Comparison of Catalytic Properties of Vanadium Centers Introduced into BEA Zeolite and Present on (010) V2O5 Surface–DFT Studies

 , ,
, ,
Abstract
:1. Introduction
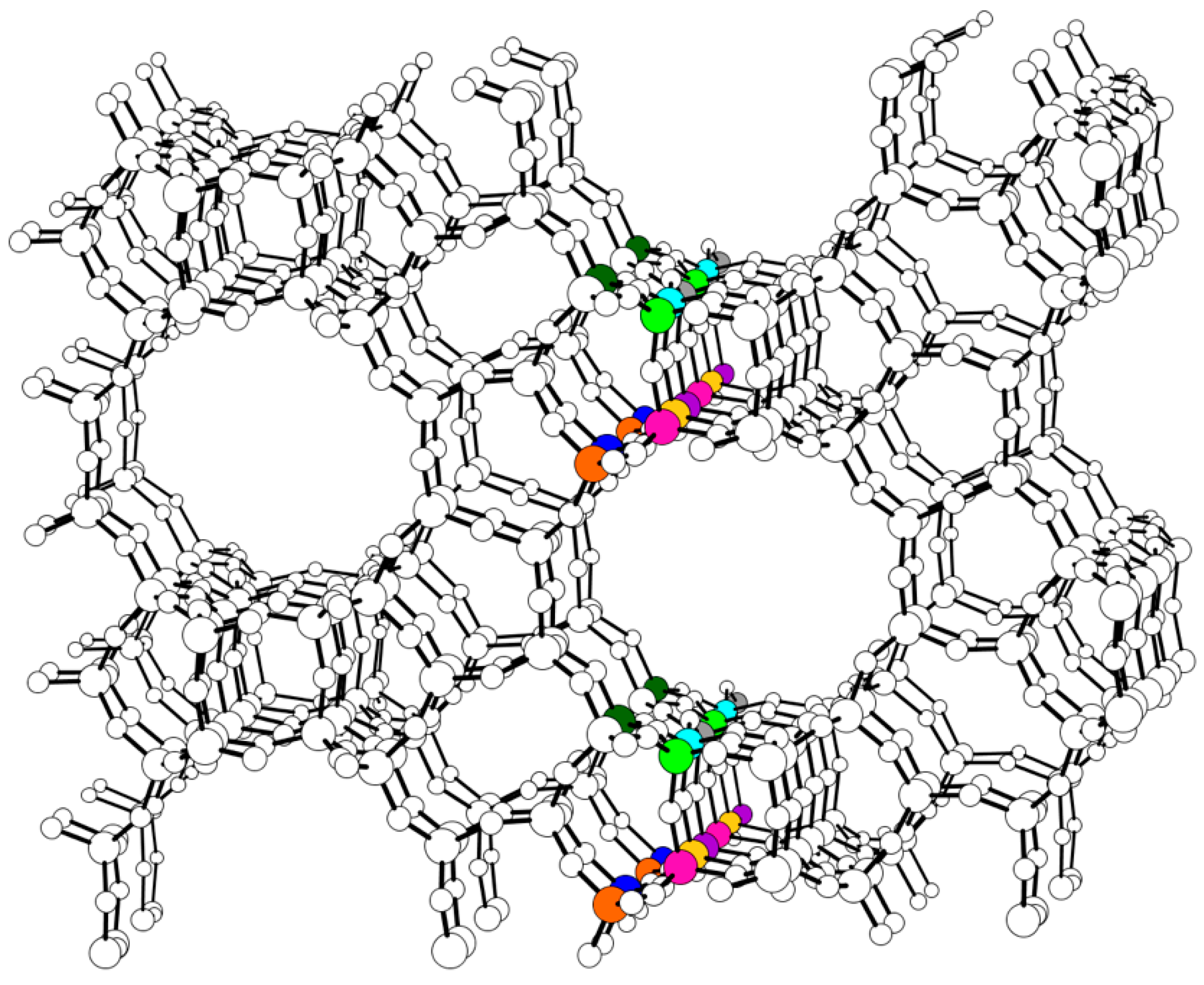
2. Results and Discussion
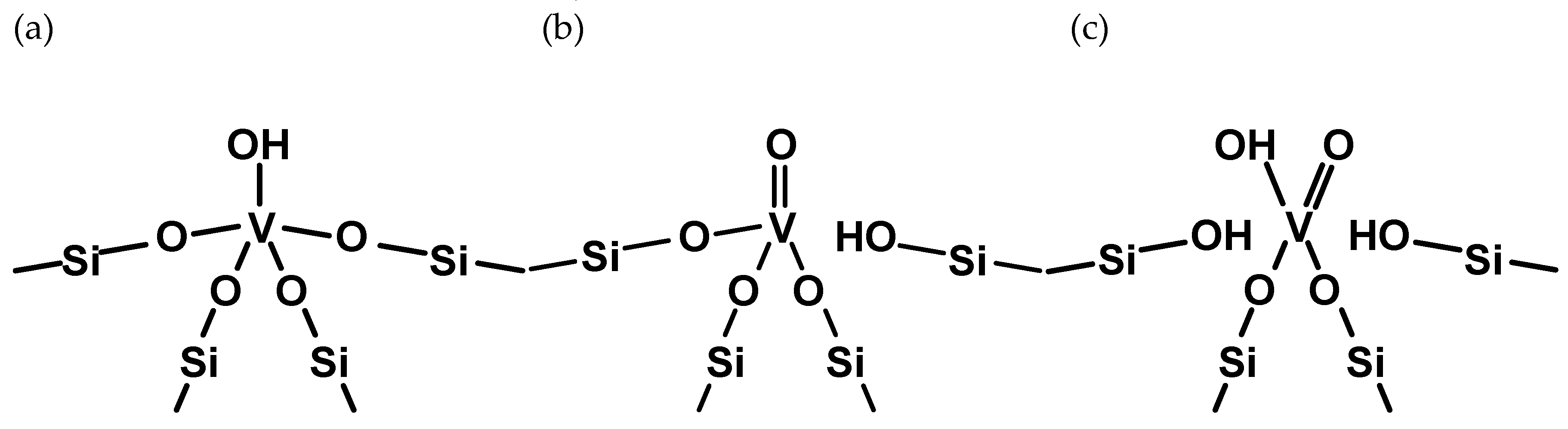
2.1. The Substitution of Si for V–OH
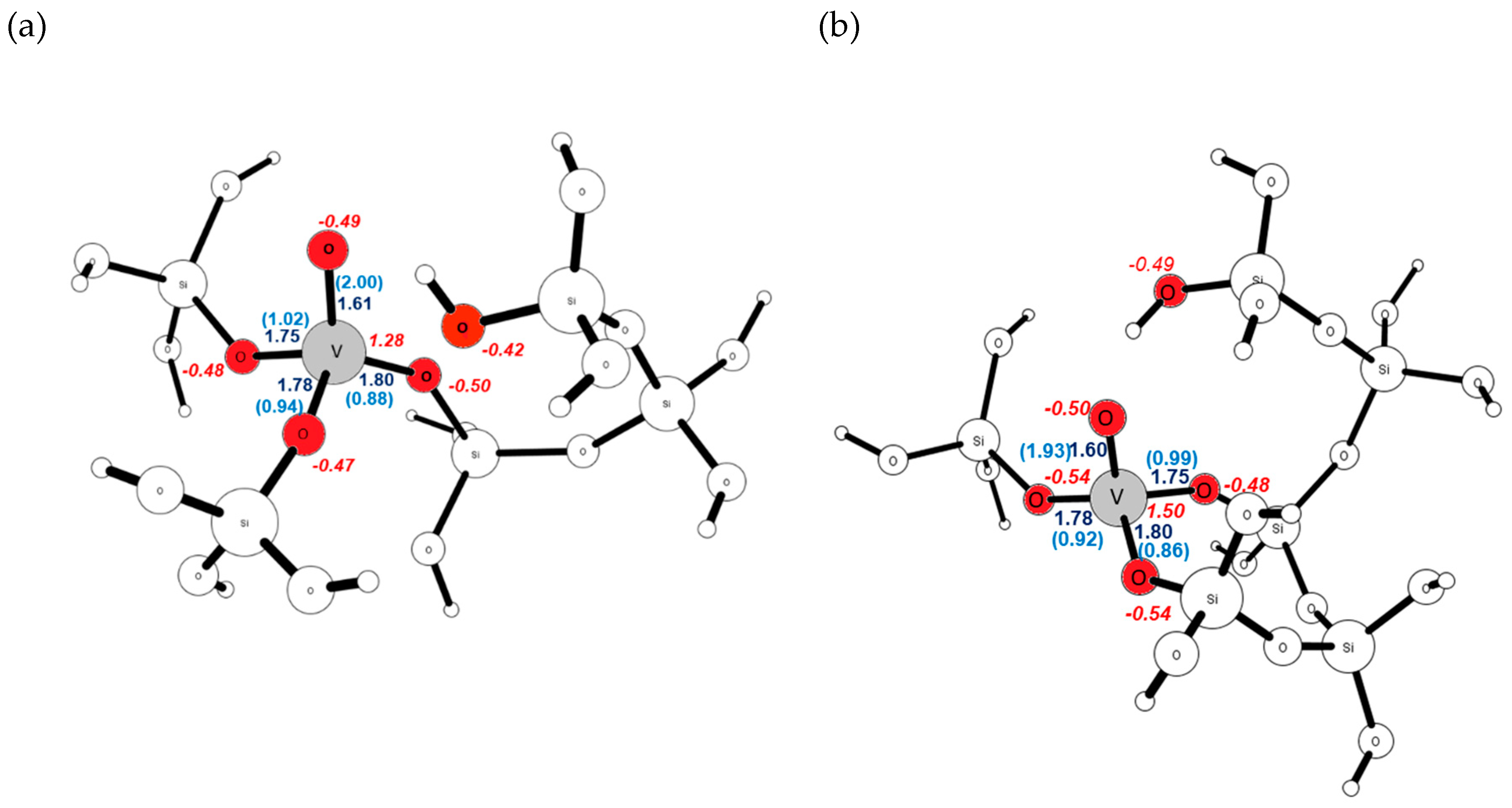
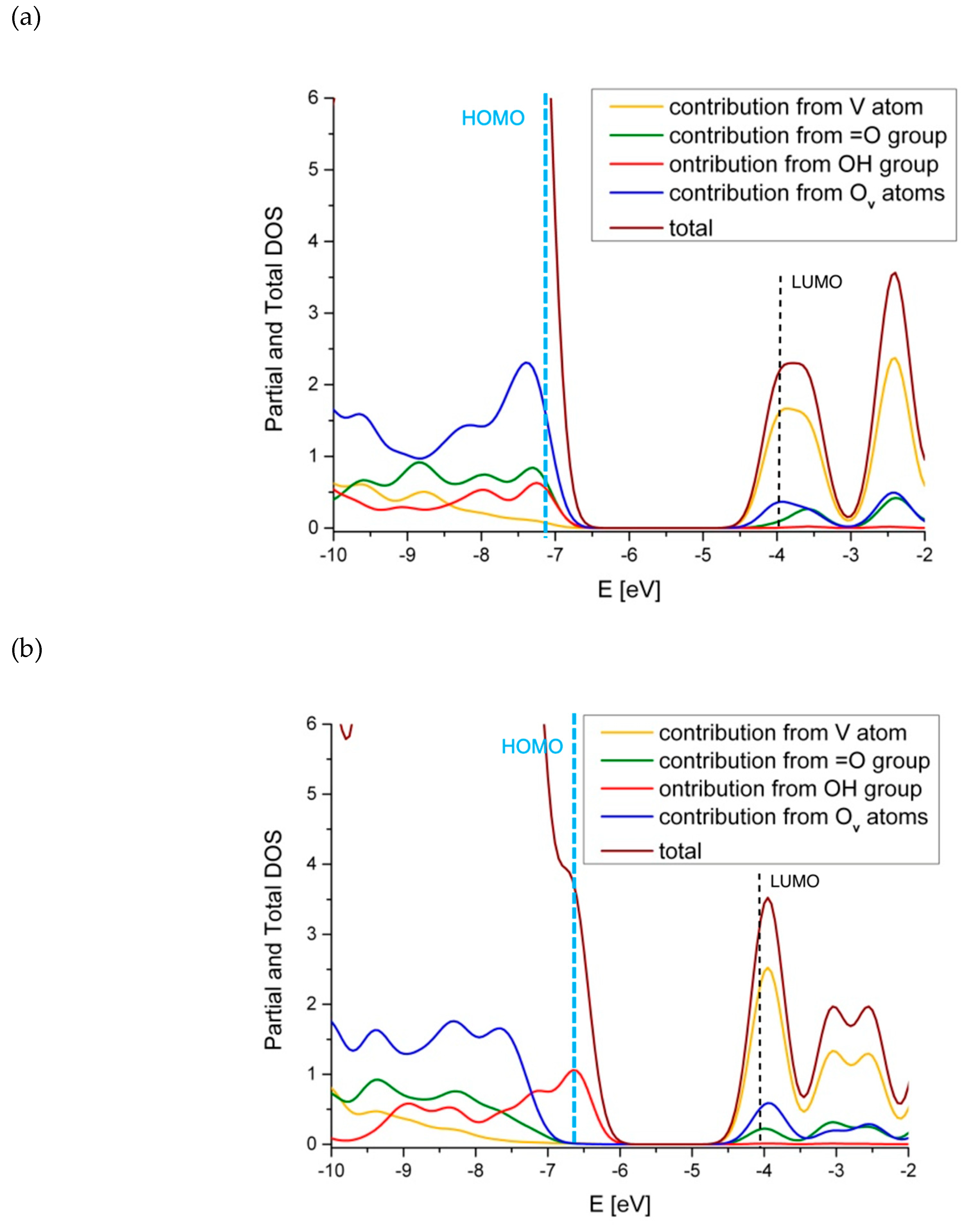
2.2. The Substitution of Si for V=O
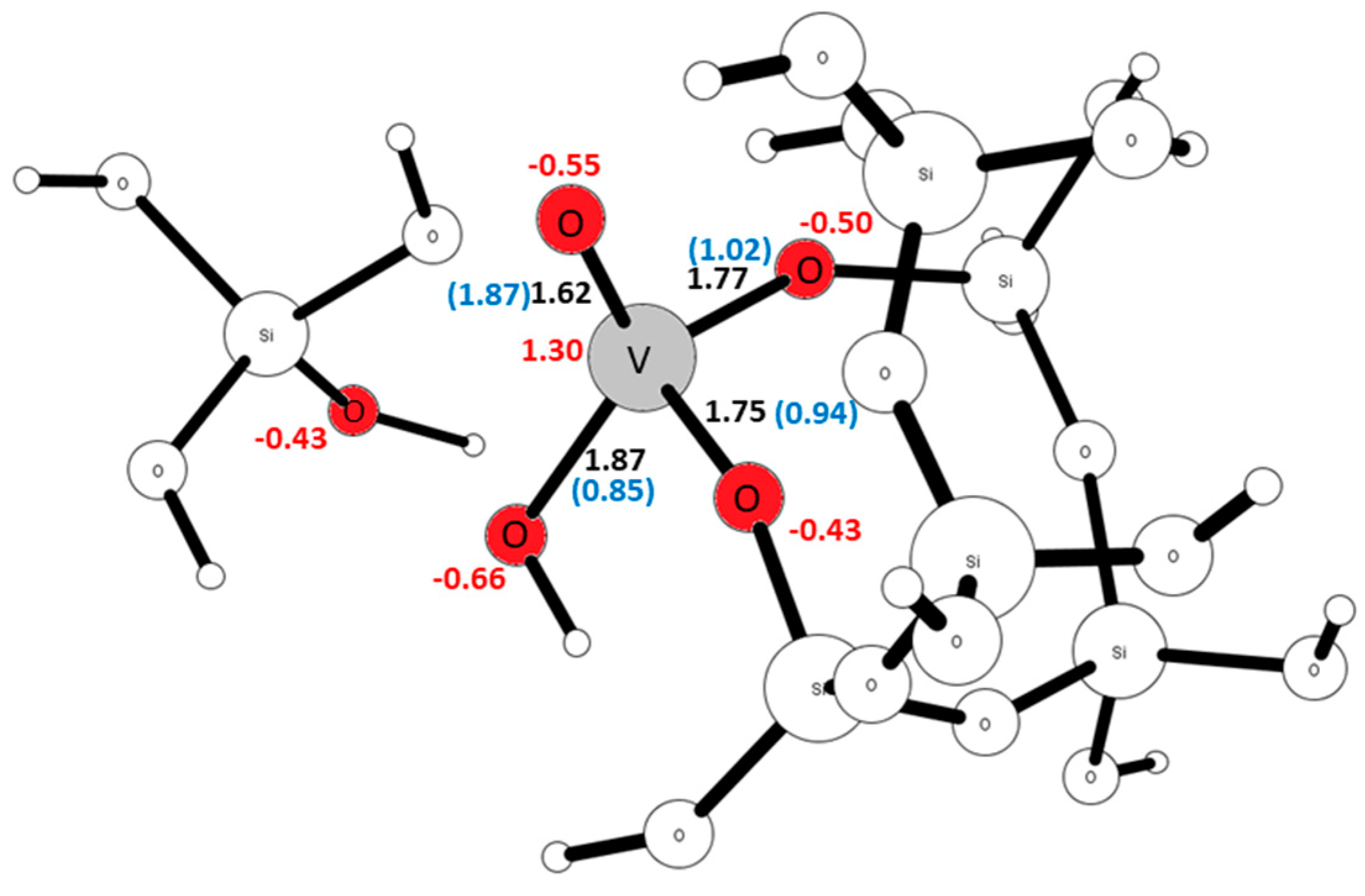
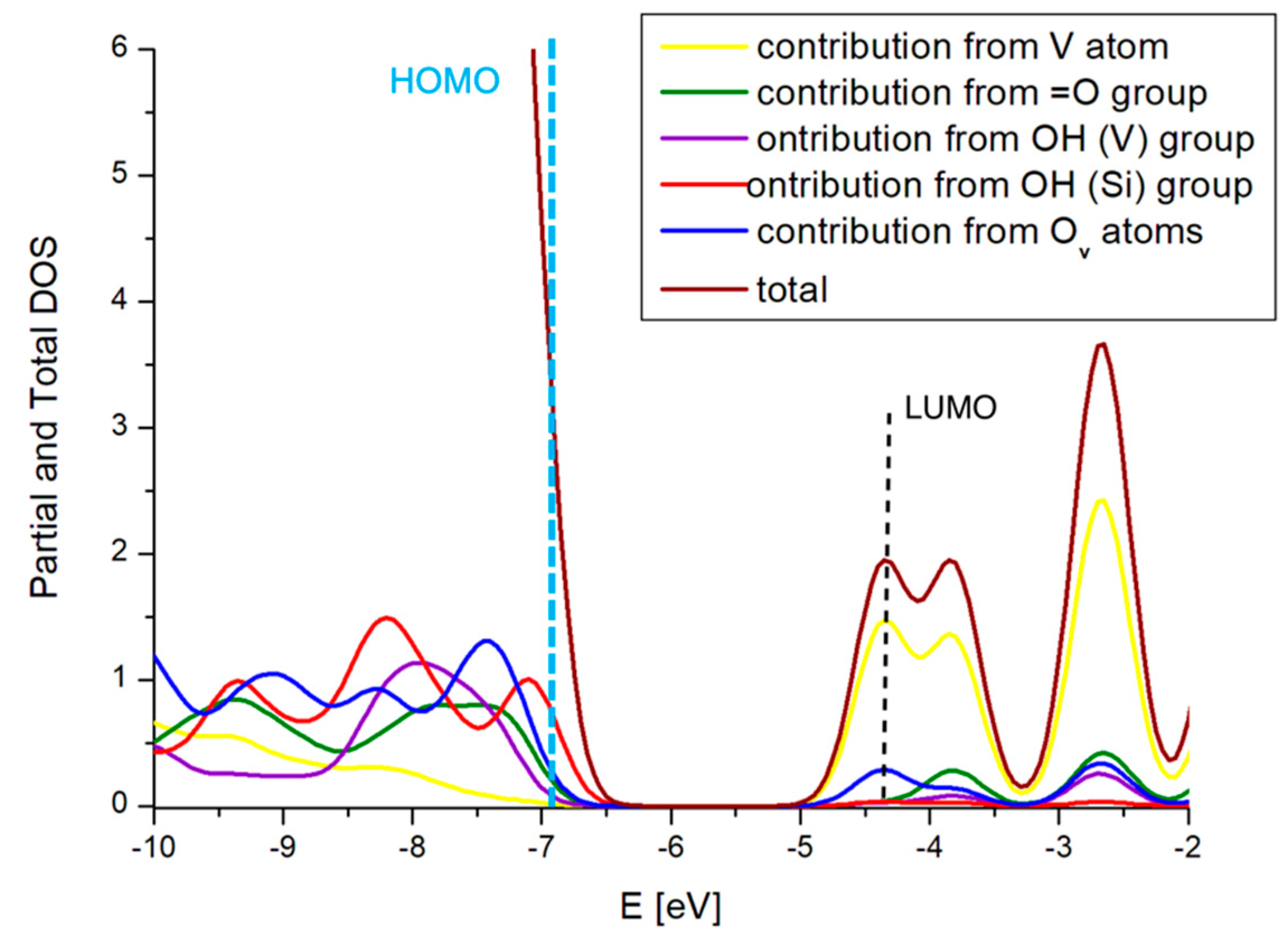
2.3. The Substitution of Si for V(=O)(–OH)
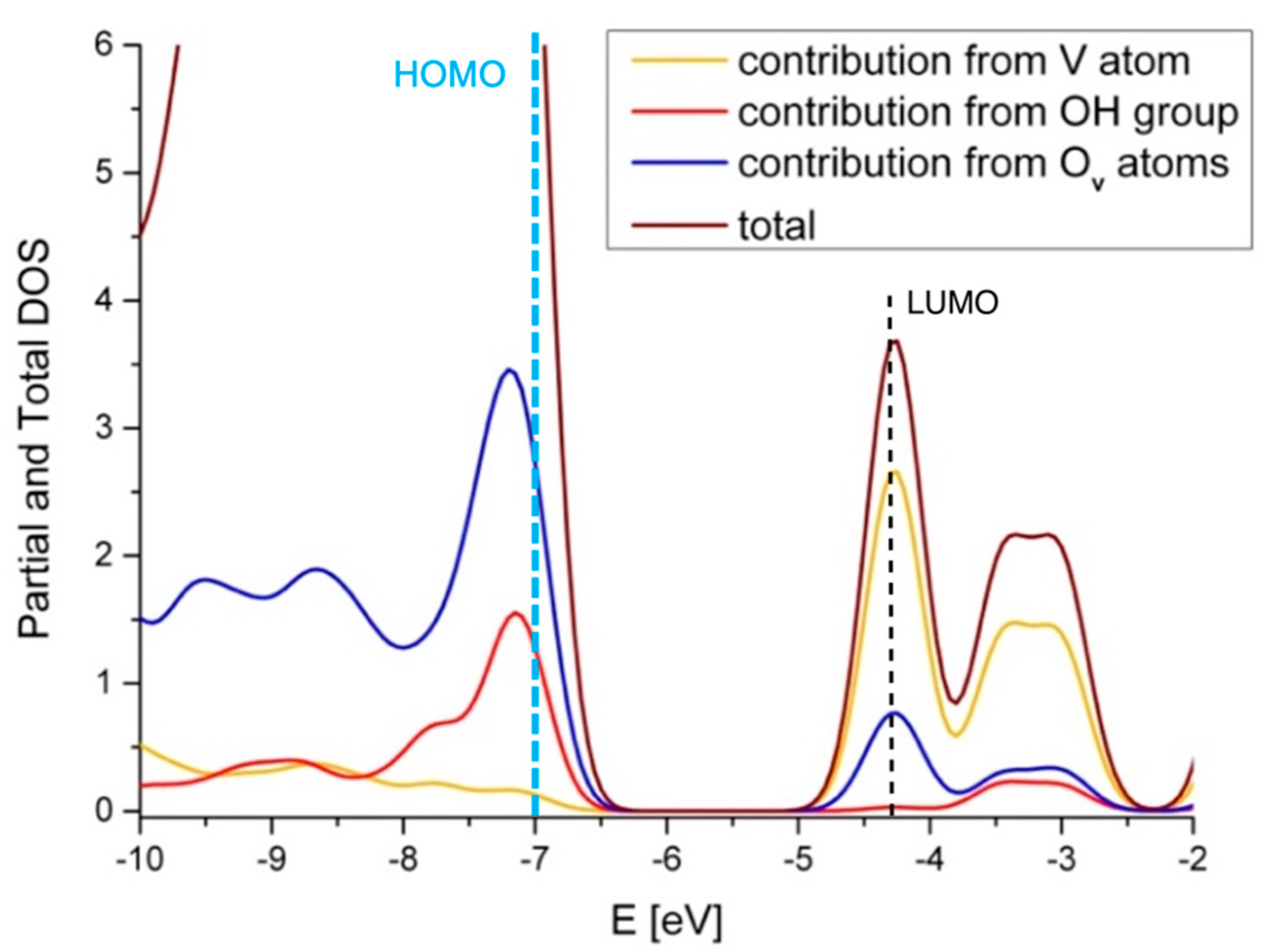
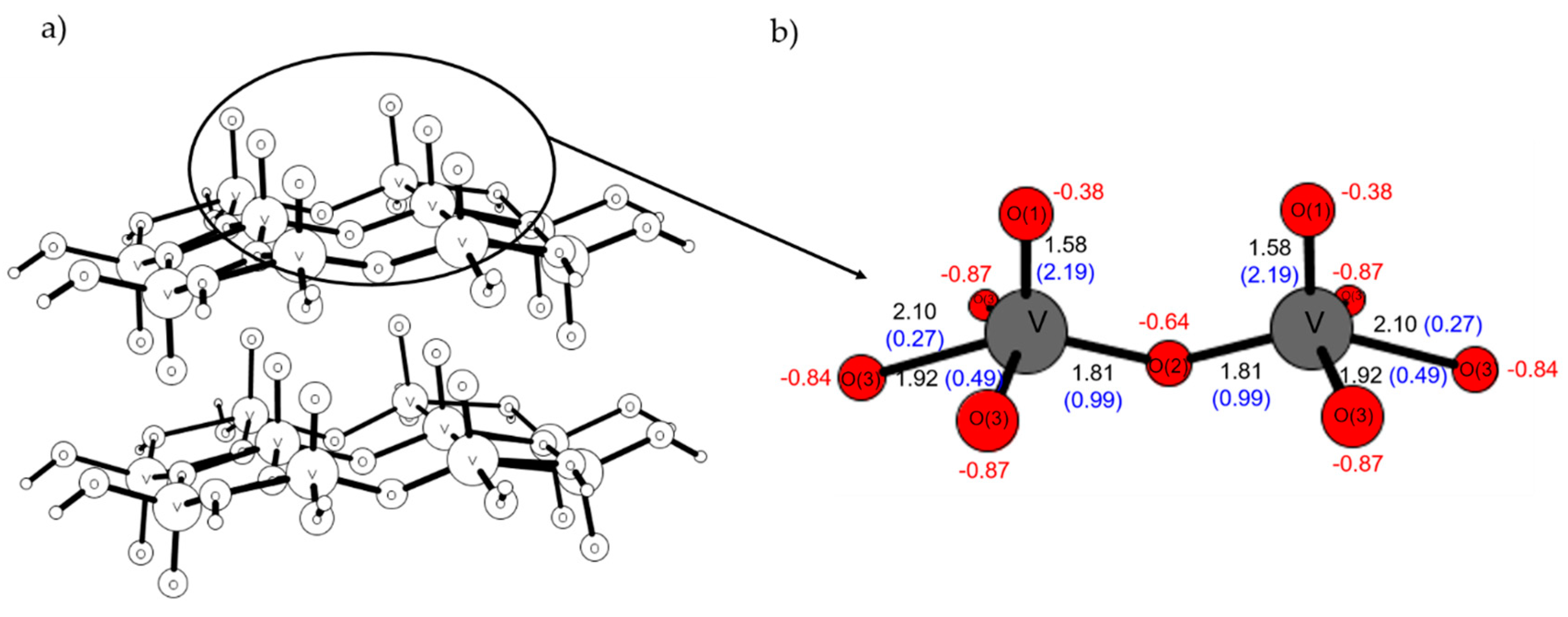
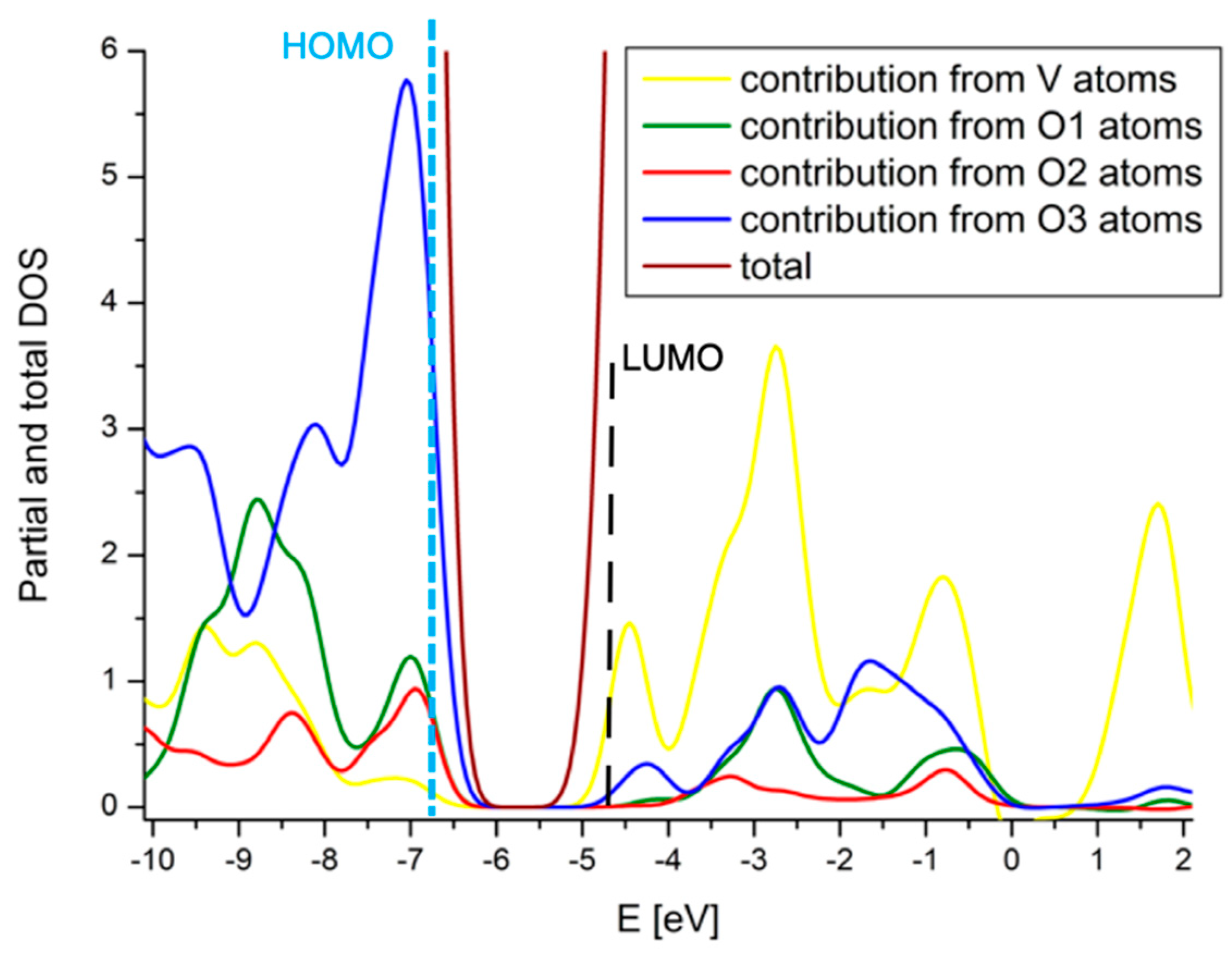
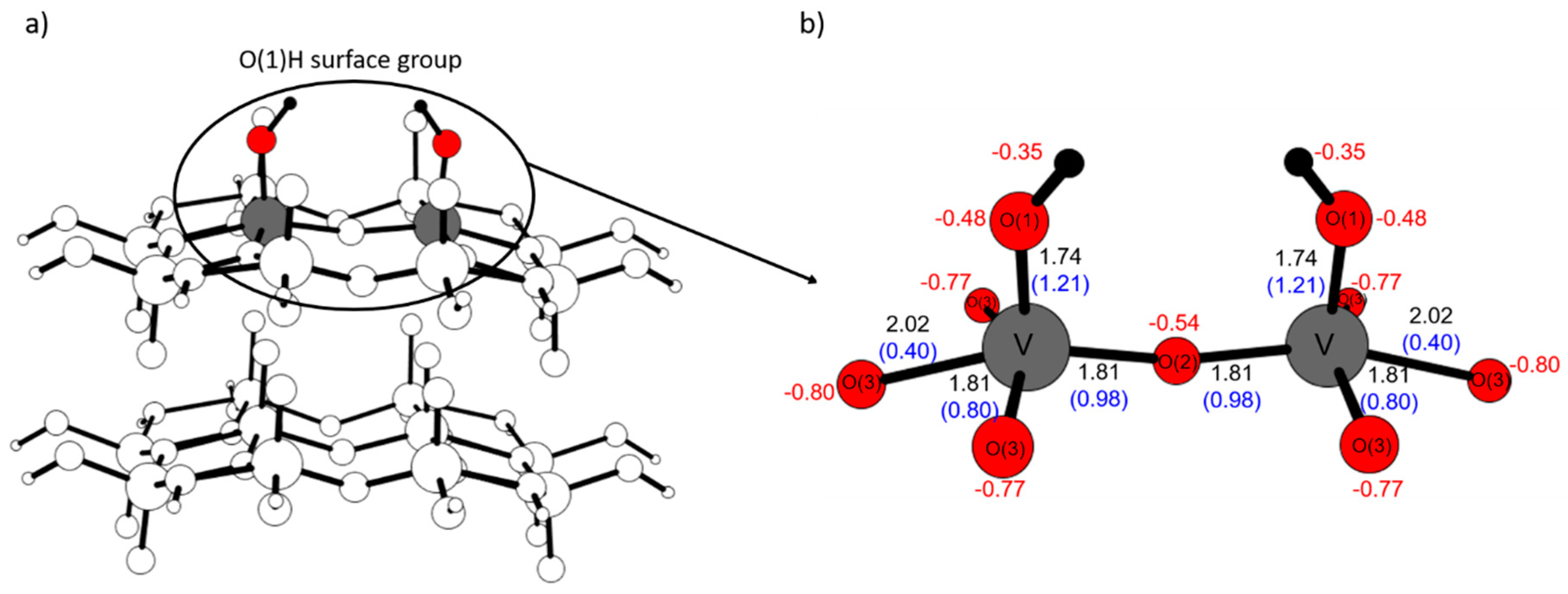
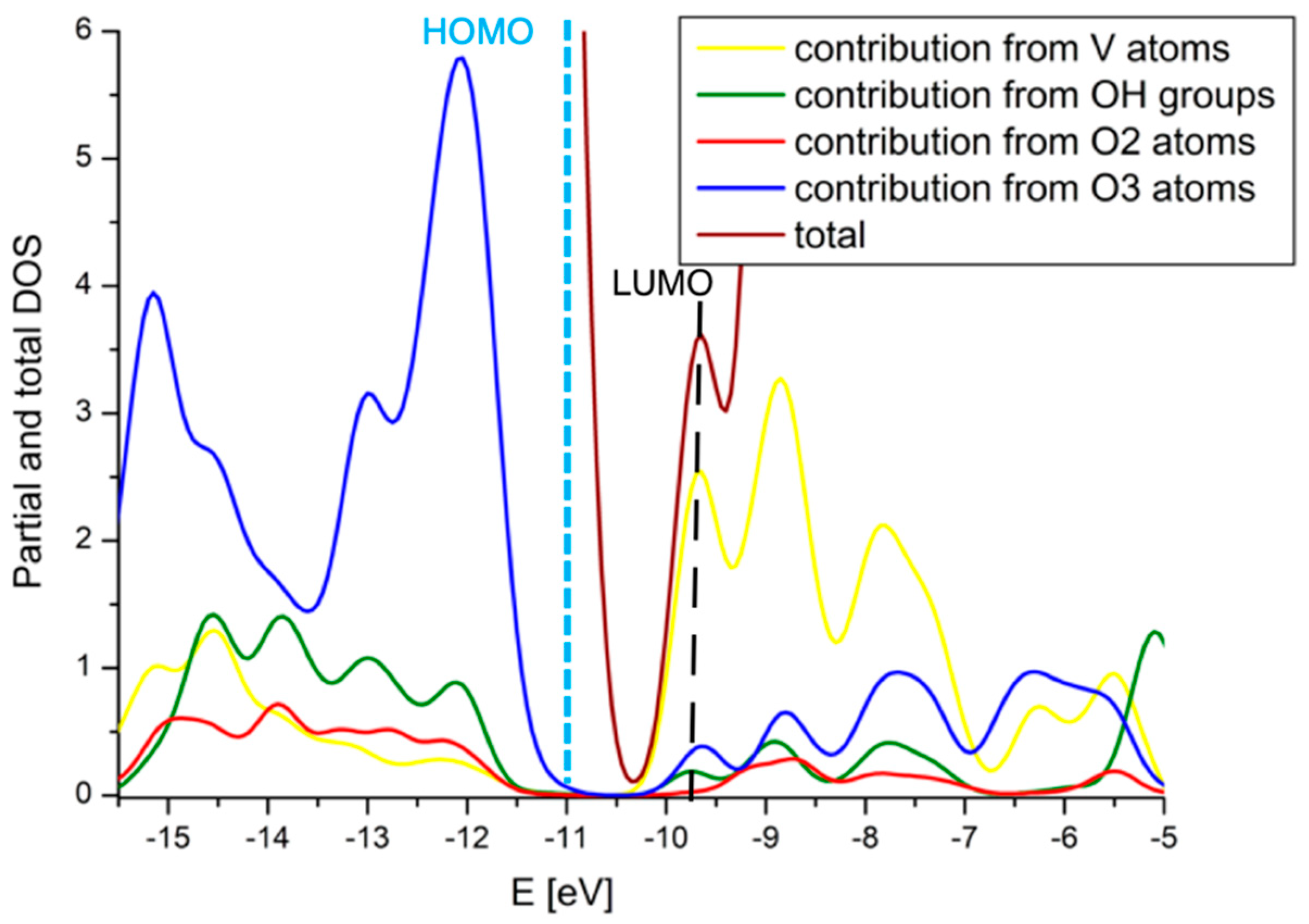
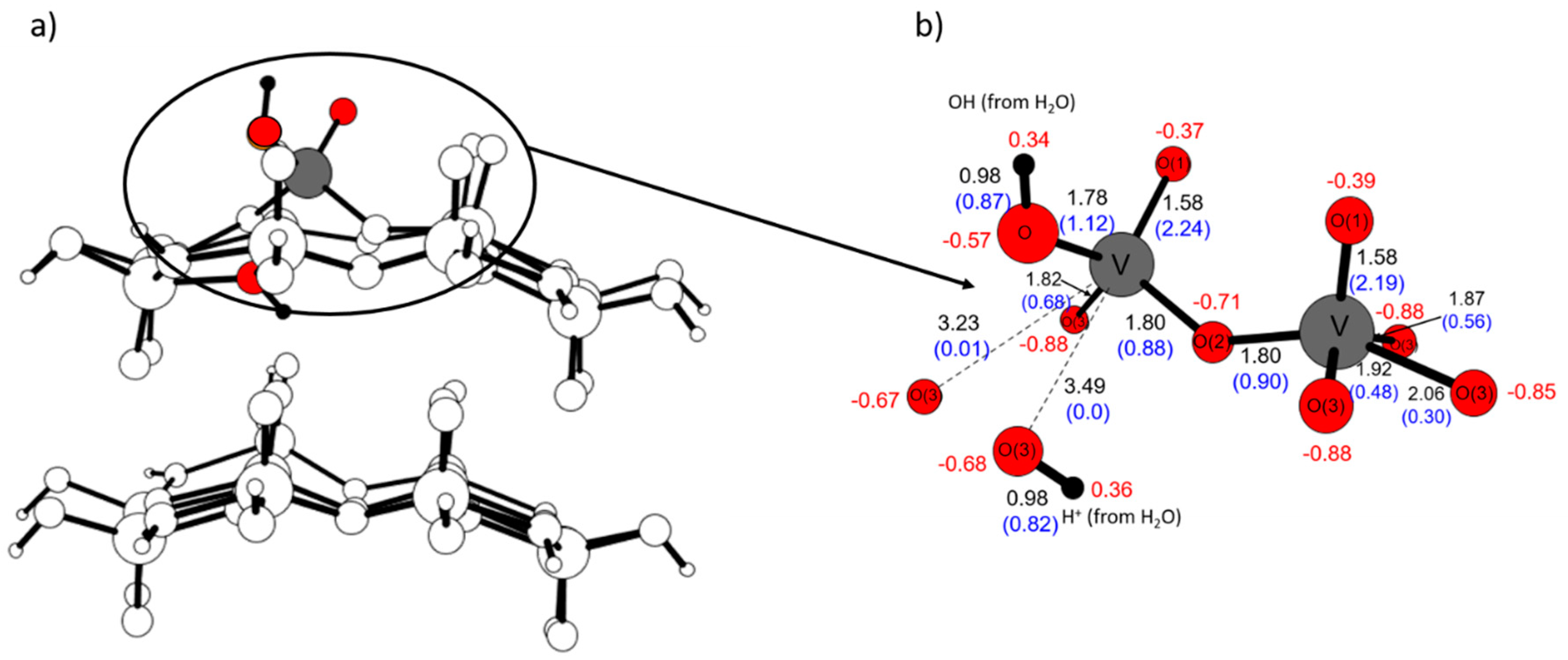
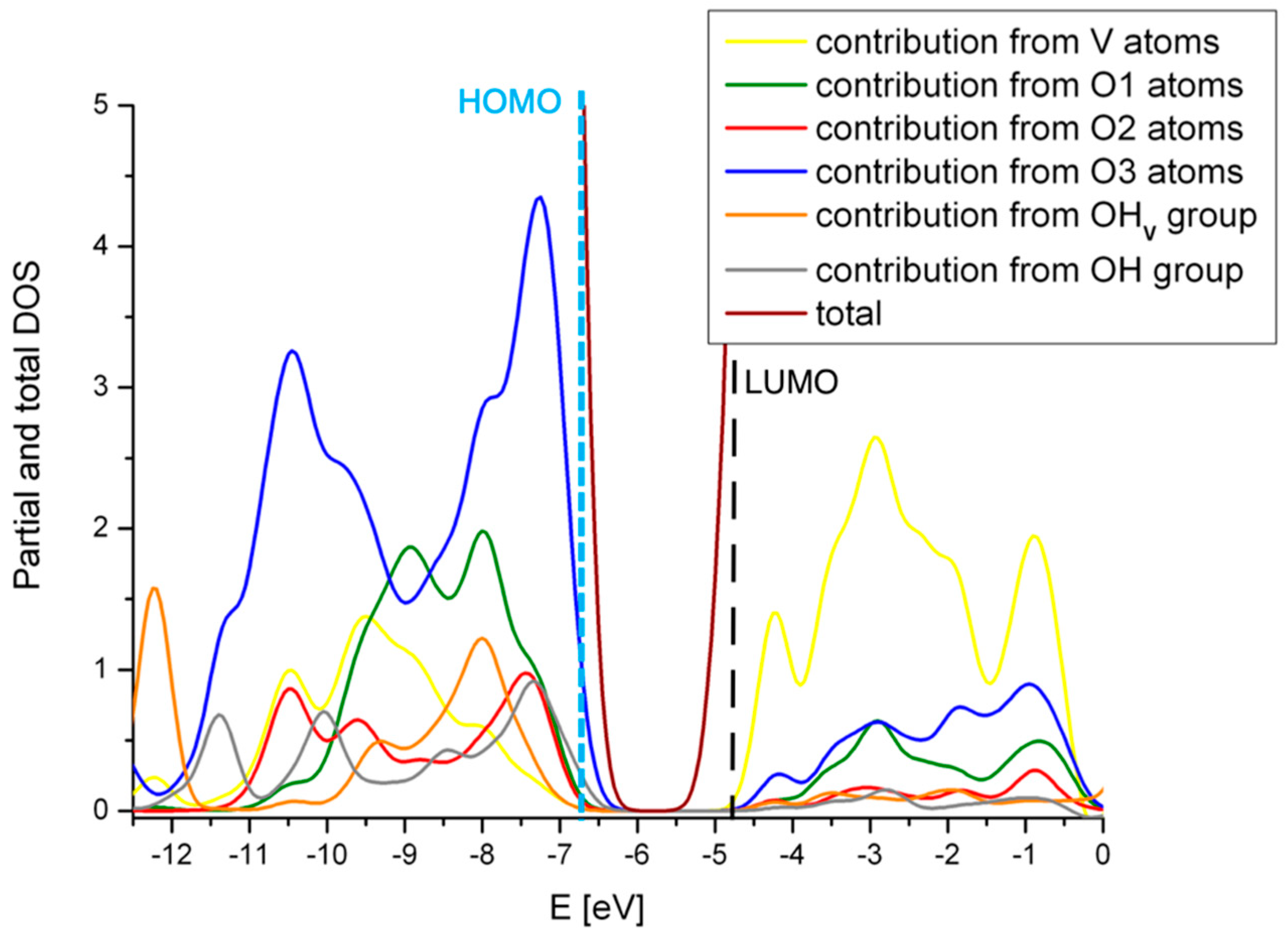
2.4. Comparison with V2O5
3. Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cornils, B.; Herrmann, W.A.; Wong, C.-H.; Zanthoff, H.-W. Catalysis from A to Z. A Concise Encyclopedia; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2013. [Google Scholar]
- Carrero, C.A.; Schloegl, R.; Wachs, I.E.; Schomaecker, R. Critical Literature Review of the Kinetics for the Oxidative Dehydrogenation of Propane over Well-Defined Supported Vanadium Oxide Catalysts. ACS Catal. 2014, 4, 3357–3380. [Google Scholar] [CrossRef]
- Langeslay, R.R.; Kaphan, D.M.; Marshall, C.L.; Stair, P.C.; Sattelberger, A.P.; Delferro, M. Catalytic Applications of Vanadium: A Mechanistic Perspective. Chem. Rev. 2019, 119, 2128–2191. [Google Scholar] [CrossRef] [PubMed]
- Cavani, F.; Trifirò, F. The oxidative dehydrogenation of ethane and propane as an alternative way for the production of light olefins. Catal. Today 1995, 24, 307–313. [Google Scholar] [CrossRef]
