CO2 Methanation on Supported Rh Nanoparticles: The combined Effect of Support Oxygen Storage Capacity and Rh Particle Size

 , , ,
, , , 
Abstract
:
1. Introduction
2. Results and Discussion
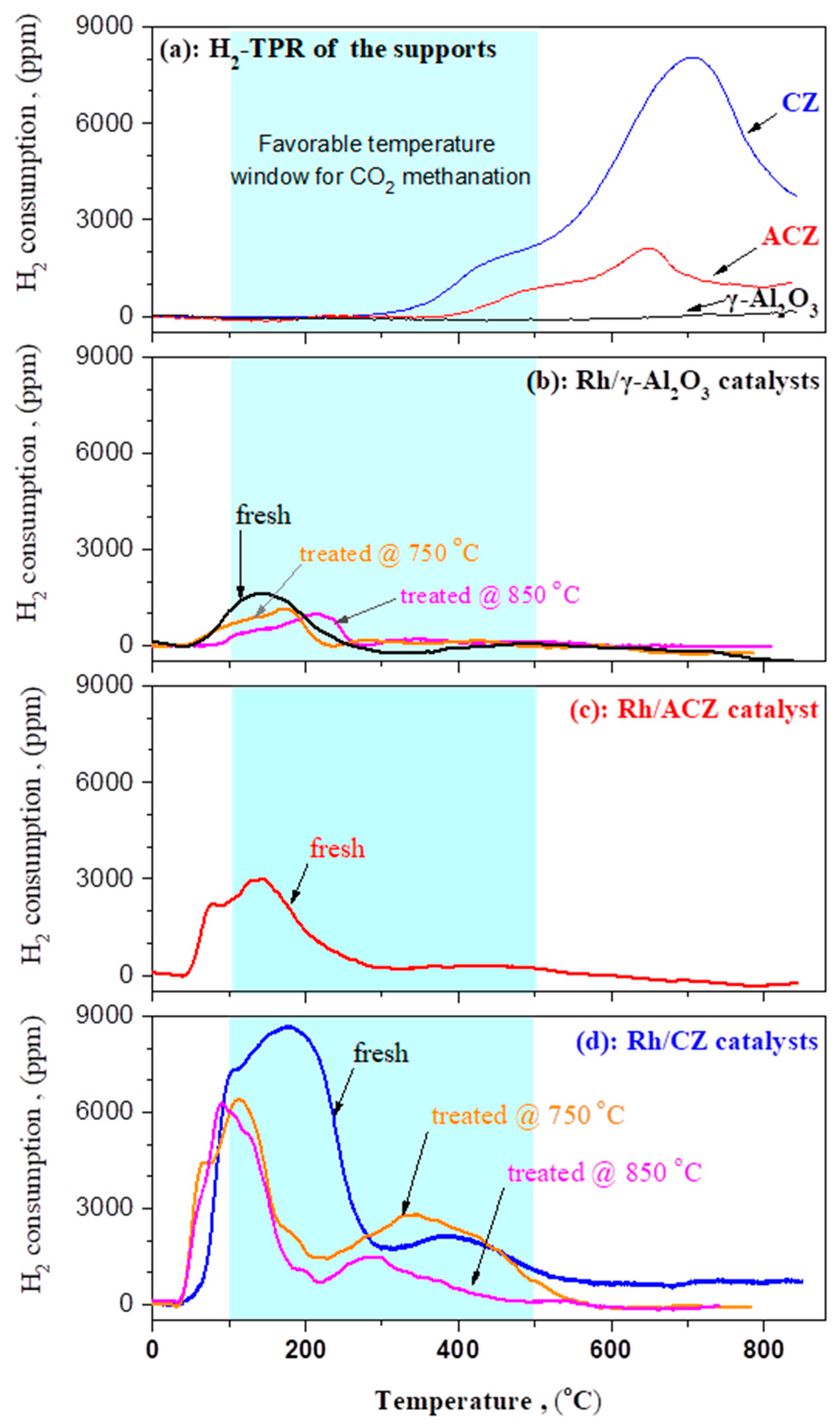
2.1. Morphological and Reducibility Characteristics of the Materials
2.2. Comparative Evaluation of Catalytic Performance: Effect of the Support
2.2.1. CO2 Hydrogenation Performance Under Integral Reaction Conditions
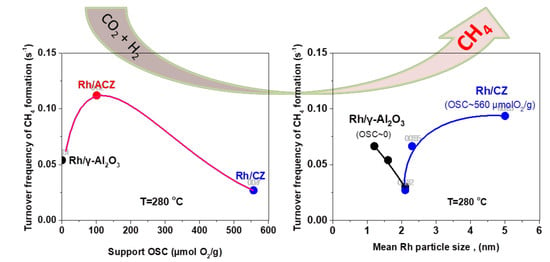
