Photocatalytic H2 Production from Naphthalene by Various TiO2 Photocatalysts: Impact of Pt Loading and Formation of Intermediates




Abstract
:
1. Introduction
2. Results and Discussion
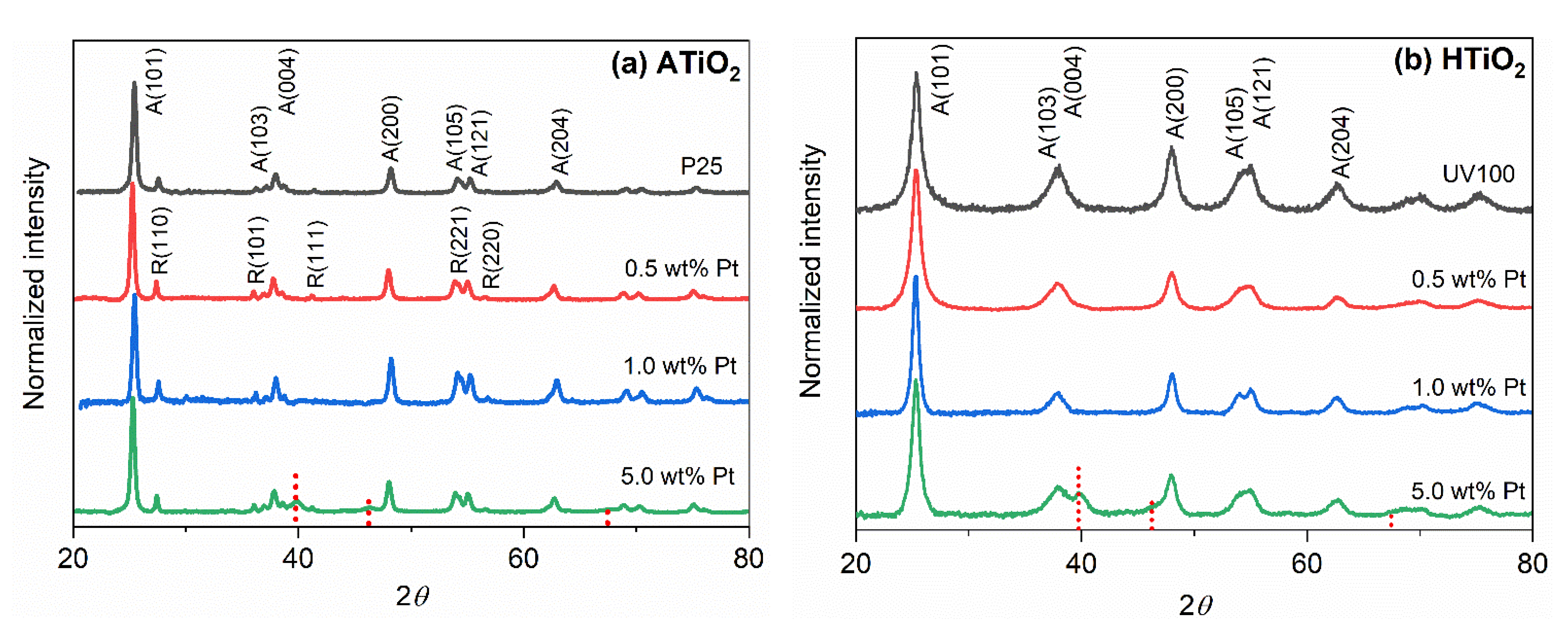
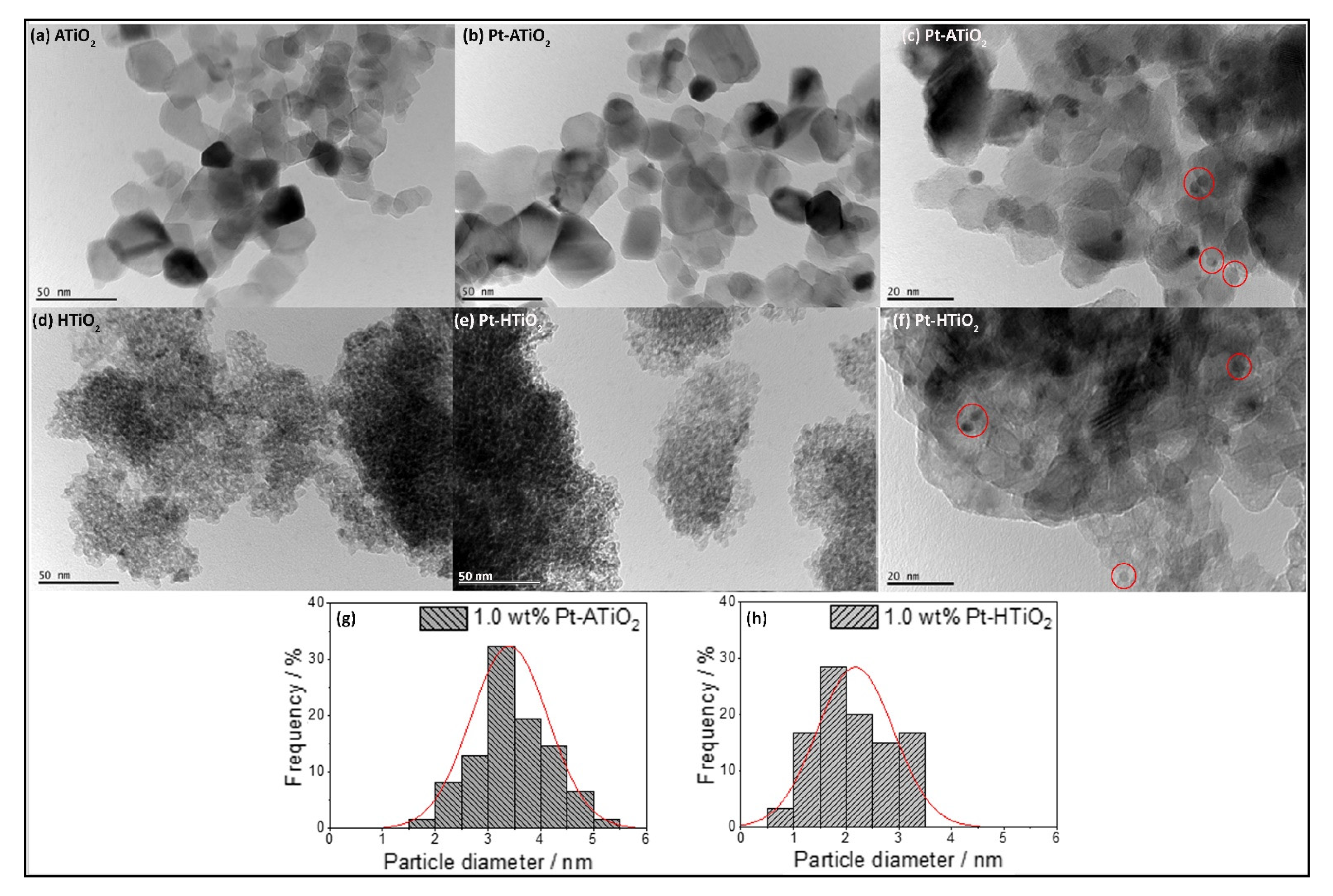
2.1. Photocatalysts Characterization
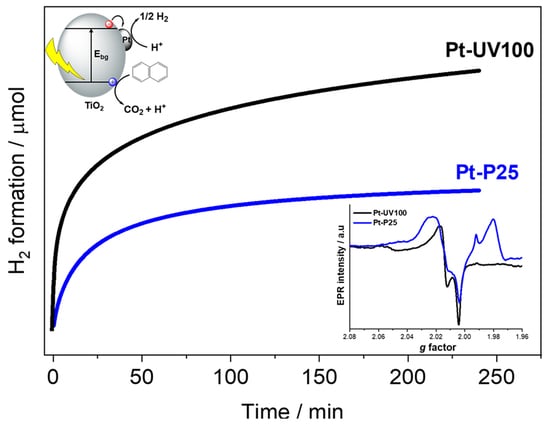
2.2. Photocatalytic Reforming of Naphthalene






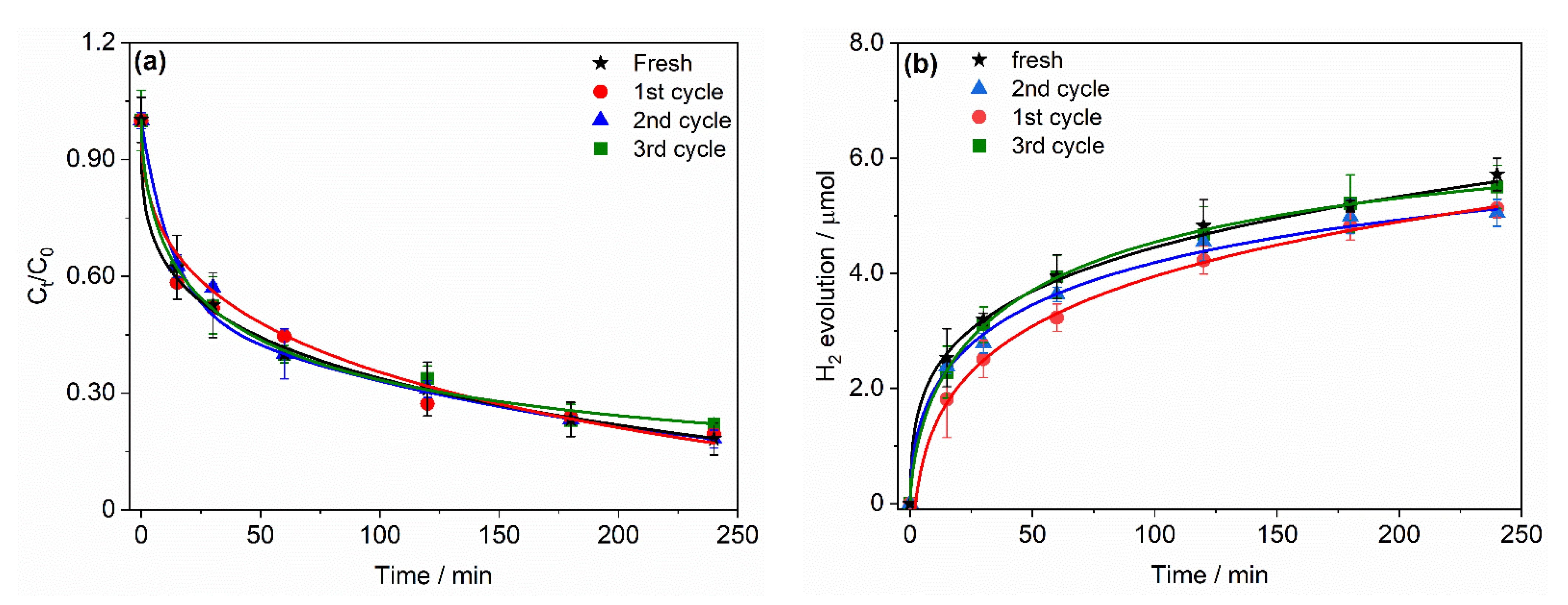
2.3. Stability of Pt Deposit
2.4. Effect of Naphthalene Oxidation Products on the H2 Evolution
2.5. EPR Study
3. Experimental
3.1. Materials
3.2. Preparation of the Pt-TiO2 Photocatalysts
3.3. Photocatalytic Experiments
3.4. Catalyst Characterization
3.5. EPR In Situ Experiments
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Møller, K.T.; Jensen, T.R.; Akiba, E.; Li, H.-W. Hydrogen—A sustainable energy carrier. Prog. Nat. Sci. 2017, 27, 34–40. [Google Scholar] [CrossRef]
- Niaz, S.; Manzoor, T.; Pandith, A.H. Hydrogen storage: Materials, methods and perspectives. Renew. Sust. Energ. Rev. 2015, 50, 457–469. [Google Scholar] [CrossRef]
- Cavendish, H., XIII. Experiments on air. Philos. Trans. R. Soc. 1784, 74, 119–153. [Google Scholar] [CrossRef]
- Kennedy, J.; Bahruji, H.; Bowker, M.; Davies, P.R.; Bouleghlimat, E.; Issarapanacheewin, S. Hydrogen generation by photocatalytic reforming of potential biofuels: Polyols, cyclic alcohols, and saccharides. J. Photochem. Photobiol. A 2018, 356, 451–456. [Google Scholar] [CrossRef]
- Fajrina, N.; Tahir, M. A critical review in strategies to improve photocatalytic water splitting towards hydrogen production. Int. J. Hydrog. Energy 2019, 44, 540–577. [Google Scholar] [CrossRef]
- Al Nasir, F.; Batarseh, M.I. Agricultural reuse of reclaimed water and uptake of organic compounds: Pilot study at Mutah University wastewater treatment plant, Jordan. Chemosphere 2008, 72, 1203–1214. [Google Scholar] [CrossRef]
- Rubio-Clemente, A.; Torres-Palma, R.A.; Penuela, G.A. Removal of polycyclic aromatic hydrocarbons in aqueous environment by chemical treatments: A review. Sci. Total Environ. 2014, 478, 201–225. [Google Scholar] [CrossRef]
- Wauchope, R.D.; Getzen, F.W. Temperature dependence of solubilities in water and heats of fusion of solid aromatic-hydrocarbons. J. Chem. Eng. Data 1972, 17, 38–41. [Google Scholar] [CrossRef]
- Ghasemi, N.; Gbeddy, G.; Egodawatta, P.; Zare, F.; Goonetilleke, A. Removal of polycyclic aromatic hydrocarbons from wastewater using dual-mode ultrasound system. Water Environ. J. 2020, 34, 425–434. [Google Scholar] [CrossRef]
- Kadri, T.; Rouissi, T.; Kaur Brar, S.; Cledon, M.; Sarma, S.; Verma, M. Biodegradation of polycyclic aromatic hydrocarbons (PAHs) by fungal enzymes: A review. J. Environ. Sci. 2017, 51, 52–74. [Google Scholar] [CrossRef]