- Grasselli, R.K. Fundamental Principles of Selective Heterogeneous Oxidation Catalysis. Top. Catal. 2002, 21, 79–88. [Google Scholar] [CrossRef]
- Knotek, P.; Čapek, L.; Bulánek, R.; Adam, J. Vanadium supported on hexagonal mesoporous silica: Active and stable catalysts in the oxidative dehydrogenation of alkanes. Top. Catal. 2007, 45, 51–55. [Google Scholar] [CrossRef]
- Han, Z.-F.; CXue, X.-L.; Wu, H.-M.; Lang, W.-Z.; Guo, Y.-J. Preparation and catalytic properties of mesoporous nV-MCM-41 for propane oxidative dehydrogenation in the presence of CO2. Chin. J. Catal. 2018, 39, 1099–1109. [Google Scholar] [CrossRef]
- Nguyen, L.D.; Loridant, S.; Launay, H.; Pigamo, A.; Dubois, J.L.; Millet, J.M.M. Study of new catalysts based on vanadium oxide supported on mesoporous silica for the partial oxidation of methane to formaldehyde: Catalytic properties and reaction mechanism. J. Catal. 2006, 237, 38–48. [Google Scholar] [CrossRef]
- Wallis, P.; Wohlrab, S.; Kelevaru, V.N.; Frank, M.; Martin, A. Impact of support pore structure and morphology on catalyst performance of VOx/SBA-15 for selective methane oxidation. Catal. Today 2016, 278, 120–126. [Google Scholar] [CrossRef]
- Baran, R.; Millot, Y.; Onfroy, T.; Averseng, F.; Krafft, J.-M.; Dzwigaj, S. Influence of the preparation procedure on the nature and environment of vanadium in VSiBEA zeolite: XRD, DR UV–vis, NMR, EPR and TPR studies. Microporous Mesoporous Mater. 2012, 161, 179–186. [Google Scholar] [CrossRef]
- El-Roz, M.; Lakiss, L.; Telegeiev, I.; Lebedev, O.I.; Bazin, P.; Vicente, A.; Fernandez, C.; Valtchev, V. High-Visible-Light Photoactivity of Plasma-Promoted Vanadium Clusters on Nanozeolites for Partial Photooxidation of Methanol. ACS Appl. Mater. Interfaces 2017, 9, 17846–17855. [Google Scholar] [CrossRef]