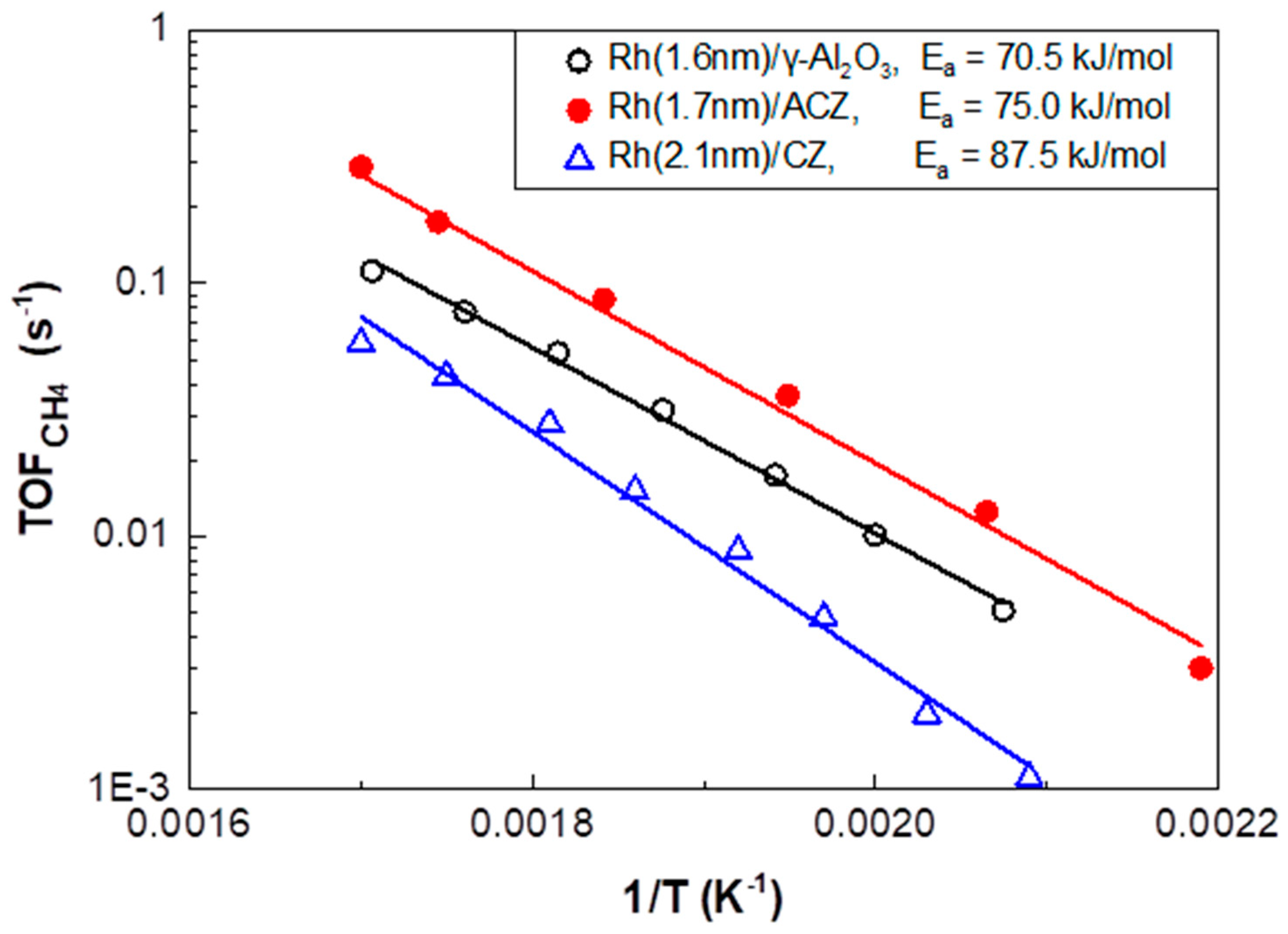
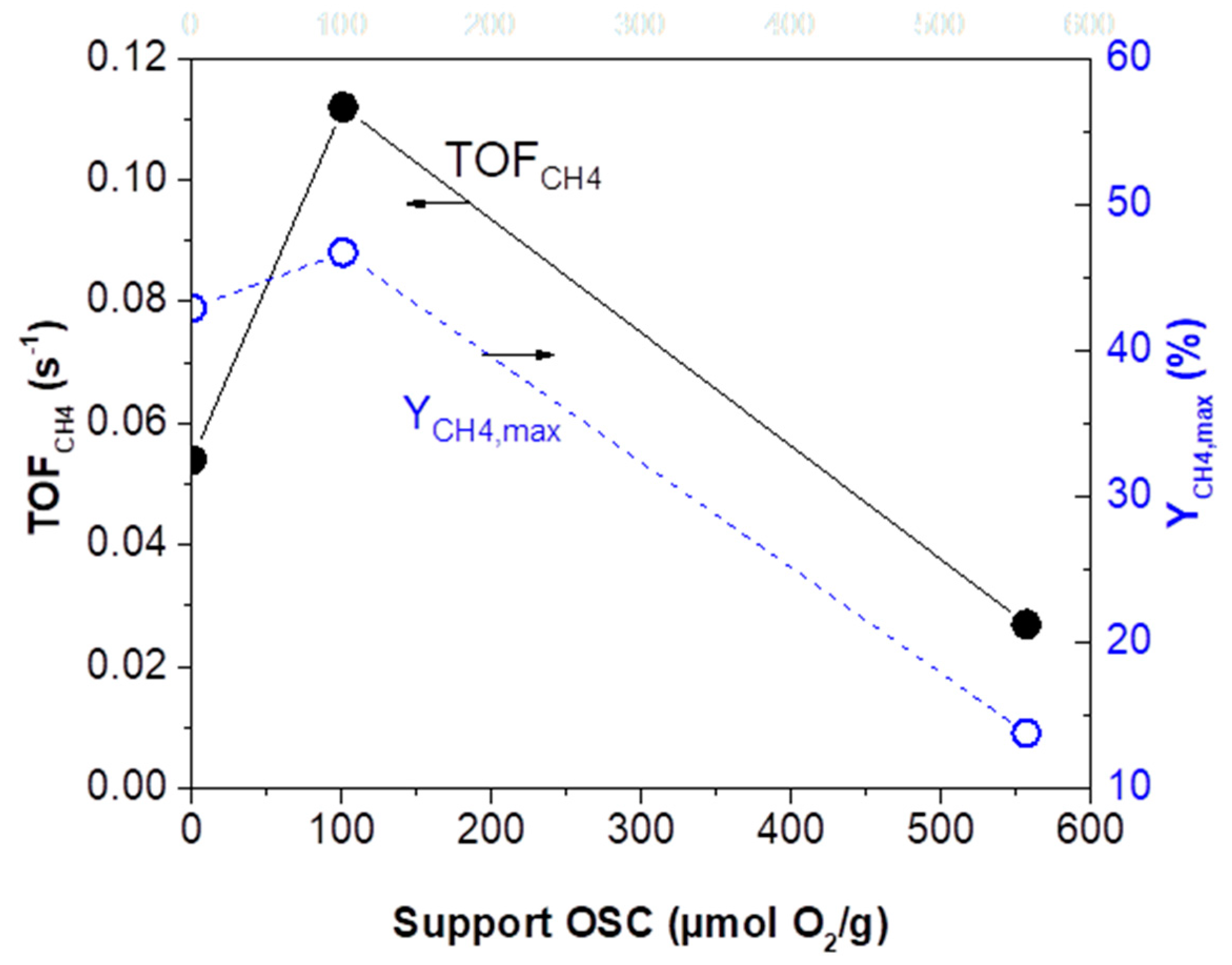
2.2.2. CO2 Hydrogenation Intrinsic Activity of Rh Nanoparticles
- (i)
- (ii)
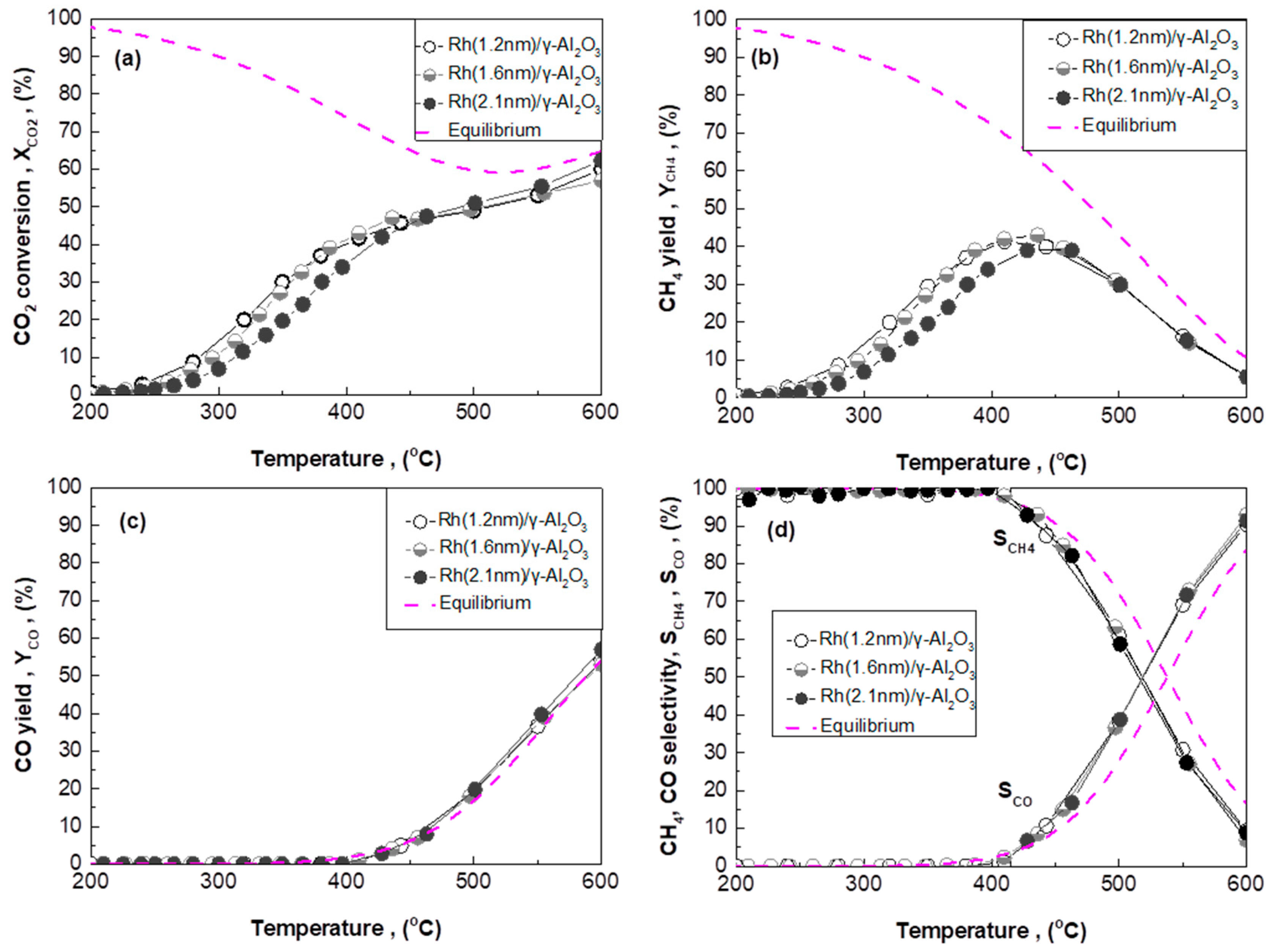
- ACZ and CZ promote the reverse water–gas shift reaction (CO formation) in a monotonic manner in respect to their OSC value; the higher the OSC, the higher the promotion of rWGS (Figure 2c). In particular, CZ also promotes the formation of additional C-containing byproducts, besides CH4 and CO (Figure 2d).
