Decomposition of Ruthenium Olefin Metathesis Catalyst


1
Centre of New Technologies, University of Warsaw, Banacha 2c, 02-097 Warsaw, Poland
2
Faculty of Chemistry, University of Warsaw, Pasteura 1, 02-093 Warsaw, Poland
*
Author to whom correspondence should be addressed.
Catalysts 2020, 10(8), 887; https://0-doi-org.brum.beds.ac.uk/10.3390/catal10080887
Submission received: 28 June 2020
/
Revised: 27 July 2020
/
Accepted: 2 August 2020
/
Published: 5 August 2020
(This article belongs to the Special Issue New Trends in Metathesis Catalysts)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
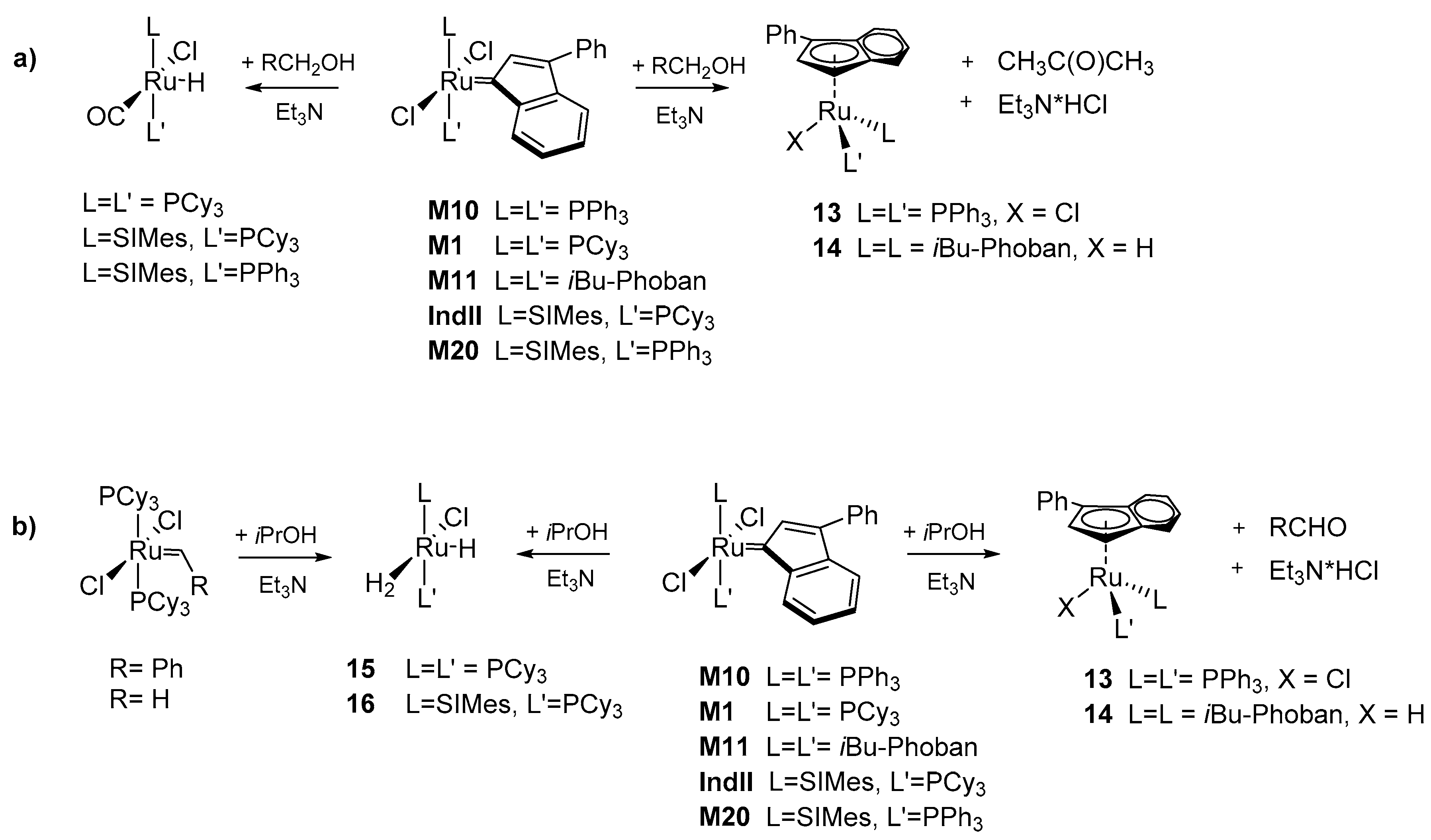{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Ruthenium olefin metathesis catalysts are one of the most commonly used class of catalysts. There are multiple reviews on their uses in various branches of chemistry and other sciences but a detailed review of their decomposition is missing, despite a large number of recent and important advances in this field. In particular, in the last five years several new mechanism of decomposition, both olefin-driven as well as induced by external agents, have been suggested and used to explain differences in the decomposition rates and the metathesis activities of both standard, N-heterocyclic carbene-based systems and the recently developed cyclic alkyl amino carbene-containing complexes. Here we present a review which explores the last 30 years of the decomposition studied on ruthenium olefin metathesis catalyst driven by both intrinsic features of such catalysts as well as external chemicals.
1. Introduction
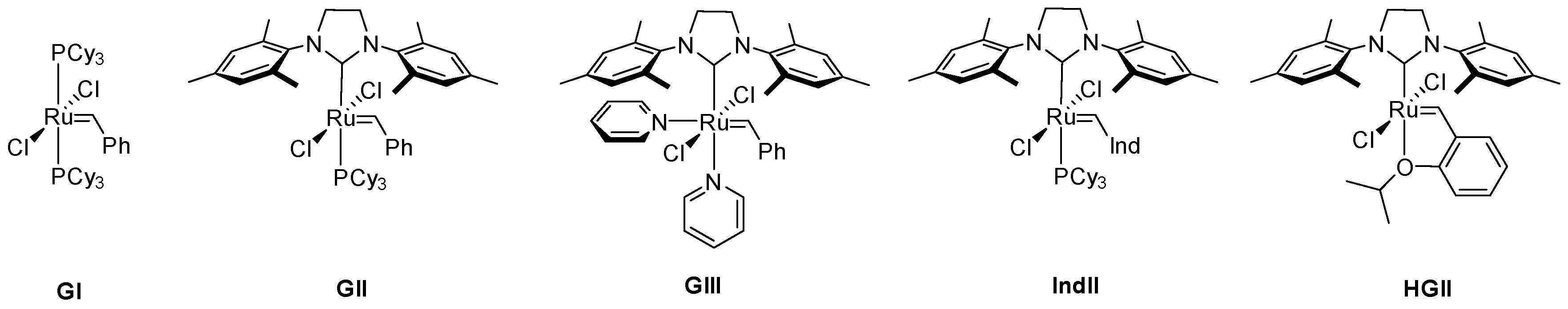
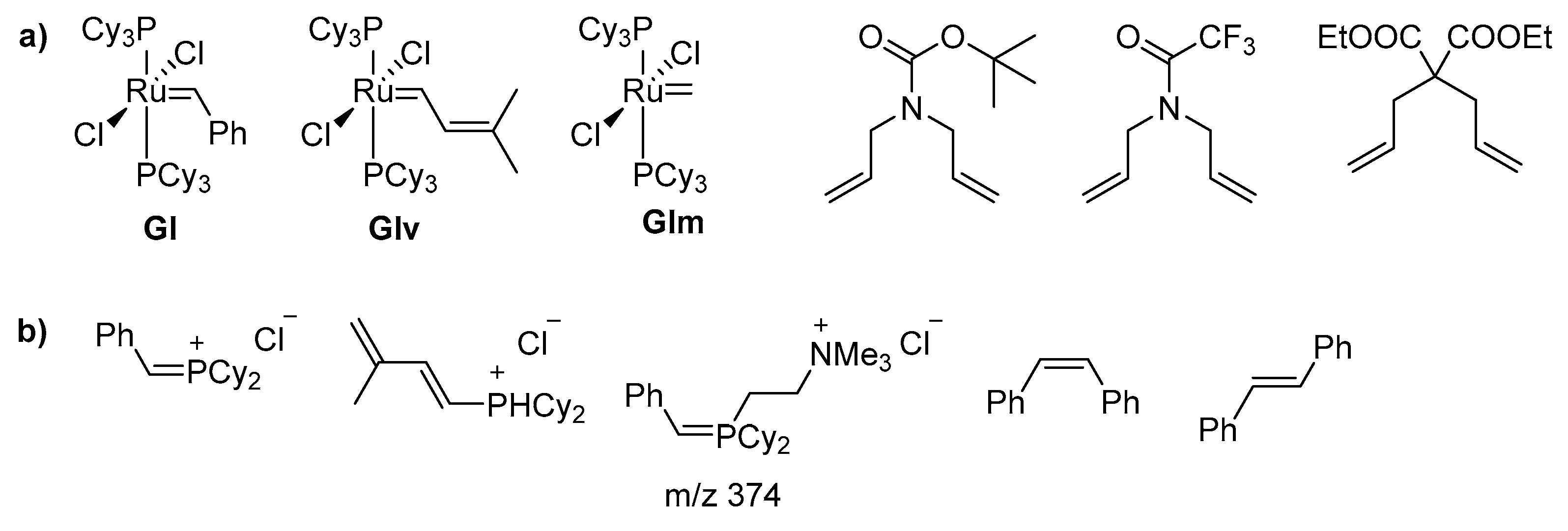
While the importance of ruthenium for olefin metathesis, one of the most prominent methods to form new C–C bonds, was realized already in the 1960s [1,2], the true dawn of the ruthenium-driven metathesis olefin catalysis came in 1992 with the synthesis of the first well-defined complex [3]. This breakthrough complex, synthesized in the Grubbs group, had many of the structural features found in modern, highly efficient and specialized metathesis catalysts. What followed were the 1st (GI), 2nd (GII) and 3rd (GIII) generation catalysts [4,5,6,7,8] and Hoveyda-Grubbs catalyst (HG) [9] which gave rise to hundreds of precatalysts with very distinct structural features but always based on the essential, 16-electron skeleton with the Ru atom at its core [10,11,12,13,14] (Figure 1). Nowadays, ruthenium metathesis catalysts are used in all branches of chemistry, ranging from pharmaceutical sciences [15,16,17,18,19] and synthesis of macrocyclic musks [20,21] to the petrochemical industry [22,23,24].
Soon after the discovery of first ruthenium metathesis catalysts it was realized that they do not easily undergo decomposition, though there are various external agents that can degrade these catalysts as well as several chemical groups that are not compatible with them. These studies were very valuable, since by proposing the probable decomposition mechanisms they inspired and guided other groups in the synthesis of new, modified catalysts with improved stabilities and less vulnerable to degradation. As a result of these over 30-year long investigations, we have today vastly improved our understanding of how structural features of ruthenium complexes affect their stabilities and reactivities as well as what are the major decomposition routes of metathesis catalysts. A good understanding of the entire catalytic cycle of metathesis, first proposed by Chauvin and presented in Figure 2, was also vital for these findings [25].
The review is organized in the following manner. First, we discuss the available scientific literature on the decomposition routes of ruthenium metathesis catalysts which are common for all catalyst, such as Ru = C double bond and do not depend on the specific structural features of such catalysts, such as different auxiliary ligands These include the olefin-driven decomposition as well as decomposition initiated by various external agents and solvents, divided by their chemical properties. Next we focus on two main classes of metathesis catalysts—phosphine-containing and n-heterocyclic carbene (NHC)-containing ones and review most known decomposition routes which are either phosphine-driven or NHC-driven. Finally we provide a summary and a future outlook for the decomposition studies of this important family of widely used catalysts.
The most recent review on ruthenium metathesis catalysts’ decomposition was reported in 2014 by Nolan group and dealt mostly with C–H activation, shortly mentioning several other mechanisms of degradation [26]. Since then a large number of new ruthenium metathesis catalysts has been designed and there have been a plethora of works presenting new results on their decomposition, therefore we believe that a new review on this topic is timely and relevant.
2. Non-Specific Decomposition Routes
In this section we describe the current state of knowledge for the non-specific decomposition routes of ruthenium metathesis catalysts. These include all the decomposition pathways which are driven by external agents and do not depend on the specific features of certain catalysts but are possible for all catalysts due to structural features and metathesis mechanisms shared by all of them.
2.1. Olefin-Driven Self-Degradations
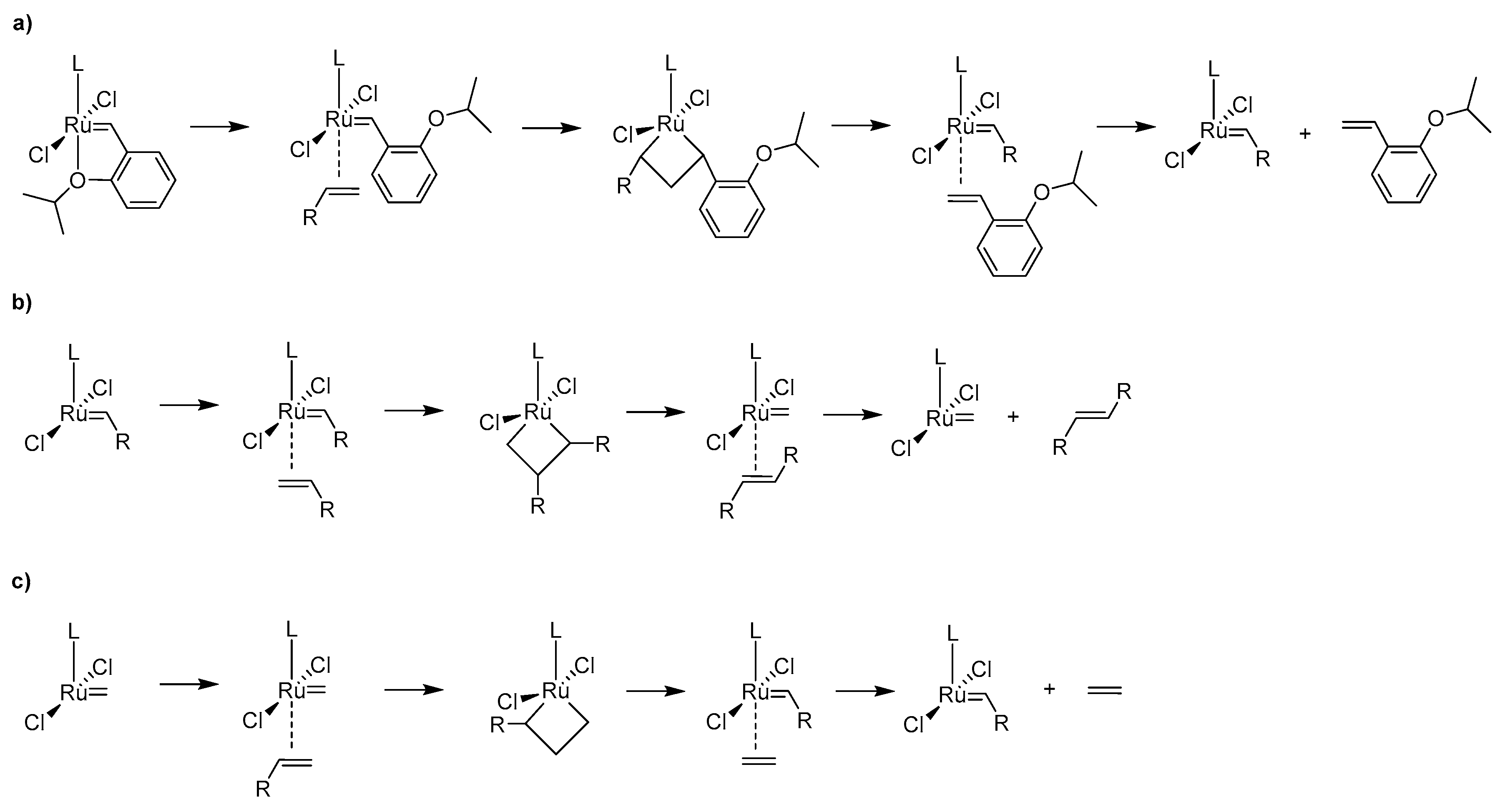
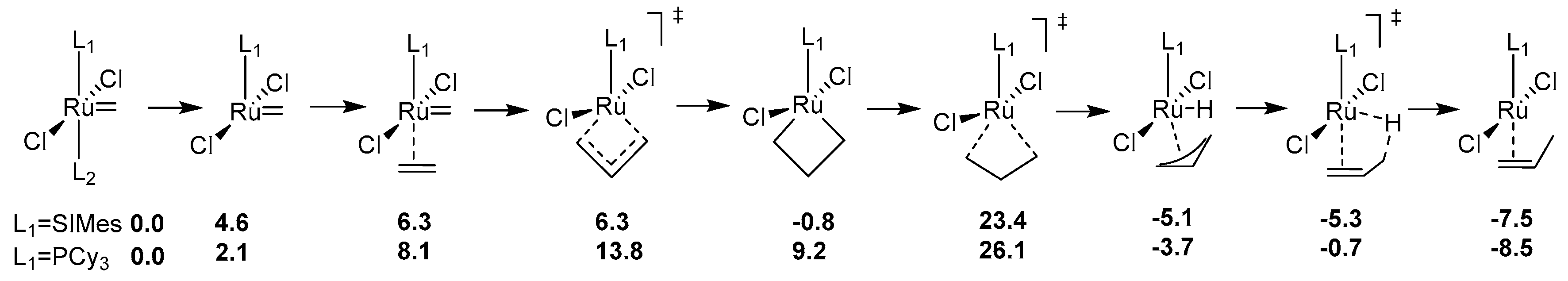
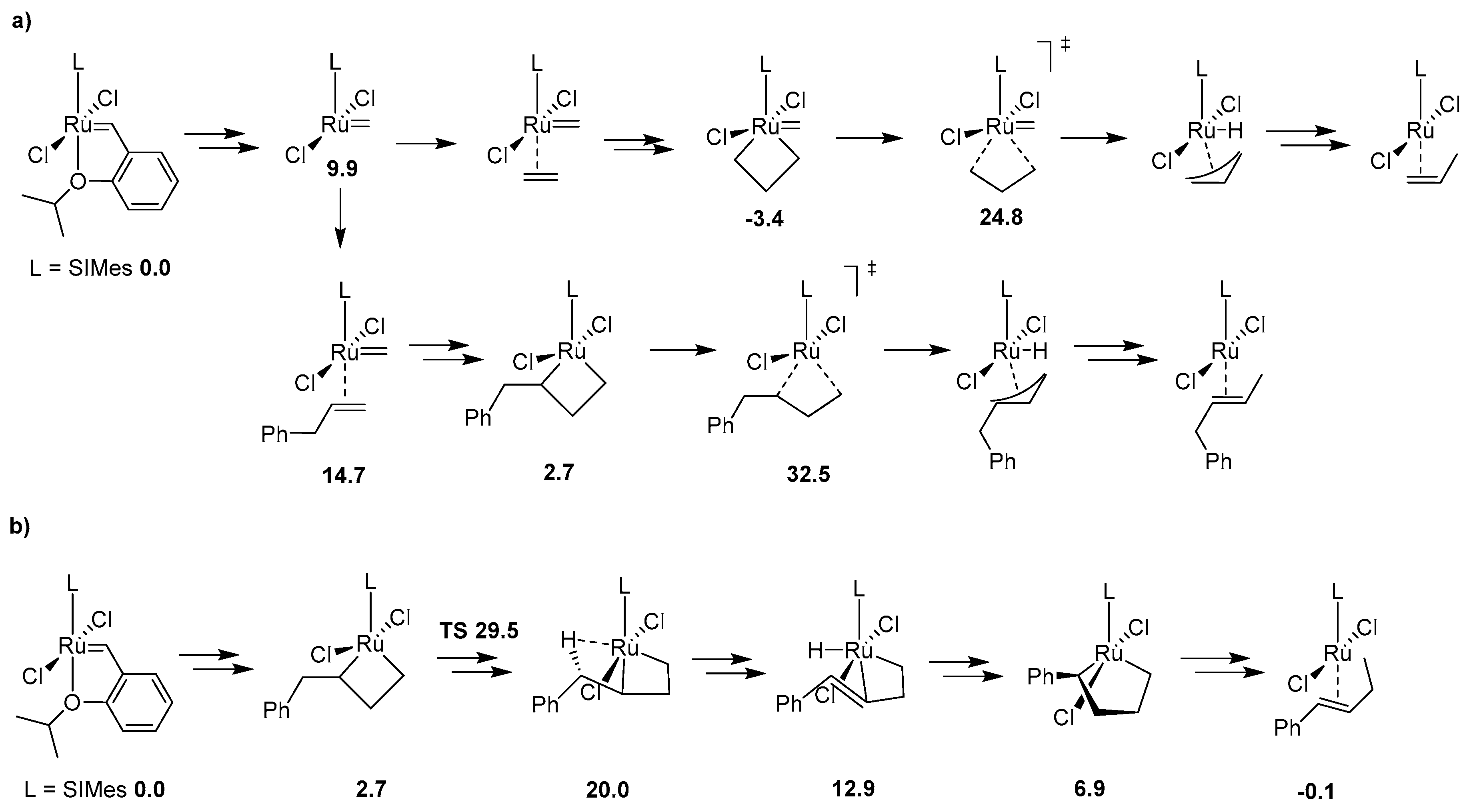
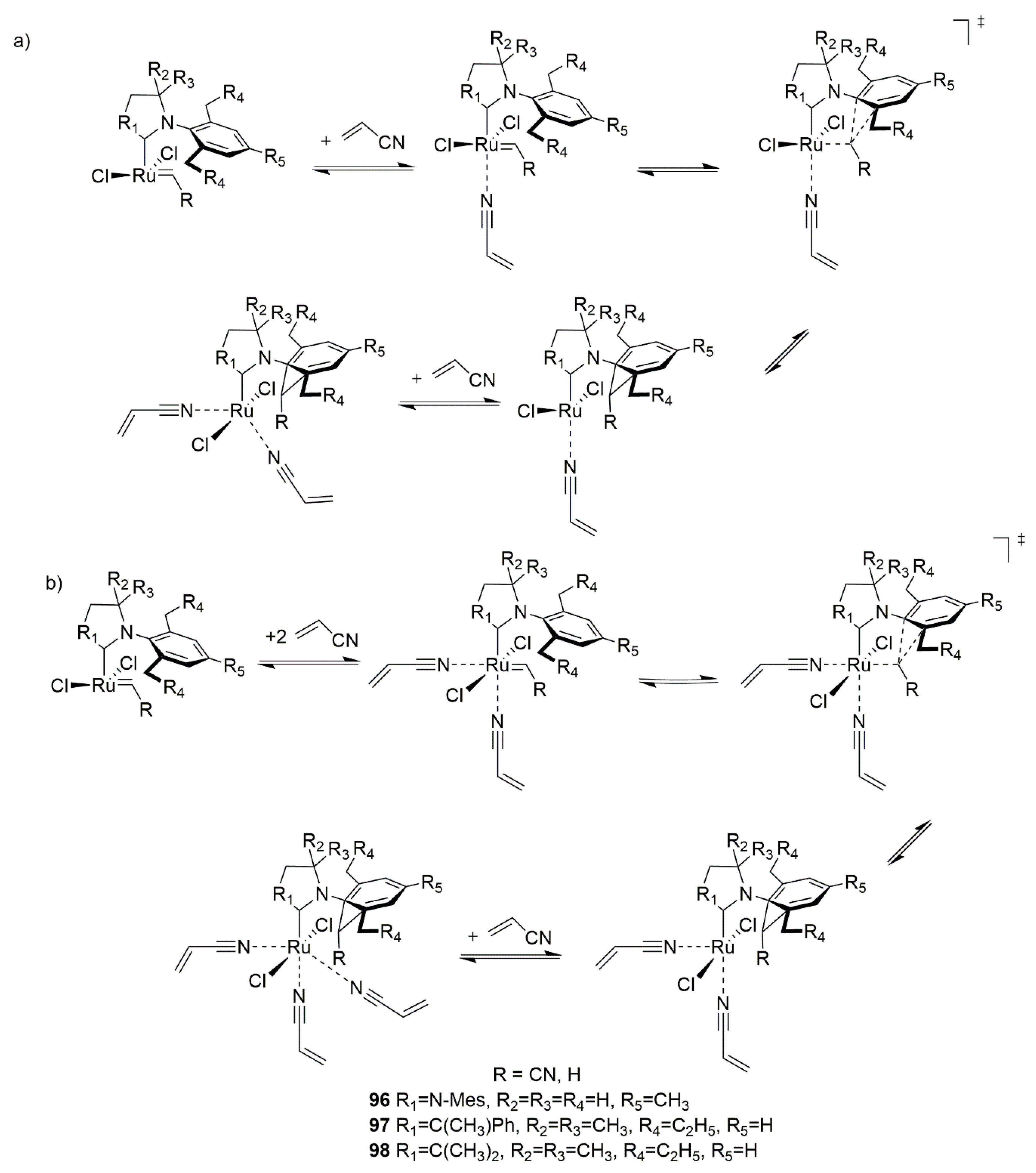
Historically the decomposition studies of Grubbs-type complexes have been initiated by Grubbs and co-workers, who investigated the phosphine-driven decomposition of 1st and 2nd generation Grubbs complexes; this is described later in this review. Soon after their studies van Rensburg et al. showed, however, that for these catalysts a substrate-induced pathway is also viable. In their seminal work authors of this study performed density functional (DFT) calculations on the 1st and 2nd generation Grubbs complexes and showed that in both cases the Gibbs free energy barriers of β-hydride transfer from metallacyclobutane are relatively low (16.9 kcal/mol for 1st generation Grubbs catalyst and 24.3 kcal/mol for 2nd generation one) [27] (Scheme 1). To confirm these findings they also performed ruthenium methylidene-catalyzed metathesis of ethylene which yielded, among other olefins, 1-propene and different isomers of butane, which is possible only via β-hydride transfer. It is worth mentioning here, that a different mechanism for olelfin isomerization (but not catalyst decomposition) was proposed 2 years earlier by Nolan and Prunet [28]. Van Rensburg et al. also found that 38% of the starting 2nd generation methylidene has decomposed after 16 h exposure to excess ethylene in 40 °C. This work marked the first study where the evidence of olefin-driven decomposition of ruthenium metathesis catalyst has been given, together with a feasible mechanistic explanation. It was followed in 2006 by a DFT study from the same group where the decomposition pathways of first-generation and second-generation Grubbs catalyst as well as two isomeric Phobcat catalysts were studied [29]. In this work the entire decomposition pathway with ethylene, starting from precatalysts and ending at the propene-associated catalyst, was explored. The results were similar to the previous study, as the transition state leading to the β-hydride transfer from metallacyclobutane was in each case the rate-limiting step of the decomposition, with values ranging from 23.4 to 26.9 kcal/mol.
In the same year an interesting experimental study performed in the continuous flow reactor by Lysenko et al. showed that ethylene pretreatment of the 1st generation Grubbs catalyst induces catalyst’s deactivation [30]. Interestingly, similar pretreatment with cis-2-butene had little effect on the catalyst activity. Authors of this investigation concluded that the methylidene species generated from the catalyst and ethylene is unstable and promotes catalyst loss, while ruthenium alkylidene derived from 2-butene is rather stable and allows to sustain catalyst integrity. This was the first indication that various olefins may deactivate/decompose metathesis catalysts with a different rate or to a different extent.
In 2007, Nizovtsev et al. perform a similar study to the original work of van Rensburg but using the Hoveyda-Grubbs catalyst [31]. They showed that in a metathesis reaction with ethylene a mixture of olefins is obtained. with a preferable formation of propylene. Based on these results they postulated that the decomposition route utilizing the β-hydride transfer is preferred, although suggested also other mechanism leading to for example, diruthenium species.
Due to the improvements in computational techniques more and more complex and sophisticated studies of ruthenium catalysts became possible in the next decade. One of the most prominent works dealing with Ru metathesis catalysts decomposition from that time is the investigation by Ashworth et al. describing the competition between the metathesis and isomerization [32]. In this work authors computationally investigated three different, complete routes of propylene isomerization using the van Rensburg mechanism, Nolan-Prunet mechanism and finally a direct hydride transfer from a ruthenium hydride species. (Scheme 2). They found very high energy barriers of hydrogen transfer to methylidene for the Nolan-Prunet mechanism (over 28 kcal/mol with respect to methylidene), which were much higher than those for the van Rensburg mechanism. They concluded that the methylidene route using the van Rensburg mechanism is the most favorable from the energy point of view and once again reaffirmed that metallacyclobutane decomposition is the rate-limiting step of this decomposition path. They also showed that for olefins (such as allylbenzene) the barrier of the van Rensburg transition state is much higher (around 8 kcal/mol higher than for ethylene). This finding was later confirmed by Trzaskowski et al., who studied the impact of structural changes in the olefin, as well as in the carbene part of the catalyst on the energetics of van Rensburg’s decomposition [33,34].
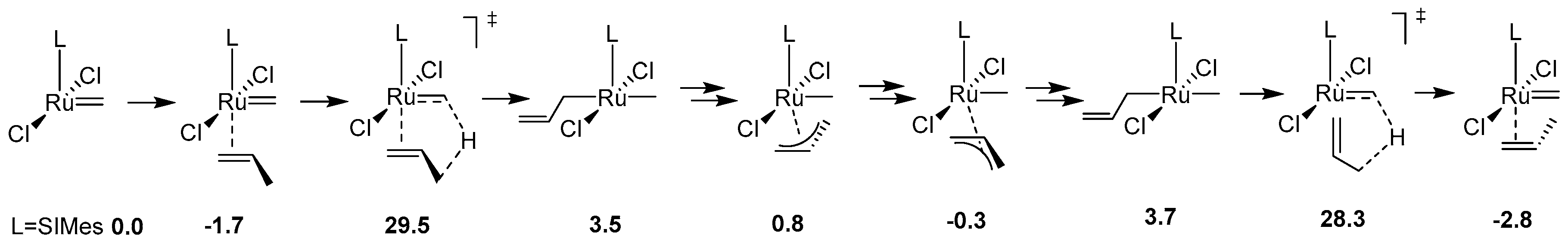
The most thorough computational analysis of many possible decomposition routes of Hoveyda-Grubbs complexes has been presented in 2017 in the work from Jensen laboratory [35]. In this study DFT approach has been used to explore all possible events during metathesis catalytic cycle, including deactivation, decomposition, regeneration, isomerization and the proper catalysis itself. From the decomposition point of view this investigation is particularly valuable, since it deals with almost all theoretically plausible degradation pathways, including those very unlikely (due to very high energy barriers) and therefore rarely/never studied. These included, for example, the previously mentioned Nolan-Prunet mechanism, the C–H activation of precatalyst and methylidene, the formation of diruthenium species and a few other mechanisms. Nevertheless, it was shown again that for ethylene the lowest Gibbs free energy barrier of decomposition is associated with the van Rensburg mechanism. Additionally, it was also demonstrated that the analogous barrier of decomposition with a larger olefin, allylbenzene, is 7.7 kcal/mol higher with respect to the precatalyst. (Scheme 3) Finally, a new mechanism of decomposition was proposed for olefins composed of at least three carbon atoms, namely 1-alkene-triggered decomposition, which proceeds via a trigonal bipyramidal metallacyclopentane intermediate (Scheme 3). The energy barrier for the rate-limiting step of this path was estimated at 29.5 kcal/mol with allylbenzene, around 3 kcal/mol lower than for the van Rensburg route, rendering this mechanism likely.
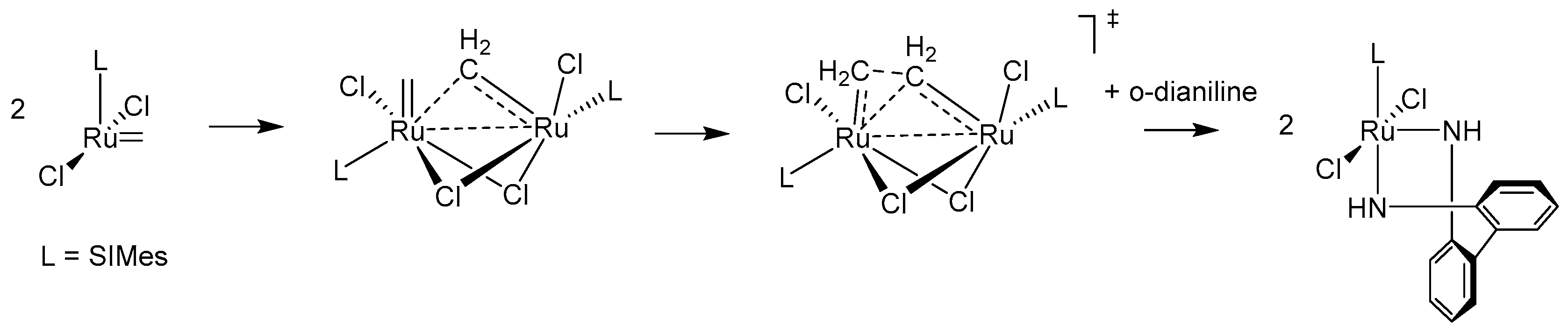
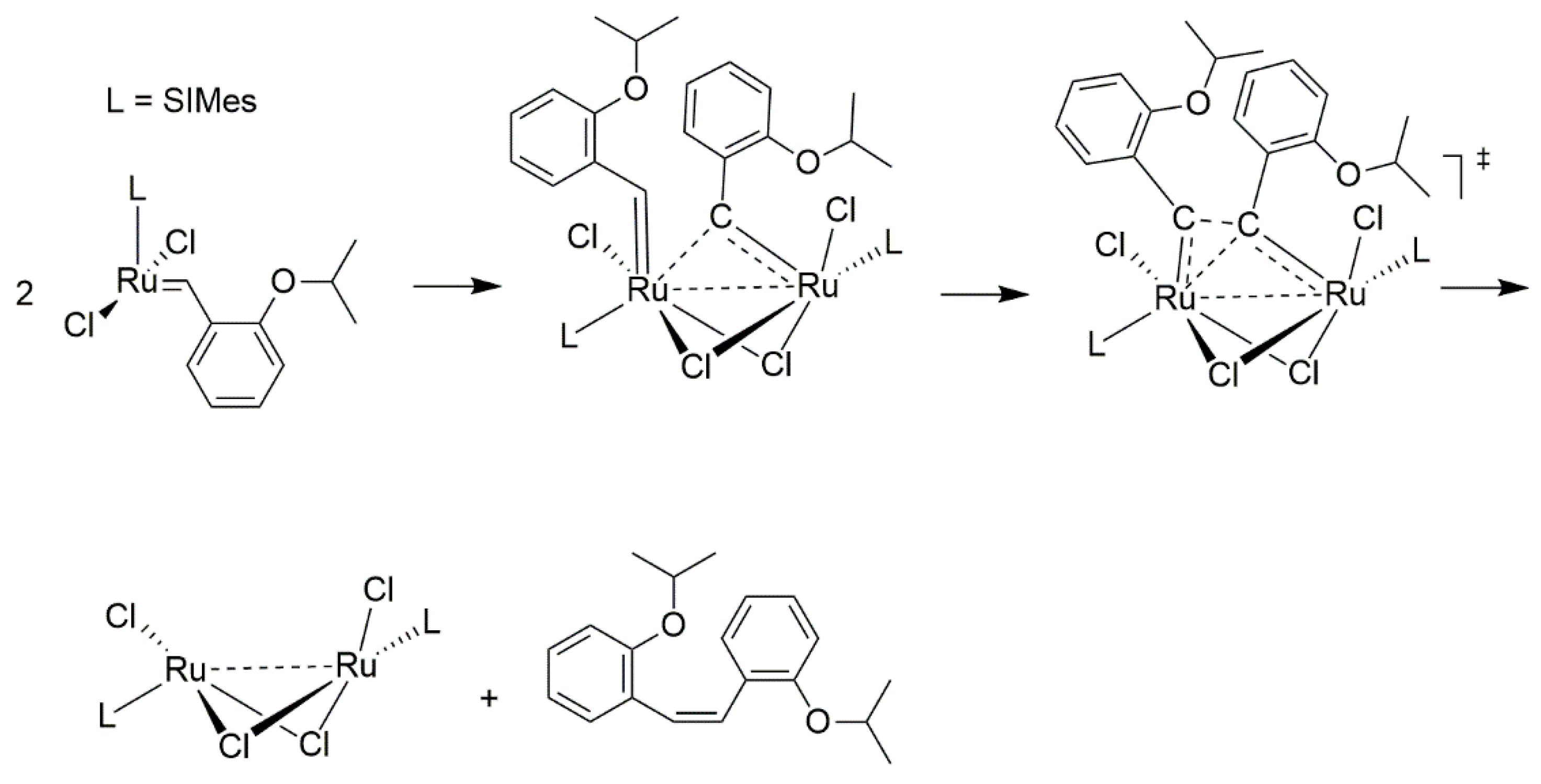
The final, for now, piece of olefin-driven decomposition puzzle was discovered in 2018 when a new mechanism involving bimolecular coupling has been suggested by Fogg and Jensen [36]. The experiments and identification of ruthenium decomposition products of Hoveyda-Grubbs catalyst allowed them first to rule out the Buchner expansion and NHC cyclometallation pathways due to the fact that the NHC remained intact. On the other hand combined kinetic and DFT mechanistic studies allowed them to suggest three different bimolecular complexes bridged either by two Cl, two ethylene or one –CH2– group. The final conclusion of this work was that the CH2–bridged complex is the only likely candidate for the further decomposition, as this transition state had a relatively low Gibbs free energy barrier (Scheme 4). These findings were further supported by experimental data from decomposition experiments of Hoveyda-Grubbs and usually more stable cyclic alkyl amino carbene (CAAC) catalysts [37]. As expected, CAAC catalysts were shown to undergo little or no β-hydride elimination via the van Rensburg mechanism, contrary to the Hoveyda-Grubbs catalyst. Surprisingly, CAAC catalysts were found to be more susceptible to bimolecular coupling than the Hoveyda-Grubbs catalyst. The final conclusion of the study was that CAAC catalyst offer a very high productivity combined with little decomposition particularly at ppm loadings, since at such low catalysts concentrations the impact of bimolecular coupling is relatively low.
2.2. Alcohol and Alkoxy-Driven Decomposition
Despite the excellent tolerance of various functional groups to ruthenium metathesis catalysts, conducting such a transformation in water or protic solvents, such as alcohols, remains a challenge. Alcohols and water can act as either Lewis acids or bases but in the presence of metathesis catalysts they usually act as a Lewis acids, abstracting a chloride from the catalyst. On the other hand simple alkoxides are strong bases and act a Lewis bases, usually attacking directly the ruthenium core bearing the formal +2 charge. Here we will describe research focused on the understanding of mechanisms governing the decomposition of metathesis catalysts.
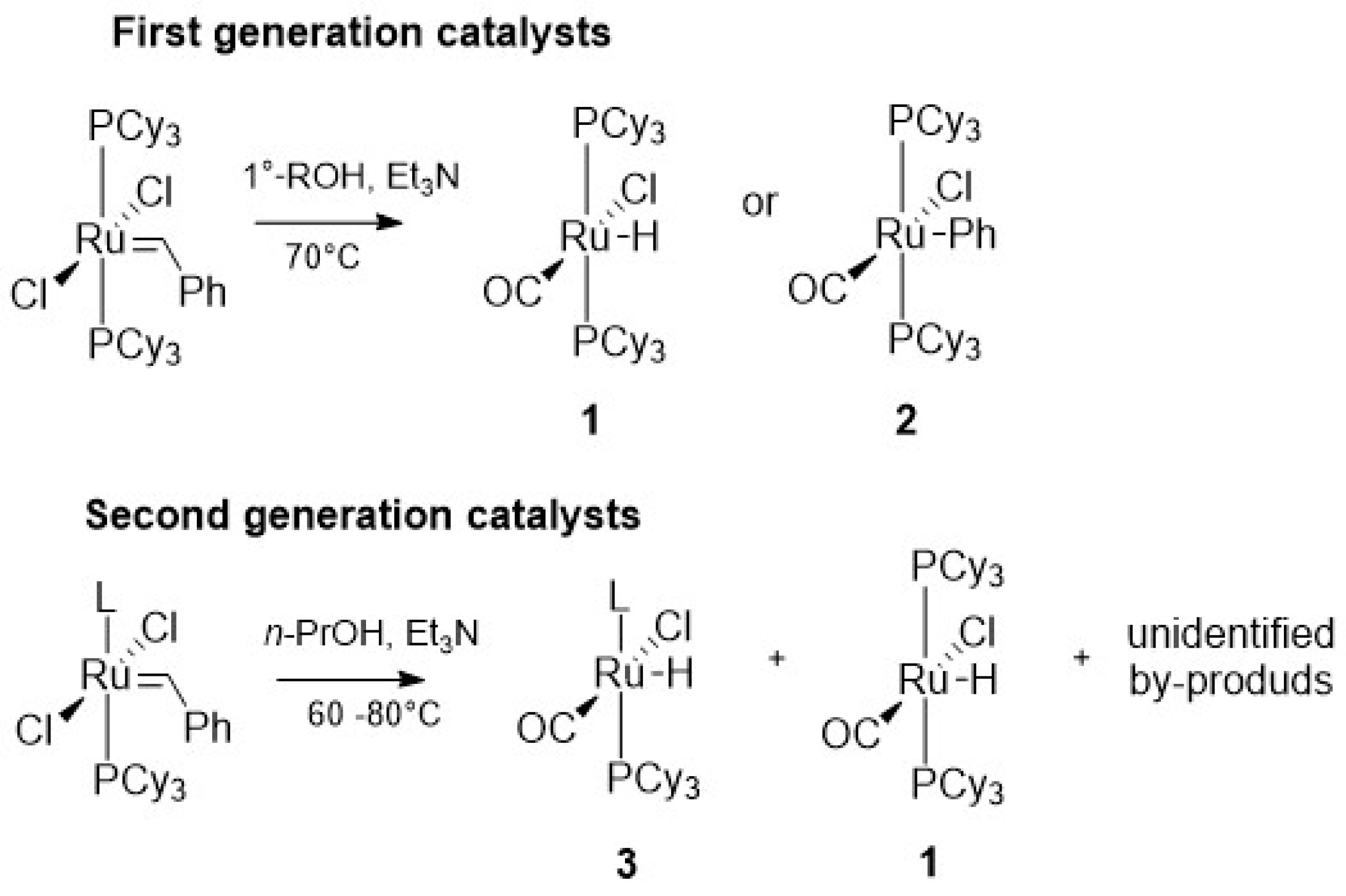
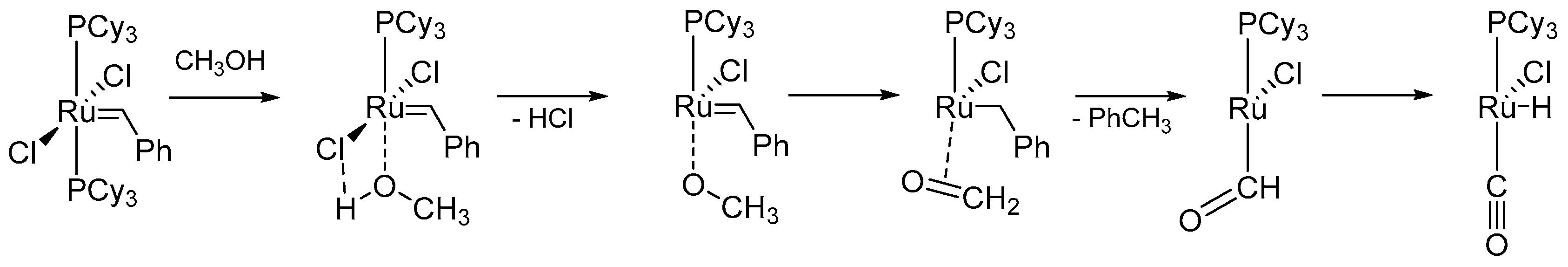
One of the first degradation mechanism of 1st generation Grubbs catalysts in the presence of the alcohols was proposed by Dinger and Mol [38]. The research was mainly dictated by the curiosity to explain the formation of ruthenium hydride 1, a potential isomerization catalyst. The authors observed the formation of 1 from GI in primary alcohol/toluene solution at 70–75 °C overnight (Scheme 5) and noticed that this reaction was accelerated by addition of appropriate bases (such as Et3N, CH3ONa, NaOH, K2CO3). Moreover, based on labeling experiments, they confirmed that the source of hydrogen in the reaction is alcohol dehydrogenation occurring in a non-catalytic manner.
The postulated mechanism suggests that the hydrochloride is eliminated as Et3N*HCl as a result of alcohol dehydrogenation. At the same time, aldehyde, formed in this stage, reacts with the metal center, which after decarbonylation gives carbonyl and ruthenium hydride species (Scheme 6). It needs to be highlighted that when reacting with longer aliphatic alcohols, beside 1, toluene was formed as the main by-product. This results implies that the above-mentioned mechanism cannot be directly extended to other alcohols with longer aliphatic chains and is valid only for small and simple alcohols. Additionally, under the same reaction conditions benzyl alcohol gave an unexpected complex 2, inconsistent with last step of the proposed mode of action (Scheme 5).
The same group extended this research to 2nd generation Grubbs (GII) complexes [39,40]. As in the case of the GII complex, the formation of the corresponding ruthenium hydride 3 was also observed but two other hydrides were also found (Scheme 5). One of them was unidentified and the other turned out to be complex 1, resulting likely from the exchange of the N-heterocyclic carbene (NHC) ligand for phosphine [39]. The authors emphasized, however, that the relatively long time needed to create this complex limited its potential role in competitive reactions. Notwithstanding, when the reaction with alcohol, including benzyl alcohol, was subjected to GII_SIPr, only the formation of an analogous product 3 was observed [40].
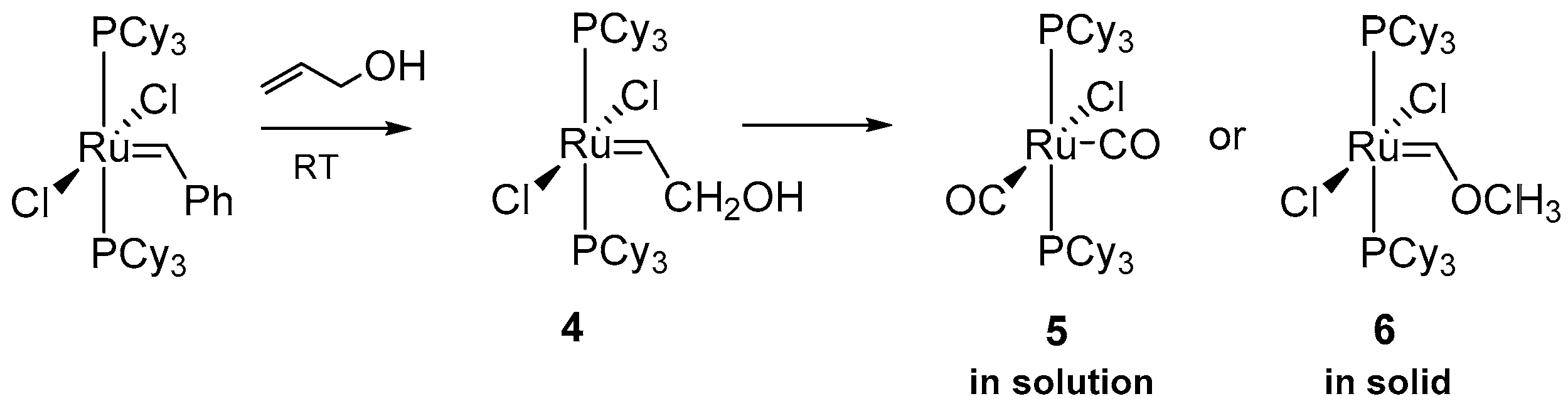
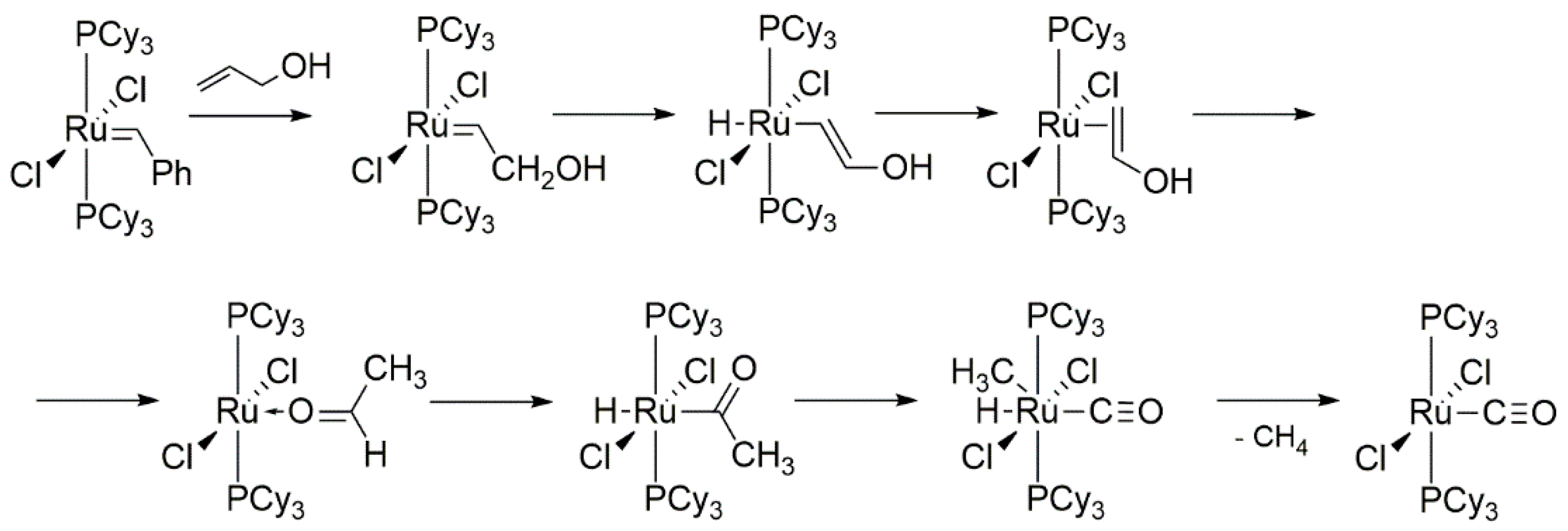
At around the same time Wolf and co-workers conducted very similar studies but with focus on the reaction of ruthenium GI with allyl alcohols at room temperature [41]. In the presence of the allylic alcohol, the GI catalyst gave first the hydroxymethyl carbene 4, which further decomposed in solution to an inactive carbonyl species 5, propanal and small amounts of ethane and propenal (Scheme 7). The mechanism of this transformation (Scheme 8) starts with the β-hydrogen transfer from CH2 of alkylidene group to the ruthenium center and the formation of an hydride intermediate which, after reductive elimination, gives a metal π-complex with enol. In the next step, such a complex isomerizes to aldehyde and, via the C-H bond oxidative addition, an (acyl)hydrideruthenium species is formed. In the final step a methyl migration and elimination yields ruthenium carbonyl complex 5.
The same ruthenium complex 4 can follow a different deactivation pathway in the solid state to rearrange into carben 6. As a possible explanation for these observations authors suggested a nucleophilic attack of the OH group of CH2OH at the carbene carbon to form a protonated oxirane, where [RuCl2(PCy3)2]- acts as a substituent of the three-membered ring. Next, opening of epoxide and hydrogen transfer to CH2 moiety occurs, which leads to the formation of complex 6 (Scheme 8).
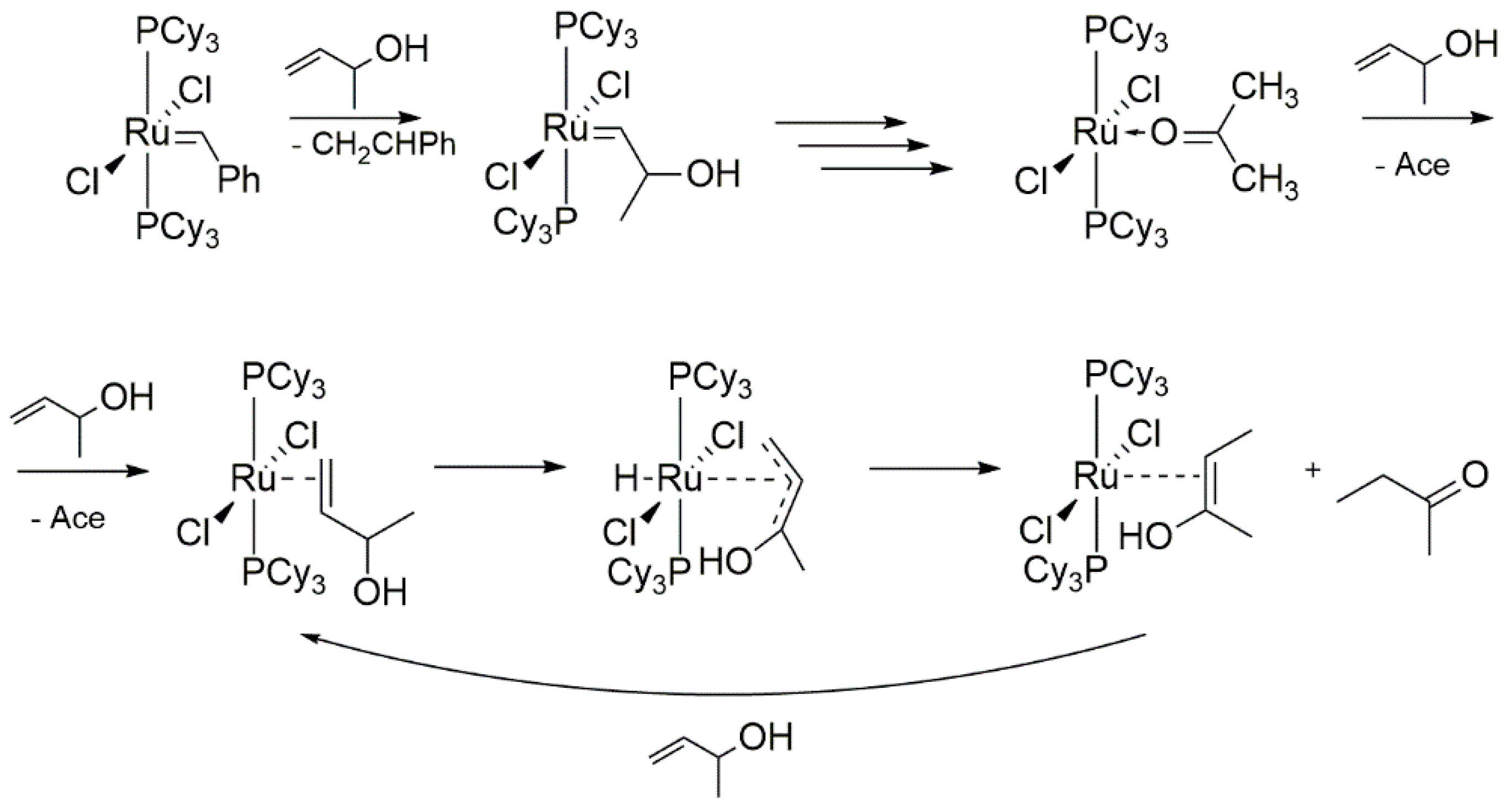
While Mol and co-workers noted that secondary alcohols were ineffective in the reaction with [RuCl2(PCy3)2], Wolf’s Group presented a full reaction pathway using 3-buten-2-ol as an example of such reaction (Scheme 9). The main difference, compared to the previously suggested primary alcohol-driven mechanism, was the inability of acetone to undergo oxidative addition, which allowed the formation of a π-complex with a substrate molecule. The following β-hydride transfer to ruthenium yields a π-allyl-derivative, which could rearrange to a Ru-enol complex. The only product observed in this reaction was butanon, which was formed as a consequence of rapid tautomerization of the starting enol molecule, coinciding with the exchange of acetone.
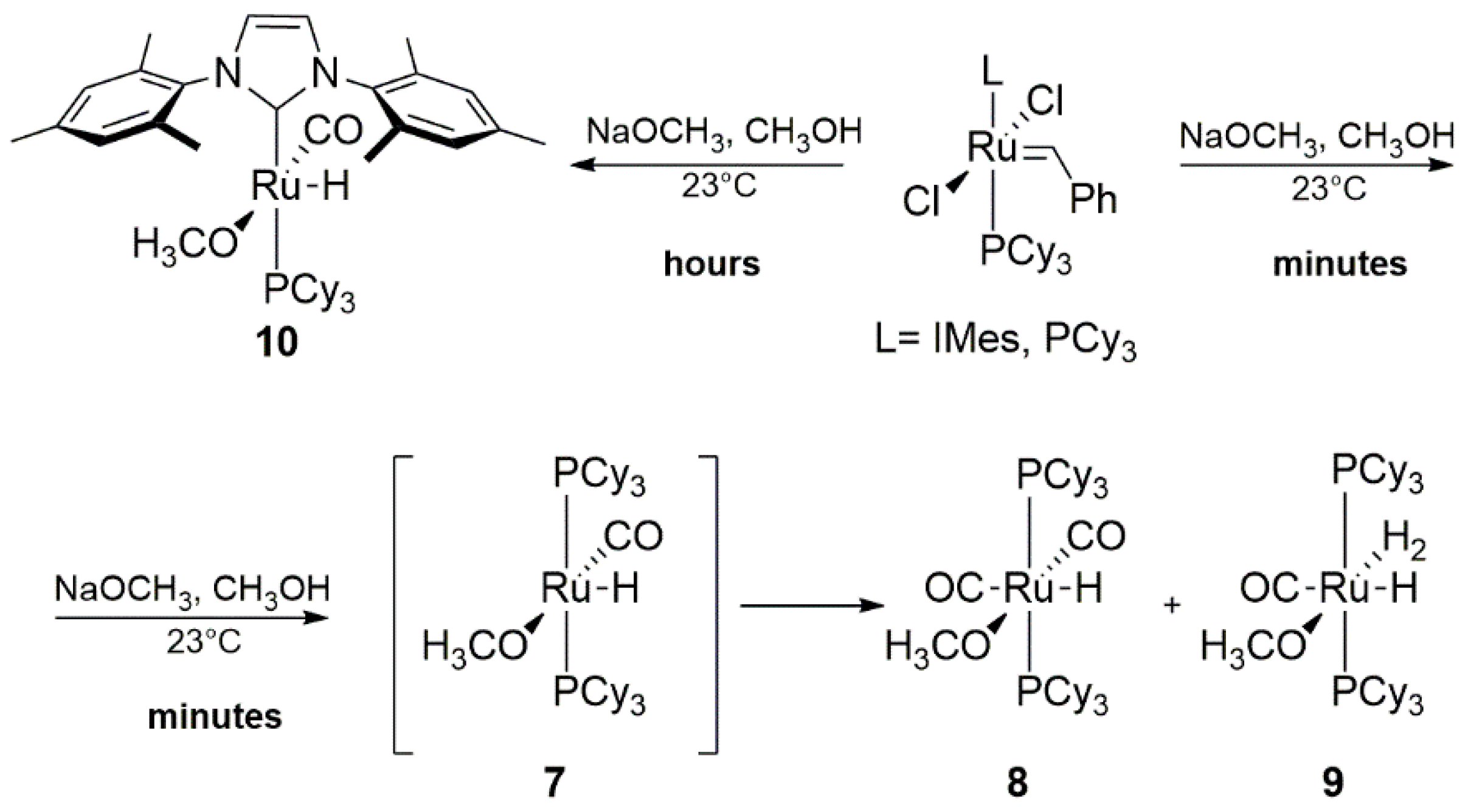
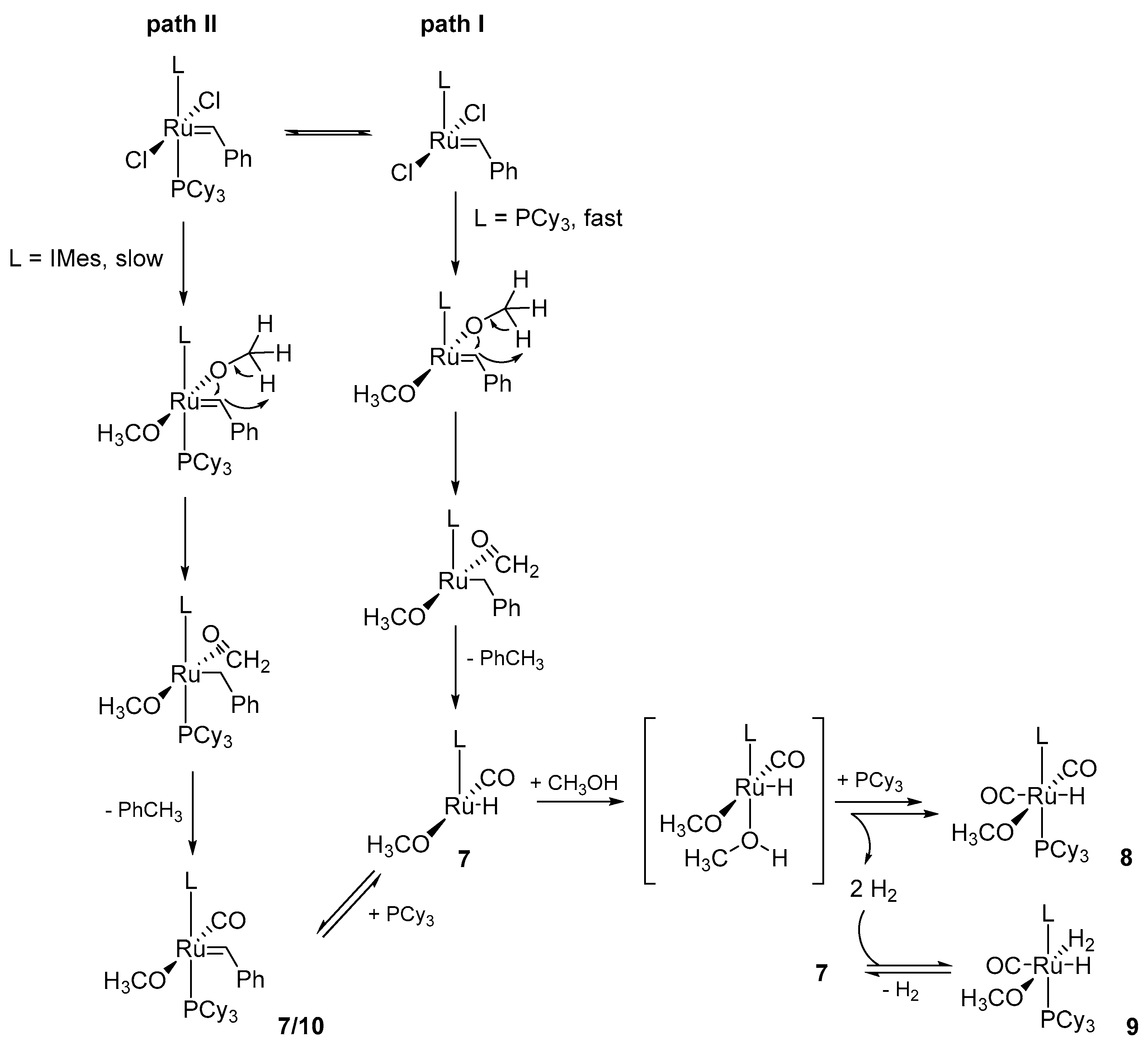
Detailed experimental studies on the conditions required for the benzylidene ligand abstraction of 1st and 2nd generation Grubbs catalysts by reacting with the excess of methoxide were investigated by Fogg and co-workers [42,43]. They showed that at ambient temperatures ruthenium catalysts treated with NaOCH3, used to eliminate nonessential byproducts derived from thermal decomposition, decompose to different hydrogen species depending on the nature of the catalyst [43]. Under these conditions, GI was transformed promptly into two hydride complexes 8 and 9 (Scheme 10), while the second generation GII_IMes gave one stable methoxyhydride species 10, in contrast with previous observations [39,40]. In this case, the relative ease of methanol undergoing β-elimination prevents the formation of carbynes and bisalkoxide complexes, in contrast with analogous reactions with other alkoxides [44,45,46,47]. Additionally, phosphine addition delayed the consumption of the GI catalyst, while the rate of the GII_IMes decomposition was not affected by it. This finding was associated with a higher lability of PCy3 moiety in 1st generation catalysts and an easier formation of a four-coordinated intermediate [Ru(Cl2)(=CHPh)L] (path 1, Scheme 11). Subsequent steps of the suggested mechanism begin with the reaction of the metathesis catalyst via exchange of the chloride ligands followed by the transfer of a hydrogen atom from attached methoxy group to the benzylidene. Next, formaldehyde coordination, insertion of CO, abstraction of benzyl moiety as toluene and rebinding of PCy3 occurs, which is in line with previous observations made by Mol et al. [38]. The formation of a four-coordinate compound as a result of phosphine dissociation is highlighted as the crucial decomposition pathway [43]. A plausible mechanism of conversion of complex 7 to a six-coordinated species 8 and 9 is multi-step and starts with the introduction of an additional methoxide ligand as shown also on Scheme 11.
