends on the atomic-scale structure of the surface—its orientation, the coordination number of the surface atoms, reconstruction, etc. However, early HER studies on Pt did not reveal HER’s rate dependence on surface orientation. It was shown in Reference [14] that the catalytic activity decreases in the order Pt(110) > Pt(100) > Pt(111). The order is the same for both acidic and alkaline solutions, but the absolute rates are quite different (Figure 2).
The dependency of the HER rate on the crystallographic orientation of the electrode was also demonstrated for the case of silver [15] and gold [16] in acid solutions. While the Tafel plots for Ag(111) were found to be linear, in the case of Ag(110) they exhibit a distinct slope change. A similar change of slope is also observed in the case of polycrystalline silver [17] and may indicate a change in HER mechanism [15]. In the case of Au surfaces, the catalytic activity follows a different sequence compared to Pt: Au(111) > Au(100) > Au(110) (Table 1), while the Tafel slope varies gradually from about −60 mV dec−1 at low overpotentials to −120 mV dec−1 at high overpotentials (in about the same way for the three mentioned Au facets) [16]. HER surface structure sensitivity was also demonstrated for less noble metals. For example, on Cu(hkl) in acidic solution, HER rate increases in the order Cu(100) > Cu(111) at low overpotentials [18]. On the other hand, at high overpotentials, the activity order is reversed, due to structure sensitive adsorption of HER inhibiting spectator species ((bi)sulfate anions) on the investigated surfaces [18]. HER is also surface-sensitive on Pt in alkaline solutions [19], Figure 2, right. It should be noted that the relatively old experimental data often suffer from inadequate preparation of the electrode surface [13], and therefore, inconsistent results can be found. However, it should also be noted that there is a very small number of studies reporting HER on metal single crystal surfaces.
The presented experimental data for HER at single metal surfaces originates from rare contributions published 20 and more years ago, most of which do not provide a deeper, atomic-level understanding of the reason why one metal single crystal facet exhibits higher HER activity than the other. In the case of Pt, an explanation was offered [11]: the difference between the mentioned Pt facets was attributed to structure-sensitive heats of adsorption of the active intermediate. Namely, hydrogen can be relatively strongly (Hupd, deposited at underpotentials) or weakly (Hopd, deposited at overpotentials) adsorbed on a Pt surface. For more details, see Reference [20]. The more weakly bound Hopd is formed on potentials around, or negative to the Nernst potential of hydrogen electrode, and represents the reactive intermediate. However, the role of Hupd was unclear. It was proposed that Hupd competes with Hopd for the same adsorption sites and that it alters its adsorption energy [11]. Of course, Eads(H) varies with coverage, mainly due to repulsion between two neighboring adsorbed H atoms. The experimental results and kinetic models were reconciled by a model according to which some unknown amount of Hupd is in deeper potential wells in the surface (in the subsurface state), making a place on top sites for Hopd adsorption. Still, the reaction mechanism cannot be definitely resolved from kinetic parameters [11]. A much larger number of studies deal with polycrystalline metals. In that case, there are many more sites available for H adsorption, and therefore, there are many choices for the reactive intermediate state. Metals that are covered by oxygen under HER electrochemical conditions exhibit amorphous structure and are not discussed here. A review on them can be found in Reference [21]. Today, it is well-known that metal catalysis generally involves d-bands with energies close to the Fermi level, and their interactions with the reactant play a key role in the theory of electrochemical reactions. A framework that allows treating d-band catalysis within the theory of electrochemical reactions is presented by Santos and Schmickler in Reference [22], correlating the d-band center with the rates of bond-breaking reactions through the activation energy.
It was mentioned above that Trasatti’s volcano curve (Figure 1) used metal hydride formation energy instead of H adsorption energy. However, EM–H trends do not always follow Eads(H) trends [13]. Therefore, modern volcano plots use adsorption energies obtained from DFT calculations, with an estimated error of ±0.1 eV [13]. A theoretical explanation of the shape of the volcano curve was given through a study of H adsorption on the fcc(111) surfaces of a variety of transition metals by Nørskov et al. [23]. The authors show that the free energy of the adsorbed state (∆GH*) can be easily estimated as ∆GH* = Eads(H) + 0.24 eV, where Eads(H) is calculated against ½H2. The results imply that on the descending branch of the volcano maximum (Figure 1), H is bound too strongly, and HER rate decreases with decreasing Eads(H) due to a lack of available sites for 2H* recombination at the surface (authors assumed that H2 is formed via Tafel step on basically all the studied surfaces). On the ascending side, H is bound too weakly, making proton transfer difficult, and HER rate decreases with increasing Eads(H). All HER steps on Pt were found to be thermo-neutral, placing Pt very close to the maximum (optimum) of the volcano curve [23]. However, the appropriate value of ∆GH* alone does not mean that metal will be a good HER catalyst, as is shown for the case of Cu in Reference [13]. Greeley et al. [24] have demonstrated the predictive power of theoretical calculations for identifying new electrocatalysts for HER. Using DFT, they have estimated the HER activity and stability of over 700 binary surface metal alloys. BiPt surface alloy was found to be a promising candidate with HER activity comparable to (or even better than) pure Pt. They have supported this claim experimentally-they have synthesized and tested the mentioned surface alloy and indeed demonstrated its superior performance compared to pure Pt [24], in agreement with the computational results. This is a great example of how theory and experiment can work together to bring faster progress and a deeper understanding of the investigated phenomena. Additionally, it illustrates the shift in the theory/experiment relationship that is becoming more pronounced with more advanced codes and more powerful computational resources: Theory used to follow and try to explain experimental data; nowadays, experiments can be guided by theoretical predictions [25]. Theoretical results have emphasized the importance of electron transfer from the metal to anti-bonding σ* orbital of H2, which induces H–H bond breaking and allows for H2 dissociation [11]. More recently, more complex and demanding DFT calculations of HER were demonstrated. These included a model of electrochemical double layer and a computational scheme which allows the potential to be kept constant during a charge transfer reaction. As a result, the Tafel–Volmer mechanism and Tafel reaction have been identified as the rate-determining step (RDS) for HER on Pt(111) [26]. The study includes 13 other metals as well, assuming the Tafel reaction as RDS. However, we must note that only Pt(111) and rhenium in acid solutions actually follow this path [13].
The volcano curve has been used for so long now that it seems that we have started taking it for granted to some extent. A fresh look at the famous volcano plot was offered by Quaino et al. [13]. They suggest that the descending part of the curve (Figure 1) is a computational artifact. Namely, metals on that side of the curve are covered by oxygen under HER conditions, which results in HER rate reduction by several orders of magnitude [13]. They demonstrate that, when these metals are excluded, the log|j00| vs. Eads(H) plot does not resemble a volcano (neither in acidic nor alkaline solutions). The investigated metals were grouped into sp metals (the worst catalysts; Cd, Hg, Pb), coinage metals (Cu, Ag, Au), Ni and Co (mediocre catalysts), and d metals (best catalysts; Rh, Ro, Ir, Pd, Pt). Several general conditions for an efficient HER catalyst are outlined: (i) ΔG should be around zero at the equilibrium potential (in accordance with Sabatier principle) [27], (ii) metal’s d band includes the Fermi level, and (iii) there should be a strong, long-range interaction between the metal’s d band and 1s orbital of hydrogen [28]. In the case of sp metals, the Volmer reaction is the RDS [29]. Due to the absence of the d-band effects, Sabatier’s volcano shape is expected and indeed found in alkaline solutions. However, in acid solutions, the situation is more complicated, with changes in the slope of current–potential curves. When it comes to coinage metals, Sabatier principle has to be superposed by the position of the Fermi level with respect to the d-band. The d-band position and the Eads(H) become more favorable in the order Au < Ag < Cu [13], without volcano-like shape. Regarding the d metals, instead of assuming Hopd as the reactive intermediate (as was discussed above), authors in Reference [13] consider Hopd and Hupd as two available states and that the reaction goes via the more energetically favorable intermediate. In such a way, a descending part of the volcano is not seen. Experiments show that on Ni and Co HER’s Volmer–Heyrovsky mechanism is operative, with Heyrovsky step as the rate-determining one [30,31], in agreement with results from Reference [13].
For some of the above-mentioned cases, the results were already given for both acidic and alkaline solutions. Generally, there are more data for HER in acid than in alkaline media, because of the widely investigated proton-exchange membrane (PEM) cells. The experimental alkaline-HER rates are, in terms of j00, about 2–3 orders of magnitude lower than those in acidic solutions [8]. Some of the main questions that arise in this area are whether the energy barrier for water dissociation affects the overall reaction rate, how does the pH value influence it, and what is the nature of HER active sites under these conditions.
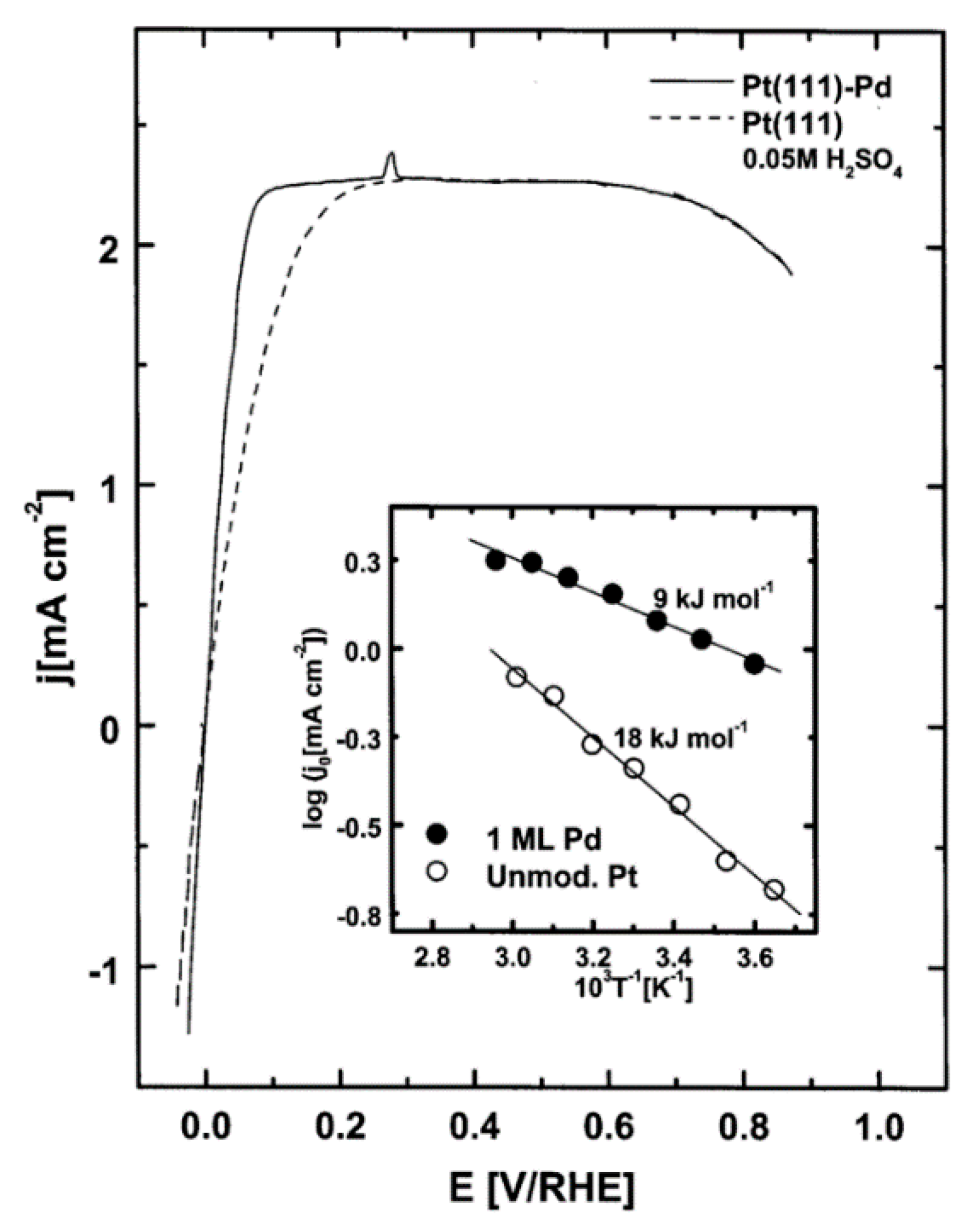
Up to now, we have overviewed some of the well-known single crystal and polycrystalline metal electrodes. However, a very interesting family of potential HER electrocatalysts is obtained when other metal’s thin layers (overlayers) are additionally spread on them. For example, it was experimentally shown that HER kinetics on Pt(111) can be improved by Pd modification of the surface, in the form of pseudomorphic Pd films [32], i.e., a uniform, epitaxial metallic layer of Pd with the lattice constant equal to the lattice constant of Pt is formed (Figure 3). It is proposed that HER mechanism on such surfaces is Volmer–Heyrovsky, with the Heyrovsky reaction as the RDS [32]. A review of the development of Pd-based catalysts can be found in Reference [33]. Clearly, metal monolayers on other metal substrates can exhibit novel catalytic properties. Such systems are stable only if strong chemical bonds are formed between the monolayer and the substrate. Therefore, chemical or ligand effect (the electronic environment of the surface layer) is always pronounced. Additionally, if there is a lattice mismatch between the two metals (the lattice constants of the two metals differ), the so-induced strain can lead to changes in the reactivity [25,34]. It was shown by Mavrikakis et al. [34] that a correlation between surface strain and adsorption energies (and activation energy barriers) exists and that it can be attributed to the metal’s d-band center shift caused by strain. For example, this was demonstrated by Soldano et al. [25] for the case of rhodium overlayers on gold and palladium substrates, where Rh/Au(111) is proposed as a good HER electrocatalyst candidate.
Naturally, pure precious metal or combinations of two such metals are not the most practical choices due to high prices. The tendency is to provide good HER activity using reduced amounts of expensive metals. Therefore, the question asked for a long time was “what can be used instead of Pt?” Transition metal carbides (TMC) were found to be similar to Pt-group metals in terms of their electronic properties and unit cell dimensions. However, their HER exchange current densities are 2–3 orders of magnitude smaller than on Pt surfaces [35,36]. Therefore, TMCs are considered only as catalyst support. Esposito et al. [36] have studied well-characterized single-phase WC, W2C, and Mo2C supports covered with a monolayer (ML) of precious metals. Both WC and W2C surfaces were shown to be excellent supports for ML Pt (good stability), with HER activities comparable to bulk Pt [36], consistent with DFT results of Eads(H) on investigated systems. Obviously, the number of possibilities grew significantly with such realizations. Different combinations of support and overlayer metal were to be investigated. That is when DFT screening shows its power—it can provide valuable information about metal adhesion to the support and interaction of such a system with hydrogen. Indeed, there is a number of DFT studies on metal overlayers on TMC supports, e.g., see References [35,37,38,39]. An exhaustive theoretical screening for cheap and stable substrate-overlayer systems for HER, when the substrate and the overlayer are pure non-precious metals, has been carried out by Bjöketurn et al. [40]. They used a pool of 39 metals, mostly transition metals, and found W-Cu as the most promising system. It was synthesized but as nanoparticles instead of overlayered structure (since this was not feasible) and tested for HER using cyclic voltammetry. An improvement compared to pure W and Cu electrodes was observed [40].
While single crystal and bulk surfaces can provide valuable fundamental insights into the HER mechanism, such systems are not practical for a large-scale production of H2. In practice, highly dispersed HER catalysts are much more attractive as the catalyst utilization is much higher.
3. HER at Supported Catalysts
Recent efforts in achieving the best possible performance of supported electrocatalysts can be summarized in several approaches: (i) modification of (established) supports in order to increase the number of active sites suitable for anchoring catalyst nanoparticles, while maintaining required electronic conductivity and stability; (ii) development of synthetic procedures that enable satisfactory catalyst dispersion; (iii) quest for the catalyst–support combinations with significant synergistic effects, beneficial for electrocatalytic activity and stability. A number of recent reviews provide extensive insights into the latest developments in the field of supported catalysts and electrocatalytic HER. Overview of general wisdom on supported catalysts and current knowledge on electrocatalytic HER is summarized in References [41,42,43]. Several fresh reviews deal with more specific catalyst and/or support materials, such as References [44,45,46] for carbon-based supports, References [47,48] for metal-organic frameworks, Reference [49] for 1D and 2D noble metals, and other specific materials in References [50,51,52]. A recent overview of heterostructured HER electrocatalysts can be found in Reference [53].
The development of computational methods enabled relatively fast and inexpensive probing of a great number of different materials [54], which lead to the immense increase in works dealing with the preparation of novel active materials for HER. Catalytic support for electrocatalysts is usually an inactive material with high electronic conductivity. However, active nanoparticles are sometimes grown or dispersed on materials that have moderate or even high catalytic activity. In all of these cases, regardless of the relative difference in activity between catalyst and support, interfacial phenomena play an important role in the HER mechanism; hence in this review, we consider supported catalysts in their broadest meaning, where support is the less active phase in contact with the active nanostructure. This section contains an overview of the most recent papers dealing with supported HER electrocatalysts, summarized in four parts: (i) supported metallic nanoparticles, dealing with nanostructured metals and alloys on different, usually inactive, supports; (ii) supported chalcogenides, hydroxides, pnictides, and carbides, covering transition metal oxides, sulfides, selenides, oxides, carbides, nitrides, and phosphides on different supports; (iii) miscellaneous catalyst–support combinations, and (iv) role of the catalyst–support interface in HER mechanism. This approach is not based solely on the chemical classification of catalysts but also on the nature of synergistic effects between catalyst and support, responsible for the promotion of the electrocatalytic activity and/or improvement of catalyst stability.
Given that platinum and platinum group metals (PGM) have unique properties required for outstanding performance and that their price limits their widespread application, all efforts in the development of supported HER electrocatalysts can be considered simply as quest for high-performance electrocatalysts with decreased PGM content or electrocatalysts where PGMs are completely replaced by other materials.
3.1. Supported Metallic Nanoparticles
Maintaining or even improving PGM-based electrocatalyst performance, whilst significantly lowering PGM content, can be achieved through several approaches, including (i) PGM nanoparticle morphology engineering, (ii) improving PGM dispersion over the support, and (iii) PGM alloying with non-noble transition metals. Bernsemier et al. reported a new synthesis approach for the preparation of ordered mesoporous carbon catalyst films (PtNP/OMC) employing colloidal size-controlled Pt nanoparticles (PtNP) as a metal precursor. The size of Pt nanoparticles changed from (2.1 ± 0.5) nm before the synthesis to (3.0 ± 1.0) nm in the final composite, and no Pt was trapped in pore walls or micropores. Such prepared catalysts reached two times higher current densities, compared to the commercial Pt/C [55]. A similar approach was employed with ruthenium nanoparticles by Creus et al. [56]. Electrocatalytic performance of impregnated pristine (pCF) and carboxylic-group-functionalized (fCF) carbon fibers with the preformed Ru nanoparticles from colloidal solution was compared with the electrocatalytic performance of Ru nanoparticles grown in situ on the pCF and fCF. According to the presented results, Ru nanoparticles impregnated on pristine carbon fiber had the best stability and activity. According to kinetic parameters (Appendix A), activities of both impregnated Ru@CF and in situ Ru@CF are to some extent affected by the functionalization of the carbon support. Considering only the onset potential, modification of carbon support seems to affect only in situ Ru@CF. However, if Ru content in the composites is taken into account, deterioration of catalytic performance in the case of impregnated Ru@CF seems plausible, particularly because overpotential at 1 A mg−1 is higher for impregnated Ru@fCF, compared to the impregnated Ru@pCP, while the former has significantly lower stability. Benefits of functionalization of carbon-based supports were demonstrated in the case of ultrasmall iridium nanoparticles supported on N-doped graphene, reported by Wu et al. [57]. They compared the electrocatalytic performance of Ir nanoparticles (1.81 ± 0.55 nm) supported on graphene (obtained by hydrothermal treatment, followed by annealing at 700 °C – Ir@G-750), Ir nanoparticles (2.18 ± 0.92 nm) supported on N-doped graphene (prepared by annealing polydopamine modified graphene oxide with Ir3+ at 750 °C – Ir@N-G-750 °C), and commercial Ir/C and Pt/C. The best hydrogen evolution catalytic performance was observed for Ir@N-G-750 and explained in terms of abundant Ir-N interactions that enhance electron density in Ir atoms. Ir nanoparticle adhesion energies and H adsorption energies were calculated by DFT on pristine graphene, graphitic N-doped graphene, and pyridinic N-doped graphene. The choice of graphene structures was based on the confirmation of these types of N-atoms in the XPS spectrum of N-G-750. Calculated Ir adsorption energy was the least positive for pyridinic N-doped graphene, which implied the favorable thermodynamics of Ir nanoparticle formation. Furthermore, three types of N atoms were resolved from the XPS spectrum of Ir@N-G-750: pyridinic, graphitic, and Ir-coordinated N atoms, that actually dominated in the spectrum, which together with calculated Ir-N distances implied the formation of Ir-N bonds. Huang et al. exploited native porosity, large surface area, uniform cavities, and polyhedral structure of zeolitic imidazolate framework-8 for preparation of N-doped porous carbon polyhedrons (NPCP) as supports of rhodium nanoparticles. The size of the obtained NPCP polyhedrons was around 120 nm, with mostly encapsulated Rh nanoparticles with the diameter of 2.8 nm. Electrocatalytic performance for hydrogen evolution reaction was evaluated in alkaline, neutral, and acidic media and compared to the activities of Pt/C and Rh-free NPCP. According to the reported results, Rh@NPCP performed better than Pt/C in alkaline electrolyte, while Rh-free NPCP showed no electrocatalytic activity. Activities of Rh@NPCP and Pt/C in acidic and neutral electrolyte were comparable below 100 mA cm−2, while Pt/C performed better at higher current densities. However, excellent performance of Rh@NPCP is even more pronounced if mass loadings of the metals (20% Pt in Pt/C and 9.84% Rh in Rh@NPCP) are compared [58]. Among different MXenes (2D early metal carbides), Ti3C2Tx (Tx = surface terminations, usually –O and –OH) is considered as a promising candidate for electrocatalytic applications, due to high electronic conductivity and hydrophilicity. Li et al. reported excellent electrocatalytic activity of intermetallic Pt3Ti nanoparticles on 2D Ti3C2Tx support, prepared by incipient wetness impregnation (IWI) and subsequent reduction in H2 at different temperatures. The size of prepared nanoparticles was in direct correlation with reduction temperature and ranged from single Pt atoms to 9.0 ± 4.1 nm. The authors also confirmed that Pt3Ti intermetallic nanoparticles cannot be formed when the reduction temperature is below 550 °C. These findings had direct reflections on the electrocatalytic activity, as can be seen in Appendix A. Although pristine MXene has poor HER activity, Pt/Ti3C2Tx have comparable to or even better activities than Pt/Vulcan with the same Pt loading: at η = 50 mV mass activity of Pt/Ti3C2Tx-550 was 1.305 mA μg−1, which was significantly higher compared to the 0.397 mA μg−1 for Pt/Vulcan. Specific activity (given per mass of surface Pt) for Pt/Ti3C2Tx-550 (17.17 mA μg−1surface Pt) was increased 10× compared to the activity of Pt/Vulcan (1.79 mA μg−1surface Pt). This increase in activity was ascribed to favorable faceting of the Pt3Ti alloy (leading to thermoneutral hydrogen adsorption). Namely nanoparticles prepared in this way have a cuboctahedral symetry (as confirmed by STEM), mainly composed of {111} and {100} facets. These faces have near-zero differential adsorption free energies: −0.01 eV and −0.05 eV for Pt3Ti(111) and Pt3Ti(100), respectively, which is improved compared to −0.03 eV for Pt(111) and −0.19 eV for Pt(100) [59].