- (iii)
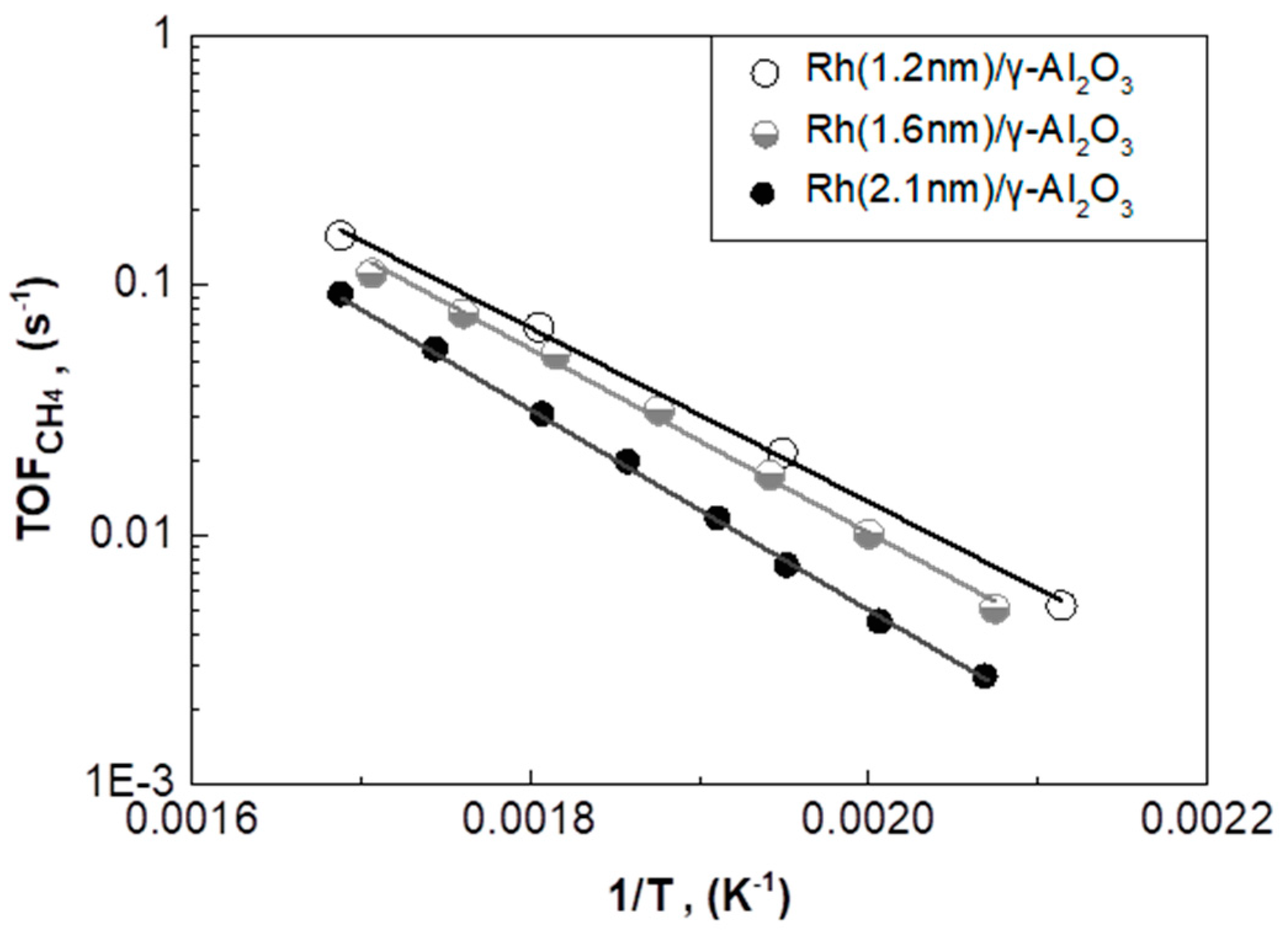
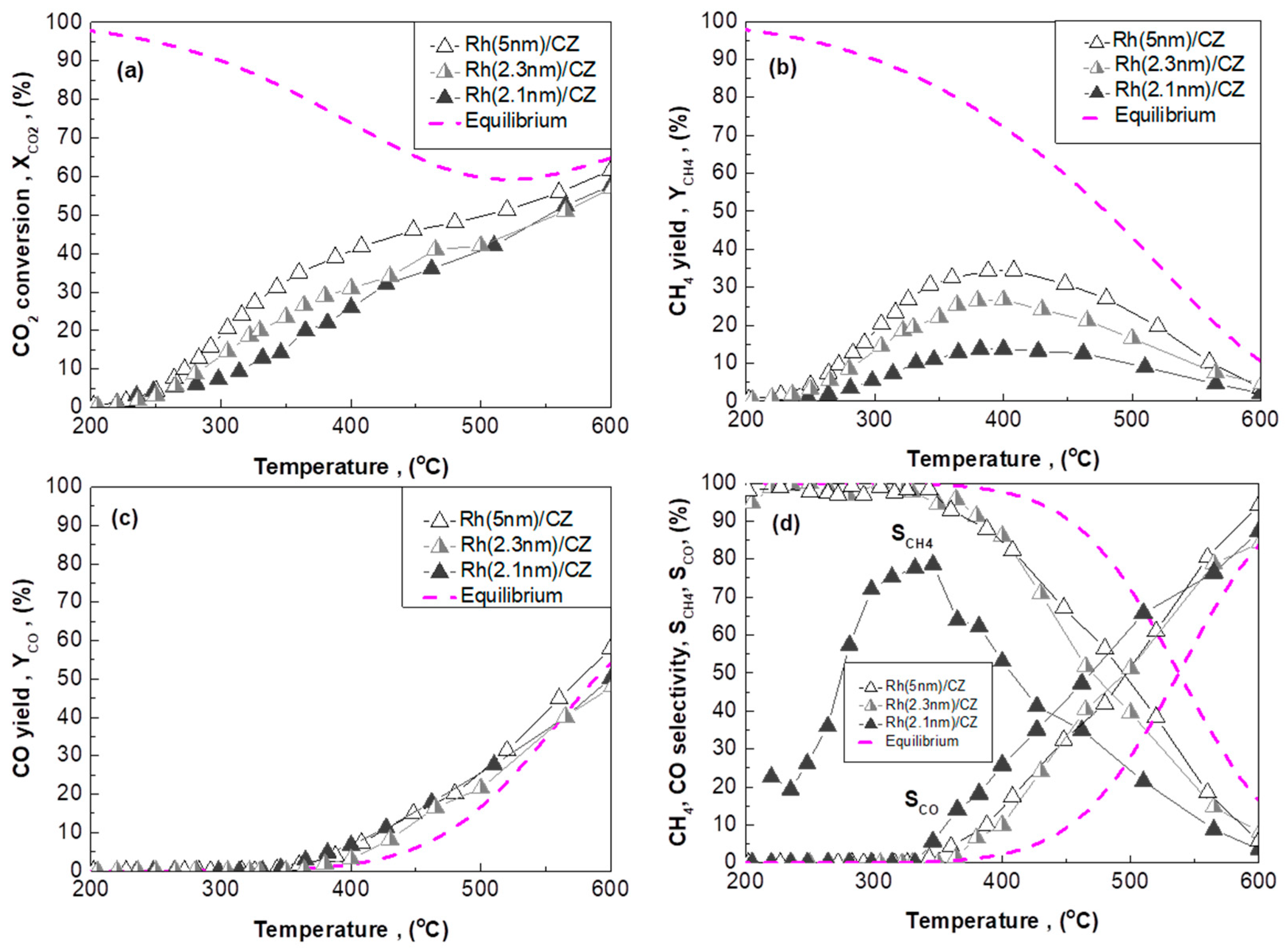
2.3. Effect of Rh Particle Size on CO2 Methanation Performance
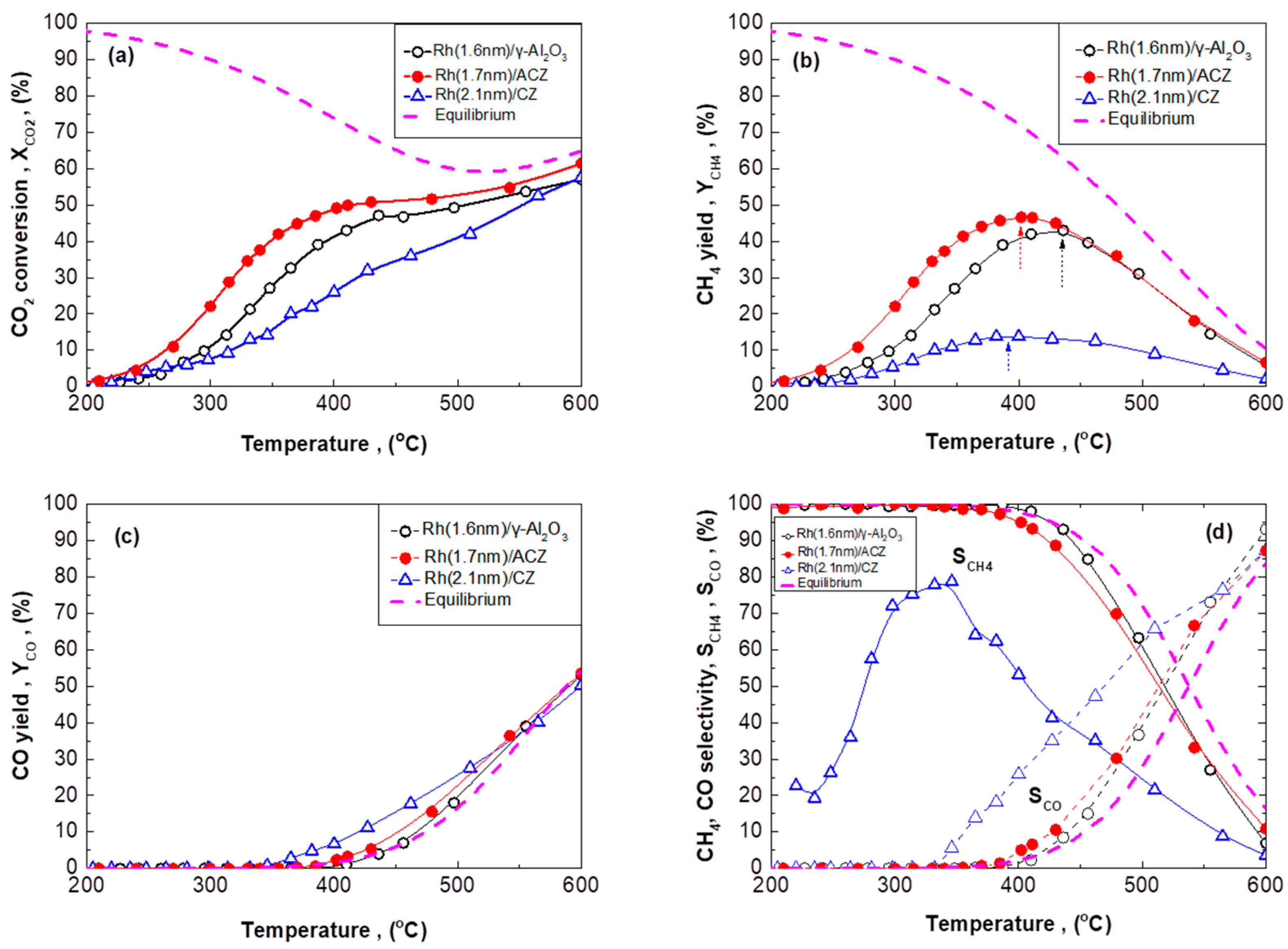
2.3.1. Rh/γ-Al2O3 Catalysts with Different Mean Rh Particle Size
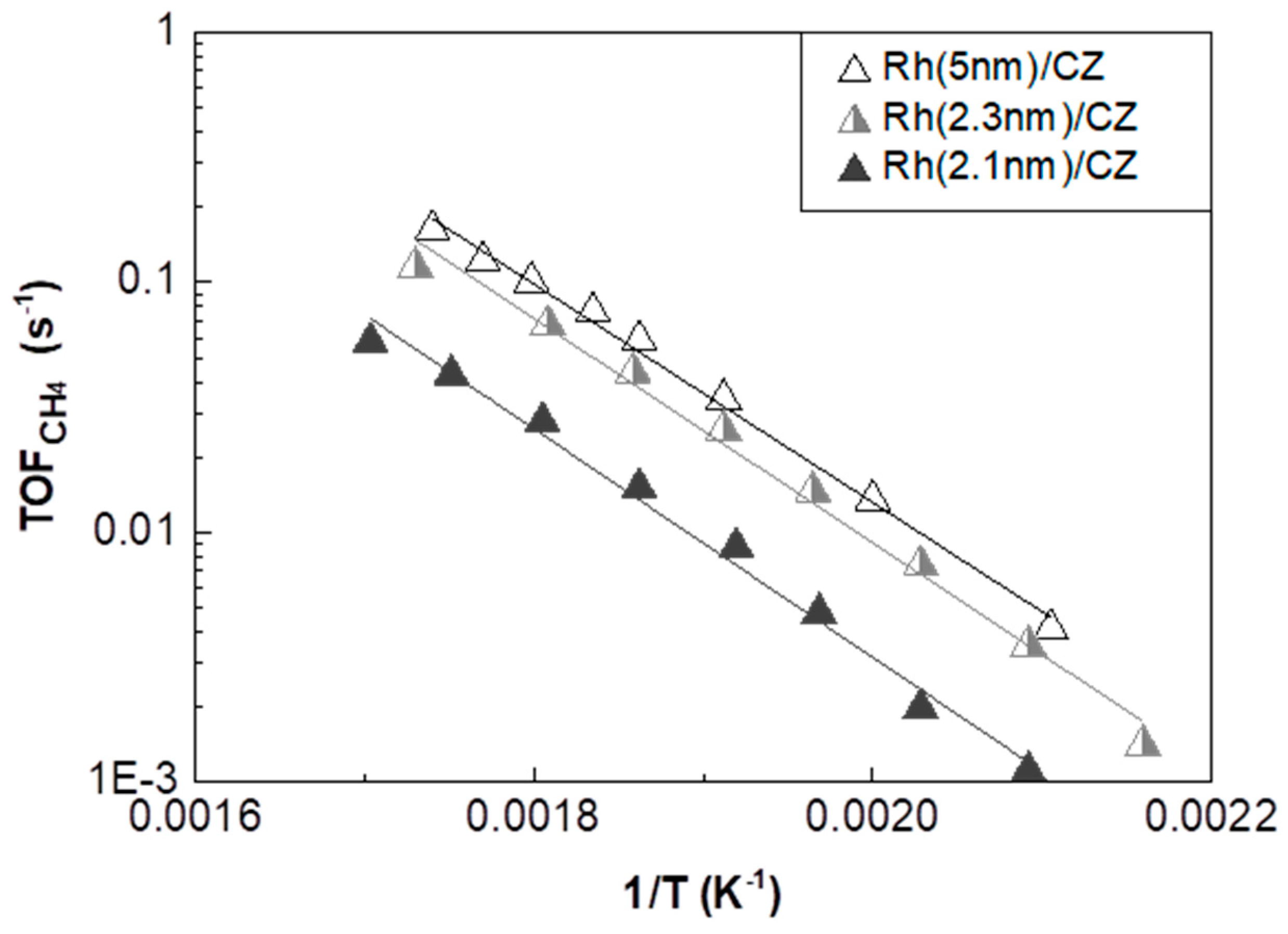
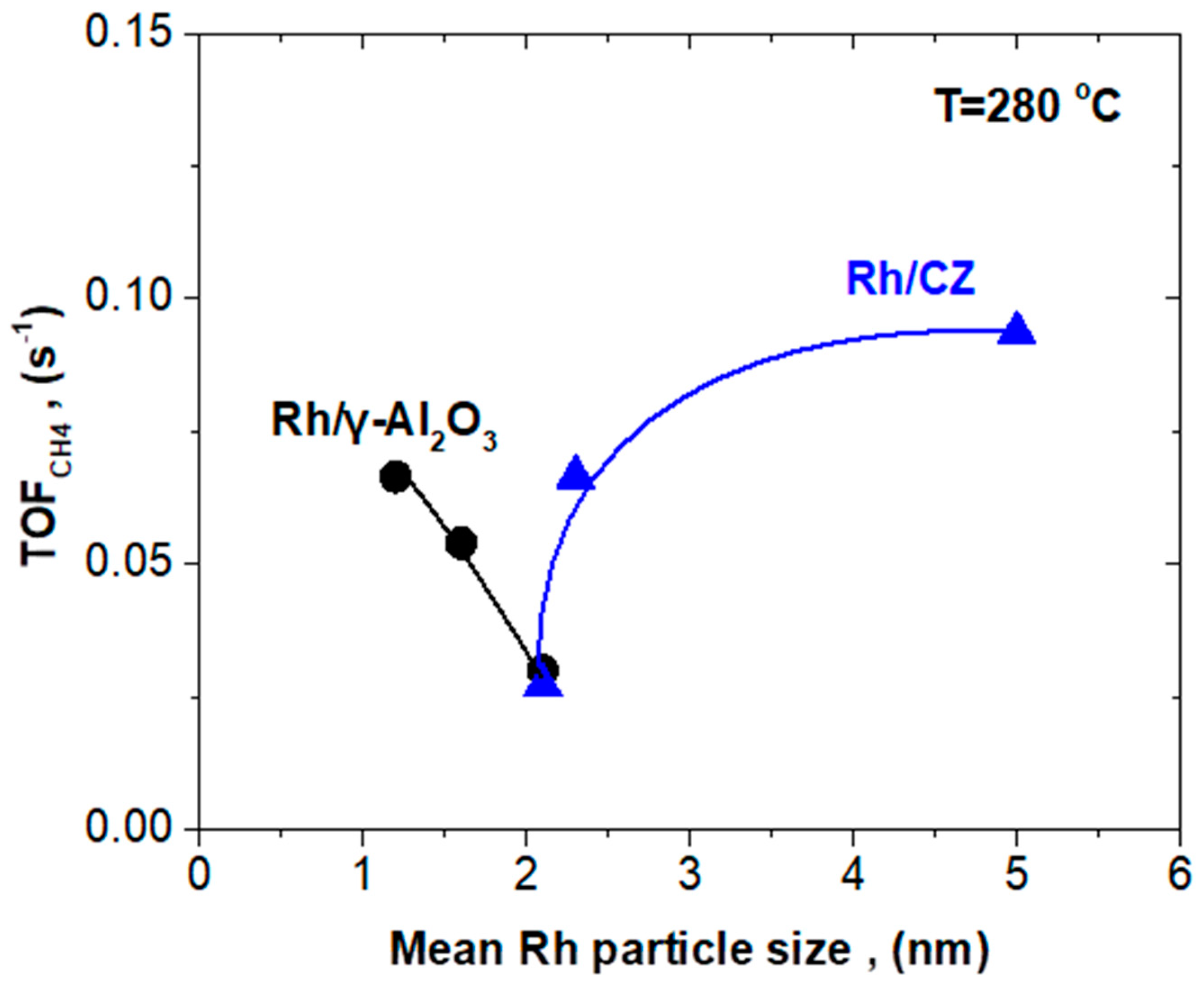
2.3.2. Rh/CZ Catalysts with Different Mean Rh Particle Sizes
3. Materials and Methods
3.1. Catalysts’ Preparation
3.1.1. Supporting Materials
3.1.2. Supported Rh Catalysts
3.1.3. Modification of Rh Particle Size
3.2. Characterization Methods
3.2.1. Materials Characterization
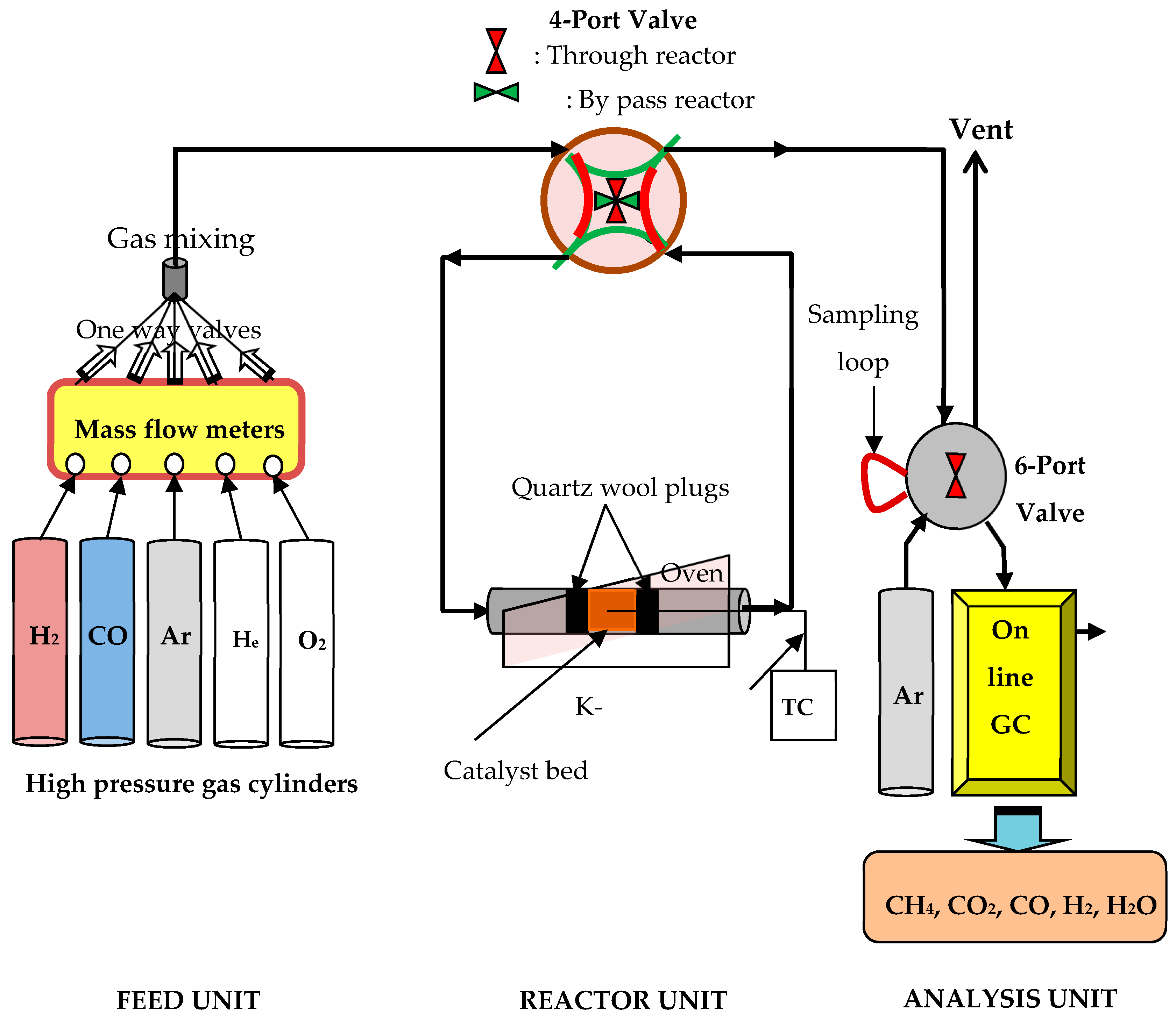
3.2.2. Catalytic Performance Evaluation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Wang, W.; Wang, S.P.; Ma, X.B.; Gong, J.L. Recent advances in catalytic hydrogenation of carbon dioxide. Chem. Soc. Rev. 2011, 40, 3703–3727. [Google Scholar] [CrossRef] [Green Version]
- Ye, R.P.; Ding, J.; Gong, W.; Argyle, M.D.; Zhong, Q.; Wang, Y.; Russell, C.K.; Xu, Z.; Russell, A.G.; Li, Q.; et al. CO2 hydrogenation to high-value products via heterogeneous catalysis. Nat. Comm. 2019, 10, 5698. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Yi, Y.; Wang, L.; Guo, H.; Bogaerts, A. Hydrogenation of Carbon Dioxide to Value-Added Chemicals by Heterogeneous Catalysis and Plasma Catalysis. Catalysts 2019, 9, 275. [Google Scholar] [CrossRef] [Green Version]
- Vogt, C.; Monai, M.; Kramer, G.J.; Weckhuysen, B.M. The renaissance of the Sabatier reaction and its applications on Earth and in space. Nat. Catal. 2019, 2, 188–197. [Google Scholar] [CrossRef]
- Tsiotsias, A.I.; Charisiou, N.D.; Yentekakis, I.V.; Goula, M.A. The Role of Alkali and Alkaline Earth Metals in the CO2 Methanation Reaction and the Combined Capture and Methanation of CO2. Catalysts 2020, 10, 812. [Google Scholar] [CrossRef]
- Frontera, P.; Macario, A.; Ferraro, M.; Antonucci, P. Supported catalysts for CO2 methanation: A review. Catalysts 2017, 7, 59. [Google Scholar] [CrossRef]
- Aziz, M.A.A.; Jalil, A.A.; Triwahyono, S.; Ahmad, A. CO2 methanation over heterogeneous catalysts: Recent progress and future prospects. Green Chem. 2015, 17, 2647–2663. [Google Scholar] [CrossRef]
- Baraj, E.; Vagaský, S.; Hlinčík, T.; Ciahotný, K.; Tekáč, V. Reaction mechanisms of carbon dioxide methanation. Chem. Pap. 2016, 70, 395–403. [Google Scholar] [CrossRef]
- Pan, S.Y.; Chiang, P.C.; Pan, W.; Kim, H. Advances in state-of-art valorization technologies for captured CO2 toward sustainable carbon cycle. Crit. Rev. Env. Sci. Technol. 2018, 48, 471–534. [Google Scholar] [CrossRef]
- Yentekakis, I.V.; Goula, G. Biogas management: Advanced utilization for production of renewable energy and added-value chemicals. Front. Environ. Sci. 2017, 5, 7. [Google Scholar] [CrossRef] [Green Version]
- Ghaiba, K.; Ben-Faresb, F.-Z. Power-to-Methane: A state-of-the-art review. Renew. Sustain. Energy Rev. 2018, 81, 433–446. [Google Scholar] [CrossRef]
- Mazza, A.; Bompard, E.; Chicco, G. Applications of power to gas technologies in emerging electrical systems. Renew. Sustain. Energy Rev. 2018, 92, 794–806. [Google Scholar] [CrossRef]
- Gotz, M.; Lefebvre, J.; Mors, F.; McDaniel Koch, A.; Graf, F.; Bajohr, S.; Reimert, R.; Kolb, T. Renewable Power-to-Gas: A technological and economic review. Renew. Energy 2016, 85, 1371–1390. [Google Scholar] [CrossRef] [Green Version]
- O’Shea, R.; Wall, D.M.; McDonagh, S.; Murphy, J.D. The potential of power to gas to provide green gas utilising existing CO2 sources from industries, distilleries and wastewater treatment facilities. Renew. Energy 2017, 114, 1090–1100. [Google Scholar] [CrossRef]
