The Role of CO2 as a Mild Oxidant in Oxidation and Dehydrogenation over Catalysts: A Review


1
Department of Chemistry, Inha University, 100 Inharo, Incheon 22212, Korea
2
Evertech Enterprise Co. Ltd., Dongtansandan 2 gil, Hwaseong 18487, Korea
*
Author to whom correspondence should be addressed.
Catalysts 2020, 10(9), 1075; https://0-doi-org.brum.beds.ac.uk/10.3390/catal10091075
Submission received: 28 July 2020
/
Revised: 5 September 2020
/
Accepted: 8 September 2020
/
Published: 17 September 2020
(This article belongs to the Special Issue New Trends in Catalysis for Sustainable CO2 Conversion)
Abstract
:Carbon dioxide (CO2) is widely used as an enhancer for industrial applications, enabling the economical and energy-efficient synthesis of a wide variety of chemicals and reducing the CO2 levels in the environment. CO2 has been used as an enhancer in a catalytic system which has revived the exploitation of energy-extensive reactions and carry chemical products. CO2 oxidative dehydrogenation is a greener alternative to the classical dehydrogenation method. The availability, cost, safety, and soft oxidizing properties of CO2, with the assistance of appropriate catalysts at an industrial scale, can lead to breakthroughs in the pharmaceutical, polymer, and fuel industries. Thus, in this review, we focus on several applications of CO2 in oxidation and oxidative dehydrogenation systems. These processes and catalytic technologies can reduce the cost of utilizing CO2 in chemical and fuel production, which may lead to commercial applications in the imminent future.
1. Introduction
Global warming is an imminent threat to our planet. It is essential to diminish the emission of greenhouse gases, especially carbon dioxide (CO2), to slow global warming. Different sources of CO2 emissions are a significant part to dictate by the ignition of liquid, solid, and gaseous chemicals. Rising atmospheric CO2 concentrations and the increasing temperature of the planet’s surface have increased public awareness of this problem [1]. CO2 is utilized in the in the manufacturing industries, which is mostly released by the combustion of fossil fuels [2]. Among different products, methanol and formic acid can be synthesized from CO2 which is used directly as fuels or to generate H2 on demand at low temperatures (<100 °C) [1]. However, CO2 can be used efficiently in various value-adding strategies and research pursuits which are converted waste emissions into valuable chemicals products, such as hydrocarbons and oxygenates [3]. Electrochemical activation technologies and conversion of CO2 and H2O into hydrocarbons has seen a marked increase in research activity over the past few years [4]. The impressive separation and utilization of CO2 technologies in a higher challenge of organizing than other gases [5]. The development of CO2 for novel approaches can add value to CO2 recycling as it may result in commercially useful carbon-based products. Today, CO2 is used commercially in the production of pharmaceuticals, air-conditioning systems, beverages, fertilizers, inert agents for food packaging, the water treatment process, fire extinguishers, and other applications. To achieve sustainable economic growth, it is crucial to study the conversion of CO2 into carbon-based chemicals and materials. Industrial companies use massive amounts of CO2 to enhance oil restoration. Biomass conversion to fuels also utilizes CO2 [6]. Recently, Drisdell et al. [7] reported that oxide-derived copper catalysts are better at making fuel products from CO2. According to Drisdell group, CO2 is initially converted into carbon monoxide under first conditions for producing fuel and then hydrocarbon chains are developed. Oxide-derived catalysts are better, not because they have oxygen remaining while they reduce carbon monoxide, but because the process of removing the oxygen creates a metallic copper structure that is better at forming ethylene. Using solar energy to convert CO2 into most needed fuels has the potential to decrease global warming impact (GWI) and produce sustainable fuels at large scale [8]. A great deal of research has focused on combining heteroatoms in the carbon structure to improve the exchangeable action of CO2 along with the adsorbent surfaces over the past few years [6].
CO2 utilization has recently become an alluring sector of research, as it will help to alleviate climate change and reduce industrial operating costs. Globally, CO2 capture and utilization are significant goals for chemicals and materials scientists [9]. Researchers are working to diminish the negative effects of CO2 by adsorption [10,11], reduction, and fixation as well as through the development of metal-organic frameworks (MOFs), zeolites, polymers and micro-porous carbons [12]. Currently, CO2 is used in an impenetrable phase under harsh conditions as an active promoter, making it a green substitute for organic compounds [13]. There are several limitations of dense phase CO2 media, including the high pressures required to assure sufficient solubility of various transition metal catalysts and low reaction rates [14]. Jessop et al. proposed, as a solution to the solubility issue, an exchangeable process using 1,8-diazabicyclo-[5.4.0]-undec-7-ene. Additionally, they were able to eliminate partition steps by adjusting polarities with the use of CO2 [15]. Another way to utilize CO2 is to use it as an oxygen source. Park et al. demonstrated the mild oxidant character of CO2 in the oxidative dehydrogenation of various types of alkyl benzene in both liquid and gaseous phases [16,17]. Using CO2 in catalytic reactions offers other advantages; for instance, absorption of hydrogen from alkanes, alkyl aromatics, and alcohols using CO2 as a reactant to create CO and oxygen species results in an expedited reaction rate, increased conversion, higher yield, and suppression of oxidation [16]. The presence of both CO2 and O2 increases the reaction rates as well as the conversion and selectivity. This process is performed under subcritical pressures of CO2 and involves CO2-promoted systems (CPS) instead of a CO2-expanded system, as evidenced by the low-pressure approach as well as catalytic CO2 activation. Recently developed CO2 use technologies require the utilization of high-energy initiators [18]. Although great progress has been made in the carbon dioxide sector, there remain innate limitations, such as high-energy requirements, and the hydrogen recession. CO2 has various benefits as a mild oxidant over several oxidizing promoters tested for oxidative dehydrogenation reaction, such as dry air, SO2, and N2O [19]. C1 products such as methanol, formic acid has become possible to produce with high initial selectivity by using CO2 over simple metal-based catalysts [4]. CO2 promotes selectivity by contaminating the non-selective species of several catalysts, preventing the production of several by-products [20]. Additionally, CO2 is used as a carbon source in the decoking process (C + CO2 = 2CO) which sustains catalytic activity [21]. Therefore, the oxidative dehydrogenation (ODH) reaction with CO2 primarily considered to be a gas-interposed adaptation of the catalyst surface. This affects the diffusion, adsorption, and red-ox characteristics of the catalyst [22]. In recent years, the CO2 conversion process has been utilized in various sectors, including thermo-chemical [23], photochemical [24], solar-chemical [25], electrochemical [26], biochemical [27] and homogenous catalysis [28] (Scheme 1).
In this review, we discuss a way to improve various technologies using CO2 as a mild oxidant and enhancer for the production of essential chemicals. The purpose of this review is to illustrate the limitations and scope of CO2 utilization and to highlight the advantages and challenges of carbon management. The use of CO2 as a feedstock is a major goal, which could have a modest impact in practice, but may impart a significant symbolic effect on worldwide carbon stability. The further impact would result from the use of CO2 as a soft oxidant and for oxidative dehydrogenation in catalytic reactions. Bartholomew et al. [29] studied the oxidizing capability of different gases in the gasification of coke. Their activities were ranked as follows: O2 (105) > H2O (3) > CO2 (1) > H2 (0.003). This demonstrates that carbon dioxide is less active than molecular oxygen and water, but still offers high oxidative capacity. However, carbon dioxide has the greatest heat capability among the commonly used alternative gases. Furthermore, CO2 can reduce the occurrence of hotspots, which cause problems, such as catalyst deactivation, runaway temperature, and undesirable product oxidation.
2. Effect of CO2 in Oxidation
2.1. Influence of CO2 on Oxidation of Cyclohexene
The impact of CO2, at various concentrations, was investigated on the oxidation of cyclohexene which is a small and symmetric molecule, similar to many starting compounds in chemical synthesis (Scheme 2) [30]. The results revealed that O2/CO2 conversion (%) was higher than O2/N2 conversion (%) rate. However, at a gas ratio of 0.066 O2:CO2/N2 (Table 1, entry 1), cyclohexene was not converted. Park et al. revealed the positive impact of carbon dioxide on mesoporous metal-free oxidation carbon nitride (MCN) catalysts [31]. These mesoporous MCN elements exhibit oxygen-carrying capabilities which are effective sites for oxidation. Additionally, the large nitrogen quantity in the CN matrix acts as a CO2-philic exterior for the incitation of CO2. Molecular oxygen promotes this synergy, allowing for the oxidation of cyclic olefins and improving the conversion of cyclic olefins with better selectivity. In-between the conversion of the O2/CO2 and the O2/N2, Park et al. observed the enhancive performance as a premier time, which can be expressed as ΔC (%) and can be calculated using the Equation (1):
where,
The efficiency of CO2 in the oxidation of cyclohexene at varying CO2 concentrations is shown in Table 1. Higher conversions were achieved by the O2/CO2 system. The results showed that the conversion of cyclohexene was nothing at a content of 0.066 O2 (entry 1). This is Possibly due to the low frictional pressure of O2, which is deficient to drive the reaction. Further, the ΔC% value was higher for higher concentrations of CO2. No meaningful change of ΔC% was demonstrated for gas ratios beyond 0.333 in the catalytic process, demonstrating the impregnation of activity.
CO2 has been used with metal-supported systems that were observed to produce a per-oxycarbonate species which are highly active in oxidation reactions. Aresta et al. were reported the composition of a metal per-oxycarbonate species, as determined by spectroscopic analysis [32]. A process for the production of per ox-carbonate has acceded in Scheme 3. Park et al. investigated the oxidation of alkyl aromatics via an EPR analysis using a metal carbonate catalyst. They demonstrated the production of metal per-oxycarbonate groups in the presence of carbon dioxide by the hyperfine cracking of manganese. Yoo et al. [20] observed the production of per-oxycarbonate on Fe/Mo/DBH (deboronated borosilicate molecular sieve); the production of per-oxycarbonate is illustrated in Figure 1. All of the catalytic schemes discussed above involve transitional metal catalysts and CO2 coupled with oxygen. The resulting enhancement over traditional metal oxide systems in O2/CO2 mixtures may occur because of an oxygen exchange between O2 and CO2, which would increase the rate of the reaction. During isotope-labeling studies, these types of exchanges have been detected by Iwata et al. [33] using different metal oxide structures.
2.2. Promotional Effect of CO2 on Oxidation of Cyclic Olefins
Park et al. demonstrated the use of CO2 as a promoter for the oxidation of cyclic olefins with mesoporous carbon nitrides (CN) as a metal-free catalyst in the presence of molecular oxygen. Analysis of the surface characteristics of the catalyst after the reaction revealed the presence of carbamate, confirmed by a new band in the FTIR spectrum at 1419 cm−1. This measurement illustrated the incitation of CO2 owing to the accumulation of surface carbamate. This surface carbamate can then react with the cyclic olefins, assisted by the catalyst. After the reaction, the IR spectra showed the presence of extra bands at 2174 and 2115 cm−1, possibly due to a gaseous CO doublet. However, these absorption bands were not present before the reaction. This analysis exposed the production of CO, which is revealed to the increased catalytic activity to credit to carbon dioxide sharing as an ‘oxygen atom’ onset [31,32]. The production of CO was previously observed in nitrogen including heterocyclic systems [34,35,36]. The positive impact of CO2 in the oxidation of cyclic olefin was quantified by measuring the catalytic performance using various reactants, cyclopentene (n = 1), cyclohexene (n = 2), cyclooctene (n = 4), and cyclododecene (n = 8) (Table 2). The epoxide selectivity was greater in O2/CO2 than O2/N2, suggesting that in the presence of CO2, the mechanism may be altered to improve the conversion and selectivity. The blend of gaseous from the autoclave was studied by IR spectroscopy to better understand the positive impact of CO2. In the reaction with no oxidant and source oxygen, it was presumed that CO2 is reduced to CO and aldehyde is oxidized to carboxylic acid in the same process. The reaction may have occurred via the addition of carbon dioxide to the quickly produced Breslow intermediate A to produce the hydroxy carboxylate B and the tautomer C (Scheme 4) [34]. Possibly, the following intermediate can lose CO and hydroxide to support benzoic acid. Additionally, it was observed that intermediate D is supplicated in the oxidative esterification of aldehydes with CO. Interestingly, phenylglyoxylic acid was revealed to nucleophilic heterocyclic carbenes (NHC) under similar experimental conditions wherein phenylglyoxylic acid was switched to benzoic acid. (Scheme 5). Under mild experimental conditions, CO2 was utilized in an NHC-intermediated conversion of the aldehyde to the carboxylic acid.
2.3. Influence of CO2 on Oxidation of p-Xylene
It was proposed that in the O2-CO2 system, metal peroxy-carbonate groups assist as oxygen transfer promoters to the oxyphilic substrate. Aresta et al. [32] also reported that the presence of O-O bonds in Rh (η2-O2) complexes imply the accumulation of metal per-oxycarbonate during the other oxidation reaction. They demonstrated that CO2 promotes the oxidative ability of O2 over the RhCl(Pet2-Ph)3 catalyst. In the presence of CO2 over the metal-based structure was found to be formation of peroxycarbonate species which are more active than hydrogen peroxide in oxidation reaction [37]. Park et al. [22] reported the performance of carbon dioxide in the liquid-phase oxidation reaction of toluene, p-tolu-aldehyde, and p-xylene with O2 over an MC-based catalyst (Co/Mn/Br). The reaction rate, selectivity, and the conversion were all enhanced by the co-presence of CO2. This enhancement was attributed to the creation of per-oxycarbonate species, as determined by electron paramagnetic resonance (EPR) analysis of the reaction with and without carbon dioxide. A hyperfine manganese arrangement was noticed in the existence of CO2, confirming the formation of a per-oxycarbonate species.
Additionally, Park et al. observed the oxidation of different alkyl aromatics applying MC-supported catalysts [22]. Oxidations were carried out using O2 as the oxidant (with N2) and compared to reactions in the presence of both O2 and CO2. The conversion of p-xylene without CO2 (Table 3) was 57.2%, whereas the conversion of p-xylene was increased to 66.8% in the presence of CO2. Moreover, in O2/CO2, the yield of terephthalic acid was improved. The Amoco Chemical Research Laboratory studied the activation of CO2 in the gas-state of p-xylene oxidation to p-tolualdehyde and terephthaldehyde over the chemical vapor deposition (CVD) of Fe/Mo/DBH [20]. The oxidation reaction was performed in two feed streams varying compositions, including p-xylene with O2/N2/He and p-xylene with O2/N2/CO2. The catalytic activity is shown in both the feeds at various temperatures in Figure 2, as shown in the figure, p-xylene conversion in the existence of CO2 in O2 was greater than the absence of CO2 in O2. This improved conversion was connected to the production of per-oxycarbonate groups over the catalyst surface. Furthermore, in the existence of CO2, the secondary reactions also emerged more remarkable, possibly due to the acidity of the CO2 molecules adsorbed onto the DBH matrix. In comparison with O2 alone, the conversion of p-xylene was higher in the co-presence of CO2 at all temperatures (Figure 2). The O2/N2/CO2 feed system, resulted in a higher conversion of p-xylene and greater selectivity towards benzaldehyde at temperatures from 300 °C to 375 °C (Table 4). It was observed that no carbon dioxide was formed by the burning of p-xylene over the catalyst at 375 °C; however, in the O2/N2/He feed system, the formation of CO2 started (10.7%) at 300 °C and significantly increased (20.2%) at 375 °C. Thus, CO2 performed as a co-oxidant for the gas-phase p-xylene oxidation reaction with oxygen. Yoo et al. [20] also reported a significant enhancement in the conversion of p-xylene, p-ethyl toluene, and o-xylene in the presence of CO2 at varying temperatures.
2.4. Oxidation of p-Toluic Acid and p-Methyl-Anisole
CO2 acts as a promoter in catalytic systems and as a co-oxidant with O2 resulting in improved reaction kinetics, more desirable product distributions, better selectivity, and higher conversion. Initially, Aresta et al. [32] reported that carbon dioxide enhanced the oxidative characteristics of dioxide in transition metal systems. Park et al. [38] studied the use of Co/Mn/Br catalysts in the fluid- phase oxidation of olefins. Interestingly, they observed the expansion effect of carbon dioxide on mesoporous carbon nitride (MCN) catalytic systems, whereas the CO2-promoted system was fabricated by them on the oxidation of alkyl-aromatics. In the presence of CO2, the conversion of p-toluic acid over the metal carbonate (MC) catalyst was increased by 12% (Table 5) compared to oxidation in O2 alone. Furthermore, the yield of terephthalic acid increased from 58.2% to 64.9%. These data demonstrate that the catalytic activity is significantly enhanced by CO2. Interestingly, over an MC-supported catalytic system, the main product of the oxidation of p-methyl-anisole is p-methoxy phenol (Table 6) along with a limited number of other products, such as p-anisaldehyde and p-anisic acid. However, the yield of p-anisaldehyde has increased the presence of CO2, again demonstrating the capacity of CO2 to sustain mono-oxygen transfer.
3. Performance of CO2 in Oxidative Dehydrogenation
3.1. Influence of CO2 on Dehydrogenation of Ethyl Benzene
Styrene is typically formed by the dehydrogenation of ethyl benzene under the steam on a metal oxide catalyst in an adiabatic reactor [39]. There are several limitations to this process, including thermodynamic drawbacks, low conversion rates, high endothermic energy (ΔHo298 = 123.6 kJ mol−1), huge energy destruction, and catalyst deactivation by coke production [40]. An alternative method of styrene production is the oxidative dehydrogenation reaction with O2; however, this results in the burning of large quantities of valuable hydrocarbons. In this context, the use of CO2 in the oxidative dehydrogenation of ethyl benzene may prove useful [39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61]. Zhang et al. [43] confirmed coke deposition using spectroscopy and reported the deactivation of a ceria catalyst without CO2 present. In a two-step, reaction mechanism for the dehydrogenation of ethyl benzene to produce styrene with H2 in the initial step and in the presence of CO2, ejection of H2 through a reverse water-gas shift (RWGS) reaction was also demonstrated [41]. Kovacevic et al. revealed the results of CeO2 catalyst morphology (i.e., rods vs. cubes vs. particles) in the presence and absence of CO2 [42]. They reported that in the presence of CO2 cubic catalysts showed higher initial benzene selectivity, and about two times more activity per m2 compared to the reaction without CO2. Interestingly, the number of oxygen species was increased by the presence of CO2. They also observed that these additional oxygen molecules were expended in the ethyl benzene conversion, demonstrating their performance as active sites for styrene formation. Periyasamy et al. reported that in the ODH reaction the conversion of ethyl benzene (EB) was 50% and the selectivity for styrene was 93% at gas hourly space velocity (GHSV) 2400 h−1. They also observed that the conversion and selectivity increased with enhancing oxidant flow ratio, up to GHSV 2400 h−1.
Park et al. [49] reported on the use of SBA-15 as a beneficial backing for a ceria-zirconium (25:75) combined oxide catalyst for oxidative dehydrogenation of ethyl benzene utilizing carbon dioxide. Ce-Mn oxide nanoparticles enclosed inside carbon nanotubes (CNTs) were used for the oxidative dehydrogenation of ethyl benzene with CO2 acting as a soft oxidant. The high diffusion and the encapsulation effect of CNTs resulted in excellent performance of the entrapped catalysts. Correlated to CeO2 support CNTs, the restriction result of CNT pathways enhanced the communication between carbon nanotube (CNT) inner walls and CeO2 particles, which is orderly, convinced the misrepresentation of CeO2 crystal lattice which is advertised CeO2 reduction and invigoration of CeO2 surface lattice oxygen. The unique process of promoting oxidative catalytic activity the addition of CO2 was reported by Zhang et al. [44]. They observed that multi-walled carbon nanotubes (MWCNTs) have a significant quantity of surface hydroxyl groups which are produced by an alkali-supported hydrothermal method after ball milling. The MWCNTs can mostly arrange the active sites for the oxidative dehydrogenation of ethyl benzene (EB) in the existence of CO2. Figure 3a shows the conversion of ethyl benzene over various types of MWCNTs at 3 hr. The HMWCNTs-OH exhibits significant catalytic activity, indicating that the surface hydroxyl groups are the active sites for the oxidative dehydrogenation of ethyl benzene. The O1s spectra of HMWCNTs-B-OH identified by XPS is shown in Figure 3b. Figure 3c demonstrates the production of carbonyl groups in the reaction. The results indicate that CO2 acts effectively as a soft oxidant, directly oxidizing -OH groups into carbonyl groups. As shown in Figure 3d, CO and H2 were also identified as byproducts for the reaction, indicating that CO2 is reduced in the RWGS reaction. CO2 activation occurs via electron donation from the surface of the catalyst to the anti-bonding orbital of CO2 [62]. However, ethyl benzene (EB) can be activated for oxidative dehydrogenation (ODH) by donating an electron to the acidic portion of the catalyst surface.
Additionally, basic sites abstract hydrogen from ethyl benzene. Thus, the aggregated effect of the basic and acidic sites of the catalyst face is the oxidative dehydrogenation reaction, resulting in high catalytic efficiency in the existence of CO2 [63]. Sato et al. reported on the use of CO2 as a mild oxidant in the oxidative dehydrogenation reaction as well as the typical dehydrogenation process in the absence of CO2. Two mechanisms utilizing acidic and basic sites were proposed, as depicted in Figure 4. Vanadium-embed catalysts also used in CO2 based oxidative dehydrogenation of oxidative dehydrogenation of ethyl benzene (ODHEB) reactions [21,45]. CO2, being a mild oxidant, cannot reproduce the active sites on the V2O5 (001) surface of the catalyst quickly enough due to the large activation energy (3.16 eV) [46]. A ceria-supported vanadium catalyst floated on a titania-zirconia combined oxide (TiO2-ZrO2) has moderate constancy which was reported by Reddy et al. [47]. XPS analysis of Ce 3d indicated the presence of Ce4+ and Ce3+ on the Ti-Zr catalyst. They also reported that CeO2-V2O5/TiO2-ZrO2 (TZ) catalysts resulted in 56% conversion of ethylbenzene and 98% selectivity of styrene. Liu et al. [48] illustrated the red-ox mechanism for the CO2-oxidative dehydrogenation of ethyl benzene (CO2-ODEB) using a ceria promoted vanadium catalyst, as shown in Figure 5. In the CO2-ODEB case, CO2 directly oxidizes Ce3+ to Ce4+, and ethylbenzene reduces of V5+ to V4+. Then, the reduction of Ce4+ to Ce3+ and the oxidation of V4+ to V5+ completes the full cycle. In the existence of CO2, modified vanadium catalysts are effective, selective, and stable for the ODEB, as reported by Park et al. [49] Rapid regeneration of active sites on a silica-assisted vanadium catalyst along in the presence of CO2 has also been reported [64]. 10% La2O3-15%V2O5/SBA-15 (wt.%) catalyst resulted in a 74% styrene yield, with La3+ resisting coke ejection [50]. The use of supporting materials, such as Aluminum mesoporous cylindrical molecular sieve (Al MCM-41) also resulted in substantial EB conversion in the ODEB using a VOx/Al MCM-41 catalyst in the presence of CO2 [51]. ZrO2-containing combined oxide catalysts for oxidative dehydrogenation of ethylbenzene with CO2 in the presence of MnO2, CeO2 and TiO2 have exhibited high activity. The styrene yield was also increased over the MnO2-ZrO2 dual oxide catalyst at a high temperature. Significant enhancement of catalytic activity was checked with increasing CO2/EB ratios [52]. A TiO2-ZrO2 catalyst was used, and the proportion of TiO2/ZrO2 determined the catalytic activity [53,54,55,65]. A 60% titania content resulted the best performance for the ODEB [65,66]. Commercial Fe-supported catalysts are unsuitable for the oxidative dehydrogenation of ethyl benzene in the existence of carbon dioxide due to the atomization of the active catalytic site [56]. However, the use of appropriate dopants’ support materials might enhance the activity by promoting re-oxidation of Fe2+ and preventing coke deposition [57,58]. High product yield stability was observed in a mesoporous silica COK12-assisted CoO3 catalyst [59]. The performance of several effective catalysts in the oxidative dehydrogenation of ethyl benzene to styrene in the existence of CO2 is shown in Table 7.
3.2. Performance of CO2 on Dehydrogenation of Ethane
Ethylene is one of the most prominent raw materials in the chemical industry. Presently, it is used to produce industrial products such as PVC, ethylene glycol, ethylbenzene, ethylene oxide, and vinyl acetate. Commercially, ethylene is formed by steam cracking dehydrogenation of hydrocarbons and fluid catalytic cracking (FCC). These conventional methods have several major limitations including the reaction endothermicity, thermodynamic drawbacks, rapid coke formation, and high energy consumption. The oxidative dehydrogenation of ethane (ODHE) to ethylene in the presence of CO2 as a mild oxidant is an environmentally friendly alternative method for the production of ethylene. A Cr-oxide catalyst with zeolite support was successfully used for the oxidative dehydrogenation of ethane in the presence of CO2 as a soft oxidant. A novel Clinoptilolite-based Cr-oxide (Cr/CLT-IA) catalyst for the ODHE in the existence of CO2 was investigated by Rahamani et al. [70]. This Cr-supported catalyst exhibits high selectivity and catalytic activity which was expected due to its acidity. Homogeneous, tunable smaller Clinoptilolite-based Cr catalyst particles with higher surface area can be generated. Thus, using a Cr/CLT-IA nano-catalyst may be feasible and favorable for the oxidative dehydrogenation of ethane to ethylene in the existence of CO2. Cr/H-ZSM-5 (SiO2/Al2O3 ≥ 190) outperformed the SiO2-based catalyst in the oxidative dehydrogenation of ethane to ethylene with CO2 [71]. CO2 is a promising soft oxidant for the ODHE reaction acting as a channel for transporting heat to the endothermic dehydrogenation. Further, CO2 improves the conversion by modifying alkanes and maintains the catalytic activity by eliminating coke from the catalyst surface. the texture of the Cr active sites and the catalyst activity are determined by the SiO2/Al2O3 ratio. The presence of more alumina amount in the zeolite negatively affected the activity of the catalyst, due to the incorporation of alumina with the Cr into the catalyst structure, affecting the red-ox properties of Cr. Mimura et al. [71] reported on the dehydrogenation of ethane on a Cr-doped HZSM-5 catalyst which is established on the redox phase of the eminent oxidation type Cr species. In their work, C2H6 was absorbed on the acidic site of CrOx and H-ZSM-5. Then, the activated C2H6 reacted with CrOx (active O species) to produce ethylene. The CrOx−1 species is then re-oxidized by the soft oxidant CO2 regenerating the active O species and eliminating coke from the surface of the catalyst. The catalytic performance of the Cr-supported mesoporous catalyst, as well as a Cr-doped silicate MSU-1 catalyst, in the ethane oxidative dehydrogenation to ethylene in the presence of CO2 was reported on by Liu et al. [72]. They initially observed high catalytic activity due to the Cr(VI) active species. However, even in the existence of CO2, the reduction of Cr(VI) to Cr(III) occurred, resulting in the deactivation of the catalyst during the dehydrogenation reaction. Shi et al. [73] reported that Cr-supported Ce/SBA-15 catalysts were comprised of hexagonally ordered mesoporous frameworks and exhibited high catalytic activity in the oxidative dehydrogenation of ethane in the existence of CO2. They confirmed the addition of Ce species using high-angle XRD, which increased the Cr species distribution in the Cr-Ce based SBA-15 zeolite. TPR results determined that Cr species in SBA-15-type zeolites are Cr6+ and Cr3+ groups. Among those two ions, Cr6+ exhibited significant activity for the oxidative dehydrogenation reaction in the existence of CO2. Including a Ce-supported in 5Cr/SBA-15 catalysts modified the red-ox properties and enhanced the activity of the catalyst. Ethane conversion was 55% and ethylene selectivity was 96% on the 5Cr-10Ce/SBA-15 catalyst in the existence of CO2 (Table 8). Cr6+ is reduced to Cr3+ during the oxidative dehydrogenation method reaction, however, in the presence of CO2, Cr3+ is re-oxidized to Cr6+. Cr2O3/ZrO2 supported catalysts with Fe, Co, Mn was also investigated in an effort to fully understand the excellent catalytic activity for the ethane dehydrogenation reaction to ethylene under CO2 treatment [64,65,74]. The Cr6+/Cr3+ red-ox cycle is crucial in the oxidative dehydrogenation reaction, as is a Fe3+/Fe2+ red-ox cycle which was removes H2 from the lattice oxygen. An SBA-15-based, Cr-modified catalyst using a Fe-Cr-Al alloy [75] also exhibited remarkable selectivity of ethylene and high ethane conversion in the oxidative dehydrogenation reaction with CO2. Wang et al. [64] observed the red-ox properties and the acidity/basicity of the Cr-supported catalyst in the oxidative dehydrogenation of ethane to ethylene with CO2. They found that Cr-supported catalysts exhibited different activities in the ODHE with CO2. Cr2O3/SiO2 showed higher ethane conversion and ethylene selectivity. The catalytic activities were ranked as follows Cr/SiO2 > Cr/ZrO2 > Cr/Al2O3 > Cr/TiO2 [76,77]. Notably, Cr2O3 interacted more with Al2O3 than with SiO2, resulting in tetrahedral Cr6+ sites and declining activity [78]. Cr is one of the vital elements of various types of nano-catalysts (Table 9). The active site of these catalysts contains both Cr3+ and Cr6+. The Cr6+/Cr3+ ratio strongly influences the reducibility of Cr/H-ZSM-5 catalysts. The red-ox performance of Cr-supported catalysts is crucial for the oxidative dehydrogenation of ethane to ethylene in the presence of CO2 as a soft oxidant. Cr6+ (or Cr5+) sites are reduced to Cr3+ as ethane is dehydrogenated. Then, the reduced Cr3+ sites are re-oxidized by carbon dioxide treatment. Mimura et al. reported that the highly active Cr-based catalysts had Cr6+ or Cr5+ species on the surface of the catalyst [79]. Apart from Cr-supported catalysts, several other effective catalysts have been used in research on ethane oxidative dehydrogenation. Among these, the Ni-Nb-mixed oxide catalyst performed very well at relatively low temperatures [80,81,82]. Additionally, a TiO2-based Ga catalyst proved applicable for oxidative dehydrogenation with CO2 [83].
Step-1
H3C―CH3 + CrOx ⇄ H2C=CH2 +H2O + CrOx−1 (oxidative dehydrogenation)
Step-2
H3C―CH3 ⇄ H2C=CH2 + H2 (Simple dehydrogenation)
CrOx + H3C―CH3 ⇄ CH4 + C + H2O + CrOx−1 (methane and coke formation)
H3C―CH3 + H2 ⇄ 2CH4 (hydrocracking)
Step-3
CrOx−1 +CO2 ⇄ CrOx + CO (reoxidizing)
C + CO2 ⇄ 2CO
3.3. Influence of CO2 on the Alkylation of Toluene Side-Chain
The dehydrogenation of ethylbenzene produces the most styrene using the Friedel-Crafts alkylation reaction [87]. However, the ethylbenzene dehydrogenation method has some limitations such as catalyst deactivation and, high energy consumption [88,89]. Alkylation of the toluene side-chain is a promising alternative process that uses basic catalysts for the formation of styrene in the existence of CO2. Another process was reported by Sindorenko et al. [90] utilizing K+ and Rb+ ion transposing Faujasite supported catalysts in 1967. However, the catalytic conversion of toluene and styrene monomer (SM) selectivity was low (Table 10) [91]. The side-chain alkylation is primarily carried out on solid base catalysts [92,93,94,95,96]. Toluene side-chain alkylation with methanol enhanced by the promotional use of alkali metal oxides. Greater catalyst acidity accelerates methanol dehydration, [97] while low concentrations of alkali metal ions prevent the decomposition of formaldehyde produced from methanol [98]. Thus, catalysts for this reaction must be optimized for their acidity and basicity [99]. Generally, catalyst sites for the side-chain alkylation are limited to alkali metal-altered zeolites [100]. One reliable, widely studied catalyst is the cesium ion-exchanged or Ce2O-impregnated zeolite-X. The advantages of a MgO-supported mesoporous catalyst for this reaction has also been reported by Park el al. [101]. Hattori et al. observed that the impregnation of Cs2O in ion-exchanged zeolite-X results in high conversion of toluene, owing to the strongly basic sites [102]. Carbon dioxide has been under consideration as a renewable, low-cost, safe, and environmentally beneficial feedstock in current years. CO2 utilization is difficult for commercial applications, owing to its high thermal stability as well as the solid oxidation phase [103]. Hence, remarkable research efforts are being directed to detect innovative technologies for the utilization of CO2. Toluene side-chain alkylation was performed to assess the efficacy of the catalytic approach with methanol over cesium-supported catalysts. Toluene and methanol conversion over the Cs-X and Cs-modified zeolites in the presence of He and CO2 are shown in Table 10. In these reactions, styrene and ethylbenzene were formed as main products. Other side-chain alkylated components, including cumin and α-methyl styrene, as well as other xylenes, tri-methylbenzene, and benzene were identified as by-products. When the catalytic reaction was carried out in the existence of CO2, methanol and toluene conversion increased. Though the styrene selectivity decreased, there was a significant increase in the conversion as well as product selectivity in the presence of He and CO2 streams. TG/DTA analysis of the used Cs-X catalyst in the presence of CO2 and He streams is shown in Figure 6. In the range of 25–200 °C, weight loss occurred owing to the desorption of adsorbed water [94]. The continued weight loss in the 200–450 °C region occurred due to the deposition of coke on the surface of the catalyst. Relatively high quantities of coke were deposited on the Cs-X catalyst in the existence of the CO2. This suggests greater deactivation of the catalyst in the presence of carbon dioxide owing to coke deposition [89]. Still the Cesium-supported catalysts performed better in the presence of CO2 than under He in terms of toluene and methanol conversion. CO2 acted as a significant performance in hydrogen skulking and enhanced the reaction rate in the decisive route. Additionally, CO2 increases alkylation to produce cumin and α-methyl styrene, which are side-chain alkylation products. Further, the increased toluene conversion enhances the aromatic yields.
3.4. Role of CO2 on Dehydrogenation of Propane
Propylene is the most prominent raw material in the chemical industries. It is primarily manufactured by steam cracking and propane oxidative dehydrogenation [104,105,106]. Oxidative dehydrogenation (ODH) is preferred due to its low energy requirements and lack of thermodynamic limitations [107,108]. However, the ODH reaction with O2 occurs under potentially flammable conditions and forms of carbon oxides due to over-oxidation with low selectivity [109,110]. This complication can be resolved using CO2 as a mild, safer oxidant. Thus, this reaction is a favorable example of CO2 utilization. Interestingly, CO2 was used as a mild oxidant to shift the equilibrium more toward the products, as well as enhance the dehydrogenation over the coupling between propane oxidative dehydrogenation to propylene and the reverse water gas shift (RWGS) reaction [111,112,113]. Dehydrogenation of propane occurred on the acid site of the catalyst. The SiO2/Al2O3 proportion is critical in determining both the catalyst physicochemical properties and its reactivity characteristics [114,115,116,117]. The HZSM-5, SBA-15, MCM-41, SBA-1 catalyst which is a two-dimensional microchannel system, has been used in the oxidative dehydrogenation of alkanes especially for the conversion of methane to propane in the existence of CO2. Various research groups have reported on the influence of catalyst acidity in the oxidative dehydrogenation reaction with CO2. The activity of the zeolites decreased with increasing Si/Al proportion in HZSM-5 based Ga2O3, although the selectivity increased, as shown is in Figure 7 [118]. Lewis acidity is present in the metal oxide (Ga2O3) catalyst, while Bronsted acidity is present in HZSM-5. Thus, extracting the aluminum from HZSM-5 declines the Bronsted acidity more than it decreases the Lewis acidity. Several transition metals, such as vanadium, molybdenum, and chromium, have been used to support catalysts for ODH of light alkanes including propane [105,112,119,120]. Among these, chromium oxide provided high catalytic performance with CO2, despite fractional deactivation by coke production. Chromium oxide enhanced propane conversion and the propylene selectivity by expelling H2 produced in the ODH reaction [112]. The catalytic performance of Cr-supported catalysts was observed by the character of chromium categories on the support surface of the catalysts [121,122,123,124,125] Park et al. found that different Cr doping of Cr-TUD-1 catalysts (3, 5, 7 and 9 wt.%) with soft oxidant (CO2) were formed by MW irradiation and investigated the propane oxidative dehydrogenation [126]. The effect of reaction temperature on the oxidative dehydrogenation of propane in the existence of CO2 as a mild oxidant over the Cr-TUD-1 catalyst (7 wt.%) was investigated thoroughly to improve the catalytic activity. The conversion of CO2 was 3.5% at 550 °C and improved to 5.5% at 650 °C. To demonstrate the importance of CO2 in the propane oxidative dehydrogenation on Cr-TUD-1 catalysts, the process was carried out at 550 °C on 7 wt.% catalyst under the same conditions in the presence of CO2 as well as He. The decline in the catalytic activity of the catalyst with helium may be due to coke production and the reduction of the Cr groups on the surface of the zeolite. The proposed mechanism of propane oxidative dehydrogenation over metal oxide surfaces with the CO2 stream is shown below [112]:
A weak exclusive propane adsorption on the lattice oxygen
C3H8 + O* → C3H8O*
C-H schism via H-abstraction from propane utilizing an abutting lattice oxygen
C3H8O* + O* → C3H7O*
Propylene desorption by hybrid expulsion from adsorbed alkoxide groups
C3H7O* → C3H6 + OH*
Reconsolidation of OH groups to produce H2O, reduced metal center (*)
OH* + OH* → H2O + O* + *
Re-oxidation of abridged M-centers by separating chemisorptions of CO2
2CO2 + * + * → 2CO + 2O*
To evaluate the deactivation of the catalyst by coke creation and the enhancement of CO2, (Equation (7)) can be used as the deactivation parameter:
Deactivation parameter (%) = Conversion of propane (initial amount − final amount)/(initial amount) * 100
The rate of Cr degradation by H2 liberated from dehydrogenation is faster than the rate at which CO2 re-oxidizes the degraded Cr species, resulting in catalytic deactivation. Selective adsorption properties can be improved by surface functional groups on activated carbons. Thus, surface treatment of activated carbon may result in more selective and efficient adsorption of the gas, liquid and the alleviation of pollution [127].
![Catalysts 10 01075 g007]()
Figure 7.
Influence of Si/Al proportion on the efficiency of (a) Ga2O3/ZSM-48 zeolites (Reproduced from [118]; copyright (2012), Elsevier), (b) ZnO-HZSM-5 zeolites in the oxidative dehydrogenation of propane along with CO2 (Reproduced from [105]; copyright (2009), Elsevier), (c) Ga2O3/M-HZSM-5 zeolites in the absence of CO2 (Reproduced from [118]; copyright (2012), Elsevier), (d) Influence of Cr substance on the effectiveness of Cr/SBA-15 in the carriage of CO2 (Reproduced from [128]; copyright (2012), Elsevier).
Figure 7.
Influence of Si/Al proportion on the efficiency of (a) Ga2O3/ZSM-48 zeolites (Reproduced from [118]; copyright (2012), Elsevier), (b) ZnO-HZSM-5 zeolites in the oxidative dehydrogenation of propane along with CO2 (Reproduced from [105]; copyright (2009), Elsevier), (c) Ga2O3/M-HZSM-5 zeolites in the absence of CO2 (Reproduced from [118]; copyright (2012), Elsevier), (d) Influence of Cr substance on the effectiveness of Cr/SBA-15 in the carriage of CO2 (Reproduced from [128]; copyright (2012), Elsevier).