- Chu, L.B.; Yu, S.Q.; Wang, J.L. Gamma radiolytic degradation of naphthalene in aqueous solution. Radiat. Phys. Chem. 2016, 123, 97–102. [Google Scholar] [CrossRef]
- Mondal, K.; Bhattacharyya, S.; Sharma, A. Photocatalytic degradation of naphthalene by electrospun mesoporous carbon-doped anatase TiO2 nanofiber mats. Ind. Eng. Chem. Res. 2014, 53, 18900–18909. [Google Scholar] [CrossRef]
- Garcia-Martinez, M.J.; Canoira, L.; Blazquez, G.; Da Riva, I.; Alcantara, R.; Llamas, J.F. Continuous photodegradation of naphthalene in water catalyzed by TiO2 supported on glass Raschig rings. Chem. Eng. J. 2005, 110, 123–128. [Google Scholar] [CrossRef]
- Al-Madanat, O.; Alsalka, Y.; Curti, M.; Dillert, R.; Bahnemann, D.W. Mechanistic insights into hydrogen evolution by photocatalytic reforming of naphthalene. ACS Catal. 2020, 10, 7398–7412. [Google Scholar] [CrossRef]
- AlSalka, Y.; Hakki, A.; Fleisch, M.; Bahnemann, D.W. Understanding the degradation pathways of oxalic acid in different photocatalytic systems: Towards simultaneous photocatalytic hydrogen evolution. J. Photochem. Photobiol. A 2018, 366, 81–90. [Google Scholar] [CrossRef]
- Weon, S.; Kim, J.; Choi, W. Dual-components modified TiO2 with Pt and fluoride as deactivation-resistant photocatalyst for the degradation of volatile organic compound. Appl. Catal. B-Environ. 2018, 220, 1–8. [Google Scholar] [CrossRef]
- Schneider, J.; Bahnemann, D.W. Undesired Role of Sacrificial Reagents in Photocatalysis. J. Phy. Chem. Lett. 2013, 4, 3479–3483. [Google Scholar] [CrossRef]
- Hashimoto, K.; Kawai, T.; Sakata, T. Photocatalytic reactions of hydrocarbons and fossil-fuels with Water—Hydrogen-production and oxidation. J. Phys. Chem. 1984, 88, 4083–4088. [Google Scholar] [CrossRef]
- Yuzawa, H.; Aoki, M.; Otake, K.; Hattori, T.; Itoh, H.; Yoshida, H. Reaction mechanism of aromatic ring hydroxylation by water over platinum-loaded titanium oxide photocatalyst. J. Phys. Chem. C 2012, 116, 25376–25387. [Google Scholar] [CrossRef]
- AlSalka, Y.; Al-Madanat, O.; Curti, M.; Hakki, A.; Bahnemann, D.W. Photocatalytic H2 evolution from oxalic acid: Effect of cocatalysts and carbon dioxide radical anion on the surface charge transfer mechanisms. ACS Appl. Energy Mater. 2020, 3, 6678–6691. [Google Scholar] [CrossRef]
- Kandiel, T.A.; Feldhoff, A.; Robben, L.; Dillert, R.; Bahnemann, D.W. Tailored titanium dioxide nanomaterials: Anatase nanoparticles and brookite nanorods as highly active photocatalysts. Chem. Mater. 2010, 22, 2050–2060. [Google Scholar] [CrossRef]