The performance of HER electrocatalysts is usually evaluated at near-equilibrium conditions or at relatively low currents. However, their activity and durability at high current densities (>1000 mA cm−2), important for large-scale industrial applications, are rarely addressed in laboratory-scale research. Jiang et al. reported IrFe nanoalloys supported on N-doped carbon layers that outperform Pt/C and Ir/C at low, as well as high current densities in both, acidic and alkaline electrolyte. Apart from high stability (>10,000 cycles), IrFe/NC needs only 850 mV to deliver 1000 mA cm−2 in 1M KOH, which surpasses all known electrocatalysts, including Pt/C which requires 1170 mV for the same current density. IrFe/NC with a mean particle diameter of IrFe amounting to 1.85 nm was prepared by pyrolyzing Ir-modified Fe-based metal-organic framework. DFT calculations revealed that the introduction of Fe atoms decreases H adsorption energy on Ir atoms through charge accumulation. Bader charges showed that Ir atoms, as the main active H adsorption sites, have a higher number of electrons if located in the vicinity of dopant Fe atom. Large specific surface area and superior conductivity, obtained by this preparation method, additionally contribute to excellent catalytic performance [60].
The joint contribution of alloying metal and catalyst support on the performance of Pt-based catalysts was also reported in the recent paper by Meng et al., describing synthesis and HER electrocatalytic properties of different PtCo alloy nanostructures supported on reduced graphene oxide (rGO). All prepared PtCo/rGO composites had slight dependence of the activity on the initial Pt/Co ratio and showed excellent electrocatalytic activities, compared to the expected performance of Pt/C and slightly inferior performance of unsupported PtCo [61].
Du et al. reported a one-step chemical reduction procedure for the preparation of Pt-Ni alloy nanoparticles supported on reduced graphene oxide. Such prepared samples also exhibited HER activity dependence on the initial ratio of Pt and Ni precursors and had better performance than Ni/rGO, Pt/rGO, and commercial Pt/C (see Appendix A). Material obtained with the initial Pt/Ni molar ratio of 1 (Pt-Ni(1:1)/rGO) had the highest specific activity of 1.09 ± 0.08 mA cm−2Pt, obtained at −0.1 V vs. RHE, which is significantly higher compared to Pt/rGO (0.21 ± 0.02 mA cm−2Pt) and commercial Pt/C (0.28 ± 0.02 mA cm−2Pt). Excellent electrocatalytic performance of rGO-supported Pt-Ni nanocatalysts could be ascribed to combined contribution of: (i) modification of platinum electronic structure by electron transfer from nickel atoms, leading to the decrease in H adsorption energy on Pt, and (ii) nickel hydroxide layer inherently formed during hydrothermal procedure, which promotes water decomposition by accepting OH− ions, previously demonstrated by Markovic group [62,63,64]. In order to test the Ni(OH)2 contribution to the HER activity, linear voltammograms for Pt-Ni(1:1)/rGO were also recorded after etching the sample in acetic acid, which caused a slight decrease in activity (see Appendix A). The excellent stability of these materials is discussed in terms of protection by Ni(OH)2 layer [65].
The importance of interfacial process in HER catalysis [66] was also demonstrated recently for supported Ni particles [67]. Change in the density of Ni/NiO interfaces, important for acceptance of OH– ions in alkaline electrolyte, has been considered as the main reason for the change in electrocatalytic activity in the recent work of Zhang et al., in which nickel nanoparticles anchored on nitrogen-doped carbon were synthesized from tetracyanoquinodimethane precursors and tested for HER electrocatalytic performance in 1 M KOH. Metal tetracyanoquinodimethane compounds have a rigid planar structure, which can hinder the metal nanoparticle aggregation during pyrolysis and thus enable size control of metal nanoparticles. This synthetic approach enabled easy control of Ni nanoparticle size through controlling the reaction temperature; sizes of anchored Ni nanoparticles were 1.3, 8.8, and 14.9 nm for 300, 400, and 500 °C, respectively. The difference in catalytic performance between obtained samples, reflected in different Tafel slopes (Appendix A), was explained in terms of different density of Ni/NiO interface in direct contact with electrolyte [67].
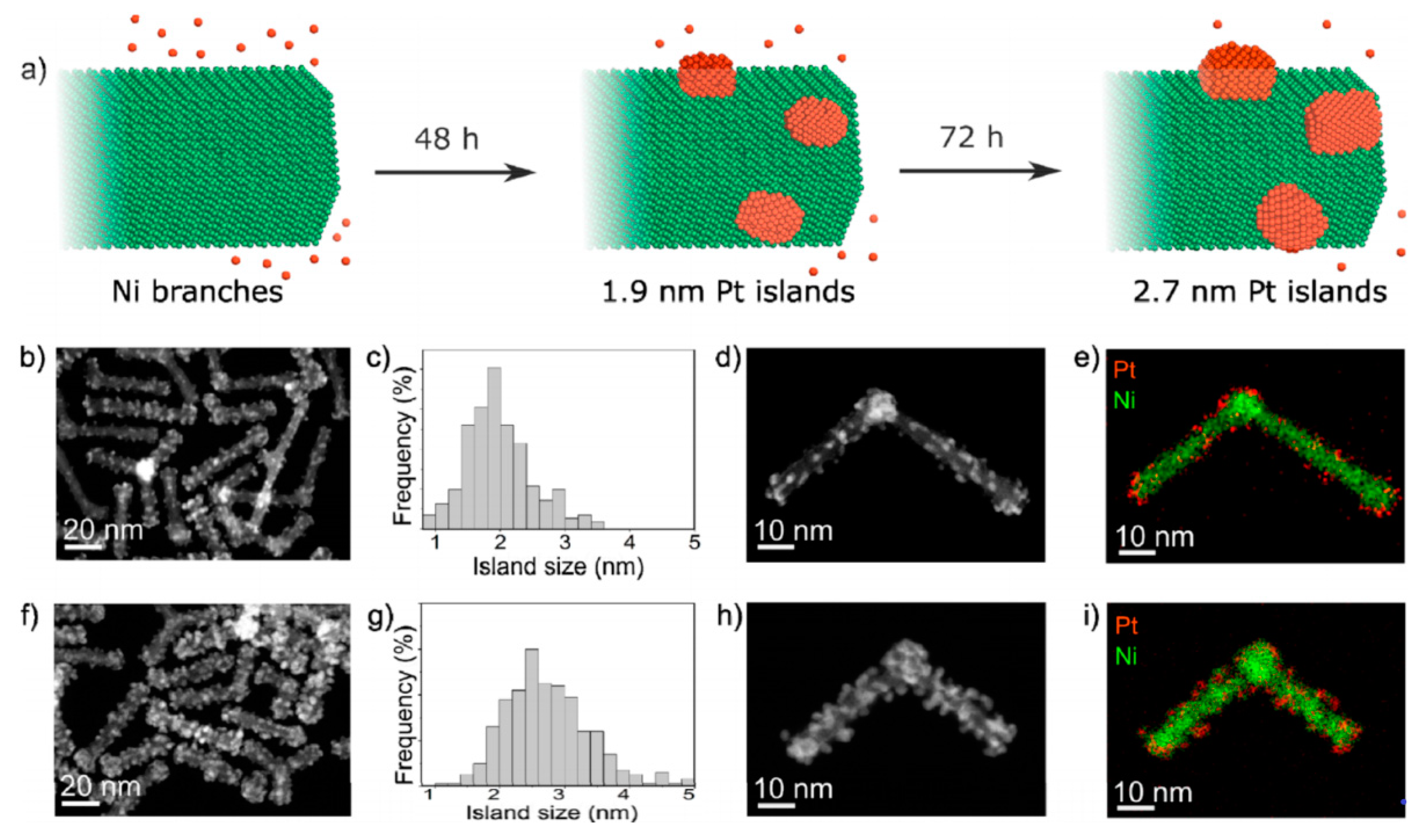
Lattice mismatched nickel nanoparticles decorated by highly strained Pt islands by slow reduction approach, under mild conditions in nonpolar solvent, yielded Pt nano-islands with the diameter of 1.9 and 2.7 nm, for the reaction time of 48 and 72 h, respectively (Figure 4). This approach enabled high control of the Pt islands size and prevented galvanic replacement. The specific activity of 1.9 nm Pt@Ni in alkaline electrolyte was 7.5 times higher than specific activity of Pt/C with Pt nanoparticle size of 1.6 ± 0.5 nm, while mass activity was 9.6 times higher (7.7 ± 0.6 mA μg−1Pt for 1.9 nm Pt@Ni; 0.8 ± 0.1 mA μg−1Pt for commercial Pt/C). Such outstanding activity is the consequence of (i) contact between Pt and Ni phases, which facilitate water dissociation through adsorption of OH– on nickel phase, thus cleaning Pt surface for easier H* recombination, and (ii) strained Pt with modified hydrogen binding energy. Strain effect is obvious from the results, as ~1.4 times increase in the compressive strain for the 1.9 nm Pt-islands, compared with the 2.7 nm Pt-islands, increased specific activity from 6.4 mA cm−2Pt to 14.7 mA cm−2Pt [68].
The effect of different carbon-based materials on HER electrocatalyst performance is also demonstrated by Cardoso et al. for SnO2-supported PdNi nanoparticles. Two highly conductive carbons (Ketjenblack EC-600JD and EC-300J, designated as KB600 and KB300, respectively), graphene, and Vulcan XC72 were composited with SnO2, in order to achieve high support conductivity and suppress aggregation and pulverization of Sn clusters. Obtained supports were decorated by PdNi (90:10) nanoparticles and tested for HER performance at different temperatures. Results obtained for the lowest (25 °C) and the highest (85 °C) are shown in Appendix A. PdNi/(SnO2-graphene) had the highest overall Tafel slopes and the highest activation energy but at the same time showed the best performance at high temperatures, at which industrial water electrolyzers usually operate. On the other hand, PdNi/(SnO2-KB600) was the best material in terms of the determined kinetic parameters, particularly below 45 °C [69].
Considering the effect of carbon support, the contribution of some specific features of the graphene-based supports to the electrocatalytic activity of carbon-supported Ni was reported recently for two series of electrochemically prepared Ni@rGO composite catalysts and tested for HER in alkaline media using two different graphene oxide (GO) samples: chemically (CGO) and electrochemically exfoliated graphene oxide (EGO). Although both materials were soluble in water and had a significant number of surface defects, based on the Raman spectra, the XRD pattern of EGO showed dominant graphitic reflection, indicating multilayer graphene material. Catalyst preparation involved single-step electrodeposition of Ni during which GO support is converted to rGO. It was observed that there is an optimal Ni deposition time for each GO which results in maximum HER activity. However, the most interesting result is the fact that the improvement of the catalytic activity of Ni is apparently not linked to the quality of GO and the exfoliation degree but to surface oxidation and disorder. It seems that relatively cheap EGO performs much better than more expensive alternatives obtained by chemical exfoliation [70]. In this case, the improvement of HER activity was governed by interfacial effects, which are addressed in Section 3.4.
Begum et al. significantly improved PdMn/CNT electrocatalytic activity by utilizing Mn-phthalocyanine as (i) the Mn source for PdMn alloying and (ii) mediator for PdMn nanoparticle anchoring onto the carbon nanotube (CNT) support, through π–π interactions. This approach enabled homogenous dispersion of Pd and Mn over the CNTs in the PdMn-N4/CNT sample, with the nanoparticle average size of 4.4 nm. Electrocatalytic activity of this sample was improved, compared with the activities of PdMn and Pd anchored directly onto the CNT (designated as PdMn/CNT and Pd/CNT), which was discussed in terms of better attachment of PdMn through N4-system onto carbon nanotubes, comparatively smaller particles formation with better dispersion and higher electrical conductivity [71] (Appendix A).
An alternative example employing support effects was reported recently for hexagonal boron nitride (h-BN) by Liu et al. Pristine h-BN is a wide bandgap semiconductor and is assumed to be electrocatalytically inert. However, Liu et al. studied h-BN supported on Au and Cu (h-BN/Au and h-BN/Cu) by scanning electrochemical droplet cell technique. Obtained results revealed improvement but still a very poor activity of h-BN on Cu and moderate activity of h-BN/Au (two orders of magnitude higher than for h-BN/Cu), which revealed a significant influence of the metallic surface [72].
Decrease in platinum content with the increase in electrocatalytic activity was also reported for platinum nanoparticles dispersed on Ni(OH)2 [73,74,75], Ni3N [76] CoP [77], nitrogen-doped carbon/Ni nanofibers [78], nitrogen-doped ordered mesoporous carbon [79], sulfur-doped graphene [80], and Fe-doped α-Ni(OH)2 [81]. Similar synthetic procedures as those described in the preceding text were used for preparation of other supported PGMs or their alloys and composites with non-noble metals, including Pd@Cu(II) phthalocyanine/MOF [82]; Ru nanoparticles supported on cobalt carbonate hydroxide nanowires [83]; PtCu anchored on N-doped carbon nanofibers [84]; and Co, Fe co-doped Ru on carbon [85]. Further improvement of electrocatalytic of supported metallic nanoparticles can also be achieved by the functionalization of Pt/CoP with ethylene glycol [77], with the specific activity of 101.2 mA μgPt−1 and Rh with polyallylamine [86]. In both cases combined surface, interface, and support engineering of materials lead to better performance of prepared materials in terms of activity, compared to Pt/C.
In addition to PGM-free works, new reports with PGM-free supported catalysts that contain metallic phase(s) include carbon encapsulated Co nanoparticles [87]; cobalt and nitrogen-doped mesoporous graphitic carbon [88]; Au-NiSx heterostructures [89]; core-shell ZnCo/N-doped carbon@MoS2 [90]; Co/CoP@N-doped carbon [91]; and Co, N, co-doped graphene [92]. It is important to note that non-noble transition metal nanoparticles are almost always oxidized to some extent, which when considering their electrocatalytic activity positions them between metal-based catalysts and (hydr)oxides, reviewed in next section.
3.2. Supported Chalcogenides, Hydroxides, Pnictides, and Carbides
The majority of papers considering transition metal-based catalysts (chalcogenides, hydroxides, carbides, pnictides), published in 2018 and 2019 deal with improvements of synthetic procedures, in order to increase electrochemical surface area and dispersion of active nanoparticles and charge engineering through introduction of nitrogen into the carbon support, thus enabling formation of metal–nitrogen bonds. Few papers also report new strategies to prepare TM sulfides, carbides, nitrides, and phosphides on the porous nickel foam substrates. Given that several recent reviews [93,94,95,96,97] deal with these types of catalysts extensively, the following text gives an overview of the most recent papers, with a focus on the optimization strategies for the preparation of high-performance HER electrocatalysts.
Several recent works dealing with supported transition metal chalcogenides are focused on the (i) improvement of electronic and catalytic properties by inserting cobalt atoms and (ii) enhancement of nanoparticle dispersion on a carbon support. In the paper by Xu et al. Co-doped Ni0.85Se on reduced graphene oxide support was obtained after one-step hydrothermal treatment at 200 °C. If HER activities (Appendix B) of unsupported Ni0.85Se, Ni0.85Se/rGO, and Co-Ni0.85Se/rGO are compared, beneficial effects of both, Co-dopant and rGO support, are clearly revealed [98]. Calcination of cobalt nitrate, ammonium molybdate, and graphene oxide at 600 °C, reported by Ma et al., yielded material with two distinct phases-CoMoO4 and MoO2-supported on reduced graphene oxide, with excellent HER performance, as can be seen in Appendix B. Improvement of electrocatalytic activity with introduction of cobalt is significant, as evidenced from kinetic parameters, given in Appendix B [99]. Hou et al. reported more extensive research on the synthetic routes for the preparation of graphene and N-doped graphene supported CoMoS2 and consequent electrocatalytic performance. They investigated the effects of H2O2, N2H4, and NaBH4 addition, with and without NH3, in the reaction mixture on the electrocatalytic activity of the final product, obtained by hydrothermal reaction between cobalt chloride, ammonium molybdate, thiourea, and graphene oxide at 140 °C. Samples designated as CoMoS2/NGO-N°, where N° = I, II, III, IV, V, VI, VII, were prepared with the addition H2O2, N2H4, NaBH4, H2O2 + NH3, N2H4 + NH3, NaBH4 + NH3 and without any of the listed additives, respectively. The lowest activity was measured for CoMoS2/NGO-VII (sample without any of the additives), which also had the highest particle size (100–200 nm) and lowest electrochemical surface area, while CoMoS2/NGO-IV (sample with H2O2 + NH3), with particle size of 5–8 nm and high electrochemically active surface area (ECSA), had the best performance. Moreover, electrocatalytic activity was higher for samples prepared with the addition of ammonia, compared to corresponding samples without NH3, and increased as additive was changed in the order NaBH4 → N2H4 → H2O2, regardless of ammonia presence (Appendix B) [100]. Heterostructured CdS@CoMo2S4/MoS2 catalyst prepared directly on indium-tin-oxide glass by the hydrothermal method was tested as photo- and electrocatalyst for HER, as reported in a recent paper by Sheng et al. [101]. CoMo2S4/MoS2 ratio, which can be tuned by reaction time or the ratio of precursors, was found to be crucial in the enhanced photo-assistant electrocatalytic hydrogen evolution: if MoS2 phase was dominant, catalyst had superior photocatalytic performance; on the other hand, composite with dominant CoMo2S4 phase had excellent photocatalytic as well as electrocatalytic activity, with η10 = 172 mV.
