α-Functionalization of Imines via Visible Light Photoredox Catalysis


1
Department of Organic Chemistry, Universidad Autónoma de Madrid, 28049 Madrid, Spain
2
Institute for Advanced Research in Chemical Sciences (IAdChem), Universidad Autónoma de Madrid, 28049 Madrid, Spain
*
Authors to whom correspondence should be addressed.
Catalysts 2020, 10(5), 562; https://doi.org/10.3390/catal10050562
Submission received: 6 May 2020
/
Revised: 14 May 2020
/
Accepted: 16 May 2020
/
Published: 19 May 2020
(This article belongs to the Special Issue New Trends in Asymmetric Catalysis)
Abstract
:The innate electrophilicity of imine building blocks has been exploited in organic synthetic chemistry for decades. Inspired by the resurgence in photocatalysis, imine reactivity has now been redesigned through the generation of unconventional and versatile radical intermediates under mild reaction conditions. While novel photocatalytic approaches have broadened the range and applicability of conventional radical additions to imine acceptors, the possibility to use these imines as latent nucleophiles via single-electron reduction has also been uncovered. Thus, multiple research programs have converged on this issue, delivering creative and practical strategies to achieve racemic and asymmetric α-functionalizations of imines under visible light photoredox catalysis.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Visible light photoredox catalysis has been at the forefront of organic chemistry research for over a decade, establishing itself as a sustainable and multifaceted synthetic tool [1]. Irradiation of catalytic amounts of polypyridyl complexes and organic sensitizers under mild conditions has proven to be an excellent activation pathway to access a wide variety of radical intermediates (Figure 1). Spurred by this resurgence, long-standing challenges in the field have been resolved, while a plethora of transformations continue to be developed in an effort to revamp organic synthesis.
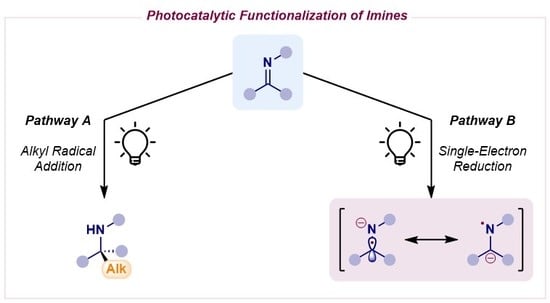
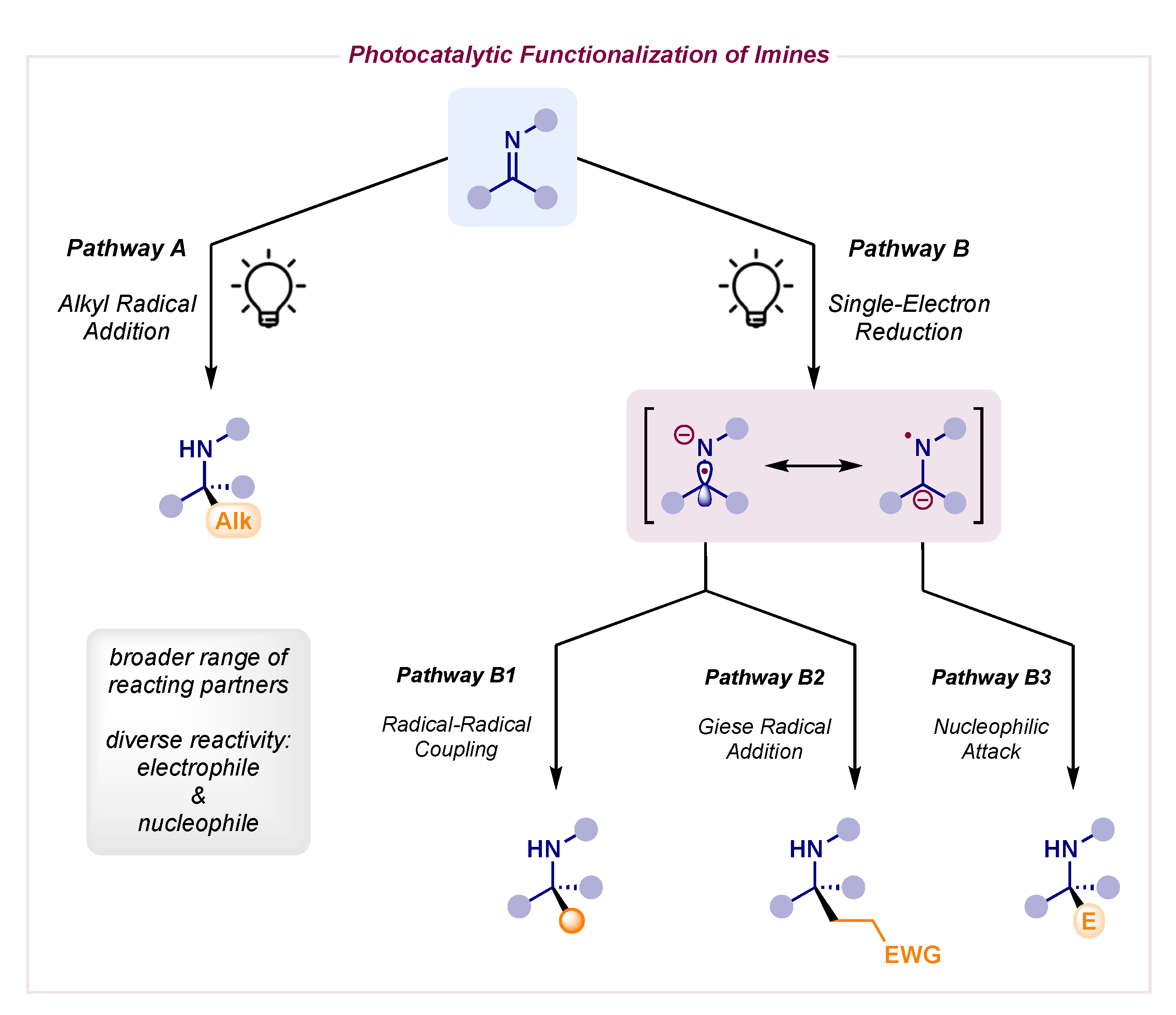
The reactivity of imines has certainly undergone a complete makeover, as different strategies involving these key building blocks have been developed (Scheme 1). The classical approach still relies on the innate electrophilic nature of imines to undergo standard alkyl radical addition (pathway A in Scheme 1, left). Thanks to photoredox catalysis, the generation of nucleophilic radicals starting from mild alkylating reagents [2] has provided a broader range to a severely limited transformation in the past due to hazardous reagents and impractical conditions.
Alternatively, the photocatalytic single-electron reduction of imines has emerged as a powerful technology to generate radical anion intermediates which exist as two different resonant forms (pathway B in Scheme 1, right) [3,4]. The α-amino radical species can engage in radical–radical couplings with a large pool of reacting partners (pathway B1 in Scheme 1), while also displaying a complementary nucleophilic behavior to their corresponding electrophilic imine precursors. Indeed, they can be trapped by electron-deficient π-systems, a combination which would not be feasible using polar chemistry (pathway B2 in Scheme 1). Interestingly, the N-centered radical species can be quickly quenched by an H atom donor to yield a stable carbanion capable of reacting with a traditional electrophile in polar fashion (pathway B3 in Scheme 1).
The single-electron reduction event (pathway B in Scheme 1, right) can be a challenging redox process which often requires assistance [4]. While some electron-poor imines can undergo a straightforward photocatalytic reduction (such as N-sulfonyl- or α-keto-imines), other neutral imines feature a reduction potential which falls out of range of most photocatalysts. The addition of an external Lewis acid can increase the reduction potential of the imine (less negative) through coordination. Moreover, hydrogen-bonding via Brønsted acid can make this reduction a thermodynamically favorable process thanks to proton-coupled electron transfer (PCET), wherein an electron transfer from the photocatalyst to the imine takes place in concert with a proton transfer from the Brønsted acid to the imine.
2. Photocatalytic Radical Additions to Imines—Pathway A
2.1. Racemic Photocatalytic Radical Additions to Imines
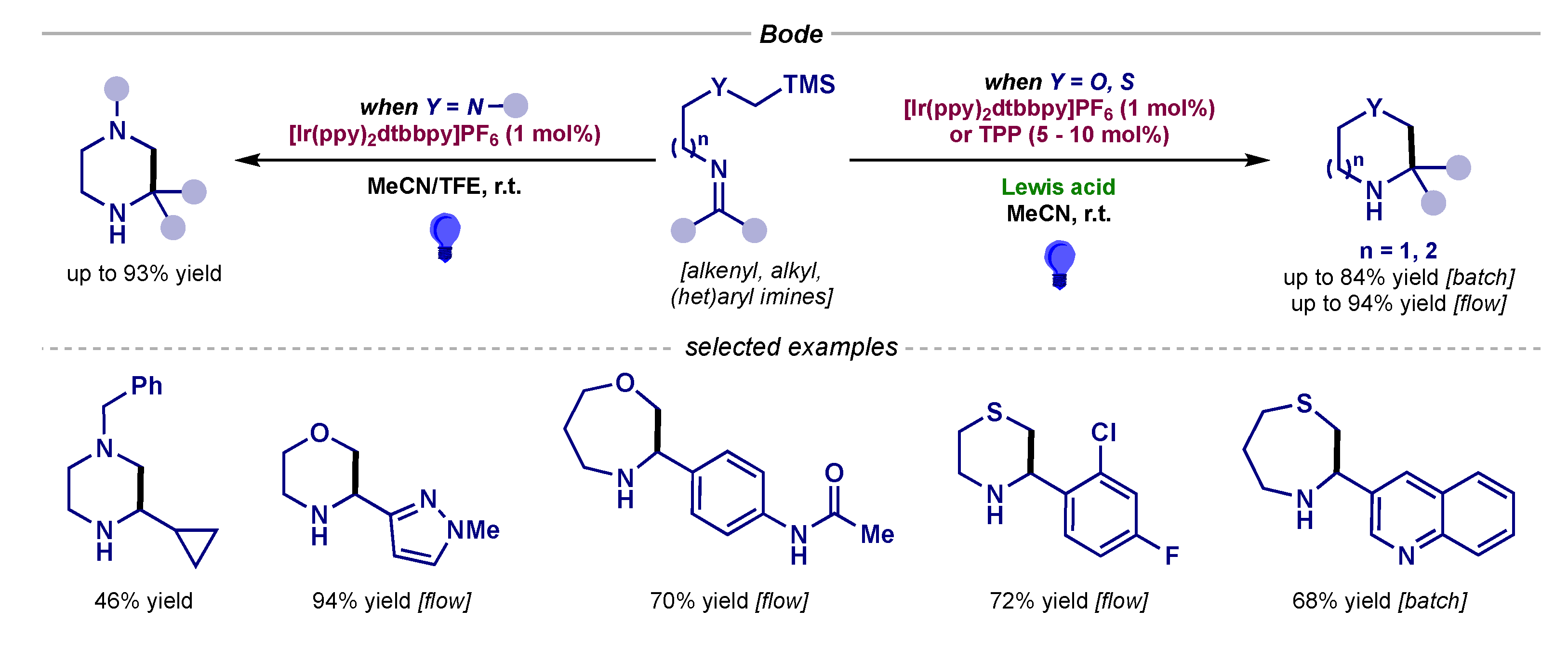
Racemic radical additions to imines under visible light photocatalysis began to appear in 2016, when Bode reported the cyclization of silicon amine protocol (SLAP) reagents with an imine moiety (Scheme 2, left) [5]. These α-silyl amine precursors could undergo mild single-electron oxidation to render α-amino radicals, which could then engage with the imine to yield a wide variety of piperazine derivatives. The protocol was expanded further with α-silyl ether and thioether precursors to access morpholines, oxazepanes, thiomorpholines and thiazepanes (Scheme 2, right) [6,7]. It should be noted that, in this case, a Lewis acid was required to activate the imine, and the photocatalytic cycle could start with an initial Lewis acid-assisted single-electron reduction of the imine.
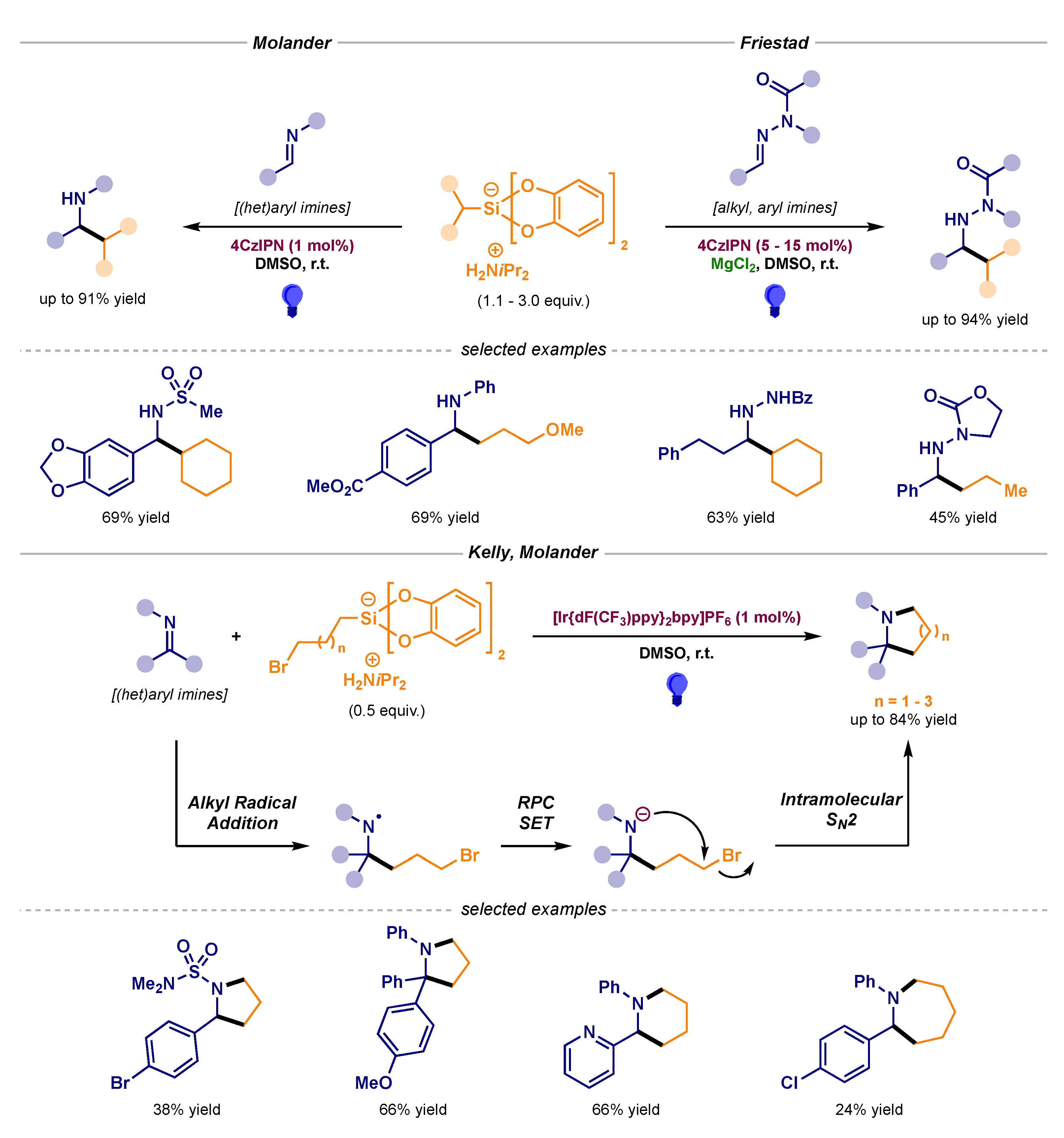