- Abdulrazzak, F.H.; Hussein, F.H.; Alkaim, A.F.; Ivanova, I.; Emeline, A.V.; Bahnemann, D.W. Sonochemical/hydration-dehydration synthesis of Pt-TiO2 NPs/decorated carbon nanotubes with enhanced photocatalytic hydrogen production activity. Photochem. Photobiol. Sci. 2016, 15, 1347–1357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colon, G.; Hidalgo, M.C.; Navio, J.A. Photocatalytic deactivation of commercial TiO2 samples during simultaneous photoreduction of Cr(VI) and photooxidation of salicylic acid. J. Photochem. Photobiol. A 2001, 138, 79–85. [Google Scholar] [CrossRef]
- Alonso-Tellez, A.; Masson, R.; Robert, D.; Keller, N.; Keller, V. Comparison of Hombikat UV100 and P25 TiO2 performance in gas-phase photocatalytic oxidation reactions. J. Photochem. Photobiol. A 2012, 250, 58–65. [Google Scholar] [CrossRef]
- Abellan, M.N.; Dillert, R.; Gimenez, J.; Bahnemann, D. Evaluation of two types of TiO2-based catalysts by photodegradation of DMSO in aqueous suspension. J. Photochem. Photobiol. A 2009, 202, 164–171. [Google Scholar] [CrossRef]
- AlSalka, Y.; Hakki, A.; Schneider, J.; Bahnemann, D.W. Co-catalyst-free photocatalytic hydrogen evolution on TiO2: Synthesis of optimized photocatalyst through statistical material science. Appl. Catal. B-Environ. 2018, 238, 422–433. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, Y.; Li, W.; Yang, Q.; Hou, Q.; Wei, L.; Liu, L.; Huang, F.; Ju, M. Enhancement of photocatalytic performance with the use of noble-metal-decorated TiO2 nanocrystals as highly active catalysts for aerobic oxidation under visible-light irradiation. Appl. Catal. B-Environ. 2017, 210, 352–367. [Google Scholar] [CrossRef]
- Diebold, U. The surface science of titanium dioxide. Surf. Sci. Rep. 2003, 48, 53–229. [Google Scholar] [CrossRef]
- Kowalska, E.; Remita, H.; Colbeau-Justin, C.; Hupka, J.; Belloni, J. Modification of titanium dioxide with platinum ions and clusters: Application in photocatalysis. J. Phys. Chem. C 2008, 112, 1124–1131. [Google Scholar] [CrossRef]
- Kozlova, E.A.; Lyubina, T.P.; Nasalevich, M.A.; Vorontsov, A.V.; Miller, A.V.; Kaichev, V.V.; Parmon, V.N. Influence of the method of platinum deposition on activity and stability of Pt/TiO2 photocatalysts in the photocatalytic oxidation of dimethyl methylphosphonate. Catal. Commun. 2011, 12, 597–601. [Google Scholar] [CrossRef]
- Kandiel, T.A.; Dillert, R.; Robben, L.; Bahnemann, D.W. Photonic efficiency and mechanism of photocatalytic molecular hydrogen production over platinized titanium dioxide from aqueous methanol solutions. Catal. Today 2011, 161, 196–201. [Google Scholar] [CrossRef]