- Bonura, G.; Cordaro, M.; Cannilla, C.; Arena, F.; Frusteri, F. The changing nature of the active site of Cu-Zn-Zr catalysts for the CO2 hydrogenation reaction to methanol. Appl. Catal. B 2014, 152–153, 152–161. [Google Scholar] [CrossRef]
- Dasireddy, V.D.B.C.; Likozar, B. The role of copper oxidation state in Cu/Zn/Al2O3 catalysts in CO2 hydrogenation and methanol productivity. Renew. Energy 2019, 140, 452–460. [Google Scholar] [CrossRef]
- Li, M.; Amari, H.; Van Veen, A.C. Metal-oxide interaction enhanced CO2 activation in methanation over ceria supported nickel nanocrystallites. Appl. Catal. B 2018, 239, 27–35. [Google Scholar] [CrossRef]
- Ye, R.P.; Li, Q.; Gong, W.; Wang, T.; Razink, J.J.; Lin, L.; Qin, Y.Y.; Zhou, Z.; Adidharma, H.; Tang, J.; et al. High-performance of nanostructured Ni/CeO2 catalyst on CO2 methanation. Appl. Catal. B 2020, 268, 118475. [Google Scholar] [CrossRef]
- Winter, L.R.; Chen, R.; Chen, X.; Chang, K.; Liu, Z.; Senenayake, S.D.; Ebrahim, A.M.; Chen, J.G. Elucidating the roles of metallic Ni and oxygen vacancies in CO2 hydrogenation over Ni/CeO2 using isotope exchange and in situ measurements. Appl. Catal. B 2019, 245, 360–366. [Google Scholar] [CrossRef]
- Winter, L.R.; Gomez, E.; Yan, B.; Yao, S.; Chen, J.G. Tuning Ni-catalyzed CO2 hydrogenation selectivity via Ni-ceria support interactions and Ni-Fe bimetallic formation. Appl. Catal. B 2018, 224, 442–450. [Google Scholar] [CrossRef]
- Cerda, M.C.; Chica, A.; Keller, S.; Rautenberg, C.; Bentrup, U. Ni-sepiolite and Ni-todorokite as efficient CO2 methanation catalysts: Mechanistic insight by operando DRIFTS. Appl. Catal. B 2020, 264, 118546. [Google Scholar] [CrossRef]
- Italiano, C.; Llorca, J.; Pino, L.; Farraro, M.; Antonucci, V.; Vita, A. CO and CO2 methanation over Ni catalysts supported on CeO2, Al2O3 and Y2O3 oxides. Appl. Catal. B 2020, 264, 118494. [Google Scholar] [CrossRef]
- Tada, S.; Shimizu, T.; Kameyama, H.; Haneda, T.; Kikuchi, R. Ni/CeO2 catalysts with high CO2 methanation activity and high CH4 selectivity at low temperatures. Int. J. Hydrog. Energy 2012, 37, 5527–5531. [Google Scholar] [CrossRef]
- Liu, H.; Zou, X.; Wang, X.; Lou, X.; Ding, W. Effect of CeO2 addition on Ni/Al2O3 catalysts for methanation of carbon dioxide with hydrogen. J. Nat. Gas Chem. 2012, 21, 703–707. [Google Scholar] [CrossRef]
- Zhu, H.; Razzaq, R.; Li, C.; Muhmmad, Y.; Zhang, S. Catalytic methanation of carbon dioxide by active oxygen material CexZr1-xO2 supported Ni-Co bimetallic nanocatalysts. AIChE J. 2013, 59, 2567–2576. [Google Scholar] [CrossRef]
- Alcalde-Santiago, V.; Davó-Quiñonero, A.; Lozano-Castelló, D.; Quindimil, A.; De-La-Torre, U.; Pereda-Ayo, B.; González-Marcos, J.A.; González-Velasco, J.R.; Bueno-López, A. Ni/LnOx catalysts (Ln = La, Ce or Pr) for CO2 methanation. ChemCatChem 2019, 11, 810–819. [Google Scholar] [CrossRef] [Green Version]
- Cárdenas, A.A.; Quindimil, A.; Davó, Q.A.; Bailón, G.E.; Lozano, C.D.; De, L.T.U.; Pereda, A.B.; González, M.J.A.; González, V.J.R.; Bueno, L.A. Design of active sites in Ni/CeO2 catalysts for the methanation of CO2: Tailoring the Ni-CeO2 contact. Appl. Mater. Today 2020, 19, 100591. [Google Scholar] [CrossRef]
- Yuan, H.; Zhu, X.; Han, J.; Wang, H.; Ge, Q. Rhenium-promoted selective CO2 methanation on Ni-based catalyst. J. CO2 Util. 2018, 26, 8–18. [Google Scholar] [CrossRef]
- Ray, K.; Deo, G. A potential descriptor for the CO2 hydrogenation to CH4 over Al2O3 supported Ni and Ni-based alloy catalysts. Appl. Catal. B 2017, 218, 525–537. [Google Scholar] [CrossRef]
- Weatherbee, G.D.; Bartholomew, C.H. Hydrogenation of CO2 on group VIII metals. IV. Specific activities and selectivities of silica-supported Co, Fe and Ru. J. Catal. 1984, 87, 352–362. [Google Scholar] [CrossRef]
