Metallophthalocyanines as Catalysts in Aerobic Oxidation

 ,
,  , , and
, , and 
Abstract
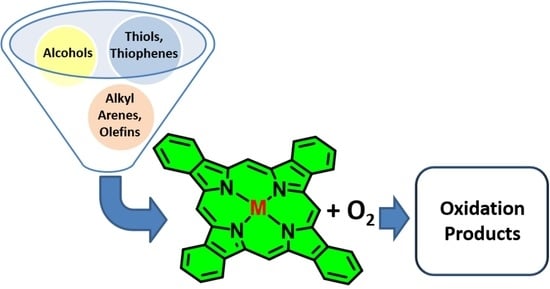
:
1. General Introduction
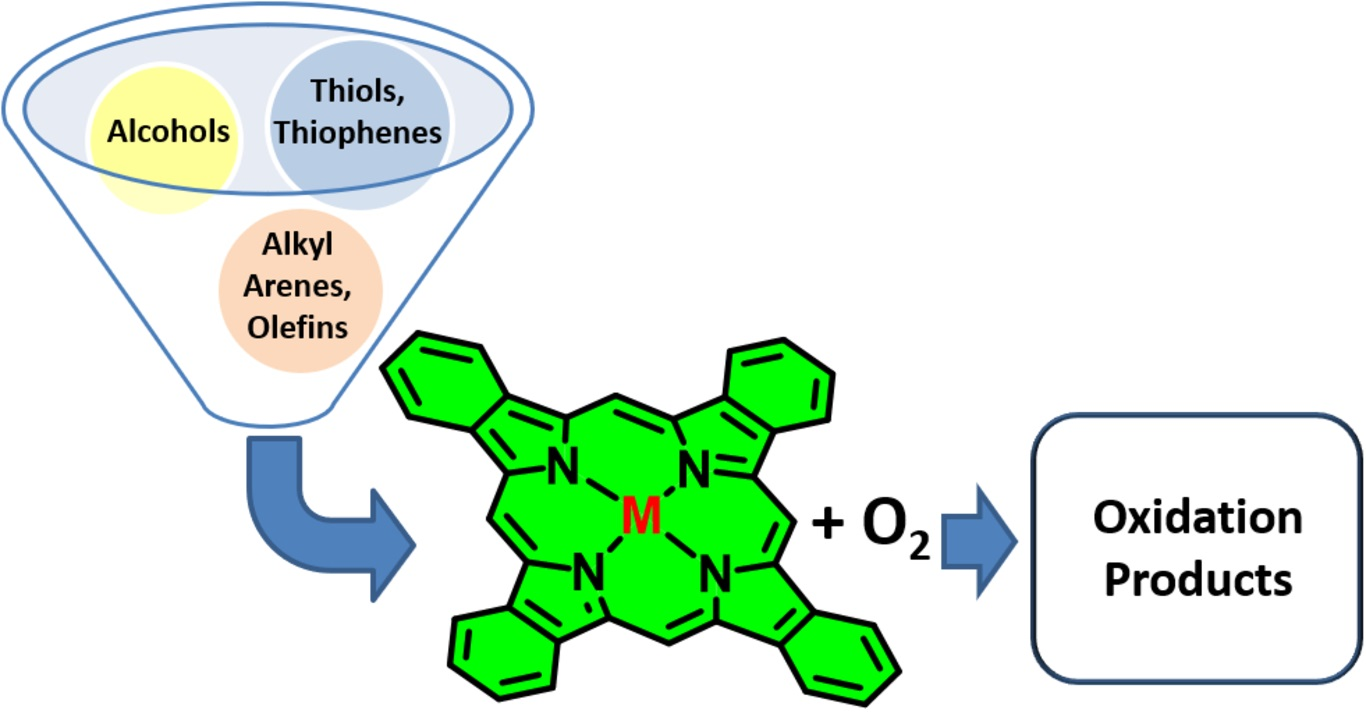
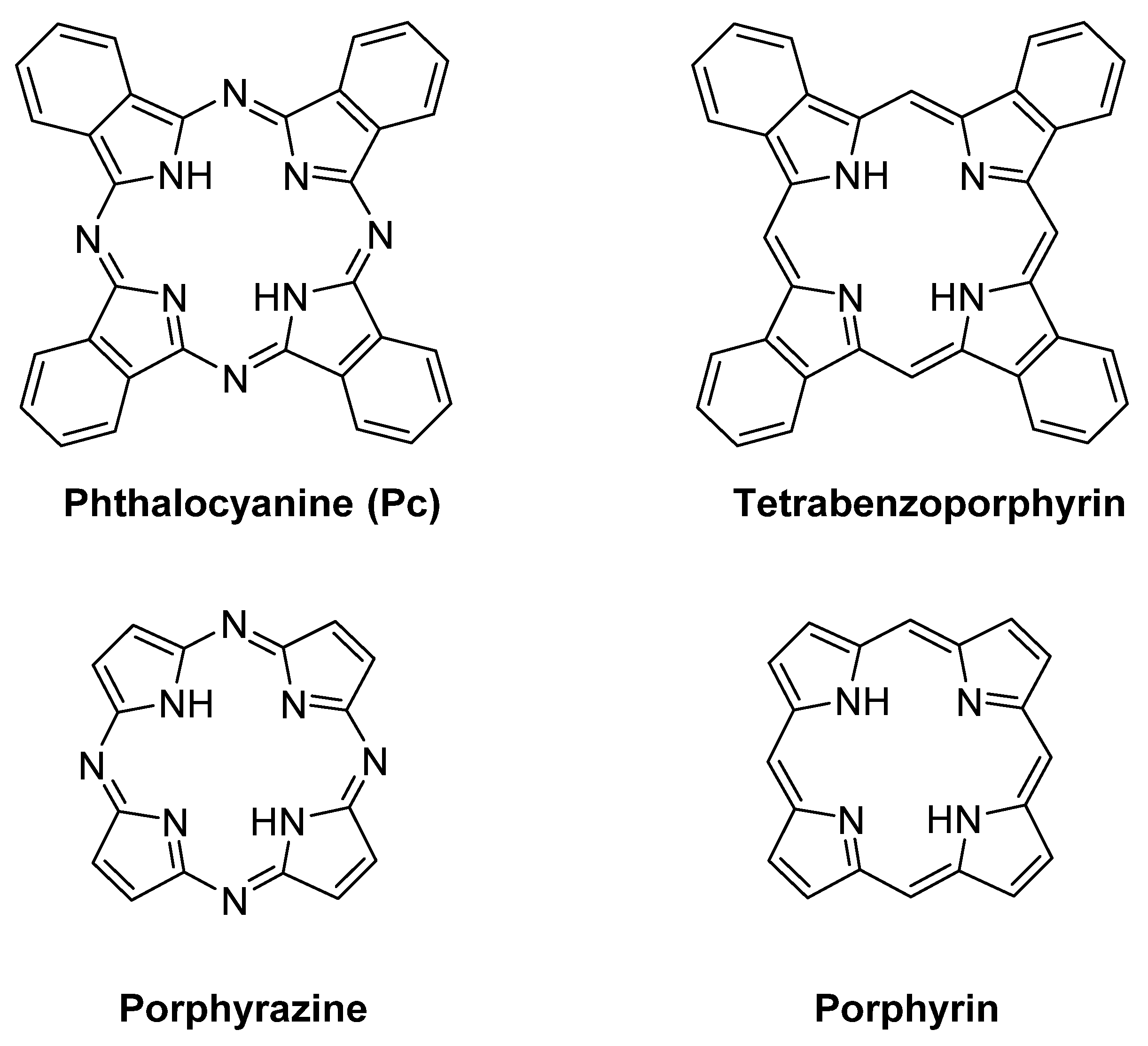