- Abdel-Azim, S.M.; Aboul-Gheit, A.K.; Ahmed, S.M.; El-Desouki, D.S.; Abdel-Mottaleb, M.S.A. Preparation and application of mesoporous nanotitania photocatalysts using different templates and PH media. Int. J. Photoenergy 2014, 2014, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Wenderich, K.; Mul, G. Methods, mechanism, and applications of photodeposition in photocatalysis: A review. Chem. Rev. 2016, 116, 14587–14619. [Google Scholar] [CrossRef]
- Al-Azri, Z.H.N.; AlOufi, M.; Chan, A.; Waterhouse, G.I.N.; Idriss, H. Metal particle size Effects on the photocatalytic hydrogen ion reduction. ACS Catal. 2019, 9, 3946–3958. [Google Scholar] [CrossRef]
- Li, H.; Yu, H.; Sun, L.; Zhai, J.; Han, X. A self-assembled 3D Pt/TiO2 architecture for high-performance photocatalytic hydrogen production. Nanoscale 2015, 7, 1610–1615. [Google Scholar] [CrossRef]
- Xing, J.; Li, Y.H.; Jiang, H.B.; Wang, Y.; Yang, H.G. The size and valence state effect of Pt on photocatalytic H2 evolution over platinized TiO2 photocatalyst. Int. J. Hydrog. Energy 2014, 39, 1237–1242. [Google Scholar] [CrossRef]
- Wang, D.; Liu, Z.P.; Yang, W.M. Revealing the size effect of platinum cocatalyst for photocatalytic hydrogen evolution on TiO2 support: A DFT study. ACS Catal. 2018, 8, 7270–7278. [Google Scholar] [CrossRef]
- Chen, H.R.; Li, P.; Umezawa, N.; Abe, H.; Ye, J.H.; Shiraishi, K.; Ohta, A.; Miyazaki, S. Bonding and electron energy-level alignment at metal/TiO2 interfaces: A density functional theory study. J. Phys. Chem. C 2016, 120, 5549–5556. [Google Scholar] [CrossRef]
- Su, R.; Bechstein, R.; So, L.; Vang, R.T.; Sillassen, M.; Esbjornsson, B.; Palmqvist, A.; Besenbacher, F. How the anatase-to-rutile ratio influences the photoreactivity of TiO2. J. Phys. Chem. C 2011, 115, 24287–24292. [Google Scholar] [CrossRef]
- Riegel, G.; Bolton, J.R. Photocatalytic efficiency variability in TiO2 particles. J. Phys. Chem. 1995, 99, 4215–4224. [Google Scholar] [CrossRef]
- Scanlon, D.O.; Dunnill, C.W.; Buckeridge, J.; Shevlin, S.A.; Logsdail, A.J.; Woodley, S.M.; Catlow, C.R.; Powell, M.J.; Palgrave, R.G.; Parkin, I.P.; et al. Band alignment of rutile and anatase TiO2. Nat. Mater. 2013, 12, 798–801. [Google Scholar] [CrossRef]
- Hurum, D.C.; Agrios, A.G.; Gray, K.A.; Rajh, T.; Thurnauer, M.C. Explaining the enhanced photocatalytic activity of Degussa P25 mixed-phase TiO2 using EPR. J. Phys. Chem. B 2003, 107, 4545–4549. [Google Scholar] [CrossRef]
- Dahl, M.; Liu, Y.; Yin, Y. Composite titanium dioxide nanomaterials. Chem. Rev. 2014, 114, 9853–9889. [Google Scholar] [CrossRef]
- Hufschmidt, D.; Bahemann, D.; Testa, J.J.; Emilio, C.A.; Litter, M.I. Enhancement of the photocatalytic activity of various TiO2 materials by platinisation. J. Photochem. Photobiol. A 2002, 148, 223–231. [Google Scholar] [CrossRef]