- Smoliło, M.; Samson, K.; Zhou, T.; Duraczyńska, D.; Ruggiero-Mikołajczyk, M.; Drzewiecka-Matuszek, A.; Rutkowska-Zbik, D. Oxidative Dehydrogenation of Propane over Vanadium-Containing Faujasite Zeolite. Molecules 2020, 25, 1961. [Google Scholar]
- Lie Bøyesen, K.; Meneau, F.; Mathisen, K. A combined in situ XAS/Raman and WAXS study on nanoparticulate V2O5 in zeolites ZSM-5 and Y. Phase Transit. 2011, 84, 675–686. [Google Scholar] [CrossRef]
- Chalupka, K.; Thomas, C.; Millot, Y.; Averseng, F.; Dzwigaj, S. Mononuclear pseudo-tetrahedral V species of VSiBEA zeolite as the active sites of the selective oxidative dehydrogenation of propane. J. Catal. 2013, 305, 46–55. [Google Scholar] [CrossRef]
- Held, A.; Kowalska-Kuś, J.; Millot, Y.; Averseng, F.; Calers, C.; Valentin, L.; Dzwigaj, S. Influence of the Preparation Procedure of Vanadium-Containing SiBEA Zeolites on Their Catalytic Activity in Propene Epoxidation. J. Phys. Chem. C 2018, 122, 18570–18582. [Google Scholar] [CrossRef]
- Tranca, D.C.; Keil, F.J.; Tranca, I.; Calatayud, M.; Dzwigaj, S.; Trejda, M.; Tielens, F. Methanol Oxidation to Formaldehyde on VSiBEA Zeolite: A Combined DFT/vdW/Transition Path Sampling and Experimental Study. J. Phys. Chem. C 2015, 119, 13619–13631. [Google Scholar] [CrossRef]
- Baran, R.; Onfroy, T.; Grzybek, T.; Dzwigaj, T. Influence of the nature and environment of vanadium in VSiBEA zeolite on selective catalytic reduction of NO with ammonia. Appl. Catal. B Environ. 2013, 136–137, 186–192. [Google Scholar] [CrossRef]
- Tielens, F. Exploring the reactivity of intraframework vanadium, niobium and tantalum sites in zeolitic materials using the molecular electrostatic potential. J. Mol. Struct. THEOCHEM 2009, 903, 23–27. [Google Scholar] [CrossRef]