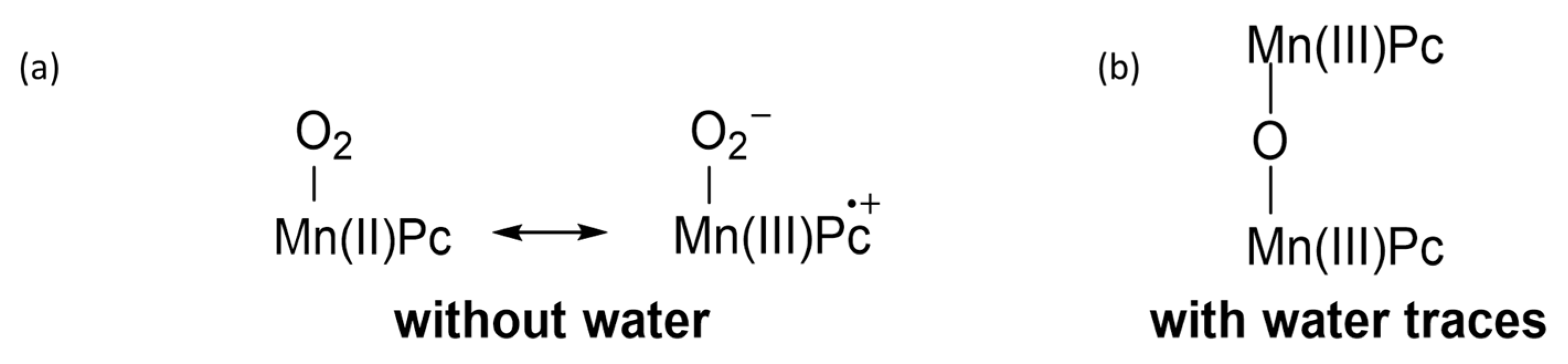
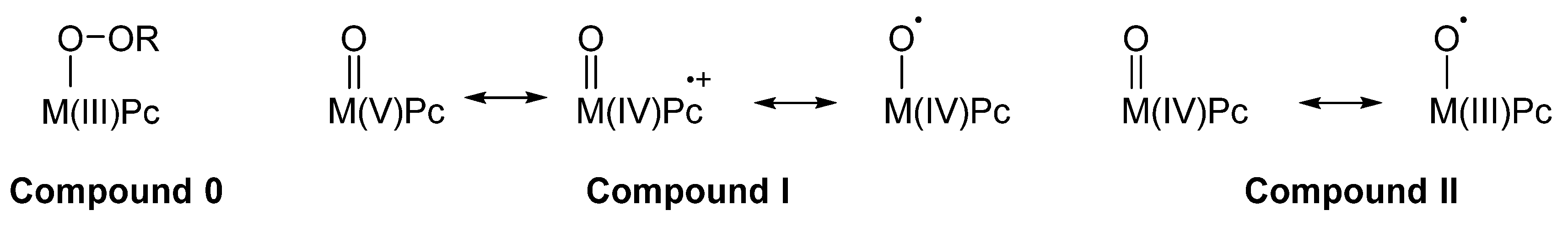
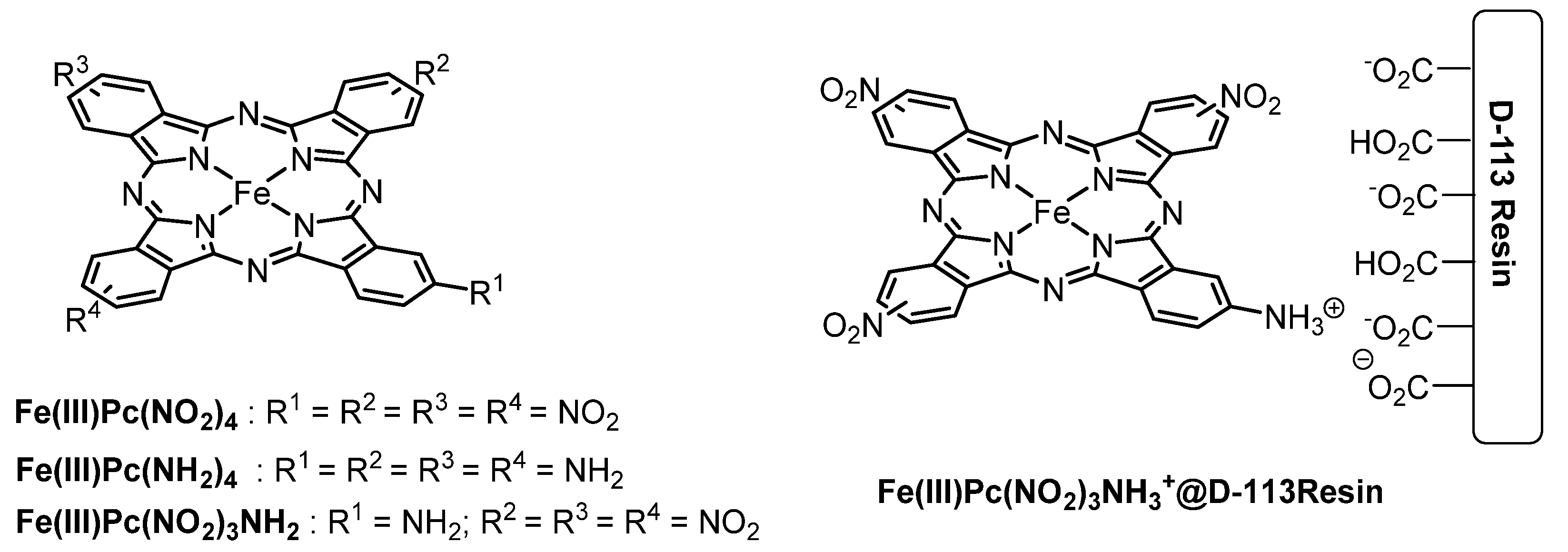
2. Metallophthalocyanines