- Sakthivel, S.; Shankar, M.V.; Palanichamy, M.; Arabindoo, B.; Bahnemann, D.W.; Murugesan, V. Enhancement of photocatalytic activity by metal deposition: Characterisation and photonic efficiency of Pt, Au and Pd deposited on TiO2 catalyst. Water Res. 2004, 38, 3001–3008. [Google Scholar] [CrossRef]
- Sun, B.; Vorontsov, A.V.; Smirniotis, P.G. Role of platinum deposited on TiO2 in phenol photocatalytic oxidation. Langmuir 2003, 19, 3151–3156. [Google Scholar] [CrossRef]
- Benz, D.; Felter, K.M.; Koser, J.; Thoming, J.; Mul, G.; Grozema, F.C.; Hintzen, H.T.; Kreutzer, M.T.; van Ommen, J.R. Assessing the role of Pt Clusters on TiO2 (P25) on the photocatalytic degradation of acid blue 9 and rhodamine B. J. Phys. Chem. C 2020, 124, 8269–8278. [Google Scholar] [CrossRef] [Green Version]
- Bamwenda, G.R.; Tsubota, S.; Nakamura, T.; Haruta, M. Photoassisted hydrogen production from a water-ethanol solution: A comparison of activities of Au-TiO2 and Pt-TiO2. J. Photochem. Photobiol. A 1995, 89, 177–189. [Google Scholar] [CrossRef]
- Khan, M.R.; Chuan, T.W.; Yousuf, A.; Chowdhury, M.N.K.; Cheng, C.K. Schottky barrier and surface plasmonic resonance phenomena towards the photocatalytic reaction: Study of their mechanisms to enhance photocatalytic activity. Catal. Sci. Technol. 2015, 5, 2522–2531. [Google Scholar] [CrossRef] [Green Version]
- Ola, O.; Maroto-Valer, M.M. Review of material design and reactor engineering on TiO2 photocatalysis for CO2 reduction. J. Photochem. Photobiol. C. 2015, 24, 16–42. [Google Scholar] [CrossRef] [Green Version]
- Bui, T.D.; Kimura, A.; Ikeda, S.; Matsumura, M. Determination of oxygen sources for oxidation of benzene on TiO2 photocatalysts in aqueous solutions containing molecular oxygen. J. Am. Chem. Soc. 2010, 132, 8453–8458. [Google Scholar] [CrossRef]
- Pang, X.; Chen, C.; Ji, H.; Che, Y.; Ma, W.; Zhao, J. Unraveling the photocatalytic mechanisms on TiO2 surfaces using the oxygen-18 isotopic label technique. Molecules 2014, 19, 16291–16311. [Google Scholar] [CrossRef] [Green Version]
- Augugliaro, V.; Bellardita, M.; Loddo, V.; Palmisano, G.; Palmisano, L.; Yurdakal, S. Overview on oxidation mechanisms of organic compounds by TiO2 in heterogeneous photocatalysis. J. Photochem. Photobiol. C 2012, 13, 224–245. [Google Scholar] [CrossRef] [Green Version]
- Montoya, J.F.; Ivanova, I.; Dillert, R.; Bahnemann, D.W.; Salvador, P.; Peral, J. Catalytic role of surface oxygens in TiO2 photooxidation reactions: Aqueous benzene photooxidation with Ti18O2 under anaerobic conditions. J. Phys. Chem. Lett. 2013, 4, 1415–1422. [Google Scholar] [CrossRef]
- Bui, T.D.; Kimura, A.; Higashida, S.; Ikeda, S.; Mafsurnura, M. Two routes for mineralizing benzene by TiO2-photocatalyzed reaction. Appl. Catal. B-Environ. 2011, 107, 119–127. [Google Scholar] [CrossRef]