- Kirchner, J.; Anolleck, J.K.; Lösch, H.; Kureti, S. Methanation of CO2 on Iron based catalysts. Appl. Catal. B 2018, 223, 47–59. [Google Scholar] [CrossRef]
- Martin, N.M.; Velin, P.; Skoglundh, M.; Bauer, M.; Carlsson, P.A. Catalytic hydrogenation of CO2 to methane over supported Pd, Rh and Ni catalysts. Catal. Sci. Technol. 2017, 7, 1086–1094. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Liu, H.; Wang, B.; Ling, L. Insights into the effect of surface hydroxyls on CO2 hydrogenation over Pd/γ-Al2O3 catalyst: A computational study. Appl. Catal. B 2012, 126, 108–120. [Google Scholar] [CrossRef]
- Janke, C.; Duyar, M.S.; Hoskins, M.; Farrauto, R. Catalytic and adsorption studies for the hydrogenation of CO2 to methane. Appl. Catal. B 2014, 152–153, 184–191. [Google Scholar] [CrossRef]
- Panagiotopoulou, P. Hydrogenation of CO2 over supported noble metal catalysts. Appl. Catal. A 2017, 542, 63–70. [Google Scholar] [CrossRef]
- Kattel, S.; Yan, B.; Chen, J.G.; Liu, P. CO2 hydrogenation on Pt, Pt/SiO2 and Pt/TiO2: Importance of synergy between Pt and oxide support. J. Catal. 2016, 343, 115–126. [Google Scholar] [CrossRef] [Green Version]
- Kwak, J.H.; Kovarik, L.; Szanyi, J. Heterogeneous catalysis on atomically dispersed supported metals: CO2 reduction on multifunctional Pd catalysts. ACS Catal. 2013, 3, 2094–2100. [Google Scholar] [CrossRef]
- Wang, X.; Shi, H.; Kwak, J.H.; Szanyi, J. Mechanism of CO2 Hydrogenation on Pd/Al2O3 Catalysts: Kinetics and Transient DRIFTS-MS Studies. ACS Catal. 2015, 5, 6337–6349. [Google Scholar] [CrossRef]
- Beuls, A.; Swalus, C.; Jacquemin, M.; Heyen, G.; Karelovic, A.; Ruiz, P. Methanation of CO2: Further insight into the mechanism over Rh/γ-Al2O3 catalyst. Appl. Catal. B 2012, 113–114, 2–10. [Google Scholar] [CrossRef]
- Karelovic, A.; Ruiz, P. CO2 hydrogenation at low temperature over Rh/γ-Al2O3 catalysts: Effect of the metal particle size on catalytic performances and reaction mechanism. Appl. Catal. B 2012, 113–114, 237–249. [Google Scholar] [CrossRef]
- Zhang, Z.; Kladi, A.; Verykios, X.E. Effects of carrier doping on kinetic parameters of CO2 hydrogenation on supported rhodium catalysts. J. Catal. 1994, 148, 737–747. [Google Scholar] [CrossRef]
- Theleritis, D.; Souentie, S.; Siokou, A.; Katsaounis, A.; Vayenas, C.G. Hydrogenation of CO2 over Ru/YSZ electropromoted catalysts. ACS Catal. 2012, 2, 770–780. [Google Scholar] [CrossRef]
- Kalaitzidou, I.; Makri, M.; Theleritis, D.; Katsaounis, A.; Vayenas, C.G. Comparative study of the electrochemical promotion of CO2 hydrogenation on Ru using Na+, K+, H+ and O2− conducting solid electrolytes. Surf. Sci. 2016, 646, 194–203. [Google Scholar] [CrossRef]
- Li, S.; Xu, Y.; Chen, Y.; Li, W.; Lin, L.; Li, M.; Deng, Y.; Wang, X.; Ge, B.; Yang, C.; et al. Tuning the Selectivity of Catalytic Carbon Dioxide Hydrogenation over Iridium/Cerium Oxide Catalysts with a Strong Metal–Support Interaction. Angew. Chem. Int. Ed. 2017, 56, 10761–10765. [Google Scholar] [CrossRef]
- Xu, J.; Su, X.; Duan, H.; Hou, B.; Lin, Q.; Liu, X.; Pan, X.; Pei, G.; Geng, H.; Huang, Y.; et al. Influence of pretreatment temperature on catalytic performance of rutile TiO2-supported ruthenium catalyst in CO2 methanation. J. Catal. 2016, 333, 227–237. [Google Scholar] [CrossRef]
- Petala, A.; Panagiotopoulou, P. Methanation of CO2 over alkali-promoted Ru/TiO2 catalysts: I. Effect of alkali additives on catalytic activity and selectivity. Appl. Catal. B 2018, 224, 919–929. [Google Scholar] [CrossRef]
- Panagiotopoulou, P.; Verykios, X.E. Mechanistic Study of the Selective Methanation of CO over Ru/TiO2 Catalysts: Effect of Metal Crystallite Size on the Nature of Active Surface Species and Reaction Pathways. J. Phys. Chem. C 2017, 121, 5058–5068. [Google Scholar] [CrossRef]