as Redox Catalysts: An Overview
3. Metallophthalocyanines and Their Catalytic Activity with Dioxygen as Oxidant
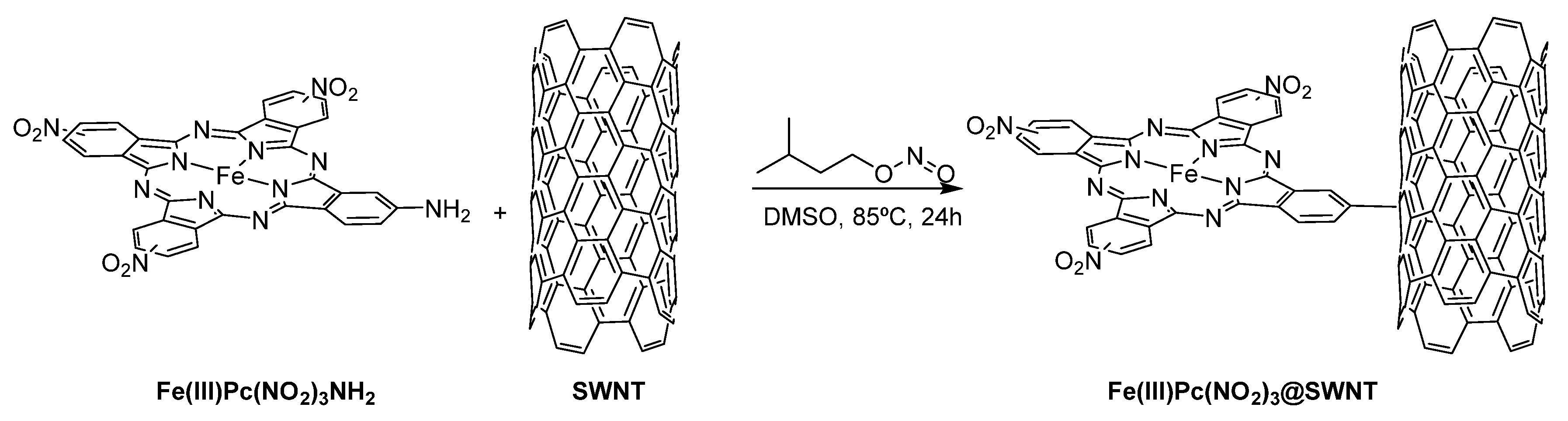
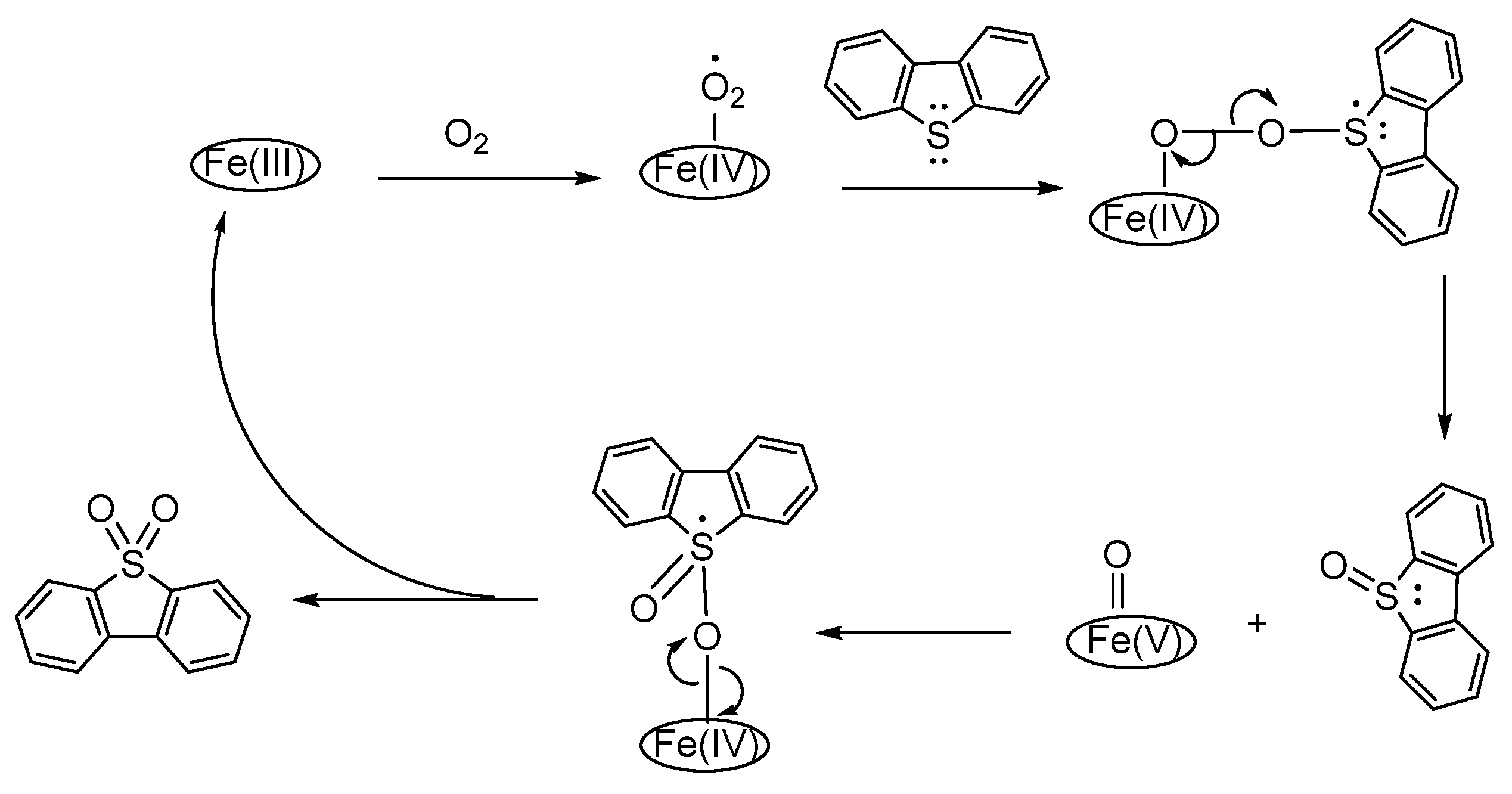
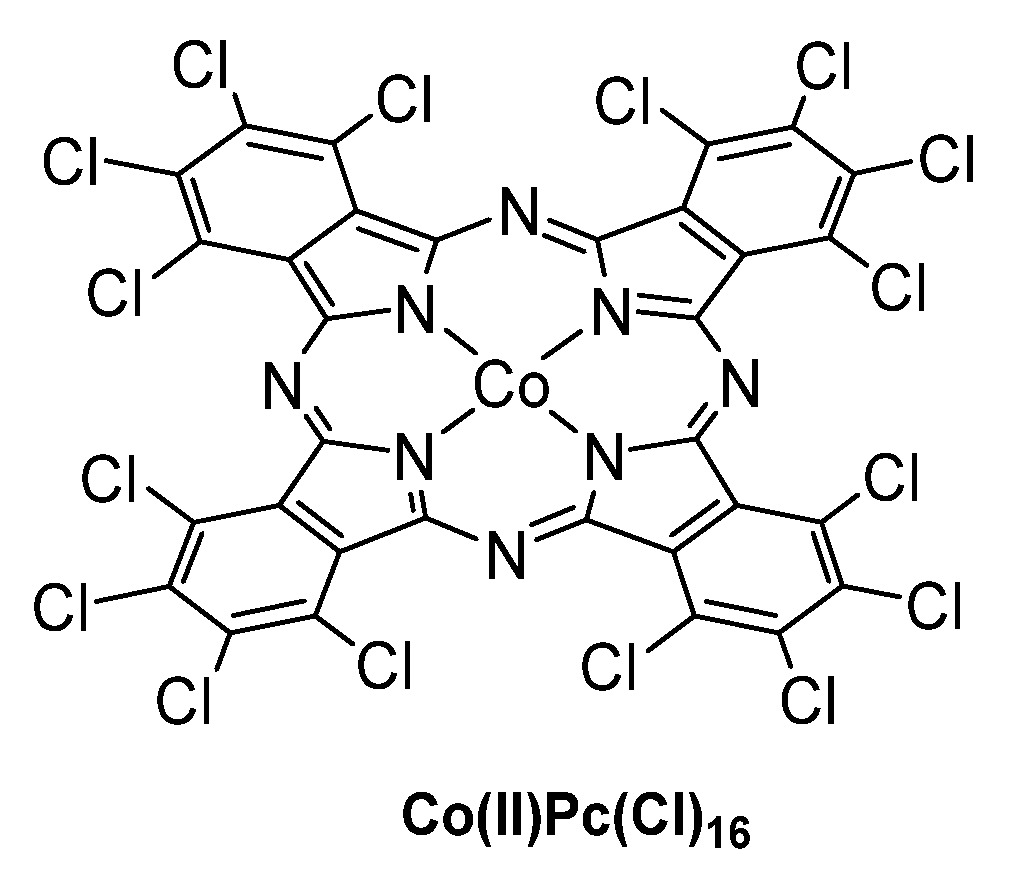