4. Conclusions
This review article has comprised a number of CO2 conversions, which are still in the research scale. These promising technologies are mitigating the continuously increasing atmospheric CO2 concentration. Among the methods employing CO2, the ethyl benzene ODH process has seen significant progress. Currently, most of the ethylbenzene dehydrogenation plants apply the oxidative dehydrogenation method, which leads to large heat losses upon compression at the gas–liquid separator. Further, this reaction is thermodynamically restrictive and energy intensive. Several industrial companies such as SABIC (Saudi Basic Industry Corporation, Saudi Arabia), Samsung General Co. in south Korea have tested the catalytic consummation for this method. The commercial implementation of such a process may support the economics of styrene monomer production. According to European Rubber Journal (ERJ), Asahi Kasei Chemical Company ‘s (Japan) 6th generation SBR (Styrene-butadiene rubber) is currently being tested by many customers in the world with positive feedback and company is planning to commercialize some grades in 2021. Moreover, Trinseo’s highly functionalized SPRINTANTM 918S Solution-Styrene Butadiene Rubber (S-SBR) has awarded second position in the elastomers for sustainability initiative of the European Rubber Journal. Based on lab indicator data confirmed by tire customers, grade 918S (compared to non-functionalized high-grip SSBR) improves fuel efficiency of the whole car approximately 1.5%. Considering in Europe alone, the benefit of this increased fuel efficiency would translate in approximately 540 tons less fuel consumed or a reduction of CO2 emissions by 1.3 million tons.
Several methods using CO2 as a mild oxidant have appeared in the technology sector. It is a long-term goal and alluring dream for chemical engineers to establish commercial industries based on the utilization of CO2. Challenges for the commercial utilization of this technology include the process rate required to ensure CO2 conversion with low coke deposition, the need to decrease energy expenditure, and the need for improved catalysts offering higher conversion. Despite the challenges, there is great room for catalyst improvement in these sectors. Recently, the carbon XPRIZE is a $20 million competition to capture and CO2 conversion which is jointly funded by COSIA (Canada’s Oil Sends Innovation Alliance) [129]. Most of the countries’ governments are concern about climate changes with a high priority. China, the world’s largest energy consumer and carbon emitter, announced USD 360 billion in renewable energy investments by 2020 to reduce carbon emissions [130]. Canada has implemented federally a carbon pricing policy with a current tax of USD 10/ton CO2 and a steady rise to USD 50/ton CO2 nationwide by 2022. However, the positive effects of CO2 in benzene hydroxylation over commercial and hierarchical zeolites in the liquid phase as well as the gas phase are under investigation by our group, wherein the byproducts are various aromatic compounds. The recycling of CO2 from the atmosphere to fuels, chemicals will lead to a real sustainable future for humanity. We expect that the use of CO2 as a promoter and as a mild oxidant at the laboratory level can be translated to the industrial scale in the future, thus contributing also to the world economy.
Author Contributions
S.T.R.: Writing original draft; J.-R.C.: Editing; J.-H.L.: Editing; S.-J.P.: Writing review & editing. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Technology Innovation Program (or Industrial Strategic Technology Development Program-Development of technology on materials and components) (20010881, Development of ACF for rigid (COG)/ flexible (COP) and secured mass production by developing core material technology for localizing latent hardener for low temperature fast curing) funded By the Ministry of Trade, Industry & Energy (MOTIE, Korea) and supported by Korea Evaluation institute of Industrial Technology (KEIT) through the Carbon Cluster Construction project [10083586, Development of petroleum based graphite fibers with ultra-high thermal conductivity] funded by the Ministry of Trade, Industry & Energy (MOTIE, Korea).
Conflicts of Interest
The authors declare that they have no conflicts of interest.
References
- Kar, S.; Goeppert, A.; Prakash, G.K.S. Integrated CO2 Capture and Conversion to Formate and Methanol: Connecting Two Threads. Acc. Chem. Res. 2019, 52, 2892–2903. [Google Scholar] [CrossRef] [PubMed]
- Smol, J.P. Climate Change: A Planet in flux. Nature 2012, 483, 12–15. [Google Scholar] [CrossRef] [PubMed]
- Ross, M.B.; De Luna, P.; Li, Y.; Dinh, C.-T.; Kim, D.; Yang, P.; Sargent, E.H. Designing materials for electrochemical carbon dioxide recycling. Nat. Catal. 2019, 2, 648–658. [Google Scholar] [CrossRef] [Green Version]
- De Luna, P.; Hahnet, C.; Higgins, D.; Jaffer, S.A.; Jaramillo, T.F.; Sargent, E.H. What would it take for renewably powered electrosynthesis to displace petrochemical processes? Science 2019, 364. [Google Scholar] [CrossRef] [Green Version]
- Nugent, P.; Belmabkhout, Y.; Burd, S.D.; Cairns, A.J.; Luebke, R.; Forrest, K.; Pham, T.; Ma, S.; Space, B.; Wojtas, L.; et al. Porous materials with optimal adsorption thermodynamics and kinetics for CO2 separation. Nature 2013, 495, 80–84. [Google Scholar] [CrossRef]
- Rehman, A.; Park, S.-J. From chitosan to urea-modified carbons: Tailoring the ultra-microporosity for enhanced CO2 adsorption. Carbon 2020, 159, 625–637. [Google Scholar] [CrossRef]
- Lee, S.H.; Sullivan, I.; Larson, D.M.; Liu, G.; Toma, F.M.; Xiang, C.; Drisdell, W.S. Correlating Oxidation State and Surface Area to Activity from Operando Studies of Copper CO Electroreduction Catalysts in a Gas Fed Device. ACS Catal. 2020, 10, 8000–8011. [Google Scholar] [CrossRef]
- Han, L.; Zhou, W.; Xiang, C. High-Rate Electrochemical Reduction of Carbon Monoxide to Ethylene Using Cu-Nanoparticles Based Gas Diffusion Electrodes. ACS Energy Lett. 2018, 3, 855–860. [Google Scholar] [CrossRef] [Green Version]
- Heo, Y.J.; Park, S.-J. Facile Synthesis of MgO-Modified Carbon Adsorbents with Microwave-Assisted Methods: Effect of MgO Particles and Porosities on CO2 Capture. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Qi, S.-C.; Wu, J.-K.; Lu, J.; Yu, G.-X.; Zhu, R.R.; Liu, Y.; Liu, X.-Q.; Sun, L.-B. Underlying mechanism of CO2 adsorption onto conjugated azacyclo-copolymers: N-doped adsorbents capture CO2 chiefly through acid-base interaction? J. Mater. Chem. A 2019, 7, 17842–17853. [Google Scholar] [CrossRef]
- Qi, S.C.; Liu, Y.; Peng, A.Z.; Xue, D.M.; Liu, X.; Liu, X.Q.; Sun, L.B. Fabrication of porous carbons from mesitylene for highly efficient CO2 capture: A rational choice improving the carbon loop. Chem. Eng. J. 2019, 361, 945–952. [Google Scholar] [CrossRef]
- Rehman, A.; Park, S.-J. Tunable nitrogen-doped microporous carbons: Delineating the role of optimum pore size for enhanced CO2 adsorption. Chem. Eng. J. 2019, 362, 731–742. [Google Scholar] [CrossRef]
- Jessop, P.G.; Ikariya, T.; Noyori, R. Homogeneous catalysis in supercritical fluids. Science 1995, 269, 1065–1069. [Google Scholar] [CrossRef]
- Musie, G.; Wei, M.; Subramaniam, B.; Busch, D.H. Catalytic oxidations in carbon dioxide-based reaction media, including novel CO2-expanded phases. Coord. Chem. Rev. 2001, 219–221, 789–820. [Google Scholar] [CrossRef]
- Heldebrant, D.J.; Jessop, P.G.; Thomas, C.A.; Eckert, C.A.; Liotta, C.L. The reaction of 1,8-diazabicyclo [5.4.0]undec-7-ene (DBU) with carbon dioxide. J. Org. Chem. 2005, 70, 5335–5338. [Google Scholar] [CrossRef]
- Chang, J.S.; Vislovskiy, V.P.; Park, M.S.; Hong, D.Y.; Yoo, J.S.; Park, S.E. Utilization of carbon dioxide as soft oxidant in the dehydrogenation of ethyl benzene over supported vanadium-antimony oxide catalysts. Green Chem. 2003, 5, 587–590. [Google Scholar] [CrossRef]
- Park, M.S.; Chang, J.S.; Kim, D.S.; Park, S.-E. Oxidative dehydrogenation of ethyl benzene with carbon dioxide over zeolite-supported iron oxide catalysts. Res. Chem. Intermed. 2002, 28, 461–469. [Google Scholar] [CrossRef]
- Sakakura, T.; Choi, J.C.; Yasuda, H. Transformation of carbon dioxide. Chem. Rev. 2007, 107, 2365–2387. [Google Scholar] [CrossRef]
- Reddy, B.M.; Lakshmanan, P.; Loridant, S.; Yamada, Y.; Kobayashi, T.; López-Cartes, C.; Rojas, T.C.; Fernández, A. Structural characterization and oxidative dehydrogenation Activity of v2O5/CexZr1-xO2/SiO2 catalysts. J. Phys. Chem. B 2006, 110, 9140–9147. [Google Scholar] [CrossRef]
- Yoo, J.S.; Lin, P.S.; Elfline, S.D. Gas-phase oxygen oxidations of alkyl aromatics over CVD Fe/Mo/borosilicate molecular sieve. II. The role of carbon dioxide as a co-oxidant. Appl. Catal. A Gen. 1993, 106, 259–273. [Google Scholar] [CrossRef]
- Sun, A.; Qin, Z.; Wang, J. Reaction coupling of ethylbenzene dehydrogenation with water-gas shift. Appl. Catal. A Gen. 2002, 234, 179–189. [Google Scholar] [CrossRef]
- Ansari, M.B.; Park, S.E. Carbon dioxide utilization as a soft oxidant and promoter in catalysis. Energy Environ. Sci. 2012, 5, 9419–9437. [Google Scholar] [CrossRef]
- Abanades, S.; Le Gal, A. CO2 splitting by thermo-chemical looping based on ZrxCe1-xO2 oxygen carriers for synthetic fuel generation. Fuel 2012, 102, 180–186. [Google Scholar] [CrossRef]
- Wang, S.; Wang, X. Imidazolium ionic liquids, imidazolylidene heterocyclic carbenes, and zeolitic imidazolate frameworks for CO2 capture and photochemical reduction. Angew. Chemie Int. Ed. 2016, 55, 2308–2320. [Google Scholar] [CrossRef] [PubMed]
- Nikulshina, V.; Hirsch, D.; Mazzotti, M.; Steinfeld, A. CO2 capture from air and co-production of H2 via the Ca(OH)2-CaCO3 cycle using concentrated solar power-Thermodynamic analysis. Energy 2006, 31, 1715–1725. [Google Scholar] [CrossRef]
- Spinner, N.S.; Vega, J.A.; Mustain, W.E. Recent progress in the electrochemical conversion and utilization of CO2. Catal. Sci. Technol. 2012, 2, 19–28. [Google Scholar] [CrossRef]
- Singh, G.; Lakhi, K.S.; Ramadass, K.; Sathish, C.I.; Vinu, A. High-Performance Biomass-Derived Activated Porous Biocarbons for Combined Pre- and Post-Combustion CO2 Capture. ACS Sustain. Chem. Eng. 2019, 7, 7412–7420. [Google Scholar] [CrossRef]
- Grice, K.A. Carbon dioxide reduction with homogenous early transition metal complexes: Opportunities and challenges for developing CO2 catalysis. Coord. Chem. Rev. 2017, 336, 78–95. [Google Scholar] [CrossRef]
- Bartholomew, C.H. Catalyst Deactivation 1991; Elsevier: Amsterdam, The Netherlands, 1991; pp. 96–112. [Google Scholar]
- Denekamp, I.M.; Antens, M.; Slot, T.K.; Rothenberg, G. Selective Catalytic Oxidation of Cyclohexene with Molecular Oxygen: Radical Versus Nonradical Pathways. ChemCatChem 2018, 10, 1035–1041. [Google Scholar] [CrossRef]
- Ansari, M.B.; Min, B.H.; Mo, Y.H.; Park, S.-E. CO2 activation and promotional effect in the oxidation of cyclic olefins over mesoporous carbon nitrides. Green Chem. 2011, 13, 1416–1421. [Google Scholar] [CrossRef]
- Aresta, M.; Tommasi, I.; Quaranta, E.; Fragale, C.; Tranquille, M.; Galan, F.; Fouassier, M. Mechanism of formation of peroxycarbonates RhOOC(O)O(Cl)(P)3 and Their Reactivity as Oxygen Transfer Agents Mimicking Monooxygenases. The First Evidence of CO2 Insertion into the O-O Bond of Rh(η2-O2) Complexes. Inorg. Chem. 1996, 35, 4254–4260. [Google Scholar] [CrossRef] [PubMed]
- Iwata, R.; Ido, T.; Fujisawa, Y.; Yamazaki, S. On-line interconversion of [15O]O2 and [15O]CO2 via metal oxide by isotopic exchange. Int. J. Radiat. Appl. Instrum. Part A. 1988, 39, 1207–1211. [Google Scholar] [CrossRef]
- Nair, V.; Varghese, V.; Paul, R.R.; Jose, A.; Sinu, C.R.; Menon, R.S. NHC catalyzed transformation of aromatic aldehydes to acids by carbon dioxide: An unexpected reaction. Org. Lett. 2010, 12, 2653–2655. [Google Scholar] [CrossRef]
- Chiang, P.C.; Bode, J.W. On the role of CO2 in NHC-catalyzed oxidation of aldehydes. Org. Lett. 2011, 13, 2422–2425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, L.; Zhang, Y. Unexpected CO2 splitting reactions to form CO with N-heterocyclic carbenes as organocatalysts and aromatic aldehydes as oxygen acceptors. J. Am. Chem. Soc. 2010, 132, 914–915. [Google Scholar] [CrossRef]
- Lane, B.S.; Vogt, M.; De Rose, V.J.; Kevin, B. Manganese-Catalyzed Epoxidations of Alkenes in Bicarbonate Solutions. J. Am. Chem. Soc. 2002, 124, 11946–11954. [Google Scholar] [CrossRef]
- Park, S.-E.; Yoo, J.S. Studies in Surface Science and Catalysis 153; Elsevier: Amsterdam, The Netherlands, 2004; pp. 303–314. [Google Scholar]
- Cavani, F.; Trifiro, F. Alternative Processes for the Production of Sponge Iron. Appl. Catal. A Gen. 1995, 133, 219–239. [Google Scholar] [CrossRef]
- Li, X.; Li, W. Effect of TiO2 loading on the activity of V/TiO2-Al2O3 in the catalytic oxidehydrogenation of ethylbenzene with carbon dioxide. Front. Chem. Eng. China 2010, 4, 142–146. [Google Scholar] [CrossRef]
- Nowicka, E.; Reece, C.; Althahban, S.M.; Mohammed, K.M.H.; Kondrat, S.A.; Morgan, D.J.; He, Q.; Willock, D.J.; Golunski, S.; Kiely, C.J.; et al. Elucidating the Role of CO2 in the Soft Oxidative Dehydrogenation of Propane over Ceria-Based Catalysts. ACS Catal. 2018, 8, 3454–3468. [Google Scholar] [CrossRef] [Green Version]
- Kovacevic, M.; Agarwal, S.; Mojet, B.L.; Van Ommen, J.G.; Lefferts, L. The effects of morphology of cerium oxide catalysts for dehydrogenation of ethylbenzene to styrene. Appl. Catal. A Gen. 2015, 505, 354–364. [Google Scholar] [CrossRef]
- Rao, R.; Zhang, Q.; Liu, H.; Yang, H.; Ling, Q.; Yang, M.; Zhang, A.; Chen, W. Enhanced catalytic performance of CeO2 confined inside carbon nanotubes for dehydrogenation of ethylbenzene in the presence of CO2. J. Mol. Catal. A Chem. 2012, 363–364, 283–290. [Google Scholar] [CrossRef]
- Rao, R.; Yang, M.; Ling, Q.; Li, C.; Zhang, Q.; Yang, H.; Zhang, A. A novel route of enhancing oxidative catalytic activity: Hydroxylation of MWCNTs induced by sectional defects. Catal. Sci. Technol. 2014, 4, 665–671. [Google Scholar] [CrossRef]
- Sakurai, Y.; Suzaki, T.; Ikenaga, N.O.; Suzuki, T. Dehydrogenation of ethylbenzene with an activated carbon-supported vanadium catalyst. Appl. Catal. A Gen. 2000, 192, 281–288. [Google Scholar] [CrossRef]
- Fan, H.; Feng, J.; Li, X.; Guo, Y.; Li, W.; Xie, K. Ethylbenzene dehydrogenation to styrene with CO2 over V2O5(001): A periodic density functional theory study. Chem. Eng. Sci. 2015, 135, 403–411. [Google Scholar] [CrossRef]
- Rao, K.N.; Reddy, B.M.; Abhishek, B.; Seo, Y.H.; Jiang, N.; Park, S.E. Effect of ceria on the structure and catalytic activity of V2O5/TiO2-ZrO2 for oxidehydrogenation of ethylbenzene to styrene utilizing CO2 as soft oxidant. Appl. Catal. B Environ. 2009, 91, 649–656. [Google Scholar] [CrossRef]
- Liu, Z.W.; Wang, C.; Fan, W.B.; Liu, Z.T.; Hao, Q.Q.; Long, X.; Lu, J.; Wang, J.G.; Qin, Z.F.; Su, D.S. V2O5/Ce0.6Zr0.4O2-Al2O3 as an efficient catalyst for the oxidative dehydrogenation of ethylbenzene with carbon dioxide. ChemSusChem 2011, 4, 341–345. [Google Scholar] [CrossRef]
- Burri, A.; Jiang, N.; Ji, M.; Park, S.-E.; Khalid, Y. Oxidative dehydrogenation of ethylbenzene to styrene with CO2 over V2O5-Sb2O5-CeO2/TiO2-ZrO2 catalysts. Top. Catal. 2013, 56, 1724–1730. [Google Scholar] [CrossRef]
- Liu, B.S.; Rui, G.; Chang, R.Z.; Au, C.T. Dehydrogenation of ethylbenzene to styrene over LaVOx/SBA-15 catalysts in the presence of carbon dioxide. Appl. Catal. A Gen. 2008, 335, 88–94. [Google Scholar] [CrossRef]
- Li, Z.; Su, K.; Cheng, B.; Shen, D.; Zhou, Y. Effects of VOx /AlMCM-41 surface structure on ethyl benzene oxydehydrogenation in the presence of CO2. Catal. Lett. 2010, 135, 135–140. [Google Scholar] [CrossRef]
- Jiang, N.; Burri, A.; Park, S.-E. Ethylbenzene to styrene over ZrO2-based mixed metal oxide catalysts with CO2 as soft oxidant. Chin. J. Catal. 2016, 37, 3–15. [Google Scholar] [CrossRef]
- Reddy, B.M.; Khan, A. Recent advances on TiO2-ZrO2 mixed oxides as catalysts and catalyst supports. Catal. Rev. Sci. Eng. 2005, 47, 257–296. [Google Scholar] [CrossRef]
- Jiang, N.; Han, D.S.; Park, S.-E. Direct synthesis of mesoporous silicalite-1 supported TiO2-ZrO2 for the dehydrogenation of EB to styrene with CO2. Catal. Today 2009, 141, 344–348. [Google Scholar] [CrossRef]
- Manríquez, M.E.; López, T.; Gómez, R.; Navarrete, J. Preparation of TiO2-ZrO2 mixed oxides with controlled acid-basic properties. J. Mol. Catal. A Chem. 2004, 220, 229–237. [Google Scholar] [CrossRef]
- Zangeneh, F.T.; Sahebdelfar, S.; Ravanchi, M.T. Conversion of carbon dioxide to valuable petrochemicals: An approach to clean development mechanism. J. Nat. Gas Chem. 2011, 20, 219–231. [Google Scholar] [CrossRef]
- Balasamy, R.J.; Tope, B.B.; Khurshid, A.; Al-Ali, A.A.S.; Atanda, L.A.; Sagata, K.; Asamoto, M.; Yahiro, H.; Nomura, K.; Sano, T.; et al. Ethylbenzene dehydrogenation over FeOx/(Mg,Zn)(Al)O catalysts derived from hydrotalcites: Role of MgO as basic sites. Appl. Catal. A Gen. 2011, 398, 113–122. [Google Scholar] [CrossRef]