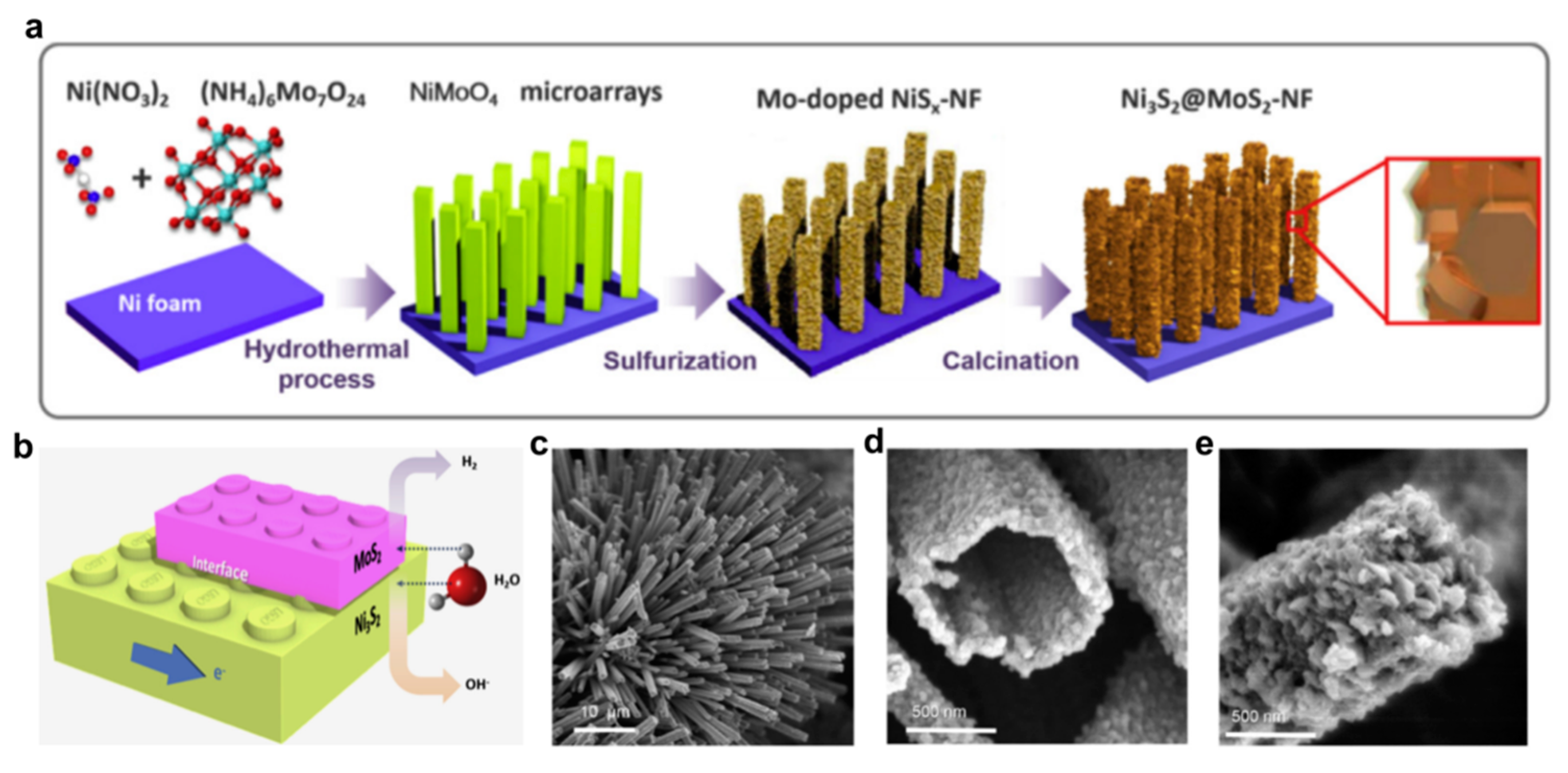
Besides different carbon-based supports, HER electrocatalysts can be prepared directly on the nickel foam, which does not serve only as conductive support with a high surface area but also as a source of nickel in the process of synthesis of different Ni-based catalysts. The use of this support provides sufficiently high electronic conductivity, very low interfacial resistance between Ni and catalysts phase, and robustness responsible for high stability. Preparation of nickel foam-supported electrocatalysts usually includes at least one solvothermal step in which nickel is replaced by other metals to some extent. This approach, using thioacetamide as the source of sulfur, reportedly yielded hierarchical Ni-Mo-S/MoOx/NF 3D frameworks with good electrocatalytic properties, as can be seen from Appendix B [102]. Solvothermal step with subsequent calcination was reported for preparation of CoMoO4 supported od nickel foam by Yan et al. [103] They investigated possible ways for optimization of the synthesis method, by varying the synthesis time, Co/Mo molar ratio, and urea content in the reaction mixture, in order to obtain desired morphology and HER activity of CoMoO4/NF. The sample obtained by optimized procedure had an excellent HER performance, particularly compared to the performance of Co3O4/NF and MoO2/NF, as listed in Appendix B. The presence of urea in reaction mixture affects the morphology of the sample, which was confirmed by SEM analysis. Cui et al. presented a similar methodology for the preparation of 3D hierarchical Ni3S2@MoS2/NF catalyst, with the inclusion of sulfurization between solvothermal and calcination step (Figure 5a). The excellent performance of this material in the alkaline electrolyte was ascribed to several factors. Hierarchical heterostructure of Ni3S2@MoS2/NF had a very high electrochemical surface area, estimated from double-layer capacitance, and was abundant in well-exposed phase interfaces (Figure 5b–e), responsible for “boosting evolution-release process of hydrogen gas bubbles” [104].
Wang et al. used a relatively simple two-step electrodeposition method for the preparation of high surface area oxygen reduction catalyst-NiFe layered double hydroxide supported on porous nickel modified nickel mesh (NiFe LDH/Ni/Ni). However, the hydrogen evolution activity of this sample was also significantly increased, compared to the activity of NiFe LDH deposited on unmodified Ni mesh in alkaline electrolyte [105]. A similar approach was reported by Yu et al. for the preparation of NiO-modified nickel nanodots supported on Ni foam (NiO/NiND@NF). Nickel nanoparticles were electrodeposited from aqueous solution, oxidized in air, and tested for electrocatalytic activity towards HER in alkaline electrolyte. According to the authors, the presence of Ni/NiO interfaces enhances electrocatalytic activity by preferential attachment of OH– ions onto the NiO surface. The authors also discuss possible improvements in conductivity caused by the presence of Ni nanoparticles [106].
Electrocatalytic activities of Titania supported PdO and Eu2O3, prepared by template-free one-step solvothermal method, were reported by Ibrahim et al. Pd or Eu salt was allowed to react with titanium butoxide in dimethyl sulfoxide (DMSO). Such preparation approach yielded PdO/TiO2 with the average particle size of 46 ± 0.78 nm and better HER activity and stability, compared to Eu2O3/TiO2 obtained by the same method with the average particle size of 39 ± 0.67 nm [107].
Other works focused on preparation of HER active chalcogenides and hydroxides include Ni-Co sulfide/Ni-Co carbonate-hydroxide grown on Ni foam [108], MoS2@Ni(OH)2/Ni foam [109], Co9S8@MoS2 [110], Te@NiTe2/NiS [111], MoS2 coated Ni3S2 [112], WS2 supported on TiO2 [113], N-doped reduced graphene oxide supported CoSe2 [114], Co9S8 supported on N,S co-doped carbon [115], MoNiS@NiS [116], Mo-incorporated Ni(OH)2 [117], Ni2P-NiSe2 [118], NiCo2S4/CdO [119], Ni3S2 decorated by Ni3Sn2S2 quantum dots [120], metallic WSe2:Sn supported on reduced graphene oxide [121], and RuO2/Co3O4-RuCo supported on N-doped carbon [122].
Ni et al. reported the difference in catalytic performance between bulk Ni3N, prepared by high-temperature reduction of Ni(OH)2 ni NH3 atmosphere, and carbon-supported nanoparticulate Ni3N, prepared from NiO via an oleylamine-assisted method. Compared to bulk Ni3N and nickel nanoparticles on the same carbon support (Vulcan-XC 72R), Ni3N/C had a significantly higher activity for HER. The difference in the mean particle size for bulk Ni3N and Ni3N/C (42.2 ± 18.5 nm and 4.6 ± 1.0 nm, respectively) reflected on the mass activity of two catalysts: 1.73 mA mg−1cat for bulk and 24.38 mA mg−1cat @ η = 50 mV for supported sample [123]. Wu et al. proposed interesting strategies for tuning the electrocatalytic performance of rGO-supported molybdenum phosphide, using NaCl as a solid template. Besides the effect of NaCl, the effects of sulfur (introduced by thiourea during one-step preparation route) and pyrolysis temperature were also investigated. According to the presented results, the NaCl template leads to noticeably increased electrocatalytic activity for HER, which is discussed in terms of increased specific surface area. Optimal values yielding the most active material were observed for all three parameters, i.e., NaCl content, sulfur content, and pyrolysis temperature: 0.5 g of NaCl in the reaction mixture (besides 0, 0.2, and 0.8 g for other samples); 0.01 g of thiourea (next to 0 and 0.019 g for other samples); and 800 °C (which was between 700 and 900 °C for other samples) [124].
Uniform dispersion of ultrasmall iron phosphide nanoparticles on carbon nanotubes was reportedly achieved by oxidation and subsequent phosphorization of precursor formed by adsorption of iron(II) phthalocyanine onto carbon nanotubes through π–π interactions. This approach yielded ultrasmall FeP nanoparticles with an average diameter of 2–4 nm, anchored on, or even impregnated into the CNTs. CNT-supported FePs, prepared from different initial FePc/CNT mass ratios (1:1, 2:1, 3:1, and 4:1, designated as FeP/CNT-1, FeP/CNT, FeP/CNT-2, and FeP/CNT-3, respectively) had significantly higher electrocatalytic activities, compared to the unsupported FeP and phosphorized CNTs [125].
Specific interactions between Mo2C and defect-rich N-doped carbon nanosheets and their effects on the electrocatalytic activity towards the HER were reported by Lei et al. Tested materials were prepared by calcination of graphitic carbon nitride, sucrose, and ammonium heptamolybdate in a nitrogen atmosphere at 800 °C. A representative sample, prepared with the g-C3N4/molybdate mass ratio of 2, consisted of Mo2C nanoparticles with the dimensions between 1–3 nm, wrapped in carbon nanosheets with thickness of 3–5 defect-rich graphene layers, and had the best HER performance (η0 = 83 mV, Appendix A, Table A2). The hydrogen evolution activity of this sample was compared to three control samples, two of which were prepared with the g-C3N4/molybdate mass ratios of 1 and 5, while the g-C3N4 was absent during the preparation of the third one (i.e., the g-C3N4/molybdate mass ratio was 0). Sample prepared without the g-C3N4 had the poorest HER performance (η0 = 391 mV) compared to MoNCs-1 (η0 = 163 mV) and MoNCs-5 (η0 = 158 mV) [126].
Formation of Mo-N bond in the molybdenum carbide supported on N-doped carbon nanotubes was also achieved by reaction between Mo(IV)-melamine polymer and oxidized carbon nanotubes, in the work by Song et al. Activity was tuned by changing the Mo(IV)-melamine/oxidized CNTs ratio in the reaction mixture, which yielded composites with different Mo atomic content (and N atomic content as well), and different activities: samples with the lowest (1.16 at %) and the highest (6.06 at %) Mo content had poorer HER activities, compared to the samples with 3.78 and 2.69 at % of Mo. However, activities of all of these samples were better compared to the activities of Mo2C/C, obtained by the same procedure but without oxidized CNTs as precursor, NCTNs, and CNTs, as can be seen in Appendix A, Table A2, which clearly demonstrate synergy between N-doped nanostructured carbons and catalyst in the improvement of electrocatalytic performance [127].
Novel HER active supported transition metal pnictides and carbides also include bimetallic phosphide; Ni2-xCoxP supported on electrospun N-doped carbon nanofibers [128]; CoP/NiCoP supported on N-doped carbon [129]; Mo nitrides supported by B,N co-doped carbon nanotubes [130]; MoC-MoP/N-doped carbon nanofibers [131]; Cu3(PO4)2/Cu-MOF [132]; biphase Ni5P4/NiP2 grown on Ni foam [133]; Co2P/CoMoPx supported on Ni foam [134]; coupled Mo2C and Ni nanoparticles encapsulated in porous carbon nanofibers [135]; Ni2P/Cu3P on carbon paper [136]; N-doped carbon coated MoP nanowires [137]; CeO2-CoP-C hybrid nanostructure [138]; Co6W6C nanocrystals anchored on N-doped carbon nanofibers [139]; FeOOH/Ni3N [140]; carbon supported CoP nanoparticles with focus on size-dependent activity [141]; Fe, Co, and Ni phosphides anchored on metal-caged graphitic carbon [142]; MoC2-doped NiFe alloy nanoparticles encapsulated with N-doped graphene [143]; C3N4@MoN [144]; CoP nanowires coupled with CoMoP nanosheets [145]; Ni-Fe-P nanoparticle embedded in N-doped carbon supported on Ni foam [146]; and Co, Al doped Fe2N/Fe3N [147].
3.3. Miscellaneous Materials
This section contains a brief overview of novel materials prepared and tested for electrocatalytic performance towards the HER, which does not fit into previously described categories. Few of these reports employ preparation methods similar to the methods overviewed in the previous section, such as synthesis of Cu3(PO4)2/Cu-MOF [132], Co(OH)F, and Co-P nanoarrays supported od Ni foam [148], CoCO3@CoSe supported also on Ni foam [149], and Ni11(HPO3)8(OH)6/Mn3O4 [150]. A comparison of HER performance for three 2D polyhalogenated Co(II)-based MOFs demonstrated the possibility to tune the activity by changing the halogen substituents [151]. Wang et al. compared HER activity of two covalent organic polymers based on Zn phthalocyanine supported on graphene oxide with the activities of pure components and their physical blends and found significant improvement caused by the covalent linkage between all of the components [152]. Another interesting material—phosphorene quantum dot modified Prussian blue porous network—with reportedly excellent HER activity was reported by Xia et al. Superb electrocatalytic activity in acidic electrolyte was explained in terms of passivation of phosphorene edges by CN-groups in Prussian blue [153]. Knowing that graphene oxide can be completely reduced at the significantly lower overpotentials compared to those needed for HER and that HER at reduced graphene oxide requires large overpotentials, results reported by Aslam et al. for HER at graphene foam seem surprising, especially because authors performed detailed characterization and excluded presence of metallic impurities [154].
3.4. Role of the Metal-Support Interfaces in HER Mechanism
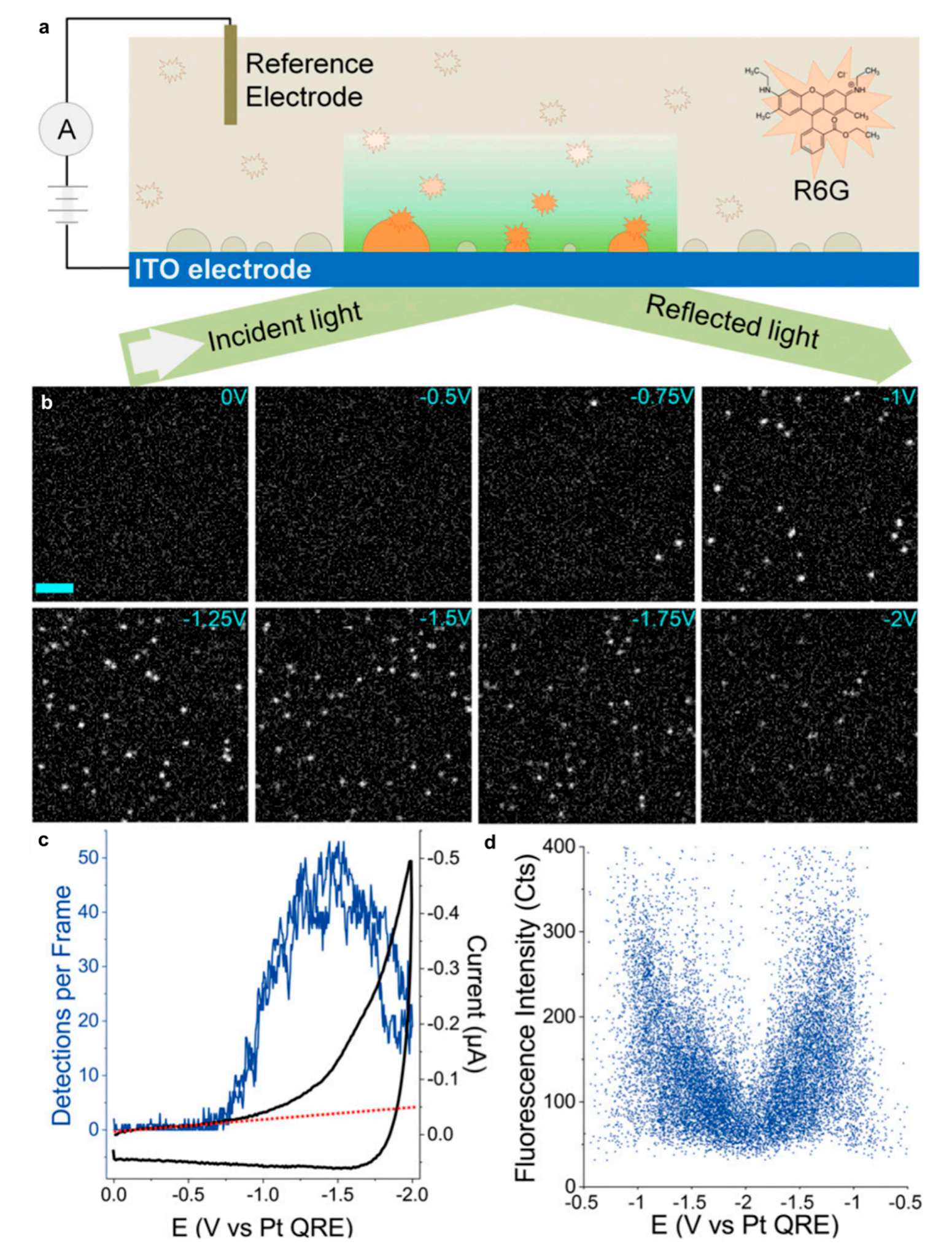
Almost all of the recently published papers, dealing with supported electrocatalysts, recognize the beneficial effects of different interfaces for catalytic activity towards the HER. However, most of them discuss these effects in vague terms of “synergistic effect” and “tailored electronic configurations”, where usually one phase “accepts OH−” and thus improves catalytic activity. Of course, the last point relies on the work of Markovic groups on the formation of complex interfaces with transition metal (oxy)hydroxides [62,63,64]. Nevertheless, only a few papers address these phenomena more extensively in terms of electronic effects. Zhdanov and Kasemo recognized four reasons responsible for unique catalytic behavior of supported nanosized catalysts: (i) diffusion-mediated communication between adjacent crystal facets at far-from-equilibrium conditions; (ii) spillover effect; (iii) adsorbate-induced reshaping of catalyst particles; (iv) electronic modification of catalyst particles by the support [155]. Spillover, being defined as “the transport of active species sorbed or formed on a first surface onto another surface that does not under the same conditions sorb or form the active species” [156], is a well-known interfacial step in mechanisms of reactions involving heterogeneous catalysts. Hydrogen atom spillover (not to be confused with H2 spillover, which includes dissociation and is an important step for hydrogenation reactions and hydrogen storage) was recognized as the phenomenon responsible for the promotion of Pt electrocatalytic activity towards the hydrogen evolution 47 years ago, for Pt supported on pyrolytic graphite [157]. Several experimental works contributed to the affirmation of Hads spillover as the important step in HER mechanism at Pt/C [158], Pt/transition-metal-oxide [159], Pt/Au [160], Pi/Ni [161], atomically-dispersed-Pt/WO3-x [162], Pd/Au [163], WO3·2H2O/WS2 [164], Rh/MoS2 [165], Rh/Si (although authors do not employ the term spillover) [166], Pd@Cu(II) phthalocyanine/MOF [82], and Ni/rGO interfaces [70,167,168]. Kinetic Monte-Carlo simulations were employed to investigate the impact of the spillover rate on the rate of H2 production at the Ni/rGO interface, using the model that was able to qualitatively reproduce the experimentally observed trends [168]. Spillover was also found to be a part of HER mechanism on nanocrystalline Ni films, where Hads transferred from Ni nanocrystals to intercrystalline non-homogenous disordered Ni [169]. However, straightforward experimental observation of Hads spillover is not an easy task, especially if quantitative data are needed. Eikerling et al. proposed a kinetic model [170] as a theoretical framework for experimental results from all-in-situ STM investigation of the local reactivity of a single Pd nanoparticle supported on Au(111) [171]. Calculated data revealed significant dependence of the kinetic parameter values on the size and geometry of Pd particle, where the contribution of Hads spillover from large Pd particle to the Au support was insignificant, compared to small particle (two layers of Pd on Au). Size (i.e., height) of large Pd particles also reduced electronic effects, making hydrogen adsorption weaker, compared to small particles [170]. This model was further developed for an array of Au-supported catalyst nanoparticles, in order to investigate the effect of particle density on the catalytic activity, where desorption rate and hydrogen diffusion coefficient on the support were found to be the key kinetic parameters that influence the current density [172]. Direct observation of Hads spillover from ultrathin gold nanocatalysts to indium tin oxide (ITO) substrate was reported recently by Hao et al., using total-internal reflection fluorescence (TIRF) microscopy (Figure 6) [173]. The results of computational studies strongly support the scenario where spillover plays a significant role in the mechanism of HER at multiphase catalysts [35,77,167,168,174].