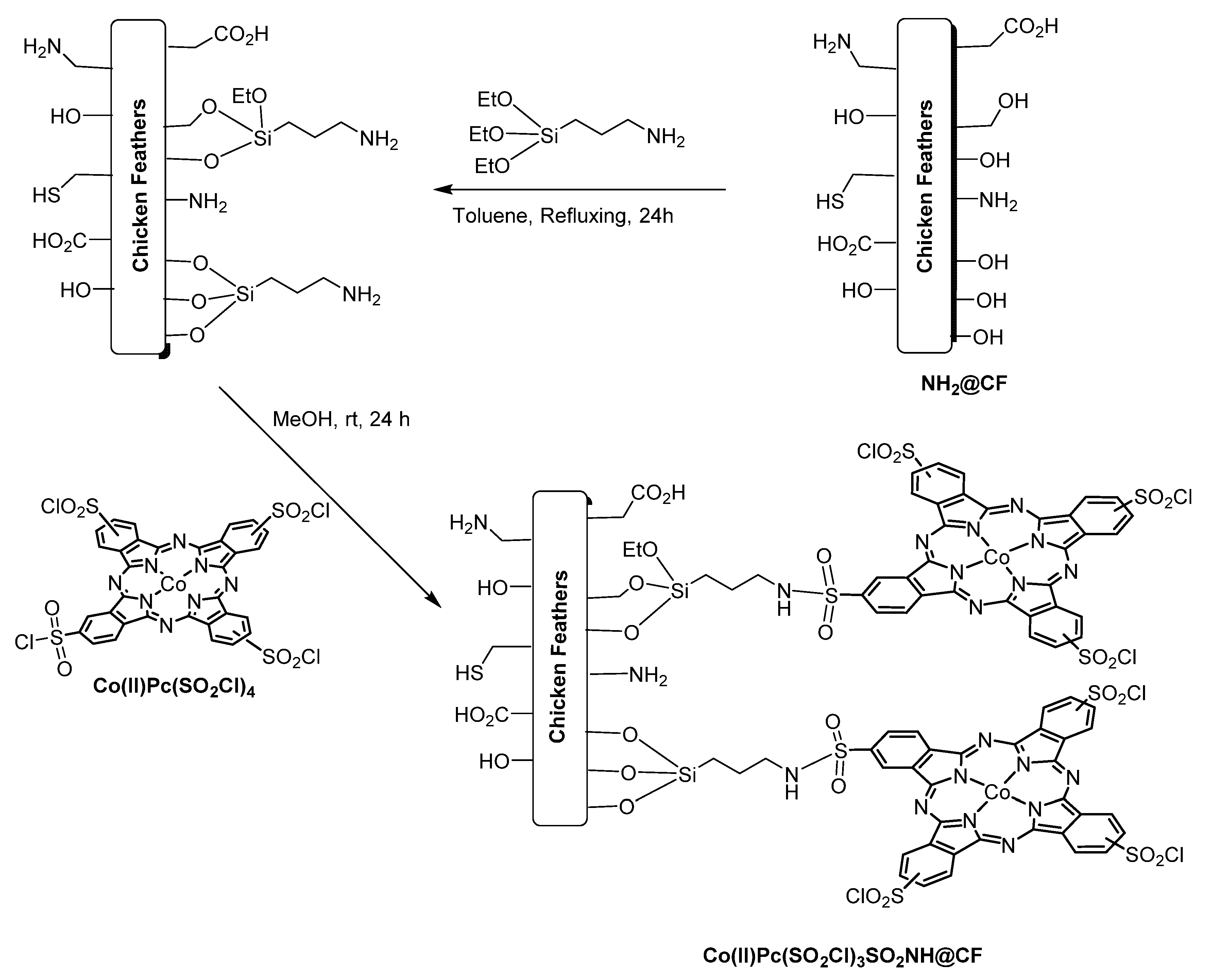
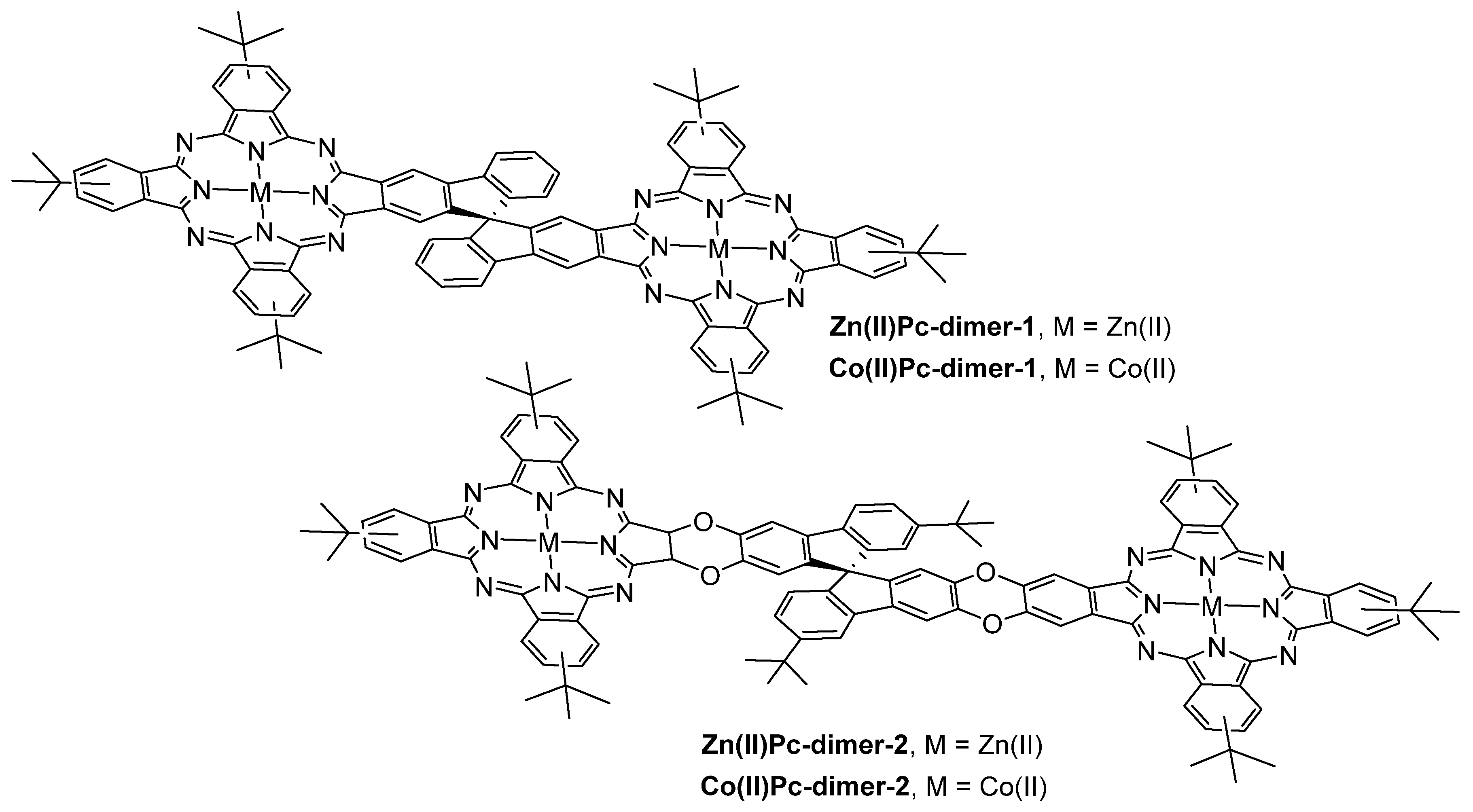
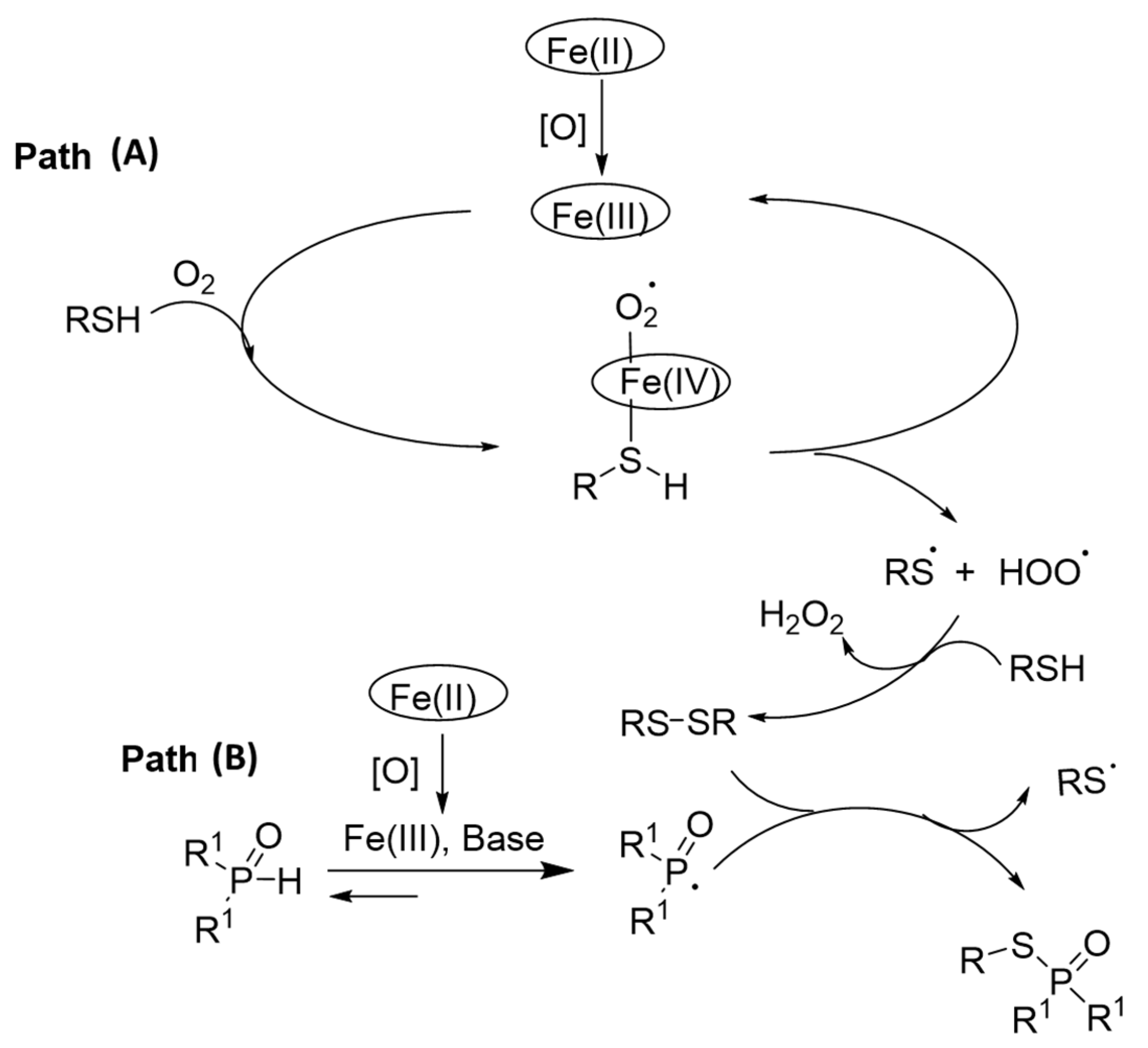
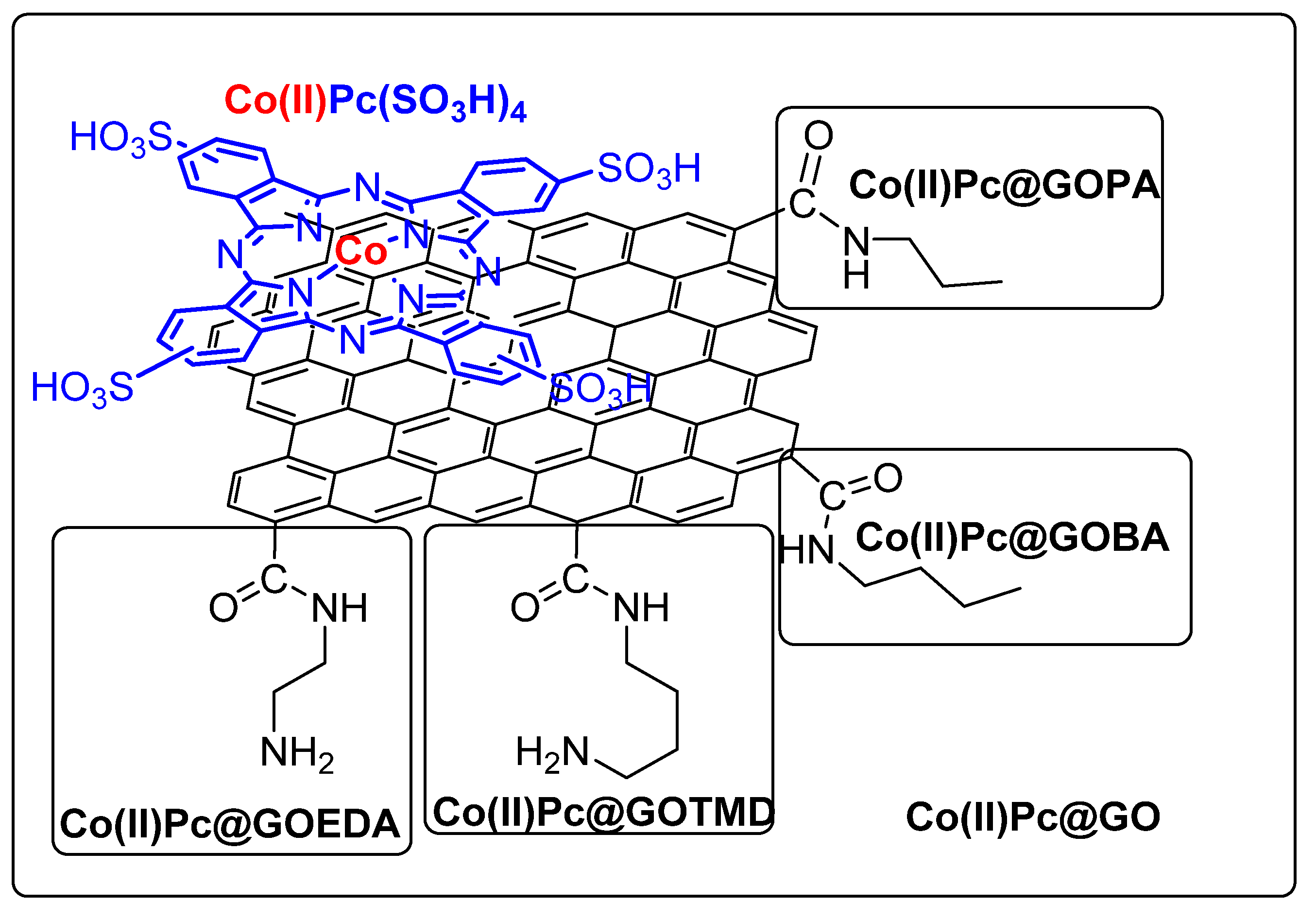