- Bui, T.D.; Kimura, A.; Ikeda, S.; Matsumura, M. Lowering of photocatalytic activity of TiO2 particles during oxidative decomposition of benzene in aerated liquid. Appl. Catal. B-Environ. 2010, 94, 186–191. [Google Scholar] [CrossRef]
- Panagiotopoulou, P.; Karamerou, E.E.; Kondarides, D.I. Kinetics and mechanism of glycerol photo-oxidation and photo-reforming reactions in aqueous TiO2 and Pt/TiO2 suspensions. Catal. Today 2013, 209, 91–98. [Google Scholar] [CrossRef]
- Imizcoz, M.; Puga, A.V. Assessment of photocatalytic hydrogen production from biomass or wastewaters depending on the metal co-catalyst and Its deposition method on TiO2. Catalysts 2019, 9, 584. [Google Scholar] [CrossRef] [Green Version]
- Schneider, J.; Matsuoka, M.; Takeuchi, M.; Zhang, J.; Horiuchi, Y.; Anpo, M.; Bahnemann, D.W. Understanding TiO2 photocatalysis: Mechanisms and materials. Chem. Rev. 2014, 114, 9919–9986. [Google Scholar] [CrossRef] [PubMed]
- King, S.M.; Leaf, P.A.; Olson, A.C.; Ray, P.Z.; Tarr, M.A. Photolytic and photocatalytic degradation of surface oil from the Deepwater Horizon spill. Chemosphere 2014, 95, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Qourzal, S.; Assabbane, A.; Ait-Ichou, Y. Synthesis of TiO2 via hydrolysis of titanium tetraisopropoxide and its photocatalytic activity on a suspended mixture with activated carbon in the degradation of 2-naphthol. J. Photochem. Photobiol. A 2004, 163, 317–321. [Google Scholar] [CrossRef]
- Brahmia, O.; Richard, C. Photochemical transformation of 1-naphthol in aerated aqueous solution. Photochem. Photobiol. Sci. 2005, 4, 454–458. [Google Scholar] [CrossRef] [PubMed]
- Weon, S.; Choi, W. TiO2 nanotubes with open channels as deactivation-resistant photocatalyst for the degradation of volatile organic compounds. Environ. Sci. Technol. 2016, 50, 2556–2563. [Google Scholar] [CrossRef] [PubMed]
- Theurich, J.; Bahnemann, D.W.; Vogel, R.; Ehamed, F.E.; Alhakimi, G.; Rajab, I. Photocatalytic degradation of naphthalene and anthracene: GC-MS analysis of the degradation pathway. Res. Chem. Intermediat. 1997, 23, 247–274. [Google Scholar] [CrossRef]
- Mills, A.; Davies, R.H.; Worsley, D. Water-purification by semiconductor photocatalysis. Chem. Soc. Rev. 1993, 22, 417–425. [Google Scholar] [CrossRef]
- Ajmal, A.; Majeed, I.; Malik, R.N.; Idriss, H.; Nadeem, M.A. Principles and mechanisms of photocatalytic dye degradation on TiO2 based photocatalysts: A comparative overview. RSC Adv. 2014, 4, 37003–37026. [Google Scholar] [CrossRef]
- Qourzal, S.; Barka, N.; Tamimi, M.; Assabbane, A.; Ait-Ichou, Y. Photodegradation of 2-naphthol in water by artificial light illumination using TiO2 photocatalyst: Identification of intermediates and the reaction pathway. Appl. Catal. A-Gen. 2008, 334, 386–393. [Google Scholar] [CrossRef]