The first intermolecular photocatalytic radical addition was published in 2017 by Molander’s group [8]. The design of a general and modular approach based on the swift single-electron oxidation of ammonium alkyl bis(catecholato)silicates enabled the alkylation of different N-sulfonyl- and N-aryl-imines (Scheme 3, top left) [9]. In addition, Friestad employed these silicon reagents to perform the alkylation of N-acyl hydrazones in the presence of a Lewis acid (Scheme 3, top right) [10]. Alkyl silicates have also been utilized by Kelly and Molander to achieve the synthesis of various saturated N-heterocycles via radical alkylation and subsequent cyclization in a radical polar crossover (RPC) process (Scheme 3, bottom) [11].
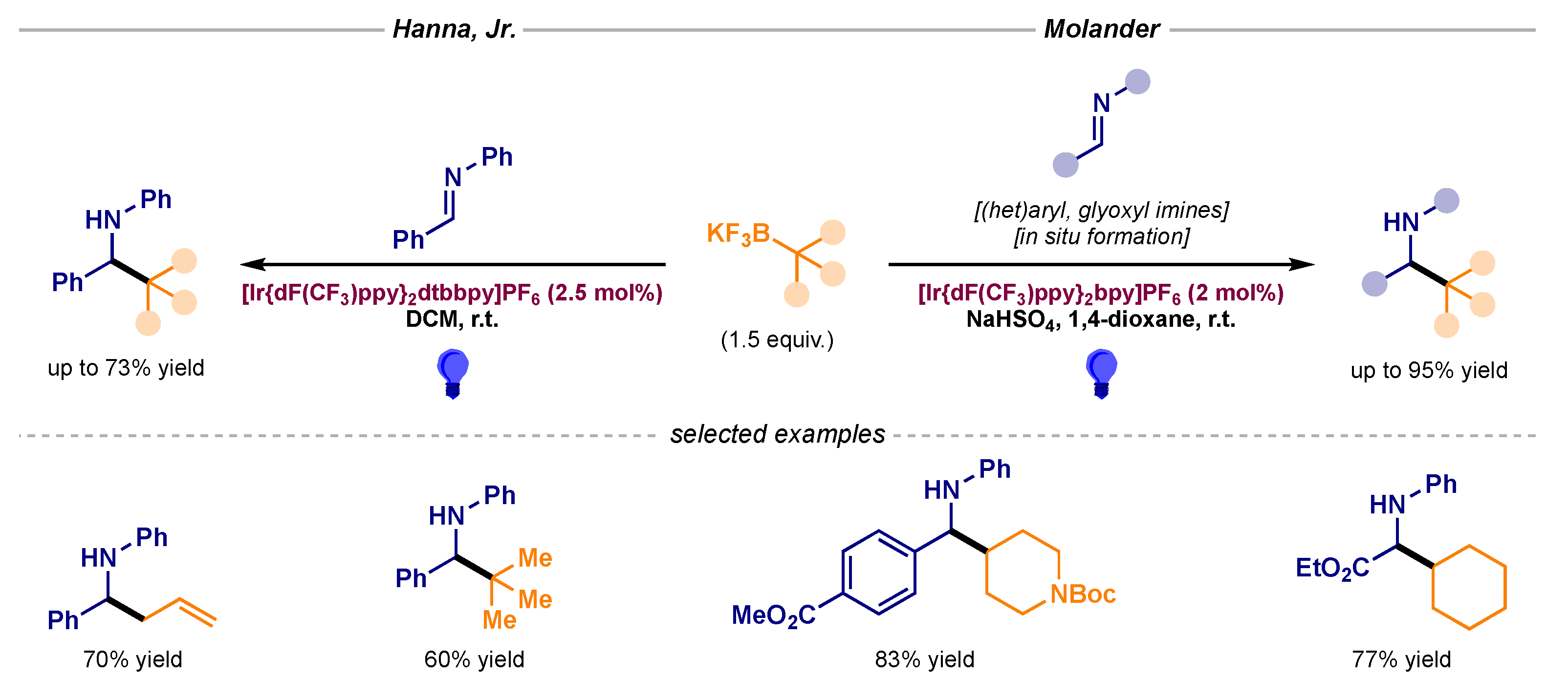
More radical precursors have also been deployed in an attempt to expand the synthetic prowess of this transformation. For instance, Hanna, Jr. and Molander disclosed the photocatalytic activation of alkyl trifluoroborates, enabling the radical alkylation of non-activated imines (Scheme 4) [12,13].
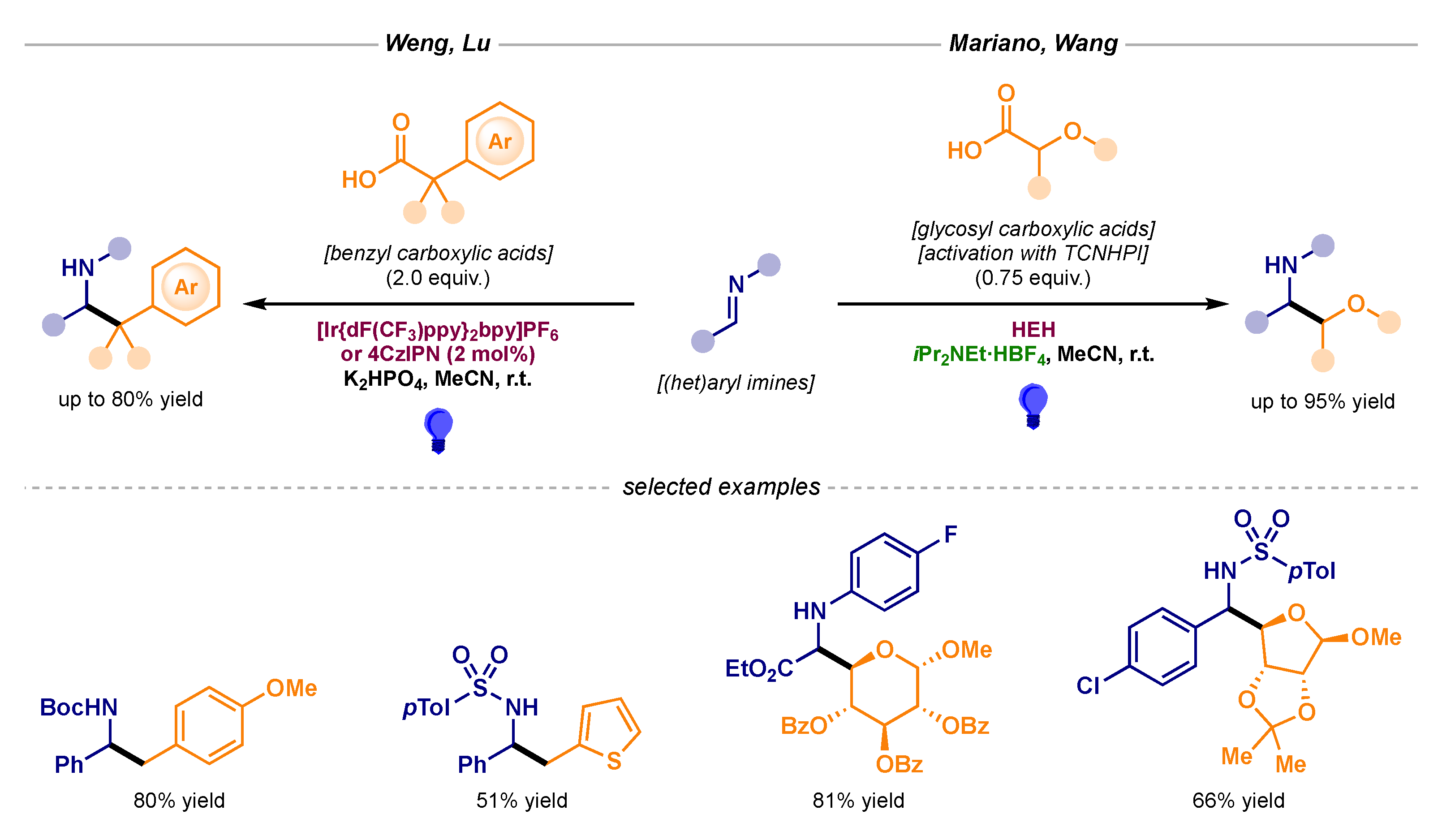
Alkyl carboxylic acids hold a preferred position among radical precursors due to their versatility and ubiquity. Indeed, these alkylating agents can be implemented into mechanistically distinct photoredox pathways. Deprotonation of the acid can render a carboxylate species which can then undergo single-electron oxidation and subsequent decarboxylation to afford the alkyl radical intermediate. Alternatively, these acids can be activated with N-hydroxyphthalimide (NHPI) or its tetrachlorinated derivative (TCNHPI) through a simple esterification process to provide redox-active esters (RAEs). In this case, single-electron reduction can deliver the alkyl radical intermediate. This flexible behavior has been exploited by several research groups attempting to perform the alkyl radical addition to imines (Scheme 5). Weng and Lu reported the decarboxylative benzylation process following the oxidative pathway (Scheme 5, left) [14,15], while Mariano and Wang published a reductive version. In this later case, the decarboxylative glycosylation of imines was featured, although a Hantzsch ester (HEH) derivative was needed as a stoichiometric photosensitizer (Scheme 5, right) [16,17].
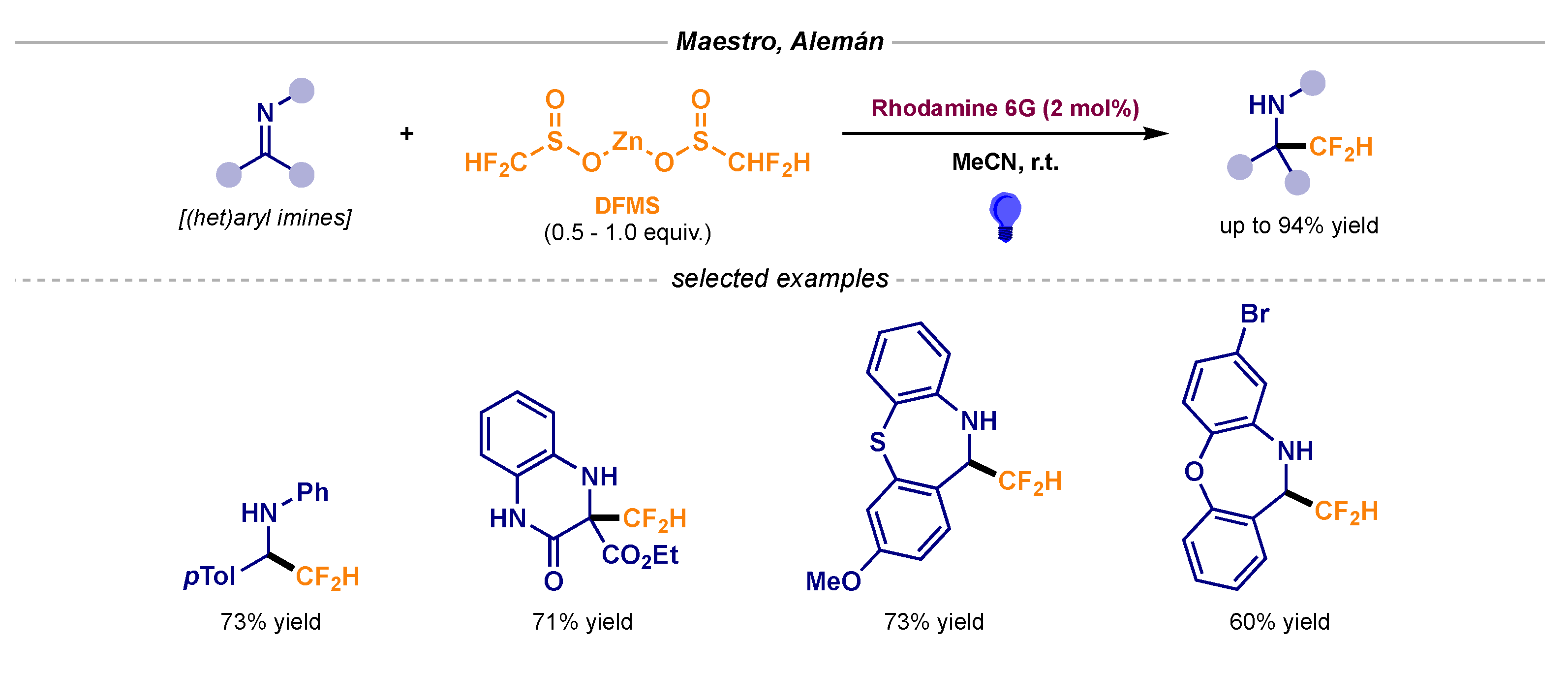
Notably, the radical fluoroalkylation of imines had remained inaccessible in the field until Maestro and Alemán recently reported the direct difluoromethylation of imine moieties (Scheme 6) [18]. This general procedure was predicated on the single-electron oxidation of readily available zinc difluoromethane sulfinate (DFMS) in the presence of an organophotoredox catalyst (Rhodamine 6G).
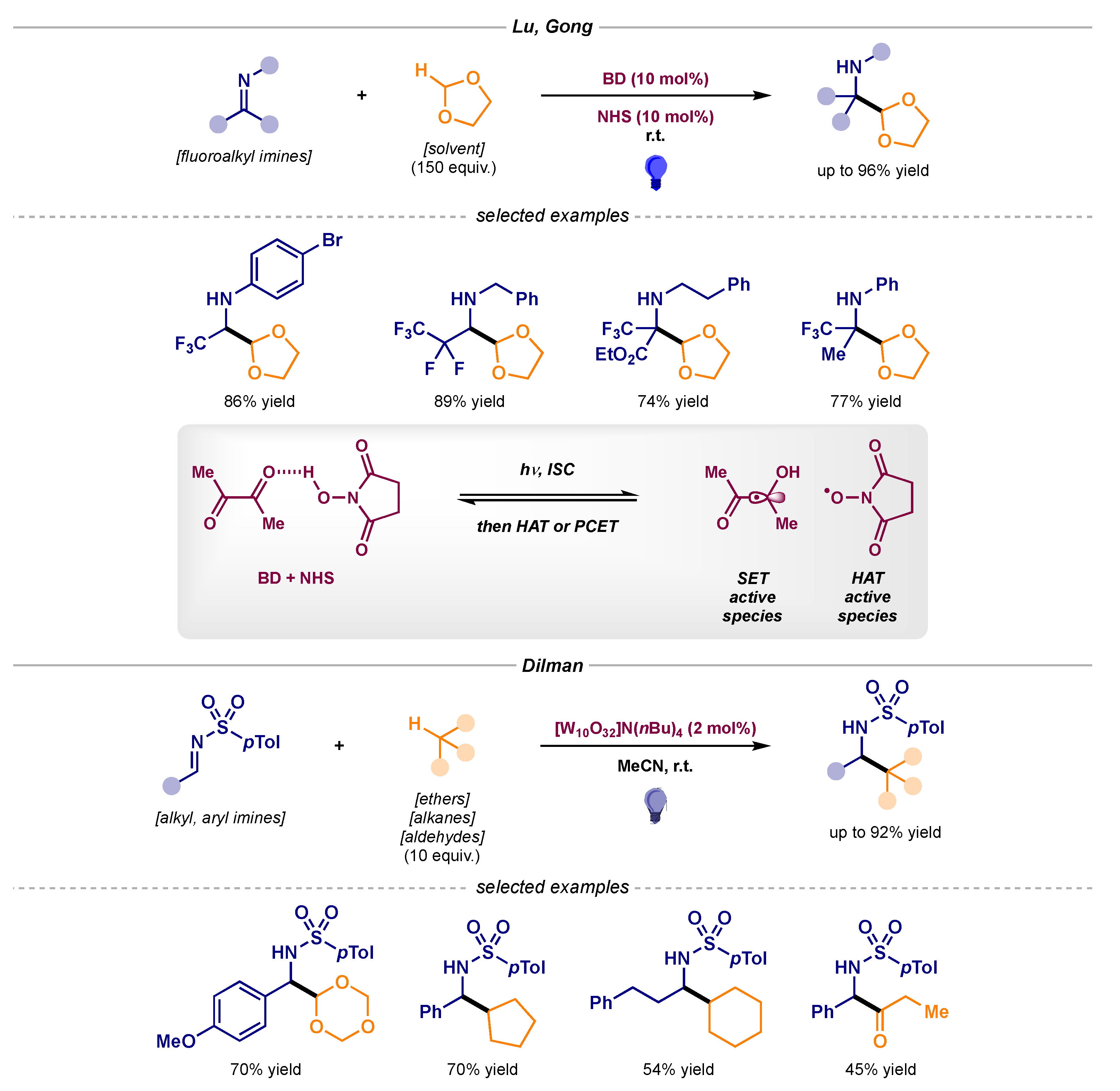
Lastly, hydrogen atom transfer (HAT) has also been used in order to perform the C-H activation of different alkyl radical precursors and perform the desired alkylation reaction with activated imines (Scheme 7). Lu and Gong reported the α-oxyalkyl radical addition of 1,3-dioxolane to fluoroalkyl imines (Scheme 7, top) [19,20], while Dilman managed to install different alkyl and acyl radicals into N-sulfonyl imines (Scheme 7, bottom) [21,22,23].
2.2. Stereoselective Photocatalytic Radical Additions to Imines
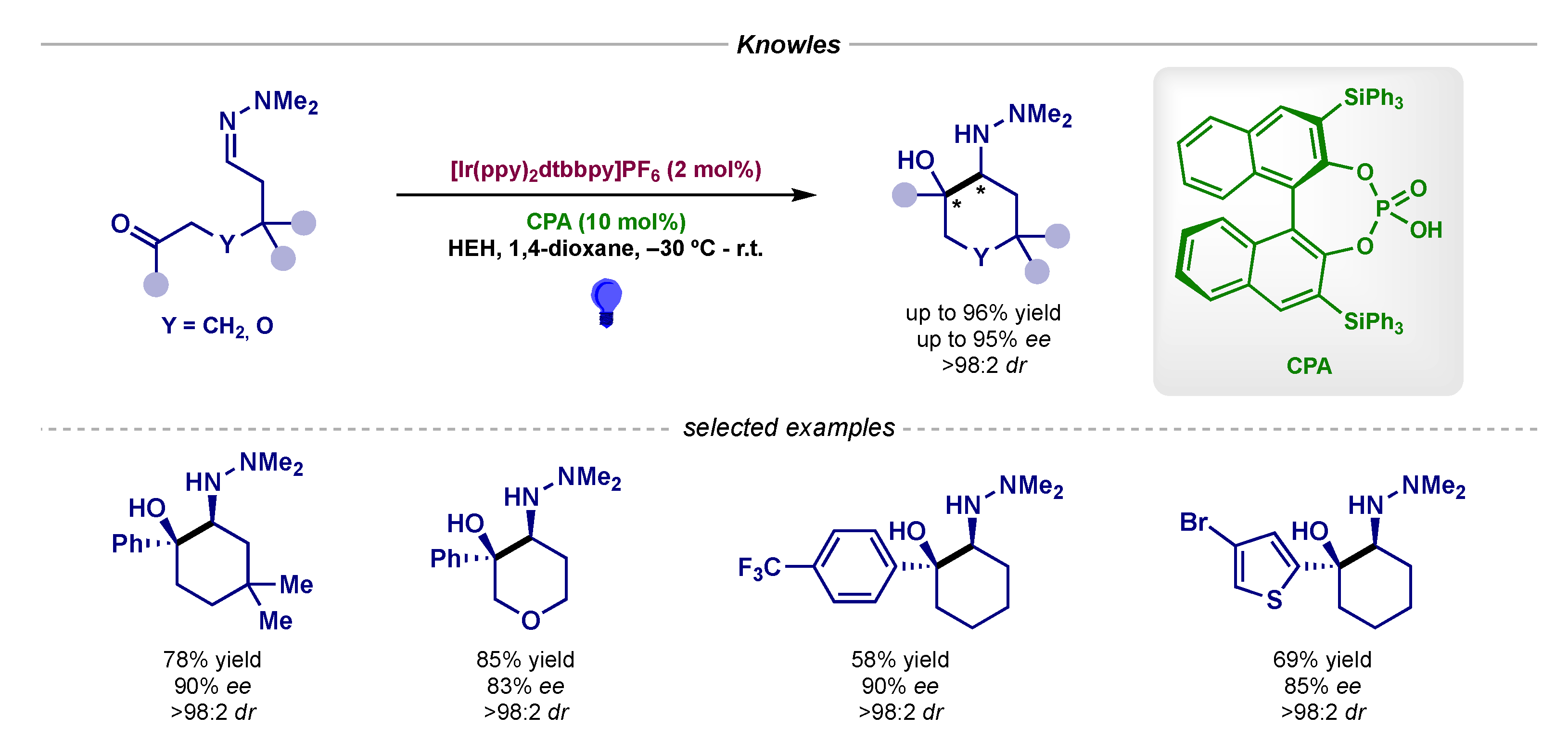
In the field of asymmetric photocatalytic additions to imines, Knowles first described in 2013 an elegant intramolecular example in which a hydrazone trapped a ketyl radical intermediate—generated by PCET—in enantioselective fashion thanks to the chiral induction exerted by a chiral phosphoric acid (Scheme 8) [24].
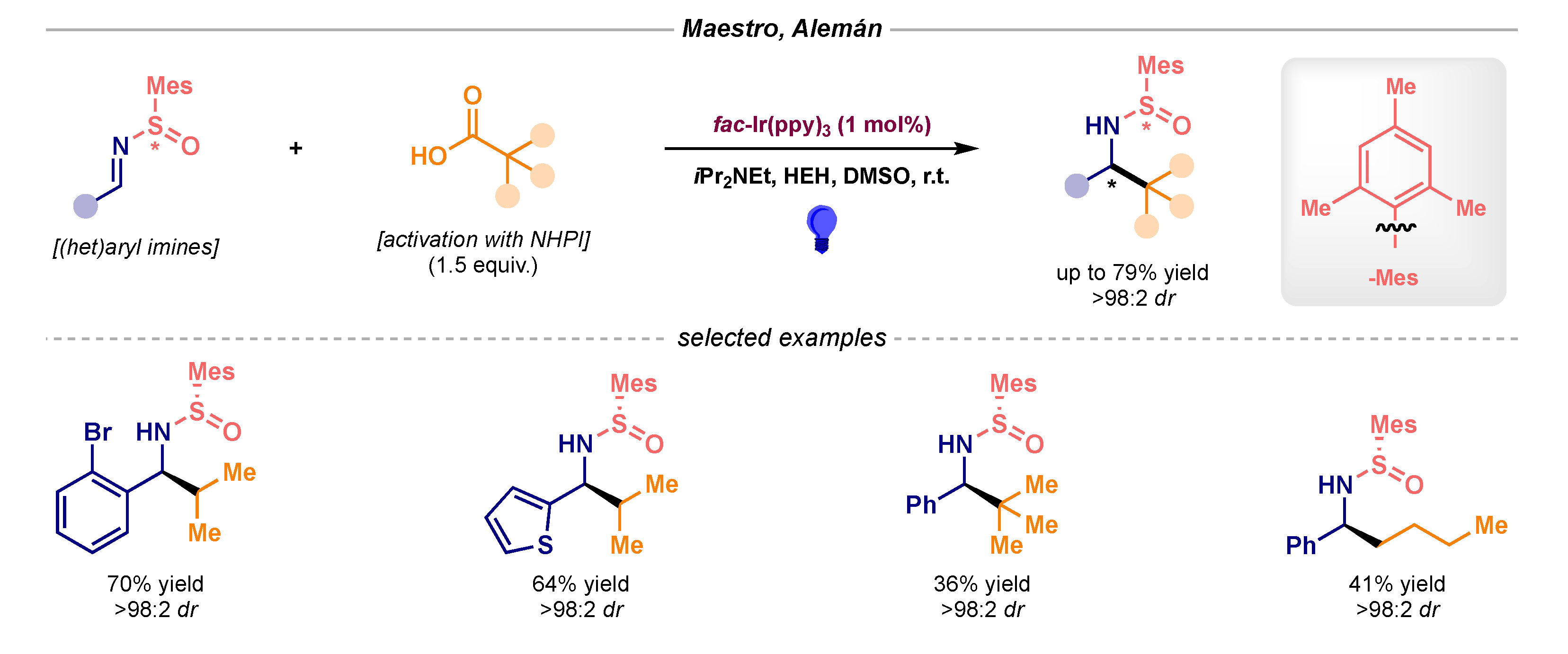
No further reports were published on this topic until Maestro and Alemán disclosed in 2017 an asymmetric intermolecular radical alkylation of imines based on the use of chiral sulfoxides (Scheme 9) [25]. The photocatalytic reduction of NHPI-derived RAEs delivered the alkyl radical, which then engaged with the enantiopure N-sulfinimine in diastereoselective fashion to afford α-branched benzyl amine derivatives.
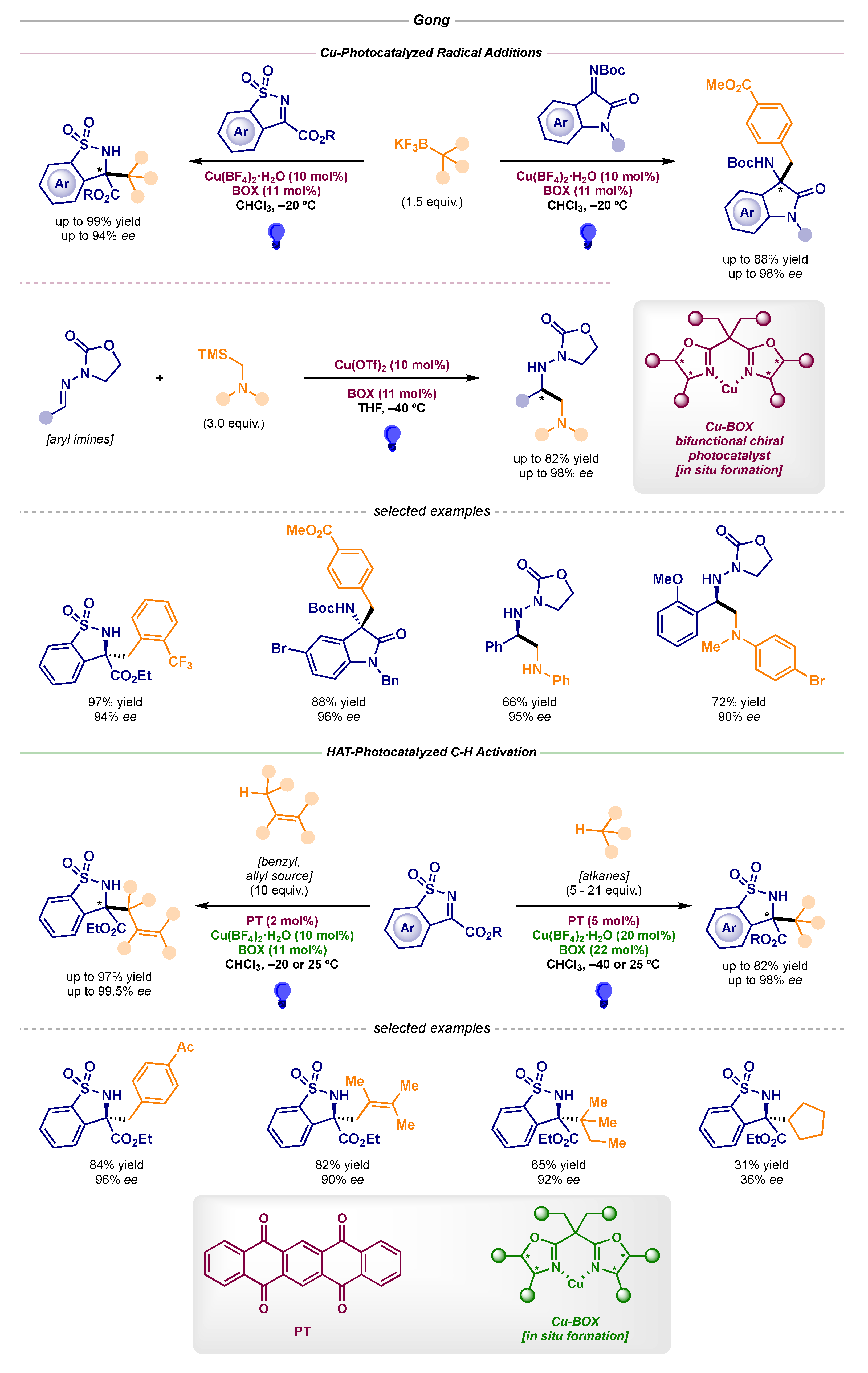
Moreover, Gong’s research group developed a series of transformations in 2018 and 2019 based on chiral Lewis acid-catalyzed radical alkylations of different imine scaffolds (Scheme 10, top) [26,27]. When using redox-active alkyl trifluoroborates and silanes, the Cu-BOX complexes acted as bifunctional chiral photocatalysts, performing both the asymmetric induction and the single-electron oxidation of the radical precursors, while suppressing the need for an external photocatalyst. In the latest report published by Gong, an HAT-photocatalyst (5,7,12,14-pentacenetetrone, PT) was required to perform the C-H activation of benzyl and allyl positions, as well as non-activated alkanes (Scheme 10, bottom) [28].
The generality observed throughout this section noticeably stands out, wherein an assortment of radical precursors has been inserted into mechanistically similar protocols based on their photocatalytic activation to render the nucleophilic radical intermediate. Most notably, the range of imine building blocks employed in these reactions is quite impressive, as both activated and non-activated substrates have proven to be suitable acceptors to the different radical additions.