- Tielens, F.; Calatayud, M.; Dzwigaj, S.; Che, M. What do vanadium framework sites look like in redox model silicate zeolites? Microporous Mesoporous Mater. 2009, 119, 137–143. [Google Scholar] [CrossRef]
- Tielens, F.; Trejda, M.; Ziolek, M.; Dzwigaj, S. Nature of vanadium species in V substituted zeolites: A combined experimental and theoretical study. Catal. Today 2008, 139, 221–226. [Google Scholar] [CrossRef]
- Tielens, F.; Dzwigaj, S. Probing acid–base sites in vanadium redox zeolites by DFT calculation and compared with FTIR results. Catal. Today 2010, 152, 66–69. [Google Scholar] [CrossRef]
- Wojtaszek, A.; Ziolek, M.; Dzwigaj, S.; Tielens, F. Comparison of competition between T=O and T–OH groups in vanadium, niobium, tantalum BEA zeolite and SOD based zeolites. Chem. Phys. Lett. 2011, 514, 70–73. [Google Scholar] [CrossRef]
- Witko, M.; Tokarz-Sobieraj, R. Surface oxygen in catalysts based on transition metal oxides—What can we learn from cluster DFT calculations? Catal. Today 2004, 91–92, 171–176. [Google Scholar] [CrossRef]
- Kerber, T.; Sierka, M.; Sauer, J. Application of semiempirical long-range dispersion corrections to periodic systems in density functional theory. J. Comput. Chem. 2008, 29, 2088–2097. [Google Scholar] [CrossRef] [PubMed]
- Jovanović, A.; Dobrota, A.S.; Rafailović, L.D.; Mentus, S.V.; Pašti, I.A.; Johansson, B.; Skorodumova, N.V. Structural and electronic properties of V2O5 and their tuning by doping with 3d elements—Modelling using the DFT+U method and dispersion correction. Phys. Chem. Chem. Phys. 2018, 20, 13934–13943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganduglia-Pirovano, M.V.; Sauer, J. Stability of reduced V2O5 (001) surfaces. Phys. Rev. B 2004, 70, 045422. [Google Scholar] [CrossRef]