- Denny, F.; Scott, J.; Chiang, K.; Teoh, W.Y.; Amal, R. Insight towards the role of platinum in the photocatalytic mineralisation of organic compounds. J. Mol. Catal. A-Chem. 2007, 263, 93–102. [Google Scholar] [CrossRef]
- Hurum, D.C.; Agrios, A.G.; Crist, S.E.; Gray, K.A.; Rajh, T.; Thurnauer, M.C. Probing reaction mechanisms in mixed phase TiO2 by EPR. J. Electron. Spectrosc. 2006, 150, 155–163. [Google Scholar] [CrossRef]
- Howe, R.F.; Gratzel, M. EPR study of hydrated anatase under UV irradiation. J. Phys. Chem. 1987, 91, 3906–3909. [Google Scholar] [CrossRef]
- Howe, R.F.; Gratzel, M. EPR observation of trapped electrons in colloidal titanium dioxide. J. Phys. Chem. 1985, 89, 4495–4499. [Google Scholar] [CrossRef]
- Kumar, C.P.; Gopal, N.O.; Wang, T.C.; Wong, M.S.; Ke, S.C. EPR investigation of TiO2 nanoparticles with temperature-dependent properties. J. Phys. Chem. B 2006, 110, 5223–5229. [Google Scholar] [CrossRef] [PubMed]
- Shapovalov, V.; Stefanovich, E.V.; Truong, T.N. Nature of the excited states of the rutile TiO2(110) surface with adsorbed water. Surf. Sci. 2002, 498, L103–L108. [Google Scholar] [CrossRef]
- Nakaoka, Y.; Nosaka, Y. ESR Investigation into the effects of heat treatment and crystal structure on radicals produced over irradiated TiO2 powder. J. Photochem. Photobiol. A 1997, 110, 299–305. [Google Scholar] [CrossRef]
- Ke, S.C.; Wang, T.C.; Wong, M.S.; Gopal, N.O. Low temperature kinetics and energetics of the electron and hole traps in irradiated TiO2 nanoparticles as revealed by EPR spectroscopy. J. Phys. Chem. B 2006, 110, 11628–11634. [Google Scholar] [CrossRef]
- Connelly, K.; Wahab, A.K.; Idriss, H. Photoreaction of Au/TiO2 for hydrogen production from renewables: A review on the synergistic effect between anatase and rutile phases of TiO2. Mater. Renew. Sustain. Energy 2012, 1, 3. [Google Scholar] [CrossRef] [Green Version]
- Arizavi, A.; Mirbagheri, N.S.; Hosseini, Z.; Chen, P.; Sabbaghi, S. Efficient removal of naphthalene from aqueous solutions using a nanoporous kaolin/Fe3O4 composite. Int. J. Environ. Sci. Technol. 2020, 17, 1991–2002. [Google Scholar] [CrossRef]
- Nesterenko-Malkovskaya, A.; Kirzhner, F.; Zimmels, Y.; Armon, R. Eichhornia crassipes capability to remove naphthalene from wastewater in the absence of bacteria. Chemosphere 2012, 87, 1186–1191. [Google Scholar] [CrossRef]
- Al-Madanat, O.; Curti, M.; Günnemann, C.; Alsalka, Y.; Dillert, R.; Bahnemann, D.W. TiO2 photocatalysis: Impact of the platinum loading method on reductive and oxidative half-reactions. Catal. Today. (under review).