3. Photocatalytic α-Amino Radical Reactivity via Single-Electron Reduction of Imine Derivatives—Pathway B
3.1. Racemic α-Amino Radical–Radical Couplings—Pathway B1
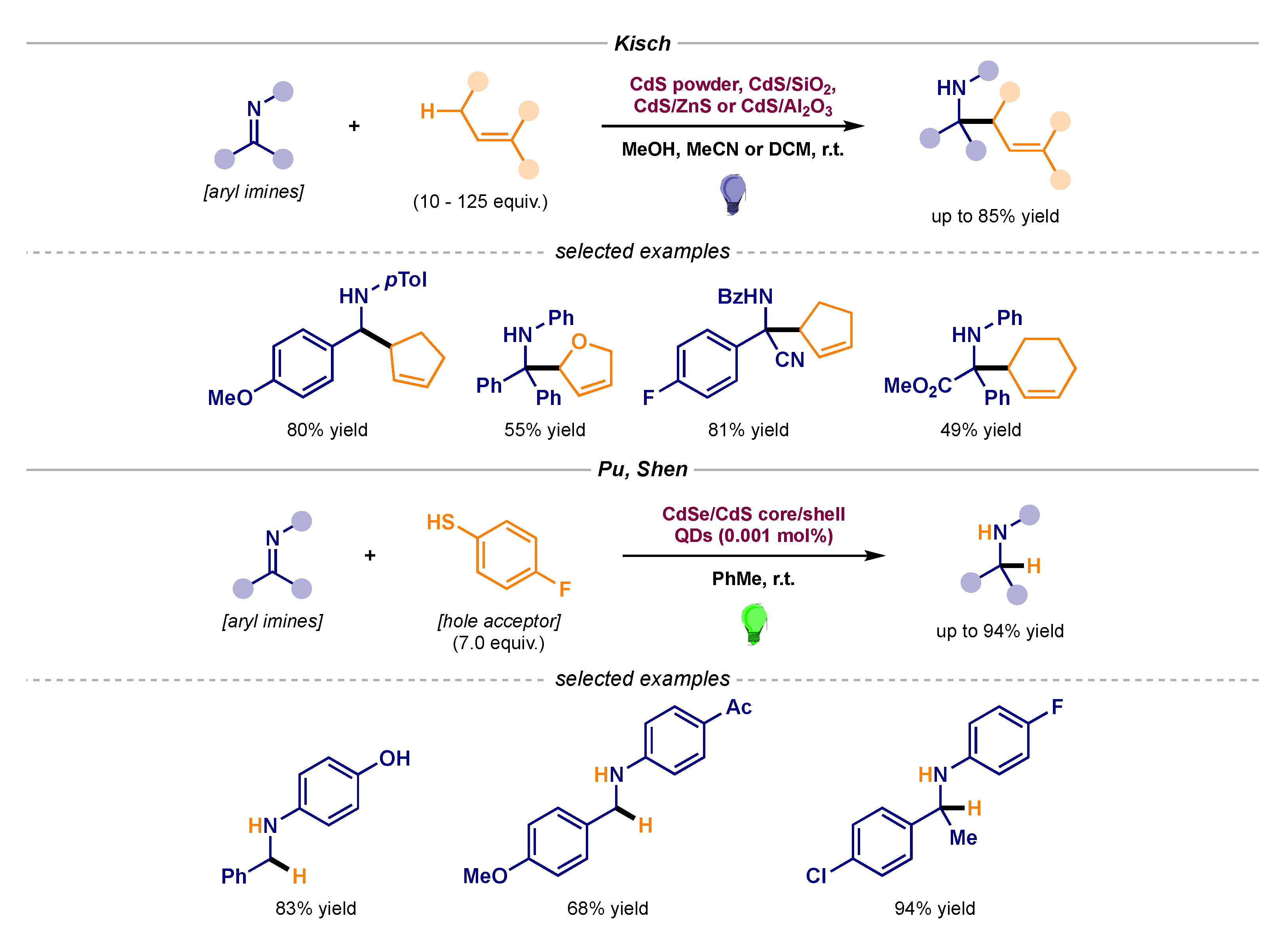
The generation of α-amino radical intermediates derived from imine building blocks was first reported by Kisch (Scheme 11, top) [29,30]. By means of a family of heterogeneous photocatalyst semiconductors, their group developed a series of transformations involving the coupling of α-amino radicals and allyl radicals (pathway B1 in Scheme 1) [31]. CdS powder as well as supported versions on SiO2, ZnS, and Al2O3 can behave as the photocatalyst semiconductor which features a surface with the ability to engage in interfacial electron transfer (IFET). Upon visible light absorption, the semiconductor can generate an electron-hole pair—essentially reducing and oxidizing surface centers. These sites can then perform IFET with the adsorbed substrates, delivering the two radical intermediates that eventually afford the recombination product (known as semiconductor photocatalysis B). Later on, Pu and Shen used CdSe/CdS core/shell quantum dots (QDs) as photocatalysts for the transfer hydrogenation of diaryl imines with a thiophenol as H atom donor (Scheme 11, bottom) [32,33].
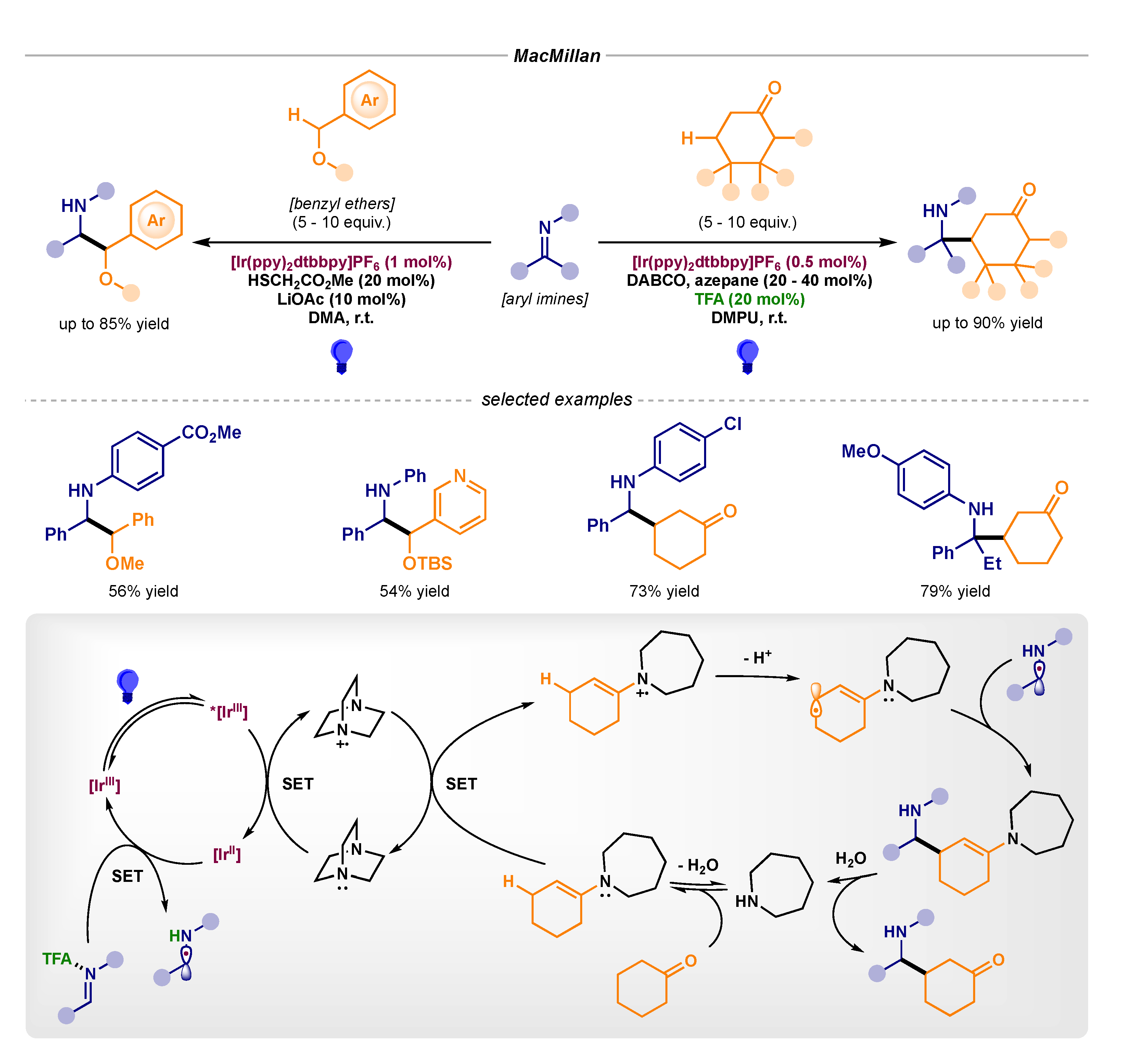
In the field of homogenous photocatalysis, MacMillan first reported the coupling of α-amino radicals with α-oxybenzyl and β-enaminyl radicals, giving access to pinacol-type products and the formal β-Mannich reaction, respectively (Scheme 12, top) [34,35]. Notably, the generation of the radical reacting partners required elegant multicatalytic approaches. The pinacol-type coupling reaction relied on the initial photocatalytic oxidation and deprotonation of methyl thioglycolate to produce a thiyl radical. Then, this S-centered radical could perform the H atom abstraction from the benzyl ether to afford the α-oxybenzyl radical and regenerate the thiol catalyst. On the other hand, the β-enaminyl radical could be accessed following: i) initial condensation of a cyclic ketone with a simple aminocatalyst (azepane), ii) subsequent oxidation of the catalytic enamine, and iii) final allylic deprotonation (Scheme 12, bottom).
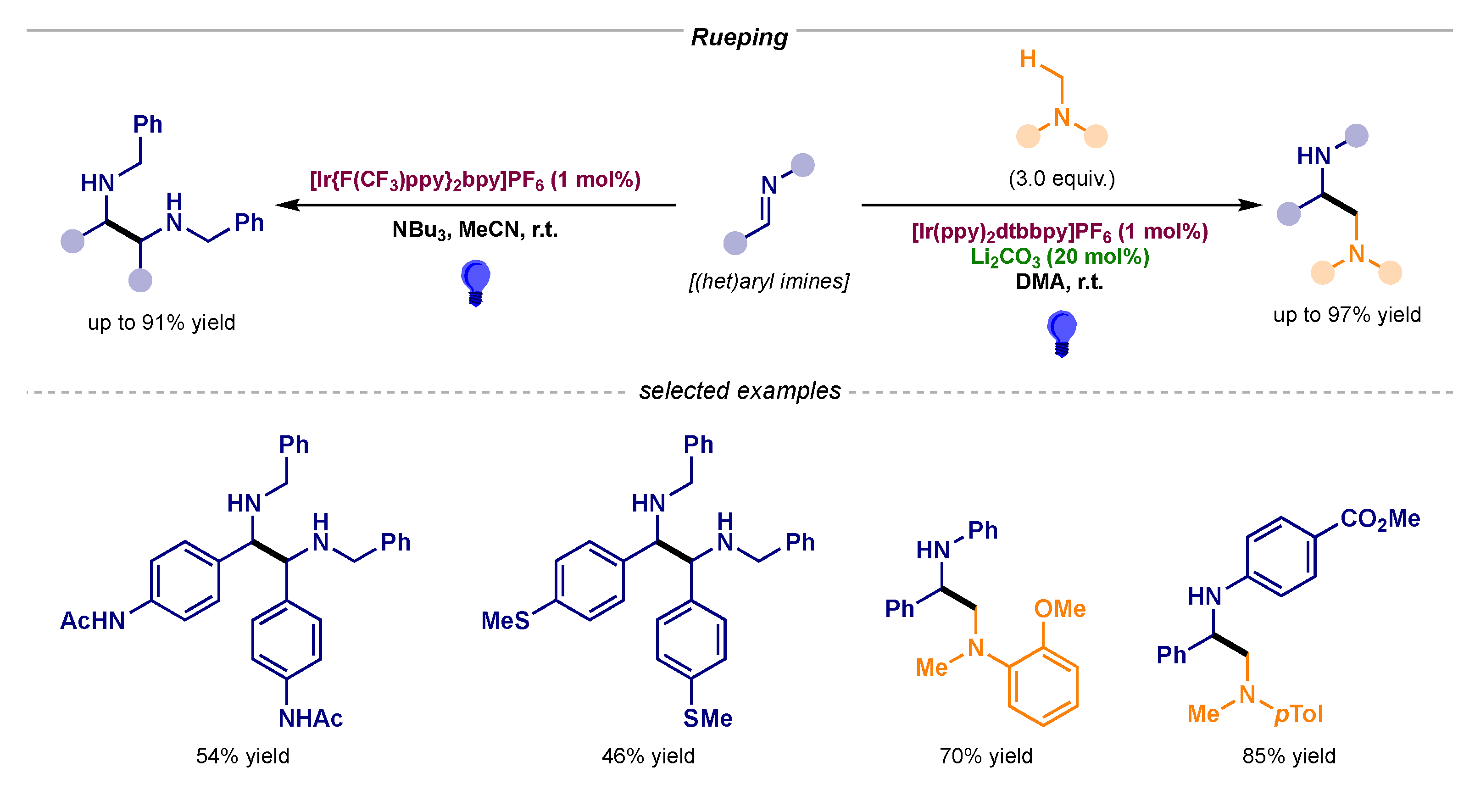
Concurrently, Rueping displayed the ability of these α-amino radicals to react with each other to render symmetrical and unsymmetrically substituted 1,2-diamines (Scheme 13) [36,37].
Following the development of these transformations, different variants of the radical–radical coupling of α-amino radicals began to appear. Sudo successfully employed an organophotoredox catalyst in a similar symmetrical coupling [38], whereas Gilmore obtained vicinal primary diamine products through in situ formation of the imines with aldehydes and ammonia [39]. Regarding unsymmetrical adducts, Wang reported the coupling of imine-derived α-amino radicals with tetrahydroisoquinoline-derived α-amino radicals [40]. In most cases, the imine reduction is believed to be assisted by coordination to an external acidic species.
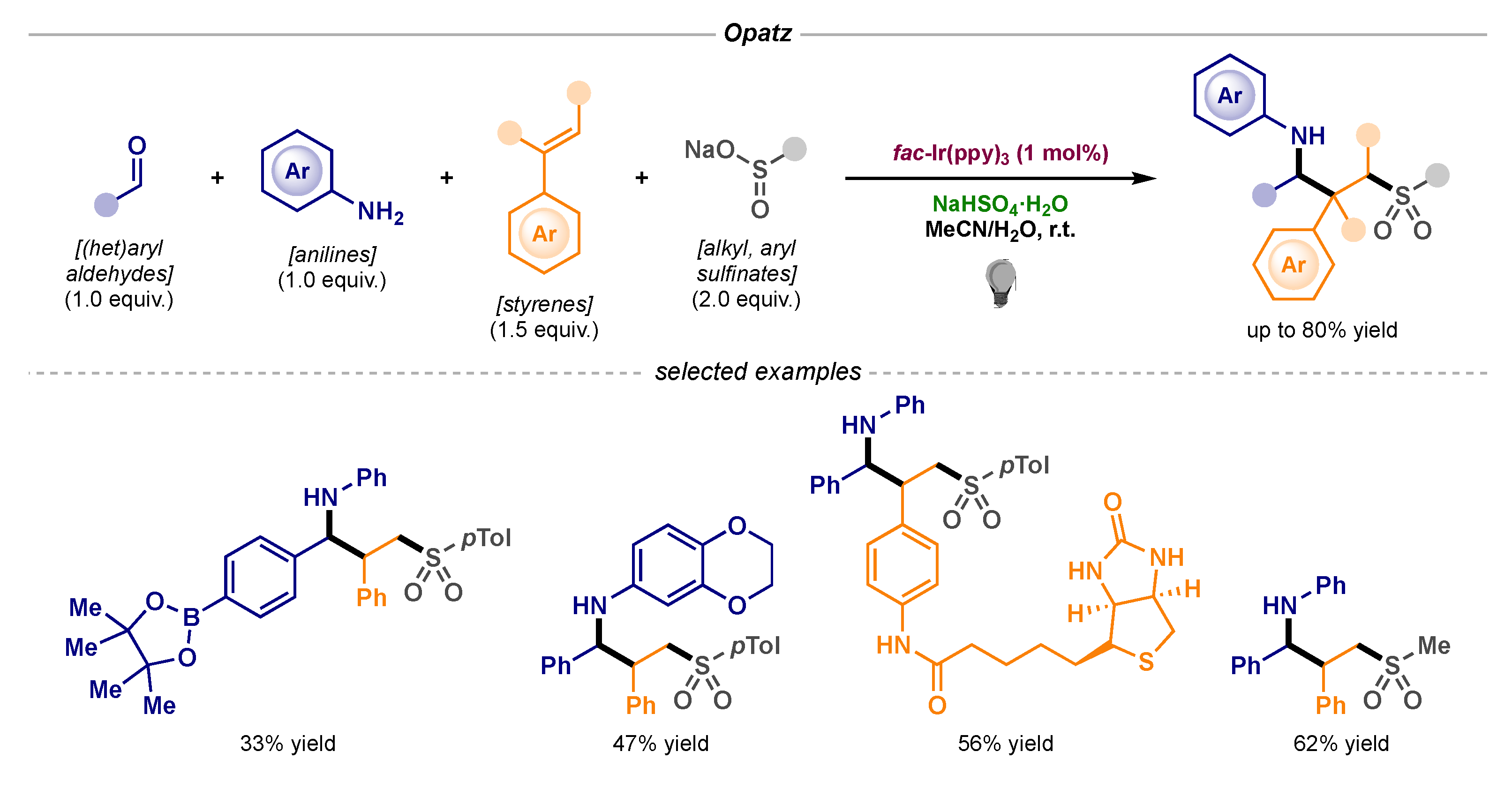
A unique example based on the reactivity of these α-amino radicals was reported by Opatz featuring a four-component reaction which gave access to structurally diverse products (Scheme 14) [41]. The protocol involved the simultaneous construction of three new bonds: C-N (via in situ formation of the imine), C-S (via sulfonyl radical addition to styrene) and C-C (via α-amino radical-benzyl radical coupling).
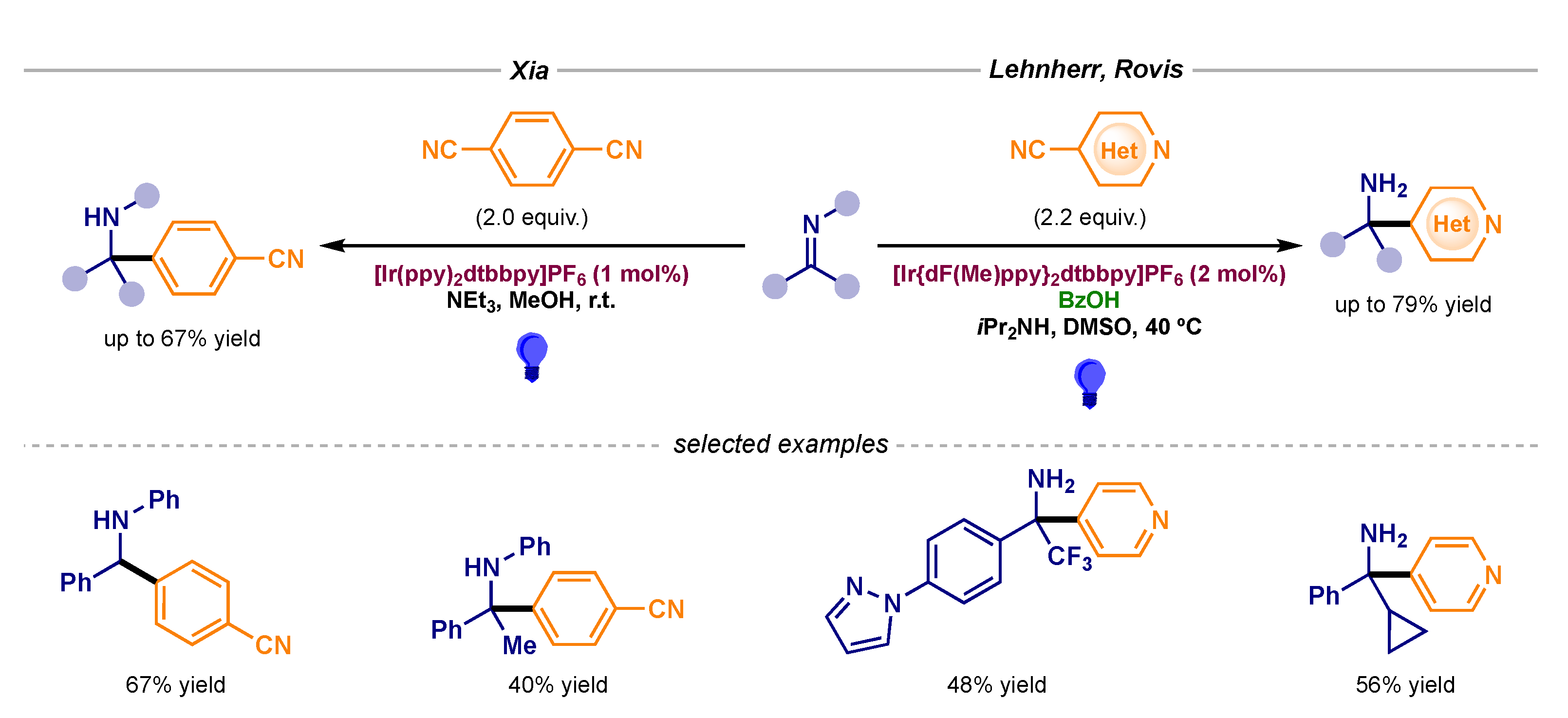
Arylation reactions, which had remained elusive in the context of radical chemistry with imines, were achieved by Xia and Lehnherr and Rovis (Scheme 15) [42,43]. Through generation of stabilized aryl radical intermediates, the radical–radical coupling became feasible with 1,4-dicyanobenzene and 4-cyanopyridine precursors, respectively.
3.2. Racemic α-Amino Radical Additions to Activated Olefins—Pathway B2
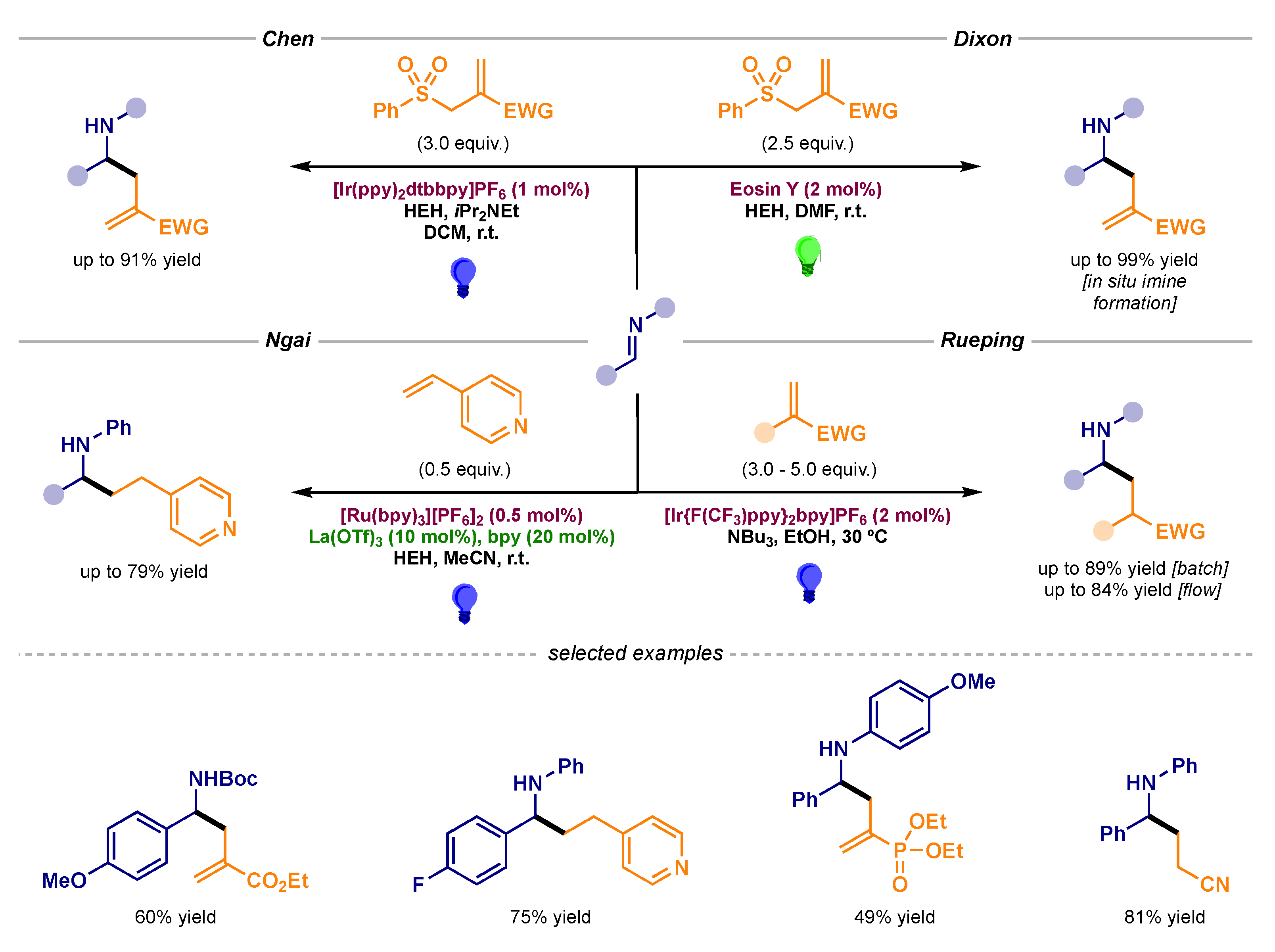
The development of the single-electron reduction of imines to produce new radical–radical couplings has undoubtedly revamped this area. Furthermore, the polarity reversal displayed in this redox process served as a platform upon which an even greater challenge could be tackled. The new nucleophilic character of the α-amino radical was exploited in a series of Giese radical additions (pathway B2 in Scheme 1) reported by several groups. Indeed, Chen [44], Dixon [45,46], Ngai [47], and Rueping [48] independently worked on the addition of these intermediates to different activated olefins (Scheme 16).
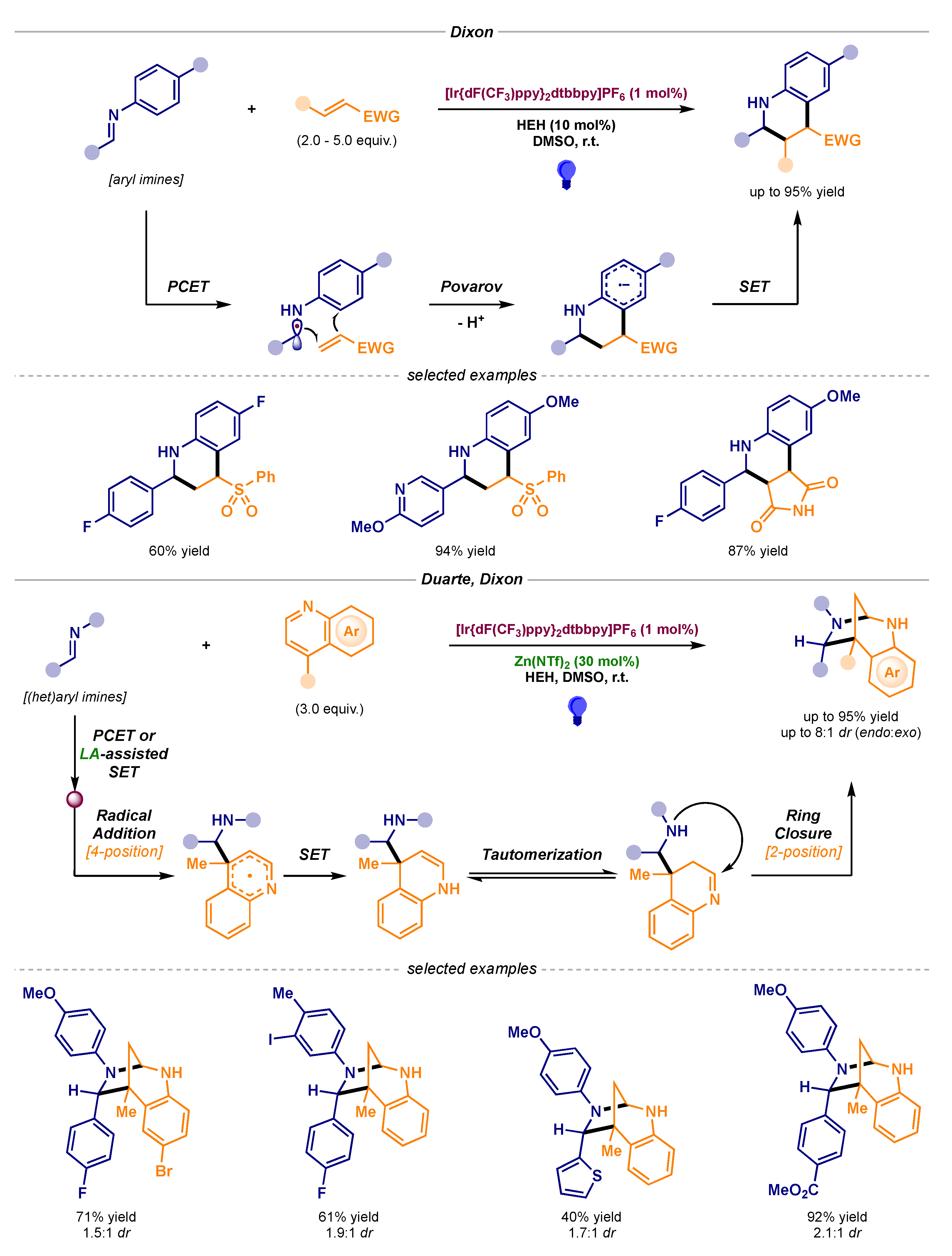
In addition, Dixon’s lab described two procedures built on these radical additions to electrophilic partners followed by cyclization events, thus granting access to molecules of higher structural complexity (Scheme 17) [49,50]. Notably, the addition of the α-amino radical to vinyl sulfones led to a reverse polarity Povarov reaction (Scheme 17, top), while bridged 1,3-diazepanes could be prepared via α-amino radical addition to the 4-position of a quinoline core and subsequent ring closure at the 2-position (Scheme 17, bottom).
3.3. Racemic α-Amino Carbanion Nucleophilic Attacks—Pathway B3
