Effect of Calcination Time on the Physicochemical Properties and Photocatalytic Performance of Carbon and Nitrogen Co-Doped TiO2 Nanoparticles

 ,
, .jpg) , , , ,
, , , ,  , ,
, ,  and
and
Abstract
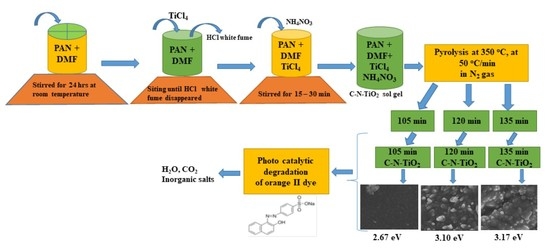
:
1. Introduction
2. Results
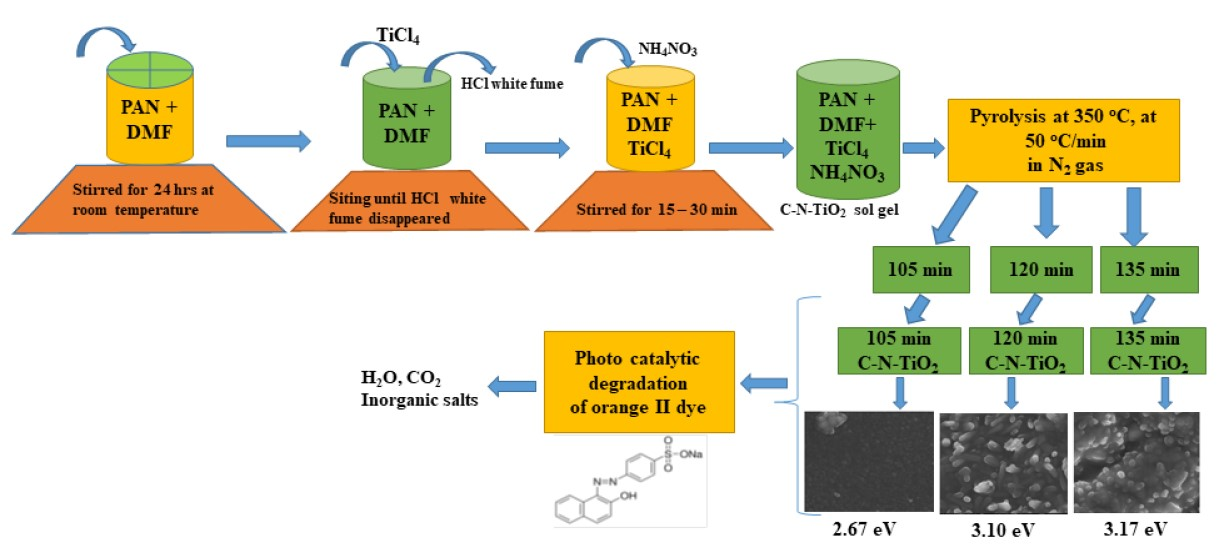
2.1. X-Ray Diffraction and Raman Spectroscopy Analysis of the Synthesized Catalysts
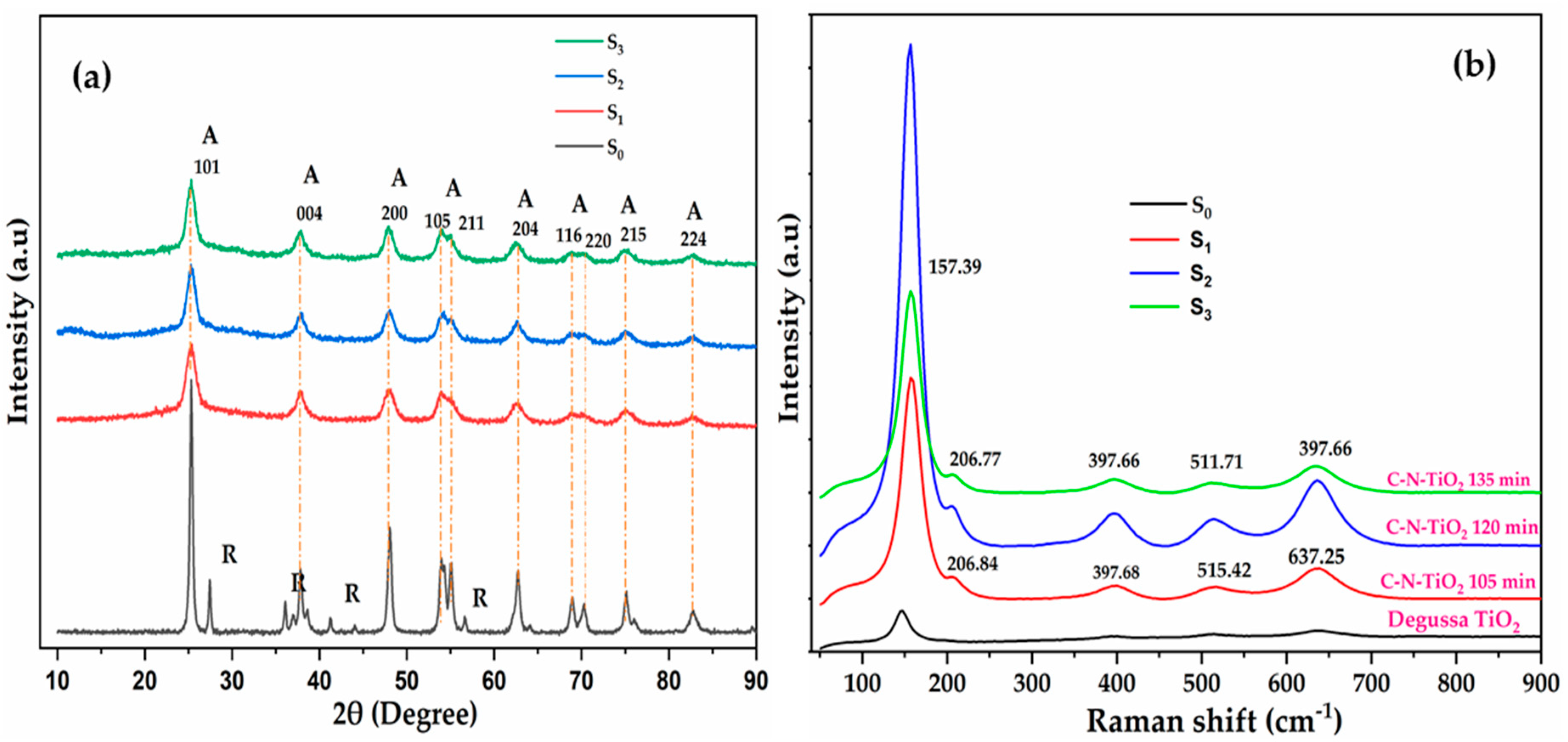
2.2. Fourier Transform Spectroscopy of the Catalysts
2.3. Thermal Gravimetric Analysis of the Synthesised Catalysts
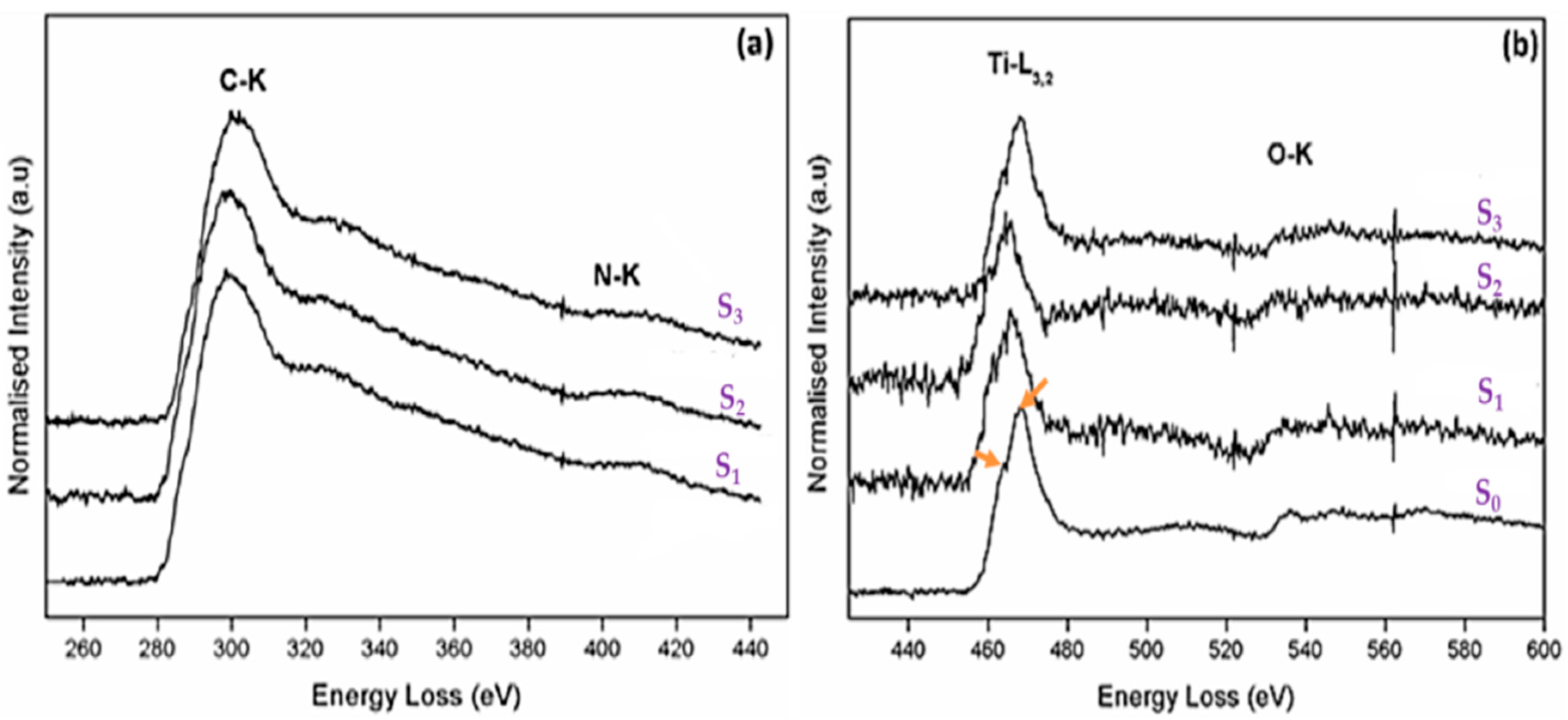
2.4. Electron Energy Loss Spectroscopy
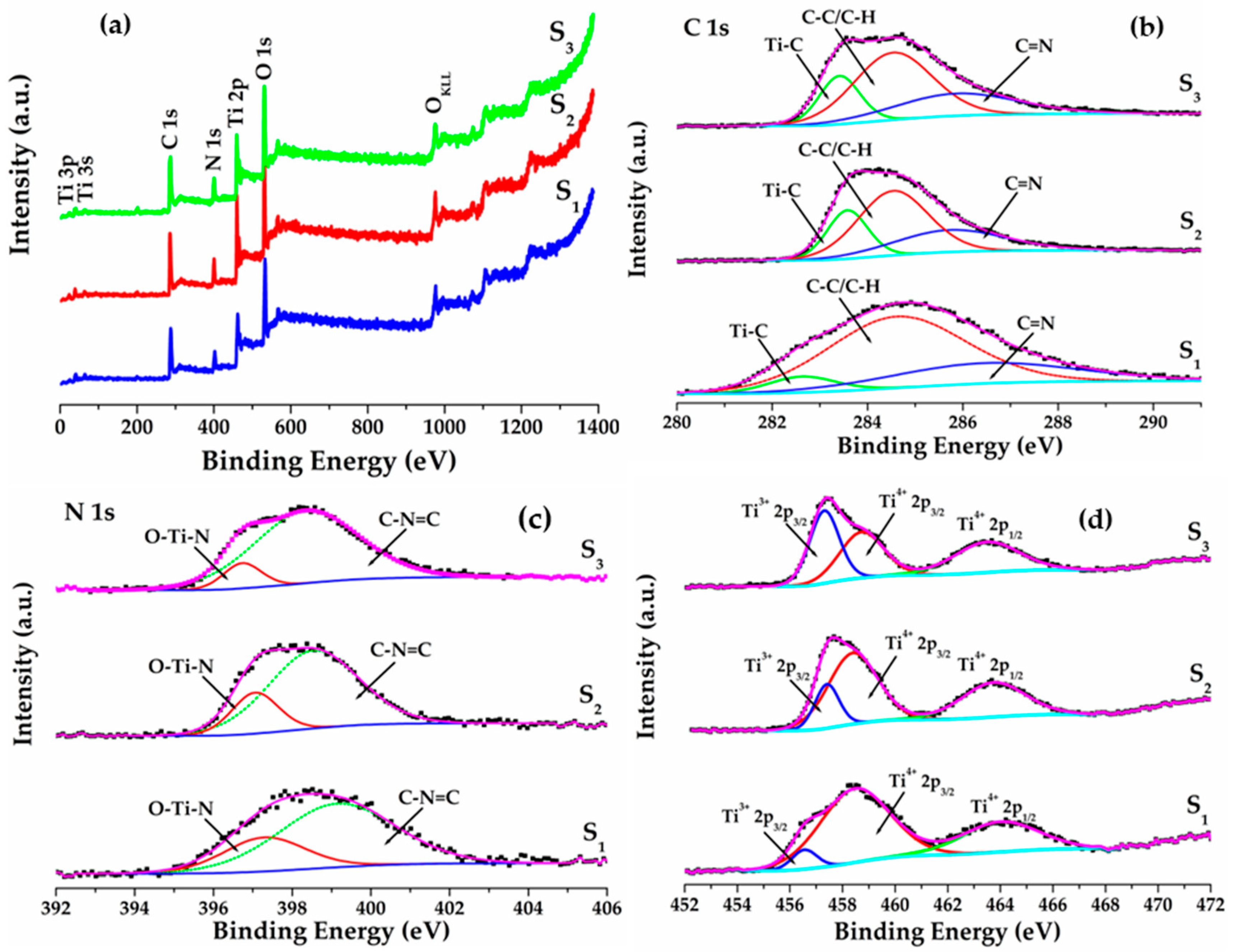
2.5. XPS Analysis of C–N–TiO2 Catalysts
2.6. Scanning Electron Microscopy and Energy-Dispersive Spectroscopy of C–N–TiO2 Pyrolysed at Different Holding Times
2.7. Transmission Electron Microscopy and Thin Film Energy-Dispersive Spectroscopy of the Synthesised C–N–TiO2 Nano-Composites
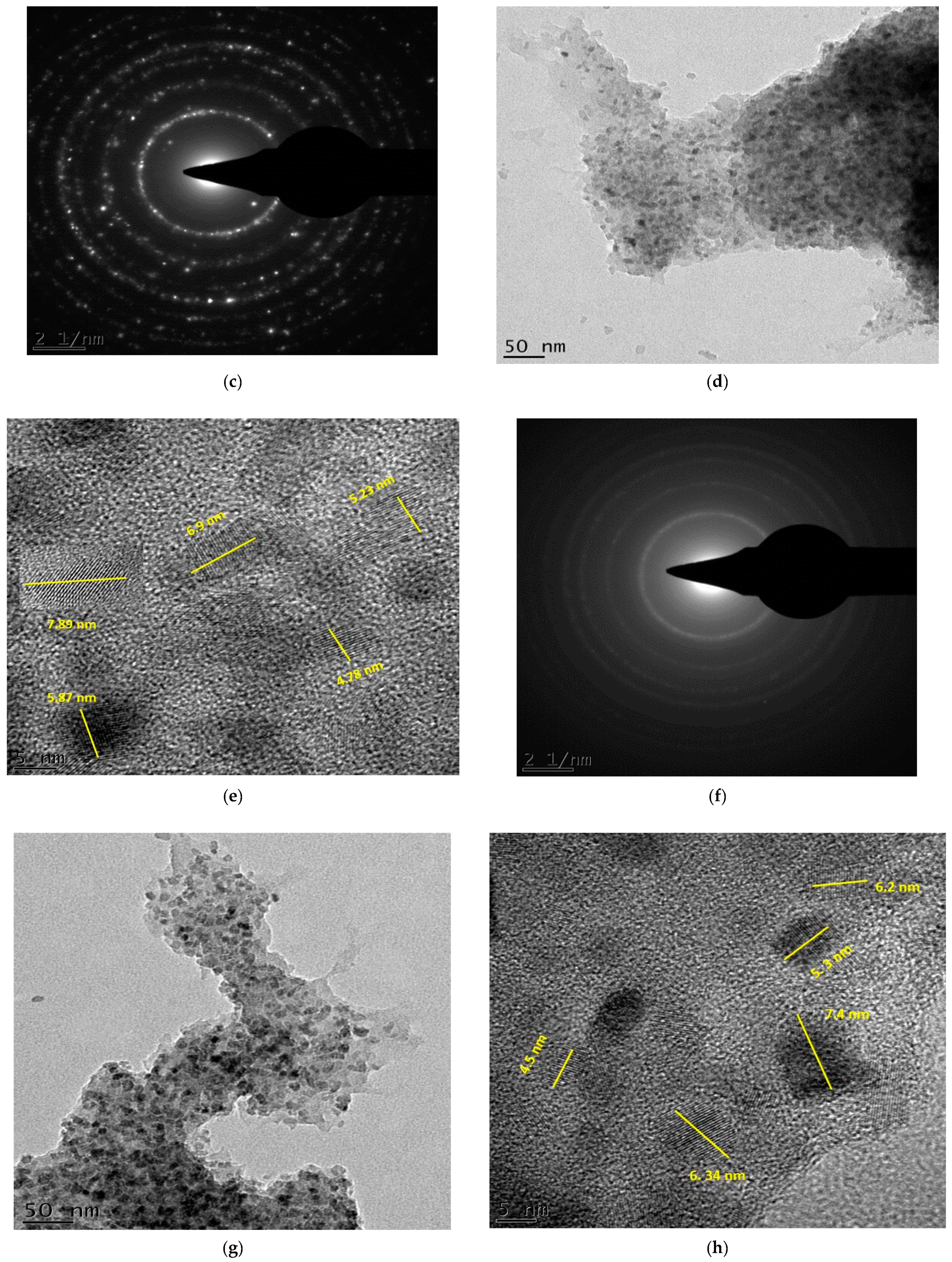
2.8. Transmission Electron Microscopy and Selected Area Electron Diffraction Analysis of TiO2 Degussa and C–N–TiO2 Nanoparticles
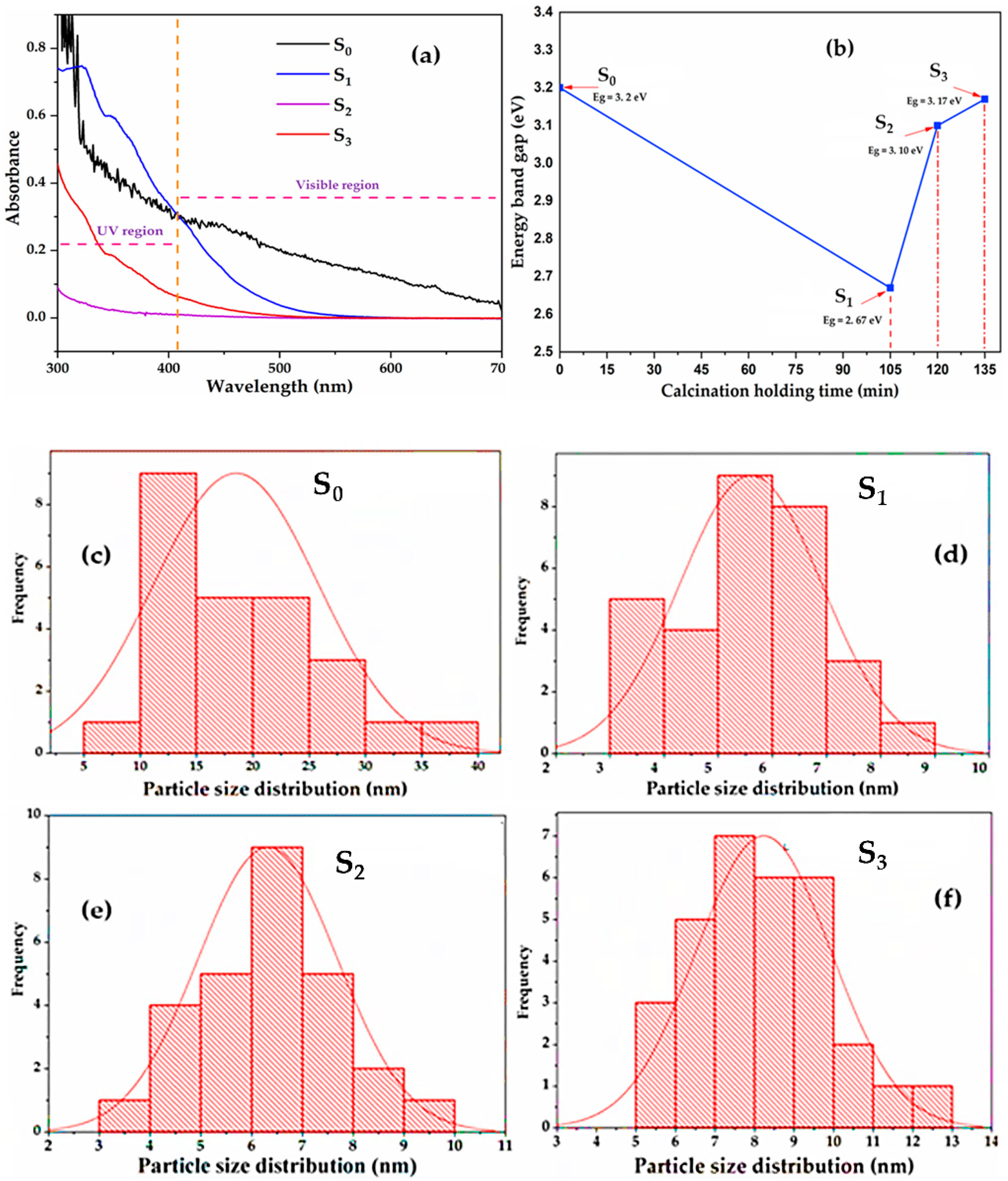
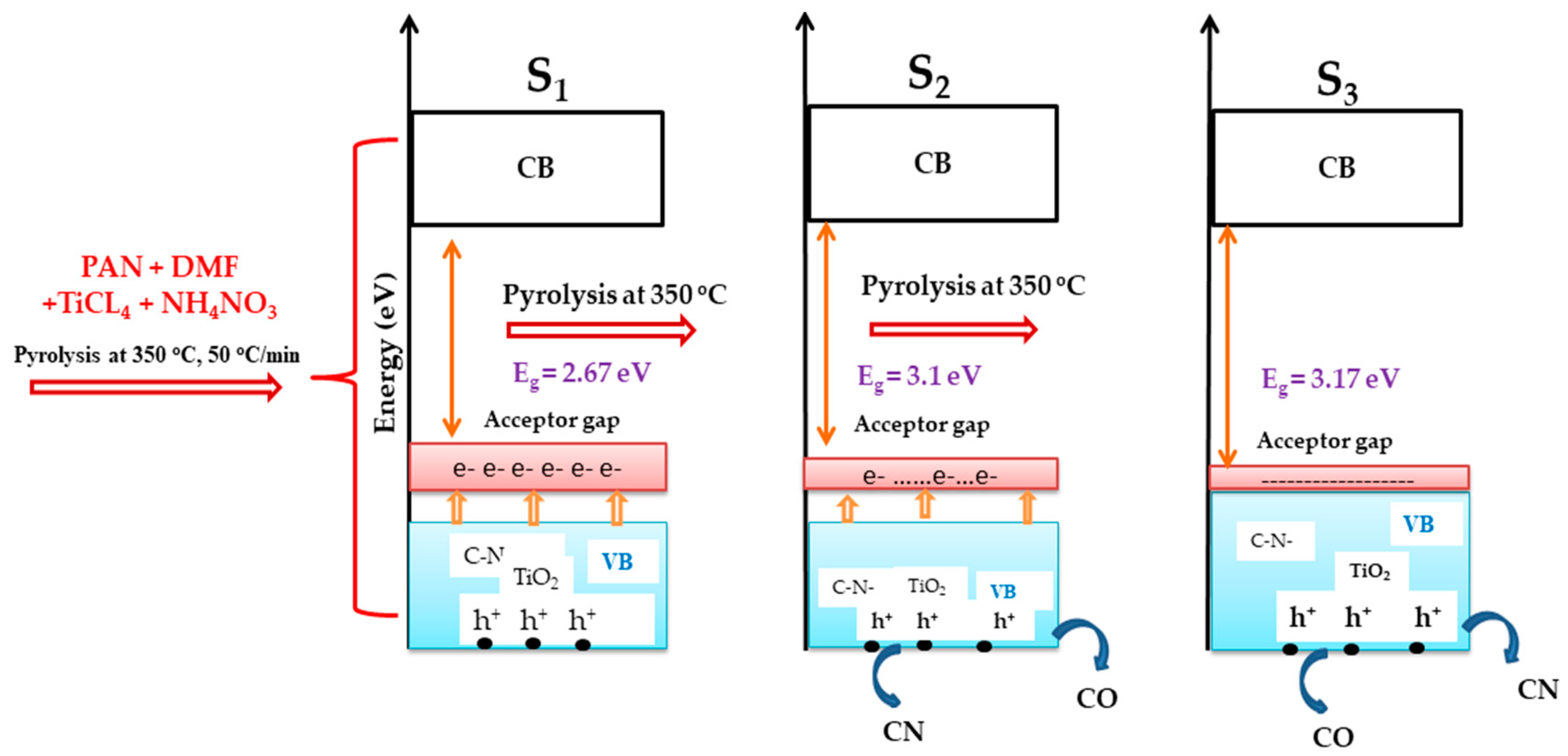
2.9. UV-Vis Spectroscopy Analysis, Determination of the Nano Catalyst Band Gap
2.10. Particles Size Distribution of TiO2 Degussa and the Synthesised C–N–TiO2 Nano Composites
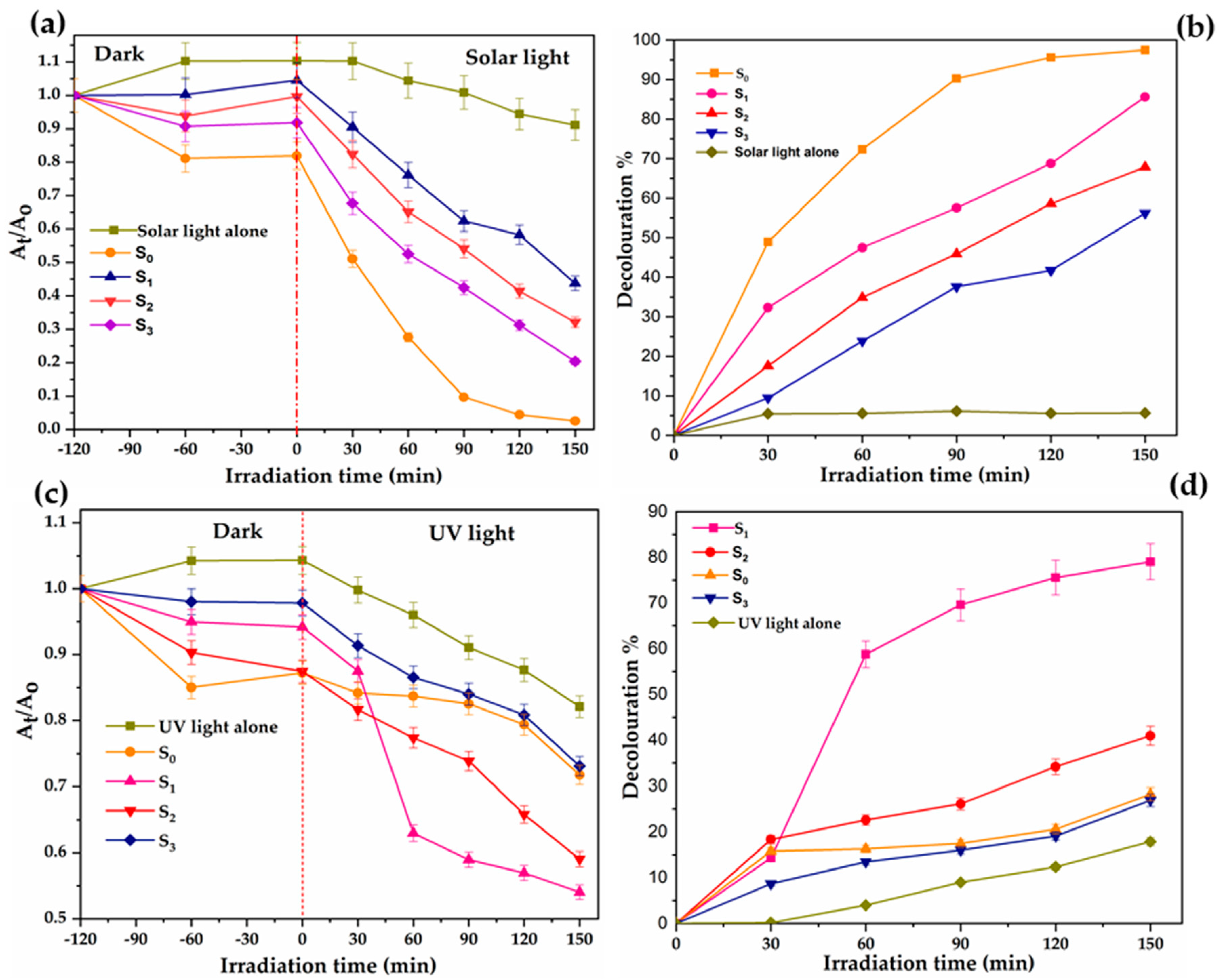
2.11. Effect of Pyrolysis Holding Time of C–N–TiO2 Nano Catalysts on Photocatalytic Decolouration of Orange II Dye
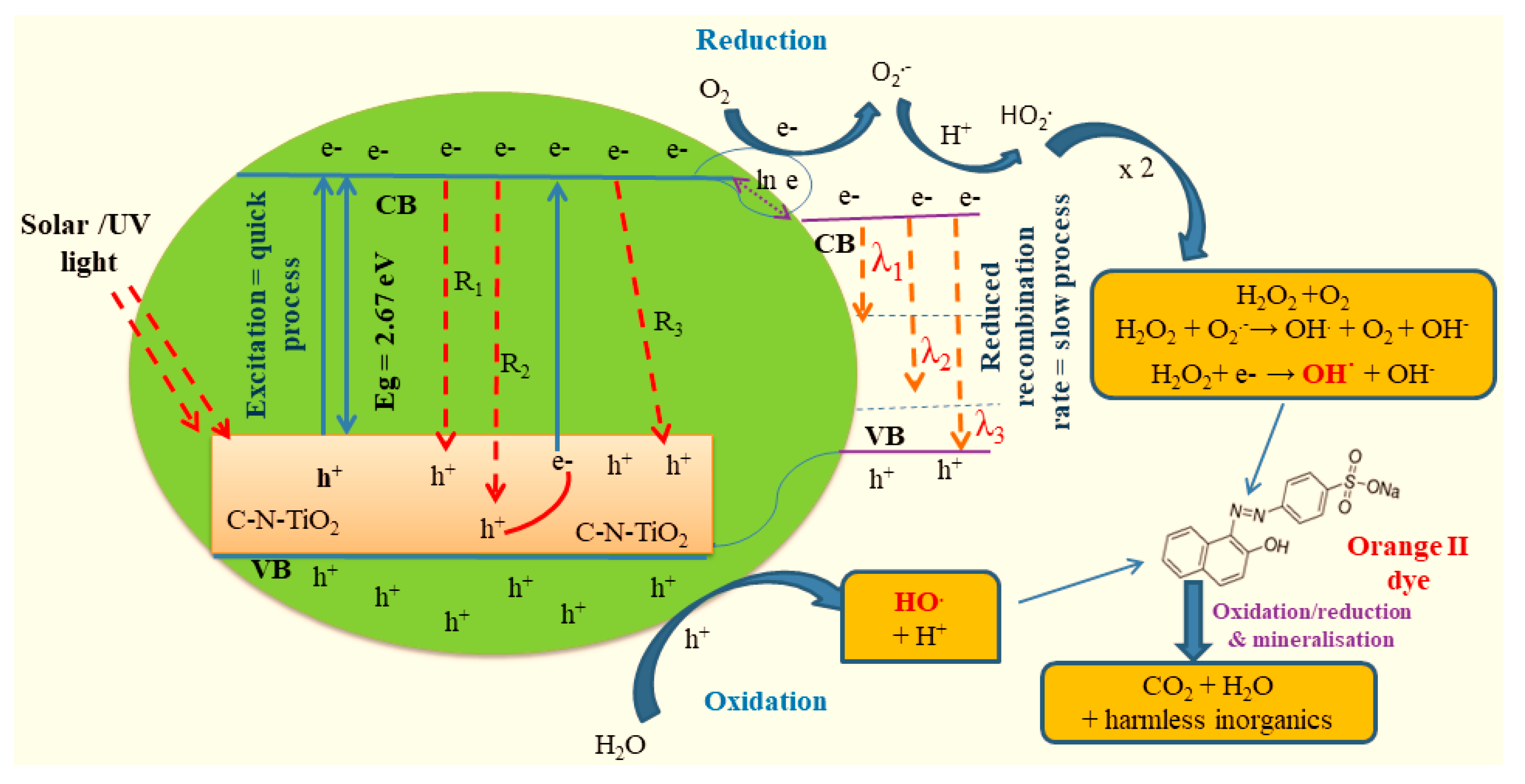
3. Discussion
4. Materials and Methods
4.1. Preparation of 5% Ammonium Nitrate Solution
4.2. Preparation of C–N–TiO2 Sol-Gel and Pyrolysis Protocol
4.3. Characterisation of Powdered C–N–TiO2 Photo Catalyst
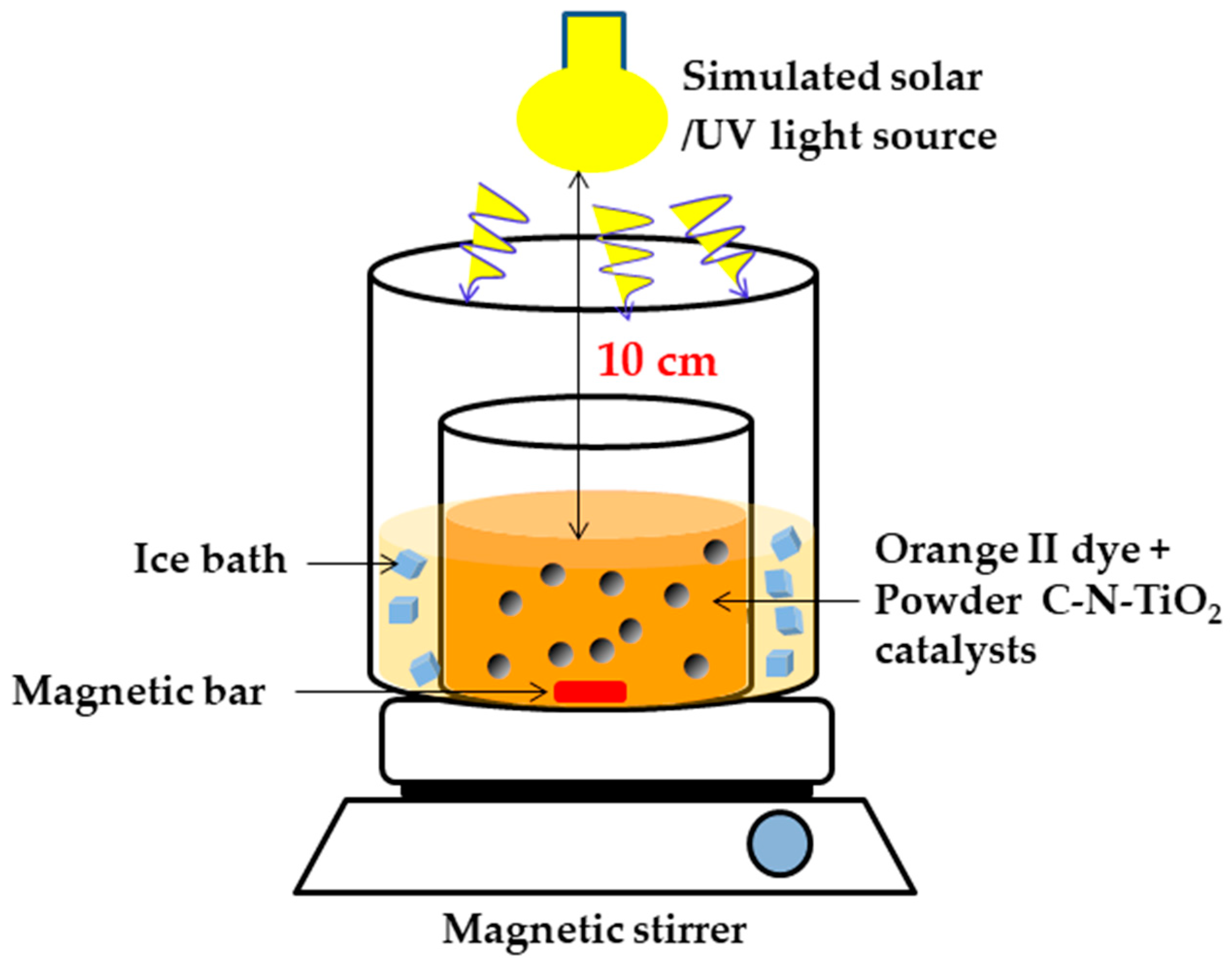
4.4. Photo Catalytic Irradiation of Orange II Dye
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Future Prospects
References
- Kümmerer, K. The presence of pharmaceuticals in the environment due to human use–present knowledge and future challenges. J. Environ. Manag. 2009, 90, 2354–2366. [Google Scholar] [CrossRef] [PubMed]
- Esplugas, S.; Bila, D.M.; Krause, L.G.T.; Dezotti, M. Ozonation and advanced oxidation technologies to remove endocrine disrupting chemicals (EDCs) and pharmaceuticals and personal care products (PPCPs) in water effluents. J. Hazard. Mater. 2007, 149, 631–642. [Google Scholar] [CrossRef] [PubMed]