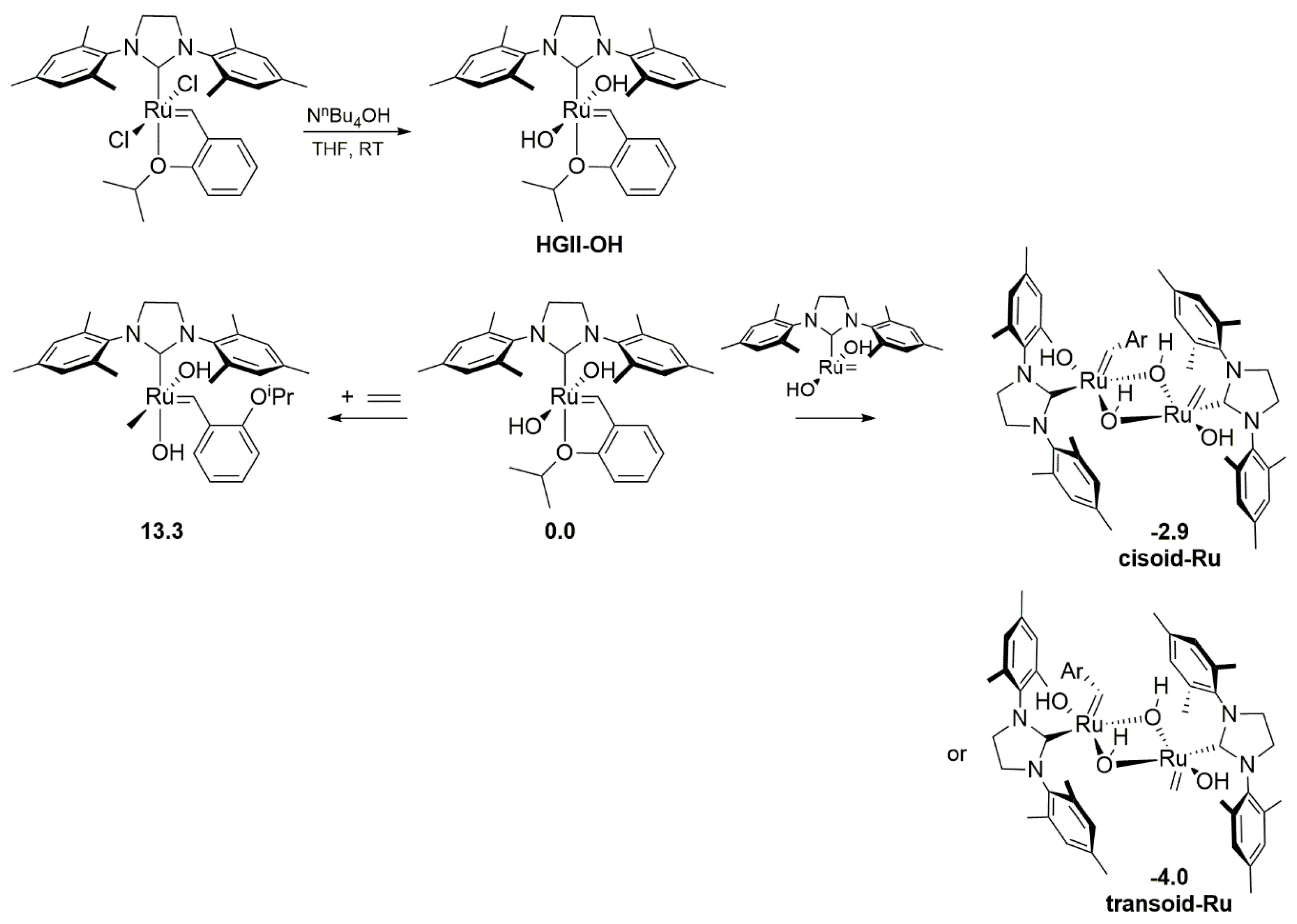
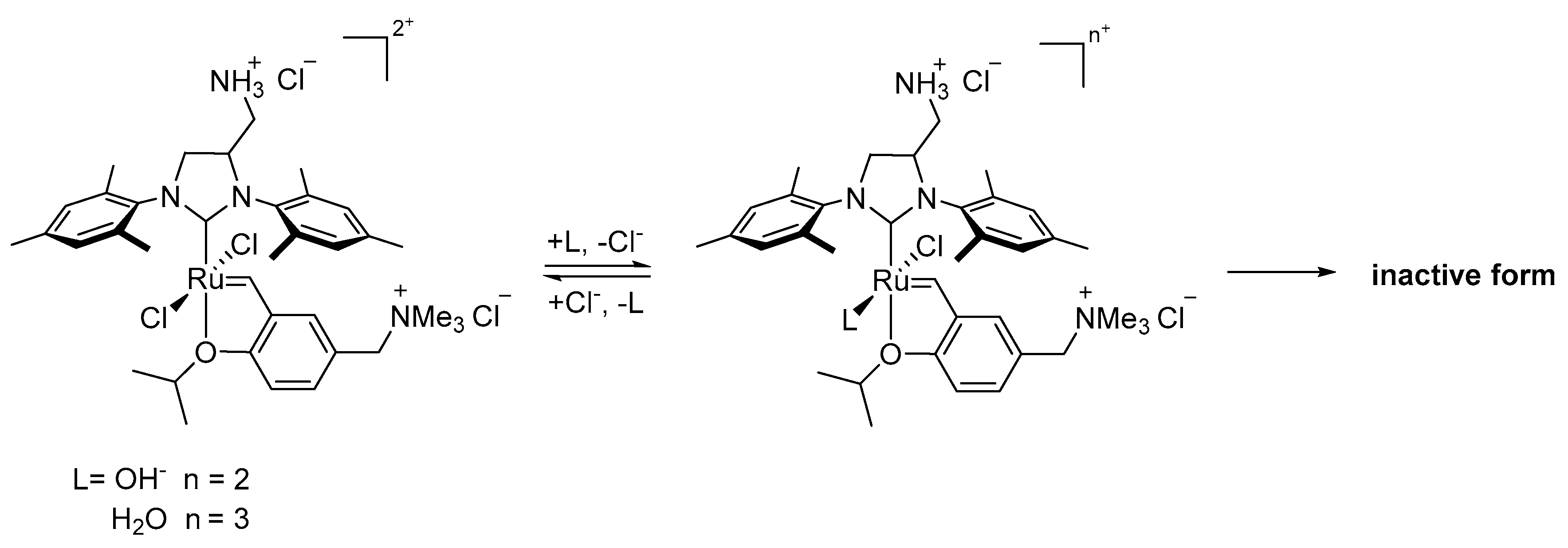
In 2020, Fogg’s group reported a study on the degradation of the 2nd generation Hoveyda-Grubbs catalyst induced by the hydroxide ion [48]. In this work a new pathway of decomposition was proposed (Scheme 12) which differed from the previously proposed decomposition driven by hydroxide in methanol [43]. In the latter work the catalysts (Scheme 10 and Scheme 11) were transformed into Ru-hydride species via β-elimination of a methoxide adduct [43]. In the present work, however, the ring-closing metathesis (RCM) reaction of diethyl diallylmalonate in the presence of [NnBu4][OH] showed a rapid decrease in the catalytic activity of HGII complex. An instant quenching of a similar experiment without the olefin suggested that hydroxide ion attacked precatalyst, yielding the product, which either decomposed during the first catalytic cycle or was inactive in metathesis. The authors synthesized the putative bis-hydroxide complex HGII-OH in 85% yield (Scheme 12) and it demonstrated an almost complete lack of reactivity (<3%) in model RCM reactions. The synthesized bis-hydroxide complex HGII-OH gave styrene ether as the product of metathesis exchange and propenes E/E’ bya β-elimination of a corresponding metallacyclobutane. Since the identified products referred to only 50% of HGII-OH used in the reaction, it was suggested that the bimolecular coupling of bis-hydroxide ruthenium methylidene adduct HGIIm-OH might be responsible for a further decomposition of this species. Computational comparison of the dimerization and the ethylene binding indicated that the hetero-coupling is indeed preferred by ca. 12.4 kcal/mol (Scheme 12). This results was in line with kinetics studies showing that the rate-limiting step of the reaction was the formation of an essential methylidene intermediate HGIIm-OH. Two stable isomers were predicted, transoid- and cisoid-Ru. Additionally, the capture and decomposition of HGII-OH by an already decomposed ruthenium species were noted and explained by a H-bonding capacity of the hydroxide ligand.
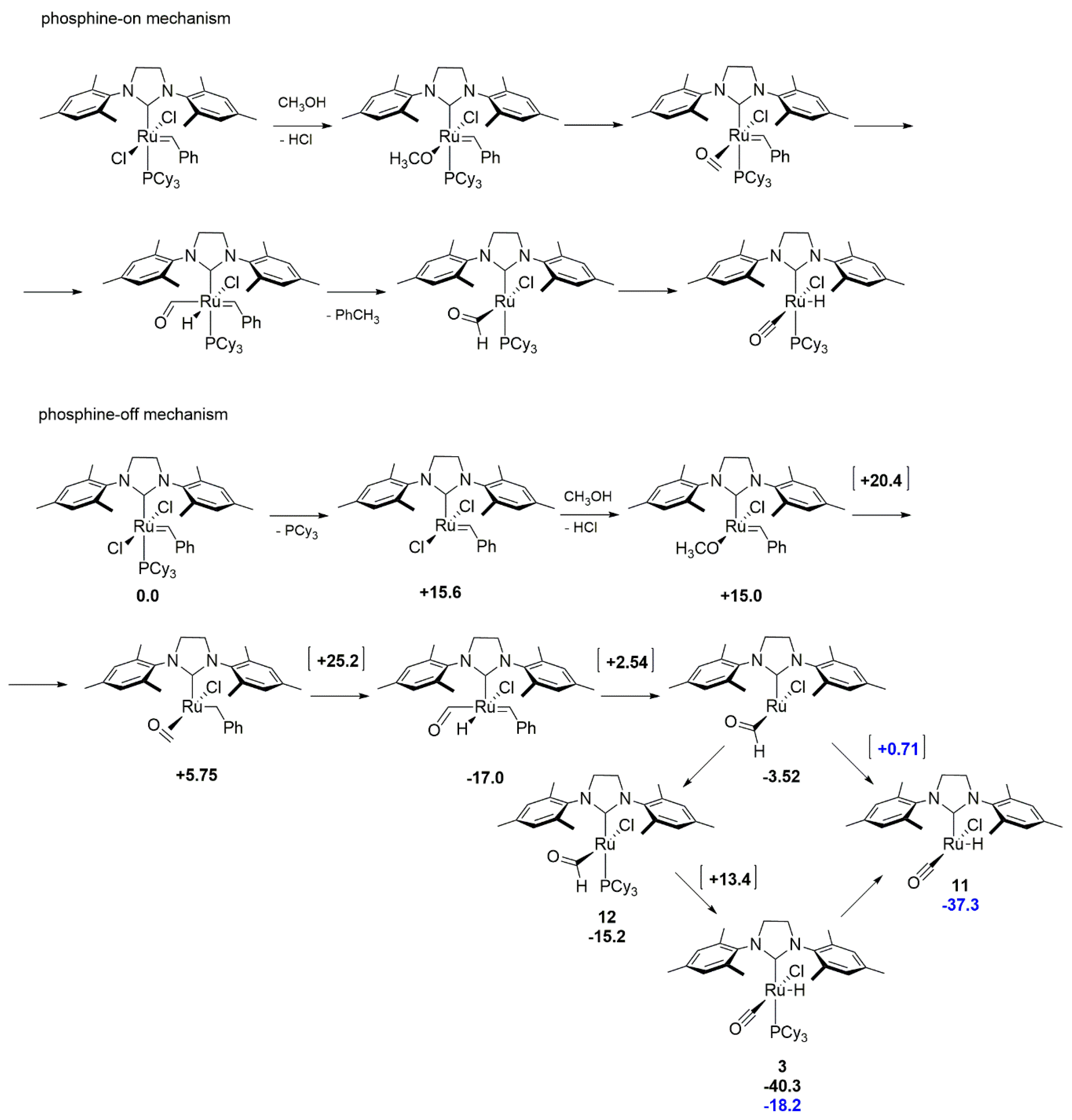
In silico studies of the alcohol-driven decomposition mechanism proposed by Mol [39] for the benzylidene 2nd generation Grubbs-type catalyst was conducted by Young et al. [49] Authors considered two possible decomposition mechanism dubbed phosphine-off and phosphine-on (Scheme 13), which correspond to either a dissociative or an associative metathesis mechanism, respectively. The initial assumption made by authors was that the associative pathway has the advantage of saving the cost of phosphine dissociation but the calculated energy barriers for individual transition states were higher than for the dissociative path, thereby indicating the dissociative mechanism as the preferred one. The value of the estimated Gibbs free energy barrier for limiting transition state (methoxide reaction with benzylidene via hydride transfer) of 55.8 kcal/mol, with respect to the initial complex is, however, at least puzzling. The authors explained this result by noting, that the reverse reaction from the –OCH3 adduct is highly unlikely, thus formally resetting the energy of this adduct to zero, resulting in the final Gibbs free energy barrier of only 25.2 kcal/mol. The last step of this pathway proceeds through an unstable intermediate 11, which converts immediately to ruthenium hydride complex 3, rather than through 12, due to a favorable position of the CHO ligand for hydrogen abstraction.
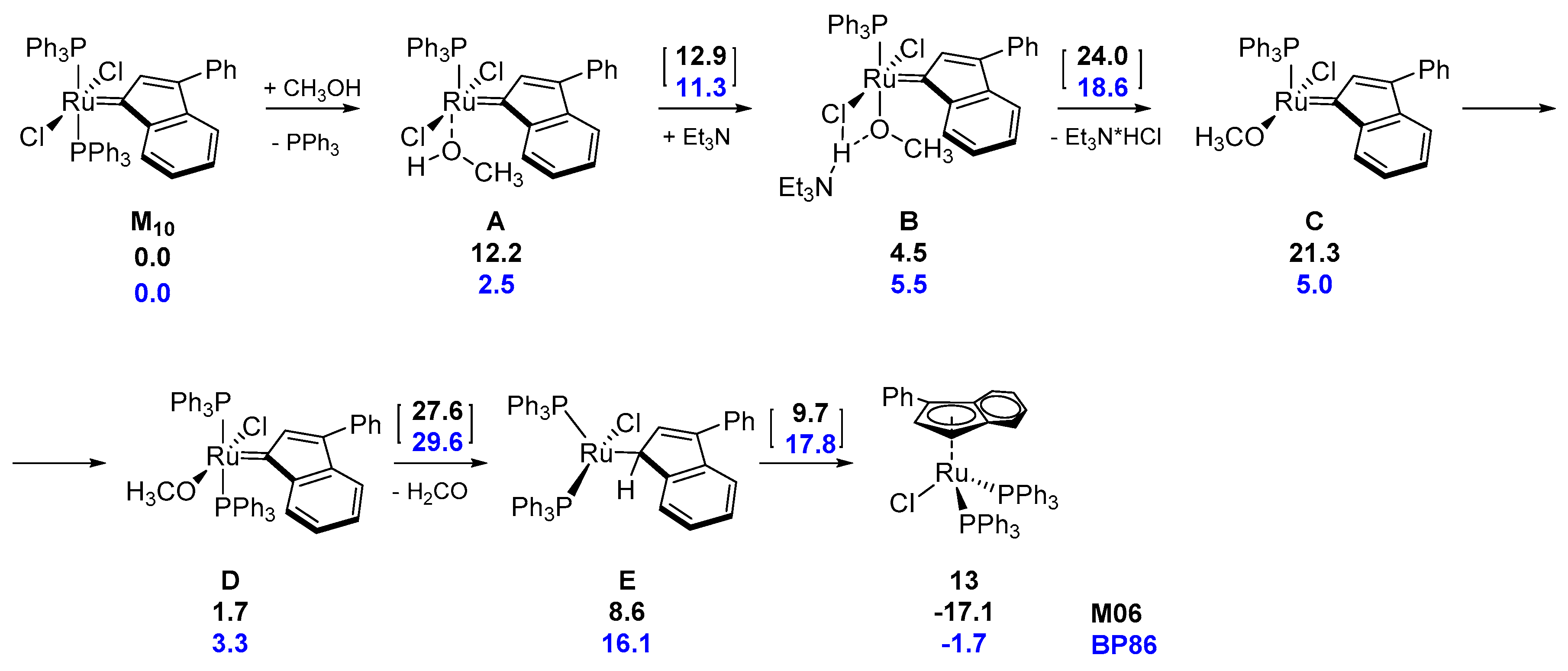
The influence of the indenylidene ligand on decomposition process was examined by Nolan’s group [50]. A quantitatively received and rather unexpected product from a reaction of M10 with a stoichiometric amount of Et3N in ethanol (Scheme 14) demonstrated different decomposition behavior compared to other metathesis catalysts investigated so far. The new η5-ruthenium complex was formed through the phosphine dissociation and rearrangement to an indenyl complex 13. The overall reaction rate, determined by means of nuclear magnetic resonance (NMR), was phosphine independent, although its excess decreases the rate of reaction.
The same group performed also theoretical DFT studies to evaluate energetic cost of such transformation (Scheme 15) using methanol as the model alcohol. The suggested mechanism starts with the substitution of the phosphine by an alcohol molecule and proton transfer from the methanol to the base through transition state TS AB (12.9 kcal/mol), yielding an intermediate with PPh3 ligand trans to MeO. Additionally, methoxy group and adjacent Cl ligand interacting via hydrogen bond with Et3NH+ force the loss of Et3NH+Cl- via TS BC (24.0 kcal/mol) transition state, giving intermediate C with the simultaneous shift of the methoxy group into the vacant cis position with respect to the phosphine. The rebinding of phosphine allows for an energy gain of 19.6 kcal/mol. Based on the analysis of the entire cycle, it was suggested that the rate-limiting step of this reaction is the final hydrogen transfer from the methoxy ligand to the ylidene carbon atom (TS D→E) and the release of formaldehyde. The last isomerization step from η1-species to a more thermodynamically stable form 13 was shown to be practically barrierless. The impact of the base on this mechanism was found to be significant, since it lowers the energy barrier of initial TS BC by ca. 16 kcal/mol, compared to direct H transfer from MeOH to Cl. This is in good agreement with experimental evidence of base-promoted alcoholysis [38,43] and the determined activation energy (25.7 kcal.mol) [50].
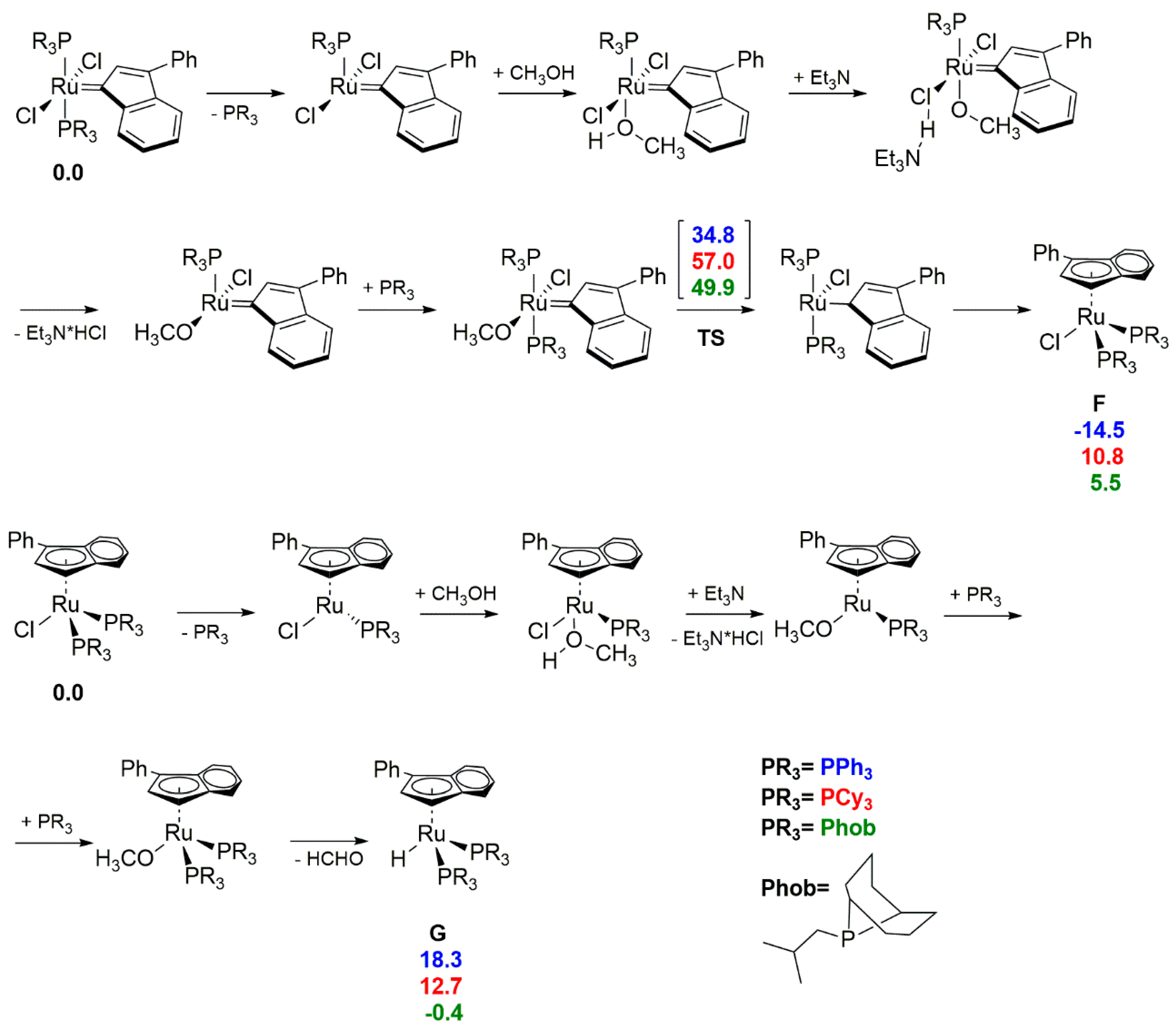
The use of an analogous methodology for obtaining ‘piano-stool’ complexes for other ruthenium catalysts containing the indenylidene fragment was further tested in later studies (Scheme 15) [51,52]. It was shown that complex M1/IndII/M20 followed an analogous decomposition pathway as previously observed for GI, while M11 gave, very similar to 13, a η5-ylidenyl complex 14 in the form of a mixture of rotamers. The ‘piano-stool’ geometry was preserved, with phosphines in the cis-orientation but it seemed that bulky Phoban-type ligands preferred the smaller hydride over the chloride. This finding appears to be confirmed by obtaining corresponding hydride [Ru(H)(η5-3-phenyl-indenyl)(PPh3)2] by prolong reaction time of 13. The final conclusion of the study is that depending on the alkylidene moiety and the phosphine ligand, different decomposition processes can occur for similar catalysts.
Extension of the initial stability research to basic solution of secondary alcohols by Nolan resulted in a corresponding dihydrogen hydride 15 for M1 and IndII catalyst [52]. The same dihydrogen decomposition product gave methylidene derivatives [RuCl2(=CH2)(PCy3)2], as well as 1st and 2nd generation metathesis precatalyst (Scheme 14), in contrary to earlier reports by Mol [38]. The fact that the characteristic decomposition product of hydrogenolysis, complex 15, is formed in an alcohol-driven decomposition reaction and the possibility of its transformation into a hydridocarbonyl species using methanol [53] implied similarities of both processes [52]. The computational modelling of the entire decomposition pathways for M1 (PCy3) and M11 (iBu-Phoban) confirmed that hydrogen transfer from the alkoxide to the indenylidene ligand TS has the highest energy barrier. It was also found that for smaller phosphines, such as PPh3, the η5-chloro complex was more stable than the starting ruthenium carbene catalyst, while for complexes containing the PCy3 or Phoban groups the formation of the intermediate G was unfavored by around 5–11 kcal/mol. Further part of the DFT calculation suggested the favorable formation of hydride 14 (intermediate F) for M11 (Scheme 16). These results were consistent with the experimentally observed formation of 14 instead of chloride complex 13 for the catalyst bearing a Phoban ligand.
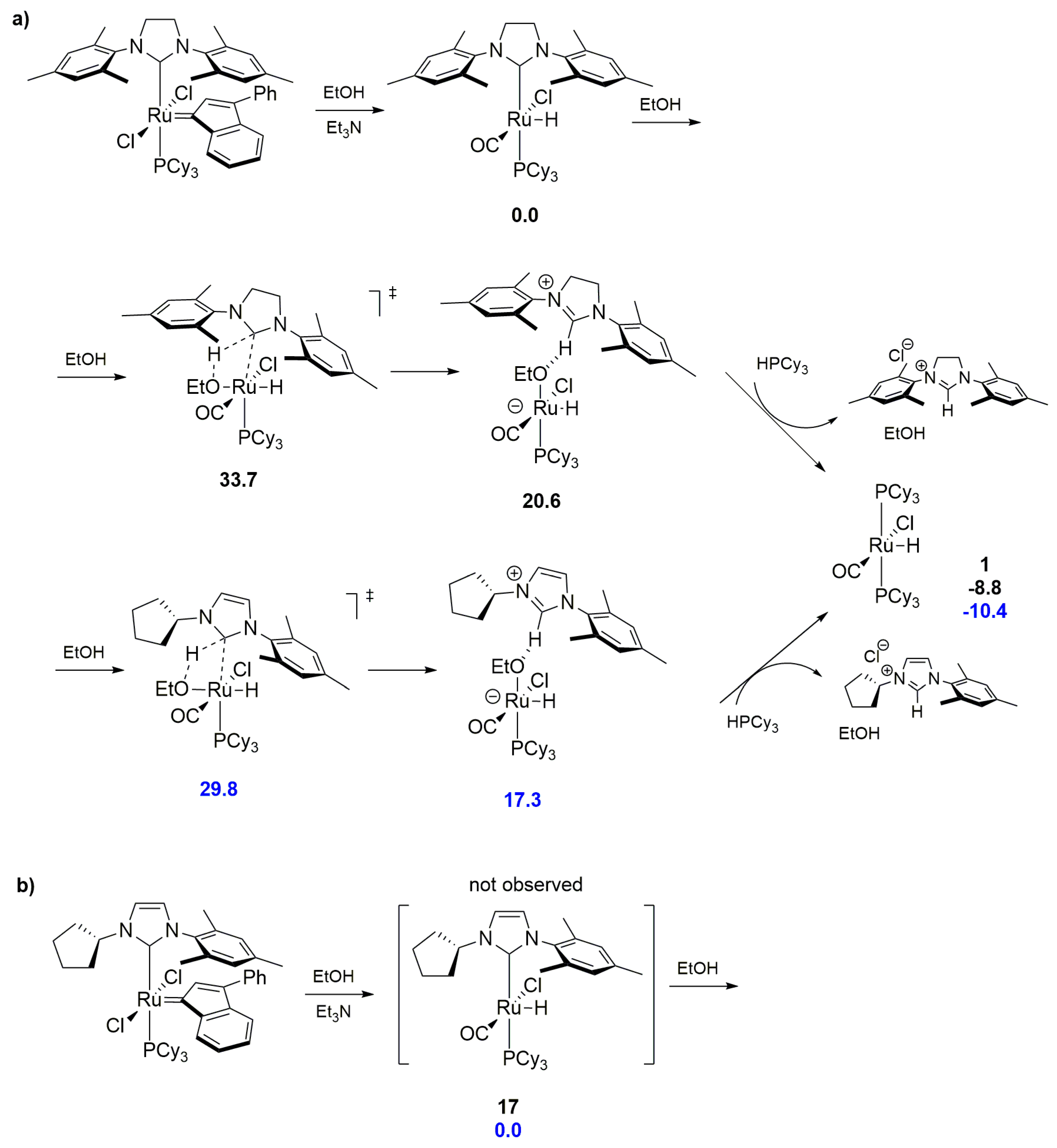
In 2016, Rouen et al. also noticed the formation of a bisphosphine hydride complexes from an unsymmetrical 2nd generation idenylidene catalysts in the presence of an alcohol [54], similar to the previous works of Cavallo and others [52]. The authors explained the formation of the complex [RuCl(H)(CO)(PCy3)2] by the protonolysis of the NHC ligand in compound 17, the formation of which in the reaction was not observed but only inferred on the basis of the identical reaction of the M1 catalyst (see Scheme 14). DFT calculations of the substitution of N-heterocyclic carbene by an ethanol molecule indicated that for a less crowded, unsymmetrical ligand, the energy barrier of the crucial transition state was 3.9 kcal/mol lower than for the standard SIMes carbene (Scheme 17).
2.3. Phenols
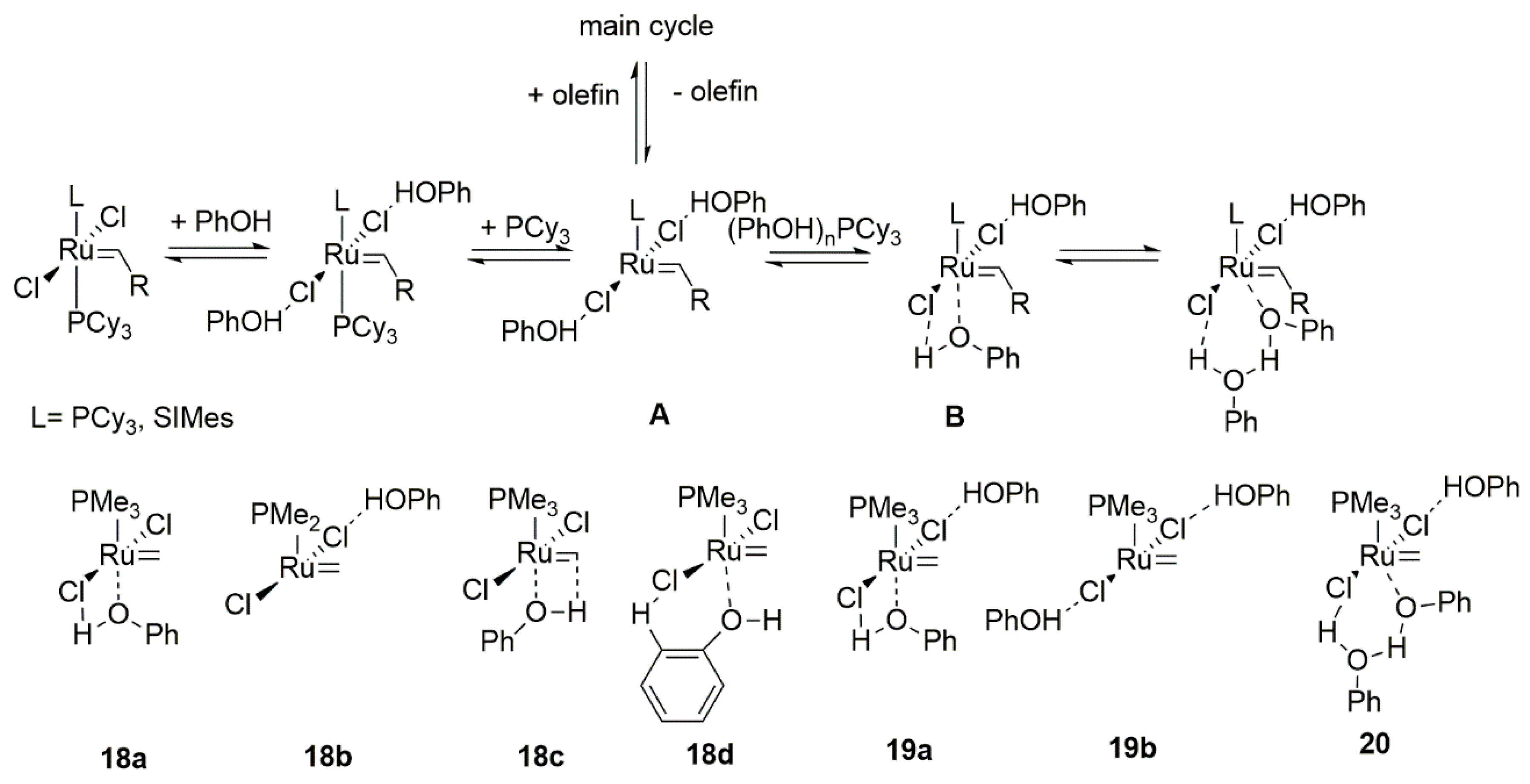
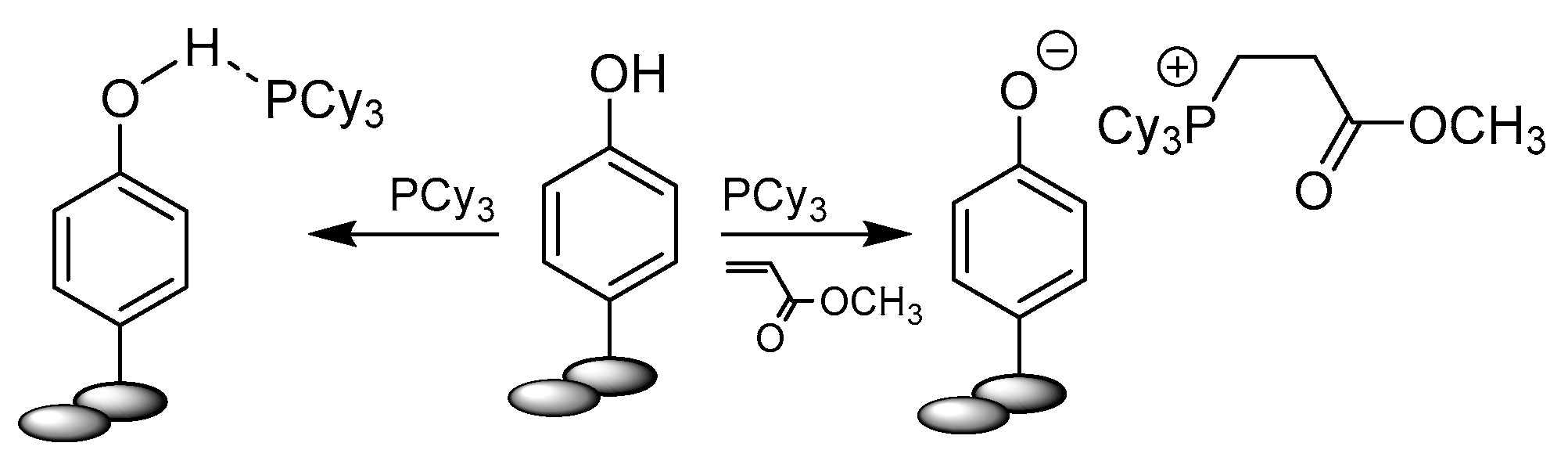
Phenols are a distinct class of compounds from alcohols due to their different chemical properties. In the case of ruthenium metathesis catalysts they mode of action is, however, similar to alcohols and based on Lewis acidity. In 2005 Forman et al. reported suppression of metathesis carbene catalyst by the addition of phenol [55]. An unexpected beneficial effect of enhanced activity and lifetime of active catalyst form was observed for the GI catalyst in the self-metathesis of 1-octene. A similar effect was observed, though to a lesser extent, for very acidic alcohols such as trifluoroethanol, perfluoro-tert-butyl alcohol or hexafluoro-2-propanol. Grubbs 2nd generation catalysts showed lower activity in the self-metathesis reaction but a significant improvement in the cross-metathesis with methyl acrylate in the presence of phenols. The NMR kinetic studies revealed that the type of phenol used alters the phosphine dissociation rate and in high concentrations it may sequester it in the form of (PhOH)nPCy3. In general, the 1st generation catalyst with less acidic phenols exhibited excellent performance in metathesis, which resulted from impeding of the phosphine rebinding to ruthenium. Based on the experimental results and additional DFT calculations, a mechanism for metathesis in the presence of phenols was proposed (Scheme 18). Computational study pointed to 18a (−11.7 kcal/mol) as the more favored mode of coordination over 18b (−3.9 kcal/mol) and at the same time showed an increase in the stability of active catalyst with a growing number of coordinated phenols molecules (20 −26.3kcal/mol > 19a −16.6kcal/mol > 19b −8.0kcal/mol). The energy barriers for the dissociation of model PMe3 for both 1st and 2nd generation catalyst was higher when phenol was coordinated. Moreover, the carbene carbon atom was activated by enhancing its electrophilicity, as demonstrated by the charge distribution analysis. The proposed equilibrium between intermediates A and B promotes the latter one and prevents the decomposition of the 14-electron species A.
2.4. Water-Mediated Degradation
Ruthenium-based olefin metathesis catalysts are usually non-soluble in water and specific modifications to their structure need to be introduced to make them water-soluble. Even in such cases water, however, can degrade these complexes. Similarly to previously described alcohols, water can act as either Lewis acid or base, though in the reported decomposition studies it usually acts via OH- ions.
In 2000 Grubbs and co-workers observed the decomposition of the 1st generation ruthenium metathesis catalysts, while studying the polymerization reaction in protic solvents. The degradations was furthermore promoted by hydroxide ions [56]. As a result of the stoichiometric reaction of the ruthenium carbene complex with NaOD in D2O, a new product without the ylidene substituent was formed, which further decomposed to a mixture of unknown products. Unfortunately, the nature of this transformation and the exact structure of the decomposition product were unknown at that time. It was also suggested that the presence of acid additives prevents the catalyst decomposition by avoiding hydroxide ions formation in the reaction of phosphines with water.
The idea of conducting metathesis in protic solvents and water was later grasped by many groups and a number of reports can be found in the literature experimenting with this approach [57,58,59,60,61,62,63,64,65,66,67,68,69]. They main focus of these studied is on introducing various modifications to the catalyst structure to improve their solubilities, testing different co-solvents [70] and checking their effectiveness in model reactions. One of the more interesting findings in this area is the observation made by Raine that ethers used as co-solvents with water give the highest RCM metathesis yields. Authors suggested that ethers are more likely to coordinate to the active intermediates and thus protecting it from water and maintaining the catalyst in the solution [70].
Hirota and Matsuo used an interesting approach to the problem of ruthenium catalysts’ degradation in water [71]. They applied a strategy consisting of suppressing the loss of chlorine ligands and thus increasing the activity of the Hoveyda-Grubbs-type ruthenium catalysts in water. Their assumption was based on a widely accepted fact that the ligand exchange modifies ruthenium metathesis catalysts performance [72,73,74,75], as well as on earlier reports indicating that the ligand replacement could lead to the formation of an inactive product or precatalyst dimer [72,74,76,77,78]. UV-Vis spectroscopy investigation of HG stability in neutral aqueous solution without KCl showed the decrease of a characteristic metal-to-ligand charge transfer absorption band (ca. 373 nm) and the formation of a product which showed no activity in RCM. There were no intensity changes of the band in the presence of the salt even after 16h. Based on these results, a mechanism of chloride ligand effect was proposed (Scheme 19), in which the presence of the salt changes the equilibrium toward chloride-bound complex, while its absence facilitates the substitution of one chloride ligand with water or hydroxide ion, ultimately leading to the deactivation of the catalyst. The structure of the final Ru-species was not determined but authors speculated that the formation of a Cl-bridged dimer or oxo-bridged form was likely. Although the exchange reaction is not a decomposition reaction, the loss of the ligand may lead to less stable intermediates and introduce new degradation pathways.
2.5. Phosphines and General Routes to Metallacyclobutane Deprotonation
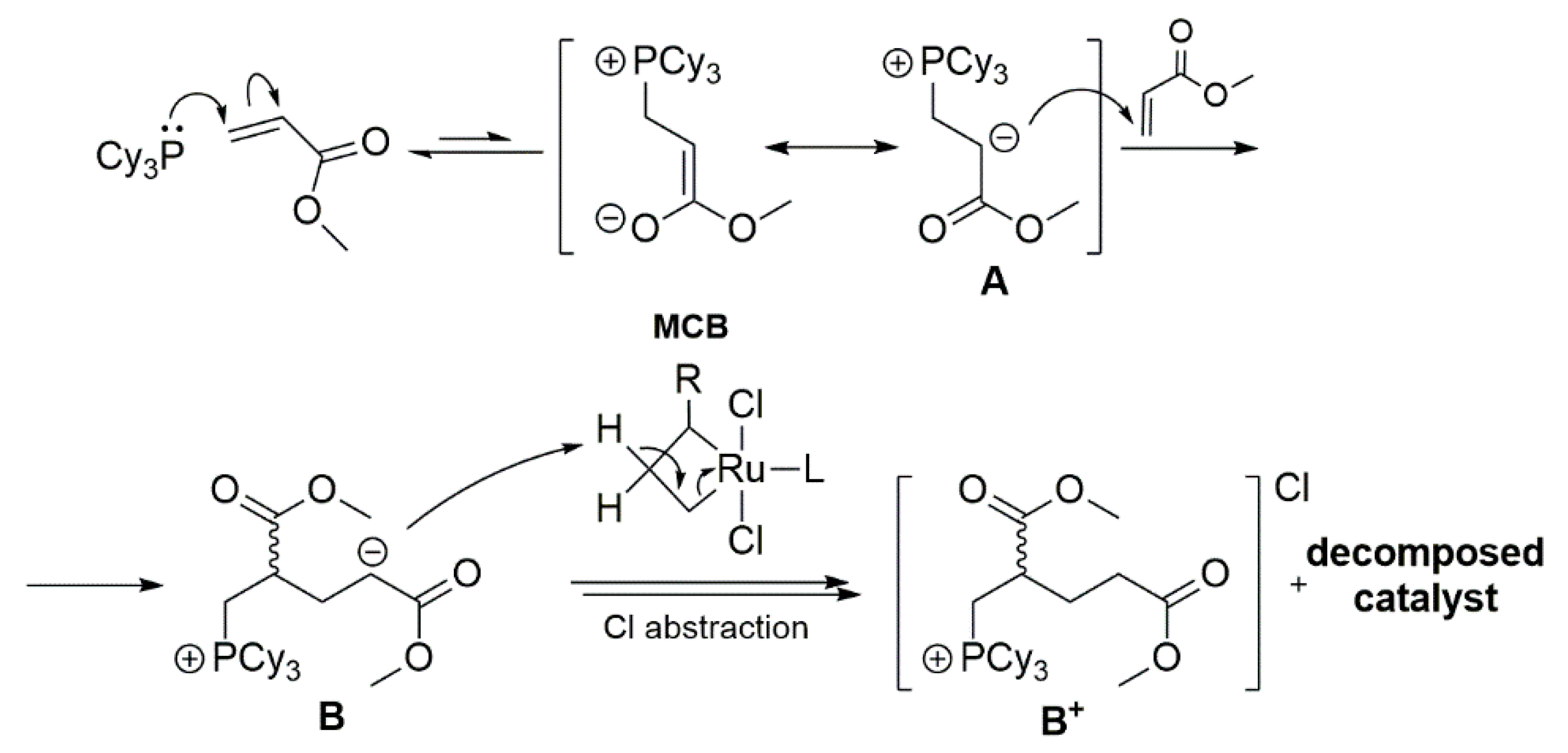
The risks associated with the use of the phosphine-containing catalysts for olefin metathesis are reviewed later. It is worth noting, however, that phosphine can be added to the Hoveyda-Grubbs catalyst as an external agent, what was reported by Bailey and Fogg [79]. They demonstrated that the phosphine is able to attack the olefin and form zwitterion A (Scheme 20), which can then undergo a number of side reactions which lead to catalyst degradation. It was observed that the main reaction is the attack on the acrylate molecule and the elimination of the B+Cl− salt, which occurs only in the presence of ruthenium species as a source of hydrogen and chloride anion. In addition, zwitterion A competes in this reaction with the proton source, as confirmed by the formation of A+. The authors suggest that the metallacyclobutane intermediate (MCB) is a likely target of phosphine attack, while the attack on the resting state intermediate is likely less preferred due to steric hindrances. These results confirm that the superiority of Hoveyda-Grubbs-type catalysts, relative to GII, in the analogous metathesis reaction results from the lack of phosphine and not from the postulated boomerang effect [79,80].
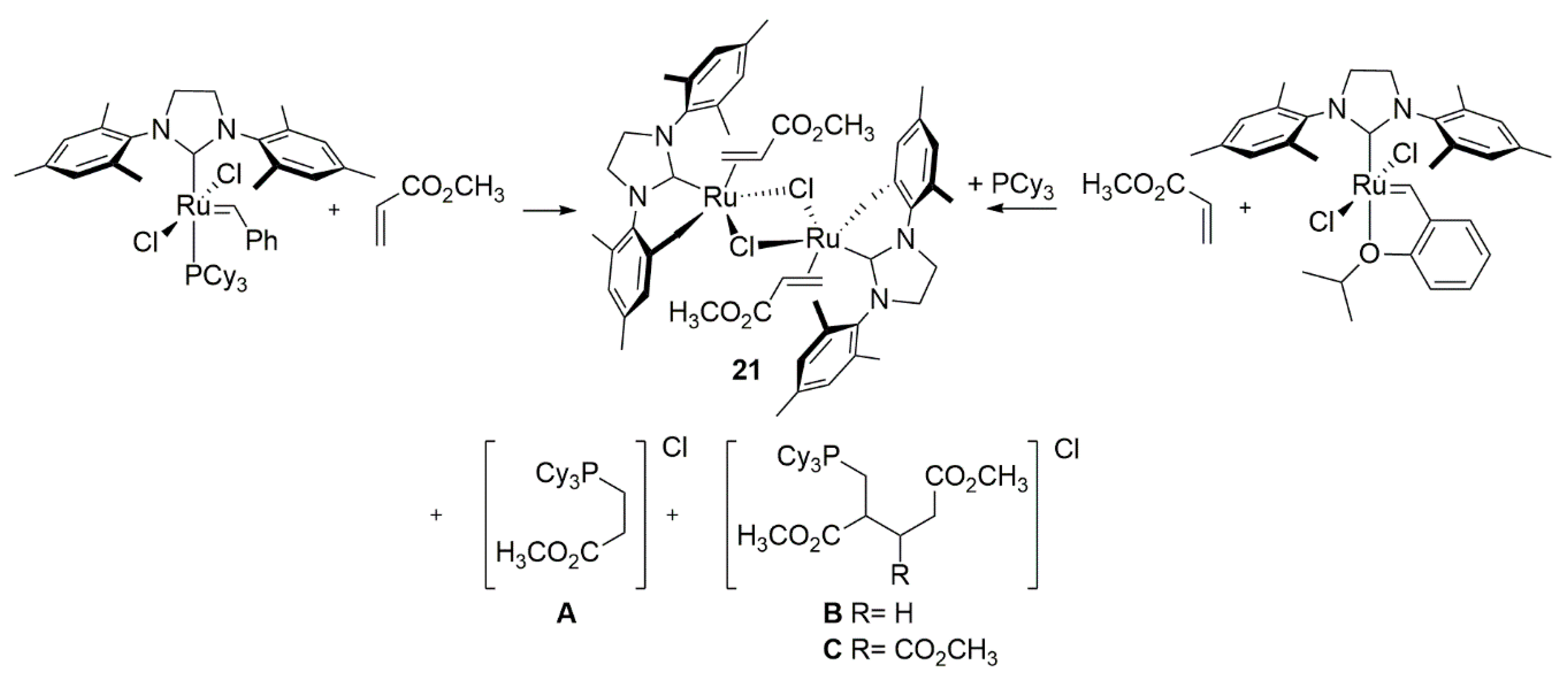
Consecutive studies of Fogg et. al. on Broensted bases—induced degradation of metathesis catalysts verified the previous hypothesis of metallacyclobutane deprotonation [81]. Dimer 21 was isolated from reaction mixtures of GII and HGII with methyl acrylate (Scheme 21), together with salts A–C. Based on the experiments with deuterated olefins and deuterated mesityl rings it was suggested that the initial deprotonation site is the metallacyclobutane intermediate rather than the NHC ligand.
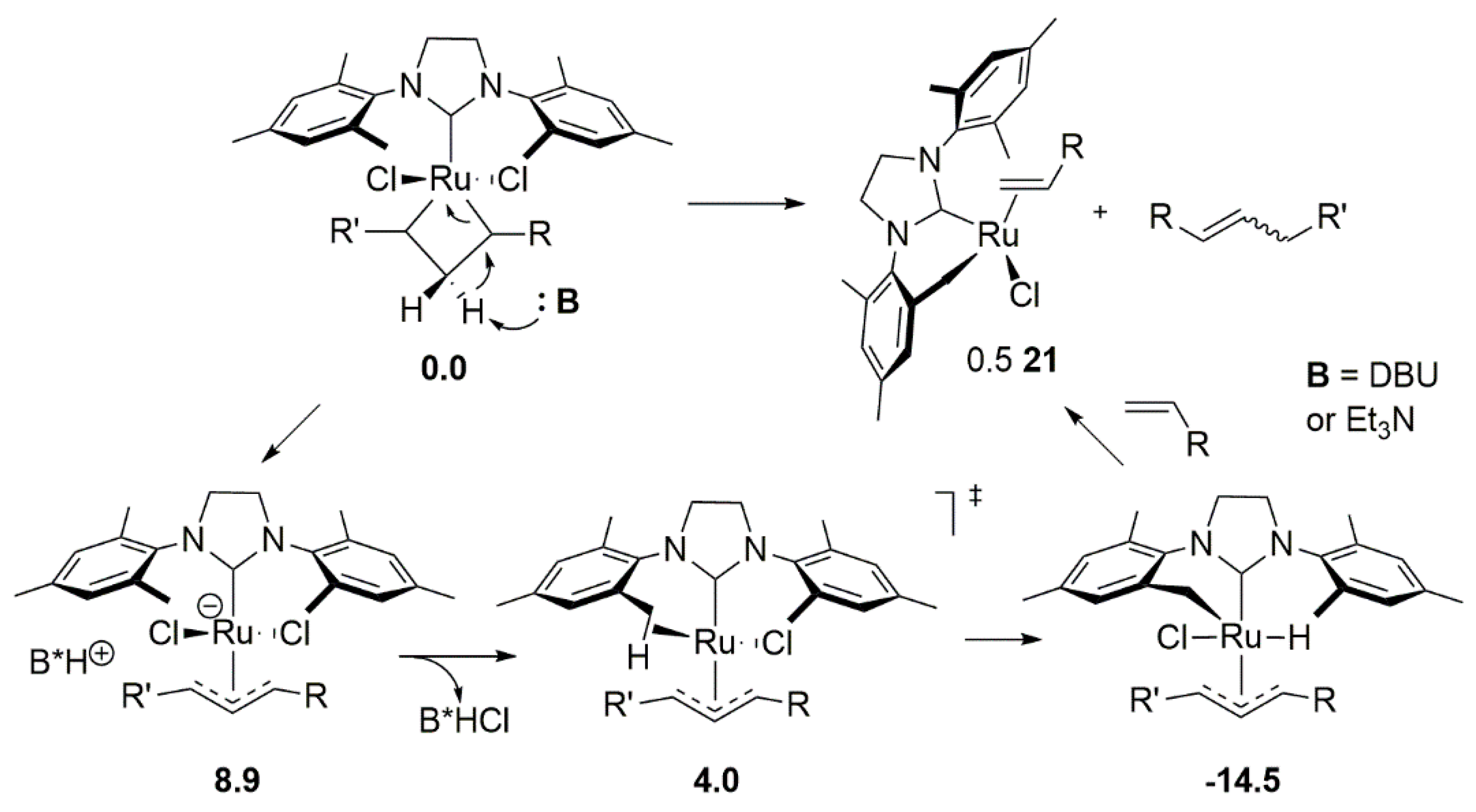
DFT calculation of acidity of protons in active intermediates MCB and methylidene species showed lower acidity of Hα compared to Hβ and, thus, excluded the deprotonation of the methylidene resting state as a competitive path. Moreover, estimated energy barriers of the further transformations after the deprotonation of MCB and the loss of chloride and allyl group to dimer 21 were low and the entire process was found to be effortless (Scheme 22). Thus, the MCB deprotonation turned out to be a general feature of Broensted bases and not only restricted to electron-deficient olefins.
In 2017 Fogg presented a method of overcoming the catalysts’ decomposition in demanding metathesis reactions with electron-deficient olefins [82]. In this new approach phenol-functionalized resins were used to quench the enolate, produced during metathesis reaction (Scheme 23). The cross metathesis of GII and IndII with acrylate at low catalyst loadings in toluene and at 70 °C indicated that the sacrificial proton source is the primary function of the resin, rather than the previously reported protonation of the phosphine [55]. Protonation of the enolate anion converts it into an unreactive phosphonium salt bound to resin, thus impeding the catalyst decomposition. Authors also suggested that in the case of the macrocyclization reaction phenol resin may remove polar impurities through hydrogen interactions. Replacing the resin with water as a source of the hydrogen leads to a further decomposition of the catalyst, most likely as a result of the formation of the hydroxide ion. The presence of the resin reduces also the harmful effects of water by competing with it in a reaction with the enolate.
2.6. Amines
Although well-defined metathesis catalysts are compatible with many functional groups, continuous reactions with nitrogen-containing olefins such as amines or heteroaromatic compounds remain a challenge [83]. There are two main modes of action of amines, which as both Lewis and Broensted bases may directly attack the ruthenium core of catalysts as well as abstract hydrogen atoms from the crucial metallacyclobutane intermediates.
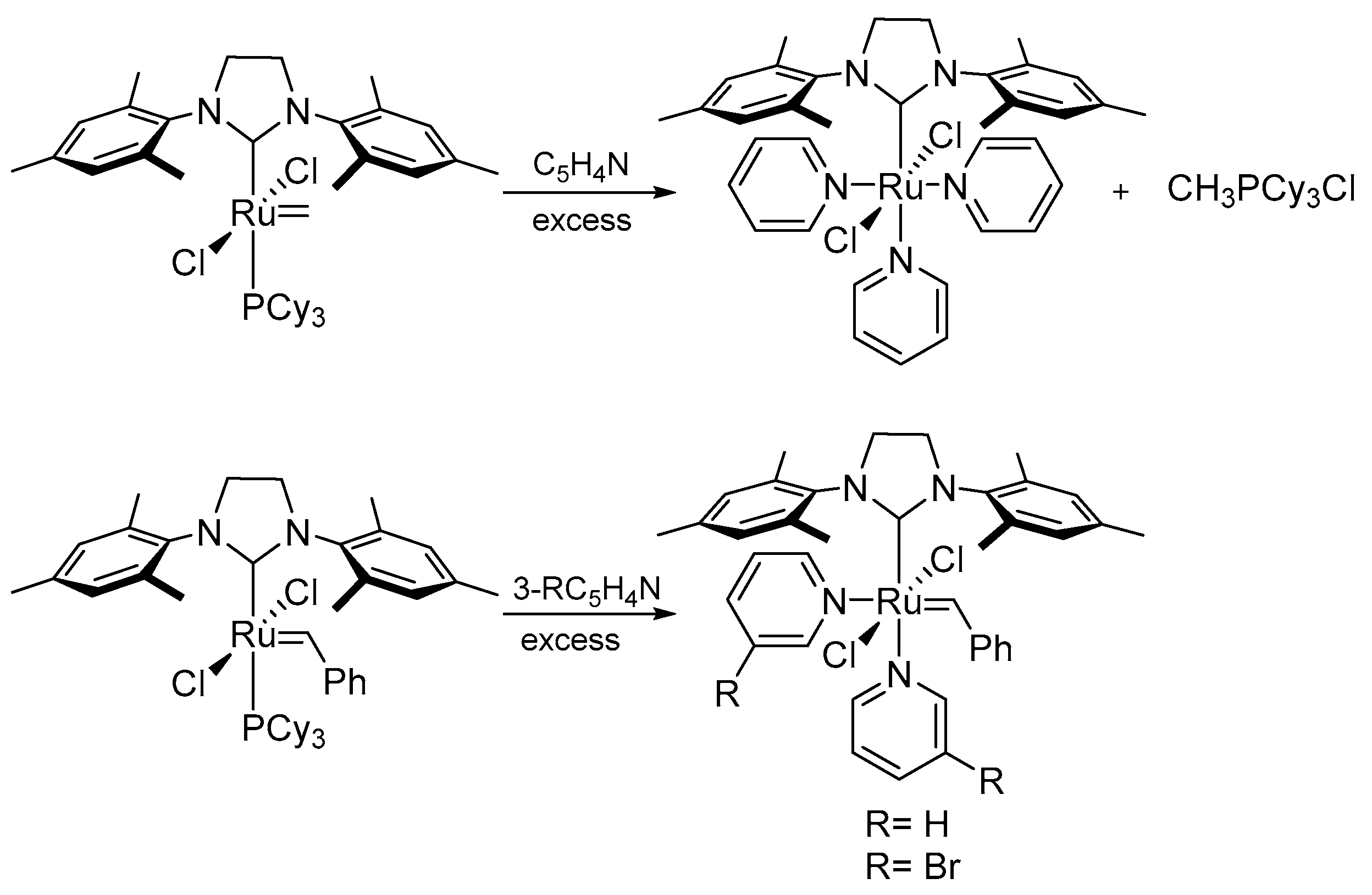
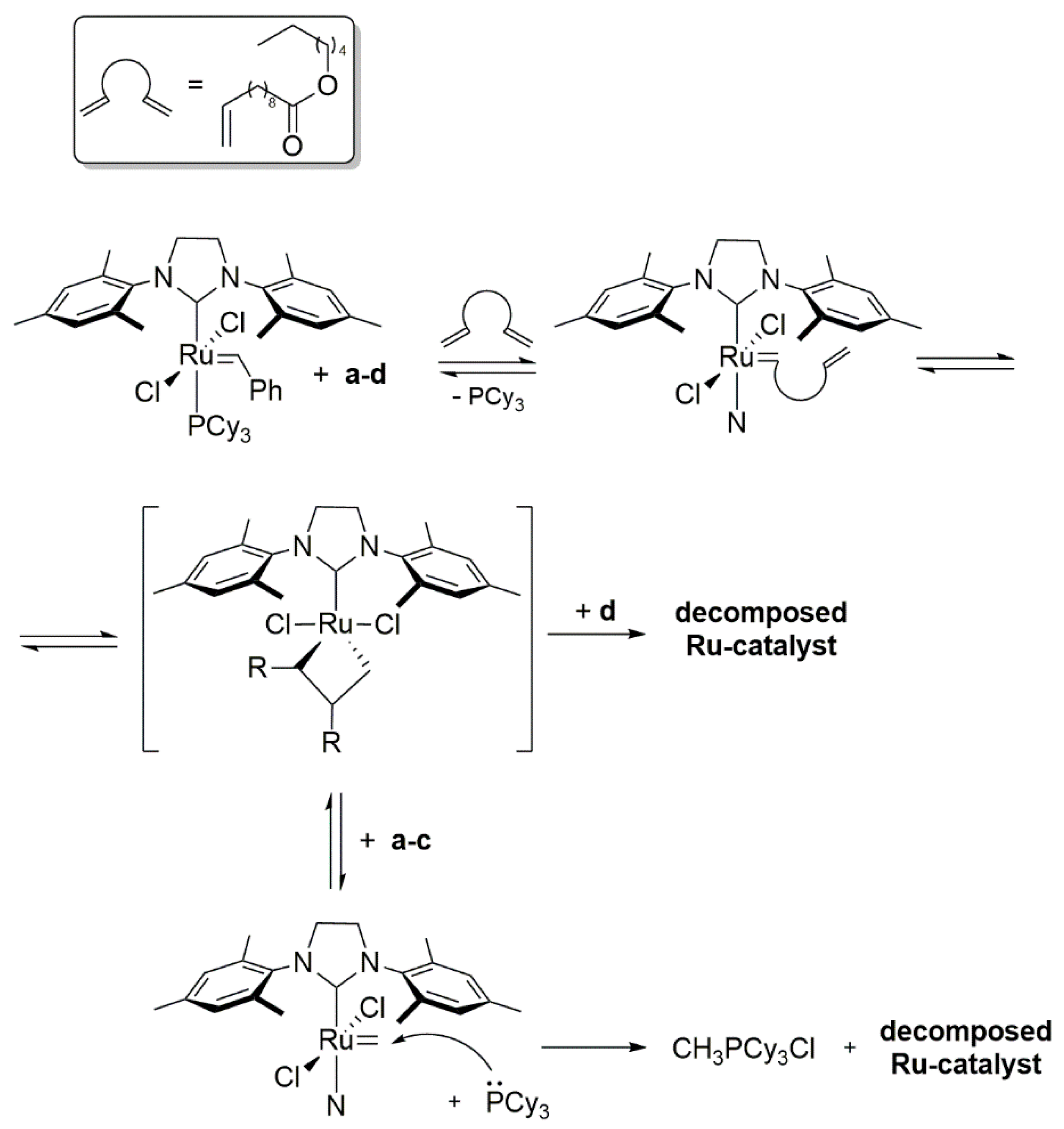
During examination of the degradation products of ruthenium methylidene complexes, Grubbs et al. noticed the formation of a tripyridine complex in the presence of amine excess [84] (Scheme 24), while the benzylidene catalyst gave a stable bispyridine adduct in previous studies [8]. Interestingly, the 3rd generation Grubbs catalyst GIII were also susceptible to the degradation and a tripyridine complex in reaction with ethylene was obtained.
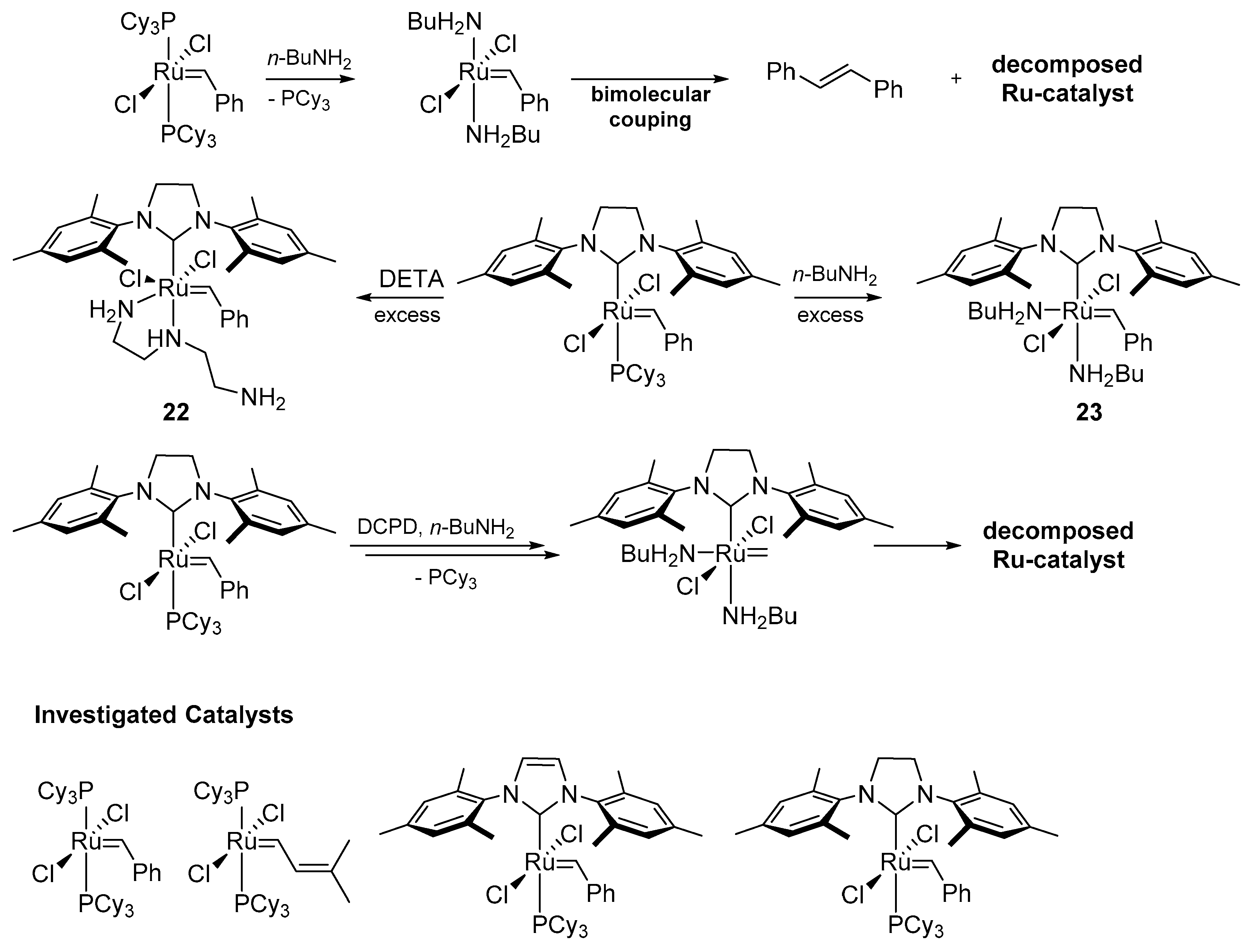