- Braga, T.P.; Pinheiro, A.N.; Teixeira, C.V.; Valentini, A. Dehydrogenation of ethylbenzene in the presence of CO2 using a catalyst synthesized by polymeric precursor method. Appl. Catal. A Gen. 2009, 366, 193–200. [Google Scholar] [CrossRef]
- Pochamoni, R.; Narani, A.; Varkolu, M.; Dhar Gudimella, M.; Prasad Potharaju, S.S.; Burri, D.R.; Rao Kamaraju, S.R. Studies on ethylbenzene dehydrogenation with CO2 as soft oxidant over Co3O4/COK-12 catalysts. J. Chem. Sci. 2015, 127, 701–709. [Google Scholar] [CrossRef] [Green Version]
- Zhao, G.; Chen, H.; Li, J.; Wang, Q.; Wang, Y.; Ma, S.; Zhu, Z. Acid-based co-catalysis for oxidative dehydrogenation of ethylbenzene to styrene with CO2 over X zeolite modified by alkali metal cation exchange. RSC Adv. 2015, 5, 75787–75793. [Google Scholar] [CrossRef]
- Periyasamy, K.; Aswathy, V.T.; Ashok Kumar, V.; Manikandan, M.; Shukla, R.; Tyagi, A.K.; Raja, T. An efficient robust fluorite CeZrO4-δ oxide catalyst for the eco-benign synthesis of styrene. RSC Adv. 2015, 5, 3619–3626. [Google Scholar] [CrossRef]
- Mukherjee, D.; Park, S.-E.; Reddy, B.M. CO2 as a soft oxidant for oxidative dehydrogenation reaction: An eco benign process for industry. J. CO2 Util. 2016, 16, 301–312. [Google Scholar] [CrossRef]
- Sato, S.; Ohhara, M.; Sodesawa, T.; Nozaki, F. Combination of ethylbenzene dehydrogenation and carbon dioxide shift-reaction over a sodium oxide/alumina catalyst. Appl. Catal. 1988, 37, 207–215. [Google Scholar] [CrossRef]
- Chen, S.; Qin, Z.; Wang, G.; Dong, M.; Wang, J. Promoting effect of carbon dioxide on the dehydrogenation of ethylbenzene over silica-supported vanadium catalysts. Fuel 2013, 109, 43–48. [Google Scholar] [CrossRef]
- Burri, D.R.; Choi, K.M.; Han, S.C.; Burri, A.; Park, S.-E. Dehydrogenation of ethylbenzene to styrene with CO2 over TiO2-ZrO2 bifunctional catalyst. Bull. Korean Chem. Soc. 2007, 28, 53–58. [Google Scholar]
- Burri, D.R.; Choi, K.M.; Han, S.C.; Burri, A.; Park, S.-E. Selective conversion of ethylbenzene into styrene over K2O/TiO2-ZrO2 catalysts: Unified effects of K2O and CO2. J. Mol. Catal. A Chem. 2007, 269, 58–63. [Google Scholar] [CrossRef]
- Burri, A.; Jiang, N.; Park, S.-E. High surface area TiO2-ZrO2 prepared by caustic solution treatment, and its catalytic efficiency in the oxidehydrogenation of para-ethyl toluene by CO2. Catal. Sci. Technol. 2012, 2, 514–520. [Google Scholar] [CrossRef]
- Burri, D.R.; Choi, K.M.; Han, D.S.; Sujandi; Jiang, N.; Burri, A.; Park, S.-E. Oxidative dehydrogenation of ethylbenzene to styrene with CO2 over SnO2-ZrO2 mixed oxide nanocomposite catalysts. Catal. Today 2008, 131, 173–178. [Google Scholar] [CrossRef]
- Burri, D.R.; Choi, K.M.; Han, D.S.; Koo, J.B.; Park, S.-E. CO2 utilization as an oxidant in the dehydrogenation of ethylbenzene to styrene over MnO2-ZrO2 catalysts. Catal. Today 2006, 115, 242–247. [Google Scholar] [CrossRef]
- Rahmani, F.; Haghighi, M.; Amini, M. The beneficial utilization of natural zeolite in preparation of Cr/clinoptilolite nanocatalyst used in CO2-oxidative dehydrogenation of ethane to ethylene. J. Ind. Eng. Chem. 2015, 31, 142–155. [Google Scholar] [CrossRef]
- Mimura, N.; Takahara, I.; Inaba, M.; Okamoto, M.; Murata, K. High-performance Cr/H-ZSM-5 catalysts for oxidative dehydrogenation of ethane to ethylene with CO2 as an oxidant. Catal. Commun. 2002, 3, 257–262. [Google Scholar] [CrossRef]
- Liu, L.; Li, H.; Zhang, Y. A comparative study on catalytic performances of chromium incorporated and supported mesoporous MSU-x catalysts for the oxidehydrogenation of ethane to ethylene with carbon dioxide. Catal. Today 2006, 115, 235–241. [Google Scholar] [CrossRef]
- Shi, X.; Ji, S.; Wang, K. Oxidative dehydrogenation of ethane to ethylene with carbon dioxide over Cr-Ce/SBA-15 catalysts. Catal. Lett. 2008, 125, 331–339. [Google Scholar] [CrossRef]
- Deng, S.; Li, S.; Li, H.; Zhang, Y. Oxidative dehydrogenation of ethane to ethylene with CO2 over Fe-Cr/ZrO2 catalysts. Ind. Eng. Chem. Res. 2009, 48, 7561–7566. [Google Scholar] [CrossRef]
- Shi, X.; Ji, S.; Li, C. Oxidative dehydrogenation of ethane with CO2 over novel Cr/SBA-15/Al2O3/FeCrAl monolithic catalysts. Energy Fuels 2008, 22, 3631–3638. [Google Scholar] [CrossRef]
- Tedeeva, M.A.; Kustov, A.L.; Pribytkov, P.V.; Leonov, A.V.; Dunaev, S.F. Dehydrogenation of Propane with CO2 on Supported CrOx/SiO2 Catalysts. Russ. J. Phys. Chem. A 2018, 92, 2403–2407. [Google Scholar] [CrossRef]
- Wang, S.; Murata, K.; Hayakawa, T.; Hamakawa, S.; Suzuki, K. Dehydrogenation of ethane with carbon dioxide over supported chromium oxide catalysts. Appl. Catal. A Gen. 2000, 196, 1–8. [Google Scholar] [CrossRef]
- Weckhuysen, B.M.; Schoonheydt, R.A.; Jehng, J.M.; Wachs, I.E.; Cho, S.J.; Ryoo, R.; Kijlstra, S.; Poels, E. Combined DRS-RS-EXAFS-XANES-TPR study of supported chromium catalysts. J. Chem. Soc. Faraday Trans. 1995, 91, 3245–3253. [Google Scholar] [CrossRef] [Green Version]
- Mimura, N.; Okamoto, M.; Yamashita, H.; Oyama, S.T.; Murata, K. Oxidative dehydrogenation of ethane over Cr/ZSM-5 catalysts using CO2 as an oxidant. J. Phys. Chem. B 2006, 110, 21764–21770. [Google Scholar] [CrossRef]
- Heracleous, E.; Lemonidou, A.A. Ni-Nb-O mixed oxides as highly active and selective catalysts for ethene production via ethane oxidative dehydrogenation. Part I: Characterization and catalytic performance. J. Catal. 2006, 237, 162–174. [Google Scholar] [CrossRef]
- Heracleous, E.; Lemonidou, A.A. Ni-Nb-O mixed oxides as highly active and selective catalysts for ethene production via ethane oxidative dehydrogenation. Part II: Mechanistic aspects and kinetic modeling. J. Catal. 2006, 237, 175–189. [Google Scholar]
- Heracleous, E.; Delimitis, A.; Nalbandian, L.; Lemonidou, A.A. HRTEM characterization of the nanostructural features formed in highly active Ni-Nb-O catalysts for ethane ODH. Appl. Catal. A Gen. 2007, 325, 220–226. [Google Scholar] [CrossRef]
- Koirala, R.; Buechel, R.; Krumeich, F.; Pratsinis, S.E.; Baiker, A. Oxidative dehydrogenation of ethane with CO2 over flame-made Ga-loaded TiO2. ACS Catal. 2015, 5, 690–702. [Google Scholar] [CrossRef]
- Cheng, Y.; Zhang, F.; Zhang, Y.; Miao, C.; Hua, W.; Yue, Y.; Gao, Z. Oxidative dehydrogenation of ethane with CO2 over Cr supported on submicron ZSM-5 zeolite. Chinese J. Catal. 2015, 36, 1242–1248. [Google Scholar] [CrossRef]
- Ramesh, Y.; Thirumala Bai, P.; Hari Babu, B.; Lingaiah, N.; Rama Rao, K.S.; Prasad, P.S.S. Oxidative dehydrogenation of ethane to ethylene on Cr2O3/Al2O3–ZrO2 catalysts: The influence of oxidizing agent on ethylene selectivity. Appl. Petrochem. Res. 2014, 4, 247–252. [Google Scholar] [CrossRef] [Green Version]
- Deng, S.; Li, H.; Li, S.; Zhang, Y. Activity and characterization of modified Cr2O3/ZrO2 nano-composite catalysts for oxidative dehydrogenation of ethane to ethylene with CO2. J. Mol. Catal. A Chem. 2007, 268, 169–175. [Google Scholar] [CrossRef]
- Yashima, T.; Sato, K.; Hayasaka, T.; Hara, N. Alkylation on synthetic zeolites. III. Alkylation of toluene with methanol and formaldehyde on alkali cation exchanged zeolites. J. Catal. 1972, 26, 303–312. [Google Scholar] [CrossRef]
- Rossetti, I.; Bencini, E.; Trentini, L.; Forni, L. Study of the deactivation of a commercial catalyst for ethylbenzene dehydrogenation to styrene. Appl. Catal. A Gen. 2005, 292, 118–123. [Google Scholar] [CrossRef]
- Meima, G.R.; Menon, P.G. Catalyst deactivation phenomena in styrene production. Appl. Catal. A Gen. 2001, 212, 239–245. [Google Scholar] [CrossRef]
- Sindorenko, L.N.; Galich, P.N.; Gutirya, V.S. Condensation of toluene and methanol upon synthetic zeolites containing ion-exchange cations of alkali metals. Dokl. Akad. Nauk. SSSR 1967, 173, 132–133. [Google Scholar]
- Seo, D.W.; Rahman, S.T.; Reddy, B.M.; Park, S.-E. Carbon dioxide assisted toluene side-chain alkylation with methanol over Cs-X zeolite catalyst. J. CO2 Util. 2018, 26, 254–261. [Google Scholar] [CrossRef]
- Hattori, H. Solid base catalysts: Fundamentals and their applications in organic reactions. Appl. Catal. A Gen. 2015, 504, 103–109. [Google Scholar] [CrossRef]
- Yoo, K.S.; Smirniotis, P.G. Zeolites-catalyzed alkylation of isobutane with 2-butene: Influence of acidic properties. Catal. Lett. 2005, 103, 249–255. [Google Scholar] [CrossRef]
- Alabi, W.O.; Tope, B.B.; Jermy, R.B.; Aitani, A.M.; Hattori, H.; Al-Khattaf, S.S. Modification of Cs-X for styrene production by side-chain alkylation of toluene with methanol. Catal. Today 2014, 226, 117–123. [Google Scholar] [CrossRef]
- Yoo, K.; Smirniotis, P.G. The deactivation pathway of one-dimensional zeolites, LTL and ZSM-12, for alkylation of isobutane with 2-butene. Appl. Catal. A Gen. 2003, 246, 243–251. [Google Scholar] [CrossRef]
- Yoo, K.; Burckle, E.C.; Smirniotis, P.G. Isobutane/2-butene alkylation using large-pore zeolites: Influence of pore structure on activity and selectivity. J. Catal. 2002, 211, 6–18. [Google Scholar] [CrossRef]
- Han, H.; Liu, M.; Nie, X.; Ding, F.; Wang, Y.; Li, J.; Guo, X.; Song, C. The promoting effects of alkali metal oxide in side-chain alkylation of toluene with methanol over basic zeolite X. Microporous Mesoporous Mater. 2016, 234, 61–72. [Google Scholar] [CrossRef]
- Itoh, H.; Hattori, T.; Suzuki, K.; Murakami, Y. Role of acid and base sites in the side-chain alkylation of alkylbenzenes with methanol on two-ion-exchanged zeolites. J. Catal. 1983, 79, 21–33. [Google Scholar] [CrossRef]
- Tope, B.B.; Alabi, W.O.; Aitani, A.M.; Hattori, H.; Al-Khattaf, S.S. Side-chain alkylation of toluene with methanol to styrene over cesium ion-exchanged zeolite X modified with metal borates. Appl. Catal. A Gen. 2012, 443–444, 214–220. [Google Scholar] [CrossRef]
- Philippou, A.; Anderson, M.W. Solid-State NMR Investigation of the Alkylation of Toluene with Methanol over Basic Zeolite X. J. Am. Chem. Soc. 1994, 116, 5774–5783. [Google Scholar] [CrossRef]
- Jiang, N.; Jin, H.; Jeong, E.-Y.; Park, S.-E. Mgo Encapsulated Mesoporous Zeolite for the Side Chain Alkylation of Toluene with Methanol. J. Nanosci. Nanotechnol. 2010, 10, 227–232. [Google Scholar] [CrossRef]
- Hattori, H.; Alabi, W.O.; Jermy, B.R.; Aitani, A.M.; Al-Khattaf, S.S. Pathway to ethylbenzene formation in side-chain alkylation of toluene with methanol over cesium ion-exchanged zeolite X. Catal. Lett. 2013, 143, 1025–1029. [Google Scholar] [CrossRef]
- Mikkelsen, M.; Jørgensen, M.; Krebs, F.C. The teraton challenge. A review of fixation and transformation of carbon dioxide. Energy Environ. Sci. 2010, 3, 43–81. [Google Scholar] [CrossRef]
- Cavani, F.; Ballarini, N.; Cericola, A. Oxidative dehydrogenation of ethane and propane: How far from commercial implementation? Catal. Today 2007, 127, 113–131. [Google Scholar] [CrossRef]
- Ren, Y.; Zhang, F.; Hua, W.; Yue, Y.; Gao, Z. ZnO supported on high silica HZSM-5 as new catalysts for dehydrogenation of propane to propene in the presence of CO2. Catal. Today 2009, 148, 316–322. [Google Scholar] [CrossRef]
- Chen, M.; Xu, J.; Liu, Y.M.; Cao, Y.; He, H.Y.; Zhuang, J.H. Supported indium oxide as novel efficient catalysts for dehydrogenation of propane with carbon dioxide. Appl. Catal. A Gen. 2010, 377, 35–41. [Google Scholar] [CrossRef]
- Schimmoeller, B.; Jiang, Y.; Pratsinis, S.E.; Baiker, A. Structure of flame-made vanadia/silica and catalytic behavior in the oxidative dehydrogenation of propane. J. Catal. 2010, 274, 64–75. [Google Scholar] [CrossRef]
- Liu, Y.M.; Cao, Y.; Yan, S.R.; Dai, W.L.; Fan, K.N. Highly effective oxidative dehydrogenation of propane over vanadia supported on mesoporous SBA-15 silica. Catal. Lett. 2003, 88, 61–67. [Google Scholar] [CrossRef]
- Santamaría-González, J.; Mérida-Robles, J.; Alcántara-Rodríguez, M.; Maireles-Torres, P.; Rodríguez-Castellón, E.; Jiménez-López, A. Catalytic behaviour of chromium supported mesoporous MCM-41 silica in the oxidative dehydrogenation of propane. Catal. Lett. 2000, 64, 209–214. [Google Scholar] [CrossRef]
- Davies, T.; Taylor, S.H. The oxidative dehydrogenation of propane using gallium-molybdenum oxide-based catalysts. J. Mol. Catal. A Chem. 2004, 220, 77–84. [Google Scholar] [CrossRef]
- Raju, G.; Reddy, B.M.; Abhishek, B.; Mo, Y.H.; Park, S.-E. Synthesis of C4 olefins from n-butane over a novel VOx/SnO2-ZrO2 catalyst using CO2 as soft oxidant. Appl. Catal. A Gen. 2012, 423–424, 168–175. [Google Scholar] [CrossRef]
- Atanga, M.A.; Rezaei, F.; Jawad, A.; Fitch, M.; Rownaghi, A.A. Oxidative dehydrogenation of propane to propylene with carbon dioxide. Appl. Catal. B Environ. 2018, 220, 429–445. [Google Scholar] [CrossRef]
- Reddy, B.M.; Lee, S.C.; Han, D.S.; Park, S.-E. Utilization of carbon dioxide as soft oxidant for oxydehydrogenation of ethylbenzene to styrene over V2O5-CeO2/TiO2-ZrO2 catalyst. Appl. Catal. B Environ. 2009, 87, 230–238. [Google Scholar] [CrossRef]
- Uy, D.; O’Neill, A.E.; Xu, L.; Weber, W.H.; McCabe, R.W. Observation of cerium phosphate in aged automotive catalysts using Raman spectroscopy. Appl. Catal. B Environ. 2003, 41, 269–278. [Google Scholar] [CrossRef]
- Armaroli, T.; Simon, L.J.; Digne, M.; Montanari, T.; Bevilacqua, M.; Valtchev, V.; Patarin, J.; Busca, G. Effects of crystal size and Si/Al ratio on the surface properties of H-ZSM-5 zeolites. Appl. Catal. A Gen. 2006, 306, 78–84. [Google Scholar] [CrossRef]
- Thakkar, H.; Eastman, S.; Hajari, A.; Rownaghi, A.A.; Knox, J.C.; Rezaei, F. 3D-Printed Zeolite Monoliths for CO2 Removal from Enclosed Environments. ACS Appl. Mater. Interfaces 2016, 8, 27753–27761. [Google Scholar] [CrossRef]
- Thakkar, H.; Eastman, S.; Al-Mamoori, A.; Hajari, A.; Rownaghi, A.A.; Rezaei, F. Formulation of Aminosilica Adsorbents into 3D-Printed Monoliths and Evaluation of Their CO2 Capture Performance. ACS Appl. Mater. Interfaces 2017, 9, 7489–7498. [Google Scholar] [CrossRef]
- Ren, Y.; Wang, J.; Hua, W.; Yue, Y.; Gao, Z. Ga2O3/HZSM-48 for dehydrogenation of propane: Effect of acidity and pore geometry of support. J. Ind. Eng. Chem. 2012, 18, 731–736. [Google Scholar] [CrossRef]
- Abello, M.C.; Gomez, M.F.; Ferretti, O. Oxidative conversion of propane over Al2O3-supported molybdenum and chromium oxides. Catal. Lett. 2003, 87, 43–49. [Google Scholar] [CrossRef]
- Cherian, M.; Rao, M.S.; Hirt, A.M.; Wachs, I.E.; Deo, G. Oxidative dehydrogenation of propane over supported chromia catalysts: Influence of oxide supports and chromia loading. J. Catal. 2002, 211, 482–495. [Google Scholar] [CrossRef]
- Rao, T.V.M.; Zahidi, E.M.; Sayari, A. Ethane dehydrogenation over pore-expanded mesoporous silica-supported chromium oxide: 2. Catalytic properties and nature of active sites. J. Mol. Catal. A Chem. 2009, 301, 159–165. [Google Scholar] [CrossRef]
- Hakuli, A.; Kytökivi, A.; Krause, A.O.I. Dehydrogenation of i-butane on CrOx/Al2O3 catalysts prepared by ALE and impregnation techniques. Appl. Catal. A Gen. 2000, 190, 219–232. [Google Scholar] [CrossRef]
- Liu, L.; Li, H.; Zhang, Y. Effect of synthesis parameters on the chromium content and catalytic activities of mesoporous Cr-MSU-x prepared under acidic conditions. J. Phys. Chem. B 2006, 110, 15478–15485. [Google Scholar] [CrossRef] [PubMed]
- Santhosh Kumar, M.; Hammer, N.; Rønning, M.; Holmen, A.; Chen, D.; Walmsley, J.C.; Øye, G. The nature of active chromium species in Cr-catalysts for dehydrogenation of propane: New insights by a comprehensive spectroscopic study. J. Catal. 2009, 261, 116–128. [Google Scholar] [CrossRef]
- Weckhuysen, B.M.; Wachs, I.E.; Schoonheydt, R.A. Surface chemistry and spectroscopy of chromium in inorganic oxides. Chem. Rev. 1996, 96, 3327–3349. [Google Scholar] [CrossRef] [Green Version]
- Burri, A.; Hasib, M.A.; Mo, Y.H.; Reddy, B.M.; Park, S.-E. An Efficient Cr-TUD-1 Catalyst for Oxidative Dehydrogenation of Propane to Propylene with CO2 as Soft Oxidant. Catal. Lett. 2018, 148, 576–585. [Google Scholar] [CrossRef]
- Park, S.-J.; Kim, K.D. Adsorption behaviors of CO2 and NH3 on chemically surface-treated activated carbons. J. Colloid Interface Sci. 1999, 212, 186–189. [Google Scholar] [CrossRef]
- Michorczyk, P.; Pietrzyk, P.; Ogonowski, J. Preparation and characterization of SBA-1-supported chromium oxide catalysts for CO2 assisted dehydrogenation of propane. Microporous Mesoporous Mater. 2012, 161, 56–66. [Google Scholar] [CrossRef]
- COSIA. $20M NRG COSIA Carbon XPRIZE Finalists Announced; Teams Ready to Test Transformative CO2 Technologies at Alberta’s Carbon Conversion Centre. Available online: www.cosia.ca/resources/newsreleases/20m-nrg-cosia-carbon-xprize-finalists-announcedteams-ready-test (accessed on 9 April 2018).
- Zhang, D.; Wang, J.; Lin, Y.; Si, Y.; Huang, C.; Yang, J.; Huang, B.; Li, W. Present situation and future prospect of renewable energy in China. Renew. Sustain. Energy Rev. 2017, 76, 865–871. [Google Scholar] [CrossRef]
Scheme 1.
The various chemical processes for CO2 conversion.