- Sirés, I.; Brillas, E.; Sadornil, I.S. Remediation of water pollution caused by pharmaceutical residues based on electrochemical separation and degradation technologies: A review. Environ. Int. 2012, 40, 212–229. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Song, Q.; Lv, W.; Liu, G. Degradation of ketoprofen by sulfate radical-based advanced oxidation processes: Kinetics, mechanisms, and effects of natural water matrices. Chemosphere 2017, 189, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Bianco, A.; Brigante, M.; Mailhot, G. UVA-UVB activation of hydrogen peroxide and persulfate for advanced oxidation processes: Efficiency, mechanism and effect of various water constituents. J. Hazard. Mater. 2018, 347, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Feng, H.; Deng, Y. Re-evaluation of sulfate radical based–advanced oxidation processes (SR-AOPs) for treatment of raw municipal landfill leachate. Water Res. 2019, 153, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Klavarioti, M.; Mantzavinos, D.; Fatta-Kassinos, D. Removal of residual pharmaceuticals from aqueous systems by advanced oxidation processes. Environ. Int. 2009, 35, 402–417. [Google Scholar] [CrossRef] [PubMed]
- Klamerth, N.; Rizzo, L.; Malato, S.; Maldonado, M.I.; Agüera, A.; Fernández-Alba, A.R. Degradation of fifteen emerging contaminants at microg l(-1) initial concentrations by mild solar photo-Fenton in MWTP effluents. Water Res. 2010, 44, 545–554. [Google Scholar] [CrossRef]
- Zhao, F.; Liu, Y.; Hammouda, S.B.; Doshi, B.; Guijarro, N.; Min, X.; Tang C-Jian Sillanpaa, M.; Sivula, K.; Wang, S. MIL-101(Fe)/g-C3N4 for enhanced visible-light-driven photocatalysis toward simultaneous reduction of Cr (VI) and oxidation of bisphenol A in aqueous media. Appl. Catal. B Environ. 2020, 272, 119033. [Google Scholar] [CrossRef]
- Alemany, L.; Bañares, M.A.; Pardo, E.; Jiménez, F.D.P.M.; Blasco, J. Morphological and Structural Characterization of a Titanium Dioxide System. Mater. Charact. 2000, 44, 271–275. [Google Scholar] [CrossRef]
- Khan, M.; Bhatti, K.; Qindeel, R.; Alonizan, N.; Althobaiti, H.S. Characterizations of multilayer ZnO thin films deposited by sol-gel spin coating technique. Results Phys. 2017, 7, 651–655. [Google Scholar] [CrossRef]
- Cheng, X.; Yu, X.; Xing, Z.; Wan, J. Enhanced Photocatalytic Activity of Nitrogen Doped TiO2 Anatase Nano-Particle under Simulated Sunlight Irradiation. Energy Procedia 2012, 16, 598–605. [Google Scholar] [CrossRef] [Green Version]
- Sambandam, B.; Surenjan, A.; Philip, L.; Pradeep, T. Rapid Synthesis of C-TiO2: Tuning the Shape from Spherical to Rice Grain Morphology for Visible Light Photocatalytic Application. ACS Sustain. Chem. Eng. 2015, 3, 1321–1329. [Google Scholar] [CrossRef]
- Nyamukamba, P.; Tichagwa, L.; Ngila, J.C.; Petrik, L. Plasmonic metal decorated titanium dioxide thin films for enhanced photodegradation of organic contaminants. J. Photochem. Photobiol. A Chem. 2017, 343, 85–95. [Google Scholar] [CrossRef] [Green Version]