In 2009 Moore and co-workers presented a study on the stability of 1st and 2nd generation Grubbs carbenes with respect to primary amines [85,86]. The influence of both n-butylamine [85] and DETA (diethylenetriamine) [86] on the catalysts were tested in ring-opening metathesis polymerization of dicyclopentadiene and monitored by means of the differential scanning calorimetry (DSC) and NMR techniques. For the 1st generation catalysts the decomposition together with the detection of a free PCy3 and other phosphine-containing products was observed within 10 min. At the same time, the 2nd generation NHC-containing catalysts (Scheme 25) formed a new carbene species accompanied by phosphine dissociation. Based on these results it was suggested that the replacement of the PCy3 by a primary amine is the first step in the bimolecular coupling degradation. It was also concluded that the greater steric hindrance of the NHC ligand and its lower propensity to exchange with amine prevents the decomposition of the 2nd generation catalysts. As an indirect evidence for the proposed degradation pathway, new ruthenium complexes were synthesized with an excess of amine. Stable complexes 22 and 23 were obtained but the structure of the second one has not been unequivocally confirmed. However, the early decomposition of catalyst 23 in the presence of n-butyloamine and diethyl diallylmalonate indicated that the methylidene intermediate is less stable compared to its benzylidene analogue.
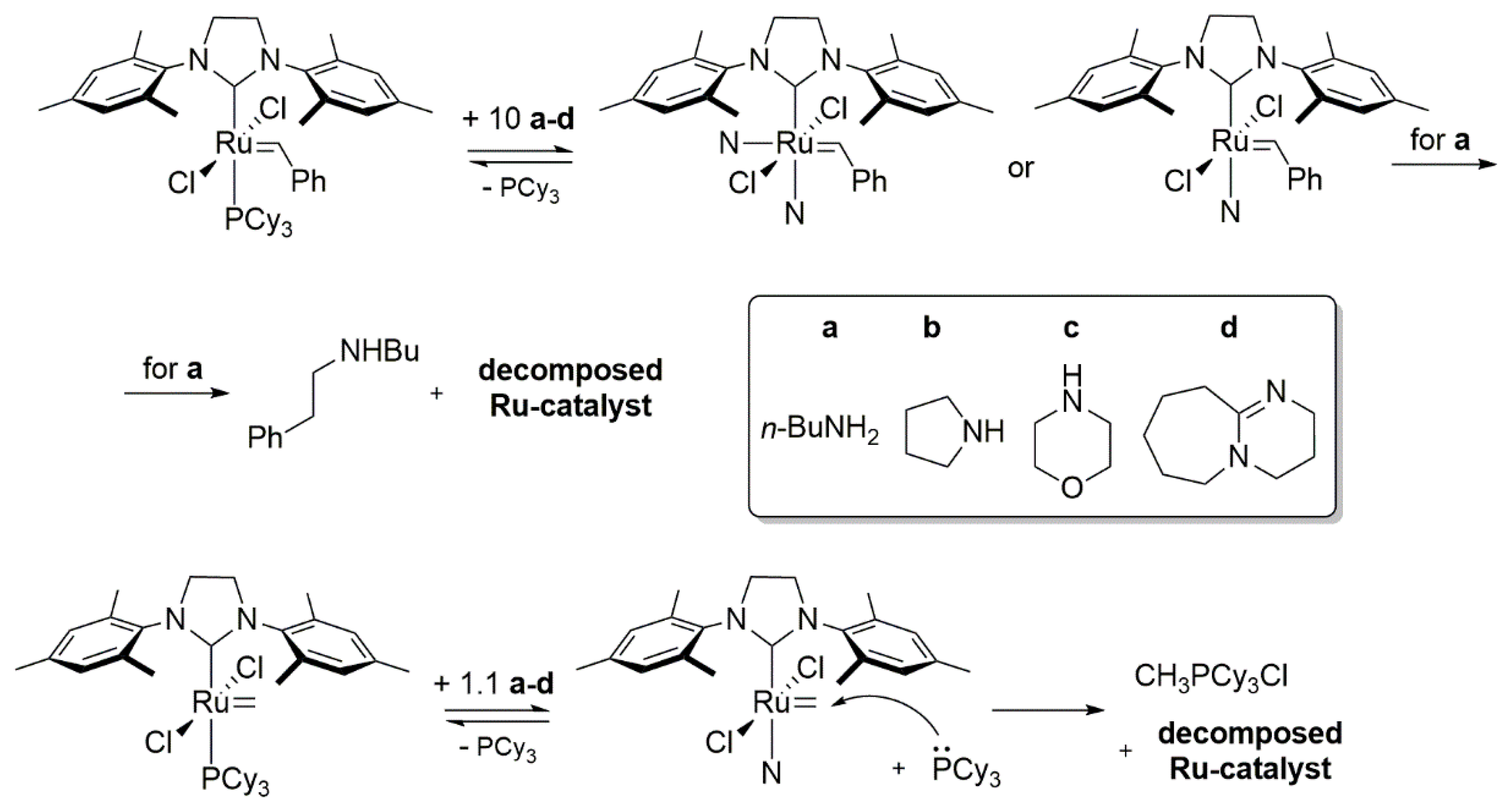
A complementary study by Fogg’s group devoted to the decomposition of metathesis catalysts was reported in 2014. The authors of this investigation showed the impact of different amines on GII and its key intermediates formed in the catalytic cycle, that is, the methylidene resting state (GIIm) and the metallacyclobutane MCB (Scheme 26) [87]. First, GII was reacted with various amines and the resulting products were examined by means of NMR. In accordance with the earlier work of Moore [85], it was found that the less sterically hindered n-buthylamine (a) yielded the N-benzyl-N-buthylamine and bisamine complex, which further decomposed via a nucleophilic attack of another amine molecule on the benzylidene carbon atom. More bulky amines, such as pyrrolidine (b), morpholine (c) and DBU (diazabicycloundecene) (d) reacted slower and formed stable mono-amine adducts at room temperature (Scheme 26).
Next, the authors investigated the sensitivity of the methylidene species to the presence of primary amines (Scheme 26). In all of these cases, reactions proceeded faster than for GII, even when using 1.1 equivalent of amine. Estimated half-lives of the resting states (a (12 min) > b (1.5 h) > c (14 h) > d (>24 h)) showed that the steric hindrance of amine is the crucial factor, as well as that DBU (d) induced virtually no decomposition. Interestingly the phosphonium chloride MePCy3+Cl−, found as one of the byproducts, indicated a degradation path through a nucleophilic attack of the PCy3 on the methylidene released by the ligand exchange with amine and not from the attack of amine itself. This was further confirmed in studies of the 13C-labeled methylidene resting state with n-butyloamine, as well as the predominance of a phosphine attack over an amine attack revealed by the 7:3 ratio of resulting products, MePCy3+Cl− and CH3NHnBu, respectively [88].
The influence of amines on the RCM reaction were also tested (Scheme 27) and the results showed an accelerated catalyst degradation compared to the control reaction. The presence of a phosphonium salt in reactions with amines a–c indicated, that the deactivation occurs through the decomposition of the resting state. In the presence of DBU an instant decomposition was observed with no trace of salt but only free PCy3. Thus, the deprotonation of the metallacyclobutane MCB by this highly basic amine was suggested as the true mechanism of decomposition. It was also concluded that the high basicity of amines together with the bulkiness of the ylidene moiety are the critical parameters in this phenomenon. These results were also in agreement with the observed ligand exchange and amine-adducts formation for secondary amines and GII [84,87] and the precatalyst degradation by unhindered primary amines.
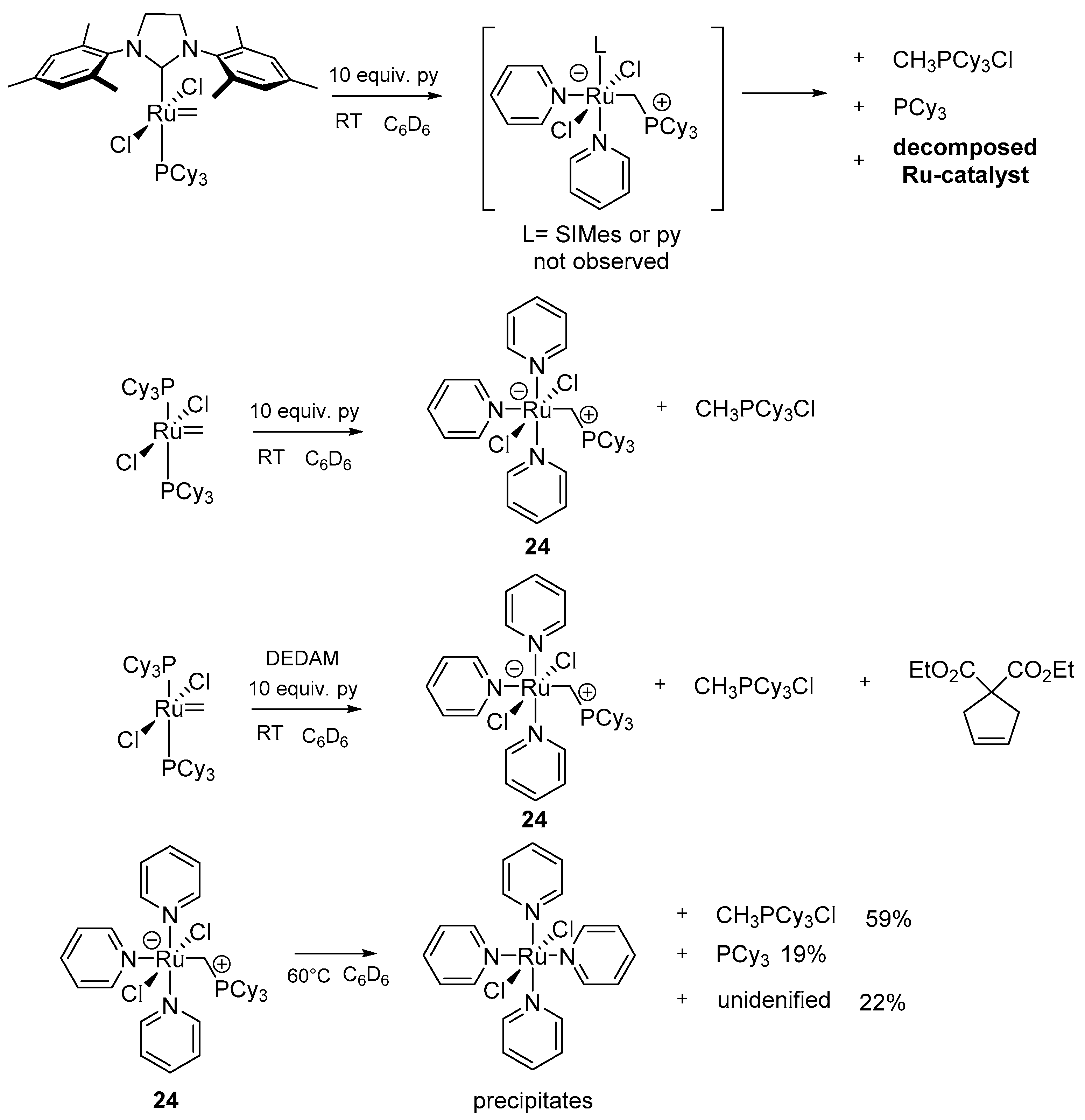
The same group carried out the reaction of the benzylidene and methylidene derivatives of Grubbs catalyst with pyridine (py). While reacting this nitrogen-containing heterocyclic base with resting states of 1st generation catalysts, a rapid disappearance of the ylidene functionality was noticed together with simultaneous formation of a new adduct bearing three pyridine molecules 24 (Scheme 28). Both the 1st and 2nd generation precatalyst provided bis-pyridine carbene complexes, as reported previously [89,90], implying the absence of the phosphine attack on benzylidene. Adduct 24 completely decomposed within 18 h at 60 °C to free PCy3, MePCy3+Cl− and three other, unidentified compounds, which probably come from other co-existing decay paths. The authors were not able to connect any of these products with the CH2 = PCy3 ylide, as proposed in the prior work of Grubbs [84]. Instead, they associated the production of MePCy3+Cl− with an intermolecular proton abstraction. Catalyst GI, under metathesis condition of diethyl diallylmalonate RCM, gave ca. 80% of 24, showing that the main attack was at the resting state and not at the metallacyclobutane. The principal conclusion of this work is that any donor able to replace the phosphine ligand and stabilize the methylidene species may cause the deactivation of the catalyst using the same route.
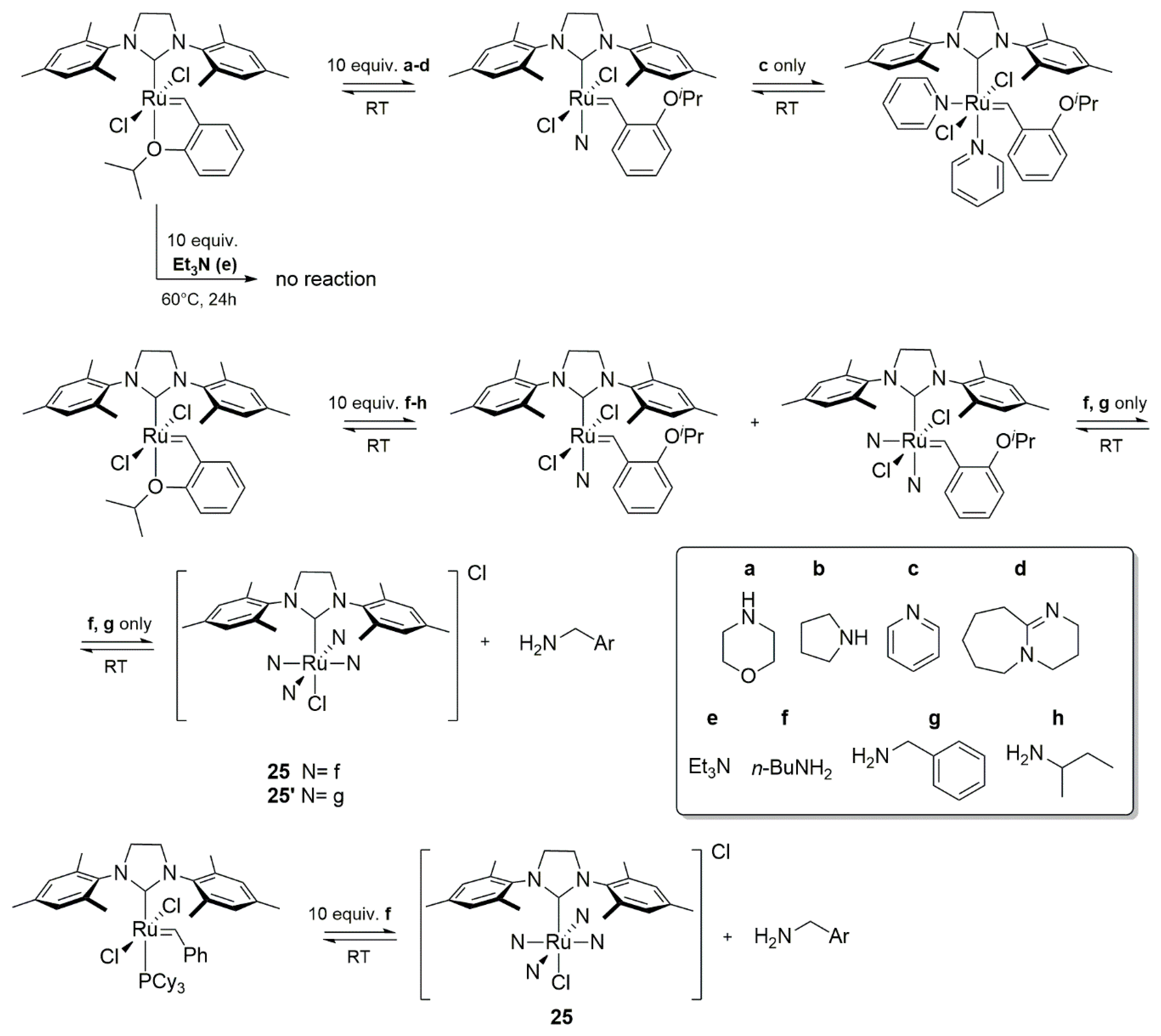
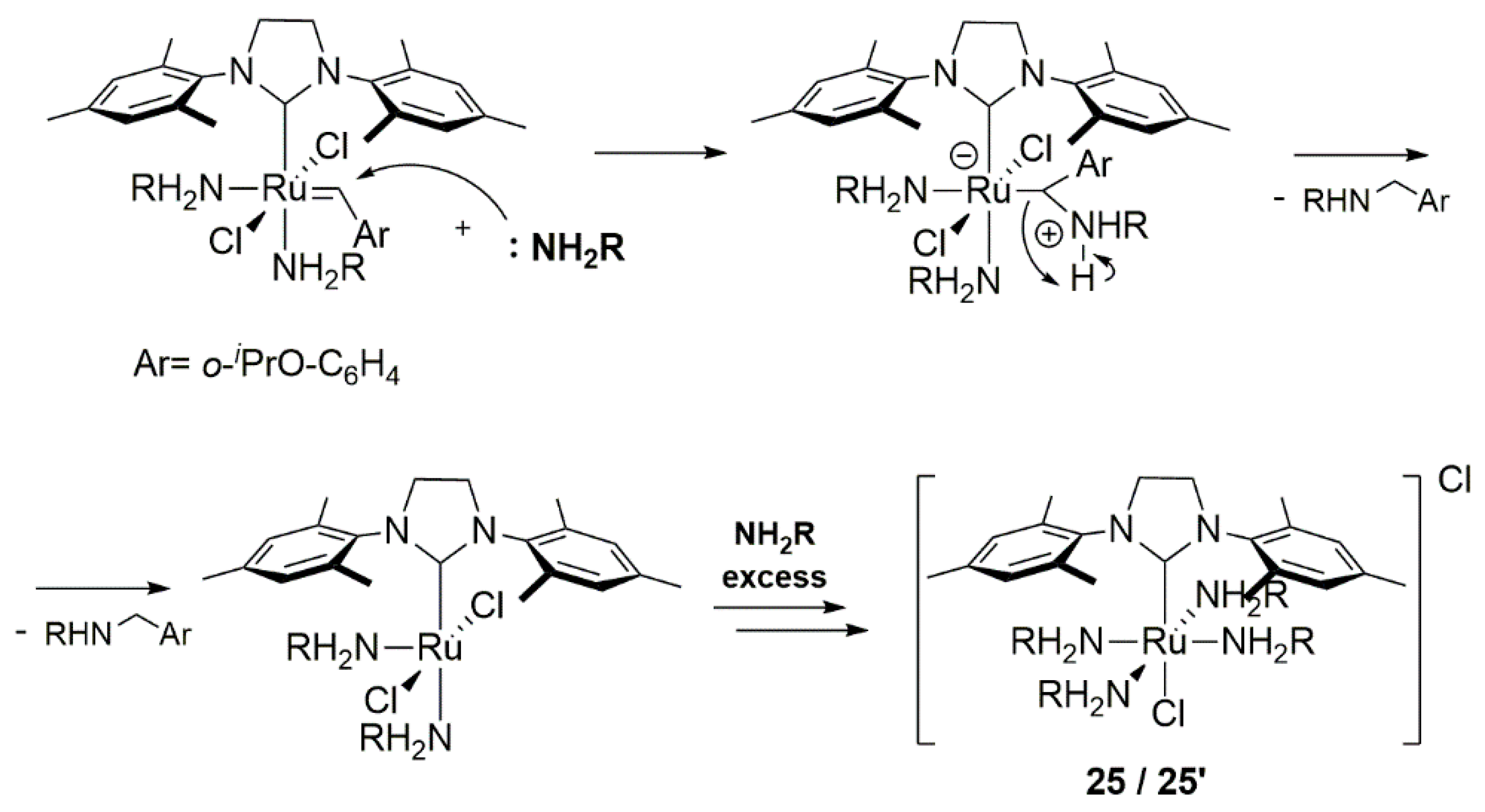
Similar results were obtained by Fogg’s group for Hoveyda-Grubbs catalyst treated with the excess of different nitrogen bases in the presence or absence of the olefin [91]. Non-bulky primary amines f,g formed mono- and bisaminobenzylidene complexes with HGII, which further decomposed to 25/25′ and NH2CH2Ar amine (Scheme 29). The n-butylamine derivative 25 was also isolated, starting from either the HGII or GII catalysts. On the other hand the sterically crowded sec-butylamine gave only the single-substituted derivative, which was resistant to further degradation (Scheme 29). Reaction with secondary amines, pyridine or DBU (a–d) led to stable complexes containing one amine molecule coordinated to ruthenium (two in the case of pyridine) for all cases, apart from Et3N, for which no reaction was observed. The dependence the on spatial hindrance (NH2nBu >> NH2CH2Ph) corresponds to the proposed loss of the benzylidene ligand as a result of a nucleophilic attack through concerted pathways presented in Scheme 30.
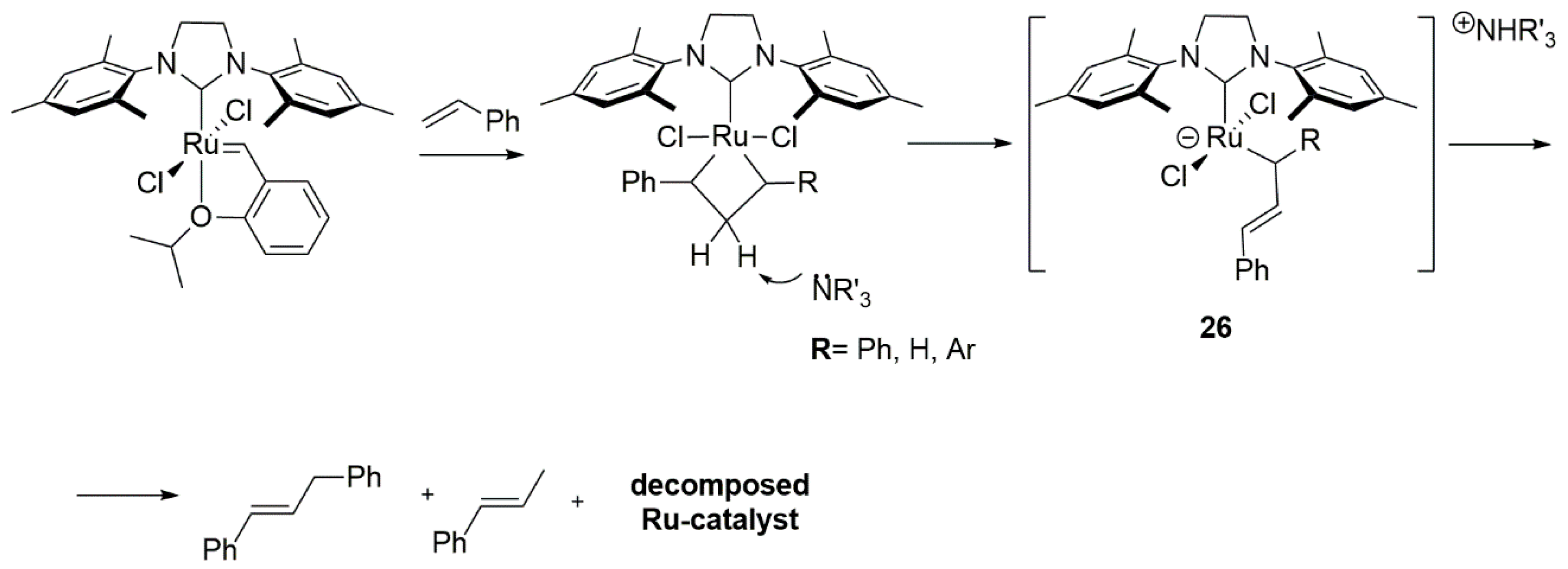
The addition of amines to styrene self-metathesis reaction resulted in the decrease of yield with the increase of Broensted basicity of the amines. The detailed decomposition pathways were studied for pyrrolidine, DBU and NEt3, which were found to be the most detrimental bases for this process. The major organic product observed during the decomposition was (E)-PhCH = CHCH2Ph, with the additional presence of (E)-PhCH = CHCH3 when NEt3 were used. To explain these observations, the authors proposed a deactivation mechanism involving the deprotonation of a sterically accessible metallacyclobutane intermediate (Scheme 31), to generate an anionic ruthenium complex 26 and then, through the subsequent protonation and elimination, the release of (E)-PhCH = CHCH2Ph. Traces of the trans-propenylbenzene suggests that, in the case of the less basic NEt3, the deprotonation of the monosubstituted metallacyclobutane occurred.
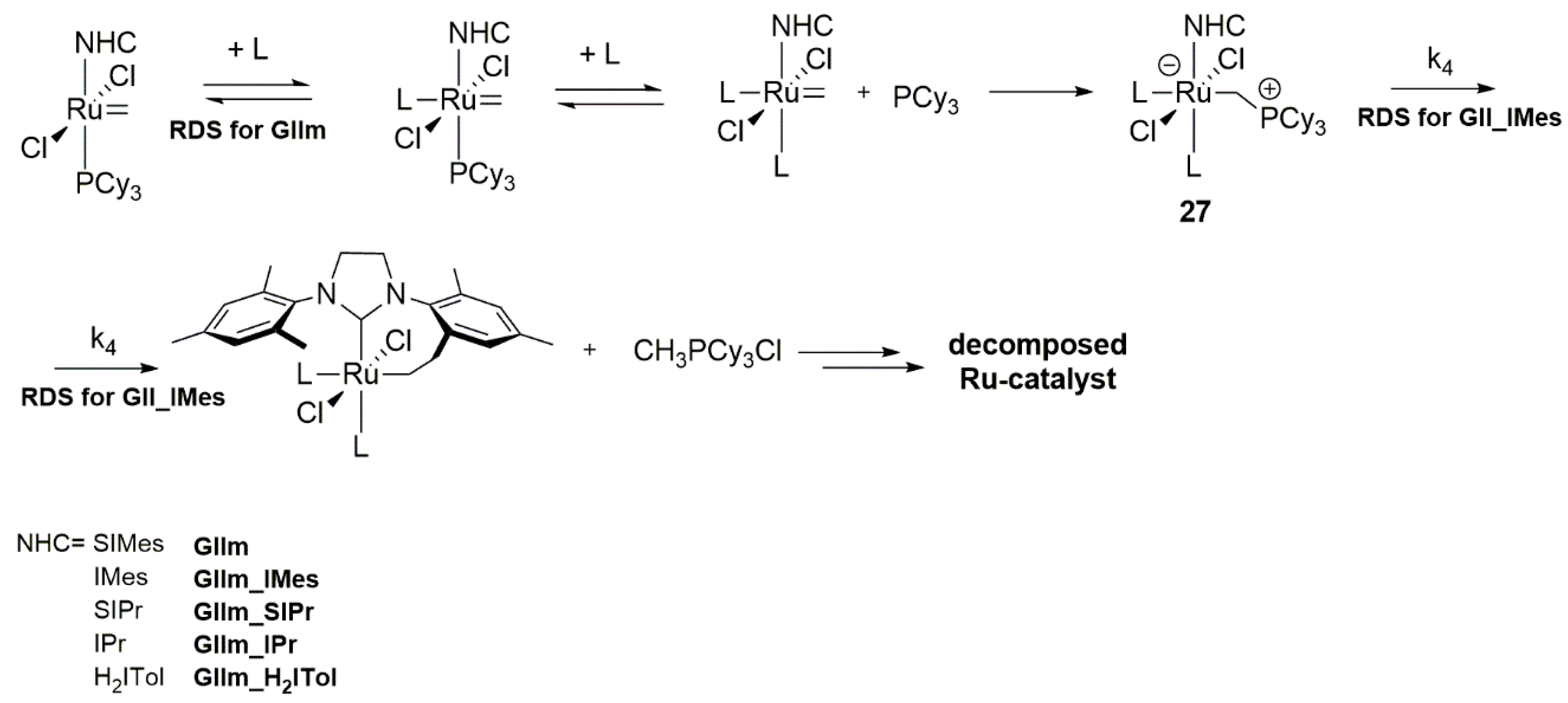
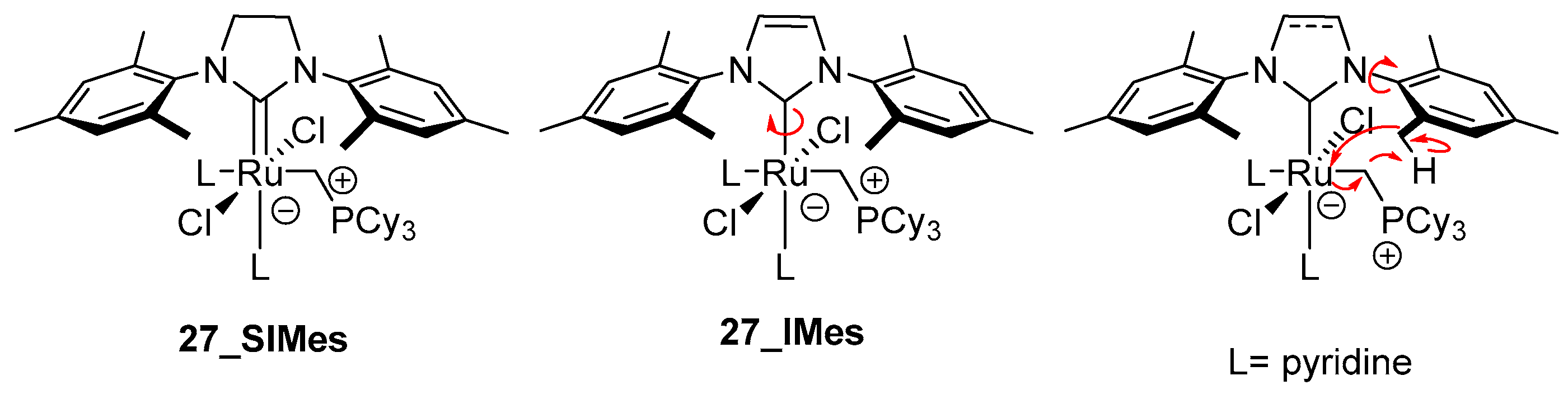
In another work devoted to metathesis catalysts’ decomposition, Fogg’s group confirmed an earlier assumption that the presence of Lewis donor accelerates the loss of methylidene from phosphine stabilized complexes [92]. The previously proposed mechanism of the nucleophilic attack of the free phosphine on the methylidene moiety, resulting in the loss of this functionality with phosphonium salt [CH3PR3]Cl release (Scheme 28), was also shown to operate for weaker Lewis donors, such as THF, H2O, MeOH, MeCN, DMSO, as well as a series of 2nd generation Grubbs catalyst, including benzylidene and indenylidene derivatives (Scheme 32). The lack of a significant effect on the catalyst degradation of such donors as NH3, urea or phosphoramide was consistent with the previously observed lack of degradation of the GII catalyst by DBU [87] and indicates that Lewis basicity is irrelevant, if steric hindrance prevents the approach to the metal center. Moreover, the formation of a hitherto undetected σ-alkyl intermediate 27 was confirmed in labelled NMR experiments with complex bearing and IMes ligand and pyridine at 50 °C. The unsuccessful attempts to detect this complex using SIMes adducts were explained by a short life of the resulting species and by effortless C–H activation of the methyl group in the ortho position of the mesityl substituent. Thus, elimination by proton abstraction from the NHC was promoted as a consequence of π-backbonding onto SIMes ligand, which was reflected in an inhibited rotation (Scheme 33) [93].
Based on the rate at which the decomposition occurs and on the influence of donors bulkiness and stoichiometry authors assumed that the observed degradation proceeds is an associative process. The established reaction rate for DMSO showed a first-order dependence on its concentration for both tested complexes, GII and the 1st generation methylidene complex GIm. Additional deuterium-labelling studies confirmed that the rate-determining step here is the ligand binding for the methylidene complex GIIm and the C-H activation for the unsaturated GIIm_IMes (Scheme 32). It was also demonstrated that ethylidene ligand introduced to GII in the reaction with 2-butene prevents the donor-induced ylidene abstraction, as evidenced by the lack of the [EtPCy3]Cl production.
2.7. N-Heterocyclic Carbenes
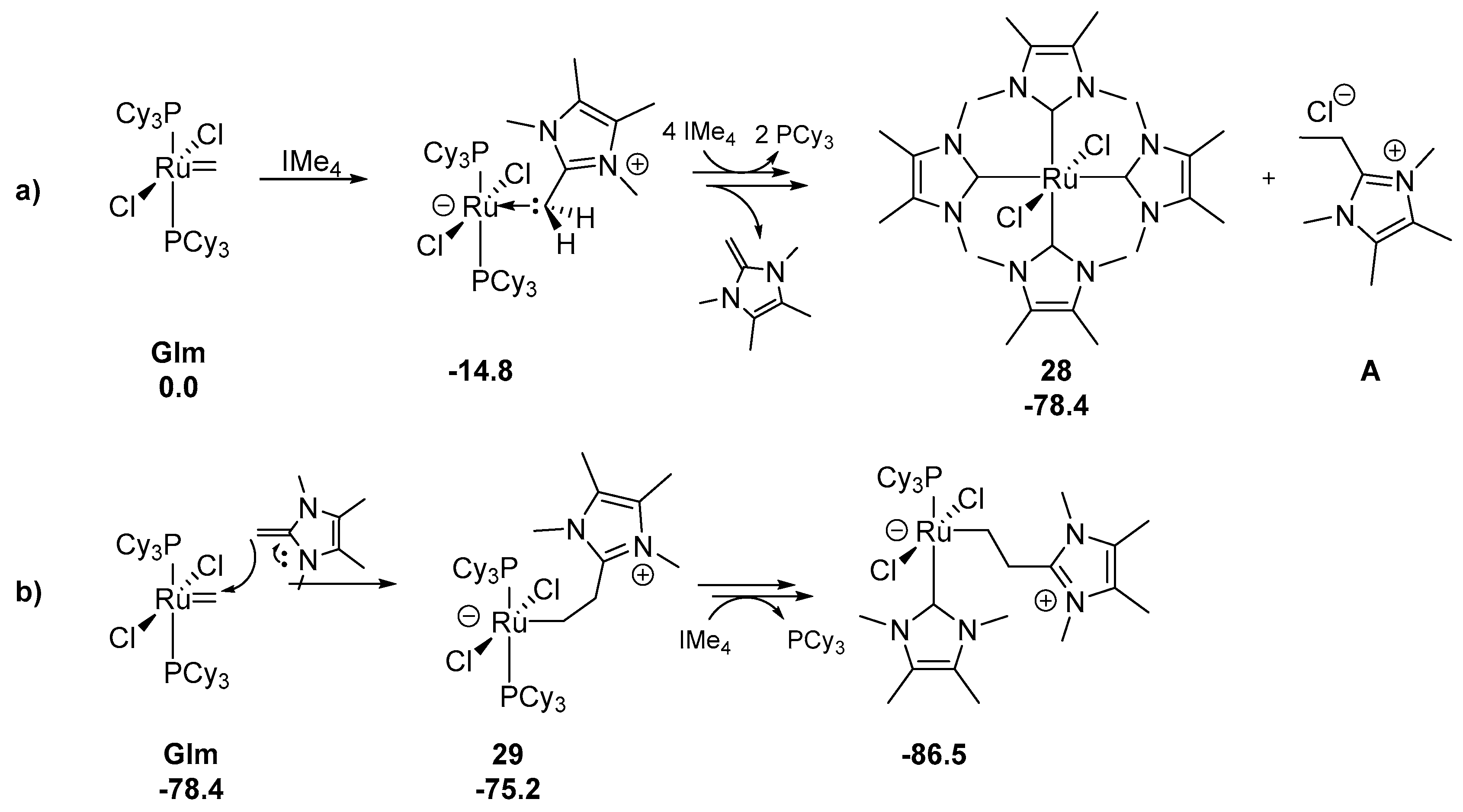
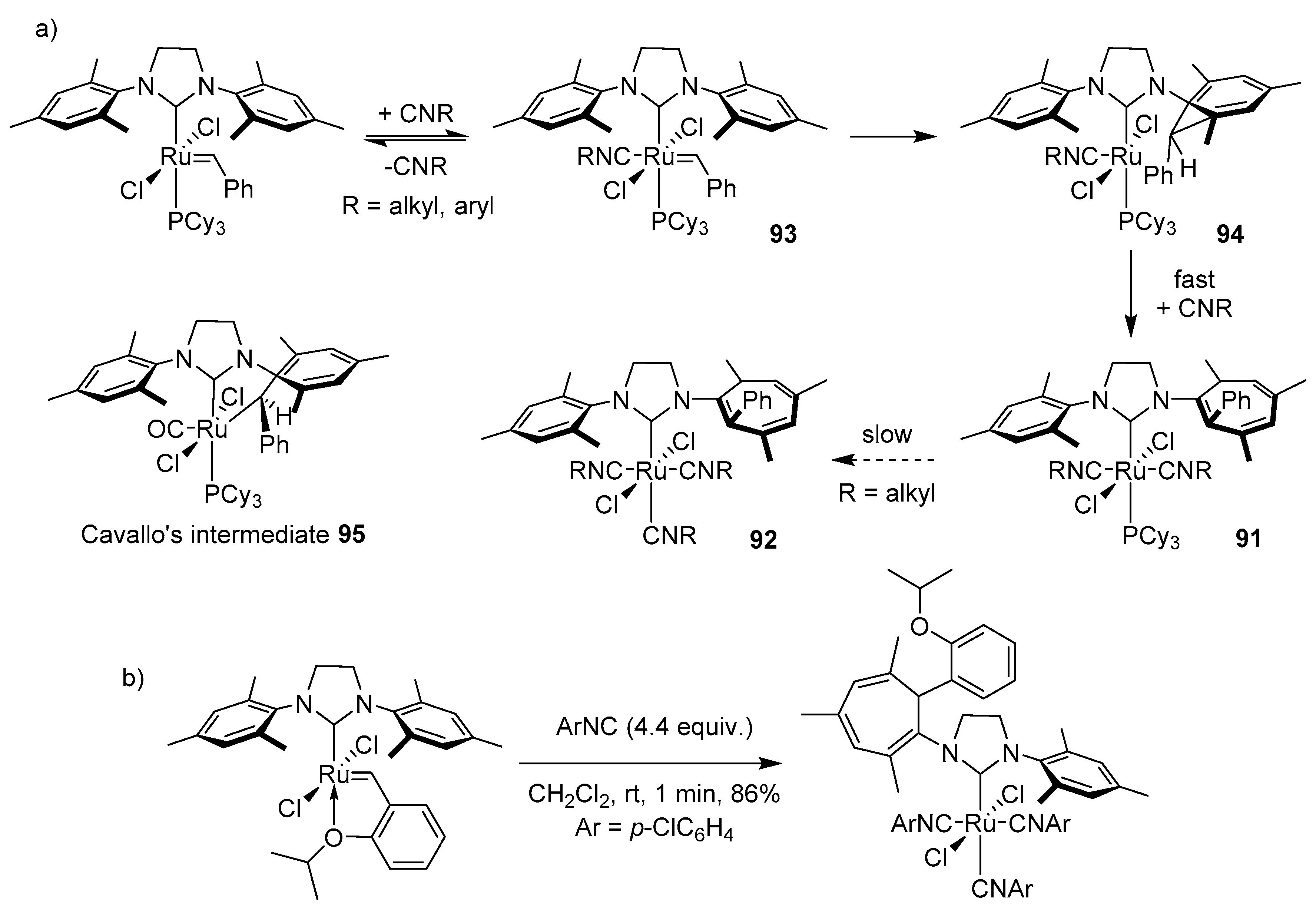
While N-heterocyclic carbenes are often used as auxiliary ligands in ruthenium metathesis catalysts, they can also act as a Lewis bases and accelerate the decomposition of such complexes when added to the reaction mixture. Recently Fogg’s group showed that small NHC ligand in ruthenium catalyst may expedite its decomposition [94]. During the attempts to synthesize the methylidene complex with the truncated IMe4 ligand from the 1st generation complex GIm, a stable four-coordinated Ru(Cl)2(IMe)4 complex 28 was observed, instead of the expected ligand exchange product (Scheme 34). This species was additionally accompanied by a free PCy3 and ethylimidazolium salt A. The reaction completed rapidly in less than 5 min at −80 °C. The formation of product A was confirmed independently by NMR experiments with labeled methylidene GIm and synthesis. Further DFT calculations indicated that complex 28 was formed by the nucleophilic IMe4 ligand attack on the methylidene carben carbon, rather than the phosphine exchange, for which the estimated energy barrier is 5–6 kcal/mol higher. In the suggested mechanism (Scheme 34) it was noted that the resulted adduct [Ru-CH2IMe4] preferred the elimination reaction which leads to N-heterocyclic olefin (NHO) over the protonolysis and the release of [CH3IMe4]Cl. Calculations provided also the evidence that the NHO may be involved in further catalyst degradation as a potential nucleophile (Scheme 34b). Complex 29, produced in this reaction and bearing highly basic CH2CH2IMe4 ligand undergoes the protonolysis to give salt A, likely proceeded by PCy3/IMe4 exchange. Additionally, analogous reaction of IMe4 with the methylidene complex of 2nd generation carbene GIIm resulted in adduct 28 instead of the expected RuCl2(SIMes)(IMe4)n product, in contrast to standard, larger NHC. The estimated energetic cost of the attack of SIMes on the methylidene was found to be higher than for a small ligand such as IMe4 and introduces significant geometric strain, therefore no such decomposition was observed for SIMes.
2.8. Acrylonitrile
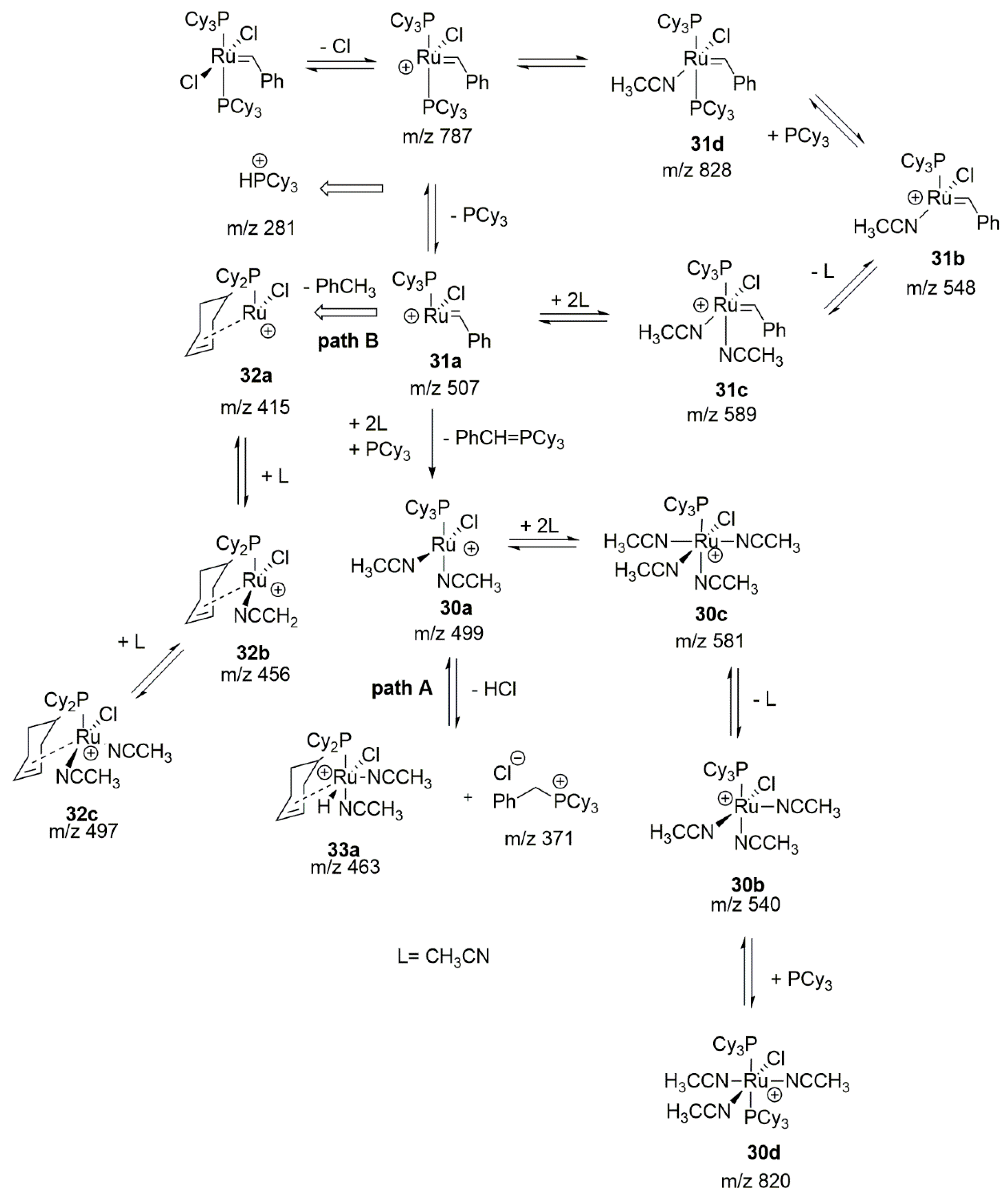
The next external agent which was found to cause ruthenium catalysts decomposition, is acrylonitrile. It is an interesting example since it is a very weak base, that is not able to abstract hydrogen atoms and can rarely form stable adducts after attack on the ruthenium core. Nevertheless it was found that at elevated temperature such adducts can be formed. In 2011 Zhao, Wang and Guo presented electrospray ionization tandem mass spectrometry (ESI-MS/MS) study with off-line and on-line detection of the degradation of the 1st generation ruthenium complexes under the influence of acrylonitrile [95]. In the first, control experiment with GI in CH2Cl2 they observed the formation of a phosphine product (PhCH2PCy3)+, which was consistent with previous work of Grubbs [96]. Shortly after the addition of acrylonitrile, signals form (HPCy3)+ ion and the relative abundance increase of (PhCH2PCy3)+ were observed (Scheme 35). Within 15 min catalyst decomposed entirely and a new signal of ruthenium decomposition products 30a–d and 33a–d arose. Complexes 30c and 30d were identified as [RuCl(CH3CN)4PCy3]+ and [RuCl(CH3CN)3(PCy3)2]+ based on their isotopic patterns. Accordingly, structures 30a and 30b in equilibrium with 30c–d were established as [RuCl(CH3CN)2PCy3]+ and [RuCl(CH3CN)3PCy3]+. Ru-species 33a–b were suggested to appear from the C-H activation but this assumption was ambiguously proved. It was further postulated that a higher concentration of acrylonitrile promoted a faster loss of chloride and phosphine ligands, as evidenced by the formation of compound 31d. Moreover, the presence of 31a–d was detected only in the early stage of the catalyst decomposition, indicating that these were transient intermediates. These results allowed to propose the predominant mechanism of degradation promoted by acrylonitrile presented on Scheme 35 path A, which was consistent with the slow degradation in CH2Cl2 observed earlier [97]. A second possible decomposition pathway (path B in Scheme 35) was associated with the formation of compound 32a through the loss of toluene from 31a and its appropriate CH3CN adducts 32b–c. The authors assumed that the C-H activation was the result of an agostic interaction between the Ru atom and one of the cyclohexyl ring, which was made possible due to a greater steric freedom of the resulting species after the loss of phosphine and chloride.
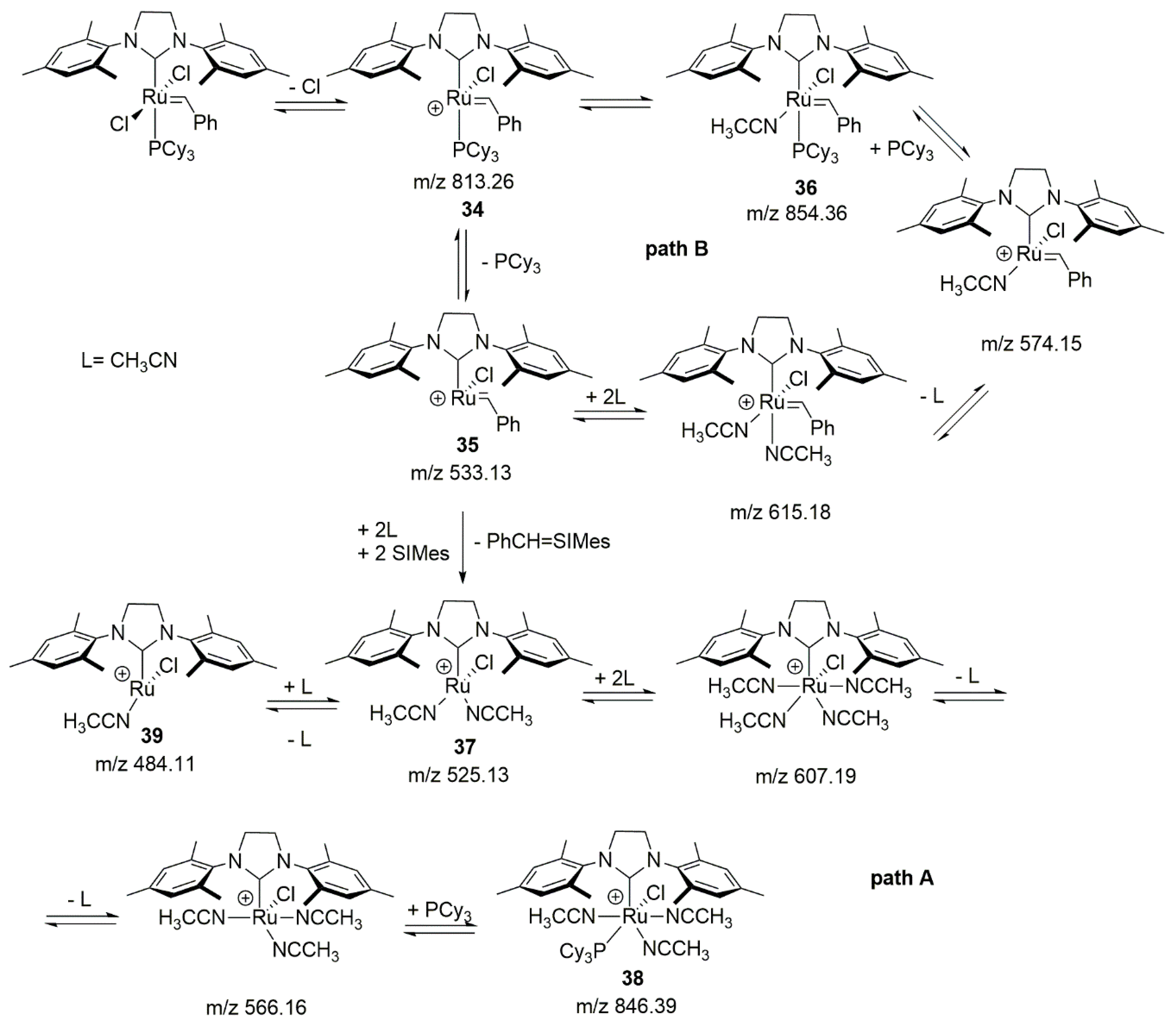
Comprehensible, real-time monitoring droplet spray ionization tandem mass spectrometry (DSI-MS) studies on the behavior of the 2nd generation catalysts in the presence of acrylonitrile were conducted by Jin in 2017 [98]. The obtained results were very similar to those for the 1st generation catalysts. The decomposition pathway proposed by authors starts with a heterolytic bond cleavage and a loss of chlorine (34) and then the dissociation of the phosphine forming complex 35. At this point, two degradation pathways are possible (Scheme 36). Path A includes Ph-CH = H2IMes ion loss with simultaneous attachment of acrylonitrile molecules and SIMes carbene to give 37. Then, a series of capture and release of CH3CN molecules followed finally by a PCy3 rebinding leads to the formation of a very stable hexacoordinated complex 38. It is worth noting that the detachment of one acrylonitrile molecule from 37 results in the formation of compound 39, which was not considered for the earlier generation of ruthenium carbenes. In the second route (Scheme 36) a series of subsequent coordination of acrylonitrile molecules combined with phosphine association leads to compound 36, which can also be formed directly from complex 34.
The latest theoretical studies on the stability of Hoveyda-Grubbs-type catalyst under the influence of acrylonitrile were presented by Trzaskowski’s group [99]. In this work the reaction of acrylonitrile self-metathesis as well as possible decomposition routes with standard Hoveyda-Grubbs catalyst and two CAAC derivatives were investigated. Three ways of degradation were considered—through β-elimination of metallacyclobutane using van Rensburg mechanism (olefin-driven), via Buchner ring expansion leading to cycloheptatriene and through the direct attack of the nitrile molecule. Since it was concluded that the most likely mechanism is via the Buchner ring expansion and depends on the structure of the catalyst, it is reviewed in the next part of this study.
2.9. CO and O2
There are two more compounds known to cause ruthenium metathesis catalysts’ decomposition. One is CO, a potent Lewis acid, commonly found in various transition metal complexes. Similarly to acrylonitrile, however, CO act as a complexing agent which enables deactivation and decomposition of ruthenium NHC complexes via Buchner-type expansion. The second species is O2 known to promote the C–H activation also mostly in the NHC-containing complexes. Due to this features, investigations on the CO-driven and O2-dependent decomposition are reviewed in the next section.
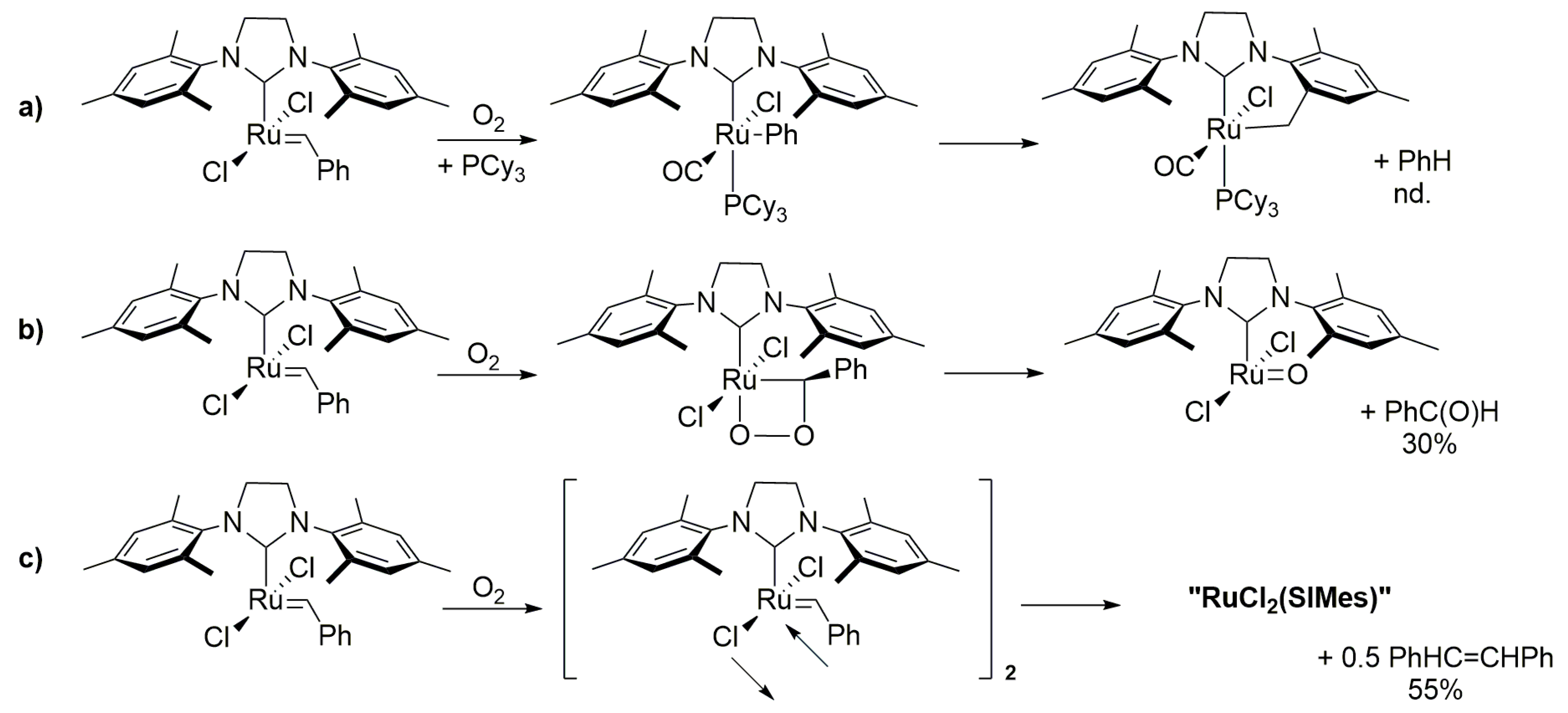
In 2003, Trnka and Grubbs observed the effect of traces of oxygen on the synthesis of the GII catalyst (Scheme 37) and they were able to isolate this compound from the reaction mixture, which was formed as a result of the C–H activation of ortho methyl group in mesityl substituent [100]. Dinger and Mol conducted similar experiment as previously with primary alcohols using water but it did not bring the expected results, since no ruthenium hydride was formed. Instead the tricyclohexylphosphane oxide was obtained together with a one-phosphorus compound that was neither hydride nor carbene and which structure was not determined [38]. They also carried out the reaction of GI with oxygen looking for a source of oxygen to form ruthenium carbonyl 5. The maximum yield in solution was around 15% for 5 (Scheme 37), while the same reaction performed overnight with a solid sample at 40 bar of O2 at 60 °C gave 75% of the desired product. Second generation Grubbs catalysts behaved in a similar manner, although they reacted more slowly (3 days, at 50 bar O2, 60 °C) and with less efficiency (29% of phenyl Ru-complex) (Scheme 37) [39]. It was suggested that the final Ru species is formed by the oxygen attack at the benzylidene group.
The decomposition of Hoveyda-type complexes under air, reported by Blechert, is mentioned later in this review in the C-H activation section. Later, Cazin’s Group studied the effect of air on the catalytic performance of commercially available ruthenium catalysts by systematically changing the reaction conditions [101]. The RCM reaction for N-tosyldiallylamine was conducted in non-degassed solvents, low catalyst loading in air and its components (N2, CO2, O2, H2O and dry air). It was shown that for all tested catalysts, the influence of oxygen and, in second place, carbon dioxide, were less harmful than expected. The most deleterious effect was assigned to water and this results was additionally confirmed by the decrease of the reaction conversion. Moreover, these results were in agreement with relatively high amounts of catalyst which need to be used in metathesis in aqueous media [67].
Recently, Ton and Fogg reported investigations on the impact of oxygen on various ruthenium catalysts, including those bearing CAACs [102]. In all cases, the decrease in ring closing metathesis productivity was observed under 8% of oxygen in Ar. A comparison with a simultaneously conducted reaction under nitrogen showed that CAAC-bearing catalysts were the most O2-resistant and gave the highest metathesis yields. Based on the inhibition of degradation by PCy3 addition authors suggested that the products of oxidative degradation operate through a four-coordinated ruthenium complex both in their experiments as well as in those described by Mol [39] and Trnka and Grubbs [100]. As such, three different transformation paths were proposed (Scheme 38)—(a) a benzylidene attack by O2, (b) a [2+2] cycloaddition of O2—proposed by analogy to molybdenum Schrock-catalysts [103] and (c) a bimolecular degradation with stilbene release.
The experimental analysis of the products led to the conclusion that the degradation through cycloaddition is the dominant reaction but with also some carbon attack on the alkylidene substituent. In addition, under the conditions of the experiment, oxygen also acts as the PCy3 scavenger, facilitating degradation via bimolecular coupling. In view of these results, the remarkable oxygen tolerance of CAAC ligand catalysts is unexpected and its origin is under further investigation.