Scheme 2.
Cyclohexene oxidation reaction over catalyst. (Redrawn from [30]; copyright (2018), WILEY-VCH). (A) = 2-cyclohexene-1-one, (B) = cyclohexene oxide, (C) = 2-cyclohexene-1-ol, (D) = 2-cyclohexene-1-hydroperoxide). Reaction conditions: 10 bar O2; 2.5 mL cyclohexene; 0.5 mL cyclohexane(IS); 10 mg catalyst; 15 mL MeCN; stirred in an autoclave (1000 rpm); 70 °C; 16 h.
Scheme 2.
Cyclohexene oxidation reaction over catalyst. (Redrawn from [30]; copyright (2018), WILEY-VCH). (A) = 2-cyclohexene-1-one, (B) = cyclohexene oxide, (C) = 2-cyclohexene-1-ol, (D) = 2-cyclohexene-1-hydroperoxide). Reaction conditions: 10 bar O2; 2.5 mL cyclohexene; 0.5 mL cyclohexane(IS); 10 mg catalyst; 15 mL MeCN; stirred in an autoclave (1000 rpm); 70 °C; 16 h.

Figure 1.
Per ox-carbonate over Fe/Mo/DBH in the O2/CO2 system (Reproduced from [20]; copyright (1993), Elsevier (Amsterdam, The Netherlands)).
Figure 1.
Per ox-carbonate over Fe/Mo/DBH in the O2/CO2 system (Reproduced from [20]; copyright (1993), Elsevier (Amsterdam, The Netherlands)).

Scheme 3.
Per-oxycarbonate production reaction mechanisms (Redrawn from [32]; copyright (1996), American Chemical Society).
Scheme 3.
Per-oxycarbonate production reaction mechanisms (Redrawn from [32]; copyright (1996), American Chemical Society).

Scheme 4.
Proposed Mechanism for aldehyde assisted CO2 to carboxylic acid process. (Reprinted from [34]; copyright (2010), American Chemical Society).
Scheme 4.
Proposed Mechanism for aldehyde assisted CO2 to carboxylic acid process. (Reprinted from [34]; copyright (2010), American Chemical Society).

Scheme 5.
Phenylglyoxylic acid to benzoic acid reaction with NHC-intermediate. (Reprinted from [34]; copyright (2010), American Chemical Society).
Scheme 5.
Phenylglyoxylic acid to benzoic acid reaction with NHC-intermediate. (Reprinted from [34]; copyright (2010), American Chemical Society).

Figure 2.
Promotional role of CO2 on Fe/Mo/DBH for the oxidation of p-xylene.

Figure 3.
(a) Conversion of EB using CO2 as a soft oxidant; (b) O1s spectra of HMWCNTs-B-OH after oxidative dehydrogenation by XPS for 3 hr; (c) Oxygen substance (mol%) of HMWCNTs-B-OH earlier and later oxidative dehydrogenation reaction at 3 h; (d) Gas derivatives of HMWCNTs-B-OH after oxidative dehydrogenation at 3 h. (Reprinted from [44]; copyright (2013), Royal Society of Chemistry).
Figure 3.
(a) Conversion of EB using CO2 as a soft oxidant; (b) O1s spectra of HMWCNTs-B-OH after oxidative dehydrogenation by XPS for 3 hr; (c) Oxygen substance (mol%) of HMWCNTs-B-OH earlier and later oxidative dehydrogenation reaction at 3 h; (d) Gas derivatives of HMWCNTs-B-OH after oxidative dehydrogenation at 3 h. (Reprinted from [44]; copyright (2013), Royal Society of Chemistry).

Figure 4.
The procedure of oxidative dehydrogenation of ethylbenzene to styrene (a) without CO2 and (b) with CO2. (Reproduced with permission from [62]; copyright (2016), Elsevier).
Figure 4.
The procedure of oxidative dehydrogenation of ethylbenzene to styrene (a) without CO2 and (b) with CO2. (Reproduced with permission from [62]; copyright (2016), Elsevier).

Figure 5.
Red-ox cycle for CO2-ODEB over the ceria-promoted vanadium catalyst. (Reproduced from [48]; copyright (2011), WILEY-VCH).
Figure 5.
Red-ox cycle for CO2-ODEB over the ceria-promoted vanadium catalyst. (Reproduced from [48]; copyright (2011), WILEY-VCH).

Figure 6.
TG/DTA results obtained for used Cs-X catalyst in the presence of CO2 and He streams (Redrawn with permission from [91]; copyright (2018), Elsevier).
Figure 6.
TG/DTA results obtained for used Cs-X catalyst in the presence of CO2 and He streams (Redrawn with permission from [91]; copyright (2018), Elsevier).