- Helali, Z.; Jedidi, A.; Syzgantseva, O.A.; Calatayud, M.; Minot, C. Scaling reducibility of metal oxides. Theor. Chem. Acc. 2017, 136, 100. [Google Scholar] [CrossRef]
- Słoczyński, J.; Grabowski, R.; Kozłowska, A.; Tokarz-Sobieraj, R.; Witko, M. Interaction of oxygen with the surface of vanadia catalysts. J. Mol. Catal. A Chem. 2007, 277, 27–34. [Google Scholar] [CrossRef]
- Goclon, J.; Grybos, R.; Witko, M.; Hafner, J. Relative stability of low-index V2O5 surfaces: A density functional investigation. J. Physics: Condens. Matter 2009, 21, 095008. [Google Scholar]
- Oshio, T.; Sakai, Y.; Ehara, S. Scanning tunneling microscopy/spectroscopy study of V2O5 surface with oxygen vacancies. J. Vac. Sci. Technol. B Microelectron. Nanometer Struct. 1994, 12, 2055–2059. [Google Scholar] [CrossRef]
- Smith, R.L.; Rohrer, G.S.; Lee, K.S.; Seo, D.-K.; Whangbo, M.-H. A scanning probe microscopy study of the (001) surfaces of V2O5 and V6O13. Surf. Sci. 1996, 367, 87–95. [Google Scholar] [CrossRef]
- Goschke, R.A.; Vey, K.; Maier, M.; Walter, U.; Goering, E.; Klemm, M.; Horn, S. Tip induced changes of atomic scale images of the vanadiumpentoxide surface. Surf. Sci. 1996, 348, 305–310. [Google Scholar] [CrossRef]
- Structure Commission of the International Zeolite Association. Zeolite Structures Database. Available online: http://www.iza-structure.org/databases/ (accessed on 21 November 2017).
- Migues, A.N.; Muskat, A.; Auerbach, S.M.; Sherman, W.; Vaitheeswaran, S. On the Rational Design of Zeolite Clusters. ACS Catal. 2015, 5, 2859–2865. [Google Scholar] [CrossRef] [Green Version]
- Migues, A.N.; Sun, Q.; Vaitheeswaran, S.; Sherman, W.; Auerbach, S.M. On the Rational Design of Zeolite Clusters for Converging Reaction Barriers: Quantum Study of Aldol Kinetics Confined in HZSM-5. J. Phys. Chem. C 2018, 122, 23230–23241. [Google Scholar] [CrossRef]