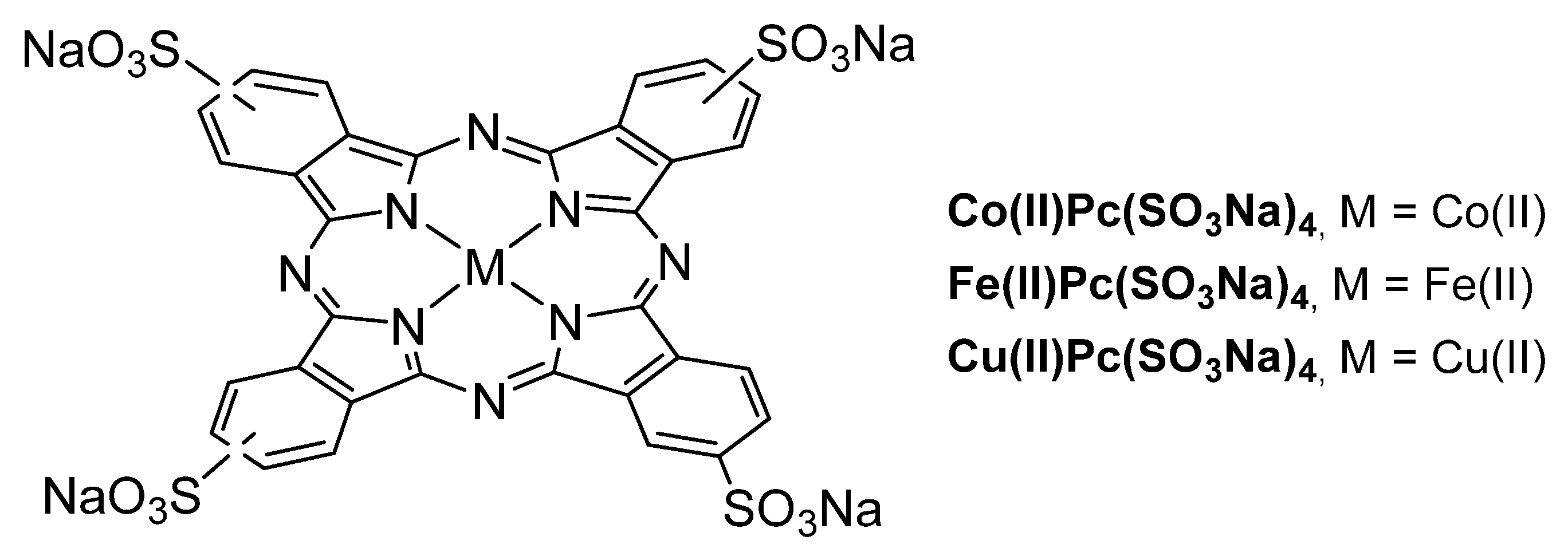
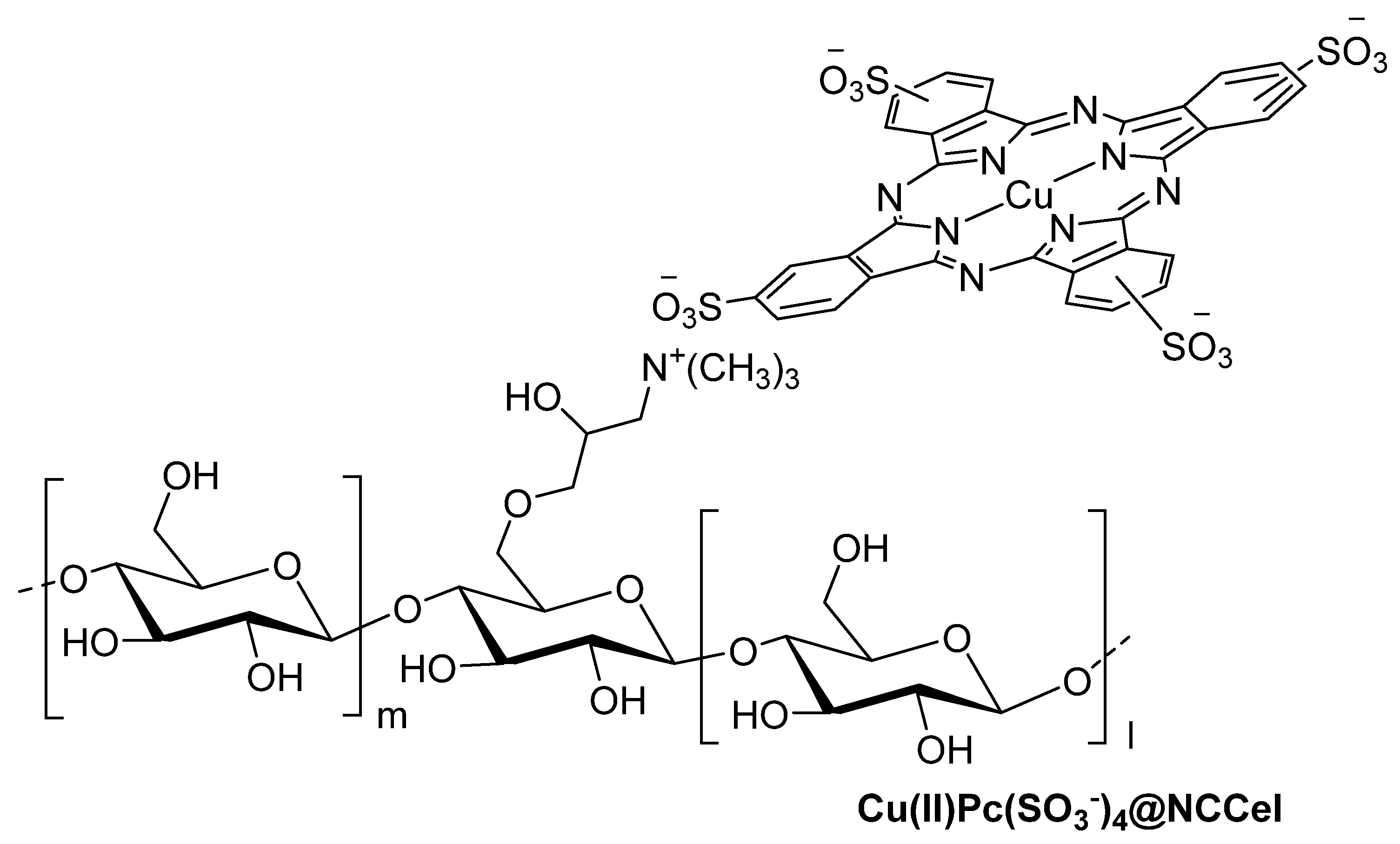
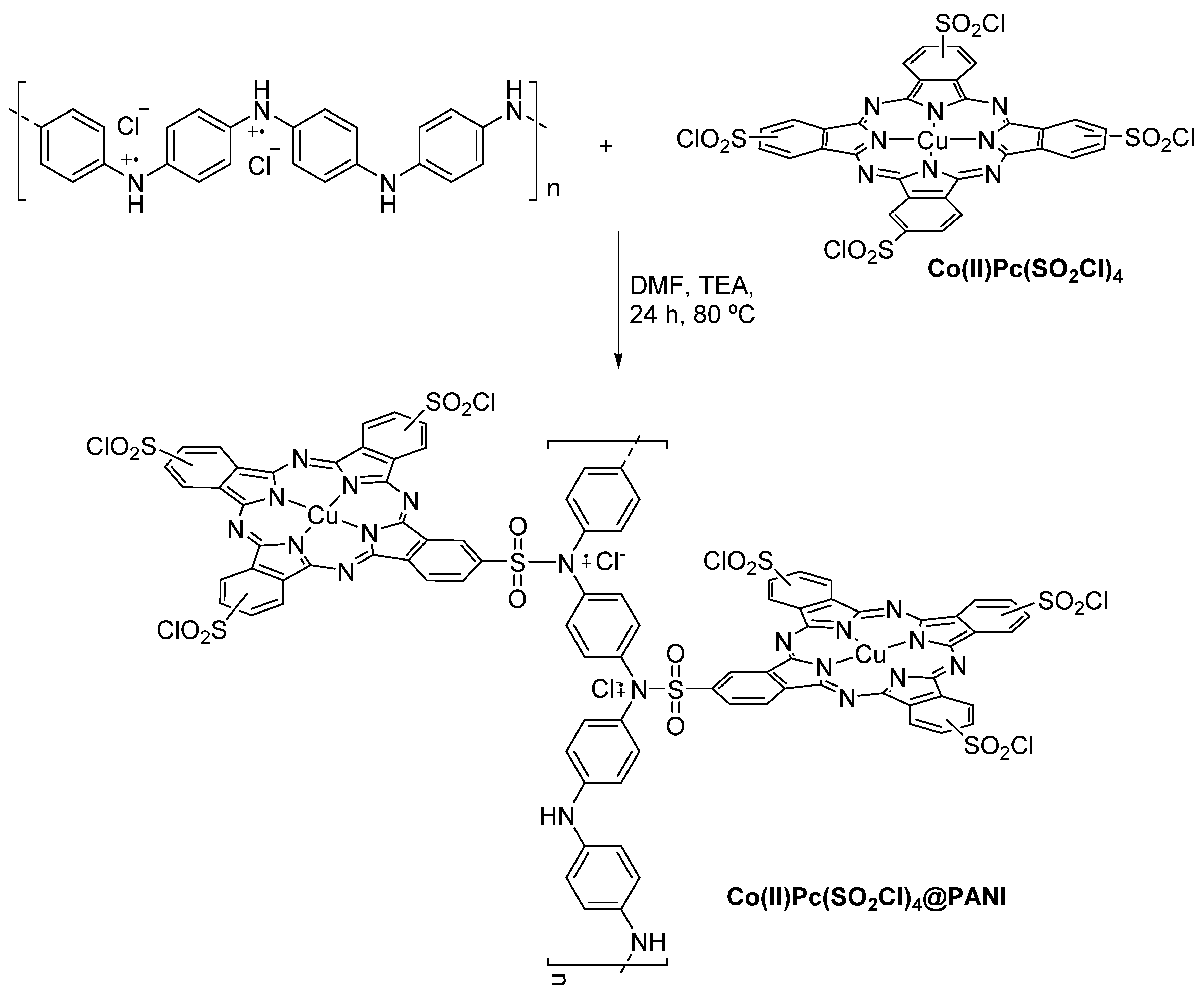