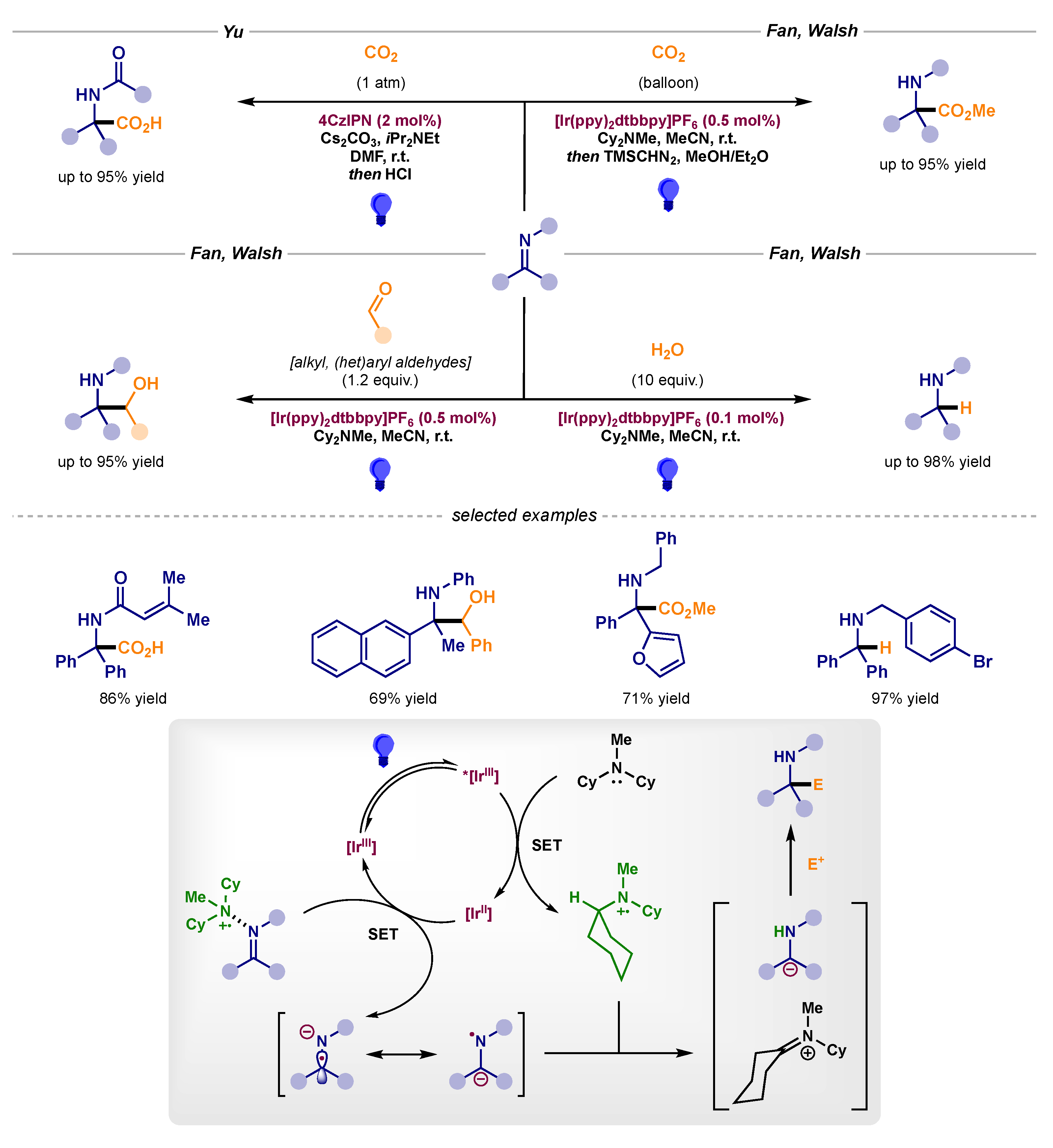
After thorough evaluation of the results outlined above, it could be assumed that the single-electron reduction of imine scaffolds usually delivers a radical anion that can exist as two different resonant forms. As mentioned previously, the N-radical resonant form can engage in an HAT to yield a stable carbanion (pathway B3 in Scheme 1). This behavior has been exploited by Yu and Fan and Walsh to achieve polar nucleophilic attacks on numerous electrophiles, such as CO2 and aldehydes, as well as the direct hydrolysis of these anionic intermediates to afford the formal reduction to benzyl amines (Scheme 18) [51,52,53,54].
3.4. Stereoselective Photocatalytic α-Amino Radical Reactivity via Single-Electron Reduction of Imine Derivatives—Pathway B
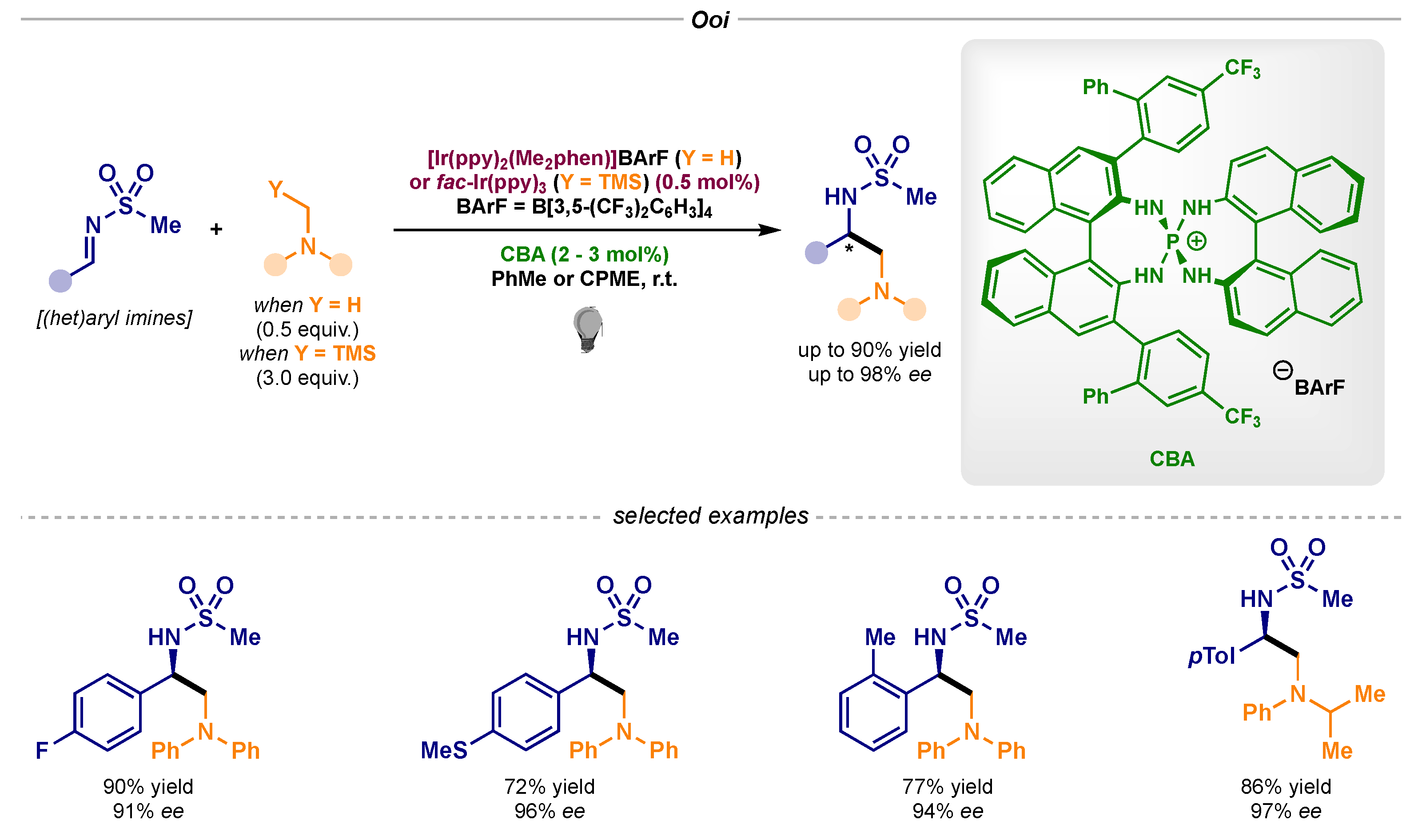
Controlling the stereochemical outcome of transformations involving the single-electron reduction of imines has proven to be an outstanding challenge. An exceptional solution to this problem was developed by Ooi’s lab in 2015 and 2016 (Scheme 19) [55,56], wherein the radical anion derived from this redox process was ion-paired with a chiral phosphonium salt, rendering an asymmetric radical–radical coupling with α-amino radicals (pathway B1 in Scheme 1).
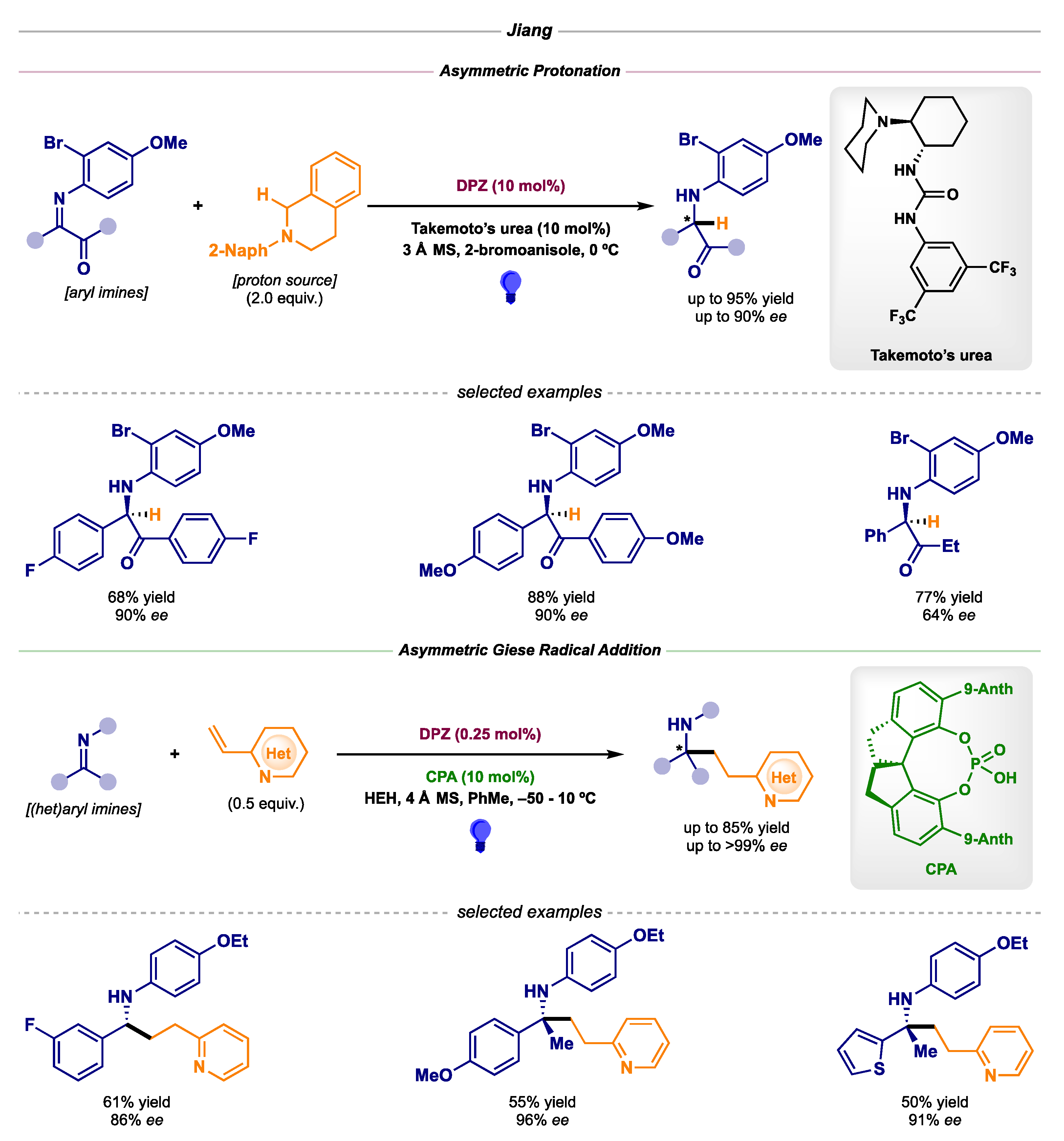
Moreover, Jiang’s lab reported two transformations involving the use of traditional organocatalysts (Scheme 20) [57,58]. In the first one, the formal reduction of activated ketimines was achieved via coordination of Takemoto’s urea catalyst to the radical anion and ensuing asymmetric HAT (pathway B1 in Scheme 1). In the second one, a chiral Brønsted acid managed to activate vinyl pyridines—acting as acceptors in a Giese radical addition—while exerting stereocontrol through H-bond interactions (pathway B2 in Scheme 1).
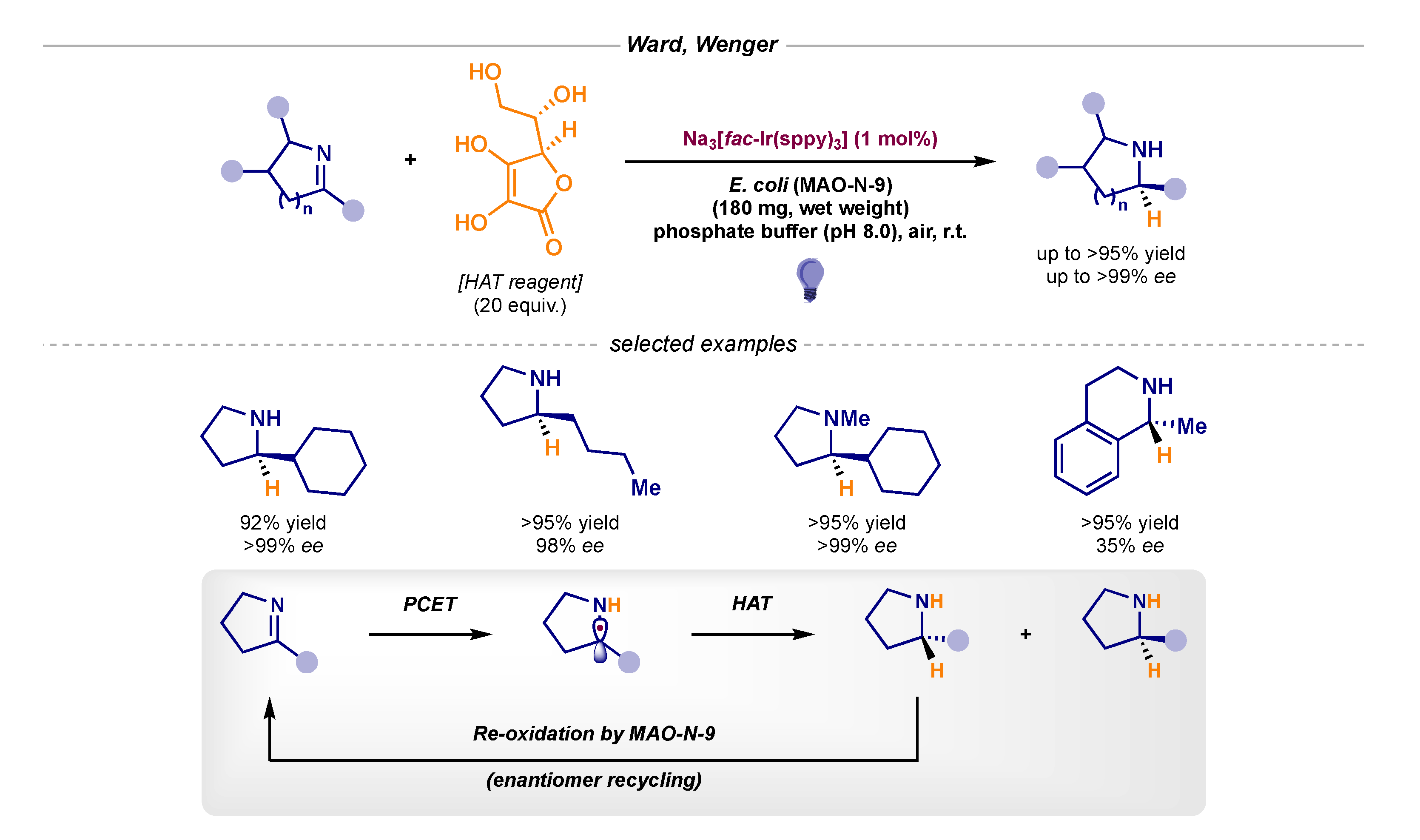
Finally, Ward and Wenger published an interesting asymmetric reduction of cyclic imines in which enzymatic catalysis played a fundamental role (Scheme 21) [59]. Initial photocatalytic reduction and HAT would deliver a racemic mixture of both amines (pathway B1 in Scheme 1), yet, in the presence of monoamine oxidase MAO-N-9, only one enantiomer could undergo subsequent re-oxidation (enantiomer recycling). Therefore, the combination of photocatalysis and biocatalysis yielded an elegant dynamic kinetic resolution (DKR) en route to chiral amines.
In this section, the pool of radical precursors employed in reactions following pathway B1 may not be as diverse when compared to conventional radical additions (pathway A). However, thanks to the nucleophilic character of the imine-derived radical anion, pathways B2 and B3 have widened the scope of reacting partners since they no longer require photocatalytic activation. Remarkably, common electrophiles used in polar methodologies find a smooth transition into these new radical processes thanks to the inspired polarity reversal enforced upon the imine substrates.
4. Conclusions
The development of new methodologies to perform the α-functionalization of imine building blocks has proven to be a subject of intense research during the past decade, as numerous approaches featuring outstanding versatility have surfaced in the context of visible light photoredox catalysis. Most importantly, the complimentary nature to the different photocatalytic strategies employed in imine chemistry has provided immense flexibility, since the imine reagent can now be used as both an electrophile and, strikingly, a nucleophile. This multifaceted behavior has delivered a wide array of racemic transformations in this area. However, asymmetric functionalization of the C=N moiety has remained a great challenge in photoredox catalysis. Nevertheless, brilliant activation strategies have been deployed to achieve stereoselectivity, although an increase in generality and modularity can be expected as the field continues to grow.
Author Contributions
Conceptualization, A.F.G.-C. and M.C.M.; literature search, A.F.G.-C. and M.C.M.; writing—original draft preparation, A.F.G.-C.; writing—review and editing, A.F.G.-C., M.C.M., and J.A.; supervision, M.C.M. and J.A.; project administration, M.C.M. and J.A.; funding acquisition, M.C.M. and J.A. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the European Research Council, grant number 647550; the Spanish Government, grant number RTI2018-095038-B-I00; the ‘‘Comunidad de Madrid’’ and European Structural Funds, grant number S2018/NMT-4367.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
For uncommon abbreviations outlined throughout the manuscript, see the following list:
| abbreviation | full description |
| 4CzIPN | 2,4,5,6-tetra(9H-carbazol-9-yl)isophthalonitrile |
| Ac | acyl |
| Alk | alkyl |
| Anth | anthracenyl |
| Ar | aryl |
| BArF | B[3,5-(CF3)2C6H3]4 |
| BD | 2,3-butanedione |
| Boc | tert-butyloxycarbonyl |
| BOX | bis(oxazoline) |
| bpy | 2,2′-bipyridine |
| Bu | butyl |
| Bz | benzoyl |
| CBA | chiral Brønsted acid |
| CPA | chiral phosphoric acid |
| CPME | cyclopentyl methyl ether |
| Cy | cyclohexyl |
| DABCO | 1,4-diazabicyclo[2.2.2]octane |
| DCM | dichloromethane |
| DFMS | zinc difluoromethanesulfinate |
| DKR | dynamic kinetic resolution |