4. HER at Single Atom Catalysts
As experimental techniques employed for studying heterogeneous catalysts mature, becoming more capable of elucidating structures at the Ångstrom scale, debates about the presence and activity of isolated sub-nanometer active sites and their comparison to NPs [175] are becoming more informed. The phase space of supported single atoms (Single Atom Heterogeneous Catalysts, SACs) expands the potential pool of candidates for HER catalysts due to the rich coordination chemistry of transition metals and plentiful supports (Figure 7). However, it is necessary to consider the traits unlocked by nanostructuring and their effect on the simple mechanism of hydrogen reduction. In comparison to OER, HER is typically not considered a bottleneck in electrochemical devices; however, SACs provide a way of increasing the economic feasibility of electrolyzers relying on more expensive (typically) Ir OER catalysts, by reducing the precious metal loading and therefore the price of the HER subassembly. Particularly, in proton exchange membrane water electrolyzers (PEMWE), even HER catalysts with state of the art catalytic performance and Pt dispersion that offer an efficiency ratio of 0.01 g Pt/kW would lead to a consumption of 1 t of Pt per year projecting an 100 GW/year PEMWE growth [176]. Therefore, SACs have a future minimizing active phase waste and ensuring the optimal catalyst atom economy [177]. Even with state-of-the-art experimental techniques such as high-resolution electron microscopies (HR-TEM, AC-STEM) and local coordination environment tools that rely on synchrotron radiation (XANES, EXAFS), the speciation of isolated atoms remains a significant challenge. Density functional calculations (DFT) often have the final say on deriving structure performance relationships. HER, due to its mechanistic simplicity, was the first candidate for deriving scaling relationships in extended metal surfaces [24,178]. However, confining the search for the optimal catalyst by optimizing a single parameter (atomic hydrogen adsorption free energy) becomes more complicated with the increasing complexity of the active site, as it is commonly the case with SACs.
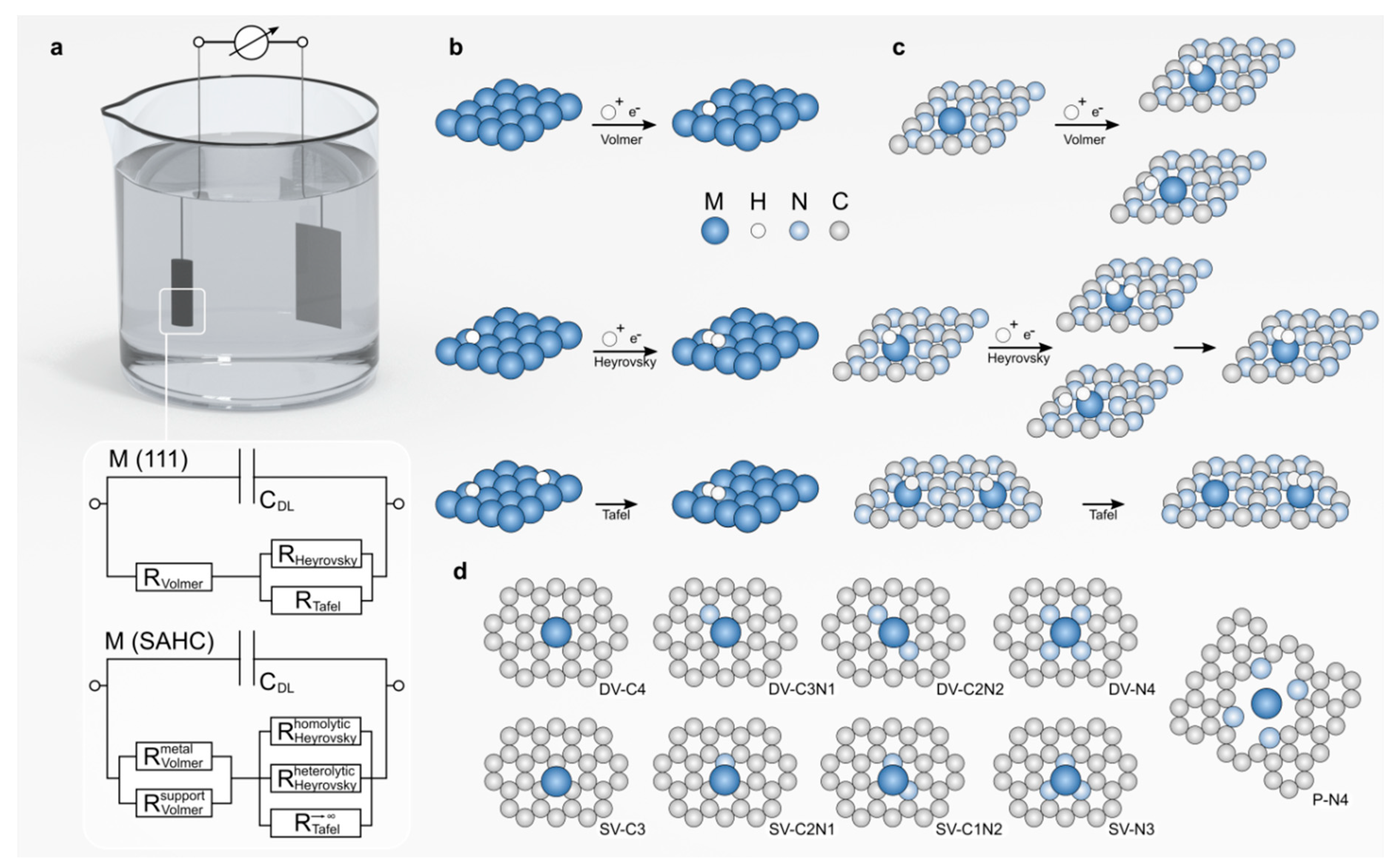
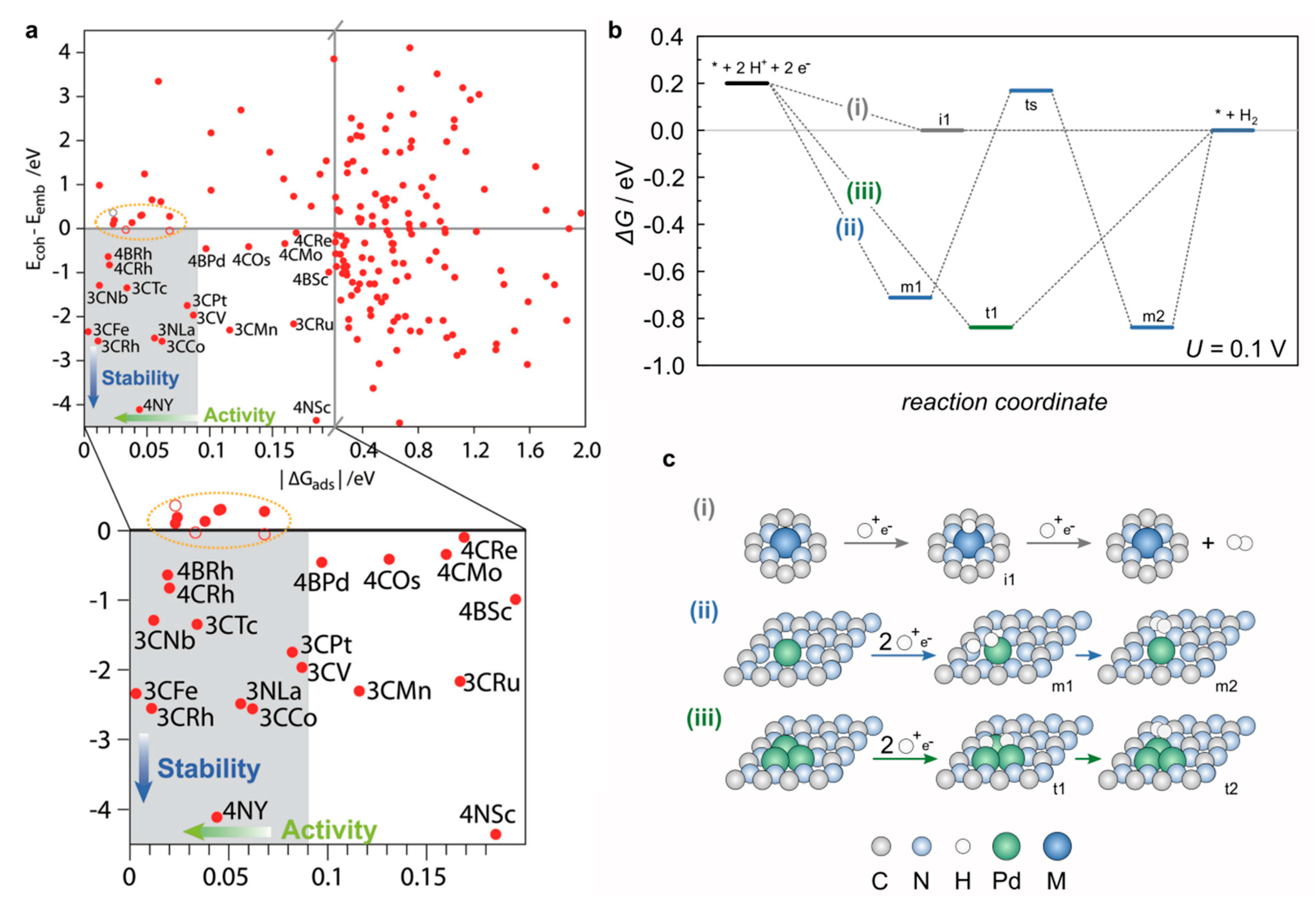
In SACs the role of the support becomes more profound as each isolated atom lies at the border between the support and metal ‘phase’. The strategy of stabilizing single atoms on carbon (and other) carriers involves providing anchoring groups or defects acting as ligands for the metal atoms [187,188]. These ligands act as Lewis bases binding the metal center and inherently poses H affinity if not saturated by bonding to the metal atom. While comparing the equivalent circuits (Figure 7a) of a single atom supported on a popular heptazinic carrier [179] to that of an extended surface of an hcp metal catalyst (Figure 7b), the conventional classification of elementary steps to Tafel/Volmer/Heyrovsky becomes more complicated. In a system where both the support and metal center adsorb H (Figure 7c) co-adsorption can occur, which can be detrimental to HER activity (trapping the active sites in off-cycle thermally modulated paths). To circumvent this possibility, SACs have been designed with saturated cavities covalently bound to the metal atom [180,181,182,183]. In this case, the preferable adsorption spot (Figure 7d) is the metal top position, where a proton (hydronium ion) can adsorb via a single coupled proton-electron transfer, with molecular hydrogen evolving recurrently trough the Heyrovsky pathway [184]. In this case, the support has a more passive role that is expressed via its influence on the electronic states of the metal center, and the existence of a shared reaction path ensures that the H* adsorption energy can be considered equivalent to the HER onset potential. The H* adsorption energy can be put into context by linearly combining the number of d electrons of the (isolated) metal atom (M), the electronegativity of M and H, and the contributions of the electronegativity of the atoms in the first coordination sphere (C or N atoms) [184]. This descriptor is universal for a wide range of metals and defects and correlates well with the experimentally determined onset potentials. The thermoneutrality condition needs to be examined in the context of SA stability (Figure 8a), as some favorable configurations may have compromised stability, in situations where scaling relations are more difficult to derive due to the active role of the support and the dynamic nature of the coordination environment. Similarly, complex behavior was observed when adjusting the nuclearity of the active site (increasing the number of metal atoms in the active site from one to two or three), with significant mechanistic differences being triggered by the existence of 3-fold sites (Figure 8 b,c) [189].
Although the effects of cluster size and (for carbon-supported SACs) the active role of the support are yet to be exploited for performance gains in HER as means of breaking the scaling relationships [190], non-Pt systems are becoming more competitive with the state-of-the-art. Particularly Co [191], Ni, Fe [192] and Ru [193] (Table 2) have shown to be comparable and even in some cases outperform conventionally accepted Pt-based catalyst (although Pt systems can be further optimized for specific activity [176]. The nanostructuring of the precious metal phase is certainly appealing from an economical perspective, but it is necessary to consider all the possible implications of providing support with good SA dispersion properties, to ensure high stability. In fact, increasing the stability of SACs can be connected to the number of defects present in the support, which can compromise electrical conductivity.
In order to drive the increasing interest in HER catalyst to the market, effective strategies are needed for catalyst preparation, and techniques that rely on complex procedures such as atomic layer deposition (ADL) [194] are in a disadvantage versus more chemical routes such as wet impregnation [179] and electro-deposition [187,195]. Considering that the expected electrolyzer power consumption is expected to be measured in GW in the near future, the scalability of catalyst preparation procedures is also highly desirable.
5. Perspective
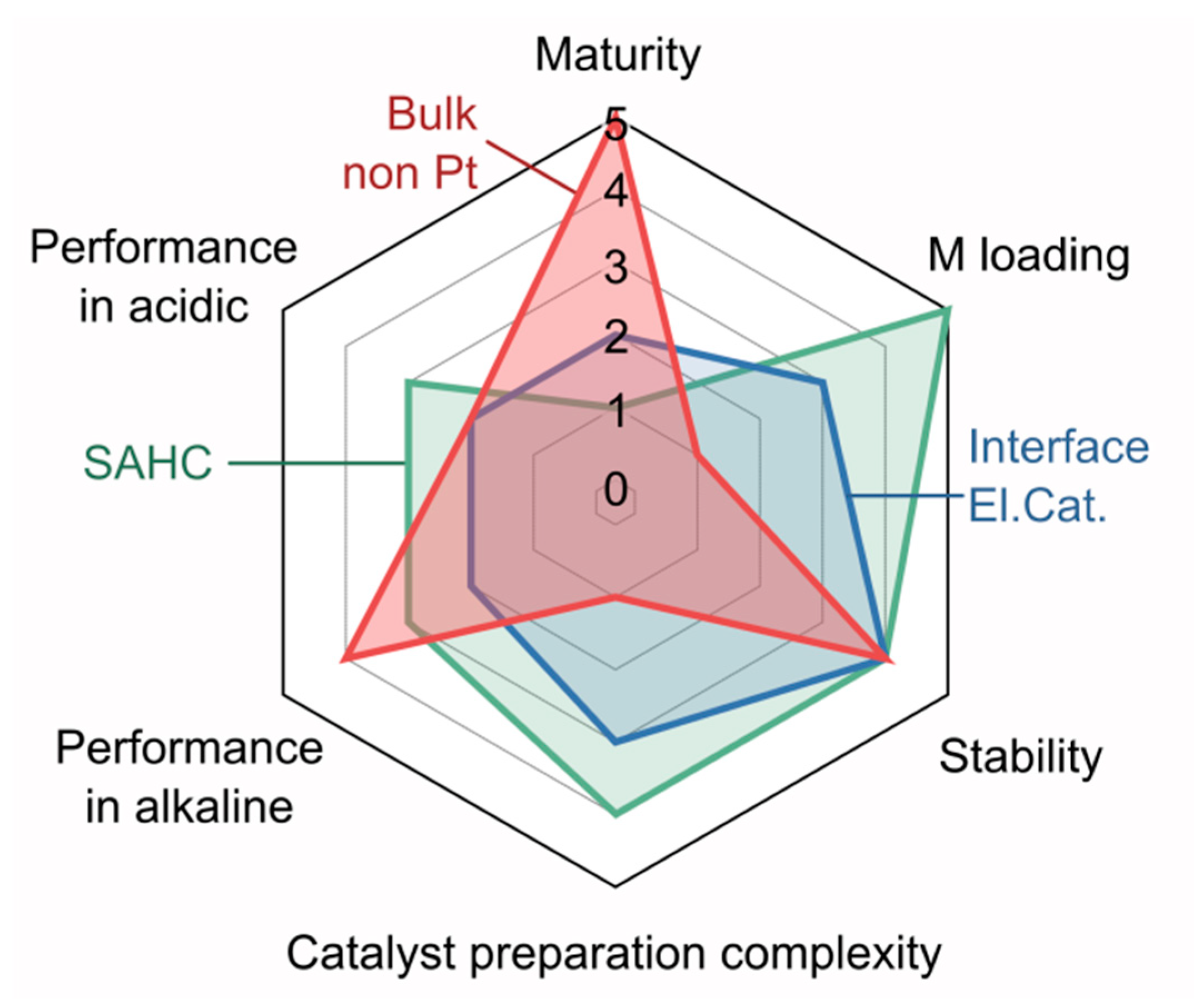
Considering the enormous number of recently published papers dealing with synthetic procedures for preparation of supported HER electrocatalysts, according to which almost every prepared material has “excellent performance”, while significant number of them even “outperforms commercial Pt/C”, as well as practically infinite number of possible synthetic approaches that can be exploited, the future of HER electrocatalysts will depend on the development of methods for fast screening of huge number of prepared materials. This is necessary in order to select those that really work, i.e., that can be applied in real systems and for which synthetic procedures can be adapted for large-scale production. As a dominant number of papers cited in this review (that describe preparation of supported electrocatalysts) does not report shortcomings of the prepared material (and there has to be some for every of them), it seems like there is nothing to improve in the field aside from tuning material a bit to get a personalized electrocatalyst with excellent HER performance. This is, of course, not the case, and there is still a lot that we have to understand when it comes to HER. If one takes just a brief look at provided Appendices, it is clear that many of the important data about catalyst performance are missing, so there is also a need to standardize testing procedures for HER catalysts. Considering the practical aspects of the use of different HER electrocatalysts, we have tried to provide our view on different properties of the main classes of HER catalysts (Figure 9). Obviously, there is a lot of space for the improvements, but the critical mass is reached in every case. Future developments will show which type will prevail as the need for efficient HER catalysts continues to grow.
Author Contributions
Data curation, S.J.G., A.S.D., and E.F.; writing—original draft preparation, S.J.G., A.S.D., E.F., and I.A.P.; writing—review and editing, N.V.S., N.L., and I.A.P.; supervision, N.V.S., N.L., and I.A.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Serbian Ministry of Education, Science and Technological Development. A.S.D. acknowledges the support provided by the Carl Tryggers Foundation (Grant No. 18:177). E. F. thanks MINECO La Caixa Severo Ochoa for a predoctoral grant through Severo Ochoa Excellence Accreditation 2014 2018 (SEV 2013 0319). This research was sponsored in part by the NATO Science for Peace and Security Programme under grant G5729.
Conflicts of Interest
The authors declare no conflict of interest.
Appendix A. Overview of Supported Metallic Nanoparticles

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table A1.
Supported metallic nanoparticles.