- Fermann, J.T.; Moniz, T.; Kiowski, O.; McIntire, T.J.; Auerbach, S.M.; Vreven, T.; Frisch, M.J. Modeling Proton Transfer in Zeolites: Convergence Behavior of Embedded and Constrained Cluster Calculations. J. Chem. Theory Comput. 2005, 1, 1232–1239. [Google Scholar] [CrossRef]
- de Vries, A.H.; Sherwood, P.; Collins, S.J.; Rigby, A.M.; Rigutto, M.; Kramer, G.J. Zeolite Structure and Reactivity by Combined Quantum-Chemical−Classical Calculations. J. Phys. Chem. B 1999, 103, 6133–6141. [Google Scholar] [CrossRef]
- Sierka, M.; Sauer, J. Finding transition structures in extended systems: A strategy based on a combined quantum mechanics–empirical valence bond approach. J. Chem. Phys. 2000, 112, 6983–6996. [Google Scholar] [CrossRef]
- Sherwood, P.; De Vries, A.H.; Collins, S.J.; Greatbanks, S.P.; Burton, N.A.; Vincent, M.A.; Hillier, I.H. Computer simulation of zeolite structure and reactivity using embedded cluster methods. Faraday Discuss. 1997, 106, 79–92. [Google Scholar] [CrossRef]
- Centi, G.; Trifiro, F. Catalytic behavior of V-containing zeolites in the transformation of propane in the presence of oxygen. Appl. Catal. A-Gen. 1996, 143, 3–16. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Hieu, N.V.; Lichtman, D. Bandgap radiation induced photodesorption from V2O5 powder and vanadium oxide surfaces. J. Vac. Sci. Technol. 1981, 18, 49–53. [Google Scholar] [CrossRef]
- Cogan, S.F.; Nguyen, N.M.; Perrotti, S.J.; Rauh, R.D. Optical properties of electrochromic vanadium pentoxide. J. Appl. Phys. 1989, 66, 1333–1337. [Google Scholar] [CrossRef]