2.10. CO
There is one more compounds known to cause ruthenium metathesis catalysts’ decomposition, namely CO, a potent Lewis acid, commonly found in various transition metal complexes. Similarly to acrylonitrile, however, CO act as a complexing agent which enables deactivation and decomposition of ruthenium NHC complexes exclusively via the Buchner-type expansion. Due to this feature, investigations on the CO-driven decomposition are reviewed in the next section.
3. Decomposition Routes Depending on the Ligands of Catalysts
Most ruthenium-based metathesis catalysts are synthesized in the form of precatalysts which require initiation and dissociation of one of the Ru-P/O/N bonds. For phosphine-containing complexes it results in the liberation of the phosphine, which can act as decomposing agent. Replacing phosphines by NHCs solves this problem but also causes a different one, since many NHCs can readily undergo either C–H activation or Buchner-type expansion of the aromatic ring. Here we will review the studied related to these subjects.
3.1. Phosphine-Driven Decomposition
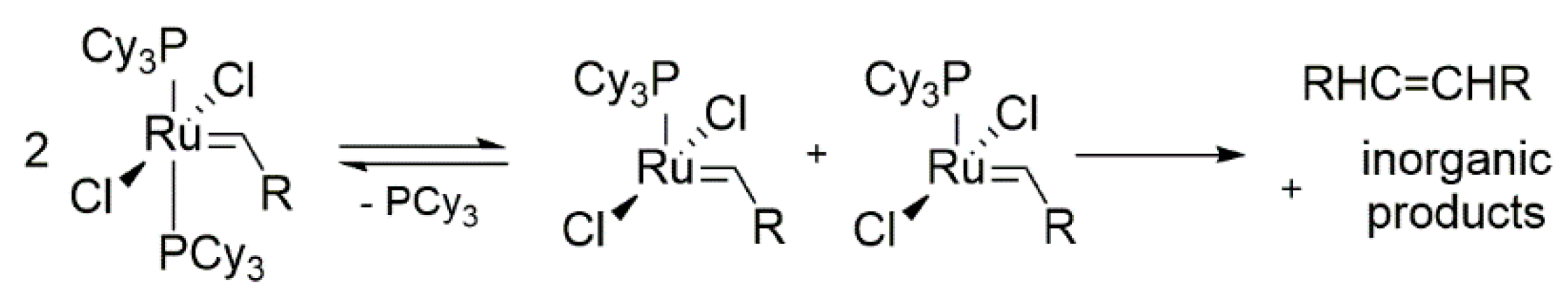
Almost as soon as the first ruthenium metathesis catalysts were synthesized, the first degradation studies of these systems were reported. In 1999 Ulman and Grubbs reported the thermolysis of the 1st generation metathesis catalysts [97]. Based on the NMR measurements, half-lives of selected complexes were estimated at 55 °C in C6D6. The decomposition product or products in this reaction were not determined due to a large number of phosphines signals arising during the course of the decomposition process (Scheme 39). Authors of this work concluded that in most cases the methylidene complexes undergo a unimolecular degradation, while alkylidene complexes a bimolecular one.
The exception to the proposed rule was observed by Forman and co-workers [104]. Thermal decomposition of deuterated complex of (PhobCy)2Cl2Ru = CD2 in benzene at 50 °C was monitored by means of the 2H NMR. Signal analysis allowed to identify the deuterated ethylene (CD2 = CD2), indicating a bimolecular decomposition pathway. Authors suggest that the phosphabicyclononane ligand was responsible for changes in the relative rates of competing degradation routs observed for other bis-phosphine catalysts.
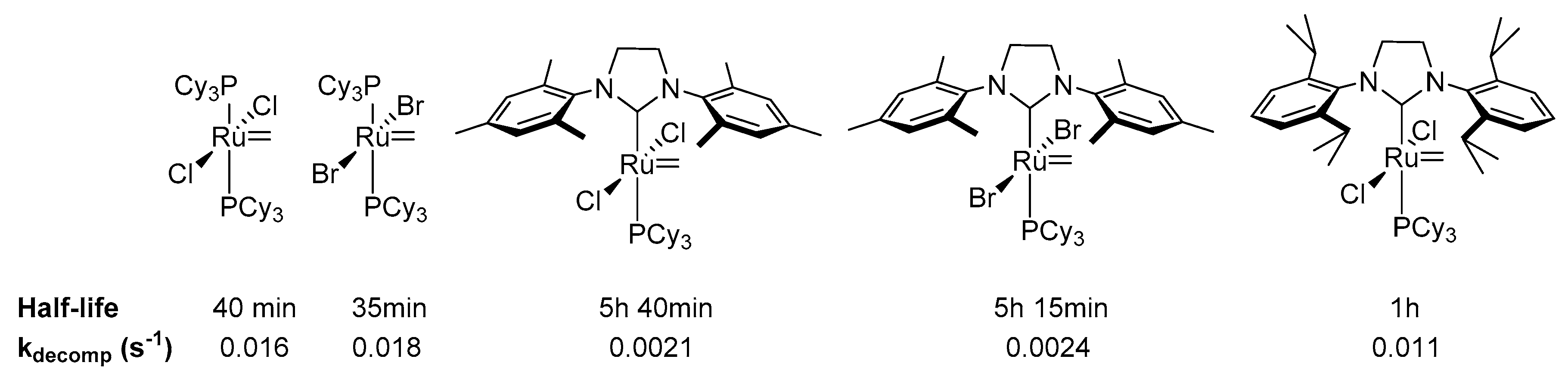
At around the same time Nolan et al. found that ruthenium complexes bearing NHC ligands exhibit higher thermal stability compared to their bis-phosphine analogs [5,105]. Toluene solution of the 1st generation Grubbs benzylidene and vinylmethylene complexes showed 75% and 90% decomposition rates, respectively, upon heating for 1 h at 100 °C, while their IMes derivatives remained intact. The higher stability of the IMes-containing complexes was attributed to an enhanced steric protection of the active 14-electron intermediates formed after the phosphine dissociation by NHC ligand.
Also during that time Grubbs and co-workers synthesized various ruthenium electron-rich carbene complexes [Ru = CHER], where ER = OR, NR, SR and characterized their thermal decomposition product as carbonyl hydride species (Scheme 40 [106,107]. The spectroscopic analysis (IR, NMR, GC/MS) of 13C-labelled and deuterated Ru(TFA)2(PPh3)2 = CHOCH2Ph complexes found signals from the carbonyl ligand and showed the formation of toluene and benzyl trifluoroacetate byproducts and, thus, supported the proposed decomposition mechanism shown on Scheme 41 [106]. In this mechanism, the phosphine dissociation and the β-alkyl elimination leads to formyl intermediates 40, which after the removal of benzyl trifluoroacetate TFACH2Ph transform into ruthenium(II) complex. Further rearrangements in this complex may result in the carbonyl species 41. Another path, with the reductive elimination of toluene from intermediate 40 to yield carbonyl adduct 42, was also proposed. Prolonged heating of the bis-phosphine ethoxy carbene complex RuCl2(PCy3)2 = CHOEt in benzene allowed Grubbs et al. to isolate a ruthenium hydride 1 in 69% yield, further characterized by means of X-ray [107]. The obtained hydride complex 1 was a subject of the previously proposed mechanism, which confirmed its validity. The remaining Fischer carbenes gave numerous signals in the NMR spectrum from the resulting degradation products, suggesting several competing degradation pathways. Based on the established half-lives, the thermal stability of the derivatives was found to be in the order of E = N > C > S > O.
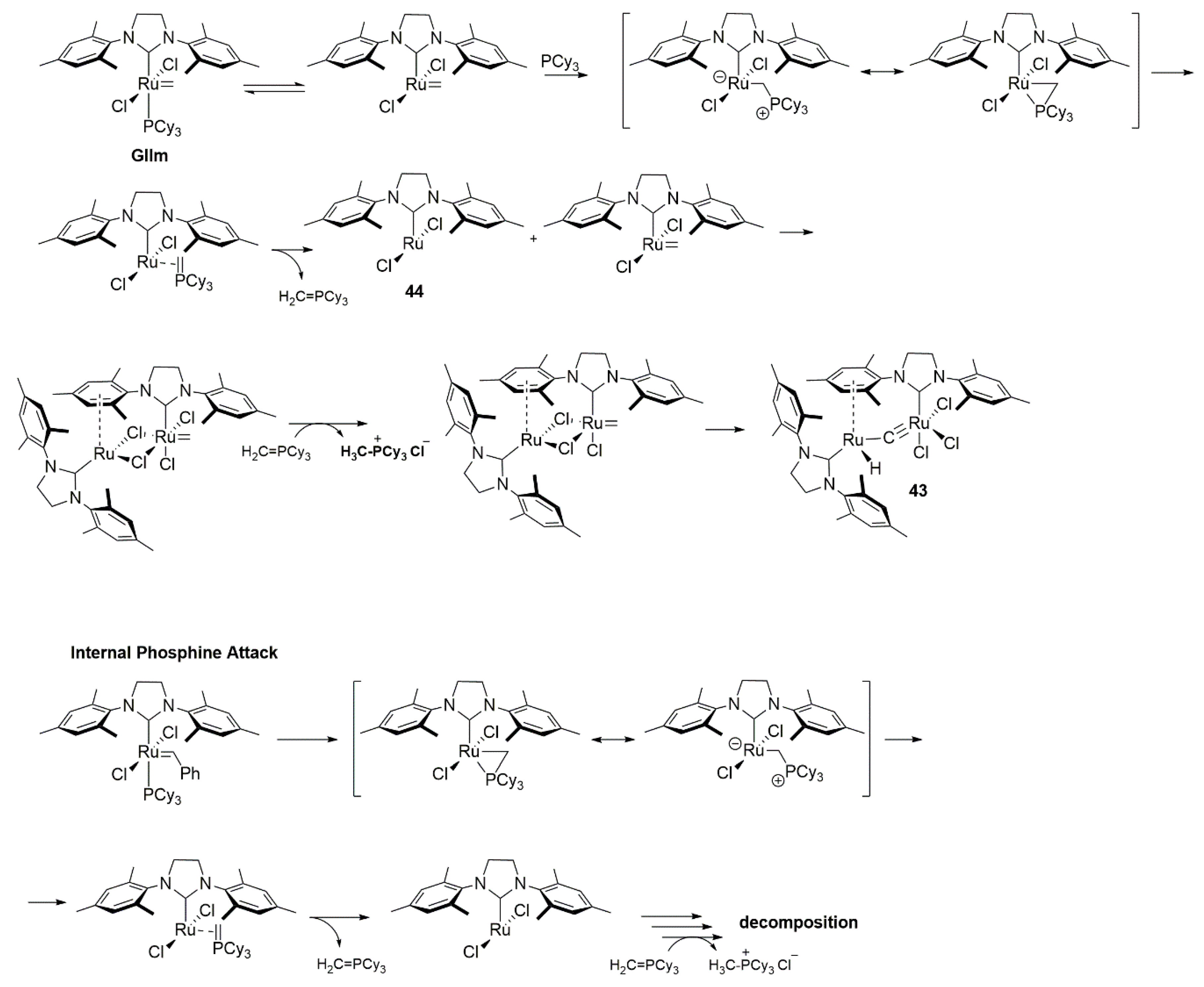
Early thermal stability studies of the 2nd generation ruthenium catalyst were reported by Hong and Grubbs [96]. The methylidene complex GIIm was heated at 55 °C in benzene and after 72 h the diruthenium compound 43 were isolated in 46% yield, together with CH3PCy3+Cl−. Complex 43 is an interesting species that can be characterized by the loss of the alkylidene functionality and the presence of the hydride ligand on one ruthenium atom as well as a η6-interaction of the second ruthenium center to one of the mesitylene moieties (Scheme 42). The authors of this study proposed a multi-step degradation pathway based on chemistry established for other ruthenium carbene complexes [108] and formation of the CH3PCy3+Cl- salt. In this pathway, a phosphine attack on the methylidene complex was suggested as the first step, which than releases the CH2 = PCy3 ylide and the 12-electron species 44. Further, it was suggested that intermediate 44 may bind one of the mesityl substituent of methylidene complex leading to a dimer with two chloride bridges, followed by HCl abstraction through ylide, forming CH3PCy3+Cl− and an alkylidyne adduct. Transformation of the latter system into the final complex 43 was elucidated by the migration of chloride ligands and an oxidative addition of alkylidyne. The formation of proposed intermediates was, however, speculative since they have not been observed experimentally. Considering the time scale of the degradation process (which usually take days) and metathesis reaction (which is usually finished in hours), it can probably be assumed that it has no significant impact on the course of the metathesis reaction. However, as later work showed, the proposed nucleophilic attack on the methylidene is the primary vector of the active methylidene species degradation [88,109].
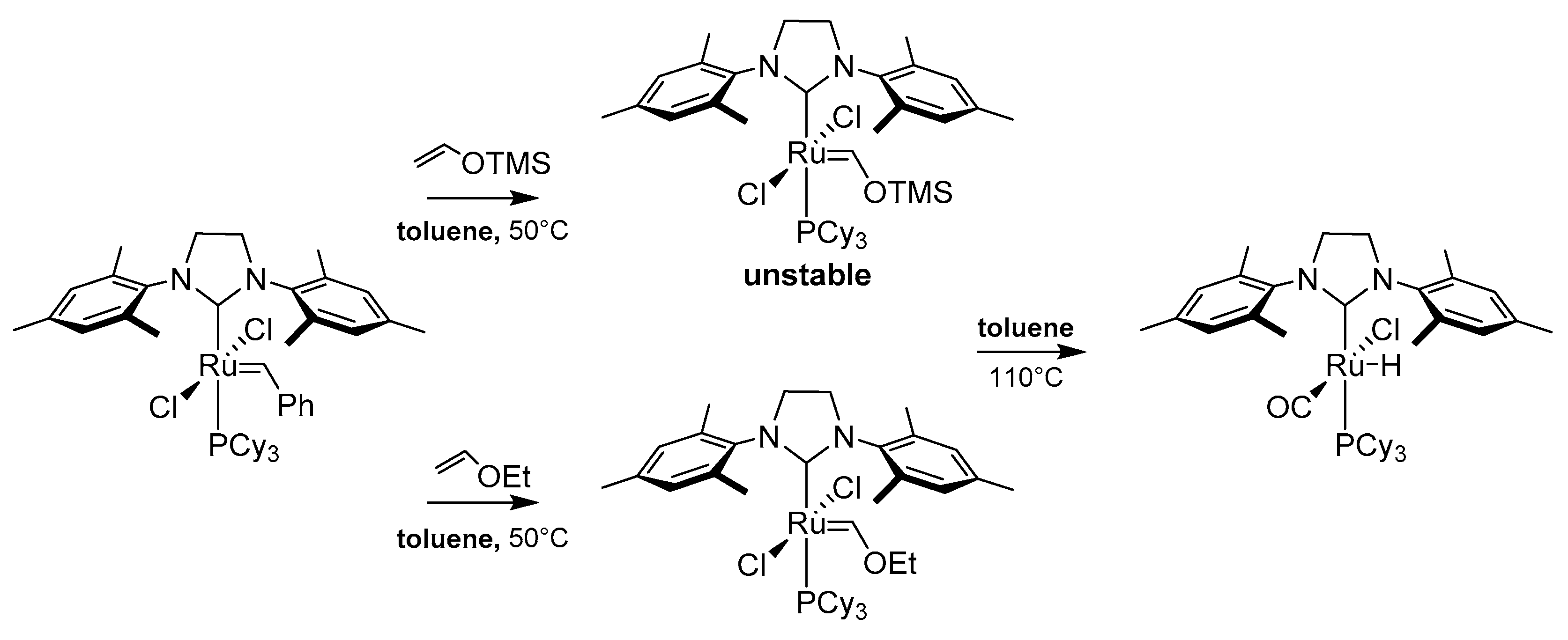
In continuation of these works Grubbs et al. screened thermal stabilities of a series of methylidene complexes (Scheme 42 and Scheme 43) [84]. The results confirmed that phosphine catalysts are subject to a first-order degradation rate, which is not affected by the excess of phosphine. Moreover, based on half-lives of NHC-catalysts authors leaned more towards a mechanism involving the dissociation followed by the phosphine attack, rather than a mechanism involving an internal attack of the phosphine on the methylidene species (Scheme 42).
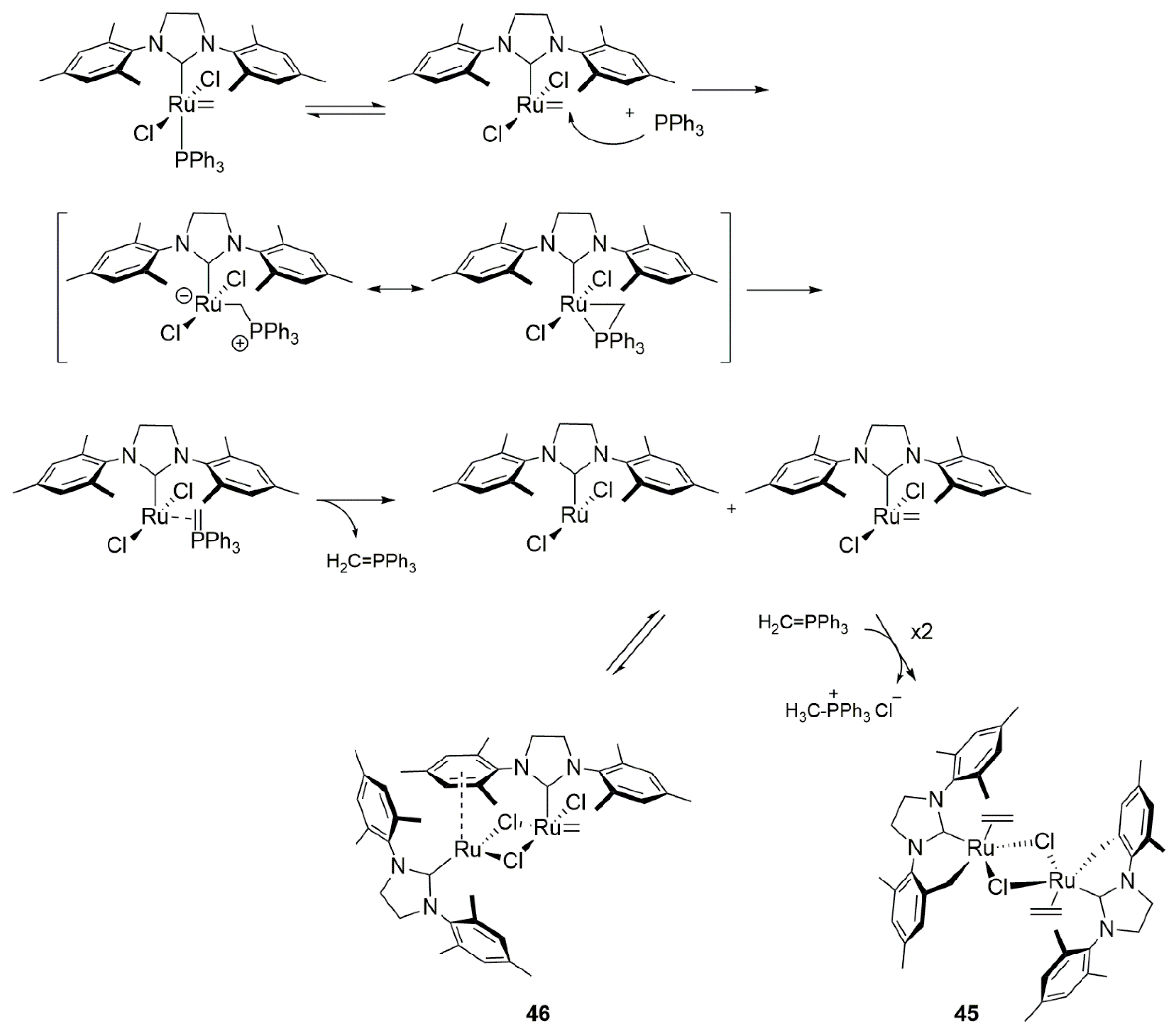
In the same work authors investigated also the degradation of an active methylidene species under metathesis reaction with ethylene in toluene. Different behavior was observed for PPh3-containing derivatives with respect to the PCy3-containing ones. Two major products were characterized as methyltriphenylphosphonium chloride CH3PCy3+Cl− and diruthenium complex 45, which structure was confirmed by the X-ray analysis. The proposed mechanism of this reaction is presented on Scheme 44. It is interesting to note that the same reaction carried out in CD2Cl2 allowed to identify the formation of complex 46, whose existence was suggested by the authors in an earlier work [96]. Interestingly, in the presence of ethylene at −40 °C this intermediate can revert to an active methylidene to give the metallocycobutane. Additionally, the decomposition of a phosphine-free Hoveyda-Grubbs catalyst with ethylene in C6D6 at 55 °C resulted in an unidentified hydride species.
Inspired by the aforementioned work, Metzger and Wang conducted a spectroscopic analysis, using electrospray ionization mass spectrometry (ESI-MS), of bis-phosphine olefin catalyst systems GI and GIv in RCM reaction with olefins, presented on Scheme 45 [110]. Although the authors failed to identify any degradation products containing ruthenium, they were able to determine both cis and trans stilbene in CH2Cl2 solution of catalyst GI, indicating a bimolecular degradation with organic dimer formation. For all investigated complexes the methylphosphonium cation CH3P+Cy3 (at m/z 295) as well as the benzylidenetrialkylphosphine (at m/z 374), which represents products of phosphine-driven degradation, were detected.
3.2. C-H Activation
Metal-promoted C-H bond activation is a very active research area and holds great potential in organic chemistry [111,112,113,114,115]. The C–H activation of complexes bearing unsubstituted ortho positions in the N-alkyl and N-aryl groups result in decomposition, which can occur even in mild conditions and is often reversible, therefore difficult to detect. It is also accompanied by the formation of transient metal hydrides affecting the catalytic activity of the catalyst.
Grubbs et al. first reported the insertion of ruthenium into one of the NHC methyl C–H bond of the second generation Grubbs catalyst [100]. This C-H oxidative addition and loss of the alkylidene ligand was promoted by air, forming a catalyst in which the C–H bond activation of the mesityl ortho-methyl group occurred (Scheme 46). The decomposition product was air-stable in solution and solid state. All bonds linked to the ruthenium in the new 16-electron complex became shorter but the mechanism, at that time, was still unknown.
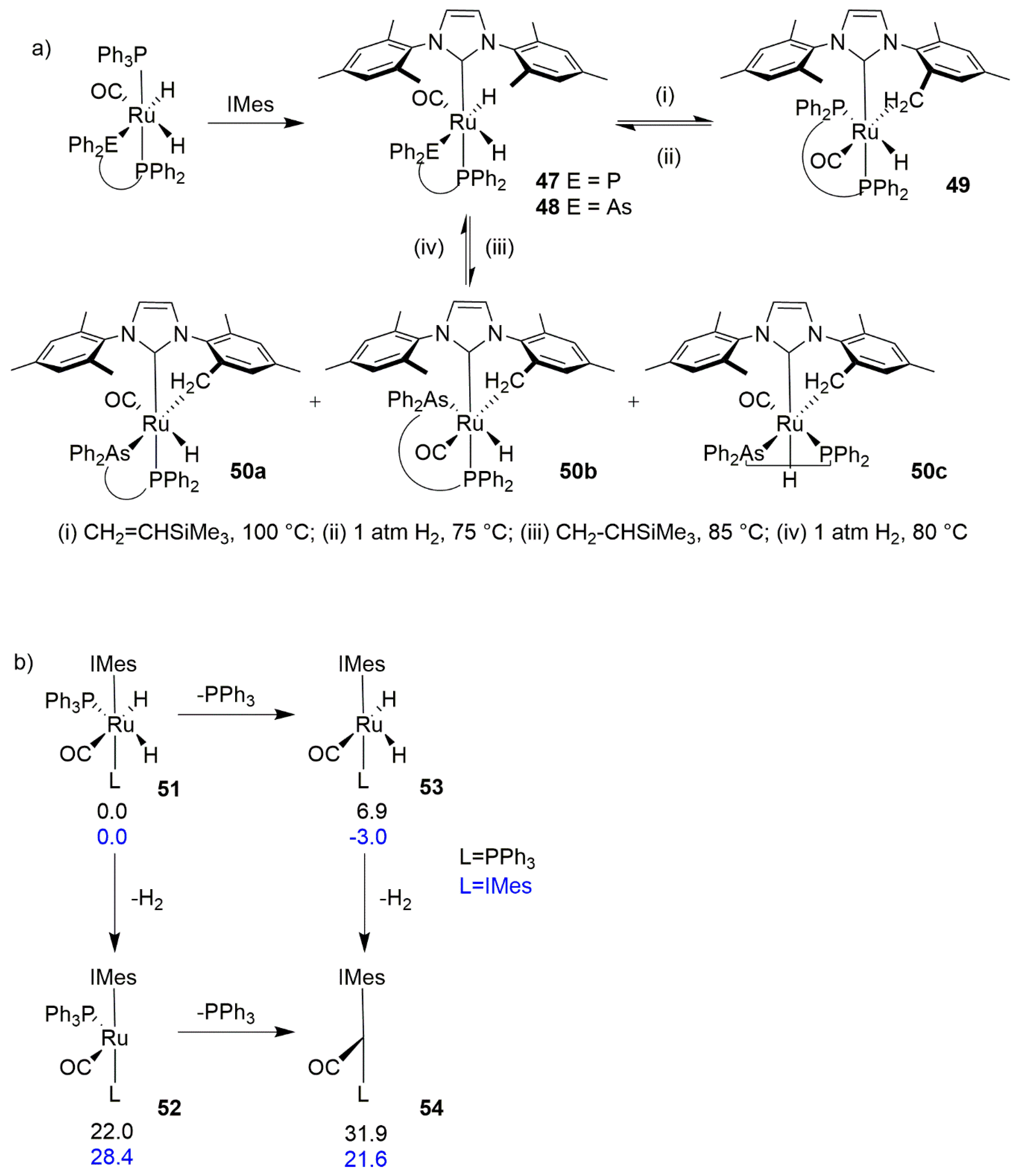
Whittlesey’s group treated the mono-carbene species Ru(IMes)(PPh3)2(CO)H2 with trimethylvinylsilane at room temperature, also observing the intramolecular C–H activation of an ortho-methyl group in the NHC ligand, reversible on the use of hydrogen [116]. The same group observed the activation of ruthenium chelating P-P and P-As complexes 47 and 48, which share many structural features with metathesis catalysts, induced by trimethylvinylsilane, which yielded complex 49 with Me3SiEt at 100 °C as well as a mixture of three regioisomers 50a–c at 85 °C, respectively (Scheme 47a) [117]. Heating under hydrogen reformed the dihydride precursors. Their later theoretical studies showed that the C–H activation occurred through a Ru(0) intermediate formed by the olefin, which acts as hydrogen acceptor [118]. The precatalyst reformed on addition of the hydrogen is useful in catalytic transfer hydrogenation involving alcohols [119,120,121]. The C–H activation of 51 took place through the thermal reductive elimination, forming reactive Ru(0) intermediates (Scheme 47b). This process may occur at the six-coordinate complex to give a four-coordinate complex 52 or at the five-coordinate transition state 53, to give the 3-coordinate complex 54. The latter can also be obtained by the loss of phosphine from 52. When any of the 52 or 54 systems are formed, they undergo a rapid C–H activation. It was also proven that the solvation by benzene does not have any impact in stabilizing species 52, 53 or 54. Similar complexes were also studied theoretically by the groups of Houk and Trzaskowski using computational methods to further elucidate the details of the reaction/decomposition mechanisms [122,123].
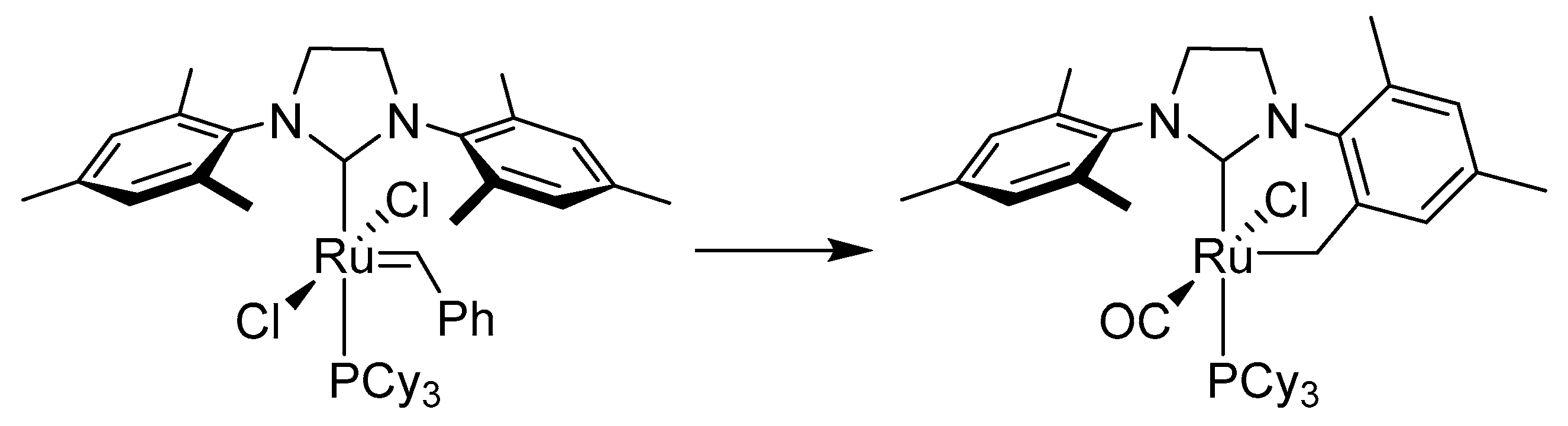
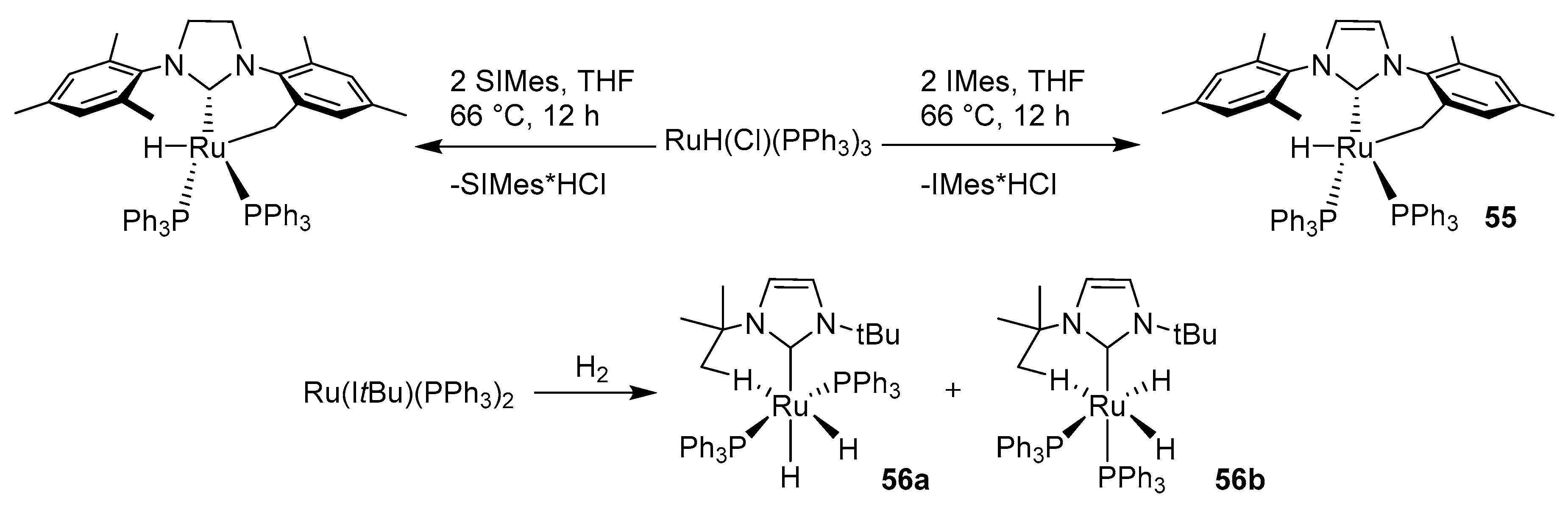
This research topic was continued by Morris et al., who observed the cyclometalation of an ortho-methyl group when reacting RuHCl(PPh3)3 with SIMes, IMes or imidazolium salt precursors (Scheme 48) [124]. The reaction of 55 with CO at 20 °C yielded a complex with an attached CO group, whereas at 68 °C the Ru(0) complex saturated with three CO groups and a reformed IMes ligand was obtained. Protonation of the cyclometalated IMes ligand by phenol, apart from associating a ɳ5-bonded phenoxide group, also caused the reformation of the IMes ligand. Reaction of RuHCl(PPh3)3 with ItBu and later with hydrogen produced a mixture of two isomers 56a and 56b of the dihydride Ru(H)2(ItBu)(PPh3)2, both with activated C-H bonds. This study proved that IMes and PPh3 ligands do not bind to 55.
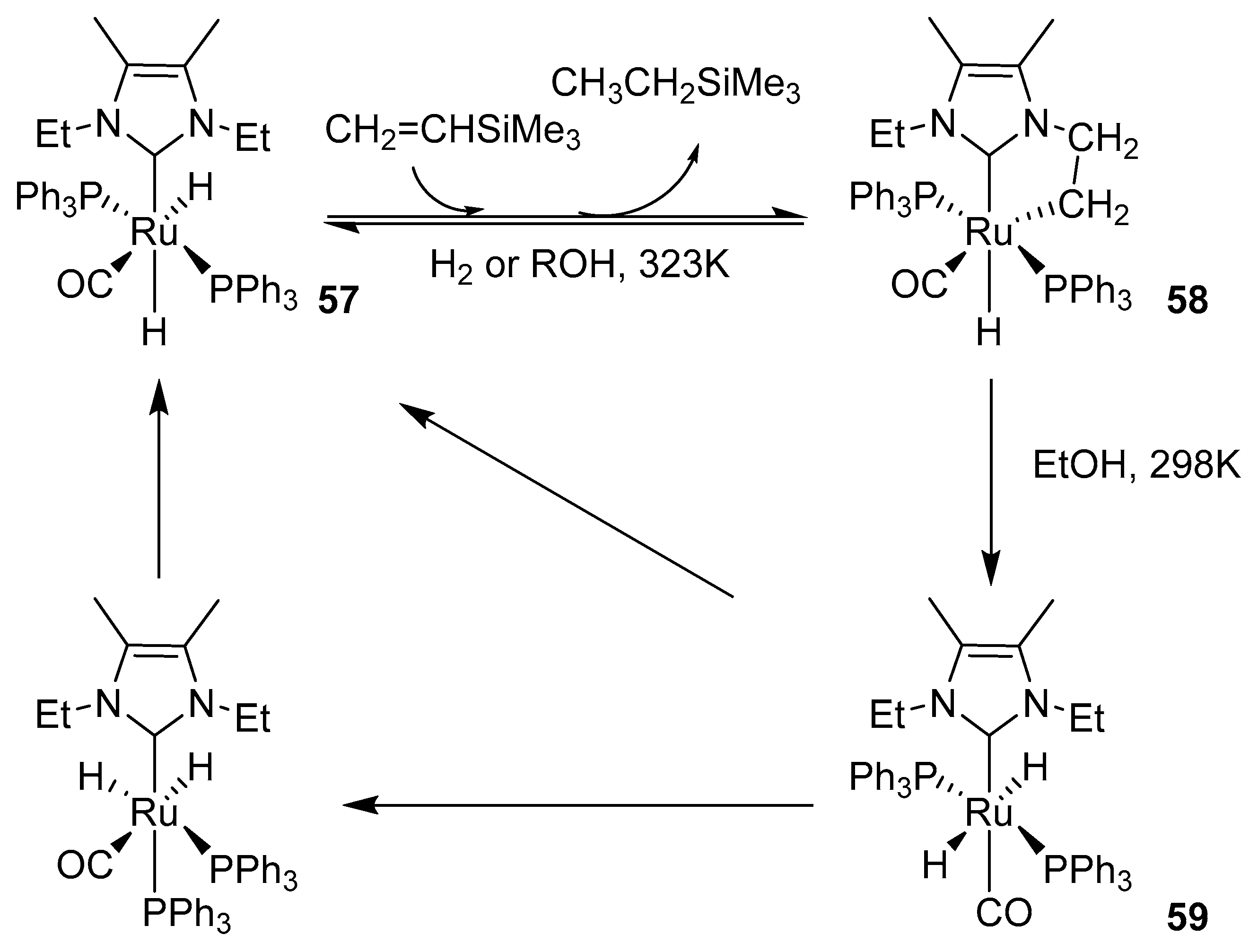
A year later, Whittlesey described trimethylvineylsilane-promoted activation of the alkyl NCH2CH2-H activation of 57 (Scheme 49) [125]. A metalated, 5-membered ring of the IEt2Me2 ligand was formed with a Ru–C–C angle similar to that of an unstrained sp3-hybridized methylene group (58). Since complexes in Morris’ work were more electron-rich, the key role in permitting the C–H activation in complexes 57 and 58 was assigned to the CO ligand. Reaction with hydrogen or benzene/alcohol solutions in room temperature caused precipitation of 59 but complex 57 was reformed in alcohol with an increasing rate in MeOH< EtOH < iPrOH.
Grubbs also reported decomposition via the C–H activation of the second generation Grubbs catalyst leading to complex 45 [84]. The decomposition occurred after 5 days in toluene at 23 °C and was initiated by the dissociation of the phosphine. The presence of ethylene facilitated its subsequent attack on the methylidene of GIIm (Scheme 44) [100,119]. The formed complex 44 in the presence of two Ph3P = CH2 molecules underwent C–H activation, producing the dinuclear catalyst 45 and methyl-trihenylphosphonium chloride.
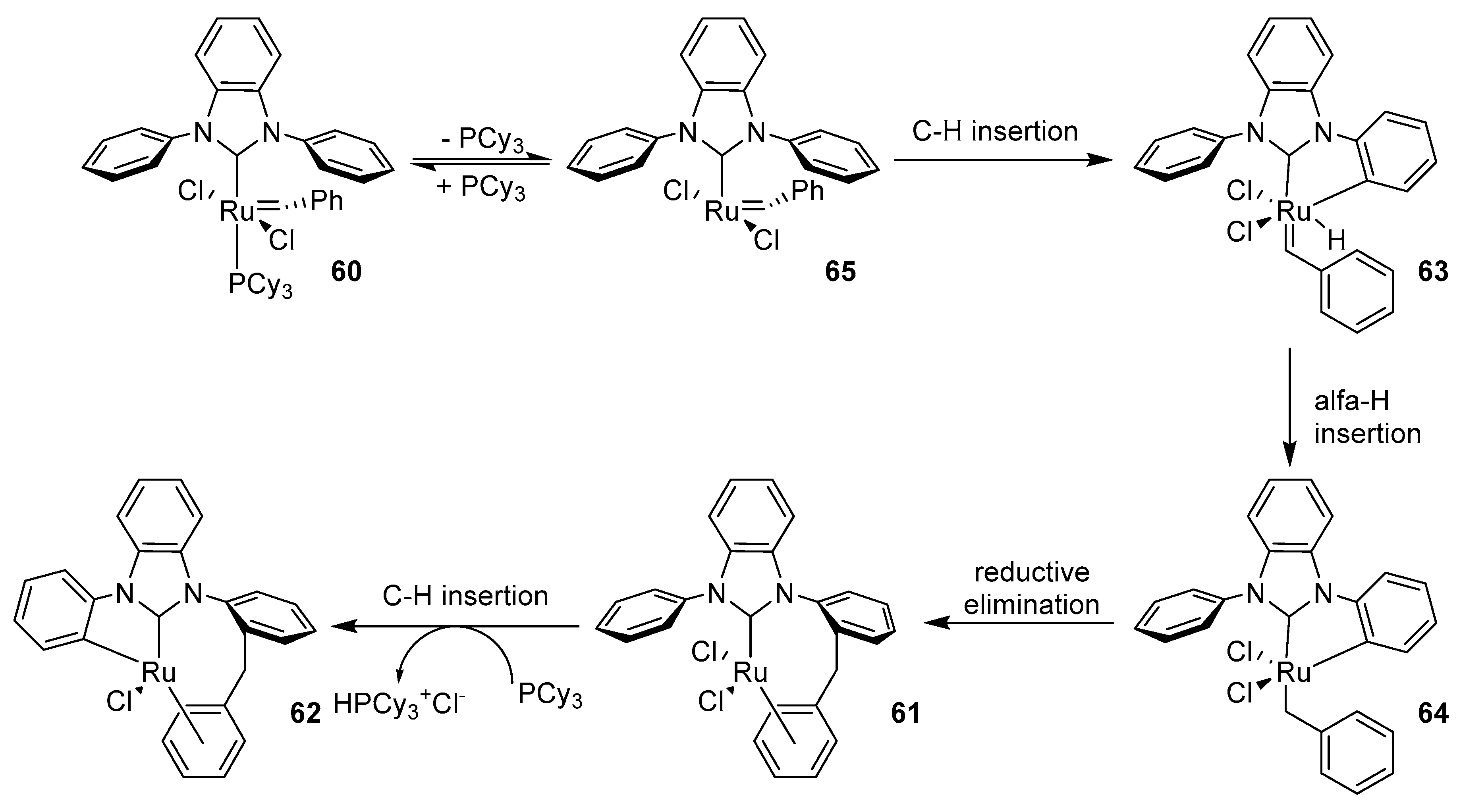
The same group reported the double C-H bond activation of a phenyl-substituted NHC in 2nd generation Grubbs catalyst 60 [126]. In complex 61 one can observe an inserted benzylidene carbon atom into the ortho C–H bond of the N-phenyl ring, as well as a ɳ6 binding of ruthenium to the benzylidene phenyl group and the loss of phosphine (Scheme 50). Catalysts with NHC ligands containing phenyl instead of mesityl groups were more prone to the C–H activation, as further insertion of ruthenium into another phenyl group and the formation of a five-membered metallacycle produced complex 62. It was suggested that the flexibility of the N-phenyl groups play the key role in the initiation and propagation of this reaction [127]. Heating of complex 60 in benzene at 60 °C under inert conditions led to product 61 after three days (58% yield) with traces of 62. Faster decomposition of this complex occurred in DCM, however, after 12 h at 40 °C the main product was 62 (38% yield) with 61 in 24% yield. After a week in 40 °C 61 turned into 62, while after adding phosphine it turned into 62 at room temperature only in three days.
Grubbs suggested a mechanism for decomposition of 60, different from the previous one due to no attack of the phosphine. In this mechanism, immediately after the loss of the phosphine (initiating the metathesis), the ortho C–H bond of the N-phenyl group adds to the ruthenium center forming a ruthenium hydride complex 63. In the next step, the hydride inserts at the benzylidene α-carbon atom giving complex 64 and a reductive elimination between the metalated N-phenyl carbon atom and benzylidene α-carbon atom generates 61. Lastly, a C-H insertion of the second N-phenyl ring and phosphine-mediated elimination of HCl yields 62. Intermediates 63-65 were not observed by spectroscopic methods, most probably due to their short lifetimes.
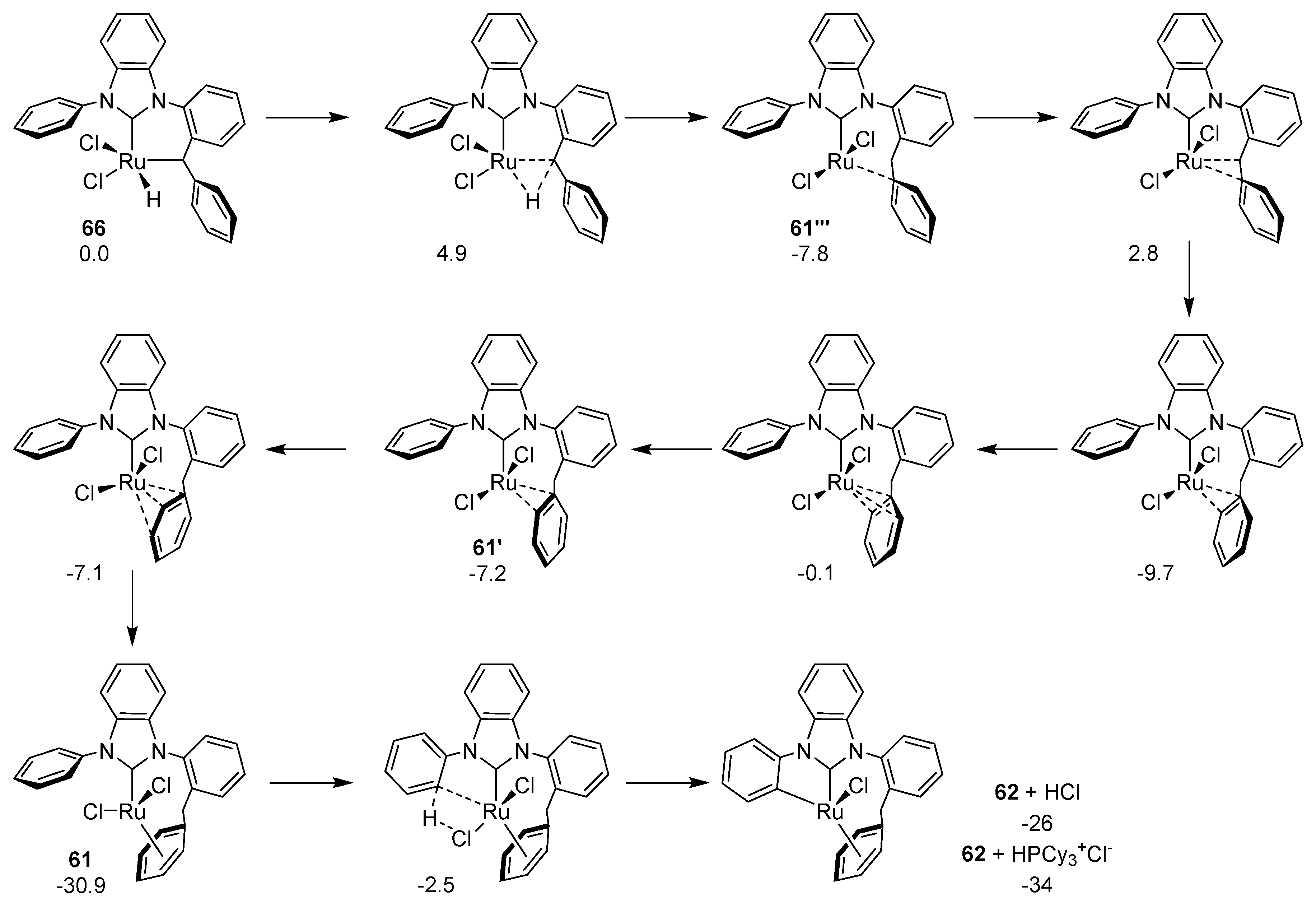
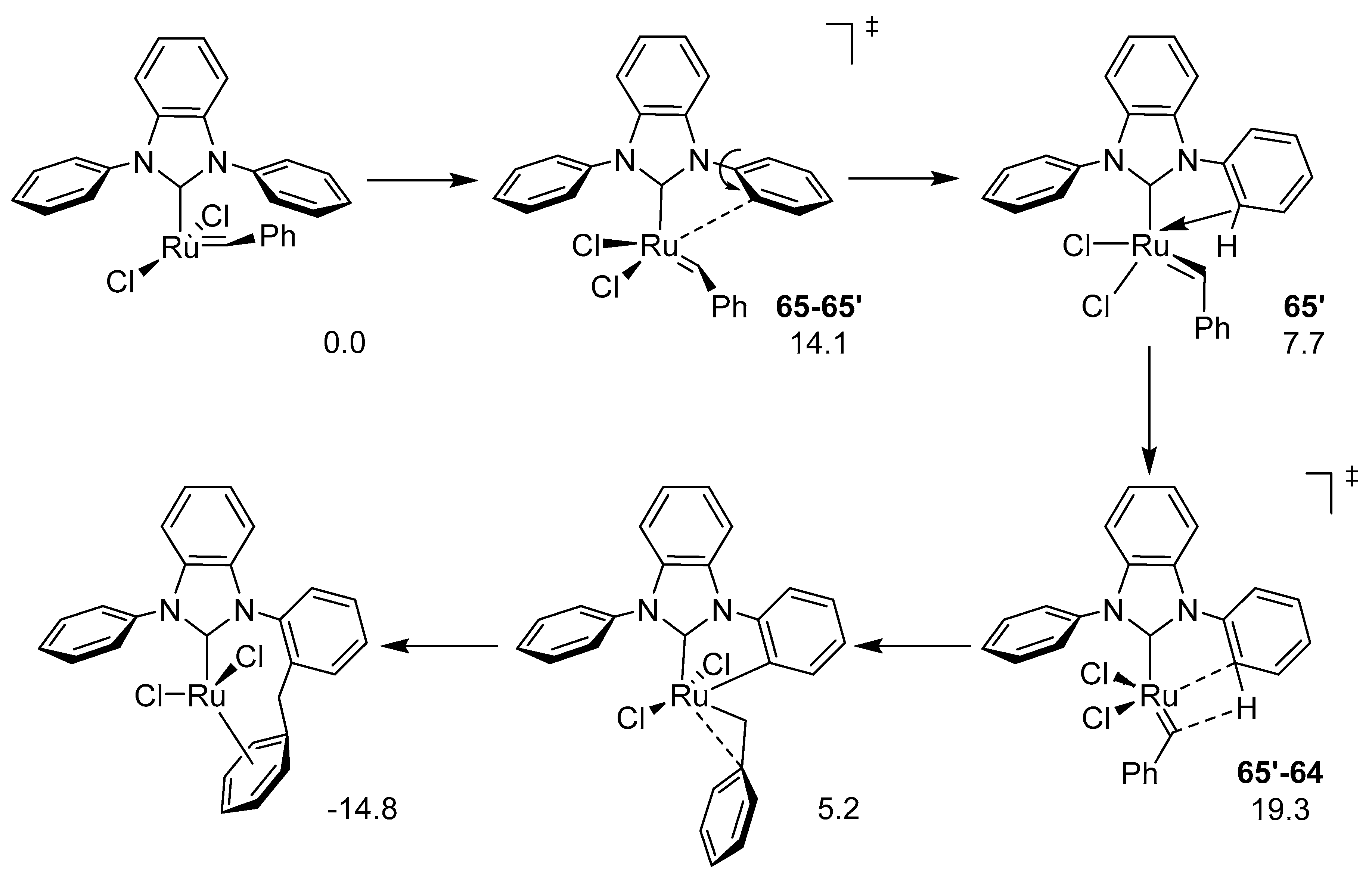
Suresh et al. further investigated this mechanism using theoretical methods and also highlighted the key role of the flexibility of N-phenyl groups in the initiation and propagation of this reaction [128]. Their work was extended later by Cavallo et al., who also theoretically studied the mechanism of this double C–H activation [129]. They presented a low energy pathway of formation of 62, displaying the importance of phosphine in the propagation of this step (Scheme 51). The first step starts from the elongation of the C–H bond in 14 electron complex 65, after dissociation of the phosphine, followed by the transfer of the agostic hydrogen to the benzylide α-carbon atom through a transition state (oxidative addition). The benzylidene group turns into a benzyl group and a new bond between ruthenium and the ortho carbon atom of the N-phenyl ring is formed. Complex 64 (suggested by Grubbs) is higher in energy than 65 and is stabilized by the double bond between ruthenium and the benzyl carbon atom. The benzyl α-hydrogen transfers to ruthenium through a transition state forming complex 63, which is unstable and prone to undergo a σ-bond insertion between the ruthenium and the N-phenyl ortho carbon atom forming ruthenium hydride 66, previously not postulated by Grubbs.
In the second part of the mechanism, the ruthenium hydride bond is broken by a reductive elimination, forming a complex stabilized by a strong interaction with the former benzylidene Cipso atom. Intermediates 61′-61′’’ were previously not postulated [128], probably due to the high stability of 61. Lastly, more stable complex 62 is formed via an interaction of one chloride atom with the other N-phenyl ortho hydrogen atom and the elimination of HCl. Experiments confirmed that complex 61 was only thermodynamically stable without a Lewis base, that is, phosphine, otherwise the equilibrium was shifted towards 62 [128]. During the C-H activation the N-phenyl rings maintain their aromaticity, being the key feature for this reaction. The benzylidene phenyl ring plays a role in stabilizing the various isomers of 61 by a strong electron donation to ruthenium center, which reduces its aromaticity but this is compensated by binding to the metal and elongating the C–C bond.
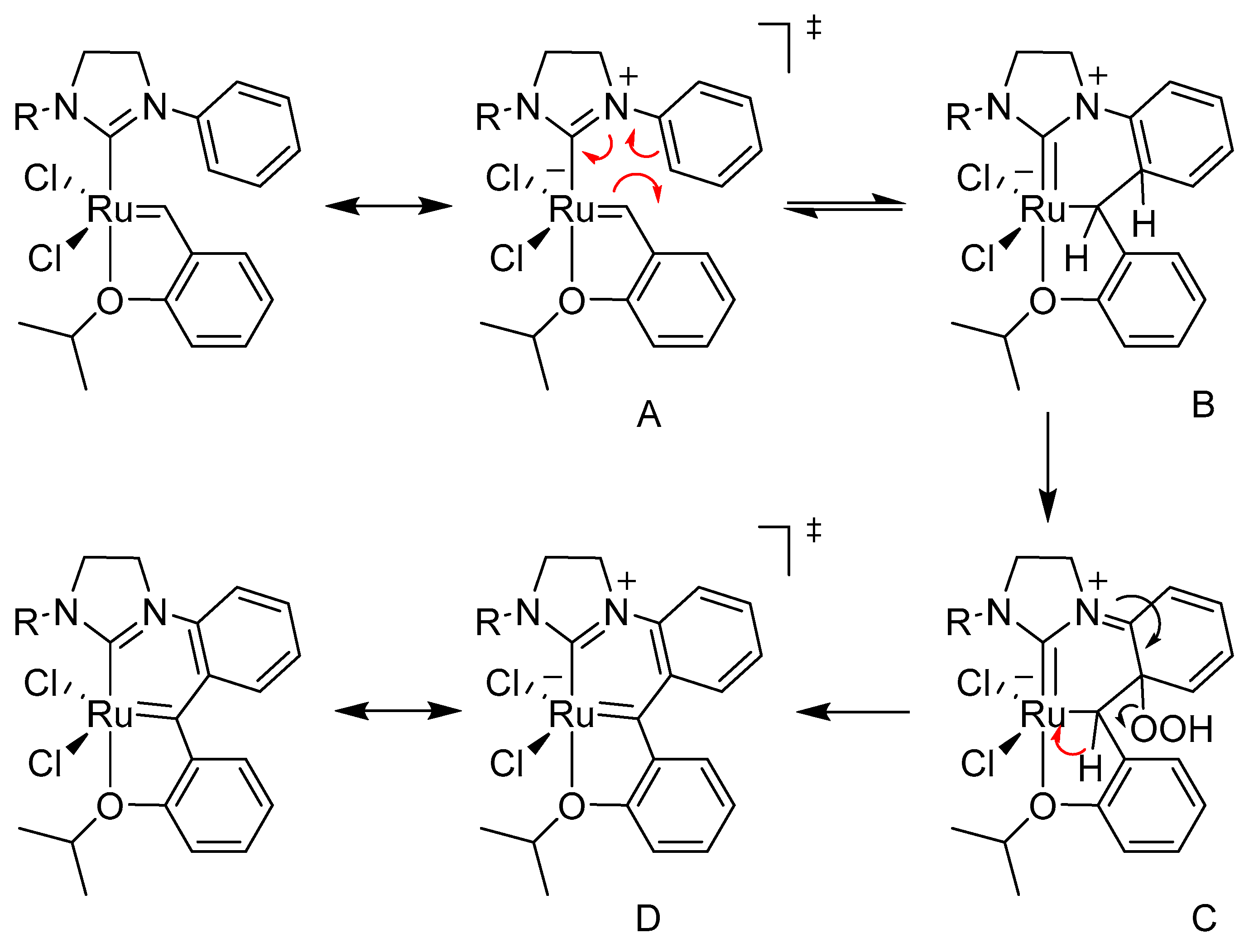
In another work devoted to the issue, Blechert et al. observed the decomposition of the Hoveyda-type complex after two weeks under air forming an intramolecular bond between the ortho position of the N-aryl ligand and the benzylidene carbon atom, the length of which was between that of a single and double bond [130]. This type of decomposition was also observed for another Hoveyda-Grubbs-type complex bearing an unsymmetrical NHC in DCM after a few hours under air, proving that presence of oxygen and less steric N-substituent allows the formation of a bond between the benzylidene carbon atom and β-position of the N-aryl ligand [131]. The postulated mechanism follows a sequence of a reversible pericyclic cyclization of A to form B, followed by an irreversible oxidation with oxygen to C. An elimination and re-aromatization finally causes the insertion, giving product D (Scheme 52).
The following year, McDonald et al. reported a thermal decomposition of phosphonium alkylidene catalysts in the presence of 1,1-dichloroethylene and B(C6F5)3, yielding a trichloro-bridged dimer (Scheme 53a) [132]. C-H activation of the mesityl ortho methyl group through the addition across the Ru = C phosphonium alkylidene bond was the rate-determining step of this process [116]. The ruthenium intermediate IV eliminates the methyl phosphonium to give the alkylidene complex V and after the cross metathesis with the trapping olefin, the dichlorocarbene and styrenic vinyl groups are formed in intermediate VI. Then, after the loss of chloride, the dimerization occurs and the ion exchange gives 67 and a methylphosphonium salt [H3CPR3][Cl]. Intermediates IV–VI were not directly observed, however the mechanism for the formation of V could be the one postulated previously by Blechert et al. [130]. After 1–2 days at room temperature, a mixed dimer 68 was observed after „self-trapping” of V by 69 (Scheme 53b). It was, however, noted that these catalysts exhibited higher initiation rates than the rate-limiting step in decomposition. In the same year, Piers et al. observed the same dimerization of this catalyst after two days at room temperature, where the C-H insertion occurred through the alkylidene moiety under air [133].