| Material | Elect. | j0 /mA cm−2 | η0 /mV | ηj /mV | b /mV dec−1 * | Mech. | RDS | Stability | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| PtNP/OMC Pt/Vulcan | - | - | - | - | 32 28 | - | - | - | [55] |
| pCF fCF Ru@pCF Ru@fCF RuPP@pCF RuPP@fCF (RuPP)other work | 1 M H2SO4 | - - - - - - - | 220 180 70 30 5 0 0 | - - (265)10 235 225 180 20 | - - 82.5 124.8 212.3 158 - | - - V/T V/T V/T V/T - | - - V V V V - | - - low low high moder. - | [56] |
| Ir@G-750 Ir/N-G-750 Ir/C Pt/C | 0.5 M H2SO4 | - - - - | - - - - | (28)10 19 33 23 | 29 26 34 28 | - V/T - - | - T - - | - | [57] |
| Ir@G-750 Ir/N-G-750 Ir/C Pt/C | 1 M KOH | - - - - | - - - - | (142)10 43 68 54 | 85 35 38 37 | - - - - | - - - - | - | |
| Rh@NPCP Pt/C | 1 M KOH | 1.67 1.34 | ~ 0 - | (20)10 35 | 33 36 | V/T - | - | excellent - | [58] |
| Ti3C2Tx Pt/Ti3C2Tx-200 Pt/Ti3C2Tx-400 Pt/Ti3C2Tx-550 Pt/Ti3C2Tx-700 Pt/Vulcan | 0.1 M HClO4 | - - - - - - | - - - - - - | 576.8 270.8 (73.0)10 32.7 122.3 55.8 | 238.0–395.2 - 46.3 32.3 77.6 43.9 | - - - - - - | - - - - - - | - - - excellent - - | [59] |
| Fe/NC IrFe/NC Ir/C Pt/C | 0.5 M H2SO4 | - - - - | - - - - | - - - - | - 22 43 29 | - - - - | - - - - | excellent | [60] |
| Fe/NC IrFe/NC Ir/C Pt/C | 1 M KOH | - - - - | - - - - | - (22)10 ca. 80 ca. 55 | - 30 97 46 | V/H | H | excellent | |
| Pt78Co22 NCs/rGO Pt29Co71 NPs/rGO Pt83Co17 NPs/rGO Pt52Co48 NPs Pt black Pt/C | 0.5 M H2SO4 | - - - - - - | 1.3 2.3 2.3 10.3 14.3 8.8 | (25.3)10 30.3 31.3 46.3 60.3 37.0 | 26 36 35 38 46 31 | V/T - - - - - | - - - - - - | excellent v. good v. good poor moder. v. good | [61] |
| Ni/rGO Pt/rGO Pt-Ni(3:1)/rGO Pt-Ni(3:2)/rGO Pt-Ni(1:1)/rGO Pt-Ni(1:1)/rGO-HAC Pt/C | - | - - - - - - - | - - - - - - - | (231)10 149 120 112 78 82 124 | 125 88 83 69 53 56 78 | - - - - - - - | - - - - - - - | - - - - excellent - - | [65] |
| NiNC-300 NiNC-400 NiNC-500 | 1 M KOH | - - - | - - - | 310 230 370 | 102 87 88 | - - - | - - - | - - - | [67] |
| 1.9 nm Pt@Ni 2.7 nm Pt@Ni Pt/C | - | - | - | - | 69 61 60 | - | - | - | [68] |
| PdNi/(SnO2-KB600) @ 25 °C PdNi/(SnO2-KB600) @ 85 °C PdNi/(SnO2-KB300) @ 25 °C PdNi/(SnO2-KB300) @ 85 °C PdNi/Vulcan @ 25 °C PdNi/Vulcan @ 85 °C PdNi/(SnO2-graphene) @ 25 °C PdNi/(SnO2-graphene) @ 85 °C | - | 0.03 0.47 < 0.01 0.07 < 0.01 0.10 0.03 1.52 | - - - - - - - - | (322)10 264 - - - - 338 201 | 129 162 105 155 131 169 149 142 | - | V/H | - | [69] |
| Pd/CNT PdMn/CNT PdMn-N4/CNT Pt/C | 1 M KOH | - - - - | 80 45 35 35 | (137)10 90 71 65 | 82.8 61.5 41.7 42.5 | - - - V/H | - - - des. | - moder. highest lowest | [71] |
| Pt/2D-Ni(OH)2 Pt@2D-Ni(OH)2 Pt/C | 0.1 M KOH | - - - | - - - | (188)5 123 210 | 119 72 129 | - - - | - - - | - - - | [73] |
| Pt@2D-Ni(OH)2 Pt/C | 0.1 M LiOH | - - | - - | (72)5 - | 50 93 | - - | - - | - - | |
| Pt/Ni(OH)2 | 0.1 M KOH | - | - | - | - | - | - | - | [74] |
| Pt-Mn/Ni(OH)2 | 0.1 M NaOH | - | - | - | ~60 | V/H | - | - | [75] |
| 0.45%Pt-Ni3N/Ni@C Ni3N/Ni@C Ni3N@C Pt/C | 0.5 M H2SO4 | - - - - | - - - - | (45)10 456 490 10 | 47.3 160.8 207.7 44.1 | - - - - | - - - - | - - - - | [76] |
| 0.45%Pt-Ni3N/Ni@C Ni3N/Ni@C Ni3N@C Pt/C | 0.2 M PBS | - - - - | - - - - | - - - - | 170.2 271.0 201.9 44.1 | - - - - | - - - - | - - - - | |
| 0.45%Pt-Ni3N/Ni@C Ni3N/Ni@C Ni3N@C Pt/C | 1 M KOH | - - - - | - - - - | 38.6 616.6 329.6 34.4 | 54.9 166.4 120.6 31.6 | - - - - | - - - - | - - - - | |
| EG/CoP EG-Pt/CoP-1.5 | 0.5 M H2SO4 | 0.28 1.67 | - - | (167)10 21 | 104.6 42.5 | - - | - - | - excellent | [77] |
| NCNFs-Pt Ni0.1-NCNFs-Pt Ni0.3-NCNFs-Pt Ni0.5-NCNFs-Pt Pt/C | 0.5 M H2SO4 | 0.275 0.279 0.303 0.326 - | 15 11 9 5 - | (64)10 62 57 47 45 | 41 40 38 31 29 | - - - - - | - - - - - | - - - - - | [78] |
| NOMC-700 NOMC-A Pt@NOMC-700 Pt@NOMC-A Pt/C | 0.5 M H2SO4 | - - - - - | - - - - - | - - (91)10 7 8 | 208 634 88 36 43 | - - - - - | - - - - - | - - - excellent - | [79] |
| 3DG 3DSG Pt/3DG 0.4% Pt/3DSG 0.9% Pt/3DSG 1.5% Pt/3DSG 2.2% Pt/3DSG 3.1% Pt/3DSG 20% Pt/C | 0.5 M H2SO4 | - - - - - - - 0.54 0.34 | - - - - - - - - - | (750)10 380 220 270 195 115 70 30 50 | 180.6 88.7 67.2 100.3 73.8 50.2 37.7 30.1 31.2 | - - - - - - - - - | - - - - - - - - - | - - - - - - - - - | [80] |
| Fe0.05Ni0.95(OH)2 1.2%Pt-Fe0.05Ni0.95(OH)2 2.3%Pt-Fe0.05Ni0.95(OH)2 3.3%Pt-Fe0.05Ni0.95(OH)2 Pt/C | - | - - - - - | - - - - - | - - (18)10 - 35 | 129.4 79.9 25.6 31.8 30.3 | - - - - - | - - - - - | - - - - - | [81] |
| Pd@CuPc/MOF | H2SO4/TBAPDMSO | 8.900 | - | - | 0.1769 | - | - | - | [82] |
| CoCH@CF Ru@CF Ru-CoCH@CF Pt/C | 1 M KOH | - - - - | - - - - | (233)10 99 66 - | 187 70 65 62 | - - - - | - - - - | - - excellent - | [83] |
| Pt/NPC PtCu1.1/NPC Pt/C | 0.5 M H2SO4 | - - - | - - - | (34)10 13 20 | 37 34 44 | - - - | - - - | - - - | [84] |
| CoFe-CDs CoFe@C Ru@C FeRu@C CoRu@C CoFeRu@C Pt/C | 1 M KOH | - - - - - - - | - - - - - - - | (334)10 232 86 18 14 4 21 | 218.4 190.2 95.7 35.5 34.6 33.7 49.1 | - - - - - - - | - - - - - - - | - - - - - - - | [85] |
| Rh-irNCs Rh-NSNSs PAH@Rh-NSNSs PAH@Rh-NSNSs/CNT Pt-JM NC | 0.5 M H2SO4 | - - - - - | - - - 1 10.4 | (22)10 12.1 9.8 4.4 - | - - - 30 31 | - - - - - | - - - - - | - - - - - | [86] |
| NG-800 Co@NG-700 Co@NG-800 Co@NG-900 Pt/C | 0.5 M H2SO4 | - - - - - | - - - - - | (470)10 309 286 363 - | 132 85 118 119 45 | - - - - - | - - - - - | - - - - - | [88] |
| Au@NiSx core@shell Au-NiSx yolk-shell Au-NiSx oligomer-like NiSx | 0.5 M H2SO4 | - - - - | - - - - | (253)10 263 283 321 | 43.7 49.4 50.6 56.1 | - - - - | - - - - | - - - - | [89] |
* Tafel constant, absolute value.
Table A2.
Supported carbides and pnictides.
| Material | Elect. | j0 /mA cm−2 | η0 /mV | η10 /mV | b /mV dec−1 * | Mech. | RDS | Stability | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| MoNCs-0 MoNCs-1 MoNCs MoNCs-5 Pt/C | 0.5 M H2SO4 | 6.20 × 10−3 - 2.65 × 10−3 - - | 391 163 83 158 - | - 233 157 223 - | 179.1 - 60.5 - 26.7 | - - V/H - - | - - - - - | - - excellent - - | [126] |
| CNT NCNT Mo2C/C Mo2C/NCNT-10 Mo2C/NCNT-20 Mo2C/NCNT-30 Mo2C/NCNT-40 Pt/C | 0.5 M H2SO4 | - - - - - - - - | 426 371 219 115 101 92 127 31 | 596 497 340 213 200 195 212 87 | - - 110 86 82 75 81 33 | - - - - - - - | - - - H ads. H ads. H ads. H ads. - | - - - excellent excellent excellent excellent - | [127] |
| P-CNT FeP FeP/CNT-1 FeP/CNT FeP/CNT-2 FeP/CNT-3 Pt/C | 0.5 M H2SO4 | 0.01 0.19 - 0.31 - - 0.76 | 171 91 - 41 - - ~ 0 | - - 100 68 85 129 - | 78 89 - 58 - - 30 | - - - V/H - - - | - - - - - - - | - - - - - - - | [125] |
| Ni-Ref Ni3N Ni3N/C Pt/C | 1 M KOH | - - - - | - - - - | - - 64 - | 88 122 48 32 | - - - - | - - - - | - - - - | [123] |
* Tafel constant, absolute value.
Appendix B. Overview of Supported Chalcogenides and Hydroxides
Table A3.
Supported chalcogenides and hydroxides.
| Material | Elect. | j0 /mA cm−2 | η0 /mV | η10/mV | b /mV dec−1 * | Mech. | RDS | Stability | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| Ni0.85Se Ni0.85Se/rGO Co-Ni0.85Se/rGO Pt/C | 0.5 M H2SO4 | - - - - | - - - - | 270 182 148 - | 81 62 49 33 | - - V/H - | - - - - | - - - | [98] |
| Ni0.85Se Ni0.85Se/rGO Co-Ni0.85Se/rGO Pt/C | 1 M KOH | - | - | 299 198 128 - | 156 118 89 36 | - - V/H - | - | - | |
| MoO2/G Co-MoO4- MoO2/G Pt/C | 0.5 M H2SO4 | - - - | 107 36 24 | 262 103 54 | 98.22 45.99 43.97 | - - - | - - - | - - - | [99] |
| CoMoS2/NGO-I CoMoS2/NGO-II CoMoS2/NGO-III CoMoS2/NGO-IV CoMoS2/NGO-V CoMoS2/NGO-VI CoMoS2/NGO-VII | 0.5 M H2SO4 | - | 116 148 173 54 76 89 204 | - | 61.77 67.05 69.97 34.13 45.33 46.03 86.67 | - | - | - | [100] |
| NF Co3O4/NF MoO2/NF CoMoO4/NF Pt/C | 1 M KOH | - | - | - 181 175 68 30 | 175 88 86 82 - | - V/H V/H V/H - | - - - excellent - | [103] | |
| NF MoS2/GC MoS2/NF Ni3S2@MoS2/NF Pt/C | 1 M KOH | - | - | - - - 60 20 | 194 120 125 48 34 | - | - | - - - excellent - | [104] |
| Ni/Ni NiFe/Ni NiFe/Ni/Ni | 1 M KOH | - | - | 221 332 132 | 122.9 122.8 107.7 | - | - | - | [105] |
| NF NiO/NiND@NF Pt/C@NF | 1 M KOH | - | - | - 119 0 | 150 113 34 | - | - | - | [106] |
| GCE TiO2 Eu2O3/TiO2 PdO/TiO2 Pt/C | 4 M NaOH | 2.5 × 10−5 4.5 × 10−3 5.0 × 10−2 26 × 10−2 71 × 10−2 | 750 390 218 110 12 | 821 490 315 195 135 | 172 146 135 125 121 | - | - | - | [107] |
* Tafel constant, absolute value.
References
- Sheng, W.; Gasteiger, H.A.; Shao-Horn, Y. Hydrogen Oxidation and Evolution Reaction Kinetics on Platinum: Acid vs. Alkaline Electrolytes. J. Electrochem. Soc. 2010, 157, B1529. [Google Scholar] [CrossRef]
- Schuldiner, S. Hydrogen overvohage on bright platinunl II. pH and salt effects in acid, neutral, and alkaline solutions. J. Electrochem. Soc. 1954, 101, 426–432. [Google Scholar] [CrossRef]
- Bockris, J.O.M.; Koch, D.F.A. Comparative rates of the electrolytic evolution of hydrogen and deuterium on iron, tungsten and platinum. J. Phys. Chem. 1961, 65, 1941–1948. [Google Scholar] [CrossRef]
- Conway, B.E.; Bai, L. Determination of adsorption of OPD H species in the cathodic hydrogen evolution reaction at Pt in relation to electrocatalysis. J. Electroanal. Chem. 1986, 198, 149–175. [Google Scholar] [CrossRef]
- Conway, B.E.; Tilak, B.V. Interfacial processes involving electrocatalytic evolution and oxidation of H2, and the role of chemisorbed H. Electrochim. Acta 2002, 47, 3571–3594. [Google Scholar] [CrossRef]
- Mahmood, N.; Yao, Y.; Zhang, J.W.; Pan, L.; Zhang, X.; Zou, J.J. Electrocatalysts for Hydrogen Evolution in Alkaline Electrolytes: Mechanisms, Challenges, and Prospective Solutions. Adv. Sci. 2018, 5, 1700464. [Google Scholar] [CrossRef] [PubMed]
- Shinagawa, T.; Garcia-Esparza, A.T.; Takanabe, K. Insight on Tafel slopes from a microkinetic analysis of aqueous electrocatalysis for energy conversion. Sci. Rep. 2015, 5, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Jiao, Y.; Vasileff, A.; Qiao, S.-Z. The Hydrogen Evolution Reaction in Alkaline Solution: From Theory, Single Crystal Models, to Practical Electrocatalysts. Angew. Chemie Int. Ed. 2018, 57, 7568–7579. [Google Scholar] [CrossRef]
- Pletcher, D.; Walsh, F.C. Industrial Electrochemistry; Springer: Berlin/Heidelberg, Germany, 1993; ISBN 978-0-7514-0148-6. [Google Scholar]
- Zieliński, P. Review of Surface Relaxation and Reconstruction Phenomena. Acta Phys. Pol. A 1996, 89, 251–263. [Google Scholar] [CrossRef]
- Markovic, N. Surface science studies of model fuel cell electrocatalysts. Surf. Sci. Rep. 2002, 45, 117–229. [Google Scholar] [CrossRef]
- Trasatti, S. Work function, electronegativity, and electrochemical behaviour of metals. J. Electroanal. Chem. Interfacial Electrochem. 1972, 39, 163–184. [Google Scholar] [CrossRef]
- Quaino, P.; Juarez, F.; Santos, E.; Schmickler, W. Volcano plots in hydrogen electrocatalysis–uses and abuses. Beilstein J. Nanotechnol. 2014, 5, 846–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marković, N.M.; Grgur, B.N.; Ross, P.N. Temperature-Dependent Hydrogen Electrochemistry on Platinum Low-Index Single-Crystal Surfaces in Acid Solutions. J. Phys. Chem. B 1997, 101, 5405–5413. [Google Scholar] [CrossRef]
- Eberhardt, D.; Santos, E.; Schmickler, W. Hydrogen evolution on silver single crystal electrodes—first results. J. Electroanal. Chem. 1999, 461, 76–79. [Google Scholar] [CrossRef]
- Perez, J.; Gonzalez, E.R.; Villullas, H.M. Hydrogen Evolution Reaction on Gold Single-Crystal Electrodes in Acid Solutions. J. Phys. Chem. B 1998, 102, 10931–10935. [Google Scholar] [CrossRef]
- Bockris, J.O.M.; Ammar, I.A.; Huq, A.K.M.S. The Mechanism of the Hydrogen Evolution Reaction on Platinum, Silver and Tungsten surfaces in Acid Solutions. J. Phys. Chem. 1957, 61, 879–886. [Google Scholar] [CrossRef]
- Brisard, G.; Bertrand, N.; Ross, P.; Marković, N. Oxygen reduction and hydrogen evolution–oxidation reactions on Cu(hkl) surfaces. J. Electroanal. Chem. 2000, 480, 219–224. [Google Scholar] [CrossRef]
- Marković, N.M.; Sarraf, S.T.; Gasteiger, H.A.; Ross, P.N. Hydrogen electrochemistry on platinum low-index single-crystal surfaces in alkaline solution. J. Chem. Soc. Faraday Trans. 1996, 92, 3719–3725. [Google Scholar] [CrossRef]
- Jerkiewicz, G. Hydrogen sorption ATIN electrodes. Prog. Surf. Sci. 1998, 57, 137–186. [Google Scholar] [CrossRef]
- Morrison, S.R. Electrochemistry at Semiconductor and Oxidized Metal Electrodes; OSTI: Oak Ridge, TN, USA, 1980. [Google Scholar]
- Santos, E.; Schmickler, W. d-Band Catalysis in Electrochemistry. ChemPhysChem 2006, 7, 2282–2285. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Bligaard, T.; Logadottir, A.; Kitchin, J.R.; Chen, J.G.; Pandelov, S.; Stimming, U. Trends in the Exchange Current for Hydrogen Evolution. J. Electrochem. Soc. 2005, 152, J23. [Google Scholar] [CrossRef] [Green Version]
- Greeley, J.; Jaramillo, T.F.; Bonde, J.; Chorkendorff, I.; Nørskov, J.K. Computational high-throughput screening of electrocatalytic materials for hydrogen evolution. Nat. Mater. 2006, 5, 909–913. [Google Scholar] [CrossRef] [PubMed]
- Soldano, G.; Schulz, E.N.; Salinas, D.R.; Santos, E.; Schmickler, W. Hydrogen electrocatalysis on overlayers of rhodium over gold and palladium substrates—more active than platinum? Phys. Chem. Chem. Phys. 2011, 13, 16437. [Google Scholar] [CrossRef] [PubMed]
- Skúlason, E.; Tripkovic, V.; Björketun, M.E.; Gudmundsdóttir, S.; Karlberg, G.; Rossmeisl, J.; Bligaard, T.; Jónsson, H.; Nørskov, J.K. Modeling the Electrochemical Hydrogen Oxidation and Evolution Reactions on the Basis of Density Functional Theory Calculations. J. Phys. Chem. C 2010, 114, 18182–18197. [Google Scholar] [CrossRef]
- Santos, E.; Quaino, P.; Schmickler, W. Theory of electrocatalysis: Hydrogen evolution and more. Phys. Chem. Chem. Phys. 2012, 14, 11224. [Google Scholar] [CrossRef]
- Santos, E.; Schmickler, W. Electrocatalysis of Hydrogen Oxidation—Theoretical Foundations. Angew. Chemie Int. Ed. 2007, 46, 8262–8265. [Google Scholar] [CrossRef]
- Kuhn, A.T.; Mortimer, C.J.; Bond, G.C.; Lindley, J. A critical analysis of correlations between the rate of the electrochemical hydrogen evolution reaction and physical properties of the elements. J. Electroanal. Chem. Interfacial Electrochem. 1972, 34, 1–14. [Google Scholar] [CrossRef]
- Peraldo Bicelli, L.; Romagnani, C.; Rosania, M. Hydrogen evolution reaction on cobalt. J. Electroanal. Chem. Interfacial Electrochem. 1975, 63, 238–244. [Google Scholar] [CrossRef]
- Gonzalez, E.; Avaca, L.; Tremiliosifilho, G.; Machado, S.; Fereirra, M. Hydrogen evolution reaction on Ni-S electrodes in alkaline solutions. Int. J. Hydrogen Energy 1994, 19, 17–21. [Google Scholar] [CrossRef]
- Marković, N.; Lucas, C.; Climent, V.; Stamenković, V.; Ross, P. Surface electrochemistry on an epitaxial palladium film on Pt(111): Surface microstructure and hydrogen electrode kinetics. Surf. Sci. 2000, 465, 103–114. [Google Scholar]
- Zhang, L.; Chang, Q.; Chen, H.; Shao, M. Recent advances in palladium-based electrocatalysts for fuel cell reactions and hydrogen evolution reaction. Nano Energy 2016, 29, 198–219. [Google Scholar] [CrossRef]
- Mavrikakis, M.; Hammer, B.; Nørskov, J.K. Effect of Strain on the Reactivity of Metal Surfaces. Phys. Rev. Lett. 1998, 81, 2819–2822. [Google Scholar] [CrossRef] [Green Version]
- Vasić, D.D.; Pašti, I.A.; Mentus, S.V. DFT study of platinum and palladium overlayers on tungsten carbide: Structure and electrocatalytic activity toward hydrogen oxidation/evolution reaction. Int. J. Hydrogen Energy 2013, 38, 5009–5018. [Google Scholar] [CrossRef]
- Esposito, D.V.; Hunt, S.T.; Kimmel, Y.C.; Chen, J.G. A New Class of Electrocatalysts for Hydrogen Production from Water Electrolysis: Metal Monolayers Supported on Low-Cost Transition Metal Carbides. J. Am. Chem. Soc. 2012, 134, 3025–3033. [Google Scholar] [CrossRef]
- Vasić Anićijević, D.D.; Nikolić, V.M.; Marčeta-Kaninski, M.P.; Pašti, I.A. Is platinum necessary for efficient hydrogen evolution?–DFT study of metal monolayers on tungsten carbide. Int. J. Hydrogen Energy 2013, 38, 16071–16079. [Google Scholar] [CrossRef]
- Vasić Anićijević, D.D.; Nikolić, V.M.; Marčeta Kaninski, M.P.; Pašti, I.A. Structure, chemisorption properties and electrocatalysis by Pd3Au overlayers on tungsten carbide-A DFT study. Int. J. Hydrogen Energy 2015, 40, 6085–6096. [Google Scholar] [CrossRef]
- Esposito, D.V.; Hunt, S.T.; Stottlemyer, A.L.; Dobson, K.D.; McCandless, B.E.; Birkmire, R.W.; Chen, J.G. Low-Cost Hydrogen-Evolution Catalysts Based on Monolayer Platinum on Tungsten Monocarbide Substrates. Angew. Chemie Int. Ed. 2010, 49, 9859–9862. [Google Scholar] [CrossRef]
- Björketun, M.E.; Bondarenko, A.S.; Abrams, B.L.; Chorkendorff, I.; Rossmeisl, J. Screening of electrocatalytic materials for hydrogen evolution. Phys. Chem. Chem. Phys. 2010, 12, 10536. [Google Scholar] [CrossRef]
- Lasia, A. Mechanism and kinetics of the hydrogen evolution reaction. Int. J. Hydrogen Energy 2019, 44, 19484–19518. [Google Scholar] [CrossRef]
- Darband, G.B.; Aliofkhazraei, M.; Shanmugam, S. Recent advances in methods and technologies for enhancing bubble detachment during electrochemical water splitting. Renew. Sustain. Energy Rev. 2019, 114, 109300. [Google Scholar] [CrossRef]
- Shiva Kumar, S.; Himabindu, V. Hydrogen production by PEM water electrolysis–A review. Mater. Sci. Energy Technol. 2019, 2, 442–454. [Google Scholar] [CrossRef]