{kind=link}
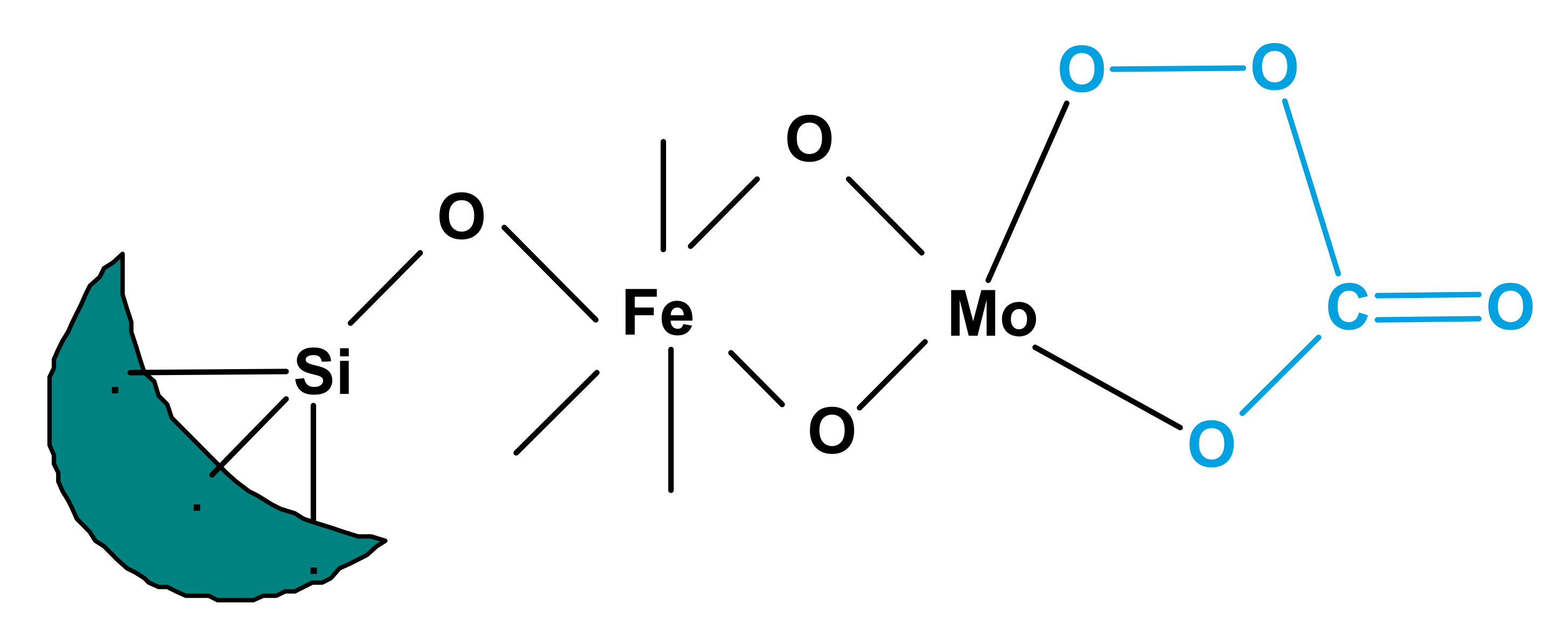{kind=link}
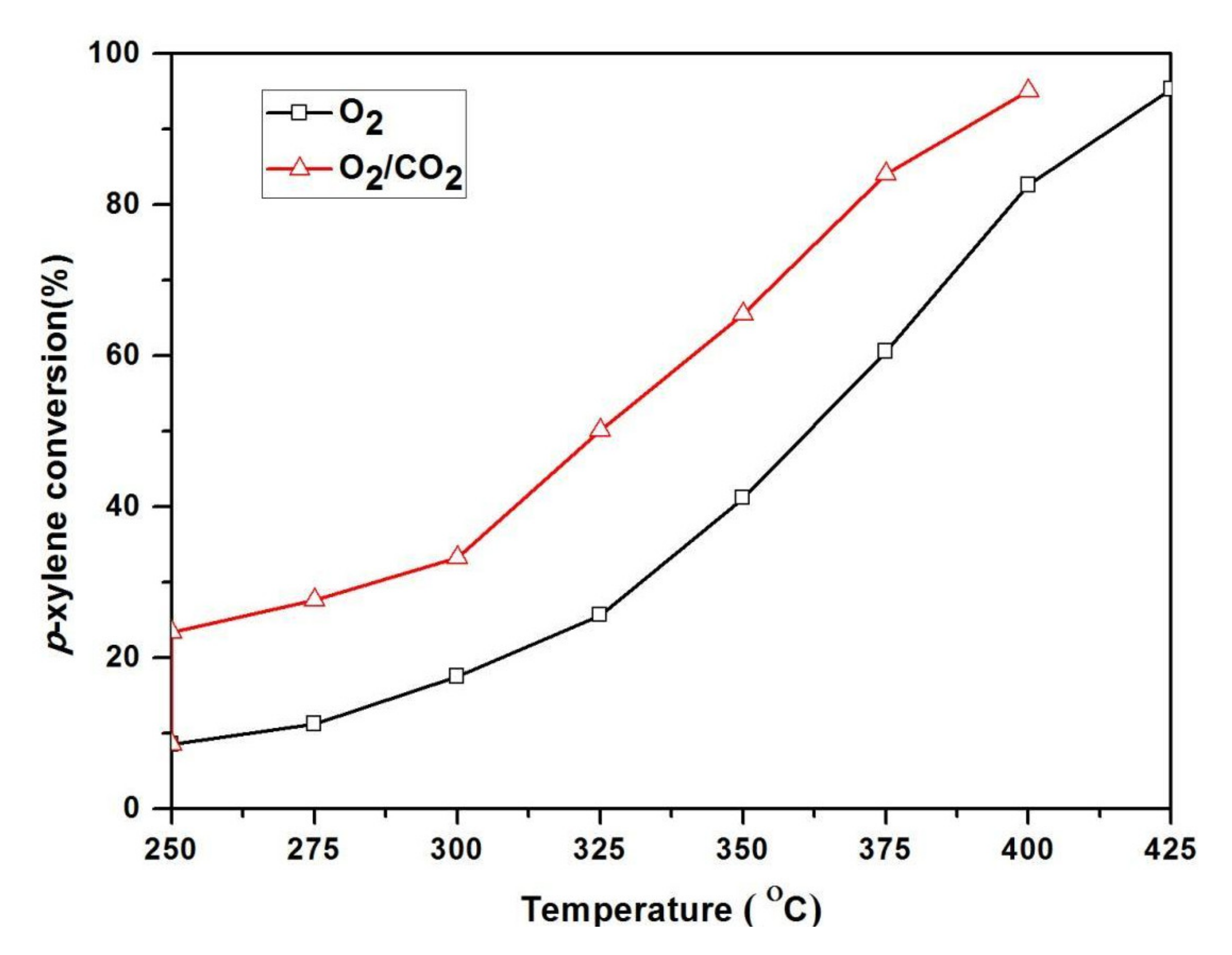{kind=link}
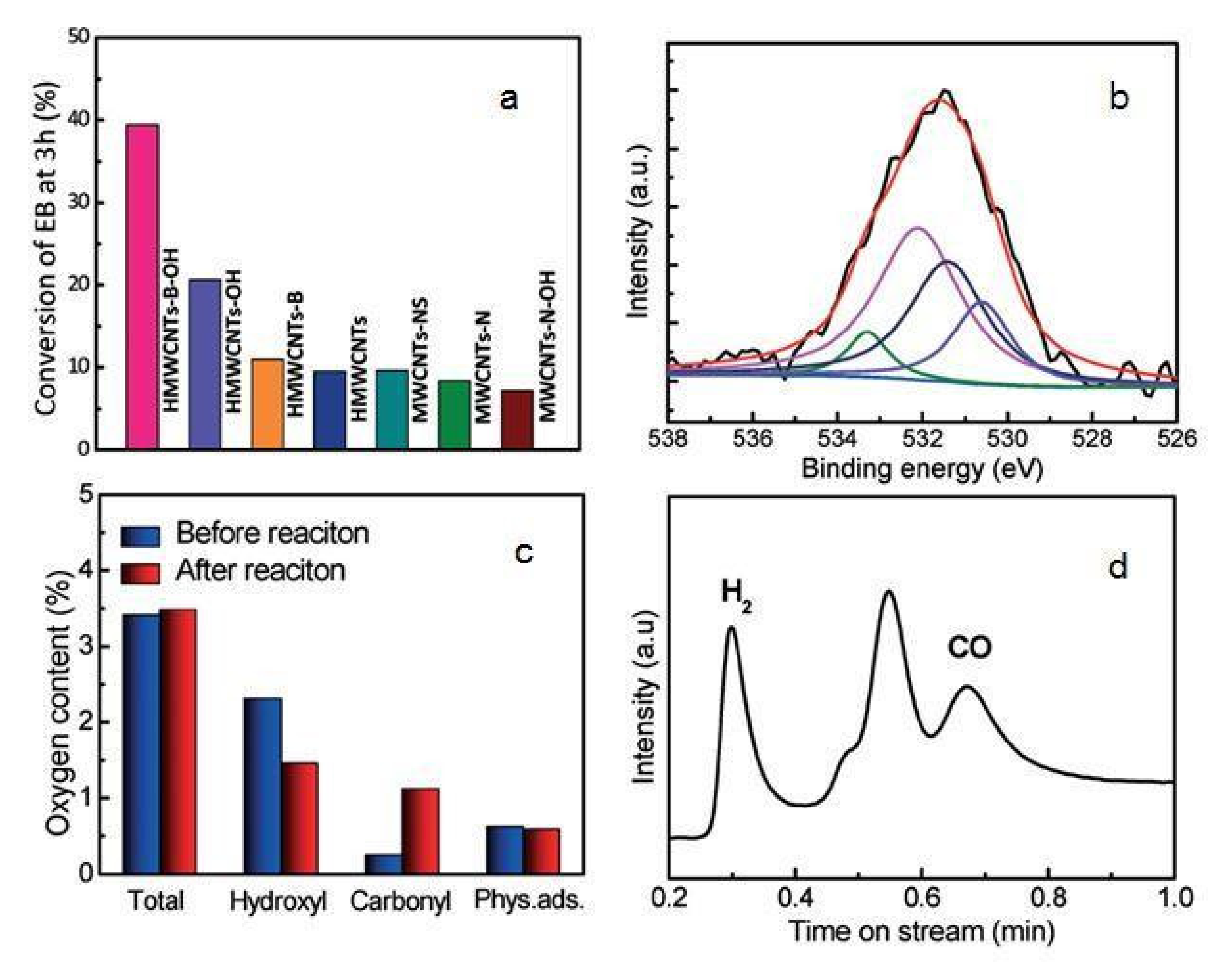{kind=link}
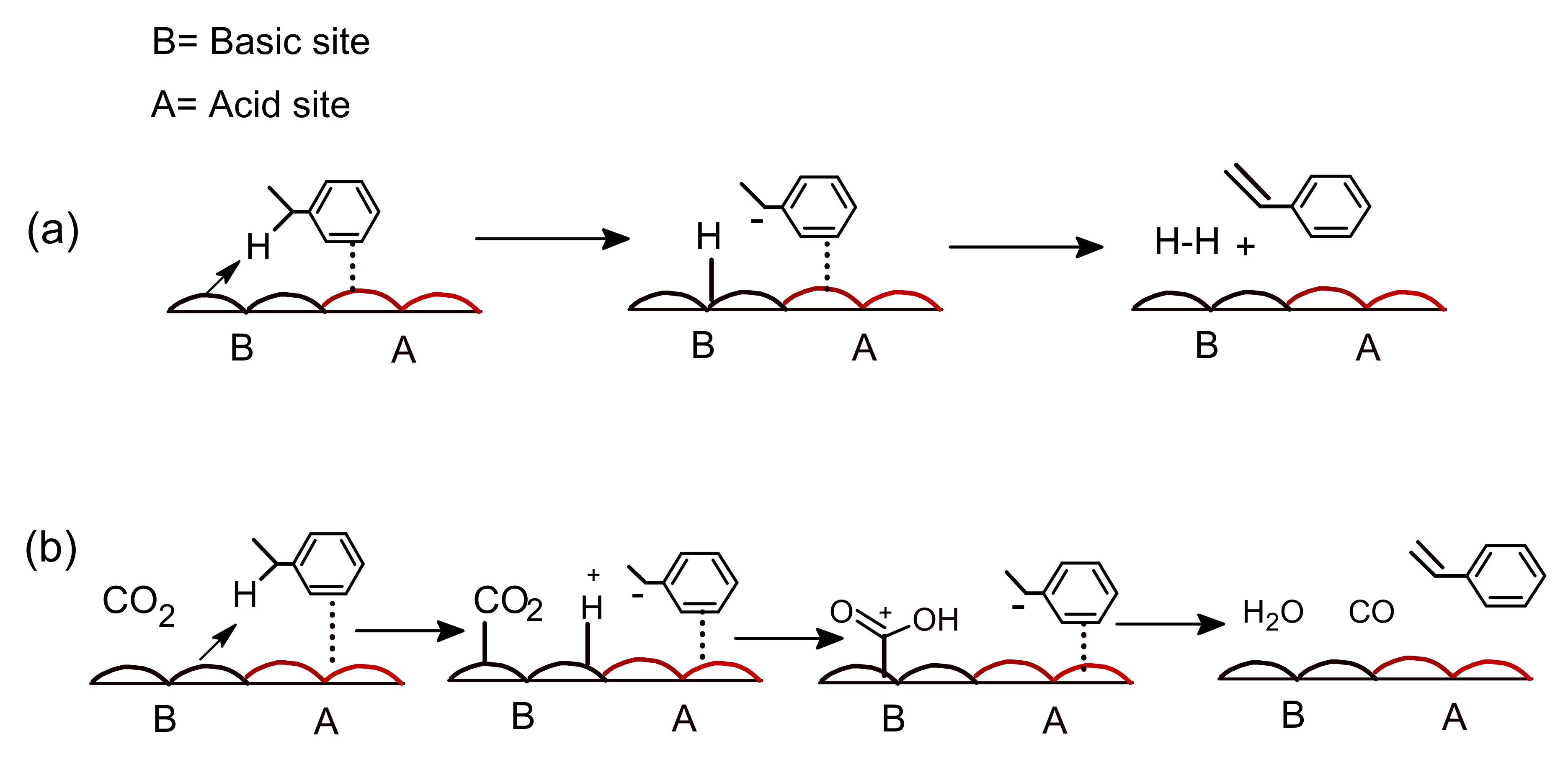{kind=link}
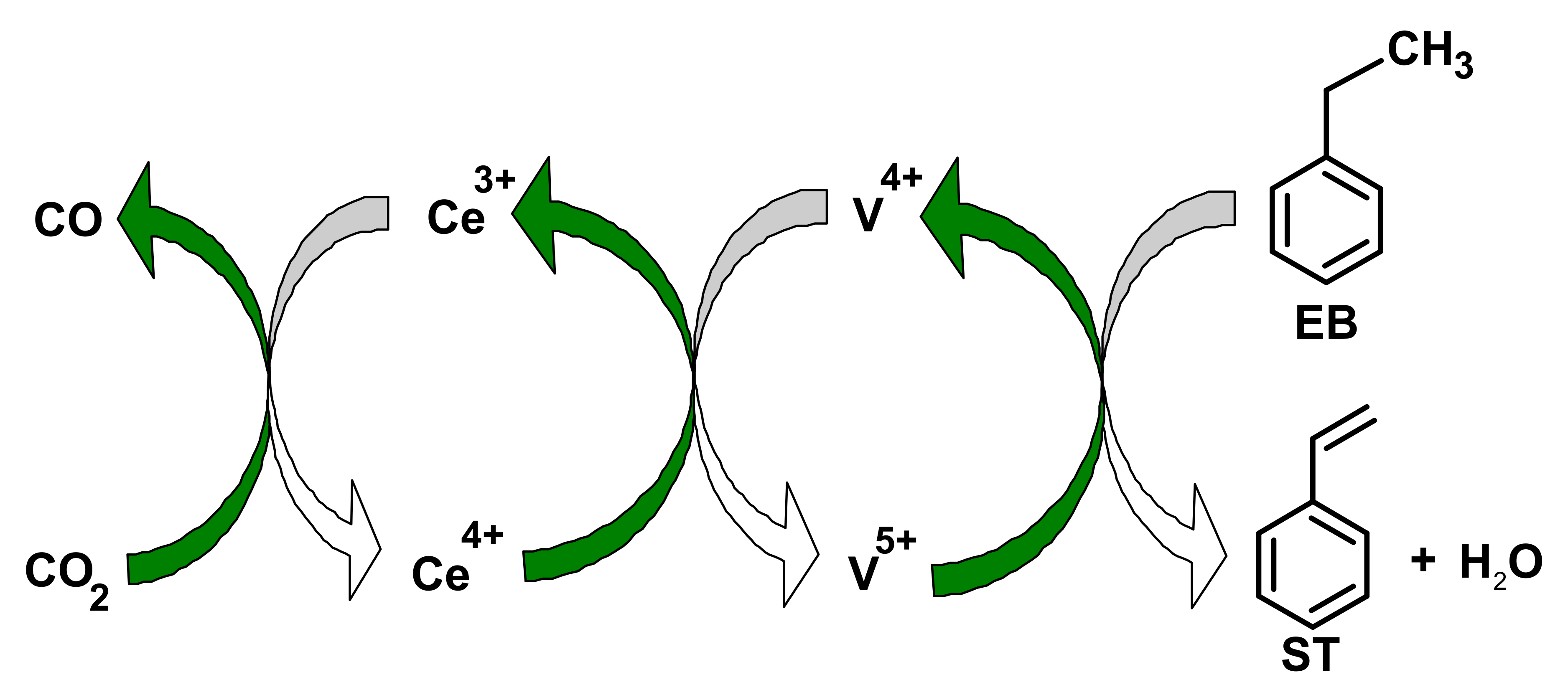{kind=link}
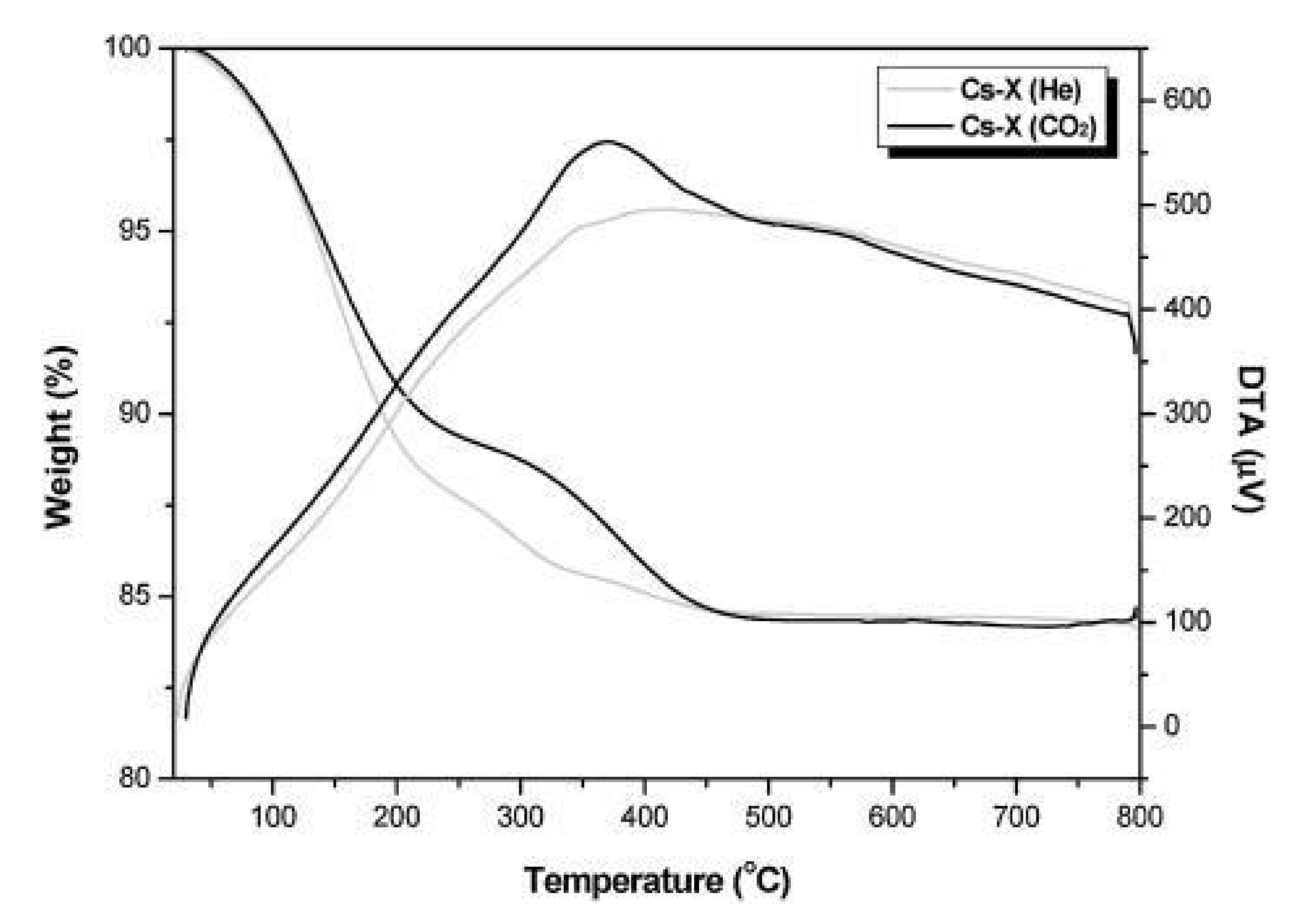{kind=link}
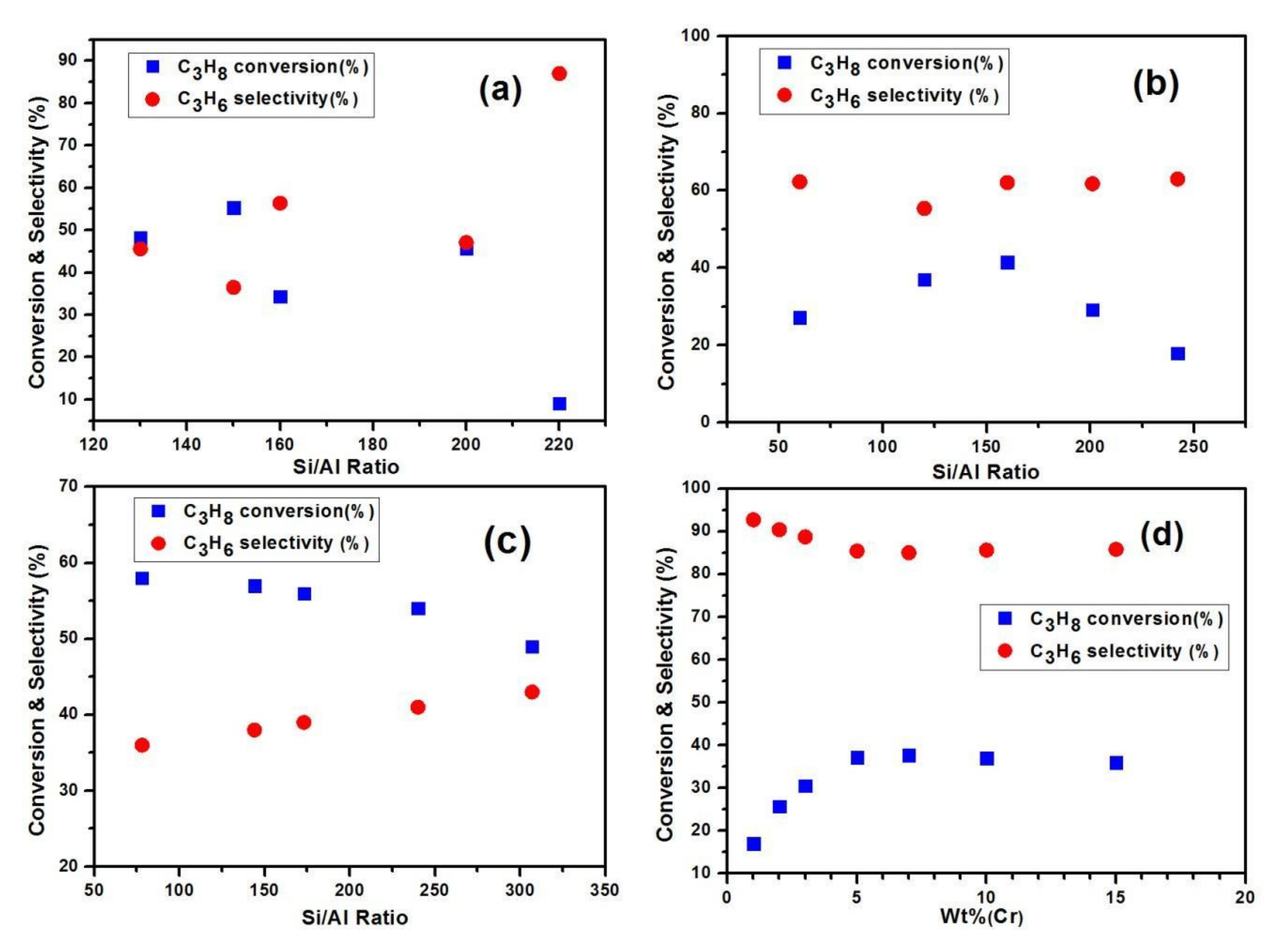{kind=link}
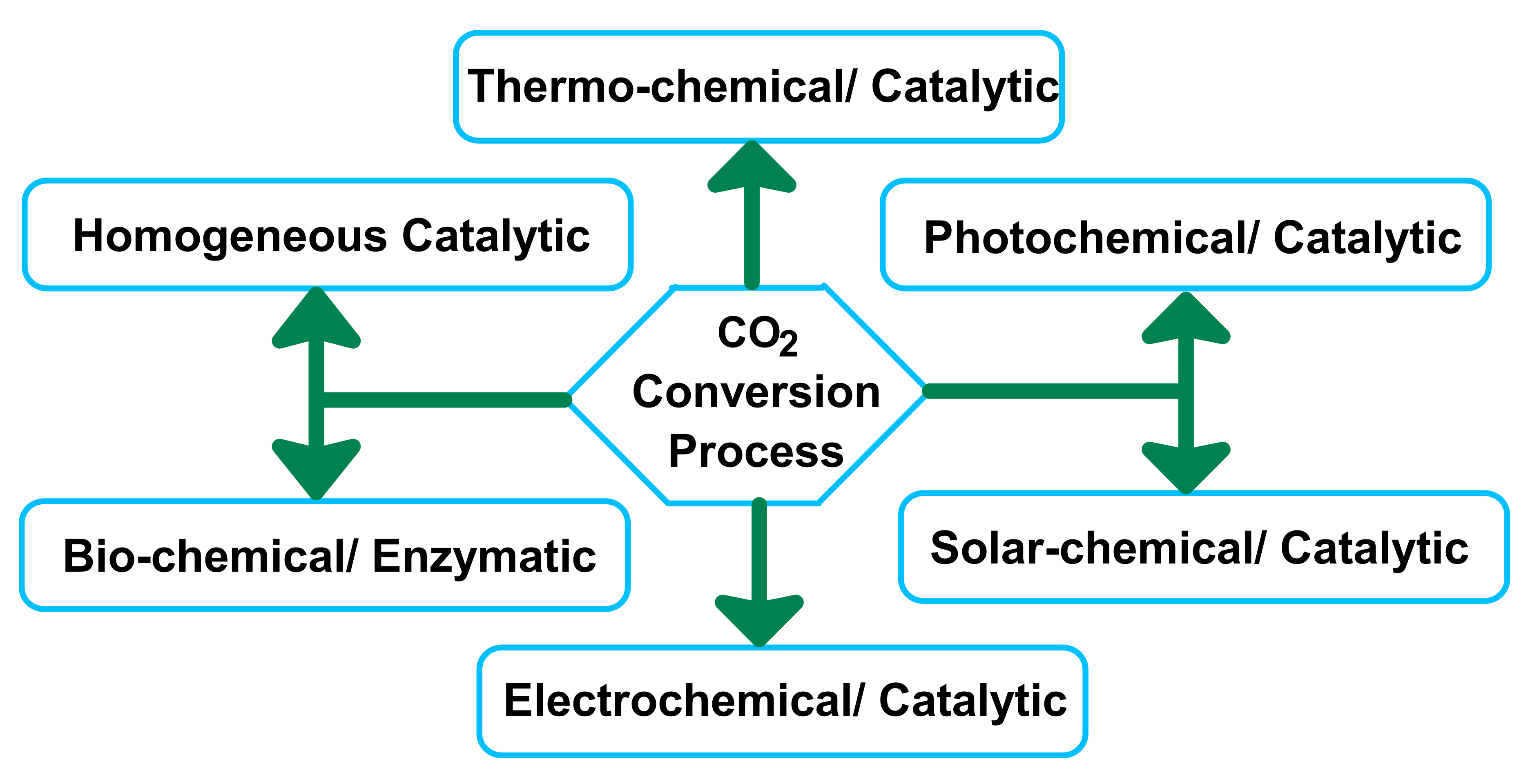{kind=link}
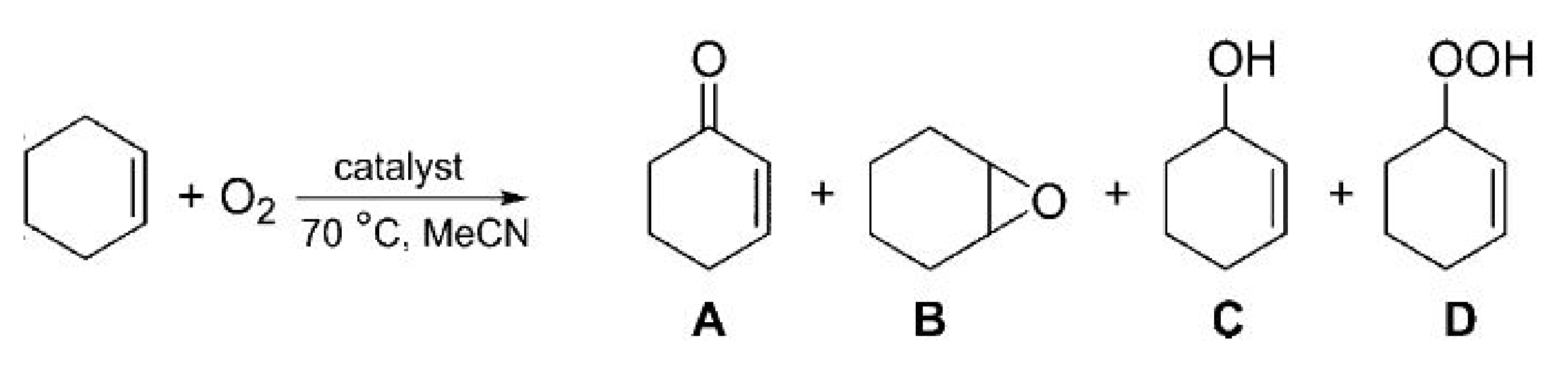{kind=link}
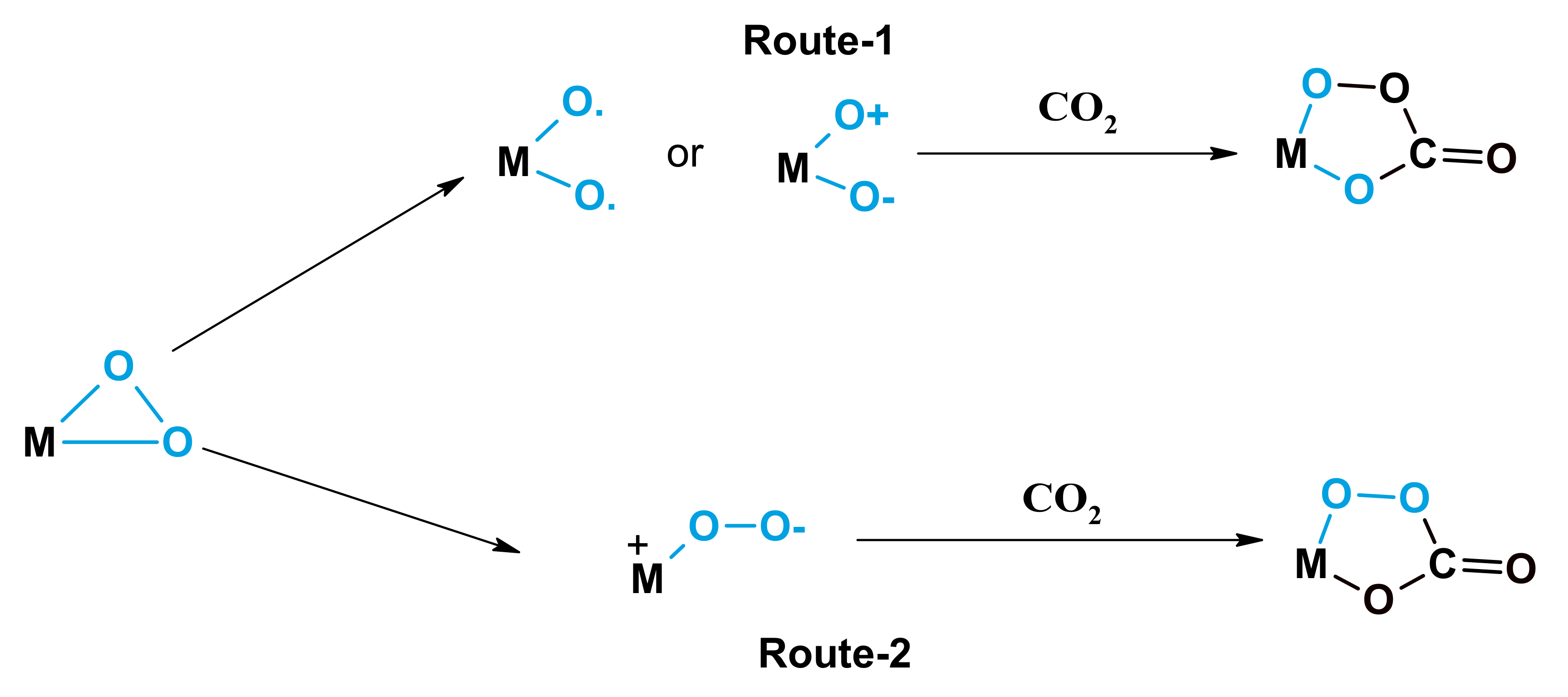{kind=link}
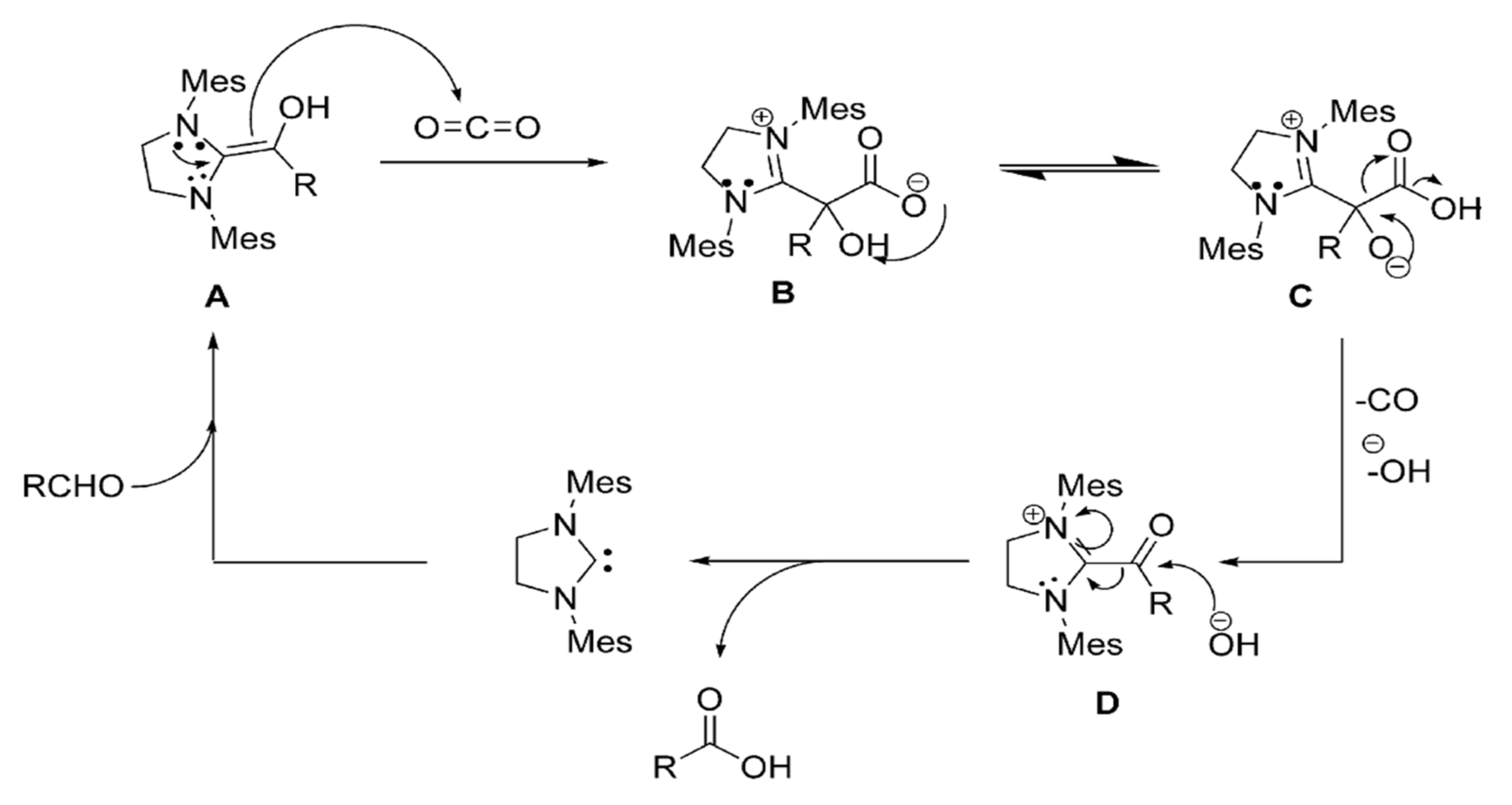{kind=link}
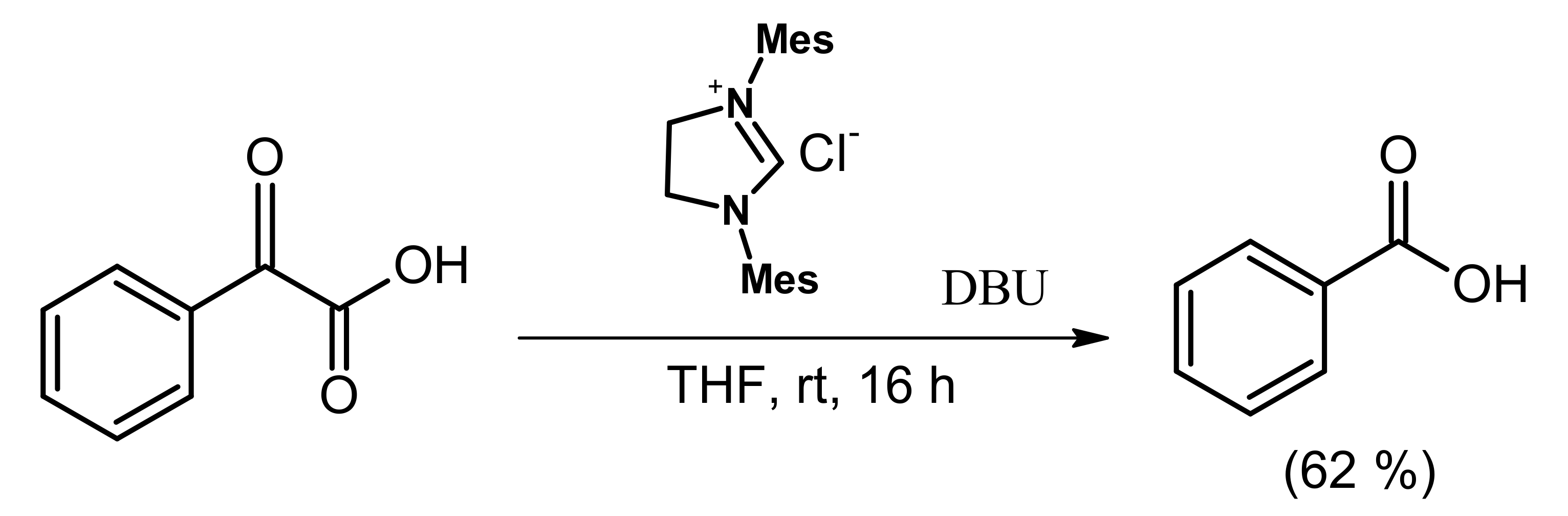{kind=link}
Table 1.
Effect of the CO2 on oxidation of cyclohexene over MCN Ref [31] (Reproduced from [31]; copyright (2011), Royal Society of Chemistry).
| Entry | Gas Ratio (PSI) a | Conversion (%) O2/CO2 | Conversion (%) O2/N2 | ΔC(%) b |
|---|---|---|---|---|
| 1 | 0.066 | 0 | 0 | 0 |
| 2 | 0.142 | 16 | 9 | 28 |
| 3 | 0.230 | 25 | 18 | 16.3 |
| 4 | 0.333 | 33 | 24 | 15.7 |
| 5 | 0.454 | 34 | 24 | 15.7 |
Reaction conditions: 20 mg Melamine mesoporous carbon nitride (M-MCN), 10 mL Dimethylformamide (DMF), temperature 373 K, Pressure 80 PSI, time 10 h; Estimated by Gas Chromatography (GC) analysis. a PSI = Pounds per Square Inch, b Conversion (%) of cyclic olefin.
Table 2.
Enhancive role of CO2 on cyclic olefins oxidation (Reproduced from [31]; copyright (2011), Royal Society of Chemistry).
Table 2.
Enhancive role of CO2 on cyclic olefins oxidation (Reproduced from [31]; copyright (2011), Royal Society of Chemistry).