In the consecutive studies, Severin et al. observed that treatment of complex 70 with one equivalent of PCy3 in dioxane at 70 °C for 3 days yielded a chiral dinuclear, chloro-bridged product 71 in which a η2-bound cyclohexene group, attained from the phosphine ligand after partial hydrogenation, was coordinated to the ruthenium center (Scheme 54) [134]. Two diastereoisomers of 71 were obtained due to chirality of the dehydrogenated PCy2(C6H9) ligand with ruthenium acting as the stereogenic center. The formation of complex 71 through the intramolecular C-H activation of the cyclohexane group proved the existence of a very reactive intermediate [(cymene)Ru(μ-Cl)3RuCl(PCy3)], with one cymene ligand replaced by phosphine, which was possible to capture by addition of the dihydrogen.
Grubbs and Houk contributed to the topic by studying the decomposition of Z-selective metathesis catalysts with chelating ligands resulting from the carboxylate-driven C-H bond insertion [135]. According to their results, the C–H activation occurred in THF at room temperature between the N-adamantyl alkyl C–H bond and the carboxylate ligand bound to ruthenium, involving a four- or six-membered transition step (Scheme 55) [136]. A subsequent hydride elimination depends on many steric and electronic factors. Compared to the oxidative addition mechanism, this carboxylate-driven process allowed for isolation of active complexes without the formation of a ruthenium hydride [127]. Treatment of 72 with the excess carbon monoxide at −78 °C led to a 1,1-insertion of CO ligand into the Ru-C bond of the NHC ligand. In the next step the alkylidene moiety decomposed and an alkyl ruthenium complex saturated with CO ligands was finally formed with the N-adamantyl group bound to the former alkylidene carbon. Substitution by the π-acidic CO ligands decreased the stabilization of the alkylidene carbon via backbonding [137].
Such alkylidene insertion could be also possible for C–H activated catalysts in the absence of CO. It was proposed that similar ruthenium-alkyl complexes could undergo a hydride elimination when not saturated with CO ligands leading to inactive catalysts. Treatment of catalyst 73 with silver pivalate (tBuCOOAg) produced the η2-bound olefin complex 74, through the ligand exchange of chlorides for pivalates and the generation of silver chloride. The C–H activation occurred through the alkylidene insertion and the β-hydride elimination (Scheme 55b). This group also studied the decomposition of complex 75 through the alkylidene insertion and the α-hydride elimination, by treatment with tBuCOOAg, resulting in a bond formation between the benzylidene carbon and ortho carbon of the N-aryl ligand, keeping the alkylidene group (Scheme 55c). The mechanism of this reaction was not carboxylate-driven and the reaction occurred only in the presence of oxygen. The initial product was complex 76 which, by treatment with methylene chloride/pentane mixture, gave product 77 with replaced pivalates for chloride ligands [138,139]. The formed pivalic acid was a proof of the C–H activation followed by the catalyst decomposition.
Cavallo performed DFT studies on the first steps of the C–H activation, the formation of bond between the ruthenium and the N-phenyl ortho methyl group (Scheme 56) [140]. The second step of this mechanism was the formation of a benzyl group through an activated proton transfer. A variety of substituents in the NHC backbone did not have a strong effect on the decomposition pathway and, in many cases, the energy of the transition state 65–65′ decreased for the ortho isopropyl groups, while the methyl substituents had no effect. For the rate limiting transition state 65′–64, the NHC substituents in intermediate 65′ had a larger effect on the decomposition.
To study the influence of the ruthenium-ylidene bond, Ru-benzylidene, Ru-indenylidene, Ru-methylidene and trimethylphosphonium Ru-alkylidene bonds were compared in the same work. Transition states 65–65′ and 65′–64 were found to be more stable for the Ru-Ind system than the Ru-benzylidene one, suggesting that the catalysts with indenylidene group were more prone to this decomposition. For the Ru-alkylidene system, the second transition state was less stable, though, on the other hand, a very stable complex 64 was formed. Catalysts with methylidene and benzylidene group showed similar results, suggesting that they undergo a similar decomposition pathway.
Recently, Fogg et al. observed a rapid decomposition of complex GIIm through the C-H activation of mesityl ortho-methyl group, by the treatment with a 10-fold excess of pyridine [92]. The saturated NHC was found to be more prone to the activation due to a π-back-donation from the ruthenium to the NHC, slowing down the rotation around this bond [141,142,143,144]. The corresponding σ-alkyl intermediate 27 was in a conformation that favored the interaction between the σ-alkyl carbon and the mesityl group [134,137], while rotating about the C–N mesityl bond [145,146]. The rapid C-H activation suggested the presence of only starting methylidene complex GIIm and the decomposition product [MePCy3]Cl (Scheme 32). The C-H activation was slower for 27_IMes due to the rotation of IMes ligand (after 15 min). The treatment with a 3-fold excess of pyridine retarded the decomposition of GIIm_IMes, while the decomposition of 27_IMes was slower than its formation. In all cases the rate-determining step of the decomposition was either pyridine binding or C–H activation, indicating that the energetic barriers for both processes were very similar. The entire decomposition was completed within 75 min and small amounts of the free phosphine suggested an additional, unknown pathway.
3.3. Buchner-Type Expansion fo NHCs
Interaction between the electrophilic carbene and the aromatic ring of the NHC may lead to carbene insertion into the aromatic ring system, also known as the Buchner ring expansion. This reaction is usually found when isocyanides or carbon monoxide, widely used as ligands in transition metal complexes due to their σ-donor strength and weak π acidity, are complexed to Ru [141,147,148,149,150].
The group led by Diver was the first to propose a decomposition pathway for 2nd generation Grubbs II catalysts involving carbene insertion into the mesityl group of NHC ligand, promoted by carbon monoxide or isocyanides, also known as Buchner reaction [151,152]. The general mechanism consists here of two steps—first, the aromatic ring undergoes a cyclopropanation which, after rearrangement by 6π electrocyclization, forms a cycloheptatriene. Two years later, Grubbs reported the loss of CH2 = PPh3 in second generation catalyst decomposition [84]. The Diver group proved that in the first generation complexes the ylide transfers from the ruthenium to the phosphorus of the phosphine [153,154]. After treatment with CO, two molecules coordinate to ruthenium already bearing three chloride ligands and the whole complex is anionic with a phosphonium counterion. The addition of an aryl isocyanide p-ClC6H4NC to the 1st generation Grubbs complex also caused loss of benzylidne resulting in a complex with three isocyanides added to the ruthenium in a mer arrangement. The first isocyanide molecule coordinates to the metal in the trans position to the benzylidene (A, Scheme 57), which causes an intramolecular migration of phosphine by a 1,2-shift and produces intermediate B. The binding of the second isocyanide ligand forms the intermediate C, which after ylide elimination produces 78 and 79.
Later, the same group synthesized polar isocyanide 80 and used it to remove Ru from metathesis reaction mixtures [131]. The isocyanide-promoted carbene insertion in the 2nd generation Grubbs catalyst (Scheme 58) resulted in a rapid quenching of the carbene reactivity. In a reaction with p-chlorophenyl isocyanide 81, complex 82 was formed, in which two molecules of the isocyanide were added to the Ru center and the benzylidene formally inserted into one of the Mes groups, similar to earlier findings with CO [151]. The same insertion was triggered by the polar isocyanide 80, which conferred greater polarity to the hexacoordinate Ru complex, facilitating the loss of catalyst’s activity and its removal from the organic products of metathesis.
In consecutive studies, the Cavallo group performed DFT calculations for the Buchner ring expansion and highlighted the significance of the π-acid CO binding to the trans position to benzylidene ligand in catalyst’s decomposition [155]. The creation of a cyclopropanone-containing complex was confirmed by other groups [156,157,158,159,160,161,162]. The group ruled out the possibility of a free carbene release as a results of CO binding on the ground of the endothermicity of the reaction. In the proposed mechanism first, the CO molecule coordinates to the trans position to the ylidene bond which elongates and pushes the phosphine almost opposite to the NHC ligand (Scheme 59). The Ru-NHC and Ru-P bonds also elongate, while the NHC-Ru = CH2 angle is reduced, causing the reactive methylidene ligand to get closer to the aromatic NHC ring. The cyclopropanation occurs in two steps—an attack of the methylidene group on the Cipso atom forming a more stable 83, still containing a Ru-CH2 bond and then the breaking of this bond with an attack of the methylidene carbon on one of the ortho carbon atoms of the mesityl ring. Intermediate 84, with a vacant trans position to CO is formed and the coordination of a second CO molecule is energetically comparable to the first binding, leading to the intermediate 85. The opening of the cyclopropane ring gives the final product 86. Noteworthy, with only one CO molecule attached to Ru, the system must overcome a larger barrier, collapsing to intermediate 87 and, after binding of second CO, the final product 86 is formed. Due to a relatively small energy barrier for the 84-to-87 step, the second CO molecule may coordinate after the ring expansion.
The first step of cyclopropanation with no ligand trans to the Ru-ylidene bond was investigated with ethylene as a model olefin, in the presence of pyridene, PMe3 or PF3 phosphine (L2, π-acids weaker than CO), also varying ligands opposite the NHC (L1). With no L2 ligand or when L2 = py, the C–H activated complex was unstable. For L2 = PMe3 or PF3, the complex with the associated L2 ligand and the C–H activated catalyst were both stable, whereas the latter was favored for more acidic PF3 than PMe3 (similarly to CO). Additional studies showed that the PH3 ligand favorably binds in the trans position to the Ru-methylidene bond, pointing out that the NHC mesityl rings shield that position. When L1 = PMe3 or pyridine, the system behaved in a similar fashion, whereas using a stronger π-acids like CO or PF3 as L1 and L2 reduced the energy barriers between the precatalyst and the C–H activated complex, the latter becoming the only stable structure. The more electrophilic the catalyst containing L2, the more reactive the methylidene group is towards the Cipso. Moreover, the results showed that when both the precatalyst and the C–H activated complex were stable, the energy barrier for the conversion was rather small.
Grela and co-workers expanded the topic by applying amine-containing isocyanides, also known as scavengers, in quenching metathesis reactions and removing ruthenium from the product mixture [163,164,165]. In one of their studies they observed a coordination of such ligands to ruthenium in the second generation complexes, initiating Buchner reaction and inserting the benzylidene group into the N-aryl moiety [163] forming a decomposed complex 88. In the absence of the NHC, the treatment with isocyanide generated an inactive complex 89 and phosphine ylide 90 (Scheme 60). Among the three new metal scavengers I-III proposed by this group which contained a polar fragment with a tertiary nitrogen atom, II decomposed much faster than the ethyl vinyl ether (EVE). Unlike EVE, CO and D1, isocyanide II seemed to be an ideal olefin metathesis quenching agent, as proposed by Fogg et al. [166].
Further work of the Diver group focused on the kinetics of the Buchner reaction, this time triggered by alkyl and aryl isocyanides [167]. For all isocyanides, product 91 was the initial reaction product (Scheme 61a). It was the only product for alkyl isocyanides at 0 °C and aryl isocyanides, whereas at higher temperatures or for slower Buchner reactions, complex 92 with three isocyanide groups was formed. Such product existing as a mixture of cycloheptatrienyl ring isomers has been obtained previously [156] in a reaction of Hoveyda-Blechert catalyst 4 with 4-chlorophenyl isocyanide (Scheme 61b). It was also shown that at higher temperatures the phosphine dissociative mechanism was favored over the associative one. When the Buchner reaction was faster than isocyanide substitution for PCy3, the reaction with the second isocyanide molecule caused Buchner insertion and binding of the third molecule, generating the final tris(isocyanide) complex. Despite faster dissociation of PPh3 with respect to PCy3, such modification of the phosphine ligand did not affect the carbene insertion. However, it was affected by the ligand trans to the NHC, since its electron donating/withdrawing properties modulated the reactivity of ruthenium towards the Buchner reaction.
In these studies it was also shown that both steric and electronic properties of the isocyanide have an impact on the decomposition of ruthenium catalysts. Due to the constrained vacant coordination, the site positioned between a mesityl and cyclohexyl group was the most accessible to less sterically-hindered ligands, which was confirmed by Fogg et al. [88,101] Aromatic isocyanides presented in this work most probably accelerated the Buchner reaction compared to highly congested aryl isocyanides, as did electron-withdrawing isocyanide substituents. Moreover, to accommodate quenching the ligand must possess strong σ-donor and π-acceptor properties. Strong σ donation only resulted in the metal binding but was not itself sufficient to cause the Buchner insertion. Lack of the π accepting properties, on the other hand, gave rise to a decomposition pathway different from Buchner reaction, as reported by Grubbs et al. [84] The electronic environment of the metal in the complex also played a key role, since previous study [156] showed that electron-rich metal carbenes, such as Fischer carbenes, do not undergo the Buchner reaction but instead, a π conjugation may take place, that is, for a vinyl carbene.
Furthermore, Diver group emphasized the most favorable mechanism in which one isocyanide molecule is reversibly coordinated, the benzylidene is added to the mesityl group (rate-limiting step), followed by next fast steps. The electrophilicity of the carbene was the rate-determining factor of the process. The isocyanide addition most probably occurred in the trans position to the carbene, forming an 18-electron species 93 and altering the electronic nature of the carbene. It was suggested that the π orbital of the benzylidene carbene carbon was destabilized, weakening the bond with ruthenium and making the carbene more electrophilic [168]. The 16-electron intermediate 94 was formed on account of proximal mesityl group cyclopropanation, followed by the addition of a second isocyanide and ring opening, forming metallocyclic intermediate 95 [155].
Recently, the Trzaskowski group performed computational studies on the decomposition of Hoveyda-Grubbs catalysts promoted by acrylonitrile and also considered Buchner ring expansion, keeping in mind the possibility of generating complexes with two acrylinitrile ligands [99]. Acrylonitrile may interact with ruthenium complexes by attacking the π-bond of benzylidene and one chloride ligand, possibly leading to chloride abstraction and catalyst decomposition. Calculations were also performed for similar systems of phosphine-containing ruthenium complex coordinating the CO molecule [155]. Removing or substitution of the CO ligand with acrylonitrile caused intermediate 83 to become unstable and form back to the original complex or the cyclopropane intermediate (Scheme 62). It was shown that the decomposition pathway with one acrylonitrile was very unlikely from the energetic point of view. When two acrylonitrile ligands were attached to the complex through Ru-N interactions, most energy barriers were lower compared to complexes with one acrylonitrile. Decomposition of HGII with two acrylonitrile moieties through Buchner ring expansion was possible in room temperature through methylidene or cyanomethylidene species. It was, however, unlikely for CAAC systems, for which energy barriers were of at least 39.0 kcal/mol. It was speculated that the opposite trends of TONs for these catalysts versus HGII could be caused by the fact that metathesis may be much faster (usually 0.5–2 h) than decomposition (usually hours/days). Grela’s group earlier showed that CAAC Hoveyda-Grubbs-like catalysts decompose by 5% in 12 h, whereas analogues of HGII decomposes much faster (45% catalyst left after 4 h, 30% after 12 h) [169]. The high/low TONs for metathesis reactions may also be caused by catalyst being stuck in a non-productive cycle with no ability to form the final product.
The results obtained by Trzaskowski et al. showed different behavior of CAAC-containing catalysts and HGII in the presence of acrylonitrile [99]. It was suggested that low barriers for HGII may be due to strong interaction between acrylonitrile ligand trans to the methylidene/cyanomethylidene and mesityl group. This interaction exists in the key transition states for system 96 and 97 but not for 98. This transition state was also stabilized by the mesityl/phenyl-acrylonitrile interactions, while the methyl-acrylonitrile interaction destabilized that intermediate in 98. Energy barriers were estimated to be very similar for catalysts 98 and 97 and lower for 96. In the case of the Buchner ring expansion, the higher stability of 98 in the presence of acrylonitrile may be caused by minimizing interactions between the NHC/CAAC side groups and the acrylonitrile molecule. It was also suggested that the NHC/CAAC electronic properties and not only steric ones, itself may have an effect on the decomposition.
4. Conclusions and Future Outlook
Over 30 years after the synthesis of the first, well-defined ruthenium metathesis catalyst [3], our knowledge and understanding of the decomposition pathways of this important family of complexes is very broad. Over 150 studies cited in this review shed more light on the relationship between the structure and decomposition mechanisms and rates of these systems, which gave rise to the synthesis of even more efficient catalysts, less prone to decomposition. While around five years ago it looked like we have a very good understanding of all the major decomposition processes of ruthenium metathesis catalyst, some newer studied in this field showed that we were and may still be, far from knowing the complete picture. Of the many interesting studied reviewed here, a few warrant a special attention of the reader. These include definitely the seminal study by the Fogg group on the general Broensted base-driven decomposition pathway, valid for a broad range of Grubbs-class catalysts [81]. This study does not simply provide new data for yet another decomposition pathway under a new external agent but addresses the decomposition problem for a broader range of external agents and catalysts, providing mechanistic insight into the action of an entire class of compounds. A second study likely to have a large impact on the catalytic society is the discovery of the new, bimolecular decomposition pathway [36]. Combined with the older studies of van Rensburg [27] this work allows to investigate the two major decomposition routes for both old and new catalysts and already made possible to explain the differences in action between the standard Hoveyda-Grubbs catalysts and newer CAAC-based ones. They also pave a way for developing new catalysts, even less prone to degradation in the presence of olefins.