- Kong, D.; Xiao, Z.; Gao, Y.; Zhang, X.; Guo, R.; Huang, X.; Li, X.; Zhi, L. Sp2-carbon dominant carbonaceous materials for energy conversion and storage. Mater. Sci. Eng. R Rep. 2019, 137, 1–37. [Google Scholar] [CrossRef]
- Murthy, A.P.; Madhavan, J.; Murugan, K. Recent advances in hydrogen evolution reaction catalysts on carbon/carbon-based supports in acid media. J. Power Sources 2018, 398, 9–26. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, X.; Zhou, Z. Carbon-Based Substrates for Highly Dispersed Nanoparticle and Even Single-Atom Electrocatalysts. Small Methods 2019, 3, 1900050. [Google Scholar] [CrossRef]
- Zhao, W.; Li, G.; Tang, Z. Metal-organic frameworks as emerging platform for supporting isolated single-site catalysts. Nano Today 2019, 27, 178–197. [Google Scholar] [CrossRef]
- Wen, X.; Guan, J. Recent progress on MOF-derived electrocatalysts for hydrogen evolution reaction. Appl. Mater. Today 2019, 16, 146–168. [Google Scholar] [CrossRef]
- Li, Y.; Guo, S. Noble metal-based 1D and 2D electrocatalytic nanomaterials: Recent progress, challenges and perspectives. Nano Today 2019, 28, 100774. [Google Scholar] [CrossRef]
- Ronchi, R.M.; Arantes, J.T.; Santos, S.F. Synthesis, structure, properties and applications of MXenes: Current status and perspectives. Ceram. Int. 2019, 45, 18167–18188. [Google Scholar] [CrossRef]
- Agarwal, T.; Kaur-Ghumaan, S. HER catalysed by iron complexes without a Fe2S2 core: A review. Coord. Chem. Rev. 2019, 397, 188–219. [Google Scholar] [CrossRef]
- Zhang, W.; Li, D.; Zhang, L.; She, X.; Yang, D. NiFe-based nanostructures on nickel foam as highly efficiently electrocatalysts for oxygen and hydrogen evolution reactions. J. Energy Chem. 2019, 39, 39–53. [Google Scholar] [CrossRef] [Green Version]
- Wei, J.; Zhou, M.; Long, A.; Xue, Y.; Liao, H.; Wei, C.; Xu, Z.J. Heterostructured Electrocatalysts for Hydrogen Evolution Reaction Under Alkaline Conditions. Nano Micro Lett. 2018, 10, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pašti, I.A.; Skorodumova, N.V.; Mentus, S.V. Theoretical studies in catalysis and electrocatalysis: From fundamental knowledge to catalyst design. React. Kinet. Mech. Catal. 2015, 115, 5–32. [Google Scholar] [CrossRef]
- Bernsmeier, D.; Sachse, R.; Bernicke, M.; Schmack, R.; Kettemann, F.; Polte, J.; Kraehnert, R. Outstanding hydrogen evolution performance of supported Pt nanoparticles: Incorporation of preformed colloids into mesoporous carbon films. J. Catal. 2019, 369, 181–189. [Google Scholar] [CrossRef]
- Creus, J.; Mallón, L.; Romero, N.; Bofill, R.; Moya, A.; Fierro, J.L.G.; Mas-Ballesté, R.; Sala, X.; Philippot, K.; García-Antón, J. Ruthenium Nanoparticles Supported on Carbon Microfibers for Hydrogen Evolution Electrocatalysis. Eur. J. Inorg. Chem. 2019, 2019, 2071–2077. [Google Scholar] [CrossRef]
- Wu, X.; Feng, B.; Li, W.; Niu, Y.; Yu, Y.; Lu, S.; Zhong, C.; Liu, P.; Tian, Z.; Chen, L.; et al. Metal-support interaction boosted electrocatalysis of ultrasmall iridium nanoparticles supported on nitrogen doped graphene for highly efficient water electrolysis in acidic and alkaline media. Nano Energy 2019, 62, 117–126. [Google Scholar] [CrossRef]
- Huang, J.; Du, C.; Nie, J.; Zhou, H.; Zhang, X.; Chen, J. Encapsulated Rh nanoparticles in N-doped porous carbon polyhedrons derived from ZIF-8 for efficient HER and ORR electrocatalysis. Electrochim. Acta 2019, 326, 134982. [Google Scholar] [CrossRef]
- Li, Z.; Qi, Z.; Wang, S.; Ma, T.; Zhou, L.; Wu, Z.; Luan, X.; Lin, F.-Y.; Chen, M.; Miller, J.T.; et al. In Situ Formed Pt3Ti Nanoparticles on a Two-Dimensional Transition Metal Carbide (MXene) Used as Efficient Catalysts for Hydrogen Evolution Reactions. Nano Lett. 2019, 19, 5102–5108. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Huang, H.; Diao, J.; Gong, S.; Chen, S.; Lu, J.; Wang, C.; Sun, Z.; Xia, G.; Yang, K.; et al. Improving electrocatalytic activity of iridium for hydrogen evolution at high current densities above 1000 mA cm−2. Appl. Catal. B Environ. 2019, 258, 117965. [Google Scholar] [CrossRef]
- Meng, H.B.; Zhang, X.F.; Pu, Y.L.; Chen, X.L.; Feng, J.J.; Han, D.M.; Wang, A.J. One-pot solvothermal synthesis of reduced graphene oxide-supported uniform PtCo nanocrystals for efficient and robust electrocatalysis. J. Colloid Interface Sci. 2019, 543, 17–24. [Google Scholar] [CrossRef]
- Subbaraman, R.; Tripkovic, D.; Strmcnik, D.; Chang, K.-C.; Uchimura, M.; Paulikas, A.P.; Stamenkovic, V.; Markovic, N.M. Enhancing Hydrogen Evolution Activity in Water Splitting by Tailoring Li+-Ni(OH)2-Pt Interfaces. Science (80-) 2011, 334, 1256–1260. [Google Scholar] [CrossRef]
- Subbaraman, R.; Tripkovic, D.; Chang, K.C.; Strmcnik, D.; Paulikas, A.P.; Hirunsit, P.; Chan, M.; Greeley, J.; Stamenkovic, V.; Markovic, N.M. Trends in activity for the water electrolyser reactions on 3d M(Ni,Co,Fe,Mn) hydr(oxy)oxide catalysts. Nat. Mater. 2012, 11, 550–557. [Google Scholar] [CrossRef] [PubMed]
- Danilovic, N.; Subbaraman, R.; Strmcnik, D.; Chang, K.C.; Paulikas, A.P.; Stamenkovic, V.R.; Markovic, N.M. Enhancing the alkaline hydrogen evolution reaction activity through the bifunctionality of Ni(OH)2/metal catalysts. Angew. Chemie Int. Ed. 2012, 51, 12495–12498. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Wang, Y.; Li, J.; Liu, J. Facile fabrication of Pt–Ni alloy nanoparticles supported on reduced graphene oxide as excellent electrocatalysts for hydrogen evolution reaction in alkaline environment. J. Nanoparticle Res. 2019, 21, 13. [Google Scholar] [CrossRef]
- Herranz, J.; Durst, J.; Fabbri, E.; Patru, A.; Cheng, X.; Permyakova, A.A.; Schmidt, T.J. Interfacial effects on the catalysis of the hydrogen evolution, oxygen evolution and CO2-reduction reactions for (co-)electrolyzer development. Nano Energy 2016, 29, 4–28. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Romeiro, F.C.; Guo, S.; Zhang, Y.; Nossol, E.; Lima, R.C.; Bond, A.M.; Zhang, J. Size Controllable Metal Nanoparticles Anchored on Nitrogen Doped Carbon for Electrocatalytic Energy Conversion. ChemElectroChem 2019, 6, 1508–1513. [Google Scholar] [CrossRef]
- Alinezhad, A.; Gloag, L.; Benedetti, T.M.; Cheong, S.; Webster, R.F.; Roelsgaard, M.; Iversen, B.B.; Schuhmann, W.; Gooding, J.J.; Tilley, R.D. Direct Growth of Highly Strained Pt Islands on Branched Ni Nanoparticles for Improved Hydrogen Evolution Reaction Activity. J. Am. Chem. Soc. 2019, 141, 16202–16207. [Google Scholar] [CrossRef]
- Cardoso, J.A.S.B.; Šljukić, B.; Kayhan, E.; Sequeira, C.A.C.; Santos, D.M.F. Palladium-nickel on tin oxide-carbon composite supports for electrocatalytic hydrogen evolution. Catal. Today 2019, 1–9. [Google Scholar] [CrossRef]
- Gutić, S.J.; Šabanović, M.; Metarapi, D.; Pašti, I.A.; Korać, F.; Mentus, S.V. Electrochemically Synthesized Ni@reduced Graphene Oxide Composite Catalysts for Hydrogen Evolution in Alkaline Media–the Effects of Graphene Oxide Support. Int. J. Electrochem. Sci. 2019, 14, 8532–8543. [Google Scholar] [CrossRef]
- Begum, H.; Ahmed, M.S.; Lee, D.-W.; Kim, Y.-B. Carbon nanotubes-based PdM bimetallic catalysts through N4-system for efficient ethanol oxidation and hydrogen evolution reaction. Sci. Rep. 2019, 9, 11051. [Google Scholar] [CrossRef]
- Liu, D.-Q.; Tao, B.; Ruan, H.-C.; Bentley, C.L.; Unwin, P.R. Metal support effects in electrocatalysis at hexagonal boron nitride. Chem. Commun. 2019, 55, 628–631. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Zhu, Y.; Zeng, Z.; Lin, C.; Giroux, M.; Jiang, L.; Han, Y.; Greeley, J.; Wang, C.; Jin, J. Platinum-nickel hydroxide nanocomposites for electrocatalytic reduction of water. Nano Energy 2017, 31, 456–461. [Google Scholar] [CrossRef] [Green Version]
- Jung, E.; Park, H.Y.; Cho, A.; Jang, J.H.; Park, H.S.; Yu, T. Aqueous-phase synthesis of metal hydroxide nanoplates and platinum/nickel hydroxide hybrid nanostructures and their enhanced electrocatalytic properties. Appl. Catal. B Environ. 2018, 225, 238–242. [Google Scholar] [CrossRef]
- Wang, Y.; Zhuo, H.; Zhang, X.; Dai, X.; Yu, K.; Luan, C.; Yu, L.; Xiao, Y.; Li, J.; Wang, M.; et al. Synergistic effect between undercoordinated platinum atoms and defective nickel hydroxide on enhanced hydrogen evolution reaction in alkaline solution. Nano Energy 2018, 48, 590–599. [Google Scholar] [CrossRef]
- Wang, C.; Sun, Y.; Tian, E.; Fu, D.; Zhang, M.; Zhao, X.; Ye, W. Easy access to trace-loading of Pt on inert Ni3N nanoparticles with significantly improved hydrogen evolution activity at entire pH values. Electrochim. Acta 2019, 320, 134597. [Google Scholar] [CrossRef]
- Li, J.; Liu, H.X.; Gou, W.; Zhang, M.; Xia, Z.; Zhang, S.; Chang, C.R.; Ma, Y.; Qu, Y. Ethylene-glycol ligand environment facilitates highly efficient hydrogen evolution of Pt/CoP through proton concentration and hydrogen spillover. Energy Environ. Sci. 2019, 12, 2298–2304. [Google Scholar] [CrossRef]
- Li, M.; Zhu, Y.; Song, N.; Wang, C.; Lu, X. Fabrication of Pt nanoparticles on nitrogen-doped carbon/Ni nanofibers for improved hydrogen evolution activity. J. Colloid Interface Sci. 2018, 514, 199–207. [Google Scholar] [CrossRef]
- Yin, Y.; Liu, T.; Liu, D.; Wang, Z.; Deng, Q.; Qu, D.; Xie, Z.; Tang, H.; Li, J. Confining nano-sized platinum in nitrogen doped ordered mesoporous carbon: An effective approach toward efficient and robust hydrogen evolution electrocatalyst. J. Colloid Interface Sci. 2018, 530, 595–602. [Google Scholar] [CrossRef]
- Zhou, J.; Wang, Z.; Liu, J.; Chen, Y. Enhanced hydrogen evolution performance by covalent-linked ultrafine, uniform Pt nanoparticles with doped sulfur atoms in three-dimensional graphene. Int. J. Hydrogen Energy 2018, 43, 23231–23238. [Google Scholar] [CrossRef]
- Yang, M.; Zhang, J.; Zhang, W.; Wu, Z.; Gao, F. Pt nanoparticles/Fe-doped α-Ni(OH)2 nanosheets array with low Pt loading as a high-performance electrocatalyst for alkaline hydrogen evolution reaction. J. Alloys Compd. 2020, 153790. [Google Scholar] [CrossRef]
- Monama, G.R.; Mdluli, S.B.; Mashao, G.; Makhafola, M.D.; Ramohlola, K.E.; Molapo, K.M.; Hato, M.J.; Makgopa, K.; Iwuoha, E.I.; Modibane, K.D. Palladium deposition on copper(II) phthalocyanine/metal organic framework composite and electrocatalytic activity of the modified electrode towards the hydrogen evolution reaction. Renew. Energy 2018, 119, 62–72. [Google Scholar] [CrossRef]
- Li, J.; Zhou, Q.; Shen, Z.; Li, S.; Pu, J.; Zhong, C.; Cao, M.; Jin, X.; Zhang, H.; Wang, Y.; et al. Synergistic effect of ultrafine nano-Ru decorated cobalt carbonate hydroxides nanowires for accelerated alkaline hydrogen evolution reaction. Electrochim. Acta 2020, 331, 135367. [Google Scholar] [CrossRef]
- Zhang, J.; Jia, W.; Dang, S.; Cao, Y. Sub-5 nm octahedral platinum-copper nanostructures anchored on nitrogen-doped porous carbon nanofibers for remarkable electrocatalytic hydrogen evolution. J. Colloid Interface Sci. 2020, 560, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Feng, T.; Chen, Y.; Liu, J.; Zhao, X.; Yang, B. Synchronously integration of Co, Fe dual-metal doping in Ru@C and CDs for boosted water splitting performances in alkaline media. Applied Catal. B Environ. 2020, 118657. [Google Scholar] [CrossRef]
- Bai, J.; Xing, S.H.; Zhu, Y.Y.; Jiang, J.X.; Zeng, J.H.; Chen, Y. Polyallylamine-Rh nanosheet nanoassemblies−carbon nanotubes organic-inorganic nanohybrids: A electrocatalyst superior to Pt for the hydrogen evolution reaction. J. Power Sources 2018, 385, 32–38. [Google Scholar] [CrossRef]
- Jin, Q.; Ren, B.; Li, D.; Cui, H.; Wang, C. In situ promoting water dissociation kinetic of Co based electrocatalyst for unprecedentedly enhanced hydrogen evolution reaction in alkaline media. Nano Energy 2018, 49, 14–22. [Google Scholar] [CrossRef]
- Yang, J.; Cai, C.; Li, Y.; Gao, L.; Guo, H.; Wang, B.; Pu, B.; Niu, X. In-situ cobalt and nitrogen doped mesoporous graphitic carbon electrocatalyst via directly pyrolyzing hyperbranched cobalt phthalocyanine for hydrogen evolution reaction. Electrochim. Acta 2018, 262, 48–56. [Google Scholar] [CrossRef]
- Lv, Y.; Duan, S.; Zhu, Y.; Guo, H.; Wang, R. Interface control and catalytic performances of Au-NiSx heterostructures. Chem. Eng. J. 2020, 382, 122794. [Google Scholar] [CrossRef]
- Feng, J.; Zhou, H.; Chen, D.; Bian, T.; Yuan, A. Core-shell structured ZnCo/NC@MoS2 electrocatalysts for tunable hydrogen evolution reaction. Electrochim. Acta 2020, 331, 135445. [Google Scholar] [CrossRef]
- Li, Y.; Li, H.; Cao, K.; Jin, T.; Wang, X.; Sun, H.; Ning, J.; Wang, Y.; Jiao, L. Electrospun three dimensional Co/CoP@nitrogen-doped carbon nanofibers network for efficient hydrogen evolution. Energy Storage Mater. 2018, 12, 44–53. [Google Scholar] [CrossRef]
- Zhang, X.; Yang, Z.; Yang, X.; Wang, Y.; Lu, Z. Engineering the activity of CoNx-graphene for hydrogen evolution. Int. J. Hydrogen Energy 2018, 43, 20573–20579. [Google Scholar] [CrossRef]
- Chen, Z.; Duan, X.; Wei, W.; Wang, S.; Ni, B.-J. Recent advances in transition metal-based electrocatalysts for alkaline hydrogen evolution. J. Mater. Chem. A 2019, 7, 14971–15005. [Google Scholar] [CrossRef]
- Hu, C.; Zhang, L.; Gong, J. Recent progress made in the mechanism comprehension and design of electrocatalysts for alkaline water splitting. Energy Environ. Sci. 2019, 12, 2620–2645. [Google Scholar] [CrossRef]
- Xie, C.; Yan, D.; Chen, W.; Zou, Y.; Chen, R.; Zang, S.; Wang, Y.; Yao, X.; Wang, S. Insight into the design of defect electrocatalysts: From electronic structure to adsorption energy. Mater. Today 2019, 31, 47–68. [Google Scholar] [CrossRef]
- Mohammed-Ibrahim, J.; Sun, X. Recent progress on earth abundant electrocatalysts for hydrogen evolution reaction (HER) in alkaline medium to achieve efficient water splitting–A review. J. Energy Chem. 2019, 34, 111–160. [Google Scholar] [CrossRef]
- Jin, W.; Maduraiveeran, G. Recent advances of porous transition metal-based nanomaterials for electrochemical energy conversion and storage applications. Mater. Today Energy 2019, 13, 64–84. [Google Scholar] [CrossRef]
- Xu, P.; Zhang, J.; Ye, Z.; Liu, Y.; Cen, T.; Yuan, D. Co doped Ni0.85Se nanoparticles on RGO as efficient electrocatalysts for hydrogen evolution reaction. Appl. Surf. Sci. 2019, 494, 749–755. [Google Scholar] [CrossRef]
- Ma, T.; Zhang, M.; Liu, H.; Wang, Y. Synthesis of CoMoO4-MoO2 nanohybrids supported on graphene as high-efficiency catalyst for hydrogen evolution. J. Electroanal. Chem. 2019, 844, 78–85. [Google Scholar] [CrossRef]
- Hou, X.; Li, Y.; Cheng, L.; Feng, X.; Zhang, H.; Han, S. Cobalt-molybdenum disulfide supported on nitrogen-doped graphene towards an efficient hydrogen evolution reaction. Int. J. Hydrogen Energy 2019, 44, 11664–11674. [Google Scholar] [CrossRef]
- Sheng, W.; Tian, Y.; Song, Y.; Ji, J.; Wang, F. Phase controlled synthesis and the phase dependent photo-and electrocatalysis of CdS@CoMo2S4/MoS2 catalyst for HER. Int. J. Hydrogen Energy 2019, 44, 19890–19899. [Google Scholar] [CrossRef]
- Han, Z.; Liang, X.; Wang, S.; Zhou, L.; Zhao, Z. One-step synthesis of Ni-Mo-S/MoOx composite self-supported on Ni foam for efficient electrocatalytic hydrogen evolution. Mater. Lett. 2019, 246, 63–66. [Google Scholar] [CrossRef]
- Yan, Q.; Yang, X.; Wei, T.; Wu, W.; Yan, P.; Zeng, L.; Zhu, R.; Cheng, K.; Ye, K.; Zhu, K.; et al. Self-supported cobalt–molybdenum oxide nanosheet clusters as efficient electrocatalysts for hydrogen evolution reaction. Int. J. Hydrogen Energy 2019, 44, 21220–21228. [Google Scholar] [CrossRef]
- Cui, B.; Zhang, M.; Zhao, Y.; Hu, S. Heterogenization of few-layer MoS2 with highly crystalline 3D Ni3S2 nanoframes effectively synergizes the electrocatalytic hydrogen generation in alkaline medium. Mater. Today Energy 2019, 13, 85–92. [Google Scholar] [CrossRef]
- Wang, P.; Wan, L.; Lin, Y.; Wang, B. NiFe Hydroxide Supported on Hierarchically Porous Nickel Mesh as a High-Performance Bifunctional Electrocatalyst for Water Splitting at Large Current Density. ChemSusChem 2019, 12, 4038–4045. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Quan, T.; Mei, S.; Kochovski, Z.; Huang, W.; Meng, H.; Lu, Y. Prompt Electrodeposition of Ni Nanodots on Ni Foam to Construct a High-Performance Water-Splitting Electrode: Efficient, Scalable, and Recyclable. Nano-Micro Lett. 2019, 11, 41. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, M.M.; Mezni, A.; Alsawat, M.; Kumeria, T.; Das, M.R.; Alzahly, S.; Aldalbahi, A.; Gornicka, K.; Ryl, J.; Amin, M.A.; et al. Enhanced hydrogen evolution reaction on highly stable titania-supported PdO and Eu 2 O 3 nanocomposites in a strong alkaline solution. Int. J. Energy Res. 2019, 43, 5367–5383. [Google Scholar] [CrossRef]
- Lu, W.; Li, X.; Wei, F.; Cheng, K.; Li, W.; Zhou, Y.; Zheng, W.; Pan, L.; Zhang, G. Fast sulfurization of nickel foam-supported nickel-cobalt carbonate hydroxide nanowire array at room temperature for hydrogen evolution electrocatalysis. Electrochim. Acta 2019, 318, 252–261. [Google Scholar] [CrossRef]
- Wei, C.; Liu, C.; Gao, L.; Sun, Y.; Liu, Q.; Zhang, X.; Guo, J. MoS2 nanosheets decorated Ni(OH)2 nanorod array for active overall water splitting. J. Alloys Compd. 2019, 796, 86–92. [Google Scholar] [CrossRef]