| Entry | n | Gas | Conversion of 3 (%) | Selectivity (%) | |||
|---|---|---|---|---|---|---|---|
| 4 | 5 | 6 | ΔC (%) | ||||
| 1 | 1 | O C | 40 | 37 | 24 | 29 | 12.6 |
| O | 31 | 30 | 22 | 40 | - | ||
| 2 | 2 | O C | 33 | 30 | 21 | 49 | 15.8 |
| O | 24 | 25 | 16 | 53 | - | ||
| 3 | 4 | O C | 21 | > 99 | - | - | 27.0 |
| - | - | O | 12 | > 99 | - | - | - |
| 4 | 8 | O C | 17 | > 99 | - | - | 30.7 |
| - | - | O | 9 | > 99 | - | - | - |
Reaction conditions: 20 mg Melamine mesoporous carbon nitride (M-MCN), 10 mL Dimethylformamide (DMF), temperature 373 K, Pressure 80 PSI, gas ratio 0.333, time 10 h; Produced analyzed by GC and GC-MS.
Table 3.
Effect of CO2 on the MC-type catalyst for oxidation of p-xylene (Reproduced from [22]; copyright (2012), Royal Society of Chemistry).
Table 3.
Effect of CO2 on the MC-type catalyst for oxidation of p-xylene (Reproduced from [22]; copyright (2012), Royal Society of Chemistry).