- Agnelli, M.; Swaan, H.M.; Marquez-Alvarez, C.; Martin, G.A.; Mirodatos, C. CO Hydrogenation on a Nickel Catalyst II. A Mechanistic Study by Transient Kinetics and Infrared Spectroscopy. J. Catal. 1998, 175, 117–128. [Google Scholar] [CrossRef]
- Charisiou, N.D.; Siakavelas, G.; Tzounis, L.; Sebastian, V.; Monzon, A.; Baker, M.A.; Hinder, S.J.; Polychronopoulou, K.; Yentekakis, I.V.; Goula, M.A. An in depth investigation of deactivation through carbon formation during the biogas dry reforming reaction for Ni supported on modified with CeO2 and La2O3 zirconia catalysts. Int. J. Hydrog. Energy 2018, 43, 18955–18976. [Google Scholar] [CrossRef]
- Goula, M.A.; Charisiou, N.D.; Siakavelas, G.; Tzounis, L.; Tsiaoussis, I.; Panagiotopoulou, P.; Goula, G.; Yentekakis, I.V. Syngas production via the biogas dry reforming reaction over Ni supported on zirconia modified with CeO2 or La2O3 catalysts. Int. J. Hydrog. Energy 2017, 42, 13724–13740. [Google Scholar] [CrossRef]
- Ashcroft, A.T.; Cheetham, A.K.; Green, M.L.H.; Vernon, P.D.F. Partial oxidation of methane to synthesis gas using carbon dioxide. Nature 1991, 352, 225–226. [Google Scholar] [CrossRef]
- Yentekakis, I.V.; Goula, G.; Panagiotopoulou, P.; Katsoni, A.; Diamadopoulos, E.; Mantzavinos, D.; Delimitis, A. Dry Reforming of Methane: Catalytic Performance and Stability of Ir Catalysts Supported on γ-Al2O3, Zr0.92Y0.08O2−δ (YSZ) or Ce0.9Gd0.1O2−δ (GDC) Supports. Top. Catal. 2015, 58, 1228–1241. [Google Scholar] [CrossRef]
- Yentekakis, I.V.; Goula, G.; Hatzisymeon, M.; Betsi-Argyropoulou, I.; Botzolaki, G.; Kousi, K.; Kondarides, D.I.; Taylor, M.J.; Parlett, C.M.A.; Osatiashtiani, A.; et al. Effect of support oxygen storage capacity on the catalytic performance of Rh nanoparticles for CO2 reforming of methane. Appl. Catal. B 2019, 243, 490–501. [Google Scholar] [CrossRef] [Green Version]
- Yentekakis, I.V.; Vernoux, P.; Goula, G.; Caravaca, A. Electropositive promotion by alkalies or alkaline earths of Pt-group metals in emission control catalysis: A status Report. Catalysts 2019, 9, 157. [Google Scholar] [CrossRef] [Green Version]
- Konsolakis, M.; Drosou, C.; Yentekakis, I.V. Support mediated promotional effects of rare earth oxides (CeO2 and La2O3) on N2O decomposition and N2O reduction by CO or C3H6 over Pt/Al2O3 structured catalysts. Appl. Catal. B 2012, 123–124, 405–413. [Google Scholar] [CrossRef]
- Yentekakis, I.V.; Tellou, V.; Botzolaki, G.; Rapakousios, I.A. A comparative study of the C3H6 + NO + O2, C3H6 + O2 and NO + O2 reactions in excess oxygen over Na-modified Pt/γ-Al2O3 catalysts. Appl. Catal. B 2005, 56, 229–239. [Google Scholar] [CrossRef]
- Vayenas, C.G.; Bebelis, S.; Yentekakis, I.V.; Lintz, H.-G. Non-faradaic electrochemical modification of catalytic activity: A status report. Catal. Today 1992, 11, 303–438. [Google Scholar] [CrossRef]
- Yang, X.-F.; Wang, A.; Qiao, B.; Li, J.; Liu, J.; Zhang, T. Single-atom catalysts: A new frontier in heterogeneous catalysis. Acc. Chem. Res. 2013, 46, 1740–1748. [Google Scholar] [CrossRef]
- Akri, M.; Zhao, S.; Li, X.; Zang, K.; Lee, A.F.; Isaacs, M.A.; Xi, W.; Gangarajula, Y.; Luo, J.; Ren, Y.; et al. Atomically dispersed nickel as coke-resistant active sites for methane dry reforming. Nat. Commun. 2019, 10, 5181. [Google Scholar] [CrossRef] [Green Version]
- Montini, T.; Melchionna, M.; Monai, M.; Forniasiero, P. Fundamentals and Catalytic Applications of CeO2-based Materials. Chem. Rev. 2016, 116, 5987–6041. [Google Scholar] [CrossRef]