- Moshfegh, A.Z.; Ignatiev, A. Formation and characterization of thin film vanadium oxides: Auger electron spectroscopy, X-ray photoelectron spectroscopy, X-ray diffraction, scanning electron microscopy, and optical reflectance studies. Thin Solid Films 1991, 198, 251–268. [Google Scholar] [CrossRef]
- Hermann, K.; Witko, M.; Druzinic, R.; Tokarz, R. Hydrogen assisted oxygen desorption from the V2O5(010) surface. Top. Catal. 2000, 11, 67–75. [Google Scholar] [CrossRef]
- Hejduk, P.; Szaleniec, M.; Witko, M. Molecular and dissociative adsorption of water at low-index V2O5 surfaces: DFT studies using cluster surface models. J. Mol. Catal. A 2010, 325, 98–104. [Google Scholar] [CrossRef]
- TURBOMOLE V6.3 adoUoKa; Forschungszentrum Karlsruhe GmbH; TURBOMOLE GmbH, saf. 2011. Available online: http://www.turbomole.com (accessed on 7 February 2011).
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Eichkorn, K.; Treutler, O.; Öhm, H.; Häser, M.; Ahlrichs, R. Auxiliary Basis Sets to Approximate Coulomb Potentials. Chem. Phys. Lett. 1995, 240, 283–289. [Google Scholar] [CrossRef]
- Eichkorn, K.; Weigend, F.; Treutler, O.; Ahlrichs, R. Auxiliary basis sets for main row atoms and transition metals and their use to approximate Coulomb potentials. Theor. Chem. Acc. 1997, 97, 119–124. [Google Scholar] [CrossRef]
- Mulliken, R.S. Electronic Population Analysis on LCAO[Single Bond]MO Molecular Wave Functions. I. J. Chem. Phys. 1955, 23, 1833–1840. [Google Scholar] [CrossRef] [Green Version]
- Gorelsky, S.I.; Lever, A.B.P. Electronic structure and spectra of ruthenium diimine complexes by density functional theory and INDO/S. Comparison of the two methods. J. Organomet. Chem. 2001, 635, 187–196. [Google Scholar] [CrossRef]
- Gorelsky, S.I. AOMix: Program for Molecular Orbital Analysis. version X.X. 2015. Available online: http://www.sg-chem.net/ (accessed on 21 May 2013).