| DMA | N,N-dimethylacetamide |
| DMF | N,N-dimethylformamide |
| DMPU | N,N’-dimethylpropyleneurea |
| DMSO | dimethylsulfoxide |
| DPZ | 5,6-bis(5-methoxythiophen-2-yl)pyrazine-2,3-dicarbonitrile |
| dr | diastereomeric ratio |
| dtbbpy | 4,4′-di-tert-butyl-2,2′-dipyridyl |
| E | electrophile |
| ee | enantiomeric excess |
| Et | ethyl |
| EWG | electron withdrawing group |
| HAT | hydrogen atom transfer |
| HEH | Hantzsch ester |
| Het | heteroaryl |
| IFET | interfacial electron transfer |
| iPr | iso-propyl |
| MAO | monoamine oxidase |
| Me | methyl |
| Mes | mesityl |
| MS | molecular sieves |
| Naph | naphthyl |
| NHPI | N-hydroxyphthalimide |
| NHS | N-hydroxysuccinimide |
| PCET | proton-coupled electron transfer |
| Ph | phenyl |
| phen | 1,10-phenanthroline |
| ppy | 2-phenylpyridine |
| PT | 5,7,12,14-pentacenetetrone |
| pTol | para-tolyl |
| QD | quantum dot |
| RAE | redox-active ester |
| RPC | radical polar crossover |
| SET | single-electron transfer |
| SLAP | silicon amine protocol |
| SN2 | bimolecular nucleophilic substitution |
| sppy | 3-(pyridin-2-yl)benzenesulfonate |
| TBS | tert-butyldimethylsilyl |
| TCNHPI | N-hydroxytetrachlorophthalimide |
| Tf | trifluoromethanesulfonyl |
| TFA | trifluoroacetic acid |
| TFE | 2,2,2-trifluoroethanol |
| THF | tetrahydrofuran |
| TMS | trimethylsilyl |
| TPP | 2,4,6-triphenylpyrylium tetrafluoroborate |
References
- Visible Light Photocatalysis in Organic Chemistry, 1st ed; Stephenson, C.R.J.; Yoon, T.P.; MacMillan, D.W.C. (Eds.) Wiley-VCH: Weinheim, Germany, 2018. [Google Scholar]
- Matsui, J.K.; Lang, S.B.; Heitz, D.R.; Molander, G.A. Photoredox-Mediated Routes to Radicals: The Value of Catalytic Radical Generation in Synthetic Methods Development. ACS Catal. 2017, 7, 2563–2575. [Google Scholar] [CrossRef]
- Lee, K.N.; Ngai, M.-Y. Recent developments in transition-metal photoredox-catalysed reactions of carbonyl derivatives. Chem. Commun. 2017, 53, 13093–13112. [Google Scholar] [CrossRef] [PubMed]
- Leitch, J.A.; Rossolini, T.; Rogova, T.; Maitland, J.A.P.; Dixon, D.J. α-Amino Radicals via Photocatalytic Single-Electron Reduction of Imine Derivatives. ACS Catal. 2020, 10, 2009–2025. [Google Scholar] [CrossRef]
- Hsieh, S.-Y.; Bode, J.W. Silicon Amine Reagents for the Photocatalytic Synthesis of Piperazines from Aldehydes and Ketones. Org. Lett. 2016, 18, 2098–2101. [Google Scholar] [CrossRef] [PubMed]
- Jackl, M.K.; Legnani, L.; Morandi, B.; Bode, J.W. Continuous Flow Synthesis of Morpholines and Oxazepanes with Silicon Amine Protocol (SLAP) Reagents and Lewis Acid Facilitated Photoredox Catalysis. Org. Lett. 2017, 19, 4696–4699. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, S.-Y.; Bode, J.W. Lewis Acid Induced Toggle from Ir(II) to Ir(IV) Pathways in Photocatalytic Reactions: Synthesis of Thiomorpholines and Thiazepanes from Aldehydes and SLAP Reagents. ACS Cent. Sci. 2017, 3, 66–72. [Google Scholar] [CrossRef]
- Patel, N.R.; Kelly, C.B.; Siegenfeld, A.P.; Molander, G.A. Mild, Redox-Neutral Alkylation of Imines Enabled by an Organic Photocatalyst. ACS Catal. 2017, 7, 1766–1770. [Google Scholar] [CrossRef]
- Prieto, A.; Bouyssi, D.; Monteiro, N. Radical-Mediated Formal C(sp2)–H Functionalization of Aldehyde-Derived N,N-Dialkylhydrazones. Eur. J. Org. Chem. 2018, 2378–2393. [Google Scholar] [CrossRef]
- Cullen, S.T.J.; Friestad, G.K. Alkyl Radical Addition to Aliphatic and Aromatic N-Acylhydrazones Using an Organic Photoredox Catalyst. Org. Lett. 2019, 21, 8290–8294. [Google Scholar] [CrossRef]
- Pantaine, L.R.E.; Milligan, J.A.; Matsui, J.K.; Kelly, C.B.; Molander, G.A. Photoredox Radical/Polar Crossover Enables Construction of Saturated Nitrogen Heterocycles. Org. Lett. 2019, 21, 2317–2321. [Google Scholar] [CrossRef]
- Plasko, D.P.; Jordan, C.J.; Ciesa, B.E.; Merrill, M.A.; Hanna, J.M., Jr. Visible light-promoted alkylation of imines using potassium organotrifluoroborates. Photochem. Photobiol. Sci. 2018, 17, 534–538. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.; Badir, S.O.; Alam, R.; Molander, G.A. Photoredox-Catalyzed Multicomponent Petasis Reaction with Alkyltrifluoroborates. Org. Lett. 2019, 21, 4853–4858. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Wu, Q.-L.; Xie, Y.; Weng, J.; Lu, G. Visible-Light-Mediated Decarboxylative Benzylation of Imines with Arylacetic Acids. J. Org. Chem. 2018, 83, 12559–12567, The reaction pathway could not be confirmed; both the radical addition and the radical–radical coupling are most likely taking place. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.-H.; Yu, S. Radical Alkylation of Imines with 4-Alkyl-1,4-dihydropyridines Enabled by Photoredox/Brønsted Acid Cocatalysis. J. Org. Chem. 2017, 82, 9995–10006, The reaction pathway could not be confirmed; both the radical addition and the radical–radical coupling are most likely taking place. [Google Scholar] [CrossRef]
- Ji, P.; Zhang, Y.; Wei, Y.; Huang, H.; Hu, W.; Mariano, P.A.; Wang, W. Visible-Light-Mediated, Chemo- and Stereoselective Radical Process for the Synthesis of C-Glycoamino Acids. Org. Lett. 2019, 21, 3086–3092. [Google Scholar] [CrossRef]
- Jia, J.; Lefebvre, Q.; Rueping, M. Reductive coupling of imines with redox-active esters by visible light photoredox organocatalysis. Org. Chem. Front. 2020, 7, 602–608. [Google Scholar] [CrossRef]