On the other hand there are still some unresolved problems and questions waiting for an answer. These include for example, the possible decomposition pathways of NHC-containing catalysts in only aprotic solutions, without any presence of olefin or external agents known to cause decomposition [169]. One possible decomposition mechanism in such setup could include a bimolecular coupling of benzylidenes, instead of methylidenes, following a pathway summarized in Scheme 63. One can also expect soon new studied on the more and more commonly used CAAC-containing catalysts’ and their susceptibility to various external agents, similar to former, analogous studies for Grubbs and Hoveyda-Grubbs complexes. We are certain, that in the next 5–10 years a new review on the decomposition pathways of ruthenium metathesis catalysts will be again timely, as there will be new, interesting results on this topic.
Author Contributions
Design, B.T.; Bibliographic research and Writing-Review, M.J., A.M. and B.T.; Editing, B.T. All authors have read and agreed to the published version of the manuscript.
Funding
The authors acknowledge research support from the National Science Centre grant UMO-2016/22/E/ST4/00573.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Rinehart, R.E.; Smith, H.P. The emulsion polymerization of the norbornene ring system catalyzed by noble metal compounds. J. Polym. Sci. B 1965, 3, 1049–1052. [Google Scholar] [CrossRef]
- Michelotti, F.W.; Keaveney, W.P. Coordinated polymerization of the bicyclo-[2.2.1]-heptene-2 ring system (norbornene) in polar media. J. Polym. Sci. A 1965, 3, 895–905. [Google Scholar] [CrossRef]
- Nguyen, S.T.; Johnson, L.K.; Grubbs, R.H.; Ziller, J.W. Ring-opening metathesis polymerization (ROMP) of norbornene by a Group VIII carbene complex in protic media. J. Am. Chem. Soc. 1992, 114, 3974–3975. [Google Scholar] [CrossRef] [Green Version]
- Schwab, P.; France, M.B.; Ziller, J.W.; Grubbs, R.H. A Series of Well-Defined Metathesis Catalysts–Synthesis of [RuCl2(=CHR′)(PR3)2] and Its Reactions. Angew. Chem. Int. Ed. Engl. 1995, 34, 2039–2041. [Google Scholar] [CrossRef]
- Huang, J.; Stevens, E.D.; Nolan, S.P.; Petersen, J.L. Olefin Metathesis-Active Ruthenium Complexes Bearing a Nucleophilic Carbene Ligand. J. Am. Chem. Soc. 1999, 121, 2674–2678. [Google Scholar] [CrossRef]
- Scholl, M.; Trnka, T.M.; Morgan, J.P.; Grubbs, R.H. Increased ring closing metathesis activity of ruthenium-based olefin metathesis catalysts coordinated with imidazolin-2-ylidene ligands. Tetrahedron Lett. 1999, 40, 2247–2250. [Google Scholar] [CrossRef]
- Ackermann, L.; Fürstner, A.; Weskamp, T.; Kohl, F.J.; Herrmann, W.A. Ruthenium carbene complexes with imidazolin-2-ylidene ligands allow the formation of tetrasubstituted cycloalkenes by RCM. Tetrahedron Lett. 1999, 40, 4787–4790. [Google Scholar] [CrossRef]
- Love, J.A.; Morgan, J.P.; Trnka, T.M.; Grubbs, R.H. A Practical and Highly Active Ruthenium-Based Catalyst that Effects the Cross Metathesis of Acrylonitrile. Angew. Chem. Int. Ed. 2002, 41, 4035–4037. [Google Scholar] [CrossRef]
- Garber, S.B.; Kingsbury, J.S.; Gray, B.L.; Hoveyda, A.H. Efficient and Recyclable Monomeric and Dendritic Ru-Based Metathesis Catalysts. J. Am. Chem. Soc. 2000, 122, 8168–8179. [Google Scholar] [CrossRef]
- Vougioukalakis, G.C.; Grubbs, R.H. Ruthenium-Based Heterocyclic Carbene-Coordinated Olefin Metathesis Catalysts. Chem. Rev. 2010, 110, 1746–1787. [Google Scholar] [CrossRef]
- Samojłowicz, C.; Bieniek, M.; Grela, K. Ruthenium-Based Olefin Metathesis Catalysts Bearing N-Heterocyclic Carbene Ligands. Chem. Rev. 2009, 109, 3708–3742. [Google Scholar] [CrossRef] [PubMed]
- Lozano-Vila, A.M.; Monsaert, S.; Bajek, A.; Verpoort, F. Ruthenium-Based Olefin Metathesis Catalysts Derived from Alkynes. Chem. Rev. 2010, 110, 4865–4909. [Google Scholar] [CrossRef] [PubMed]
- Hamad, F.B.; Sun, T.; Xiao, S.; Verpoort, F. Olefin metathesis ruthenium catalysts bearing unsymmetrical heterocylic carbenes. Coord. Chem. Rev. 2013, 257, 2274–2292. [Google Scholar] [CrossRef]
- Nolan, S.P.; Clavier, H. Chemoselective olefin metathesis transformations mediated by ruthenium complexes. Chem. Soc. Rev. 2010, 39, 3305. [Google Scholar] [CrossRef]
- Higman, C.S.; Lummiss, J.A.M.; Fogg, D.E. Olefin Metathesis at the Dawn of Implementation in Pharmaceutical and Specialty-Chemicals Manufacturing. Angew. Chem. Int. Ed. 2016, 55, 3552–3565. [Google Scholar] [CrossRef]
- Hughes, D.; Wheeler, P.; Ene, D. Olefin Metathesis in Drug Discovery and Development—Examples from Recent Patent Literature. Org. Process Res. Dev. 2017, 21, 1938–1962. [Google Scholar] [CrossRef]
- Yu, M.; Lou, S.; Gonzalez-Bobes, F. Ring-Closing Metathesis in Pharmaceutical Development: Fundamentals, Applications, and Future Directions. Org. Process Res. Dev. 2018, 22, 918–946. [Google Scholar] [CrossRef]
- Turczel, G.; Kovács, E.; Merza, G.; Coish, P.; Anastas, P.T.; Tuba, R. Synthesis of Semiochemicals via Olefin Metathesis. ACS Sustain. Chem. Eng. 2019, 7, 33–48. [Google Scholar] [CrossRef]
- Flick, A.C.; Leverett, C.A.; Ding, H.X.; McInturff, E.; Fink, S.J.; Helal, C.J.; O’Donnell, C.J. Synthetic Approaches to the New Drugs Approved During 2017. J. Med. Chem. 2019, 62, 7340–7382. [Google Scholar] [CrossRef] [Green Version]
- Sytniczuk, A.; Milewski, M.; Kajetanowicz, A.; Grela, K. Preparation of macrocyclic musks via olefin metathesis: Comparison with classical syntheses and recent advances. Russ. Chem. Rev. 2020, 89, 469–490. [Google Scholar] [CrossRef]
- Montgomery, T.P.; Johns, A.M.; Grubbs, R.H. Recent Advancements in Stereoselective Olefin Metathesis Using Ruthenium Catalysts. Catalysts 2017, 7, 87. [Google Scholar] [CrossRef] [Green Version]
- Chikkali, S.; Mecking, S. Refining of Plant Oils to Chemicals by Olefin Metathesis. Angew. Chem. Int. Ed. 2012, 51, 5802–5808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winkler, M.; Meier, M.A.R. Olefin cross-metathesis as a valuable tool for the preparation of renewable polyesters and polyamides from unsaturated fatty acid esters and carbamates. Green Chem 2014, 16, 3335–3340. [Google Scholar] [CrossRef] [Green Version]
- Vignon, P.; Vancompernolle, T.; Couturier, J.-L.; Dubois, J.-L.; Mortreux, A.; Gauvin, R.M. Cross-Metathesis of Biosourced Fatty Acid Derivatives: A Step Further Toward Improved Reactivity. ChemSusChem 2015, 8, 1143–1146. [Google Scholar] [CrossRef]
- Jean-Louis Hérisson, P.; Chauvin, Y. Catalyse de transformation des olefines par les complexes du tungstene II. Telomerisation des oleffines cycliques en presence d’olefines acycliques. Makromol. Chem. 1971, 141, 161–176. [Google Scholar] [CrossRef]
- Nelson, D.J.; Manzini, S.; Urbina-Blanco, C.A.; Nolan, S.P. Key processes in ruthenium-catalysed olefin metathesis. Chem. Commun. 2014, 50, 10355. [Google Scholar] [CrossRef] [Green Version]
- Janse van Rensburg, W.; Steynberg, P.J.; Meyer, W.H.; Kirk, M.M.; Forman, G.S. DFT Prediction and Experimental Observation of Substrate-Induced Catalyst Decomposition in Ruthenium-Catalyzed Olefin Metathesis. J. Am. Chem. Soc. 2004, 126, 14332–14333. [Google Scholar] [CrossRef]
- Bourgeois, D.; Pancrazi, A.; Nolan, S.P.; Prunet, J. The Cl2(PCy3)(IMes)Ru(=CHPh) catalyst: Olefin metathesis versus olefin isomerization. J. Organomet. Chem. 2002, 643–644, 247–252. [Google Scholar] [CrossRef]
- van Rensburg, W.J.; Steynberg, P.J.; Kirk, M.M.; Meyer, W.H.; Forman, G.S. Mechanistic comparison of ruthenium olefin metathesis catalysts: DFT insight into relative reactivity and decomposition behavior. J. Organomet. Chem. 2006, 691, 5312–5325. [Google Scholar] [CrossRef]
- Lysenko, Z.; Maughon, B.R.; Mokhtar-Zadeh, T.; Tulchinsky, M.L. Stability of the first-generation Grubbs metathesis catalyst in a continuous flow reactor. J. Organomet. Chem. 2006, 691, 5197–5203. [Google Scholar] [CrossRef]
- Bespalova, N.B.; Nizovtsev, A.V.; Afanasiev, V.V.; Shutko, E.V. Metathesis Catalysts Stability and Decomposition Pathway. In Metathesis Chemistry; Imamoglu, Y., Dragutan, V., Karabulut, S., Eds.; Springer: Dordrecht, The Netherlands, 2007; pp. 125–135. [Google Scholar]
- Ashworth, I.W.; Hillier, I.H.; Nelson, D.J.; Percy, J.M.; Vincent, M.A. Searching for the Hidden Hydrides: The Competition between Alkene Isomerization and Metathesis with Grubbs Catalysts. Eur. J. Org. Chem. 2012, 2012, 5673–5677. [Google Scholar] [CrossRef]
- Jawiczuk, M.; Młodzikowska-Pieńko, K.; Trzaskowski, B. Impact of the olefin structure on the catalytic cycle and decomposition rates of Hoveyda–Grubbs metathesis catalysts. Phys. Chem. Chem. Phys. 2020, 22, 13062–13069. [Google Scholar] [CrossRef] [PubMed]
- Jawiczuk, M.; Marczyk, A.; Młodzikowska-Pieńko, K.; Trzaskowski, B. Impact of the Carbene Derivative Charge on the Decomposition Rates of Hoveyda–Grubbs-like Metathesis Catalysts. J. Phys. Chem. A 2020, acs.jpca.0c03096. [Google Scholar] [CrossRef] [PubMed]
- Engel, J.; Smit, W.; Foscato, M.; Occhipinti, G.; Törnroos, K.W.; Jensen, V.R. Loss and Reformation of Ruthenium Alkylidene: Connecting Olefin Metathesis, Catalyst Deactivation, Regeneration, and Isomerization. J. Am. Chem. Soc. 2017, 139, 16609–16619. [Google Scholar] [CrossRef]
- Bailey, G.A.; Foscato, M.; Higman, C.S.; Day, C.S.; Jensen, V.R.; Fogg, D.E. Bimolecular Coupling as a Vector for Decomposition of Fast-Initiating Olefin Metathesis Catalysts. J. Am. Chem. Soc. 2018, 140, 6931–6944. [Google Scholar] [CrossRef]
- Nascimento, D.L.; Fogg, D.E. Origin of the Breakthrough Productivity of Ruthenium–Cyclic Alkyl Amino Carbene Catalysts in Olefin Metathesis. J. Am. Chem. Soc. 2019, 141, 19236–19240. [Google Scholar] [CrossRef]
- Dinger, M.B.; Mol, J.C. Degradation of the First-Generation Grubbs Metathesis Catalyst with Primary Alcohols, Water, and Oxygen. Formation and Catalytic Activity of Ruthenium(II) Monocarbonyl Species. Organometallics 2003, 22, 1089–1095. [Google Scholar] [CrossRef]
- Dinger, M.B.; Mol, J.C. Degradation of the Second-Generation Grubbs Metathesis Catalyst with Primary Alcohols and Oxygen—Isomerization and Hydrogenation Activities of Monocarbonyl Complexes. Eur. J. Inorg. Chem. 2003, 2827–2833. [Google Scholar] [CrossRef]
- Banti, D.; Mol, J.C. Degradation of the ruthenium-based metathesis catalyst [RuCl2(CHPh)(H2IPr)(PCy3)] with primary alcohols. J. Organomet. Chem. 2004, 689, 3113–3116. [Google Scholar] [CrossRef]
- Werner, H.; Grünwald, C.; Stüer, W.; Wolf, J. Deactivation of the Grubbs Carbene Complex [RuCl2(CHPh)(PCy3)2] by Allylic Alcohols. Organometallics 2003, 22, 1558–1560. [Google Scholar] [CrossRef]
- Beach, N.J.; Camm, K.D.; Fogg, D.E. Hydrogenolysis versus Methanolysis of First- and Second-Generation Grubbs Catalysts: Rates, Speciation, and Implications for Tandem Catalysis. Organometallics 2010, 29, 5450–5455. [Google Scholar] [CrossRef]
- Beach, N.J.; Lummiss, J.A.M.; Bates, J.M.; Fogg, D.E. Reactions of Grubbs Catalysts with Excess Methoxide: Formation of Novel Methoxyhydride Complexes. Organometallics 2012, 31, 2349–2356. [Google Scholar] [CrossRef]
- Coalter, J.N.; Bollinger, J.C.; Eisenstein, O.; Caulton, K.G. R-Group reversal of isomer stability for RuH(X)L2(CCHR) vs. Ru(X)L2(CCH2R): Access to four-coordinate ruthenium carbenes and carbynes. New J. Chem. 2000, 24, 925–927. [Google Scholar] [CrossRef]
- Sanford, M.S.; Henling, L.M.; Day, M.W.; Grubbs, R.H. Ruthenium-Based Four-Coordinate Olefin Metathesis Catalysts. Angew. Chem. Int. Ed. 2000, 39, 3451–3453. [Google Scholar] [CrossRef]
- Conrad, J.C.; Amoroso, D.; Czechura, P.; Yap, G.P.A.; Fogg, D.E. The First Highly Active, Halide-Free Ruthenium Catalyst for Olefin Metathesis. Organometallics 2003, 22, 3634–3636. [Google Scholar] [CrossRef]
- Caskey, S.R.; Stewart, M.H.; Ahn, Y.J.; Johnson, M.J.A.; Kampf, J.W. Dehydrohalogenation by a Germylene: Conversion of Carbene Ligands into Carbynes at Ruthenium. Organometallics 2005, 24, 6074–6076. [Google Scholar] [CrossRef]
- Goudreault, A.Y.; Walden, D.M.; Nascimento, D.L.; Botti, A.G.; Steinmann, S.N.; Michel, C.; Fogg, D.E. Hydroxide-Induced Degradation of Olefin Metathesis Catalysts: A Challenge for Metathesis in Alkaline Media. ACS Catal. 2020, 3838–3843. [Google Scholar] [CrossRef]
- Young, A.; Vincent, M.A.; Hillier, I.H.; Percy, J.M.; Tuttle, T. Forming a ruthenium isomerisation catalyst from Grubbs II: A DFT study. Dalton Trans. 2014, 43, 8493–8498. [Google Scholar] [CrossRef]
- Manzini, S.; Urbina-Blanco, C.A.; Poater, A.; Slawin, A.M.Z.; Cavallo, L.; Nolan, S.P. From Olefin Metathesis Catalyst to Alcohol Racemization Catalyst in One Step. Angew. Chem. Int. Ed. 2012, 51, 1042–1045. [Google Scholar] [CrossRef]
- Manzini, S.; Nelson, D.J.; Lebl, T.; Poater, A.; Cavallo, L.; Slawin, A.M.Z.; Nolan, S.P. From ruthenium olefin metathesis catalyst to (η5-3-phenylindenyl)hydrido complex via alcoholysis. Chem. Commun. 2014, 50, 2205–2207. [Google Scholar] [CrossRef]
- Manzini, S.; Poater, A.; Nelson, D.J.; Cavallo, L.; Slawin, A.M.Z.; Nolan, S.P. Insights into the Decomposition of Olefin Metathesis Precatalysts. Angew. Chem. Int. Ed. 2014, 53, 8995–8999. [Google Scholar] [CrossRef]
- Beach, N.J.; Blacquiere, J.M.; Drouin, S.D.; Fogg, D.E. Carbonyl-Amplified Catalyst Performance: Balancing Stability against Activity for Five-Coordinate Ruthenium Hydride and Hydridocarbonyl Catalysts. Organometallics 2009, 28, 441–447. [Google Scholar] [CrossRef]
- Rouen, M.; Queval, P.; Borré, E.; Falivene, L.; Poater, A.; Berthod, M.; Hugues, F.; Cavallo, L.; Baslé, O.; Olivier-Bourbigou, H.; et al. Selective Metathesis of α-Olefins from Bio-Sourced Fischer–Tropsch Feeds. ACS Catal. 2016, 6, 7970–7976. [Google Scholar] [CrossRef]
- Forman, G.S.; McConnell, A.E.; Tooze, R.P.; Janse van Rensburg, W.; Meyer, W.H.; Kirk, M.M.; Dwyer, C.L.; Serfontein, D.W. A Convenient System for Improving the Efficiency of First-Generation Ruthenium Olefin Metathesis Catalysts. Organometallics 2005, 24, 4528–4542. [Google Scholar] [CrossRef]
- Lynn, D.M.; Mohr, B.; Grubbs, R.H.; Henling, L.M.; Day, M.W. Water-Soluble Ruthenium Alkylidenes: Synthesis, Characterization, and Application to Olefin Metathesis in Protic Solvents. J. Am. Chem. Soc. 2000, 122, 6601–6609. [Google Scholar] [CrossRef] [Green Version]
- Dowden, J.; Savović, J. Olefin metathesis in non-degassed solvent using a recyclable, polymer supported alkylideneruthenium. Chem. Commun. 2001, 37–38. [Google Scholar] [CrossRef]
- Van Veldhuizen, J.J.; Garber, S.B.; Kingsbury, J.S.; Hoveyda, A.H. A Recyclable Chiral Ru Catalyst for Enantioselective Olefin Metathesis. Efficient Catalytic Asymmetric Ring-Opening/Cross Metathesis in Air. J. Am. Chem. Soc. 2002, 124, 4954–4955. [Google Scholar] [CrossRef]
- Connon, S.J.; Rivard, M.; Zaja, M.; Blechert, S. Practical Olefin Metathesis in Protic Media under an Air Atmosphere. Adv. Synth. Catal. 2003, 345, 572–575. [Google Scholar] [CrossRef]
- Binder, J.B.; Guzei, I.A.; Raines, R.T. Salicylaldimine Ruthenium Alkylidene Complexes: Metathesis Catalysts Tuned for Protic Solvents. Adv. Synth. Catal. 2007, 349, 395–404. [Google Scholar] [CrossRef] [Green Version]
- Vila, A.M.L.; Monsaert, S.; Drozdzak, R.; Wolowiec, S.; Verpoort, F. New Indenylidene-Schiff Base-Ruthenium Complexes for Cross-Metathesis and Ring-Closing Metathesis. Adv. Synth. Catal. 2009, 351, 2689–2701. [Google Scholar] [CrossRef]
- Burtscher, D.; Grela, K. Aqueous Olefin Metathesis. Angew. Chem. Int. Ed. 2009, 48, 442–454. [Google Scholar] [CrossRef] [PubMed]
- Kniese, M.; Meier, M.A.R. A simple approach to reduce the environmental impact of olefin metathesis reactions: A green and renewable solvent compared to solvent-free reactions. Green Chem. 2010, 12, 169–173. [Google Scholar] [CrossRef]
- Skowerski, K.; Szczepaniak, G.; Wierzbicka, C.; Gułajski, Ł.; Bieniek, M.; Grela, K. Highly active catalysts for olefin metathesis in water. Catal. Sci. Technol. 2012, 2, 2424–2427. [Google Scholar] [CrossRef]
- Skowerski, K.; Kasprzycki, P.; Bieniek, M.; Olszewski, T.K. Efficient, durable and reusable olefin metathesis catalysts with high affinity to silica gel. Tetrahedron 2013, 69, 7408–7415. [Google Scholar] [CrossRef]
- Occhipinti, G.; Hansen, F.R.; Törnroos, K.W.; Jensen, V.R. Simple and Highly Z-Selective Ruthenium-Based Olefin Metathesis Catalyst. J. Am. Chem. Soc. 2013, 135, 3331–3334. [Google Scholar] [CrossRef]
- Tomasek, J.; Schatz, J. Olefin metathesis in aqueous media. Green Chem. 2013, 15, 2317–2338. [Google Scholar] [CrossRef] [Green Version]
- Skowerski, K.; Białecki, J.; Tracz, A.; Olszewski, T.K. An attempt to provide an environmentally friendly solvent selection guide for olefin metathesis. Green Chem. 2014, 16, 1125–1130. [Google Scholar] [CrossRef]
- Ablialimov, O.; Kędziorek, M.; Malińska, M.; Woźniak, K.; Grela, K. Synthesis, Structure, and Catalytic Activity of New Ruthenium(II) Indenylidene Complexes Bearing Unsymmetrical N-Heterocyclic Carbenes. Organometallics 2014, 33, 2160–2171. [Google Scholar] [CrossRef]
- Binder, J.B.; Blank, J.J.; Raines, R.T. Olefin Metathesis in Homogeneous Aqueous Media Catalyzed by Conventional Ruthenium Catalysts. Org. Lett. 2007, 9, 4885–4888. [Google Scholar] [CrossRef] [Green Version]
- Matsuo, T.; Yoshida, T.; Fujii, A.; Kawahara, K.; Hirota, S. Effect of Added Salt on Ring-Closing Metathesis Catalyzed by a Water-Soluble Hoveyda–Grubbs Type Complex to Form N-Containing Heterocycles in Aqueous Media. Organometallics 2013, 32, 5313–5319. [Google Scholar] [CrossRef]
- Tanaka, K.; Böhm, V.P.W.; Chadwick, D.; Roeper, M.; Braddock, D.C. Anionic Ligand Exchange in Hoveyda−Grubbs Ruthenium(II) Benzylidenes. Organometallics 2006, 25, 5696–5698. [Google Scholar] [CrossRef]
- Zirngast, M.; Pump, E.; Leitgeb, A.; Albering, J.H.; Slugovc, C. Pyridine as trigger for chloride isomerisation in chelated ruthenium benzylidene complexes: Implications for olefin metathesis. Chem. Commun. 2011, 47, 2261–2263. [Google Scholar] [CrossRef]
- Keitz, B.K.; Endo, K.; Patel, P.R.; Herbert, M.B.; Grubbs, R.H. Improved Ruthenium Catalysts for Z-Selective Olefin Metathesis. J. Am. Chem. Soc. 2012, 134, 693–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falivene, L.; Poater, A.; Cazin, C.S.J.; Slugovc, C.; Cavallo, L. Energetics of the ruthenium—Halide bond in olefin metathesis (pre)catalysts. Dalton Trans. 2013, 42, 7312–7317. [Google Scholar] [CrossRef] [PubMed]
- Wappel, J.; Urbina-Blanco, C.A.; Abbas, M.; Albering, J.H.; Saf, R.; Nolan, S.P.; Slugovc, C. Halide exchanged Hoveyda-type complexes in olefin metathesis. Beilstein J. Org. Chem. 2010, 6, 1091–1098. [Google Scholar] [CrossRef] [PubMed]
- Krause, J.O.; Nuyken, O.; Wurst, K.; Buchmeiser, M.R. Synthesis and Reactivity of Homogeneous and Heterogeneous Ruthenium-Based Metathesis Catalysts Containing Electron-Withdrawing Ligands. Chem. Eur. J. 2004, 10, 777–784. [Google Scholar] [CrossRef]
- Pump, E.; Fischer, R.C.; Slugovc, C. Halide Exchange in Second-Generation cis-Dihalo Ruthenium Benzylidene Complexes. Organometallics 2012, 31, 6972–6979. [Google Scholar] [CrossRef]
- Bailey, G.A.; Fogg, D.E. Acrylate Metathesis via the Second-Generation Grubbs Catalyst: Unexpected Pathways Enabled by a PCy3-Generated Enolate. J. Am. Chem. Soc. 2015, 137, 7318–7321. [Google Scholar] [CrossRef]
- Bates, J.M.; Lummiss, J.A.M.; Bailey, G.A.; Fogg, D.E. Operation of the Boomerang Mechanism in Olefin Metathesis Reactions Promoted by the Second-Generation Hoveyda Catalyst. ACS Catal. 2014, 4, 2387–2394. [Google Scholar] [CrossRef]
- Bailey, G.A.; Lummiss, J.A.M.; Foscato, M.; Occhipinti, G.; McDonald, R.; Jensen, V.R.; Fogg, D.E. Decomposition of Olefin Metathesis Catalysts by Brønsted Base: Metallacyclobutane Deprotonation as a Primary Deactivating Event. J. Am. Chem. Soc. 2017, 139, 16446–16449. [Google Scholar] [CrossRef]
- Santos, A.G.; Bailey, G.A.; Dos Santos, E.N.; Fogg, D.E. Overcoming Catalyst Decomposition in Acrylate Metathesis: Polyphenol Resins as Enabling Agents for PCy3-Stabilized Metathesis Catalysts. ACS Catal. 2017, 7, 3181–3189. [Google Scholar] [CrossRef]
- Compain, P. Olefin Metathesis of Amine-Containing Systems: Beyond the Current Consensus. Adv. Synth. Catal. 2007, 349, 1829–1846. [Google Scholar] [CrossRef]
- Soon, H.H.; Wenzel, A.G.; Salguero, T.T.; Day, M.W.; Grubbs, R.H. Decomposition of ruthenium olefin metathesis catalysts. J. Am. Chem. Soc. 2007, 129, 7961–7968. [Google Scholar] [CrossRef] [Green Version]
- Wilson, G.O.; Porter, K.A.; Weissman, H.; White, S.R.; Sottos, N.R.; Moore, J.S. Stability of Second Generation Grubbs’ Alkylidenes to Primary Amines: Formation of Novel Ruthenium-Amine Complexes. Adv. Synth. Catal. 2009, 351, 1817–1825. [Google Scholar] [CrossRef]