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Site | ΔE V–OH [eV] | ΔE V=O [eV] | ΔE V(=O)(–OH) [eV] |
|---|---|---|---|
| T1 | 0.23 | −0.69 | 1.43 |
| T2 | −2.16 | −2.24 | −1.51 |
| T3 | −1.69 | −2.21 | −2.19 |
| T4 | −0.42 | −1.14 | −1.29 |
| T5 | −0.41 | −0.85 | −1.09 |
| T6 | 0.10 | 0.00 | −0.40 |
| T7 | −0.48 | −1.12 | −0.14 |
| T8 | 0.06 | −1.42 | −1.38 |
| T9 | −1.19 | −1.79 | −1.41 |
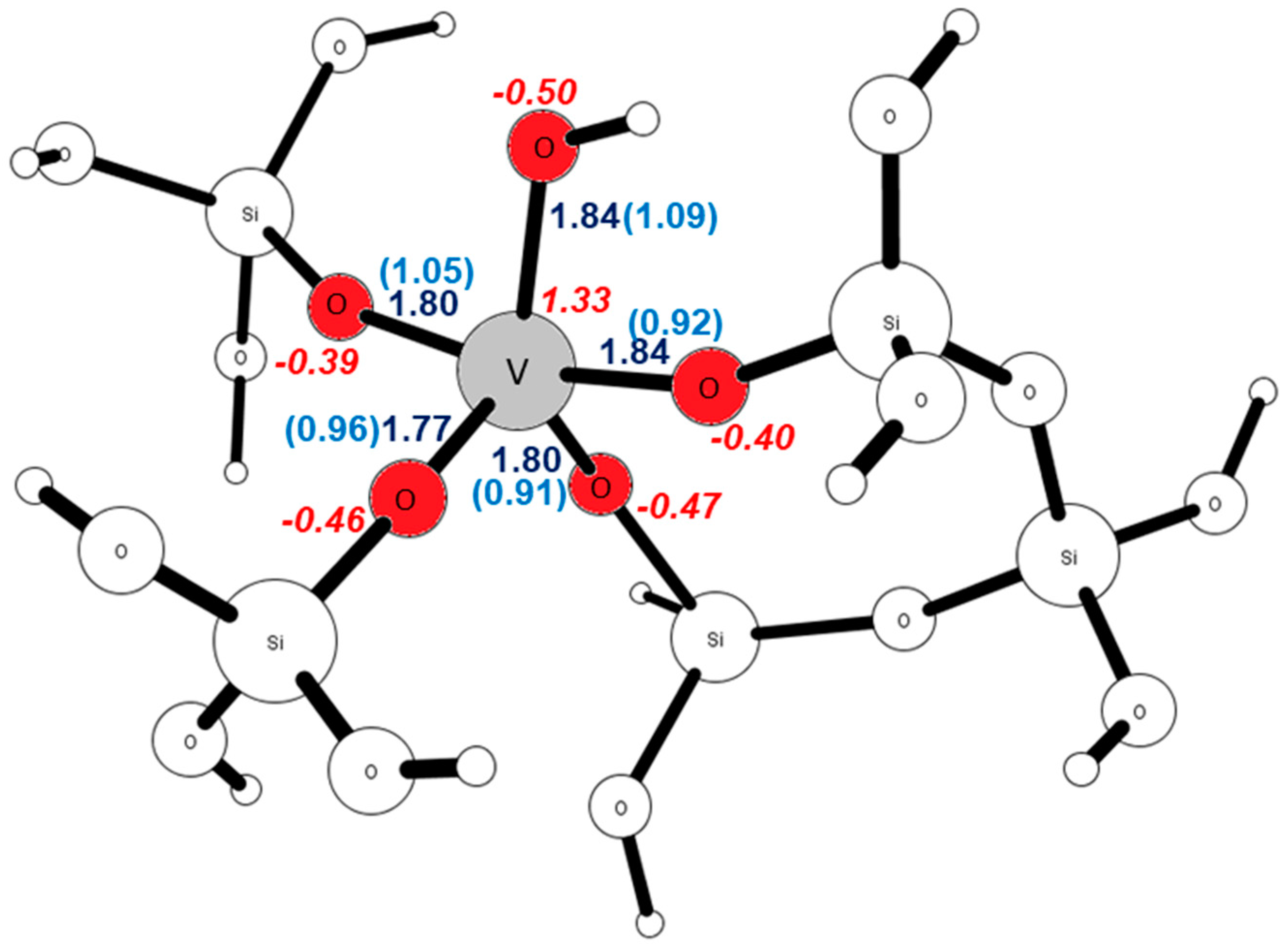
| Site | R(V–OH) [Å] | B.O. | qOOH | qOH | qV |
|---|---|---|---|---|---|
| T1 | 1.85 | 1.12 | −0.48 | −0.16 | 1.25 |
| T2 | 1.84 | 1.09 | −0.50 | −0.17 | 1.33 |
| T3 | 1.82 | 1.12 | −0.49 | −0.16 | 1.37 |
| T4 | 1.83 | 1.09 | −0.50 | −0.17 | 1.39 |
| T5 | 1.83 | 1.09 | −0.49 | −0.16 | 1.38 |
| T6 | 1.84 | 1.14 | −0.48 | −0.16 | 1.31 |
| T7 | 1.84 | 1.09 | −0.50 | −0.17 | 1.39 |
| T8 | 1.81 | 0.94 | −0.46 | −0.13 | 1.31 |
| T9 | 1.82 | 1.22 | −0.44 | −0.12 | 1.27 |
| Site | R(V–O) [Å] | B.O. | qO | qV |
|---|---|---|---|---|
| T1 | 1.60 | 2.09 | −0.46 | 1.23 |
| T2 | 1.61 | 2.00 | −0.49 | 1.28 |
| T3 | 1.60 | 1.93 | −0.50 | 1.50 |
| T4 | 1.60 | 1.83 | −0.54 | 1.58 |
| T5 | 1.61 | 1.87 | −0.54 | 1.49 |
| T6 | 1.59 | 1.91 | −0.50 | 1.53 |
| T7 | 1.61 | 1.89 | −0.52 | 1.49 |
| T8 | 1.60 | 1.91 | −0.51 | 1.48 |
| T9 | 1.60 | 1.91 | −0.51 | 1.60 |
| Site | R(V=O) [Å] | B.O. V=O | R(V–OH) [Å] | B.O. V–OH | qOO | qOOH | qOH | qV |
|---|---|---|---|---|---|---|---|---|
| T1 | 1.62 | 2.18 | 1.89 | 1.03 | −0.34 | −0.58 | −0.27 | 0.76 |
| T2 | 162 | 2.17 | 1.83 | 1.03 | −0.51 | −0.65 | −0.33 | 1.06 |
| T3 | 1.62 | 1.87 | 1.87 | 0.85 | −0.55 | −0.66 | −0.35 | 1.30 |
| T4 | 1.62 | 1.84 | 1.85 | 0.90 | −0.57 | −0.64 | −0.32 | 1.30 |
| T5 | 1.61 | 1.99 | 1.89 | 0.82 | −0.49 | −0.70 | −0.38 | 1.24 |
| T6 | 1.58 | 2.17 | 1.80 | 1.03 | −0.41 | −0.61 | −0.26 | 1.40 |
| T7 | 1.62 | 1.90 | 1.86 | 0.82 | −0.54 | −0.69 | −0.38 | 1.24 |
| T8 | 1.60 | 1.98 | 1.78 | 1.14 | −0.51 | −0.54 | −0.21 | 1.33 |
| T9 | 1.62 | 1.78 | 1.76 | 1.19 | −0.61 | −0.54 | −0.20 | 1.40 |
| Parameter | Site on (010) V2O5 | Site in V-BEA |
|---|---|---|
| V(–OH) group | ||
| R (V–OH) | 1.74 | 1.84 a |
| B.O. (V–OH) | 1.21 | 1.09 a |
| qO | −0.48 | −0.50 a |
| qOH | −0.13 | −0.17 a |
| qV | 1.70 | 1.33 a |
| V(=O) group | ||
| R (V=O) | 1.58 | 1.60 a, 1.60 b |
| B.O. (V=O) | 2.19 | 2.00 a, 1.93 b |
| qO | −0.38 | −0.49 a, −0.50 b |
| qV | 1.70 | 1.28 a, 1.50 b |
| V(=O)(–OH) group | ||
| R (V–OH) | 1.78 | 1.83 b |
| R (V=O) | 1.58 | 1.62 b |
| B.O. (V–OH) | 1.12 | 0.85 b |
| B.O. (V=O) | 2.24 | 1.87 b |
| qOOH | −0.57 | −0.66 b |
| qOH | −0.23 | −0.35 b |
| qOO | −0.37 | −0.55 b |
| qV | 1.41 | 1.30 b |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Drzewiecka-Matuszek, A.; Tokarz-Sobieraj, R.; Witko, M.; Rutkowska-Zbik, D. Comparison of Catalytic Properties of Vanadium Centers Introduced into BEA Zeolite and Present on (010) V2O5 Surface–DFT Studies. Catalysts 2020, 10, 1080. https://0-doi-org.brum.beds.ac.uk/10.3390/catal10091080
Drzewiecka-Matuszek A, Tokarz-Sobieraj R, Witko M, Rutkowska-Zbik D. Comparison of Catalytic Properties of Vanadium Centers Introduced into BEA Zeolite and Present on (010) V2O5 Surface–DFT Studies. Catalysts. 2020; 10(9):1080. https://0-doi-org.brum.beds.ac.uk/10.3390/catal10091080
Chicago/Turabian StyleDrzewiecka-Matuszek, Agnieszka, Renata Tokarz-Sobieraj, Małgorzata Witko, and Dorota Rutkowska-Zbik. 2020. "Comparison of Catalytic Properties of Vanadium Centers Introduced into BEA Zeolite and Present on (010) V2O5 Surface–DFT Studies" Catalysts 10, no. 9: 1080. https://0-doi-org.brum.beds.ac.uk/10.3390/catal10091080