- Morikawa, T.; Asahi, R.; Ohwaki, T.; Aoki, K.; Taga, Y. Band-Gap Narrowing of Titanium Dioxide by Nitrogen Doping. Jpn. J. Appl. Phys. 2001, 40, L561–L563. [Google Scholar] [CrossRef]
- Palanivelu, K.; Im, J.S.; Lee, Y.-S. Carbon Doping of TiO2 for Visible Light Photo Catalysis—A review. Carbon Lett. 2007, 8, 214–224. [Google Scholar] [CrossRef] [Green Version]
- Kanade, K.; Kale, B.; Baeg, J.-O.; Lee, S.M.; Lee, C.W.; Moon, S.-J.; Chang, H. Self-assembled aligned Cu doped ZnO nanoparticles for photocatalytic hydrogen production under visible light irradiation. Mater. Chem. Phys. 2007, 102, 98–104. [Google Scholar] [CrossRef]
- Wang, J.; Huang, B.; Wang, Z.; Qin, X.; Zhang, X. Synthesis and characterization of C, N-codoped TiO2 nanotubes/nanorods with visible-light activity. Rare Met. 2011, 30, 161–165. [Google Scholar] [CrossRef]
- Cheng, X.; Yu, X.; Xing, Z.; Yang, L. Synthesis and characterization of N-doped TiO2 and its enhanced visible-light photocatalytic activity. Arab. J. Chem. 2016, 9 (Suppl. 2), S1706–S1711. [Google Scholar] [CrossRef]
- Tijani, J.O.; Fatoba, O.O.; Totito, T.C.; Roos, W.D.; Petrik, L.F.; Info, A. Synthesis and characterization of carbon doped TiO2 photo-catalysts supported on stainless steel mesh by sol-gel method Original Articles. Carbon Lett. 2017, 22, 48–59. [Google Scholar] [CrossRef]
- Yu, C.; Yu, J.C.-M. A Simple Way to Prepare C–N-Codoped TiO2 Photocatalyst with Visible-Light Activity. Catal. Lett. 2009, 129, 462–470. [Google Scholar] [CrossRef]
- Li, F.; Zhou, J.; Du, C.; Li, W.; Wang, Y.; He, G.; He, Q. Preparation and photocatalytic properties of porous C and N co-doped TiO2 deposited on brick by a fast, one-step microwave irradiation method. J. Environ. Sci. 2017, 60, 24–32. [Google Scholar] [CrossRef]
- Mohamed, M.A.; Othman, M.H.D.; Zain, M.M.; Minggu, L.J.; Kassim, M.B.; Salehmin, M.N.I.; Rosmi, M.S.; Salleh, W.N.W.; Othman, M.H.D. Concurrent growth, structural and photocatalytic properties of hybridized C, N co-doped TiO2 mixed phase over g-C3N4 nanostructured. Scr. Mater. 2018, 142, 143–147. [Google Scholar] [CrossRef]
- Osin, O.A.; Yu, T.; Cai, X.; Jiang, Y.; Peng, G.; Cheng, X.; Li, R.; Qin, Y.; Lin, S. Photocatalytic Degradation of 4-Nitrophenol by C, N-TiO2: Degradation Efficiency vs. Embryonic Toxicity of the Resulting Compounds. Front. Chem. 2018, 6, 192. [Google Scholar] [CrossRef] [Green Version]
- Thamaphat, K.; Limsuwan, P.; Ngotawornchai, B. Phase Characterization of TiO2 Powder by XRD and TEM. Nat. Sci. 2008, 42, 357–361. [Google Scholar]
- Xie, C.; Yang, S.; Li, B.; Wang, H.; Shi, J.-W.; Li, G.; Niu, C. C-doped mesoporous anatase TiO2 comprising 10 nm crystallites. J. Colloid Interface Sci. 2016, 476, 1–8. [Google Scholar] [CrossRef]