- Diao, L.; Zhang, B.; Sun, Q.; Wang, N.; Zhao, N.; Shi, C.; Liu, E.; He, C. An in-plane Co9S8@MoS2 heterostructure for the hydrogen evolution reaction in alkaline media. Nanoscale 2019, 11, 21479–21486. [Google Scholar] [CrossRef]
- Mingli, F.; Xue, L.; Dandan, W.; Yinling, W.; Maoguo, L. Fabrication of Te@NiTe2/NiS heterostructures for electrocatalytic hydrogen evolution reaction. Electrochim. Acta 2019, 328, 135075. [Google Scholar] [CrossRef]
- Song, S.; Wang, Y.; Li, W.; Tian, P.; Zhou, S.; Gao, H.; Tian, X.; Zang, J. Amorphous MoS2 coated Ni3S2 nanosheets as bifunctional electrocatalysts for high-efficiency overall water splitting. Electrochim. Acta 2020, 332, 135454. [Google Scholar] [CrossRef]
- Liu, S.; Xu, Y.; Chanda, D.; Tan, L.; Xing, R.; Li, X.; Mao, L.; Kazuya, N.; Fujishima, A. Ultrathin WS2 nanosheets vertically aligned on TiO2 nanobelts as efficient alkaline hydrogen evolution electrocatalyst. Int. J. Hydrogen Energy 2019, 45, 1697–1705. [Google Scholar] [CrossRef]
- Fang, X.; Wang, Z.; Kang, S.; Zhao, L.; Jiang, Z.; Dong, M. Hexagonal CoSe2 nanosheets stabilized by nitrogen-doped reduced graphene oxide for efficient hydrogen evolution reaction. Int. J. Hydrogen Energy 2019, 45, 1738–1747. [Google Scholar] [CrossRef]
- Yu, H.; Sun, X.; Tang, D.; Huang, Y.; Zhang, W.; Miao, S.; Qiao, Z.A.; Wang, J.; Zhao, Z. Molten salt strategy to synthesize alkali metal-doped Co9S8 nanoparticles embedded, N, S co-doped mesoporous carbon as hydrogen evolution electrocatalyst. Int. J. Hydrogen Energy 2020, 45, 6006–6014. [Google Scholar] [CrossRef]
- Xie, Y.; Liu, Y.; Yang, Z. Interfaces engineering of MoNi-based sulfides electrocatalysts for hydrogen evolution reaction in both acid and alkaline media. Int. J. Hydrogen Energy 2020, 45, 6500–6507. [Google Scholar] [CrossRef]
- Zhang, W.; Tang, Y.; Yu, L.; Yu, X.Y. Activating the alkaline hydrogen evolution performance of Mo-incorporated Ni(OH)2 by plasma-induced heterostructure. Appl. Catal. B Environ. 2020, 260, 118154. [Google Scholar] [CrossRef]
- Liu, C.; Gong, T.; Zhang, J.; Zheng, X.; Mao, J.; Liu, H.; Li, Y.; Hao, Q. Engineering Ni2P-NiSe2 heterostructure interface for highly efficient alkaline hydrogen evolution. Appl. Catal. B Environ. 2020, 262, 1–8. [Google Scholar] [CrossRef]
- Anwer, H.; Lee, H.; Kim, H.R.; Kim, H.K.; Park, J.W. Selective transport and separation of charge–carriers by an electron transport layer in NiCo2S4/CdO@CC for excellent water splitting. Appl. Catal. B Environ. 2020, 265, 118564. [Google Scholar] [CrossRef]
- Lu, S.Y.; Li, S.; Jin, M.; Gao, J.; Zhang, Y. Greatly boosting electrochemical hydrogen evolution reaction over Ni3S2 nanosheets rationally decorated by Ni3Sn2S2 quantum dots. Appl. Catal. B Environ. 2020, 267, 118675. [Google Scholar] [CrossRef]
- He, H.Y. Metallic WSe2: Sn nanosheets assembled on graphene by a modified hydrothermal process for hydrogen evolution reaction. Colloids Surfaces A Physicochem. Eng. Asp. 2020, 589, 1–7. [Google Scholar] [CrossRef]
- Fan, Z.; Jiang, J.; Ai, L.; Shao, Z.; Liu, S. Rational Design of Ruthenium and Cobalt-Based Composites with Rich Metal-Insulator Interfaces for Efficient and Stable Overall Water Splitting in Acidic Electrolyte. ACS Appl. Mater. Interfaces 2019, 11, 47894–47903. [Google Scholar] [CrossRef]
- Ni, W.; Krammer, A.; Hsu, C.-S.; Chen, H.M.; Schüler, A.; Hu, X. Ni3N as an Active Hydrogen Oxidation Reaction Catalyst in Alkaline Medium. Angew. Chemie Int. Ed. 2019, 58, 7445–7449. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Song, M.; Zhang, Z.; Wang, J.; Liu, X. Various strategies to tune the electrocatalytic performance of molybdenum phosphide supported on reduced graphene oxide for hydrogen evolution reaction. J. Colloid Interface Sci. 2019, 536, 638–645. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Chen, Z.; Wu, H.; Xiao, F.; Du, S.; He, C.; Wu, Y.; Ren, Z. In Situ Catalytic Etching Strategy Promoted Synthesis of Carbon Nanotube Inlaid with Ultrasmall FeP Nanoparticles as Efficient Electrocatalyst for Hydrogen Evolution. ACS Sustain. Chem. Eng. 2019, 7, 12741–12749. [Google Scholar] [CrossRef]
- Lei, C.; Zhou, W.; Feng, Q.; Lei, Y.; Zhang, Y.; Chen, Y.; Qin, J. Charge Engineering of Mo2C@Defect-Rich N-Doped Carbon Nanosheets for Efficient Electrocatalytic H2 Evolution. Nano-Micro Lett. 2019, 11, 45. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.-J.; Ren, J.-T.; Yuan, G.; Yao, Y.; Liu, X.; Yuan, Z.-Y. Facile synthesis of Mo2C nanoparticles on N-doped carbon nanotubes with enhanced electrocatalytic activity for hydrogen evolution and oxygen reduction reactions. J. Energy Chem. 2019, 38, 68–77. [Google Scholar] [CrossRef] [Green Version]
- Mo, Q.; Zhang, W.; He, L.; Yu, X.; Gao, Q. Bimetallic Ni2-xCoxP/N-doped carbon nanofibers: Solid-solution-alloy engineering toward efficient hydrogen evolution. Appl. Catal. B Environ. 2019, 244, 620–627. [Google Scholar] [CrossRef]
- Boppella, R.; Tan, J.; Yang, W.; Moon, J. Homologous CoP/NiCoP Heterostructure on N-Doped Carbon for Highly Efficient and pH-Universal Hydrogen Evolution Electrocatalysis. Adv. Funct. Mater. 2019, 29, 1–9. [Google Scholar] [CrossRef]
- Miao, J.; Lang, Z.; Zhang, X.; Kong, W.; Peng, O.; Yang, Y.; Wang, S.; Cheng, J.; He, T.; Amini, A.; et al. Polyoxometalate-Derived Hexagonal Molybdenum Nitrides (MXenes) Supported by Boron, Nitrogen Codoped Carbon Nanotubes for Efficient Electrochemical Hydrogen Evolution from Seawater. Adv. Funct. Mater. 2019, 29, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Chen, N.N.; Mo, Q.; He, L.; Huang, X.; Yang, L.; Zeng, J.; Gao, Q. Heterostructured MoC-MoP/N-doped carbon nanofibers as efficient electrocatalysts for hydrogen evolution reaction. Electrochim. Acta 2019, 299, 708–716. [Google Scholar] [CrossRef]
- Rong, J.; Qiu, F.; Zhang, T.; Fang, Y.; Xu, J.; Zhu, Y. Self-directed hierarchical Cu3(PO4)2/Cu-BDC nanosheets array based on copper foam as an efficient and durable electrocatalyst for overall water splitting. Electrochim. Acta 2019, 313, 179–188. [Google Scholar] [CrossRef]
- Liu, S.; Hu, C.; Lv, C.; Cai, J.; Duan, M.; Luo, J.; Song, J.; Shi, Y.; Chen, C.; Luo, D.; et al. Facile preparation of large-area self-supported porous nickel phosphide nanosheets for efficient electrocatalytic hydrogen evolution. Int. J. Hydrogen Energy 2019, 44, 17974–17984. [Google Scholar] [CrossRef]
- Chen, X.; Li, Q.; Che, Q.; Chen, Y.; Xu, X. Interface Engineering of Crystalline/Amorphous Co2P/CoMoPx Nanostructure as Efficient Electrocatalysts for Hydrogen Evolution Reaction. ACS Sustain. Chem. Eng. 2019, 7, 2437–2445. [Google Scholar] [CrossRef]
- Sun, J.; Liu, J.; Chen, H.; Han, X.; Wu, Y.; He, J.; Han, C.; Yang, G.; Shan, Y. Strongly coupled Mo2C and Ni nanoparticles with in-situ formed interfaces encapsulated by porous carbon nanofibers for efficient hydrogen evolution reaction under alkaline conditions. J. Colloid Interface Sci. 2019, 558, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Yan, Y.; Xu, T.; Cao, J.; Zheng, X.; Feng, J.; Qi, J. Rich P vacancies modulate Ni2P/Cu3P interfaced nanosheets for electrocatalytic alkaline water splitting. J. Colloid Interface Sci. 2020, 564, 37–42. [Google Scholar] [CrossRef]
- Pi, C.; Huang, C.; Yang, Y.; Song, H.; Zhang, X.; Zheng, Y.; Gao, B.; Fu, J.; Chu, P.K.; Huo, K. In situ formation of N-doped carbon-coated porous MoP nanowires: A highly efficient electrocatalyst for hydrogen evolution reaction in a wide pH range. Appl. Catal. B Environ. 2020, 263, 118358. [Google Scholar] [CrossRef]
- Xiong, L.; Bi, J.; Wang, L.; Yang, S. Improving the electrocatalytic property of CoP for hydrogen evolution by constructing porous ternary CeO2-CoP-C hybrid nanostructure via ionic exchange of MOF. Int. J. Hydrogen Energy 2018, 43, 20372–20381. [Google Scholar] [CrossRef]
- Fang, Y.; Zhang, T.; Wu, Y.; Liu, Y.; Guan, J.; Du, X.; Wang, L.; Zhang, M. Stacked Co6W6C nanocrystals anchored on N-doping carbon nanofibers with excellent electrocatalytic performance for HER in wide-range pH. Int. J. Hydrogen Energy 2019, 45, 1901–1910. [Google Scholar] [CrossRef]
- Guan, J.; Li, C.; Zhao, J.; Yang, Y.; Zhou, W. The schematic illustration of FeOOH as the promoter for promoting water dissociation ur na l P of. Applied Catal. B Environ. 2020, 118600. [Google Scholar] [CrossRef]
- Li, X.; Jiang, L.; Liu, J.; Hua, Q.; Wang, E.; Xie, G. Size-dependent catalytic activity of cobalt phosphides for hydrogen evolution reaction. J. Energy Chem. 2020, 43, 121–128. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Ma, W.; Xu, Z.; Wang, H.; Fan, W.; Zong, X.; Li, C. Metal phosphide catalysts anchored on metal-caged graphitic carbon towards efficient and durable hydrogen evolution electrocatalysis. Nano Energy 2018, 48, 500–509. [Google Scholar] [CrossRef]
- Hu, Q.; Liu, X.; Zhu, B.; Fan, L.; Chai, X.; Zhang, Q.; Liu, J.; He, C.; Lin, Z. Crafting MoC2-doped bimetallic alloy nanoparticles encapsulated within N-doped graphene as roust bifunctional electrocatalysts for overall water splitting. Nano Energy 2018, 50, 212–219. [Google Scholar] [CrossRef]
- Jin, H.; Liu, X.; Jiao, Y.; Vasileff, A.; Zheng, Y.; Qiao, S.Z. Constructing tunable dual active sites on two-dimensional C 3 N 4 @MoN hybrid for electrocatalytic hydrogen evolution. Nano Energy 2018, 53, 690–697. [Google Scholar] [CrossRef]
- Huang, X.; Xu, X.; Luan, X.; Cheng, D. CoP nanowires coupled with CoMoP nanosheets as a highly efficient cooperative catalyst for hydrogen evolution reaction. Nano Energy 2020, 68, 104332. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, S.; Zhu, Y.; Qiu, R.; Gengenbach, T.; Liu, Y.; Zu, L.; Mao, H.; Wang, H.; Tang, J.; et al. Three-Dimensional Hierarchical Porous Nanotubes Derived from Metal-Organic Frameworks for Highly Efficient Overall Water Splitting. iScience 2020, 23, 100761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.; Huang, D.; Zhang, J.; Huang, Y.; Balogun, M.S.J.T.; Tong, Y. Dual Doping Induced Interfacial Engineering of Fe2N/Fe3N Hybrids with Favorable d-Band towards Efficient Overall Water Splitting. ChemCatChem 2019, 11, 6051–6060. [Google Scholar] [CrossRef]
- Song, M.; Zhang, Z.; Li, Q.; Jin, W.; Wu, Z.; Fu, G.; Liu, X. Ni-foam supported Co(OH)F and Co-P nanoarrays for energy-efficient hydrogen production: Via urea electrolysis. J. Mater. Chem. A 2019, 7, 3697–3703. [Google Scholar] [CrossRef]
- Que, R.; Ji, G.; Liu, D.; Li, M.; Wang, X.; Jiang, S.P. Nickel Foam-Supported CoCO3 @CoSe Nanowires with a Heterostructure Interface for Overall Water Splitting with Low Overpotential and High Efficiency. Energy Technol. 2019, 7, 1–7. [Google Scholar] [CrossRef]
- Tan, Y.; Che, Q.; Li, Q. Generating highly active Ni11(HPO3)8(OH)6/Mn3O4 catalyst for electrocatalytic hydrogen evolution reaction by electrochemical activation. J. Colloid Interface Sci. 2020, 560, 714–721. [Google Scholar] [CrossRef]
- Li, Y.S.; Yi, J.W.; Wei, J.H.; Wu, Y.P.; Li, B.; Liu, S.; Jiang, C.; Yu, H.G.; Li, D.S. Three 2D polyhalogenated Co(II)-based MOFs: Syntheses, crystal structure and electrocatalytic hydrogen evolution reaction. J. Solid State Chem. 2020, 281, 1–7. [Google Scholar] [CrossRef]
- Wang, A.; Li, C.; Zhang, J.; Chen, X.; Cheng, L.; Zhu, W. Graphene-oxide-supported covalent organic polymers based on zinc phthalocyanine for efficient optical limiting and hydrogen evolution. J. Colloid Interface Sci. 2019, 556, 159–171. [Google Scholar] [CrossRef]
- Xia, X.; Liu, L.; Li, X.; Gao, S.; Yang, T. Highly efficient electrocatalytic hydrogen evolution over edge-modified phosphorene quantum dot/prussian blue skeleton structure. J. Catal. 2019, 374, 401–408. [Google Scholar] [CrossRef]
- Aslam, S.; Sagar, R.U.R.; Kumar, H.; Zhang, G.; Nosheen, F.; Namvari, M.; Mahmood, N.; Zhang, M.; Qiu, Y. Mixed-dimensional heterostructures of hydrophobic/hydrophilic graphene foam for tunable hydrogen evolution reaction. Chemosphere 2020, 245, 125607. [Google Scholar] [CrossRef] [PubMed]
- Zhdanov, V.P.; Kasemo, B. Simulations of the reaction kinetics on nanometer supported catalyst particles. Surf. Sci. Rep. 2000, 39, 25–104. [Google Scholar] [CrossRef]
- Conner, W.C.; Falconer, J.L. Spillover in Heterogeneous Catalysis. Chem. Rev. 1995, 95, 759–788. [Google Scholar] [CrossRef]
- Burwell, R.L.; Read, J.F.; Taylor, K.C.; Haller, G.L. Adsorptive and Catalytic Properties of Chromia. Zeitschrift für Phys. Chemie 1969, 64, 18–25. [Google Scholar] [CrossRef]
- Liu, W.J.; Wu, B.L.; Cha, C.S. Surface diffusion and the spillover of H-adatoms and oxygen-containing surface species on the surface of carbon black and Pt/C porous electrodes. J. Electroanal. Chem. 1999, 476, 101–108. [Google Scholar] [CrossRef]
- Sata, S.; Awad, M.I.; El-Deab, M.S.; Okajima, T.; Ohsaka, T. Hydrogen spillover phenomenon: Enhanced reversible hydrogen adsorption/desorption at Ta2O5-coated Pt electrode in acidic media. Electrochim. Acta 2010, 55, 3528–3536. [Google Scholar] [CrossRef]
- Guo, R.; Xu, X.; Xia, Y.; Huang, W.; Li, Z.; Teng, B. Insights into electrocatalytic hydrogen evolution reaction in acidic medium at in-situ dispersed Pt atoms on nanoporous gold films. J. Catal. 2018, 368, 379–388. [Google Scholar] [CrossRef]
- Abbas, S.A.; Kim, S.H.; Iqbal, M.I.; Muhammad, S.; Yoon, W.S.; Jung, K.D. Synergistic effect of nano-Pt and Ni spine for HER in alkaline solution: Hydrogen spillover from nano-Pt to Ni spine. Sci. Rep. 2018, 8, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Lee, S.; Kim, H.E.; Cho, A.; Kim, S.; Ye, Y.; Han, J.W.; Lee, H.; Jang, J.H.; Lee, J. Investigation of the Support Effect in Atomically Dispersed Pt on WO3−x for Utilization of Pt in the Hydrogen Evolution Reaction. Angew. Chemie Int. Ed. 2019, 58, 16038–16042. [Google Scholar] [CrossRef]
- Pandelov, S.; Stimming, U. Reactivity of monolayers and nano-islands of palladium on Au(111) with respect to proton reduction. Electrochim. Acta 2007, 52, 5548–5555. [Google Scholar] [CrossRef]
- Yang, L.; Zhu, X.; Xiong, S.; Wu, X.; Shan, Y.; Chu, P.K. Synergistic WO3·2H2O Nanoplates/WS2 Hybrid Catalysts for High-Efficiency Hydrogen Evolution. ACS Appl. Mater. Interfaces 2016, 8, 13966–13972. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Lu, S.; Liao, F.; Liu, L.; Li, Y.; Shao, M. Rh—MoS2 Nanocomposite Catalysts with Pt-Like Activity for Hydrogen Evolution Reaction. Adv. Funct. Mater. 2017, 27, 2–7. [Google Scholar] [CrossRef]
- Zhu, L.; Lin, H.; Li, Y.; Liao, F.; Lifshitz, Y.; Sheng, M.; Lee, S.T.; Shao, M. A rhodium/silicon co-electrocatalyst design concept to surpass platinum hydrogen evolution activity at high overpotentials. Nat. Commun. 2016, 7, 12272. [Google Scholar] [CrossRef] [Green Version]
- Chanda, D.; Hnát, J.; Dobrota, A.S.; Pašti, I.A.; Paidar, M.; Bouzek, K. The effect of surface modification by reduced graphene oxide on the electrocatalytic activity of nickel towards the hydrogen evolution reaction. Phys. Chem. Chem. Phys. 2015, 17, 26864–26874. [Google Scholar] [CrossRef] [Green Version]
- Gutić, S.J.; Dobrota, A.S.; Leetmaa, M.; Skorodumova, N.V.; Mentus, S.V.; Pašti, I.A. Improved catalysts for hydrogen evolution reaction in alkaline solutions through the electrochemical formation of nickel-reduced graphene oxide interface. Phys. Chem. Chem. Phys. 2017, 19, 13281–13293. [Google Scholar] [CrossRef]
- Grubač, Z.; Metikoš-Huković, M.; Babić, R. Nanocrystalline and coarse grained polycrystalline nickel catalysts for the hydrogen evolution reaction. Int. J. Hydrogen Energy 2013, 38, 4437–4444. [Google Scholar] [CrossRef]
- Eikerling, M.; Meier, J.; Stimming, U. Hydrogen evolution at a single supported nanoparticle: A kinetic model. Zeitschrift fur Phys. Chemie 2003, 217, 395–414. [Google Scholar] [CrossRef]
- Meier, J.; Friedrich, K.A.; Stimming, U. Novel method for the investigation of single nanoparticle reactivity. Faraday Discuss. 2002, 121, 365–372. [Google Scholar] [CrossRef]
- Wang, L.; Stimming, U.; Eikerling, M. Kinetic Model of Hydrogen Evolution at an Array of Au-Supported Catalyst Nanoparticles. Electrocatalysis 2010, 1, 60–71. [Google Scholar] [CrossRef]
- Hao, R.; Fan, Y.; Howard, M.D.; Vaughan, J.C.; Zhang, B. Imaging nanobubble nucleation and hydrogen spillover during electrocatalytic water splitting. Proc. Natl. Acad. Sci. USA 2018, 115, 5878–6588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pašti, I.A.; Leetmaa, M.; Skorodumova, N.V. General principles for designing supported catalysts for hydrogen evolution reaction based on conceptual Kinetic Monte Carlo modeling. Int. J. Hydrogen Energy 2016, 41, 2526–2538. [Google Scholar] [CrossRef]
- Thomas, J.M. Tens of thousands of atoms replaced by one. Nature 2015, 525, 325–326. [Google Scholar] [CrossRef] [PubMed]
- Bernt, M.; Siebel, A.; Gasteiger, H.A. Analysis of Voltage Losses in PEM Water Electrolyzers with Low Platinum Group Metal Loadings. J. Electrochem. Soc. 2018, 165, F305–F314. [Google Scholar] [CrossRef]
- Sheldon, R.A. The E factor 25 years on: The rise of green chemistry and sustainability. Green Chem. 2017, 19, 18–43. [Google Scholar] [CrossRef]
- Koper, M.T.M. Thermodynamic theory of multi-electron transfer reactions: Implications for electrocatalysis. J. Electroanal. Chem. 2011, 660, 254–260. [Google Scholar] [CrossRef]