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
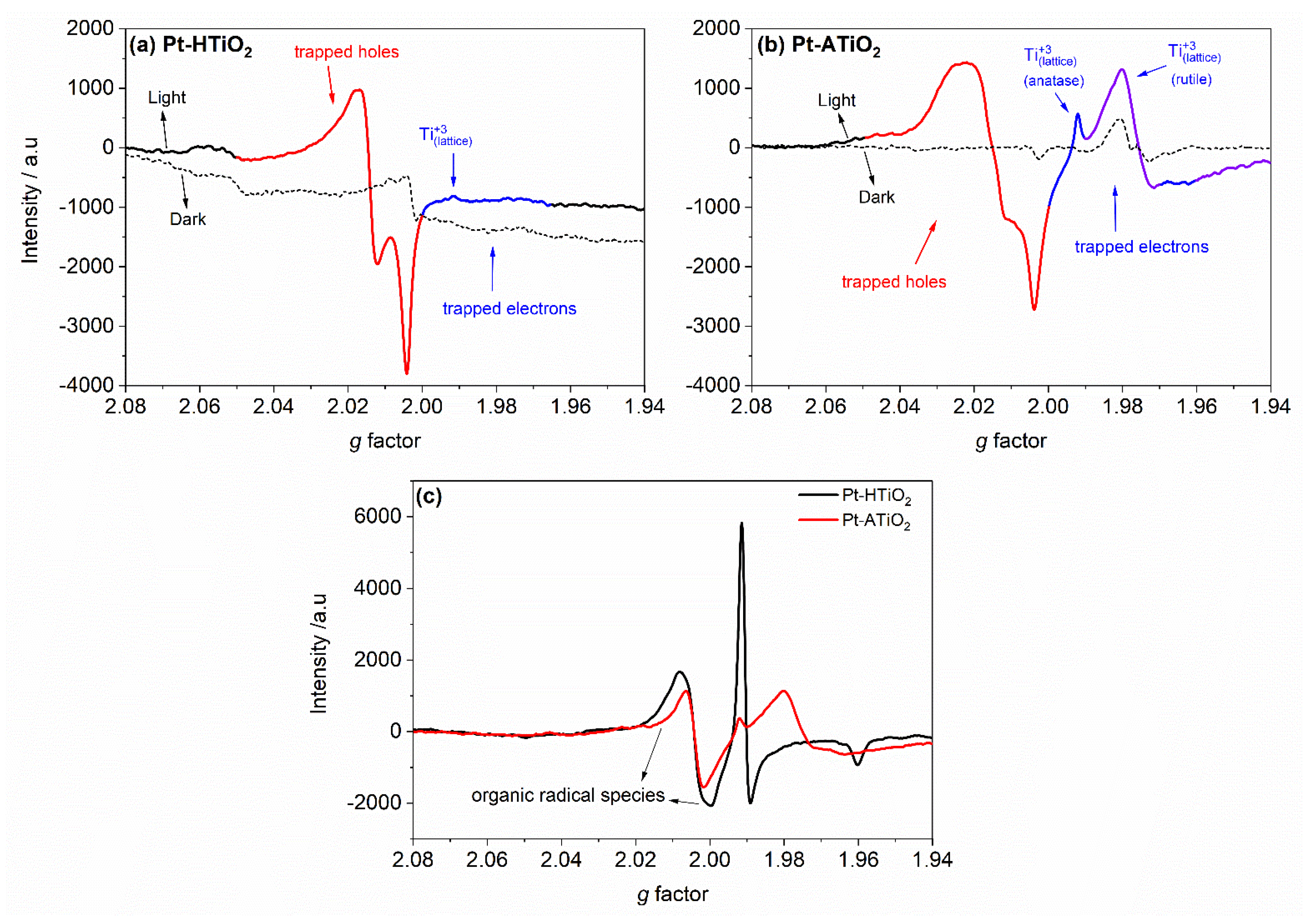{kind=link}
| Photocatalyst | Measured Pt wt% | BET Surface Area g m−2 | Crystallite Size a nm |
|---|---|---|---|
| HTiO2 (UV100) | - | 295 ± 3 | 8.0 ± 0.7 |
| 0.5 wt% Pt-HTiO2 | 0.46 ± 0.02 | 290 ± 2 | 8.4 ± 1.1 |
| 1.0 wt% Pt-HTiO2 | 0.88 ± 0.03 | 287 ± 2 | 8.9 ± 2.8 |
| 5.0 wt% Pt-HTiO2 | 4.75 ± 0.07 | 278 ± 2 | 9.2 ± 2.1 |
| ATiO2 (P25) | - | 52.3 ± 0.8 | 17.5 ± 6.3 |
| 0.5 wt% Pt-ATiO2 | 0.45± 0.05 | 50.0 ± 0.2 | 18.5 ± 4.7 |
| 1.0 wt% Pt-ATiO2 | 0.90 ± 0.05 | 48.0 ± 0.3 | 19.3 ± 5.5 |
| 5.0 wt% Pt-ATiO2 | 4.50 ± 0.10 | 43.8 ± 1.2 | 20.6 ± 6.1 |
| Photocatalyst | HTiO2 | ATiO2 | |||||
|---|---|---|---|---|---|---|---|
| Property | 0.5 wt% Pt | 1.0 wt% Pt | 5.0 wt% Pt | 0.5 wt% Pt | 1.0 wt% Pt | 5.0 wt% Pt | |
| Total naphthalenols selectivity % | 22.4 ± 1.3 | 34.7 ± 2.4 | 46.2 ± 8.5 | 21.3 ± 1.2 | 27.3 ± 2.4 | 45.1 ± 4.4 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Madanat, O.; AlSalka, Y.; Dillert, R.; Bahnemann, D.W. Photocatalytic H2 Production from Naphthalene by Various TiO2 Photocatalysts: Impact of Pt Loading and Formation of Intermediates. Catalysts 2021, 11, 107. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11010107
Al-Madanat O, AlSalka Y, Dillert R, Bahnemann DW. Photocatalytic H2 Production from Naphthalene by Various TiO2 Photocatalysts: Impact of Pt Loading and Formation of Intermediates. Catalysts. 2021; 11(1):107. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11010107
Chicago/Turabian StyleAl-Madanat, Osama, Yamen AlSalka, Ralf Dillert, and Detlef W. Bahnemann. 2021. "Photocatalytic H2 Production from Naphthalene by Various TiO2 Photocatalysts: Impact of Pt Loading and Formation of Intermediates" Catalysts 11, no. 1: 107. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11010107