- Zhang, Q. Effects of calcination on the photocatalytic properties of nanosized TiO2 powders prepared by TiCl4 hydrolysis. Appl. Catal. B Environ. 2000, 26, 207–215. [Google Scholar] [CrossRef]
- Geng, J.; Yang, N.; Zhu, J.; Chen, D.; Jiang, Z. Nitrogen-doped TiO2 nanotubes with enhanced photocatalytic activity synthesized by a facile wet chemistry method. Mater. Res. Bull. 2009, 44, 146–150. [Google Scholar] [CrossRef]
- Wu, S.; Gao, A.; Wang, Y.; Xu, L. Modification of polyacrylonitrile stabilized fibers via post-thermal treatment in nitrogen prior to carbonization and its effect on the structure of carbon fibers. J. Mater. Sci. 2018, 53, 8627–8638. [Google Scholar] [CrossRef]
- Cagnina, S.; Rotureau, P.; Singh, S.; Turcotte, R.; Fayet, G.; Adamo, C. Theoretical and Experimental Study of the Reaction between Ammonium Nitrate and Sodium Salts. Ind. Eng. Chem. Res. 2016, 55, 12183–12190. [Google Scholar] [CrossRef] [Green Version]
- Shao, G.-S.; Zhang, X.-J.; Yuan, Z.-Y. Preparation and photocatalytic activity of hierarchically mesoporous-macroporous TiO2−xNx. Appl. Catal. B Environ. 2008, 82, 208–218. [Google Scholar] [CrossRef]
- Sánchez-Soto, P.J.; Avilés, M.; Del Río, J.; Ginés, J.; Pascual, J.; Pérez-Rodríguez, J. Thermal study of the effect of several solvents on polymerization of acrylonitrile and their subsequent pyrolysis. J. Anal. Appl. Pyrolysis 2001, 58, 155–172. [Google Scholar] [CrossRef]
- Darányi, M.; Sarusi, I.; Sápi, A.; Kukovecz, Á.; Konya, Z.; Erdőhelyi, A. Characterization of carbon thin films prepared by the thermal decomposition of spin coated polyacrylonitrile layers containing metal acetates. Thin Solid Films 2011, 520, 57–63. [Google Scholar] [CrossRef]
- Laffont, L.; Monthioux, M.; Serin, V.; Mathur, R.; Guimon, C.; Guimon, M. An EELS study of the structural and chemical transformation of PAN polymer to solid carbon. Carbon 2004, 42, 2485–2494. [Google Scholar] [CrossRef]
- Huang, Y.; Ho, W.; Lee, S.-C.; Zhang, L.; Li, G.; Yu, J.C.-M. Effect of Carbon Doping on the Mesoporous Structure of Nanocrystalline Titanium Dioxide and Its Solar-Light-Driven Photocatalytic Degradation of NOx. Langmuir 2008, 24, 3510–3516. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Guo, T.; Chen, J.; Wei, J.; Bai, L.; Ye, X.; Ding, Z.; Xu, W.; Zhou, Z. Synthesis of C–N–S co-doped TiO2 mischcrystal with an isobandgap characteristic and its photocatalytic activity under visible light. Catal. Sci. Technol. 2018, 8, 4108–4121. [Google Scholar] [CrossRef]