- Garrido-Castro, A.F.; Gini, A.; Maestro, M.C.; Alemán, J. Unlocking the direct photocatalytic difluoromethylation of C=N bonds. Chem. Commun. 2020, 56, 3769–3772. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Zhu, S.; Lu, D.; Gong, Y. Formylation of Fluoroalkyl Imines through Visible-Light-Enabled H-Atom Transfer Catalysis: Access to Fluorinated α-Amino Aldehydes. Org. Lett. 2019, 21, 2019–2024. [Google Scholar] [CrossRef]
- For a photoinduced α-oxyalkylation of imines generated in situ, see: Zhang, L.; Deng, Y.; Shi, F. Light promoted aqueous phase amine synthesis via three-component coupling reactions. Tetrahedron Lett. 2013, 54, 5217–5219. [Google Scholar]
- Supranovich, V.I.; Levin, V.V.; Dilman, A.D. Radical Addition to N-Tosylimines via C−H Activation Induced by Decatungstate Photocatalyst. Org. Lett. 2019, 21, 4271–4274. [Google Scholar] [CrossRef]
- For an inverse hydroboration of imines wherein the boryl radical is generated from N-Heterocyclic Carbene (NHC) boranes via HAT, see: Zhou, N.; Yuan, X.-A.; Zhao, Y.; Xie, J.; Zhu, C. Synergistic Photoredox Catalysis and Organocatalysis for Inverse Hydroboration of Imines. Angew. Chem., Int. Ed. 2018, 57, 3990–3994, The reaction pathway could not be confirmed; both the radical addition and the radical–radical coupling are most likely taking place. [Google Scholar]
- Zhang, H.-H.; Yu, S. Visible-Light-Induced Radical Acylation of Imines with α-Ketoacids Enabled by Electron-Donor−Acceptor Complexes. Org. Lett. 2019, 21, 3711–3715, A radical–radical coupling is most likely taking place. [Google Scholar] [CrossRef] [PubMed]
- Rono, L.J.; Yayla, H.G.; Wang, D.Y.; Armstrong, M.F.; Knowles, R.R. Enantioselective Photoredox Catalysis Enabled by Proton-Coupled Electron Transfer: Development of an Asymmetric Aza-Pinacol Cyclization. J. Am. Chem. Soc. 2013, 135, 17735–17738. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Castro, A.F.; Choubane, H.; Daaou, M.; Maestro, M.C.; Alemán, J. Asymmetric radical alkylation of N-sulfinimines under visible light photocatalytic conditions. Chem. Commun. 2017, 53, 7764–7767. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Zhou, K.; Wen, Z.; Cao, S.; Shen, X.; Lei, M.; Gong, L. Copper(II)-Catalyzed Asymmetric Photoredox Reactions: Enantioselective Alkylation of Imines Driven by Visible Light. J. Am. Chem. Soc. 2018, 140, 15850–15858. [Google Scholar] [CrossRef]
- Han, B.; Li, Y.; Yu, Y.; Gong, L. Photocatalytic enantioselective α-aminoalkylation of acyclic imine derivatives by a chiral copper catalyst. Nat. Commun. 2019, 10, 3804–3812. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Lei, M.; Gong, L. Photocatalytic regio- and stereoselective C(sp3)–H functionalization of benzylic and allylic hydrocarbons as well as unactivated alkanes. Nat. Catal. 2019, 2, 1016–1026, The reaction pathway could not be confirmed; both the radical addition and the radical–radical coupling are most likely taking place. [Google Scholar] [CrossRef]
- Schindler, W.; Knoch, F.; Kisch, H. Semiconductor-Catalysed Photoaddition: γ,δ-Unsaturated Amines from Cyclopentene and Schiff Bases. Chem. Ber. 1996, 129, 925–932. [Google Scholar] [CrossRef]
- Keck, H.; Schindler, W.; Knoch, F.; Kisch, H. Type B Semiconductor Photocatalysis: The Synthesis of Homoallyl Amines by Cadmium Sulfide-Catalyzed Linear Photoaddition of Olefins and Enol/Allyl Ethers to N-Phenylbenzophenone Imine. Chem. Eur. J. 1997, 3, 1638–1645. [Google Scholar] [CrossRef]
- Kisch, H. Semiconductor Photocatalysis for Chemoselective Radical Coupling Reactions. Acc. Chem. Res. 2017, 50, 1002–1010. [Google Scholar] [CrossRef]
- Xi, Z.-W.; Yang, L.; Wang, D.-Y.; Pu, C.-D.; Shen, Y.-M.; Wu, C.-D.; Peng, X.-G. Visible-Light Photocatalytic Synthesis of Amines from Imines via Transfer Hydrogenation Using Quantum Dots as Catalysts. J. Org. Chem. 2018, 83, 11886–11895. [Google Scholar] [CrossRef] [PubMed]
- Van As, D.J.; Connell, T.U.; Brzozowski, M.; Scully, A.D.; Polyzos, A. Photocatalytic and Chemoselective Transfer Hydrogenation of Diarylimines in Batch and Continuous Flow. Org. Lett. 2018, 20, 905–908. [Google Scholar] [CrossRef] [PubMed]
- Hager, D.; MacMillan, D.W.C. Activation of C−H Bonds via the Merger of Photoredox and Organocatalysis: A Coupling of Benzylic Ethers with Schiff Bases. J. Am. Chem. Soc. 2014, 136, 16986–16989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeffrey, J.L.; Petronijević, F.R.; MacMillan, D.W.C. Selective Radical−Radical Cross-Couplings: Design of a Formal β-Mannich Reaction. J. Am. Chem. Soc. 2015, 137, 8404–8407. [Google Scholar] [CrossRef] [Green Version]
- Nakajima, M.; Fava, E.; Loescher, S.; Jiang, Z.; Rueping, M. Photoredox-Catalyzed Reductive Coupling of Aldehydes, Ketones, and Imines with Visible Light. Angew. Chem., Int. Ed. 2015, 54, 8828–8832. [Google Scholar] [CrossRef]
- Fava, E.; Millet, A.; Nakajima, M.; Loescher, S.; Rueping, M. Reductive Umpolung of Carbonyl Derivatives with Visible-Light Photoredox Catalysis: Direct Access to Vicinal Diamines and Amino Alcohols via α-Amino Radicals and Ketyl Radicals. Angew. Chem., Int. Ed. 2016, 55, 6776–6779. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, S.; Kojiyama, K.; Tsujioka, H.; Sudo, A. Metal-free reductive coupling of C=O and C=N bonds driven by visible light: Use of perylene as a simple photoredox catalyst. Chem. Commun. 2016, 52, 11339–11342. [Google Scholar] [CrossRef]