- Vilé, G.; Albani, D.; Nachtegaal, M.; Chen, Z.; Dontsova, D.; Antonietti, M.; López, N.; Pérez-Ramírez, J. A Stable Single-Site Palladium Catalyst for Hydrogenations. Angew. Chemie Int. Ed. 2015, 54, 11265–11269. [Google Scholar] [CrossRef]
- Chung, H.T.; Cullen, D.A.; Higgins, D.; Sneed, B.T.; Holby, E.F.; More, K.L.; Zelenay, P. Direct atomic-level insight into the active sites of a high-performance PGM-free ORR catalyst. Science (80-) 2017, 357, 479–484. [Google Scholar] [CrossRef] [Green Version]
- Fei, H.; Dong, J.; Feng, Y.; Allen, C.S.; Wan, C.; Volosskiy, B.; Li, M.; Zhao, Z.; Wang, Y.; Sun, H.; et al. General synthesis and definitive structural identification of MN4C4 single-atom catalysts with tunable electrocatalytic activities. Nat. Catal. 2018, 1, 63–72. [Google Scholar] [CrossRef]
- Liu, J.; Sun, X.; Song, P.; Zhang, Y.; Xing, W.; Xu, W. High-Performance Oxygen Reduction Electrocatalysts based on Cheap Carbon Black, Nitrogen, and Trace Iron. Adv. Mater. 2013, 25, 6879–6883. [Google Scholar] [CrossRef]
- Zitolo, A.; Goellner, V.; Armel, V.; Sougrati, M.; Mineva, T.; Stievano, L.; Fonda, E.; Jaouen, F. Identification of catalytic sites for oxygen reduction in iron- and nitrogen-doped graphene materials. Nat. Mater. 2015, 14, 937–942. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Cheng, D.; Cao, D.; Zeng, X.C. A universal principle for a rational design of single-atom electrocatalysts. Nat. Catal. 2018, 1, 339–348. [Google Scholar] [CrossRef]
- Kaiser, S.K.; Lin, R.; Mitchell, S.; Fako, E.; Krumeich, F.; Hauert, R.; Safonova, O.V.; Kondratenko, V.A.; Kondratenko, E.V.; Collins, S.M.; et al. Controlling the speciation and reactivity of carbon-supported gold nanostructures for catalysed acetylene hydrochlorination. Chem. Sci. 2019, 10, 359–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Prslja, P.; Shinagawa, T.; Martin Fernandez, A.J.; Krumeich, F.; Artyushkova, K.; Atanassov, P.; Zitolo, A.; Zhou, Y.; García-Muelas, R.; et al. The Volcano Trend in Electrocatalytic CO2 Reduction Activity over Atomically-Dispersed Metal Sites on Nitrogen-Doped Carbon. ACS Catal. 2019, 9, 10426–10439. [Google Scholar] [CrossRef]
- Tavakkoli, M.; Holmberg, N.; Kronberg, R.; Jiang, H.; Sainio, J.; Kauppinen, E.I.; Kallio, T.; Laasonen, K. Electrochemical Activation of Single-Walled Carbon Nanotubes with Pseudo-Atomic-Scale Platinum for the Hydrogen Evolution Reaction. ACS Catal. 2017, 7, 3121–3130. [Google Scholar] [CrossRef]
- Langer, R.; Fako, E.; Błoński, P.; Vavrečka, M.; Bakandritsos, A.; Otyepka, M.; López, N. Anchoring of single-platinum-adatoms on cyanographene: Experiment and theory. Appl. Mater. Today 2019, 100462. [Google Scholar] [CrossRef]
- Vorobyeva, E.; Fako, E.; Chen, Z.; Collins, S.M.; Johnstone, D.; Midgley, P.A.; Hauert, R.; Safonova, O.V.; Vilé, G.; López, N.; et al. Atom-by-Atom Resolution of Structure–Function Relations over Low-Nuclearity Metal Catalysts. Angew. Chemie-Int. Ed. 2019, 58, 8724–8729. [Google Scholar] [CrossRef]
- Pérez-Ramírez, J.; López, N. Strategies to break linear scaling relationships. Nat. Catal. 2019, 2, 971–976. [Google Scholar] [CrossRef]
- Fei, H.; Dong, J.; Arellano-Jiménez, M.J.; Ye, G.; Dong Kim, N.; Samuel, E.L.G.; Peng, Z.; Zhu, Z.; Qin, F.; Bao, J.; et al. Atomic cobalt on nitrogen-doped graphene for hydrogen generation. Nat. Commun. 2015, 6, 8668. [Google Scholar] [CrossRef]
- Xue, Y.; Huang, B.; Yi, Y.; Guo, Y.; Zuo, Z.; Li, Y.; Jia, Z.; Liu, H.; Li, Y. Anchoring zero valence single atoms of nickel and iron on graphdiyne for hydrogen evolution. Nat. Commun. 2018, 9, 1460. [Google Scholar] [CrossRef]
- Lu, B.; Guo, L.; Wu, F.; Peng, Y.; Lu, J.E.; Smart, T.J.; Wang, N.; Finfrock, Y.Z.; Morris, D.; Zhang, P.; et al. Ruthenium atomically dispersed in carbon outperforms platinum toward hydrogen evolution in alkaline media. Nat. Commun. 2019, 10, 631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, N.; Stambula, S.; Wang, D.; Banis, M.N.; Liu, J.; Riese, A.; Xiao, B.; Li, R.; Sham, T.-K.; Liu, L.-M.; et al. Platinum single-atom and cluster catalysis of the hydrogen evolution reaction. Nat. Commun. 2016, 7, 13638. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Han, L.; Liu, H.; Liu, X.; Luo, J. Potential-Cycling Synthesis of Single Platinum Atoms for Efficient Hydrogen Evolution in Neutral Media. Angew. Chemie Int. Ed. 2017, 56, 13694–13698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, W.I.; Wood, B.C.; Schwegler, E.; Ogitsu, T. Combinatorial Search for High-Activity Hydrogen Catalysts Based on Transition-Metal-Embedded Graphitic Carbons. Adv. Energy Mater. 2015, 5, 1501423. [Google Scholar] [CrossRef]
Figure 1.
Trasatti’s volcano plot for hydrogen evolution in acid solutions: dependence of the common logarithm of the exchange current density (j00) from the energy of hydride formation (EM–H). Reprinted from Reference [13], © 2020 Quaino et al.; licensee, Beilstein Institut.
Figure 1.
Trasatti’s volcano plot for hydrogen evolution in acid solutions: dependence of the common logarithm of the exchange current density (j00) from the energy of hydride formation (EM–H). Reprinted from Reference [13], © 2020 Quaino et al.; licensee, Beilstein Institut.

Figure 2.
Polarization curves for hydrogen evolution/oxidation on Pt(hkl), with potential sweep rate of 20 mV s−1, in (a) acid and (b) alkaline environments. Adapted from Reference [11], Copyright (2002), with permission from Elsevier.
Figure 2.
Polarization curves for hydrogen evolution/oxidation on Pt(hkl), with potential sweep rate of 20 mV s−1, in (a) acid and (b) alkaline environments. Adapted from Reference [11], Copyright (2002), with permission from Elsevier.

Figure 3.
Main: polarization curves for hydrogen evolution/oxidation on Pt(111) and on Pt(111) modified by Pd, in acid solution (rotation rate 1600 rpm). Inset: Arrhenius plots for hydrogen evolution/oxidation on Pt(111) and on Pt(111) modified by Pd. Reprinted from Reference [32], copyright (2000), with permission from John Wiley & Sons.
Figure 3.
Main: polarization curves for hydrogen evolution/oxidation on Pt(111) and on Pt(111) modified by Pd, in acid solution (rotation rate 1600 rpm). Inset: Arrhenius plots for hydrogen evolution/oxidation on Pt(111) and on Pt(111) modified by Pd. Reprinted from Reference [32], copyright (2000), with permission from John Wiley & Sons.

Figure 4.
(a) Scheme illustrating the growth of Pt atoms (red) directly on a Ni branch (green). (b) TEM image of the Ni-branched Pt-islands nanoparticles after 48 h (1.9 nm Pt-island). (c) Pt island size distribution. (d) HAADF-STEM image of a Ni-branched 1.9 Pt-island nanoparticle. (e) STEM-EDX map showing Pt (red) islands on Ni (green) nanoparticle. (f) TEM image and (g) Pt island size distribution after 72 h (Ni-branched 2.7 nm Pt-island). (h) HAADF-STEM image of a Ni-branched 2.7 nm Pt-island. (i) STEM-EDX map with Pt (red) and Ni (green). Reprinted (adapted) with permission from [68]. Copyright (2019) American Chemical Society.
Figure 4.
(a) Scheme illustrating the growth of Pt atoms (red) directly on a Ni branch (green). (b) TEM image of the Ni-branched Pt-islands nanoparticles after 48 h (1.9 nm Pt-island). (c) Pt island size distribution. (d) HAADF-STEM image of a Ni-branched 1.9 Pt-island nanoparticle. (e) STEM-EDX map showing Pt (red) islands on Ni (green) nanoparticle. (f) TEM image and (g) Pt island size distribution after 72 h (Ni-branched 2.7 nm Pt-island). (h) HAADF-STEM image of a Ni-branched 2.7 nm Pt-island. (i) STEM-EDX map with Pt (red) and Ni (green). Reprinted (adapted) with permission from [68]. Copyright (2019) American Chemical Society.

Figure 5.
(a) Schematic illustration of the synthetic procedures for Ni3S2@MoS2eNF heterostructure. (b) Schematic diagram of hybridization between MoS2 and Ni3S2 for HER. (c–e) SEM images of 3D Ni3S2@MoS2eNF composite with different magnification. Reprinted from [104], Copyright (2019), with permission from Elsevier.
Figure 5.
(a) Schematic illustration of the synthetic procedures for Ni3S2@MoS2eNF heterostructure. (b) Schematic diagram of hybridization between MoS2 and Ni3S2 for HER. (c–e) SEM images of 3D Ni3S2@MoS2eNF composite with different magnification. Reprinted from [104], Copyright (2019), with permission from Elsevier.

Figure 6.
(a) A scheme of the experimental setup used for imaging H2 nanobubbles in electrocatalytic water splitting. H2 nanobubbles generated on an ITO surface are labeled by single R6G molecules and imaged by TIRF microscopy. (b) A series of TIRF images of a 22.8 × 22.8 μm2 area on an ITO electrode taken from a potential scan from 0 V to −2.0 V at 100 mV/s vs. Pt QRE in water containing 1 M Na2SO4 and 10 nM R6G. Fluorescence images were recorded at 19.81 frames per second with a 50-ms exposure time. (Scale bar, 5 μm.) (c) A comparison between the rate of nanobubble detection (detections per frame) (blue) and the current-voltage response recorded on the same electrode (black). The red dashed line indicates the baseline of the faradaic current. (d) A scatter plot showing the fluorescence intensity (counts) of individual H2 nanobubbles in the potential range of interest from −0.5 V to −2.0 V from the same recording. The photon counts of each fluorescent burst on one frame were converted from the total integrated fluorescence signal under the fitted 2D Gaussian function using Thunder STORM. Adapted from Reference [173]. Copyright 2018 National Academy of Sciences.
Figure 6.
(a) A scheme of the experimental setup used for imaging H2 nanobubbles in electrocatalytic water splitting. H2 nanobubbles generated on an ITO surface are labeled by single R6G molecules and imaged by TIRF microscopy. (b) A series of TIRF images of a 22.8 × 22.8 μm2 area on an ITO electrode taken from a potential scan from 0 V to −2.0 V at 100 mV/s vs. Pt QRE in water containing 1 M Na2SO4 and 10 nM R6G. Fluorescence images were recorded at 19.81 frames per second with a 50-ms exposure time. (Scale bar, 5 μm.) (c) A comparison between the rate of nanobubble detection (detections per frame) (blue) and the current-voltage response recorded on the same electrode (black). The red dashed line indicates the baseline of the faradaic current. (d) A scatter plot showing the fluorescence intensity (counts) of individual H2 nanobubbles in the potential range of interest from −0.5 V to −2.0 V from the same recording. The photon counts of each fluorescent burst on one frame were converted from the total integrated fluorescence signal under the fitted 2D Gaussian function using Thunder STORM. Adapted from Reference [173]. Copyright 2018 National Academy of Sciences.

Figure 7.
Equivalent circuit of the anodic part of an electrolyzer depending on the speciation of the active metal phase (a). Schematic representation of crucial hydrogen evolution steps in protic acidic medium on an extended (111) surface of an fcc metal (b) and a typical [179] Single Atom Heterogeneous Catalyst (SAC) (Pd atoms supported on heptazinic carbon nitride) (c). Coordination environments (d) of successful single atom HER catalyst described in the literature [180,181,182,183,184,185,186].
Figure 7.
Equivalent circuit of the anodic part of an electrolyzer depending on the speciation of the active metal phase (a). Schematic representation of crucial hydrogen evolution steps in protic acidic medium on an extended (111) surface of an fcc metal (b) and a typical [179] Single Atom Heterogeneous Catalyst (SAC) (Pd atoms supported on heptazinic carbon nitride) (c). Coordination environments (d) of successful single atom HER catalyst described in the literature [180,181,182,183,184,185,186].

Figure 8.
(a) Descriptor-based screening results of transition metal embedded graphene (TMEG) catalyst candidates based on activity (|ΔGads|) and stability (Ecoh – Eemb). The lower-left quadrant of (a) contains materials that passed both criteria. For comparison, the gray shaded region covers the range of |ΔGads| that a Pt (111) surface could have depending on the coverage of hydrogen. Note the different scales in the left and right sides of the horizontal axis. Adapted from Reference [196], copyright (2015), with permission from John Wiley & Sons. (b,c) influence of active site nuclearity of single Pd atoms (ii) and Pd trimmers (iii) in the thermal part of HER versus an idealized single atom active site (i). (Values for (ii) and (iii) are originally computed as hydrogen activation and taken from reference [189].
Figure 8.
(a) Descriptor-based screening results of transition metal embedded graphene (TMEG) catalyst candidates based on activity (|ΔGads|) and stability (Ecoh – Eemb). The lower-left quadrant of (a) contains materials that passed both criteria. For comparison, the gray shaded region covers the range of |ΔGads| that a Pt (111) surface could have depending on the coverage of hydrogen. Note the different scales in the left and right sides of the horizontal axis. Adapted from Reference [196], copyright (2015), with permission from John Wiley & Sons. (b,c) influence of active site nuclearity of single Pd atoms (ii) and Pd trimmers (iii) in the thermal part of HER versus an idealized single atom active site (i). (Values for (ii) and (iii) are originally computed as hydrogen activation and taken from reference [189].

Figure 9.
Comparison of the analyzed groups of electrocatalytic systems presented in this review (Authors’ view). We consider maturity, metal loading (M loading), stability (under operating conditions), the complexity of the catalyst preparation, the overall performance in alkaline solutions, and the overall performance in acidic solutions. Note that in the case of the catalyst preparation complexity case, higher grade means more complex preparation.
Figure 9.
Comparison of the analyzed groups of electrocatalytic systems presented in this review (Authors’ view). We consider maturity, metal loading (M loading), stability (under operating conditions), the complexity of the catalyst preparation, the overall performance in alkaline solutions, and the overall performance in acidic solutions. Note that in the case of the catalyst preparation complexity case, higher grade means more complex preparation.

Table 1.
Hydrogen evolution reaction (HER) on pure metal single crystal surfaces, in acid solutions, and the corresponding available data: Tafel slopes (b), exchange current densities (j0) at given temperatures (T), activation energies (Ea), and identified mechanism and rate-determining step (RDS).
Table 1.
Hydrogen evolution reaction (HER) on pure metal single crystal surfaces, in acid solutions, and the corresponding available data: Tafel slopes (b), exchange current densities (j0) at given temperatures (T), activation energies (Ea), and identified mechanism and rate-determining step (RDS).
| Single Crystal | b /mV dec−1 | j0 /mA cm−2 (T) | Ea /kJ mol−1 | Mechanism and RDS | Ref. |
|---|---|---|---|---|---|
| Pt(100) | 2(2.3RT/F) | 0.36 (274 K) 0.60 (303 K) 0.76 (333 K) | 12 | Heyrovsky-Volmer | [14] |
| Pt(110) | 2.3RT/2F | 0.65 (274 K) 0.98 (303 K) 1.35 (333 K) | 9.5 | Tafel-Volmer | |
| Pt(111) | ≈2.3RT/F | 0.21 (274 K) 0.45 (303 K) 0.83 (333 K) | 18 | Tafel-Volmer, Heyrovsky-Volmer | |
| Au(100) | varies gradually from −60 to −120 mV dec−1 | 0.5 × 10−4 | - | - | [16] |
| Au(110) | 0.3 × 10−4 | - | - | ||
| Au(111) | 2.5 × 10−4 | - | - |
Table 2.
Comparison of experimentally measured kinetic and thermodynamic parameters of recently developed carbon-supported HER SACs in comparison to state-of-the-art Pt systems.
Table 2.
Comparison of experimentally measured kinetic and thermodynamic parameters of recently developed carbon-supported HER SACs in comparison to state-of-the-art Pt systems.
| M | M Speciation | η (V) | Tafel Slope (mV/dec) ** | Conditions (Acid /Base) | Reference |
|---|---|---|---|---|---|
| Pt | SOA/NP | −0.01 | 31.0 | Acid | [191] |
| Co | SAHC | −0.03 | 82.0 | Acid | [191] |
| Pt | SOA/NP | −0.00 | 33.9 | Acid | [193] |
| Fe | SAHC | −0.04 | 37.8 | Acid | [193] |
| Ni | SAHC | −0.05 | 45.8 | Acid | [193] |
| Pt | SOA/NP | −0.03 | 39.0 | Base | [192] |
| Ru | SAHC | −0.01 | 24.0 | Base | [192] |
M = metal; SA = single atoms; SOA = state-of-the-art, ** - absolute value.
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Gutić, S.J.; Dobrota, A.S.; Fako, E.; Skorodumova, N.V.; López, N.; Pašti, I.A. Hydrogen Evolution Reaction-From Single Crystal to Single Atom Catalysts. Catalysts 2020, 10, 290. https://0-doi-org.brum.beds.ac.uk/10.3390/catal10030290
AMA Style
Gutić SJ, Dobrota AS, Fako E, Skorodumova NV, López N, Pašti IA. Hydrogen Evolution Reaction-From Single Crystal to Single Atom Catalysts. Catalysts. 2020; 10(3):290. https://0-doi-org.brum.beds.ac.uk/10.3390/catal10030290
Chicago/Turabian StyleGutić, Sanjin J., Ana S. Dobrota, Edvin Fako, Natalia V. Skorodumova, Núria López, and Igor A. Pašti. 2020. "Hydrogen Evolution Reaction-From Single Crystal to Single Atom Catalysts" Catalysts 10, no. 3: 290. https://0-doi-org.brum.beds.ac.uk/10.3390/catal10030290
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.