- Abdullah, A.M.; Al-Thani, N.J.; Tawbi, K.; Al-Kandari, H. Carbon/nitrogen-doped TiO2: New synthesis route, characterization and application for phenol degradation. Arab. J. Chem. 2016, 9, 229–237. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Han, J.; Hu, Y.; Rong, G. Mesoporous C, N-codoped TiO2 hybrid shells with enhanced visible light photocatalytic performance. RSC Adv. 2017, 7, 15513–15520. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Xing, Z.; Cui, J.; Li, Z.; Tan, S.; Yin, J.; Zou, J.; Zhu, Q.; Zhou, W. C,N co-doped porous TiO2 hollow sphere visible light photocatalysts for efficient removal of highly toxic phenolic pollutants. Dalton Trans. 2018, 47, 4877–4884. [Google Scholar] [CrossRef] [PubMed]
- Ghazzal, M.N.; Wojcieszak, R.; Raj, G.; Gaigneaux, E. Study of mesoporous CdS-quantum-dot-sensitized TiO2 films by using X-ray photoelectron spectroscopy and AFM. Beilstein J. Nanotechnol. 2014, 5, 68–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, T.; Fu, F.; Yu, D.; Cao, J.; Sun, G. Facile synthesis and characterization of N-doped TiO2/C nanocomposites with enhanced visible-light photocatalytic performance. Appl. Surf. Sci. 2018, 430, 438–447. [Google Scholar] [CrossRef]
- Igathinathane, C.; Pordesimo, L.; Columbus, E.; Batchelor, W.; Methuku, S.; Cannayen, I. Shape identification and particles size distribution from basic shape parameters using ImageJ. Comput. Electron. Agric. 2008, 63, 168–182. [Google Scholar] [CrossRef]
- Igathinathane, C.; Pordesimo, L.; Batchelor, W.; Cannayen, I. Major orthogonal dimensions measurement of food grains by machine vision using ImageJ. Food Res. Int. 2009, 42, 76–84. [Google Scholar] [CrossRef]
- Chen, T.; Xia, Y.; Jia, Z.; Liu, Z.; Zhang, H. Synthesis, Characterization, and Tribological Behavior of Oleic Acid Capped Graphene Oxide. J. Nanomater. 2014, 2014, 1–8. [Google Scholar] [CrossRef]
- Carp, O.; Huisman, C.; Reller, A. Photoinduced reactivity of titanium dioxide. Prog. Solid State Chem. 2004, 32, 33–177. [Google Scholar] [CrossRef]
- Li, S.; Gong, Y.; Yang, Y.; He, C.; Hu, L.; Zhu, L.; Sun, L.; Shu, D. Recyclable CNTs/Fe3O4 magnetic nanocomposites as adsorbents to remove bisphenol A from water and their regeneration. Chem. Eng. J. 2015, 260, 231–239. [Google Scholar] [CrossRef]
- Gilmour, C.R.; Ray, A.; Zhu, J.; Ray, M.B. Photocatalytic Performance of Titanium Dioxide Thin Films from Polymer-Encapsulated Titania. Ind. Eng. Chem. Res. 2013, 52, 17800–17811. [Google Scholar] [CrossRef]
- Sakthivel, S.; Kisch, H. Daylight Photocatalysis by Carbon-Modified Titanium Dioxide. Angew. Chem. Int. Ed. 2003, 42, 4908–4911. [Google Scholar] [CrossRef] [PubMed]