- Rong, J.; Seeberger, P.H.; Gilmore, K. Chemoselective Photoredox Synthesis of Unprotected Primary Amines Using Ammonia. Org. Lett. 2018, 20, 4081–4085. [Google Scholar] [CrossRef]
- Xia, Q.; Tian, H.; Dong, J.; Qu, Y.; Li, L.; Song, H.; Liu, Y.; Wang, Q. N-Arylamines Coupled with Aldehydes, Ketones, and Imines by Means of Photocatalytic Proton-Coupled Electron Transfer. Chem. Eur. J. 2018, 24, 9269–9273. [Google Scholar] [CrossRef]
- Kammer, L.M.; Krumb, M.; Spitzbarth, B.; Lipp, B.; Kühlborn, J.; Busold, J.; Mulina, O.M.; Terentev, A.O.; Opatz, T. Photoredox-Catalyzed Four-Component Reaction for the Synthesis of Complex Secondary Amines. Org. Lett. 2020, 22, 3318–3322. [Google Scholar] [CrossRef]
- Chen, M.; Zhao, X.; Yang, C.; Xia, W. Visible-Light-Triggered Directly Reductive Arylation of Carbonyl/Iminyl Derivatives through Photocatalytic PCET. Org. Lett. 2017, 19, 3807–3810. [Google Scholar] [CrossRef] [PubMed]
- Nicastri, M.C.; Lehnherr, D.; Lam, Y.; DiRocco, D.A.; Rovis, T. Synthesis of Sterically Hindered Primary Amines by Concurrent Tandem Photoredox Catalysis. J. Am. Chem. Soc. 2020, 142, 987–998. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.; Chen, Y. Polarity-Reversed Allylations of Aldehydes, Ketones, and Imines Enabled by Hantzsch Ester in Photoredox Catalysis. Angew. Chem., Int. Ed. 2016, 55, 13312–13315. [Google Scholar] [CrossRef] [PubMed]
- Fuentes de Arriba, A.L.; Urbitsch, F.; Dixon, D.J. Umpolung synthesis of branched α-functionalized amines from imines via photocatalytic three-component reductive coupling reactions. Chem. Commun. 2016, 52, 14434–14437. [Google Scholar] [CrossRef] [Green Version]
- Rossolini, T.; Leitch, J.A.; Grainger, R.; Dixon, D.J. Photocatalytic Three-Component Umpolung Synthesis of 1,3-Diamines. Org. Lett. 2018, 20, 6794–6798. [Google Scholar] [CrossRef]
- Lee, K.N.; Lei, Z.; Ngai, M.-Y. β-Selective Reductive Coupling of Alkenylpyridines with Aldehydes and Imines via Synergistic Lewis Acid/Photoredox Catalysis. J. Am. Chem. Soc. 2017, 139, 5003–5006. [Google Scholar] [CrossRef]
- Lefebvre, Q.; Porta, R.; Millet, A.; Jia, J.; Rueping, M. One Amine–3 Tasks: Reductive Coupling of Imines with Olefins in Batch and Flow. Chem. Eur. J. 2020, 26, 1363–1367. [Google Scholar] [CrossRef]
- Leitch, J.A.; Fuentes de Arriba, A.L.; Tan, J.; Hoff, O.; Martínez, C.M.; Dixon, D.J. Photocatalytic reverse polarity Povarov reaction. Chem. Sci. 2018, 9, 6653–6658. [Google Scholar] [CrossRef] [Green Version]
- Leitch, J.A.; Rogova, T.; Duarte, F.; Dixon, D.J. Dearomative Photocatalytic Construction of Bridged 1,3-Diazepanes. Angew. Chem., Int. Ed. 2020, 59, 4121–4130. [Google Scholar] [CrossRef]
- Ju, T.; Fu, Q.; Ye, J.-H.; Zhang, Z.; Liao, L.-L.; Yan, S.-S.; Tian, X.-Y.; Luo, S.-P.; Li, J.; Yu, D.-G. Selective and Catalytic Hydrocarboxylation of Enamides and Imines with CO2 to Generate α,α-Disubstituted α-Amino Acids. Angew. Chem., Int. Ed. 2018, 57, 13897–13901. [Google Scholar] [CrossRef]
- Fan, X.; Gong, X.; Ma, M.; Wang, R.; Walsh, P.J. Visible light-promoted CO2 fixation with imines to synthesize diaryl α-amino acids. Nat. Commun. 2018, 9, 4936–4943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.; Ma, M.; Gong, X.; Fan, X.; Walsh, P.J. Reductive Cross-Coupling of Aldehydes and Imines Mediated by Visible Light Photoredox Catalysis. Org. Lett. 2019, 21, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Ma, M.; Gong, X.; Panetti, G.B.; Fan, X.; Walsh, P.J. Visible-Light-Mediated Umpolung Reactivity of Imines: Ketimine Reductions with Cy2NMe and Water. Org. Lett. 2018, 20, 2433–2436. [Google Scholar] [CrossRef] [PubMed]
- Uraguchi, D.; Kinoshita, N.; Kizu, T.; Ooi, T. Synergistic Catalysis of Ionic Brønsted Acid and Photosensitizer for a Redox Neutral Asymmetric α-Coupling of N-Arylaminomethanes with Aldimines. J. Am. Chem. Soc. 2015, 137, 13768–13771. [Google Scholar] [CrossRef]
- Kizu, T.; Uraguchi, D.; Ooi, T. Independence from the Sequence of Single-Electron Transfer of Photoredox Process in Redox-Neutral Asymmetric Bond-Forming Reaction. J. Org. Chem. 2016, 81, 6953–6958. [Google Scholar] [CrossRef]
- Lin, L.; Bai, X.; Ye, X.; Zhao, X.; Tan, C.-H.; Jiang, Z. Organocatalytic Enantioselective Protonation for Photoreduction of Activated Ketones and Ketimines Induced by Visible Light. Angew. Chem., Int. Ed. 2017, 56, 13842–13846. [Google Scholar] [CrossRef]
- Cao, K.; Tan, S.M.; Lee, R.; Yang, S.; Jia, H.; Zhao, X.; Qiao, B.; Jiang, Z. Catalytic Enantioselective Addition of Prochiral Radicals to Vinylpyridines. J. Am. Chem. Soc. 2019, 141, 5437–5443. [Google Scholar] [CrossRef]
- Guo, X.; Okamoto, Y.; Schreier, M.R.; Ward, T.R.; Wenger, O.S. Enantioselective synthesis of amines by combining photoredox and enzymatic catalysis in a cyclic reaction network. Chem. Sci. 2018, 9, 5052–5056. [Google Scholar] [CrossRef] [Green Version]
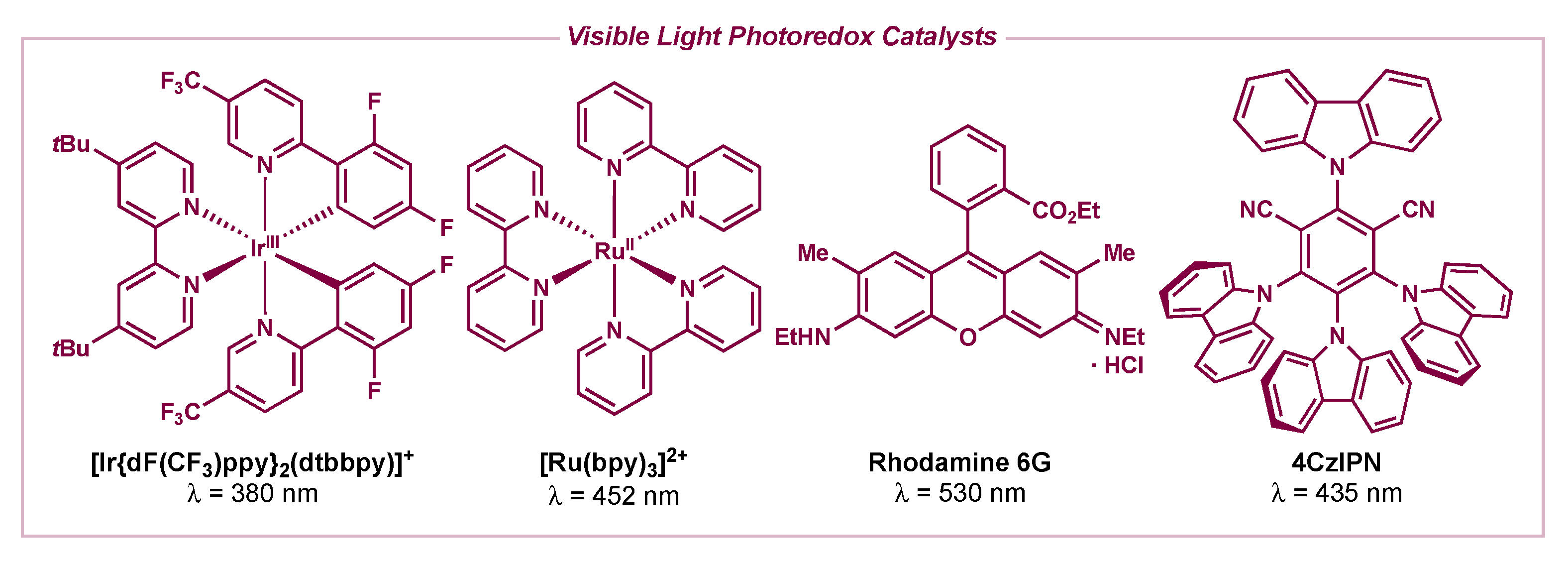
Figure 1.
Visible light photoredox catalysts (λ = local absorbance maximum for lowest energy absorption).
Figure 1.
Visible light photoredox catalysts (λ = local absorbance maximum for lowest energy absorption).