- He, H.; Dai, H.X.; Au, C.T. Defective structure, oxygen mobility, oxygen storage capacity, and redox properties of RE-based (RE= Ce, Pr) solid solutions. Catal. Today 2004, 90, 245–254. [Google Scholar] [CrossRef]
- Yentekakis, I.V.; Konsolakis, M. Three-way Catalysis (Book Chapter). In Perovskites and Related Mixed Oxides: Concepts and Applications; Wiley-VCH, Vergal GmbH & Co. KGaA: Weinheim, Germany, 2016; pp. 559–586. [Google Scholar]
- Bunluesin, T.; Gorte, R.J.; Graham, G.W. Studies of the Water-Gas-Shift Reaction on Ceria-Supported Pt, Pd, and Rh: Implications for Oxygen-Storage Properties. Appl. Catal. B 1998, 15, 107–114. [Google Scholar] [CrossRef]
- Yentekakis, I.V.; Goula, G.; Panagiotopoulou, P.; Kampouri, S.; Taylor, M.J.; Kyriakou, G.; Lambert, R.M. Stabilization of catalyst particles against sintering on oxide supports with high oxygen ion lability exemplified by Ir-catalyzed decomposition of N2O. Appl. Catal. B 2016, 192, 357–364. [Google Scholar] [CrossRef]
- Goula, G.; Botzolaki, G.; Osatiashtiani, A.; Parlett, C.M.A.; Kyriakou, G.; Lambert, R.M.; Yentekakis, I.V. Oxidative thermal sintering and redispersion of Rh nanoparticles on supports with high oxygen ion lability. Catalysts 2019, 9, 541. [Google Scholar] [CrossRef] [Green Version]
- Vayenas, C.G.; Brosda, S.; Pliangos, C. The double-layer approach to promotion, electrocatalysis, electrochemical promotion, and metal–support interactions. J. Catal. 2003, 216, 487–504. [Google Scholar] [CrossRef]
- Yentekakis, I.V.; Lambert, R.M.; Konsolakis, M.; Kiousis, V. The effect of sodium on the Pd-catalyzed reduction of NO by methane. Appl. Catal. B 1998, 18, 293–305. [Google Scholar] [CrossRef] [Green Version]
- Fornasiero, P.; Dimonte, R.; Ranga Rao, G.; Kaspar, J.; Meriani, S.; Trovarelli, A.; Graziani, M. Rh-loaded CeO2-ZrO2 solid-solutions as highly efficient oxygen exchangers: Dependence of the reduction behavior and the oxygen storage capacity on the structural properties. J. Catal. 1995, 151, 168–177. [Google Scholar] [CrossRef]
- Ozawa, M.; Takahashi-Morita, M.; Kobayashi, K.; Haneda, M. Core-shell type ceria zirconia support for platinum and rhodium three way catalysts. Catal. Today 2017, 281, 482–489. [Google Scholar] [CrossRef]
- Zhao, B.; Ran, R.; Cao, Y.; Wu, X.; Weng, D.; Fan, J.; Wu, X. Insight into the effects of different ageing protocols on Rh/Al2O3 catalysts. Appl. Surf. Sci. 2014, 308, 230–236. [Google Scholar] [CrossRef]
- Hori, C.E.; Permana, H.; Simon Ng, K.Y.; Brenner, A.; More, K.; Rahmoeller, K.M.; Belton, D. Thermal stability of oxygen storage properties in a mixed CeO2-ZrO2 system. Appl. Catal. B 1998, 16, 105–117. [Google Scholar] [CrossRef]
- Trovarelli, A.; Mustazza, C.; Dolcetti, G. Carbon dioxide hydrogenation on rhodium supported on transition metal oxides. Effect of reduction temperature on product distribution. Appl. Catal. 1990, 65, 129–142. [Google Scholar] [CrossRef]
- Solymosi, F.; Erdöhelyi, A.; Bánsági, T. Methanation of CO2 on supported rhodium catalyst. J. Catal. 1981, 68, 371–382. [Google Scholar] [CrossRef] [Green Version]
- Pliangos, A.; Yentekakis, I.V.; Papadakis, V.G.; Vayenas, C.G.; Verykios, X.E. Support-induced promotional effects on the activity of automotive exhaust catalysts: 1. The case of oxidation of light hydrocarbons (C2H4). Appl. Catal. B 1997, 14, 161–173. [Google Scholar] [CrossRef]
- Vayenas, C.G.; Brosda, S. Electron donation–backdonation and the rules of catalytic promotion. Top. Catal. 2014, 57, 1287–1301. [Google Scholar] [CrossRef]
- Lizarraga, L.; Souentie, S.; Mazri, L.; Billard, A.; Vernoux, P. Investigation of the CO oxidation rate oscillations using electrochemical promotion of catalysis over sputtered-Pt films interfaced with YSZ. Electroch. Commun. 2020, 12, 1310–1313. [Google Scholar] [CrossRef]