| Gas | Conversion of 1 (%) | Yield mol (%) | ||||
|---|---|---|---|---|---|---|
| 2 | 3 | 4 | 5 | 6 | ||
| O2 | 57.2 | 17.7 | 47.9 | 2.8 | 1.7 | 29.2 |
| O2/CO2 | 66.8 | 34.8 | 36.9 | 1.7 | 2.4 | 24.2 |
Reaction conditions: Temperature 170 °C, time 3 h, Mesoporus carbon (MC) type catalyst with transition metal additive (Co/Mn/Br), Co 100 ppm, Mn 200 ppm, Br 300 ppm [38].
Table 4.
Enhancive effect of CO2 on oxidation of p-xylene (Reproduced from [20]; copyright (1993), Elsevier). Reaction conditions: WHSV: 0.22 h−1, contact time: 0.21 s, gas flowrate: 400 sccm, Feed gas 1: 0.1% p-xylene, 1% O2, 1% N2 in He. Feed gas 2: 0.1% p-xylene, 1% O2, 1% N2 in commercial grade CO2.
Table 4.
Enhancive effect of CO2 on oxidation of p-xylene (Reproduced from [20]; copyright (1993), Elsevier). Reaction conditions: WHSV: 0.22 h−1, contact time: 0.21 s, gas flowrate: 400 sccm, Feed gas 1: 0.1% p-xylene, 1% O2, 1% N2 in He. Feed gas 2: 0.1% p-xylene, 1% O2, 1% N2 in commercial grade CO2.
| Temperature (°C) Feed a | 300 | 350 | 375 | |||
|---|---|---|---|---|---|---|
| 1 | 2 | 1 | 2 | 1 | 2 | |
| p-Xylene (Con.%) | 17.6 | 33.3 | 41.2 | 65.5 | 60.7 | 84.1 |
| Product selectivity (mol%) | - | - | - | - | - | - |
| p-Tolu-aldehyde | 57.9 | 57.2 | 50.2 | 40.9 | 40.6 | 40.6 |
| Terephthaldehyde | 16.4 | 27.5 | 23.5 | 33.6 | 32.6 | 30.2 |
| Benzaldehyde | 1.3 | 1.5 | 2.4 | 2.7 | 2.7 | 3.1 |
| Maleic anhydride | 0.0 | 0.0 | 2.4 | 6.0 | 5.8 | 13.7 |
| Toluene | 6.2 | 6.9 | 3.5 | 4.8 | 3.1 | 4.8 |
| Trimethyl biphenyl methane | 7.5 | 6.8 | 1.7 | 0.6 | 0.4 | 0.0 |
| CO | 0.0 | 0.0 | 0.6 | 3.4 | 4.7 | 7.5 |
| CO2 | 10.7 | 0.0 | 15.6 | 0.0 | 20.2 | 0.0 |
a Feed gas 1: O2/N2/He, Feed gas 2: O2/N2/CO2.
Table 5.
p-toluic acid oxidation with CO2 on an MC-type catalyst (Reproduced from [22]; copyright (2012), Royal Society of Chemistry).
Table 5.
p-toluic acid oxidation with CO2 on an MC-type catalyst (Reproduced from [22]; copyright (2012), Royal Society of Chemistry).

| Gas | Conversion of 1 (1%) | Yield mol (%) | |
|---|---|---|---|
| 2 | 3 | ||
| O2 | 60.9 | 58.2 | 3.7 |
| O2/CO2 | 72.7 | 64.9 | 10.6 |
Reaction conditions: 6 mL p-toluic acid, 0.1183 g CoBr2, 0.1587 g Mn(OAc)2·4H2O in 24 mL HOAc, temperature 190 °C, time 3 h; PCO2 = 0-6 atm, PO2 = 2 atm [38].
Table 6.
Performance of CO2 on oxidation of p-methylanisole (Reproduced from [22]; copyright (2012), Royal Society of Chemistry).
Table 6.
Performance of CO2 on oxidation of p-methylanisole (Reproduced from [22]; copyright (2012), Royal Society of Chemistry).

| Gas | Conversion of 1 (%) | Yield mol (%) | ||
|---|---|---|---|---|
| 2 | 3 | 4 | ||
| O2 | 94.9 | 2.05 | 0.83 | 92 |
| O2/CO2 | 98 | 7.7 | 0.38 | 90 |
Reaction conditions: 43.5 mmol p-methylanisole, 0.6 mmol CoBr2, 0.6 mmol Co(OAc)2, 0.6 mmol Mn(OAc)2·4H2O in 30 g HOAc, total pressure 12 atm (PCO2 = 0-2 atm, PO2 = 2,3,6 atm, PN2 balance). temperature 120 °C, time 3 h [38].
Table 7.
Performance of CO2 on oxidative dehydrogenation of ethylbenzene to styrene.
| Catalyst | Reaction Temperature (°C) | EB Conversion (%) | ST Selectivity (%) | ST Yield (%) | Ref. |
|---|---|---|---|---|---|
| Co3O4/COK-12 | 600 | 57.5 | 95.5 | 54.9 | [59] |
| CeZrO4-δ | 550 | 7 | 97 | 6.8 | [61] |
| Na X zeolite | 545 | 9.4 | 89.6 | 8.4 | [60] |
| K X zeolite | 545 | 10.5 | 92.1 | 9.6 | [60] |
| VOx/Al MCM-41 | 550 | 52.3 | 96.7 | 50.6 | [51] |
| TiO2-ZrO2 | 600 | 69.3 | 96.2 | 66.6 | [67] |
| V2O5/SiO2 | 550 | 50.5 | 96.8 | 48.8 | [64] |
| SnO2-ZrO2 | 600 | 61.1 | 97.6 | 59.6 | [68] |
| MnO2-ZrO2 | 600 | 51.1 | 99.1 | 50.9 | [69] |
Table 8.
Catalytic activity for the dehydrogenation of ethane (Reproduced from [73]; copyright (2008), Springer (Berlin, Germany)).
Table 8.
Catalytic activity for the dehydrogenation of ethane (Reproduced from [73]; copyright (2008), Springer (Berlin, Germany)).
| Catalyst | In the Presence of CO2 | In the Presence of Ar | |||||
|---|---|---|---|---|---|---|---|
| Conv. (%) | Selectivity (%) | Conv. (%) | Selectivity (%) | ||||
| C2H6 | CO2 | C2H4 | CH4 | C2H6 | C2H4 | CH4 | |
| SBA-15 | 2.7 | 0.04 | 93.5 | 6.5 | 2.4 | 93.0 | 7.0 |
| 2.5Cr/SBA-15 | 39.6 | 15.9 | 95.5 | 4.5 | 30.2 | 89.7 | 10.3 |
| 5.0Cr/SBA-15 | 46.3 | 16.6 | 94.7 | 5.3 | 34.1 | 91.4 | 8.6 |
| 7.5Cr/SBA-15 | 45.3 | 18.8 | 92.2 | 7.8 | 33.9 | 92.8 | 7.2 |
| 10Cr/SBA-15 | 44.2 | 18.9 | 92.0 | 8.0 | 31.2 | 90.9 | 9.1 |
| 5Cr-5Ce/SBA-15 | 48.4 | 17.9 | 96.4 | 4.6 | 35.8 | 87.6 | 12.4 |
| 5Cr-7.5Ce/SBA-15 | 50.0 | 20.9 | 96.0 | 4.0 | 37.9 | 88.2 | 11.8 |
| 5Cr-10Ce/SBA-15 | 55.0 | 21.9 | 96.0 | 4.0 | 40.8 | 83.1 | 16.9 |
| 5Cr-15Ce/SBA-15 | 52.2 | 21.2 | 95.5 | 4.5 | 40.1 | 82.4 | 17.6 |
Reaction conditions: GHSV = 3600 mL/g h, T = 700 °C.
Table 9.
Influence of CO2 on oxidative dehydrogenation of ethane.
| Catalyst | Ethane Conversion (%) | Ethylene Selectivity (%) | Ethylene Yield (%) | Ref. |
|---|---|---|---|---|
| Cr2O3 (5 wt.%) CLT-IA | 39.7 | 98.8 | 39.2 | [70] |
| 3Cr/NaZSM-5-160 | 65.5 | 75.4 | 49.3 | [84] |
| Cr2O3/Al2O3-ZrO2 | 36.0 | 56.2 | 20.2 | [85] |
| Cr/MSU-1 | 68.1 | 81.6 | 55.6 | [72] |
| Cr2O3/ZrO2 | 77.4 | 46.3 | 35.8 | [86] |
| 2.5 Cr/SBA-15 | 46.3 | 94.7 | 43.8 | [75] |
| 5 Cr-10Ce/SBA-15 | 55.0 | 96.0 | 52.8 | [73] |
| 5% Cr2O3/Al2O3 | 19.2 | 56.5 | 10.8 | [77] |
Table 10.
Performance of CO2 in the toluene side-chain alkylation (Reproduced with permission from [91]; copyright (2018), Elsevier).
Table 10.
Performance of CO2 in the toluene side-chain alkylation (Reproduced with permission from [91]; copyright (2018), Elsevier).
| Catalyst | Carrier Gas | MeOH Conv. (%) | Toluene Conv. (%) | Selectivity (%) | ||
|---|---|---|---|---|---|---|
| SM | EB | Others | ||||
| Ce-X | He | 12.54 | 1.42 | 78.61 | 15.32 | 6.07 |
| CO2 | 35.35 | 3.48 | 45.83 | 33.36 | 20.81 | |
| Cs2O/ | He | 46.48 | 3.59 | 28.76 | 68.02 | 3.22 |
| Cs-X | CO2 | 39.16 | 2.52 | 36.02 | 43.02 | 20.78 |
Reaction conditions: WHSV = 2.1 h−1, Reaction temperature = 425 °C, Toluene/MeOH molar ratio = 2, SM = Styrene Monomer and other byproducts = Cumene, Xylenes, TMB and Benzene.
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Rahman, S.T.; Choi, J.-R.; Lee, J.-H.; Park, S.-J. The Role of CO2 as a Mild Oxidant in Oxidation and Dehydrogenation over Catalysts: A Review. Catalysts 2020, 10, 1075. https://0-doi-org.brum.beds.ac.uk/10.3390/catal10091075
AMA Style
Rahman ST, Choi J-R, Lee J-H, Park S-J. The Role of CO2 as a Mild Oxidant in Oxidation and Dehydrogenation over Catalysts: A Review. Catalysts. 2020; 10(9):1075. https://0-doi-org.brum.beds.ac.uk/10.3390/catal10091075
Chicago/Turabian StyleRahman, Sheikh Tareq, Jang-Rak Choi, Jong-Hoon Lee, and Soo-Jin Park. 2020. "The Role of CO2 as a Mild Oxidant in Oxidation and Dehydrogenation over Catalysts: A Review" Catalysts 10, no. 9: 1075. https://0-doi-org.brum.beds.ac.uk/10.3390/catal10091075
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.