- Wilson, G.O.; Caruso, M.M.; Reimer, N.T.; White, S.R.; Sottos, N.R.; Moore, J.S. Evaluation of Ruthenium Catalysts for Ring-Opening Metathesis Polymerization-Based Self-Healing Applications. Chem. Mater. 2008, 20, 3288–3297. [Google Scholar] [CrossRef]
- Lummiss, J.A.M.; Ireland, B.J.; Sommers, J.M.; Fogg, D.E. Amine-mediated degradation in olefin metathesis reactions that employ the second-generation Grubbs catalyst. ChemCatChem 2014, 6, 459–463. [Google Scholar] [CrossRef]
- Lummiss, J.A.M.; Botti, A.G.G.; Fogg, D.E. Isotopic probes for ruthenium-catalyzed olefin metathesis. Catal. Sci. Technol. 2014, 4, 4210–4218. [Google Scholar] [CrossRef]
- Sanford, M.S.; Love, J.A.; Grubbs, R.H. A Versatile Precursor for the Synthesis of New Ruthenium Olefin Metathesis Catalysts. Organometallics 2001, 20, 5314–5318. [Google Scholar] [CrossRef] [Green Version]
- Getty, K.; Delgado-Jaime, M.U.; Kennepohl, P. Assignment of pre-edge features in the Ru K-edge X-ray absorption spectra of organometallic ruthenium complexes. Inorganica Chim. Acta 2008, 361, 1059–1065. [Google Scholar] [CrossRef] [Green Version]
- Ireland, B.J.; Dobigny, B.T.; Fogg, D.E. Decomposition of a Phosphine-Free Metathesis Catalyst by Amines and Other Bronsted Bases: Metallacyclobutane Deprotonation as a Major Deactivation Pathway. ACS Catal. 2015, 5, 4690–4698. [Google Scholar] [CrossRef]
- McClennan, W.L.; Rufh, S.A.; Lummiss, J.A.M.; Fogg, D.E. A General Decomposition Pathway for Phosphine-Stabilized Metathesis Catalysts: Lewis Donors Accelerate Methylidene Abstraction. J. Am. Chem. Soc. 2016, 138, 14668–14677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lummiss, J.A.M.; Higman, C.S.; Fyson, D.L.; McDonald, R.; Fogg, D.E. The divergent effects of strong NHC donation in catalysis. Chem. Sci. 2015, 6, 6739–6746. [Google Scholar] [CrossRef] [PubMed]
- Rufh, S.A.; Goudreault, A.Y.; Foscato, M.; Jensen, V.R.; Fogg, D.E. Rapid decomposition of olefin metathesis catalysts by a truncated N-heterocyclic carbene: Efficient catalyst quenching and n-heterocyclic carbene vinylation. ACS Catal. 2018, 8, 11822–11826. [Google Scholar] [CrossRef]
- Zhao, Z.-X.; Wang, H.-Y.; Guo, Y.-L. Studies on CH3CN-assisted decomposition of 1st Grubbs catalyst by electrospray ionization tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2011, 25, 3401–3410. [Google Scholar] [CrossRef]
- Hong, S.H.; Day, M.W.; Grubbs, R.H. Decomposition of a Key Intermediate in Ruthenium-Catalyzed Olefin Metathesis Reactions. J. Am. Chem. Soc. 2004, 126, 7414–7415. [Google Scholar] [CrossRef] [Green Version]
- Ulman, M.; Grubbs, R.H. Ruthenium Carbene-Based Olefin Metathesis Initiators: Catalyst Decomposition and Longevity. J. Org. Chem. 1999, 64, 7202–7207. [Google Scholar] [CrossRef]
- Jiang, J.; Zhao, D.; Zhang, H.; He, J.; Li, N. In situ analysis and real-time monitoring of the decomposition of the 2nd Grubbs catalyst in CH3CN by droplet spray ionization tandem mass spectrometry. Anal. Methods 2017, 9, 4201–4206. [Google Scholar] [CrossRef]
- Jawiczuk, M.; Młodzikowska-Pieńko, K.; Osella, S.; Trzaskowski, B. Molecular Modeling of Mechanisms of Decomposition of Ruthenium Metathesis Catalysts by Acrylonitrile. Organometallics 2020, 39, 239–246. [Google Scholar] [CrossRef]
- Trnka, T.M.; Morgan, J.P.; Sanford, M.S.; Wilhelm, T.E.; Scholl, M.; Choi, T.L.; Ding, S.; Day, M.W.; Grubbs, R.H. Synthesis and activity of ruthenium alkylidene complexes coordinated with phosphine and N-heterocyclic carbene ligands. J. Am. Chem. Soc. 2003, 125, 2546–2558. [Google Scholar] [CrossRef] [Green Version]
- Guidone, S.; Songis, O.; Nahra, F.; Cazin, C.S.J. Conducting Olefin Metathesis Reactions in Air: Breaking the Paradigm. ACS Catal. 2015, 5, 2697–2701. [Google Scholar] [CrossRef] [Green Version]
- Ton, S.J.; Fogg, D.E. The Impact of Oxygen on Leading and Emerging Ru-Carbene Catalysts for Olefin Metathesis: An Unanticipated Correlation Between Robustness and Metathesis Activity. ACS Catal. 2019, 9, 11329–11334. [Google Scholar] [CrossRef]
- Feast, W.J.; Gibson, V.C.; Khosravi, E.; Marshall, E.L.; Mitchell, J.P. Bimolecular termination in living ring opening metathesis polymerization. Polymer 1992, 33, 872–873. [Google Scholar] [CrossRef]
- Forman, G.S.; McConnell, A.E.; Hanton, M.J.; Slawin, A.M.Z.; Tooze, R.P.; van Rensburg, W.J.; Meyer, W.H.; Dwyer, C.; Kirk, M.M.; Serfontein, D.W. A Stable Ruthenium Catalyst for Productive Olefin Metathesis. Organometallics 2004, 23, 4824–4827. [Google Scholar] [CrossRef]
- Huang, J.; Schanz, H.-J.; Stevens, E.D.; Nolan, S.P. Influence of Sterically Demanding Carbene Ligation on Catalytic Behavior and Thermal Stability of Ruthenium Olefin Metathesis Catalysts. Organometallics 1999, 18, 5375–5380. [Google Scholar] [CrossRef]
- Wu, Z.; Nguyen, S.T.; Grubbs, R.H.; Ziller, J.W. Reactions of Ruthenium Carbenes of the Type (PPh3)2(X)2Ru:CH-CH:CPh2 (X = Cl and CF3COO) with Strained Acyclic Olefins and Functionalized Olefins. J. Am. Chem. Soc. 1995, 117, 5503–5511. [Google Scholar] [CrossRef]
- Louie, J.; Grubbs, R.H. Metathesis of Electron-Rich Olefins: Structure and Reactivity of Electron-Rich Carbene Complexes. Organometallics 2002, 21, 2153–2164. [Google Scholar] [CrossRef] [Green Version]
- Hansen, S.M.; Rominger, F.; Metz, M.; Hofmann, P. The first grubbs-type metathesis catalyst with cis stereochemistry: Synthesis of [(n2-dtbpm)Cl2Ru=CH-CH=CMe2] from a novel, coordinatively unsaturated dinuclear ruthenium dihydride. Chem. Eur. J. 1999, 5, 557–566. [Google Scholar] [CrossRef]
- Lummiss, J.A.M.; McClennan, W.L.; McDonald, R.; Fogg, D.E. Donor-induced decomposition of the Grubbs catalysts: An intercepted intermediate. Organometallics 2014, 33, 6738–6741. [Google Scholar] [CrossRef]
- Wang, H.; Metzger, J.O. ESI-MS Study on First-Generation Ruthenium Olefin Metathesis Catalysts in Solution: Direct Detection of the Catalytically Active 14-Electron Ruthenium Intermediate. Organometallics 2008, 27, 2761–2766. [Google Scholar] [CrossRef]
- Labinger, J.A.; Bercaw, J.E. Understanding and exploiting C–H bond activation. Nature 2002, 417, 507–514. [Google Scholar] [CrossRef]
- Bergman, R.G. C–H activation. The stability of the chemical bonds in saturated hydrocarbons makes them generally unreactive. But the invention of processes in which carbon–hydrogen (C–H) bonds in hydrocarbons can be activated is allowing chemists to exploit organic compounds in previously unimaginable ways. Nature 2007, 446, 391–393. [Google Scholar] [PubMed]
- Forsman, Å.; Grimvall, A. Reduced models for efficient simulation of spatially integrated outputs of one-dimensional substance transport models. Environ. Model. Softw. 2003, 18, 319–327. [Google Scholar] [CrossRef]
- Dyker, G. Transition Metal Catalyzed Coupling Reactions under C-H Activation. Angew. Chem. Int. Ed. 1999, 38, 1698–1712. [Google Scholar] [CrossRef]
- Guari, Y.; Sabo-Etienne, S.; Chaudret, B. Catalytic Formation of Carbon-Carbon Bonds by Activation of Carbon-Hydrogen Bonds. Eur. J. Inorg. Chem. 1999, 1999, 1047–1055. [Google Scholar] [CrossRef]
- Jazzar, R.F.R.; Macgregor, S.A.; Mahon, M.F.; Richards, S.P.; Whittlesey, M.K. C-C and C-H bond activation reactions in N-heterocyclic carbene complexes of ruthenium. J. Am. Chem. Soc. 2002, 124, 4944–4945. [Google Scholar] [CrossRef]
- Chilvers, M.J.; Jazzar, R.F.R.; Mahon, M.F.; Whittlesey, M.K. Reversible C-H Bond Activation Reactions of theN-Heterocyclic Carbene Ligands in Ru(Ph2PCH2CH2CH2PPh2)(IMes)(CO)H2 and Ru(Ph2AsCH2CH2PPh2)(IMes)(CO)H2 (IMes=1,3-Dimesityl-1,3-dihydro-2H-imidazol-2-ylidene). Adv. Synth. Catal. 2003, 345, 1111–1114. [Google Scholar] [CrossRef]
- Diggle, R.A.; Macgregor, S.A.; Whittlesey, M.K. Computational study of C-C activation of 1,3-dimesitylimidazol-2-ylidene (IMes) at ruthenium: The role of Ligand bulk in accessing reactive intermediates. Organometallics 2008, 27, 617–625. [Google Scholar] [CrossRef]
- Edwards, M.G.; Jazzar, R.F.R.; Paine, B.M.; Shermer, D.J.; Whittlesey, M.K.; Williams, J.M.J.; Edney, D.D. Borrowing hydrogen: A catalytic route to C–C bond formation from alcohols. Chem. Commun. 2004, 4, 90–91. [Google Scholar] [CrossRef]
- Burling, S.; Whittlesey, M.K.; Williams, J.M.J. Direct and transfer hydrogenation of ketones and imines with a ruthenium N-heterocyclic carbene complex. Adv. Synth. Catal. 2005, 347, 591–594. [Google Scholar] [CrossRef]
- Burling, S.; Paine, B.M.; Nama, D.; Brown, V.S.; Mahon, M.F.; Prior, T.J.; Pregosin, P.S.; Whittlesey, M.K.; Williams, J.M.J. C-H activation reactions of ruthenium N-heterocyclic carbene complexes: Application in a catalytic tandem reaction involving C-C bond formation from alcohols. J. Am. Chem. Soc. 2007, 129, 1987–1995. [Google Scholar] [CrossRef]
- Wenz, K.M.; Liu, P.; Houk, K.N. Intramolecular C-H Activation Reactions of Ru(NHC) Complexes Combined with H2 Transfer to Alkenes: A Theoretical Elucidation of Mechanisms and Effects of Ligands on Reactivities. Organometallics 2017, 36, 3613–3623. [Google Scholar] [CrossRef]
- Młodzikowska-Pieńko, K.; Trzaskowski, B. Rate-limiting Steps in the Intramolecular C-H Activation of Ruthenium N-Heterocyclic Carbene Complexes. J. Phys. Chem. A 2020, 124, 3609–3617. [Google Scholar] [CrossRef] [PubMed]
- Abdur-Rashid, K.; Fedorkiw, T.; Lough, A.J.; Morris, R.H. Coordinatively unsaturated hydridoruthenium(II) complexes of N-heterocyclic carbenes. Organometallics 2004, 23, 86–94. [Google Scholar] [CrossRef]
- Burling, S.; Mahon, M.F.; Paine, B.M.; Whittlesey, M.K.; Williams, J.M.J. Reversible intramolecular Alkyl C-H bond activation, alcohol dehydrogenation, and trans-Cis dihydride isomerization in ruthenium N-heterocyclic carbene complexes. Organometallics 2004, 23, 4537–4539. [Google Scholar] [CrossRef]
- Hong, S.H.; Chlenov, A.; Day, M.W.; Grubbs, R.H. Double C-H activation of an N-heterocyclic carbene ligand in a ruthenium olefin metathesis catalyst. Angew. Chem. Int. Ed. 2007, 46, 5148–5151. [Google Scholar] [CrossRef]
- Sandford, M.S.; Love, J.A.; Grubbs, R.H. Mechanism and activity of ruthenium olefin metathesis catalysts. J. Am. Chem. Soc. 2001, 123, 6543–6554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathew, J.; Koga, N.; Suresh, C.H. C-H bond activation through σ-bond metathesis and agostic interactions: Deactivation pathway of a Grubbs second-generation catalyst. Organometallics 2008, 27, 4666–4670. [Google Scholar] [CrossRef]
- Poater, A.; Cavallo, L. Mechanistic insights into the double C-H (de)activation route of a Ru-based olefin metathesis catalyst. J. Mol. Catal. Chem. 2010, 324, 75–79. [Google Scholar] [CrossRef]
- Vehlow, K.; Gessler, S.; Blechert, S. Deactivation of ruthenium olefin metathesis catalysts through intramolecular carbene-arene bond formation. Angew. Chem. Int. Ed. 2007, 46, 8082–8085. [Google Scholar] [CrossRef]
- Galan, B.R.; Kalbarczyk, K.P.; Szczepankiewicz, S.; Keister, J.B.; Diver, S.T. A rapid and simple cleanup procedure for metathesis reactions. Org. Lett. 2007, 9, 1203–1206. [Google Scholar] [CrossRef]
- Leitao, E.M.; Dubberley, S.R.; Piers, W.E.; Wu, Q.; McDonald, R. Thermal decomposition modes for four-coordinate ruthenium phosphonium alkylidene olefin metathesis catalysts. Chem. Eur. J. 2008, 14, 11565–11572. [Google Scholar] [CrossRef]
- Van Der Eide, E.F.; Romero, P.E.; Piers, W.E. Generation and spectroscopic characterization of ruthenacyclobutane and ruthenium olefin carbene intermediates relevant to ring closing metathesis catalysis. J. Am. Chem. Soc. 2008, 130, 4485–4491. [Google Scholar] [CrossRef]
- Solari, E.; Gauthier, S.; Scopelliti, R.; Severin, K. Multifaceted chemistry of [(Cymene)RuCl2]2 and PCy3. Organometallics 2009, 28, 4519–4526. [Google Scholar] [CrossRef]
- Herbert, M.B.; Lan, Y.; Keitz, B.K.; Liu, P.; Endo, K.; Day, M.W.; Houk, K.N.; Grubbs, R.H. Decomposition pathways of Z-selective ruthenium metathesis catalysts. J. Am. Chem. Soc. 2012, 134, 7861–7866. [Google Scholar] [CrossRef] [Green Version]
- Endo, K.; Grubbs, R.H. Chelated ruthenium catalysts for Z-selective olefin metathesis. J. Am. Chem. Soc. 2011, 133, 8525–8527. [Google Scholar] [CrossRef] [Green Version]
- Weng, W.; Parkin, S.; Ozerov, O.V. Double C-H activation results in ruthenium complexes of a neutral PCP ligand with a central carbene moiety. Organometallics 2006, 25, 5345–5354. [Google Scholar] [CrossRef]
- Oliván, M.; Caulton, K.G. C-(halide) oxidative addition routes to ruthenium carbenes. Inorg. Chem. 1999, 38, 566–570. [Google Scholar] [CrossRef]
- Prechtl, M.H.G.; Ben-David, Y.; Giunta, D.; Busch, S.; Taniguchi, Y.; Wisniewski, W.; Görls, H.; Mynott, R.J.; Theyssen, N.; Milstein, D.; et al. Synthesis and characterisation of nonclassical ruthenium hydride complexes containing chelating bidentate and tridentate phosphine ligands. Chem. Eur. J. 2007, 13, 1539–1546. [Google Scholar] [CrossRef] [Green Version]
- Poater, A.; Bahri-Laleh, N.; Cavallo, L. Rationalizing current strategies to protect N-heterocyclic carbene-based ruthenium catalysts active in olefin metathesis from C-H (de)activation. Chem. Commun. 2011, 47, 6674–6676. [Google Scholar] [CrossRef]
- Fernández, I.; Lugan, N.; Lavigne, G. Effects of attractive through space π-π* Interactions on the structure, reactivity, and activity of Grubbs II complexes. Organometallics 2012, 31, 1155–1160. [Google Scholar] [CrossRef]
- Comas-Vives, A.; Harvey, J.N. How important is backbonding in metal complexes containing N-heterocyclic carbenes? Structural and NBO analysis. Eur. J. Inorg. Chem. 2011, 5025–5035. [Google Scholar] [CrossRef]
- Vummaleti, S.V.C.; Nelson, D.J.; Poater, A.; Gómez-Suárez, A.; Cordes, D.B.; Slawin, A.M.Z.; Nolan, S.P.; Cavallo, L. What can NMR spectroscopy of selenoureas and phosphinidenes teach us about the π-accepting abilities of N-heterocyclic carbenes? Chem. Sci. 2015, 6, 1895–1904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Credendino, R.; Falivene, L.; Cavallo, L. π-face donation from the aromatic N-substituent of N-heterocyclic carbene ligands to metal and its role in catalysis. J. Am. Chem. Soc. 2012, 134, 8127–8135. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, M.M.; Rooney, A.D.; Rooney, J.J. Variable temperature 1H NMR studies on Grubbs catalysts. J. Organomet. Chem. 2008, 693, 1252–1260. [Google Scholar] [CrossRef] [Green Version]
- Kotyk, M.W.; Gorelsky, S.I.; Conrad, J.C.; Carra, C.; Fogg, D.E. Geometric and electronic structure of a C1-symmetric ruthenium-Aryloxide metathesis catalyst: An experimental and computational study. Organometallics 2009, 28, 5424–5431. [Google Scholar] [CrossRef]
- Song, B.; Xu, B. Metal-catalyzed C-H functionalization involving isocyanides. Chem. Soc. Rev. 2017, 46, 1103–1123. [Google Scholar] [CrossRef]
- Boyarskiy, V.P.; Bokach, N.A.; Luzyanin, K.V.; Kukushkin, V.Y. Metal-mediated and metal-catalyzed reactions of isocyanides. Chem. Rev. 2015, 115, 2698–2779. [Google Scholar] [CrossRef]
- Chakrabarty, S.; Choudhary, S.; Doshi, A.; Liu, F.Q.; Mohan, R.; Ravindra, M.P.; Shah, D.; Yang, X.; Fleming, F.F. Catalytic isonitrile insertions and condensations initiated by RNC-X complexation. Adv. Synth. Catal. 2014, 356, 2135–2196. [Google Scholar] [CrossRef] [Green Version]
- Vlaar, T.; Ruijter, E.; Maes, B.U.W.; Orru, R.V.A. Palladium-catalyzed migratory insertion of isocyanides: An emerging platform in cross-coupling chemistry. Angew. Chem. Int. Ed. 2013, 52, 7084–7097. [Google Scholar] [CrossRef]
- Galan, B.R.; Gembicky, M.; Dominiak, P.M.; Keister, J.B.; Diver, S.T. Carbon monoxide-promoted carbene insertion into the aryl substituent of an N-heterocyclic carbene ligand: Buchner reaction in a ruthenium carbene complex. J. Am. Chem. Soc. 2005, 127, 15702–15703. [Google Scholar] [CrossRef]
- Buchner, E.; Curtius, T. Ueber die Einwirkung von Diazoessigäther auf aromatische Kohlenwasserstoffe. Berichte Dtsch. Chem. Ges. 1885, 18, 2377–2379. [Google Scholar] [CrossRef] [Green Version]
- Galan, B.R.; Pitak, M.; Keister, J.B.; Diver, S.T. Isocyanide-promoted ylidene transfer to phosphorus: Rearrangement in the first-generation grubbs complex. Organometallics 2008, 27, 3630–3632. [Google Scholar] [CrossRef]
- Johnson, L.K.; Frey, M.; Ulibarri, T.A.; Virgil, S.C.; Grubbs, R.H.; Ziller, J.W. Alkylidene Transfer from Phosphoranes to Tungsten(IV) Imido Complexes. J. Am. Chem. Soc. 1993, 115, 8167–8177. [Google Scholar] [CrossRef]
- Poater, A.; Ragone, F.; Correa, A.; Cavallo, L. Exploring the reactivity of ru-based metathesis catalysts with a π-acid ligand trans to the Ru-ylidene bond. J. Am. Chem. Soc. 2009, 131, 9000–9006. [Google Scholar] [CrossRef] [PubMed]
- Galan, B.R.; Pitak, M.; Gembicky, M.; Keister, J.B.; Diver, S.T. Ligand-promoted carbene insertion into the aryl substituent of an n-heterocyclic carbene ligand in ruthenium-based metathesis catalysts. J. Am. Chem. Soc. 2009, 131, 6822–6832. [Google Scholar] [CrossRef] [PubMed]
- Monnier, F.; Castillo, D.; Dérien, S.; Toupet, L.; Dixneuf, P.H. Addition of Diazoalkanes to Enynes Promoted by a Ruthenium Catalyst: Simple Synthesis of Alkenyl Bicyclo[3.1.0]hexane Derivatives. Angew. Chem. Int. Ed. 2003, 42, 5474–5477. [Google Scholar] [CrossRef]
- Peppers, B.P.; Diver, S.T. Tandem cyclopropanation/ring-closing metathesis of dienynes. J. Am. Chem. Soc. 2004, 126, 9524–9525. [Google Scholar] [CrossRef]
- Kitamura, T.; Sato, Y.; Mori, M. Unexpected results of enyne metathesis using a ruthenium complex containing an N-heterocyclic carbene ligand. Chem. Commun. 2001, 1258–1259. [Google Scholar] [CrossRef]
- Nishiyama, H.; Itoh, Y.; Matsumoto, H.; Park, S.B.; Itoh, K. New Chiral Ruthenium Bis(oxazolinyl)pyridine Catalyst. Efficient Asymmetric Cyclopropanation of Olefins with Diazoacetates. J. Am. Chem. Soc. 1994, 116, 2223–2224. [Google Scholar] [CrossRef]
- Anciaux, A.J.; Demonceau, A.; Noels, A.F.; Hubert, A.J.; Warin, R.; Teyssié, P. Transition-Metal-Catalyzed Reactions of Diazo Compounds. 2.1 Addition to Aromatic Molecules: Catalysis of Buchner’s Synthesis of Cycloheptatrienes. J. Org. Chem. 1981, 46, 873–876. [Google Scholar] [CrossRef] [Green Version]
- Basato, M.; Tubaro, C.; Biffis, A.; Bonato, M.; Buscemi, G.; Lighezzolo, F.; Lunardi, P.; Yianini, C.; Benetollo, F.; Del Zotto, A. Reactions of diazo compounds with alkenes catalysed by [RuCl(cod)(Cp)]: Effect of the substituents in the formation of cyclopropanation or metathesis products. Chem. Eur. J. 2009, 15, 1516–1526. [Google Scholar] [CrossRef] [PubMed]
- Szczepaniak, G.; Urbaniak, K.; Wierzbicka, C.; Kosiński, K.; Skowerski, K.; Grela, K. High-Performance Isocyanide Scavengers for Use in Low-Waste Purification of Olefin Metathesis Products. ChemSusChem 2015, 8, 4139–4148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szczepaniak, G.; Nogaś, W.; Piatkowski, J.; Ruszczyńska, A.; Bulska, E.; Grela, K. Semiheterogeneous Purification Protocol for the Removal of Ruthenium Impurities from Olefin Metathesis Reaction Products Using an Isocyanide Scavenger. Org. Process. Res. Dev. 2019, 23, 836–844. [Google Scholar] [CrossRef]
- Szczepaniak, G.; Ruszczyńska, A.; Kosiński, K.; Bulska, E.; Grela, K. Highly efficient and time economical purification of olefin metathesis products from metal residues using an isocyanide scavenger. Green Chem. 2018, 20, 1280–1289. [Google Scholar] [CrossRef]
- Blacquiere, J.M.; Jurca, T.; Weiss, J.; Fogg, D.E. Time as a dimension in high-throughput homogeneous catalysis. Adv. Synth. Catal. 2008, 350, 2849–2855. [Google Scholar] [CrossRef]
- Griffiths, J.R.; Hofman, E.J.; Keister, J.B.; Diver, S.T. Kinetics and Mechanism of Isocyanide-Promoted Carbene Insertion into the Aryl Substituent of an N-Heterocyclic Carbene Ligand in Ruthenium-Based Metathesis Catalysts. Organometallics 2017, 36, 3043–3052. [Google Scholar] [CrossRef]
- Klose, A.; Solari, E.; Hesschenbrouck, J.; Floriani, C.; Re, N.; Geremia, S.; Randaccio, L. Ruthenium-carbene functionality bonded to dibenzotetramethyltetraaza[14]annulene: Metal-to-macrocycle ligand-induced carbene migration. Organometallics 1999, 18, 360–372. [Google Scholar] [CrossRef]
- Małecki, P.; Gajda, K.; Gajda, R.; Woźniak, K.; Trzaskowski, B.; Kajetanowicz, A.; Grela, K. Specialized ruthenium olefin metathesis catalysts bearing bulky unsymmetrical NHC Ligands: Computations, synthesis, and application. ACS Catal. 2019, 9, 587–598. [Google Scholar] [CrossRef]
Figure 1.
Common ruthenium metathesis catalysts.