- Tijani, J.; Mouele, M.E.S.; Fatoba, O.; O Babajide, O.; Petrik, L.F. Degradation of bisphenol-A by dielectric barrier discharge system: Influence of polyethylene glycol stabilized nano zero valent iron particles. Adv. Nat. Sci. Nanosci. Nanotechnol. 2017, 8, 035013. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Zhang, J.Z. Optical properties and applications of hybrid semiconductor nanomaterials. Coord. Chem. Rev. 2009, 253, 3015–3041. [Google Scholar] [CrossRef]
- Chen, Y.; Dionysiou, D.D. A comparative study on physicochemical properties and photocatalytic behavior of macroporous TiO2-P25 composite films and macroporous TiO2 films coated on stainless steel substrate. Appl. Catal. A Gen. 2007, 317, 129–137. [Google Scholar] [CrossRef]
- Wu, Y.-C.; Ju, L.-S. Annealing-free synthesis of CN co-doped TiO2 hierarchical spheres by using amine agents via microwave-assisted solvothermal method and their photocatalytic activities. J. Alloy Compd. 2014, 604, 164–170. [Google Scholar] [CrossRef]
- Saravanan, R.; Gupta, V.K.; Narayanan, V.; Stephen, A. Comparative study on photocatalytic activity of ZnO prepared by different methods. J. Mol. Liq. 2013, 181, 133–141. [Google Scholar] [CrossRef]
- Khan, M.M.; Ansari, S.A.; Pradhan, D.; Ansari, M.O.; Han, D.H.; Lee, J.; Cho, M.H. Band gap engineered TiO2 nanoparticles for visible light induced photoelectrochemical and photocatalytic studies. J. Mater. Chem. A 2014, 2, 637–644. [Google Scholar] [CrossRef]
- Khan, M.M.; Adil, S.F.; Al-Mayouf, A.M. Metal oxides as photocatalysts. J. Saudi Chem. Soc. 2015, 19, 462–464. [Google Scholar] [CrossRef] [Green Version]
- Gnanasekaran, L.; Hemamalini, R.; Ravichandran, K. Synthesis and characterization of TiO2 quantum dots for photocatalytic application. J. Saudi Chem. Soc. 2015, 19, 589–594. [Google Scholar] [CrossRef] [Green Version]
- Shriver, D.F.; Atkins, P.W.; Langford, C.H. Inorganic Chemistry, 2nd ed.; International Student Edition; Oxford University Press: Oxford, UK; Melbourne, Australia; Tokyo, Japan, 1994. [Google Scholar]
- Mu, Y.; Yu, H.-Q.; Zheng, J.-C.; Zhang, S. TiO2-mediated photocatalytic degradation of Orange II with the presence of Mn2+ in solution. J. Photochem. Photobiol. A Chem. 2004, 163, 311–316. [Google Scholar] [CrossRef]
- Yin, W.-J.; Tang, H.; Wei, S.-H.; Al-Jassim, M.; Turner, J.; Yan, Y. Band structure engineering of semiconductors for enhanced photo electrochemical water splitting: The case of TiO2. Phys. Rev. B 2010, 82, 045106. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Wu, Y.; Lu, Y.; Xu, J.; Wang, Y.; Zhu, Y. Effect of Compensated Codoping on the Photo electrochemical Properties of Anatase TiO2 Photocatalyst. J. Phys. Chem. C 2011, 115, 16963–16969. [Google Scholar] [CrossRef]
- Fujishima, A.; Zhang, X.; Tryk, D. TiO2 photocatalysis and related surface phenomena. Surf. Sci. Rep. 2008, 63, 515–582. [Google Scholar] [CrossRef]