Scheme 1.
Photocatalytic functionalization of imines: pathway A, alkyl radical addition (left); pathway B, single-electron reduction (right).
Scheme 1.
Photocatalytic functionalization of imines: pathway A, alkyl radical addition (left); pathway B, single-electron reduction (right).

Scheme 2.
Cyclization of SLAP reagents developed by Bode for the synthesis of piperazines (left) and morpholines, oxazepanes, thiomorpholines and thiazepanes (right).
Scheme 2.
Cyclization of SLAP reagents developed by Bode for the synthesis of piperazines (left) and morpholines, oxazepanes, thiomorpholines and thiazepanes (right).

Scheme 3.
Intermolecular photocatalytic radical additions to imines using alkyl silicates developed by Molander (top left), Friestad (top right) and Kelly and Molander (bottom).
Scheme 3.
Intermolecular photocatalytic radical additions to imines using alkyl silicates developed by Molander (top left), Friestad (top right) and Kelly and Molander (bottom).

Scheme 4.
Photocatalytic radical additions to imines using alkyl trifluoroborates developed by Hanna, Jr (left) and Molander (right).
Scheme 4.
Photocatalytic radical additions to imines using alkyl trifluoroborates developed by Hanna, Jr (left) and Molander (right).

Scheme 5.
Photocatalytic radical additions to imines using alkyl carboxylic acids developed by Weng and Lu (left) and Mariano and Wang (right).
Scheme 5.
Photocatalytic radical additions to imines using alkyl carboxylic acids developed by Weng and Lu (left) and Mariano and Wang (right).

Scheme 6.
Photocatalytic difluoromethyl radical addition to imines using DFMS developed by Maestro and Alemán.
Scheme 6.
Photocatalytic difluoromethyl radical addition to imines using DFMS developed by Maestro and Alemán.

Scheme 7.
Photocatalytic radical additions to imines using HAT developed by Lu and Gong (top) and Dilman (bottom).
Scheme 7.
Photocatalytic radical additions to imines using HAT developed by Lu and Gong (top) and Dilman (bottom).

Scheme 8.
Asymmetric intramolecular photocatalytic radical addition to hydrazones developed by Knowles.
Scheme 8.
Asymmetric intramolecular photocatalytic radical addition to hydrazones developed by Knowles.

Scheme 9.
Asymmetric intermolecular photocatalytic radical addition to N-sulfinimines developed by Maestro and Alemán.
Scheme 9.
Asymmetric intermolecular photocatalytic radical addition to N-sulfinimines developed by Maestro and Alemán.

Scheme 10.
Asymmetric intermolecular photocatalytic reactions developed by Gong: Cu-photocatalyzed radical additions (top) and HAT-photocatalyzed C-H activation (bottom).
Scheme 10.
Asymmetric intermolecular photocatalytic reactions developed by Gong: Cu-photocatalyzed radical additions (top) and HAT-photocatalyzed C-H activation (bottom).

Scheme 11.
Heterogeneous photocatalytic α-amino radical–radical couplings developed by Kisch (top) and Pu and Shen (bottom).
Scheme 11.
Heterogeneous photocatalytic α-amino radical–radical couplings developed by Kisch (top) and Pu and Shen (bottom).

Scheme 12.
Photocatalytic α-amino radical–radical couplings (top) and mechanistic proposal for the formal β-Mannich reaction (bottom) developed by MacMillan.
Scheme 12.
Photocatalytic α-amino radical–radical couplings (top) and mechanistic proposal for the formal β-Mannich reaction (bottom) developed by MacMillan.

Scheme 13.
Photocatalytic α-amino radical–radical couplings developed by Rueping.

Scheme 14.
Photocatalytic α-amino radical-benzyl radical coupling developed by Opatz.

Scheme 15.
Photocatalytic α-amino radical-aryl radical couplings developed by Xia (left) and Lehnherr and Rovis (right).
Scheme 15.
Photocatalytic α-amino radical-aryl radical couplings developed by Xia (left) and Lehnherr and Rovis (right).

Scheme 16.
Photocatalytic α-amino radical additions to activated olefins developed by Chen (top left), Dixon (top right), Ngai (bottom left), and Rueping (bottom right).
Scheme 16.
Photocatalytic α-amino radical additions to activated olefins developed by Chen (top left), Dixon (top right), Ngai (bottom left), and Rueping (bottom right).

Scheme 17.
Photocatalytic reactions of α-amino radicals for the synthesis of polycyclic structures developed by Dixon: reverse polarity Povarov reaction (top) and bridged 1,3-diazepane construction (bottom).
Scheme 17.
Photocatalytic reactions of α-amino radicals for the synthesis of polycyclic structures developed by Dixon: reverse polarity Povarov reaction (top) and bridged 1,3-diazepane construction (bottom).

Scheme 18.
Photocatalytic carbanion additions to electrophiles developed by Yu and Fan and Walsh: carboxylation (top), reaction with aldehydes (middle left) and hydrolysis (middle right).
Scheme 18.
Photocatalytic carbanion additions to electrophiles developed by Yu and Fan and Walsh: carboxylation (top), reaction with aldehydes (middle left) and hydrolysis (middle right).

Scheme 19.
Asymmetric photocatalytic α-amino radical–radical couplings developed by Ooi.

Scheme 20.
Asymmetric photocatalytic α-amino radical-mediated reactions developed by Jiang: asymmetric protonation (top) and asymmetric Giese radical addition (bottom).
Scheme 20.
Asymmetric photocatalytic α-amino radical-mediated reactions developed by Jiang: asymmetric protonation (top) and asymmetric Giese radical addition (bottom).

Scheme 21.
Asymmetric bio- and photo-catalytic reduction of cyclic imines developed by Ward and Wenger.
Scheme 21.
Asymmetric bio- and photo-catalytic reduction of cyclic imines developed by Ward and Wenger.

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Garrido-Castro, A.F.; Maestro, M.C.; Alemán, J. α-Functionalization of Imines via Visible Light Photoredox Catalysis. Catalysts 2020, 10, 562. https://0-doi-org.brum.beds.ac.uk/10.3390/catal10050562
AMA Style
Garrido-Castro AF, Maestro MC, Alemán J. α-Functionalization of Imines via Visible Light Photoredox Catalysis. Catalysts. 2020; 10(5):562. https://0-doi-org.brum.beds.ac.uk/10.3390/catal10050562
Chicago/Turabian StyleGarrido-Castro, Alberto F., M. Carmen Maestro, and José Alemán. 2020. "α-Functionalization of Imines via Visible Light Photoredox Catalysis" Catalysts 10, no. 5: 562. https://0-doi-org.brum.beds.ac.uk/10.3390/catal10050562
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.