Figure 2.
Schematic representation of the metathesis catalytic cycle; (a) initiation, (b,c) propagation.
Figure 2.
Schematic representation of the metathesis catalytic cycle; (a) initiation, (b,c) propagation.

Scheme 1.
The van Rensburg decomposition mechanism for the 1st generation and 2nd generation Grubbs catalysts. L2 = PCy3, numbers represent relative Gibbs free energies at the density functional (DFT) level of theory in kcal/mol.
Scheme 1.
The van Rensburg decomposition mechanism for the 1st generation and 2nd generation Grubbs catalysts. L2 = PCy3, numbers represent relative Gibbs free energies at the density functional (DFT) level of theory in kcal/mol.

Scheme 2.
The Nolan-Prunet mechanism of propylene isomerization with relative Gibbs free energies based on DFT results (in kcal/mol).
Scheme 2.
The Nolan-Prunet mechanism of propylene isomerization with relative Gibbs free energies based on DFT results (in kcal/mol).

Scheme 3.
(a) The ethylene-driven and allylbenzene-driven decomposition of metathesis catalysts using van Rensburg decomposition pathway and (b) 1-alkene-triggered catalyst decomposition for allylbenzene with DFT-based relative Gibbs free energies (in kcal/mol).
Scheme 3.
(a) The ethylene-driven and allylbenzene-driven decomposition of metathesis catalysts using van Rensburg decomposition pathway and (b) 1-alkene-triggered catalyst decomposition for allylbenzene with DFT-based relative Gibbs free energies (in kcal/mol).

Scheme 4.
The bimolecular coupling mechanism.

Scheme 5.
Catalysts degradation in primary alcohols. L = SIMes or SIPr.

Scheme 6.
Postulated alcohol-driven degradation mechanism by Dinger and Mol.

Scheme 7.
Reaction of GI catalyst with the allyl alcohol in solution or in solid state.

Scheme 8.
Primary allyl alcohol driven degradation in solution postulated by Werner and Wolf.

Scheme 9.
The secondary allyl alcohol-driven degradation in solution postulated by Werner and Wolf.

Scheme 10.
The reaction of Grubbs-type catalyst with methoxides.

Scheme 11.
The proposed mechanism of formation of ruthenium complexes 7–10 due to a methoxide attack on Grubbs-type complexes.
Scheme 11.
The proposed mechanism of formation of ruthenium complexes 7–10 due to a methoxide attack on Grubbs-type complexes.

Scheme 12.
The degradation mechanism in presence of hydroxide ion proposed by Fogg et al. with DFT-based relative Gibbs free energies (in kcal/mol).
Scheme 12.
The degradation mechanism in presence of hydroxide ion proposed by Fogg et al. with DFT-based relative Gibbs free energies (in kcal/mol).

Scheme 13.
Two mechanisms of alcoholysis and Gibbs free energy profile (in kcal/mol) for the phosphine-off mechanism obtained at the DFT level of theory (blue route via 11).
Scheme 13.
Two mechanisms of alcoholysis and Gibbs free energy profile (in kcal/mol) for the phosphine-off mechanism obtained at the DFT level of theory (blue route via 11).

Scheme 14.
The alcoholysis decomposition products of the Grubbs-type indenylidene precatalyst in (a) RCH2OH and (b) iPrOH.
Scheme 14.
The alcoholysis decomposition products of the Grubbs-type indenylidene precatalyst in (a) RCH2OH and (b) iPrOH.

Scheme 15.
The energy profile of M10 formation from the two different DFT methods (in kcal/mol).

Scheme 16.
The energy profile of M11, M10, M1 formation from the DFT calculations (values in kcal/mol).
Scheme 16.
The energy profile of M11, M10, M1 formation from the DFT calculations (values in kcal/mol).

Scheme 17.
Comparison of the alcohol-driven decomposition of (a) symmetric and (b) asymmetric 2nd generation indenylidene catalysts with the Gibbs free energies (in kcal/mol) calculated at the DFT level of theory.
Scheme 17.
Comparison of the alcohol-driven decomposition of (a) symmetric and (b) asymmetric 2nd generation indenylidene catalysts with the Gibbs free energies (in kcal/mol) calculated at the DFT level of theory.

Scheme 18.
The mechanism of metathesis in the presence of phenols with suggested PhOH coordination structures to activated catalyst.
Scheme 18.
The mechanism of metathesis in the presence of phenols with suggested PhOH coordination structures to activated catalyst.

Scheme 19.
A schematic representation of the equilibrium step with salt during conversion to the inactive form of the catalyst.
Scheme 19.
A schematic representation of the equilibrium step with salt during conversion to the inactive form of the catalyst.

Scheme 20.
The formation of phosphonium salt and the mechanism of acrylate-induced decomposition.

Scheme 21.
The methyl-acrylate induced formation of ruthenium dimers.

Scheme 22.
The amine-driven decomposition of metallacyclobutane with the Gibbs free energy profile obtained at the DFT level of theory.
Scheme 22.
The amine-driven decomposition of metallacyclobutane with the Gibbs free energy profile obtained at the DFT level of theory.

Scheme 23.
A schematic representation of role of phenol during olefin metathesis reaction.

Scheme 24.
An amine-driven decomposition product obtained with the excess of pyridine.

Scheme 25.
The structure of investigated catalysts and obtained amine adducts in the study of ruthenium complexes stability towards amines.
Scheme 25.
The structure of investigated catalysts and obtained amine adducts in the study of ruthenium complexes stability towards amines.

Scheme 26.
The amine-driven decomposition of ruthenium complexes studied by Fogg.

Scheme 27.
The proposed mechanism of amine-induced decomposition in conditions of ring-closing metathesis (RCM).
Scheme 27.
The proposed mechanism of amine-induced decomposition in conditions of ring-closing metathesis (RCM).

Scheme 28.
The donor-induced decomposition of 1st generation ruthenium carbene catalysts.

Scheme 29.
HGII and GII reactions with secondary amines and DBU.

Scheme 30.
The reaction of HGII with primary amines.

Scheme 31.
The proposed mechanism of deactivation of HGII by amine.

Scheme 32.
The rate-limiting step for the donor-accelerated loss of methylidene.

Scheme 33.
The rate limiting step for the donor-accelerated loss of methylidene based on the inhibition of the carbene rotation.
Scheme 33.
The rate limiting step for the donor-accelerated loss of methylidene based on the inhibition of the carbene rotation.

Scheme 34.
The decomposition pathway of the methylidene active species by small ligands: (a) tetramethylimidazol-2-ylidene IMe4 and (b) N-heterocyclic olefin CH2 = IMe4, with the Gibbs free energy profile from DFT calculations.
Scheme 34.
The decomposition pathway of the methylidene active species by small ligands: (a) tetramethylimidazol-2-ylidene IMe4 and (b) N-heterocyclic olefin CH2 = IMe4, with the Gibbs free energy profile from DFT calculations.

Scheme 35.
The mechanism of acrylonitrile driven decomposition proposed by Guo et al.

Scheme 36.
The mechanism of decomposition of GII by CH3CN based on mass spectrometry analysis.

Scheme 37.
The reactions of ruthenium Grubbs carbenes in presence of O2.

Scheme 38.
Three proposed reaction of Ru-catalyst under oxygen, n.d. = not determined; (a) a benzylidene attack by O2, (b) a [2+2] cycloaddition of O2, (c) a bimolecular degradation with stilbene release.
Scheme 38.
Three proposed reaction of Ru-catalyst under oxygen, n.d. = not determined; (a) a benzylidene attack by O2, (b) a [2+2] cycloaddition of O2, (c) a bimolecular degradation with stilbene release.

Scheme 39.
The temperature-induced 1st generation Grubbs catalyst decomposition route.

Scheme 40.
The thermal degradation of Fisher carbenes generated from GII catalyst.

Scheme 41.
The thermal decomposition of the Fischer-type carbene.

Scheme 42.
The proposed mechanism of the thermolysis of the 2nd generation Grubbs carbenes.

Scheme 43.
The decomposition rates of ruthenium methylidene complexes.

Scheme 44.
The suggested paths of the decomposition of the methylene complex under metathesis reaction with ethylene.
Scheme 44.
The suggested paths of the decomposition of the methylene complex under metathesis reaction with ethylene.

Scheme 45.
The structures of (a) investigated ruthenium 1st generation catalysts and (b) identified decomposition species.
Scheme 45.
The structures of (a) investigated ruthenium 1st generation catalysts and (b) identified decomposition species.

Scheme 46.
The C–H activation of Grubbs catalyst.

Scheme 47.
(a) The C-H activation of the P-P and P-As complexes and (b) the proposed mechanism for the C-H activation of six-coordinate complex with additional PPh3 (blue) and IMes ligand (red), with the Gibbs free energies estimated at the DFT level of theory (in kcal/mol).
Scheme 47.
(a) The C-H activation of the P-P and P-As complexes and (b) the proposed mechanism for the C-H activation of six-coordinate complex with additional PPh3 (blue) and IMes ligand (red), with the Gibbs free energies estimated at the DFT level of theory (in kcal/mol).

Scheme 48.
The C-H activation of ruthenium hydride complexes.

Scheme 49.
The C–H activation of CO-containing complexes.

Scheme 50.
The C–H activation of the second generation Grubbs catalysts containing phenyl-substituted NHC ligand.
Scheme 50.
The C–H activation of the second generation Grubbs catalysts containing phenyl-substituted NHC ligand.

Scheme 51.
The mechanism of the C–H activation of catalysts containing phenyl-substituted NHC ligands with the relative Gibbs free energies (in kcal/mol), as proposed by Cavallo.
Scheme 51.
The mechanism of the C–H activation of catalysts containing phenyl-substituted NHC ligands with the relative Gibbs free energies (in kcal/mol), as proposed by Cavallo.

Scheme 52.
The mechanism for insertion reaction proposed by Blechert.

Scheme 53.
(a) The proposed mechanism of the decomposition of phosphonium complexes and (b) the self-trapping of the proposed intermediate V.
Scheme 53.
(a) The proposed mechanism of the decomposition of phosphonium complexes and (b) the self-trapping of the proposed intermediate V.

Scheme 54.
The decomposition of the diruthenium chloro-bridged complex.

Scheme 55.
(a) The synthesis of the C-H activated catalyst 72 and its decomposition via exposure to an excess CO; (b,c) the C-H activation via tBuCOOAg.
Scheme 55.
(a) The synthesis of the C-H activated catalyst 72 and its decomposition via exposure to an excess CO; (b,c) the C-H activation via tBuCOOAg.

Scheme 56.
The initial step of the deactivation pathway for complexes containing phenyl-substituted NHC ligands with relative Gibbs free energies from the DFT calculations (in kcal/mol).
Scheme 56.
The initial step of the deactivation pathway for complexes containing phenyl-substituted NHC ligands with relative Gibbs free energies from the DFT calculations (in kcal/mol).

Scheme 57.
The mechanism of isocyanide-promoted ylidene transfer to phosphorus proposed by Diver.

Scheme 58.
The isocyanide-promoted ligand insertion into GII complex.

Scheme 59.
The CO-driven deactivation pathway of GII with L = PMe3 or CO, with Gibbs free energies (in kcal/mol) based on DFT calculations.
Scheme 59.
The CO-driven deactivation pathway of GII with L = PMe3 or CO, with Gibbs free energies (in kcal/mol) based on DFT calculations.

Scheme 60.
The reactivity of isocyanides towards the first and second generation metathesis catalysts.
Scheme 60.
The reactivity of isocyanides towards the first and second generation metathesis catalysts.

Scheme 61.
The mechanism for the CNR-promoted Buchner insertion proposed by Diver for (a) GII and (b) HGII.
Scheme 61.
The mechanism for the CNR-promoted Buchner insertion proposed by Diver for (a) GII and (b) HGII.

Scheme 62.
The Gibbs free energy profile of decomposition of HGII and CAAC-containing catalysts via Buchner ring expansion with either (a) one or (b) two acrylonitrile ligands.
Scheme 62.
The Gibbs free energy profile of decomposition of HGII and CAAC-containing catalysts via Buchner ring expansion with either (a) one or (b) two acrylonitrile ligands.

Scheme 63.
The hypothetical bimolecular coupling mechanism for activated benzylidenes.

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Jawiczuk, M.; Marczyk, A.; Trzaskowski, B. Decomposition of Ruthenium Olefin Metathesis Catalyst. Catalysts 2020, 10, 887. https://0-doi-org.brum.beds.ac.uk/10.3390/catal10080887
AMA Style
Jawiczuk M, Marczyk A, Trzaskowski B. Decomposition of Ruthenium Olefin Metathesis Catalyst. Catalysts. 2020; 10(8):887. https://0-doi-org.brum.beds.ac.uk/10.3390/catal10080887
Chicago/Turabian StyleJawiczuk, Magdalena, Anna Marczyk, and Bartosz Trzaskowski. 2020. "Decomposition of Ruthenium Olefin Metathesis Catalyst" Catalysts 10, no. 8: 887. https://0-doi-org.brum.beds.ac.uk/10.3390/catal10080887
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.