- Ikoma, T.; Zhang, Q.; Saito, F.; Akiyama, K.; Tero-Kubota, S.; Kato, T. Radicals in the Mechanochemical Dechlorination of Hazardous Organochlorine Compounds Using CaO Nanoparticles. Bull. Chem. Soc. Jpn. 2001, 74, 2303–2309. [Google Scholar] [CrossRef]
- Haque, S.A.; Tachibana, Y.; Willis, R.L.; Moser, J.-E.; Grätzel, M.; Klug, D.R.; Durrant, J.R. Parameters Influencing Charge Recombination Kinetics in Dye-Sensitized Nanocrystalline Titanium Dioxide Films. J. Phys. Chem. B 2000, 104, 538–547. [Google Scholar] [CrossRef] [Green Version]
- Prokes, S.; Gole, J.L.; Chen, X.; Burda, C.; Carlos, W.E. Defect-Related Optical Behavior in Surface Modified TiO2 Nanostructures. Adv. Funct. Mater. 2005, 15, 161–167. [Google Scholar] [CrossRef]
- MacNeil, J.H.; Berseth, P.A.; Bruner, E.L.; Perkins, T.L.; Wadia, Y.; Westwood, G.; Trogler, W.C. Mechanism of Nitrous Oxide Formation by Metal-Catalyzed Reduction of Nitric Oxide in Aqueous Solution. J. Am. Chem. Soc. 1997, 119, 1668–1675. [Google Scholar] [CrossRef]
- Kogelschatz, U.; Eliasson, B.; Egli, W. Dielectric-Barrier Discharges. Principle and Applications. Le J. de Phys. Colloq. 1997, 7, C4-47–C4-66. [Google Scholar] [CrossRef]
- Kogelschatz, U. Dielectric-Barrier Discharges: Their History, Discharge Physics, and Industrial Applications. Plasma Chem. Plasma Process. 2003, 23, 1–46. [Google Scholar] [CrossRef]
- Nehra, V.; Kumar, A.; Dwivedi, H.K. Atmospheric Non-Thermal Plasma Sources. Int. J. Eng. 2008, 2, 53–68. [Google Scholar]
- Lopez, J.L. Dielectric Barrier Discharge, Ozone Generation, and Their Applications; Complex Plasmas Summer Institute: Jersey City, NJ, USA, 2008. [Google Scholar]
- Valinčius, V.; Grigaitienė, V.; Tamošiūnas, A. Report on the Different Plasma Modules for Pollution Removal; Plasma for Environ Protect, Lithuanian Energy Institute: Kaunas, Lithuania, 2012; pp. 1–49. [Google Scholar]
- Totito, T.C. Photocatalytic Activity of Supported TiO2 Nanocrystals. Master’s Thesis, University of the Western Cape, Cape Town, South Africa, 2013. [Google Scholar]
- Kuang, L.; Zhao, Y.; Liu, L. Photodegradation of Orange II by mesoporous TiO2. J. Environ. Monit. 2011, 13, 2496. [Google Scholar] [CrossRef]
- Sayed, E.; El-Ashtoukhy, Z. Removal of Indigo Carmine Dye from Synthetic Wastewater by Electrochemical Oxidation in a New Cell with Horizontally Oriented Electrodes. Int. J. Electrochem. Sci. 2013, 8, 846–858. [Google Scholar]
- Mouele, E.S.M.; Dinu, M.; Parau, A.C.; Missengue, R.; Vladescu, A.; Petrik, L.F.; Braic, M. Dinu Evaluation of Photocatalysis Effect of Stainless Steel Mesh Coated with Nitrides, Oxynitrides and Transition Metals Cr and Ti on the Degradation of Orange II Dye. Proceedings 2019, 29, 14. [Google Scholar] [CrossRef] [Green Version]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Photo Catalysts | Catalyst Size (nm) XRD (Scherrer Equation) |