- Amsler, J.; Sarma, B.B.; Agostini, G.; Prieto, G.; Plessow, P.N.; Studt, F. Prospects of heterogeneous hydroformylation with supported single atom catalysts. J. Am. Chem. Soc. 2020, 142, 5087–5096. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Supports and Catalysts | Rh Content (wt%) a | SBET (m2/g) | Reducibility Characteristics | Mean Rh Particle Size (nm) b | |
|---|---|---|---|---|---|
| Total OSC (μmol O2/g) | Redox Temperature Region and (Main Peaks) (°C) b | ||||
| Supports | |||||
| γ-Al2O3 | 178 | 0 | - | - | |
| ACZ | 149 | 101 | 400–750 (500, 650) | - | |
| CZ | 22 | 557 | 300–850 (425, 710) | - | |
| Rh nanoparticle Catalysts on γ-Al2O3 | |||||
| Rh(1.2nm)/γ-Al2O3 | |||||
| (fresh) | 1.0 | 160 | 69 | 50–600 (150, 500) | 1.2 |
| Rh(1.6nm)/γ-Al2O3 | |||||
| (treated at 750 °C) | 1.0 | 159 | 70 | 50–600 (175, 400) | 1.6 |
| Rh(2.1nm)/γ-Al2O3 | |||||
| (treated at 850 °C) | 1.0 | 140 | 65 | 50–600 (215, 400) | 2.1 |
| Rh nanoparticle Catalysts on ACZ (ACZ: 80 wt%Al2O3–20 wt%Ce0.5Zr0.5O2-δ) | |||||
| Rh(1.7nm)/ACZ | |||||
| (fresh) | 0.8 | 136 | 146 | 30–550 (75, 150, 450) | 1.7 |
| Rh nanoparticle Catalysts on CZ (CZ: Ce0.5Zr0.5O2-δ) | |||||
| Rh(5nm)/CZ | |||||
| (fresh) | 0.8 | 17 | 589 | 35–600 (105, 175, 380) | 5.0 |
| Rh(2.3nm)/CZ | |||||
| (treated at 750 °C) | 0.8 | 16 | 476 | 30–550 (65, 115, 340) | 2.3 |
| Rh(2.1nm)/CZ | |||||
| (treated at 850 °C) | 0.8 | 15 | 327 | 30–450 (55, 90, 290) | 2.1 |
| Catalyst | Support OSC (μmol O2/g) | YCH4,max (%) | T@YCH4,max (°C) | TOFCH4 Apparent Activation Energy (Ea) (kJ/mol) | Pre-Exponential Factor (s−1) |
|---|---|---|---|---|---|
| Rh(1.6nm)/γ-Al2O3 | 0 | 43.3 | 436 | 70.5 ± 1.9 | 12.4 ± 0.4 |
| Rh(1.7nm)/ACZ | 101 | 46.8 | 402 | 75.0 ± 2.8 | 14.1 ± 0.6 |
| Rh(2.1nm)/CZ | 557 | 14.2 | 388 | 87.5 ± 3.5 | 15.3 ± 0.8 |
| Catalyst | Mean Rh Particle Size (nm) | TOFCH4 Apparent Activation Energy (Ea) (kJ/mol) | Pre-Exponential Factor (s−1) | TOFCH4 at T = 280 °C (s−1) |
|---|---|---|---|---|
| Rh(1.2nm)/γ-Al2O3 | 1.2 | 67.5 ± 1.5 | 12.0 ± 0.3 | 66.5∙10−3 |
| Rh(1.6nm)/γ-Al2O3 | 1.6 | 70.5 ± 1.9 | 12.4 ± 0.4 | 54.0∙10−3 |
| Rh(2.1nm)/γ-Al2O3 | 2.1 | 77.9 ± 0.7 | 13.4 ± 0.2 | 30.0∙10−3 |
| Catalyst | Mean Rh Particle Size (nm) | TOFCH4 Apparent Activation Energy (Ea) (kJ/mol) | Pre-Exponential Factor (s−1) | TOFCH4 at T = 280 °C (s−1) |
|---|---|---|---|---|
| Rh(5nm)/CZ | 5 | 83.6 ± 2.5 | 15.8 ± 0.6 | 93.6∙10−3 |
| Rh(2.3nm)/CZ | 2.3 | 85.8 ± 3.5 | 16.0 ± 0.8 | 66.5∙10−3 |
| Rh(2.1nm)/CZ | 2.1 | 87.5 ± 3.5 | 15.3 ± 0.8 | 27.5∙10−3 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Botzolaki, G.; Goula, G.; Rontogianni, A.; Nikolaraki, E.; Chalmpes, N.; Zygouri, P.; Karakassides, M.; Gournis, D.; Charisiou, N.; Goula, M.; et al. CO2 Methanation on Supported Rh Nanoparticles: The combined Effect of Support Oxygen Storage Capacity and Rh Particle Size. Catalysts 2020, 10, 944. https://0-doi-org.brum.beds.ac.uk/10.3390/catal10080944
Botzolaki G, Goula G, Rontogianni A, Nikolaraki E, Chalmpes N, Zygouri P, Karakassides M, Gournis D, Charisiou N, Goula M, et al. CO2 Methanation on Supported Rh Nanoparticles: The combined Effect of Support Oxygen Storage Capacity and Rh Particle Size. Catalysts. 2020; 10(8):944. https://0-doi-org.brum.beds.ac.uk/10.3390/catal10080944
Chicago/Turabian StyleBotzolaki, Georgia, Grammatiki Goula, Anatoli Rontogianni, Ersi Nikolaraki, Nikolaos Chalmpes, Panagiota Zygouri, Michalis Karakassides, Dimitrios Gournis, Nikolaos Charisiou, Maria Goula, and et al. 2020. "CO2 Methanation on Supported Rh Nanoparticles: The combined Effect of Support Oxygen Storage Capacity and Rh Particle Size" Catalysts 10, no. 8: 944. https://0-doi-org.brum.beds.ac.uk/10.3390/catal10080944