|---|---|
| S0 | 21.0 |
| S1 | 5.5 |
| S2 | 6.3 |
| S3 | 6.4 |
| Photo Catalysts and Pyrolysis Time (Min) | Absorption Bands (cm−1) | Functional Groups/Stretching/Vibrations |
|---|---|---|
| S0 | 3800–3000, 275–1000 | OH stretching, Ti–O, Ti –O–Ti |
| S1 | 3800–3600 | OH/N-H stretching, etc. |
| 1900–1800, 1800–1200 | –N–H, –C–C–, C–O and C–N, OH, stretching, etc. | |
| 1200–700 | TiO, Ti–O–Ti, N–Ti–O2, C–Ti–O, C–Ti, C–O–Ti–O, N–Ti–O, N=O, etc. | |
| S2 | 3800–3600 | OH/N–H stretching, etc. |
| 1900–1800, 1800–1200 | –N–H, –C–C–, C–O and C–N, OH, stretching, etc. | |
| 1200–700 | Ti–O, Ti–O–Ti, N–Ti–O2, C–O–Ti–O, C–Ti, N–Ti–O, N=O, etc. | |
| S3 | 3800–3600 | OH/N–H stretching, etc. |
| 1900–1800, 1800–1200 | –N–H, –C–C–, C–O and C–N, OH, stretch, etc. | |
| 1200–700 | Ti–O, Ti–O–Ti, N–Ti–O2, C–O–Ti–O, C–Ti, N–Ti–O N=O, etc. |
| Elements | Weight Percentage (%) | |||
|---|---|---|---|---|
| S0 | S1 | S2 | S3 | |
| C | NA | ±72.2 | ±64 | ±40 |
| N | NA | ±7.1 | ±5.0 | ±3.6 |
| Ti | 63.4 | ±10 | ±21.8 | ±49.6 |
| O | 34.6 | ±8.3 | ±12 | ±2.9 |
| Cl | NA | ±1.6 | ±2.1 | ±3.7 |
| C to Ti ratio | NA | 7:1 | 3:1 | 1:1 |
| Photo Catalysts | Pyrolysis Holding Time (min) | UV-Vis Absorption Wavelength (nm) | Band Gap (eV) |
|---|---|---|---|
| S0 | NA | 408 | 3.2 |
| S1 | 105 | 462 | 2.67 |
| S2 | 120 | 393 | 3.10 |
| S3 | 135 | 333 | 3.17 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Massima Mouele, E.S.; Dinu, M.; Cummings, F.; Fatoba, O.O.; Zar Myint, M.T.; Kyaw, H.H.; Parau, A.C.; Vladescu, A.; Francesconi, M.G.; Pescetelli, S.; et al. Effect of Calcination Time on the Physicochemical Properties and Photocatalytic Performance of Carbon and Nitrogen Co-Doped TiO2 Nanoparticles. Catalysts 2020, 10, 847. https://0-doi-org.brum.beds.ac.uk/10.3390/catal10080847
Massima Mouele ES, Dinu M, Cummings F, Fatoba OO, Zar Myint MT, Kyaw HH, Parau AC, Vladescu A, Francesconi MG, Pescetelli S, et al. Effect of Calcination Time on the Physicochemical Properties and Photocatalytic Performance of Carbon and Nitrogen Co-Doped TiO2 Nanoparticles. Catalysts. 2020; 10(8):847. https://0-doi-org.brum.beds.ac.uk/10.3390/catal10080847
Chicago/Turabian StyleMassima Mouele, Emile Salomon, Mihaela Dinu, Franscious Cummings, Ojo O. Fatoba, Myo Tay Zar Myint, Htet Htet Kyaw, Anca C. Parau, Alina Vladescu, M. Grazia Francesconi, Sara Pescetelli, and et al. 2020. "Effect of Calcination Time on the Physicochemical Properties and Photocatalytic Performance of Carbon and Nitrogen Co-Doped TiO2 Nanoparticles" Catalysts 10, no. 8: 847. https://0-doi-org.brum.beds.ac.uk/10.3390/catal10080847