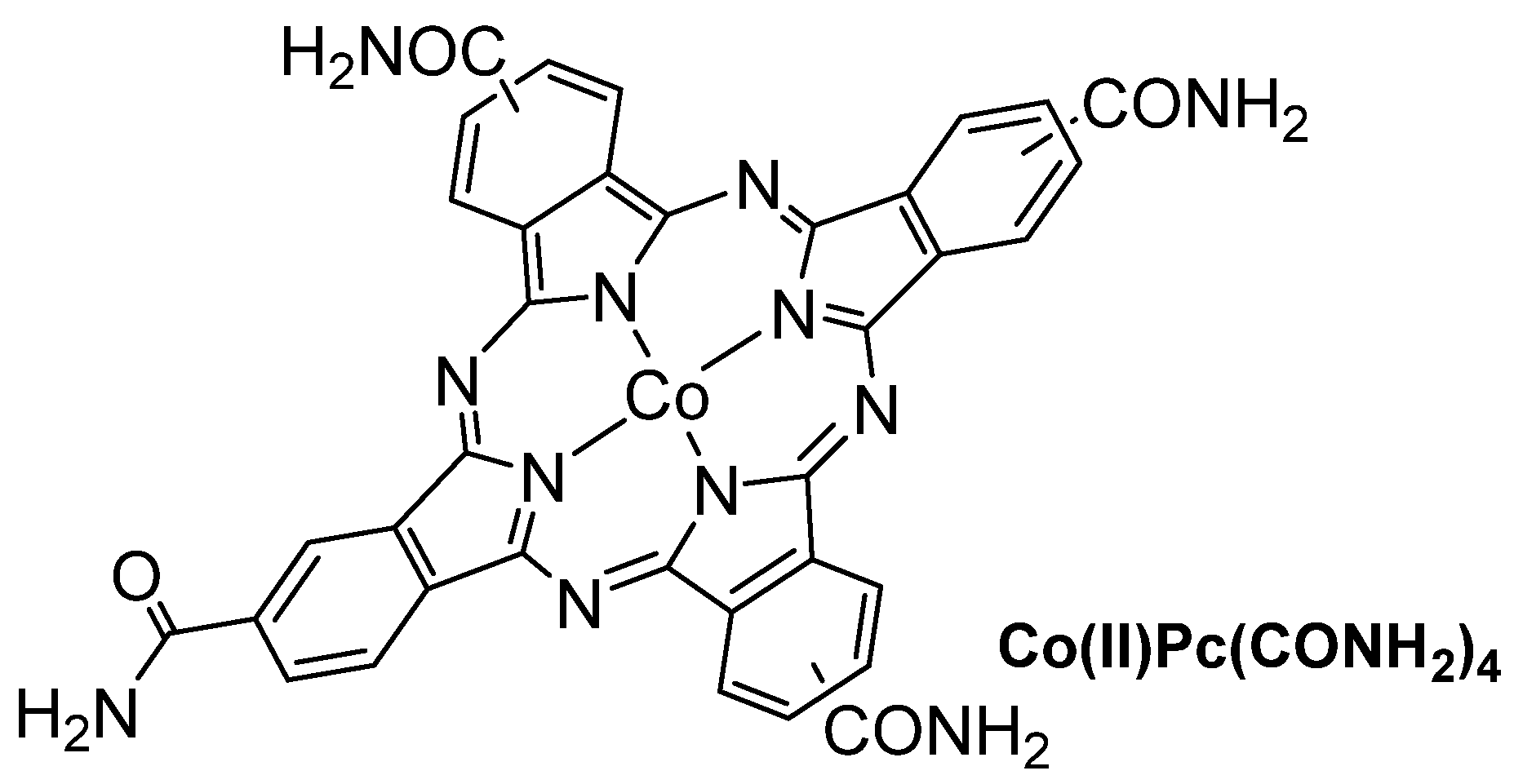
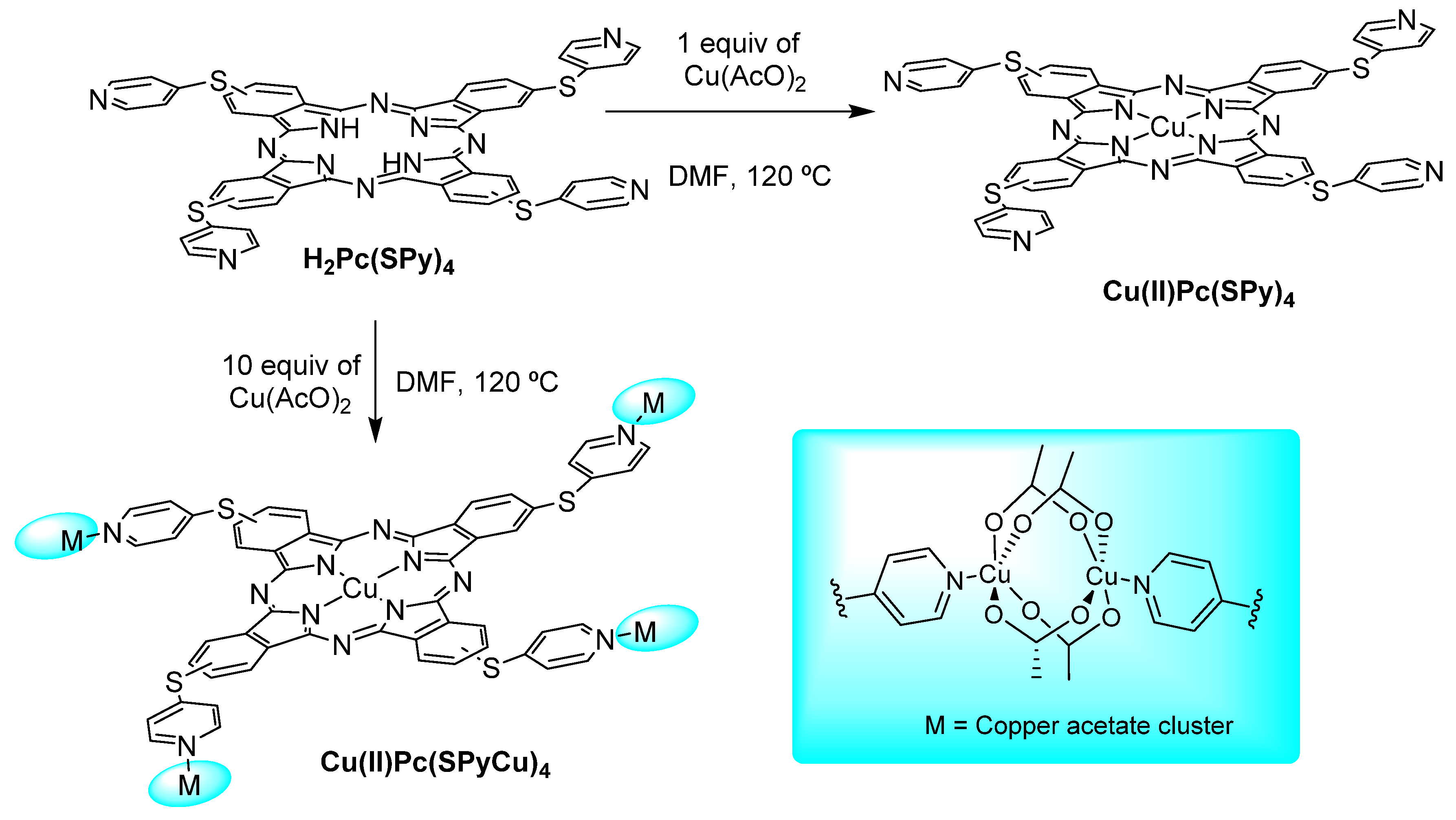
3.1. Thiols and Thiophenes




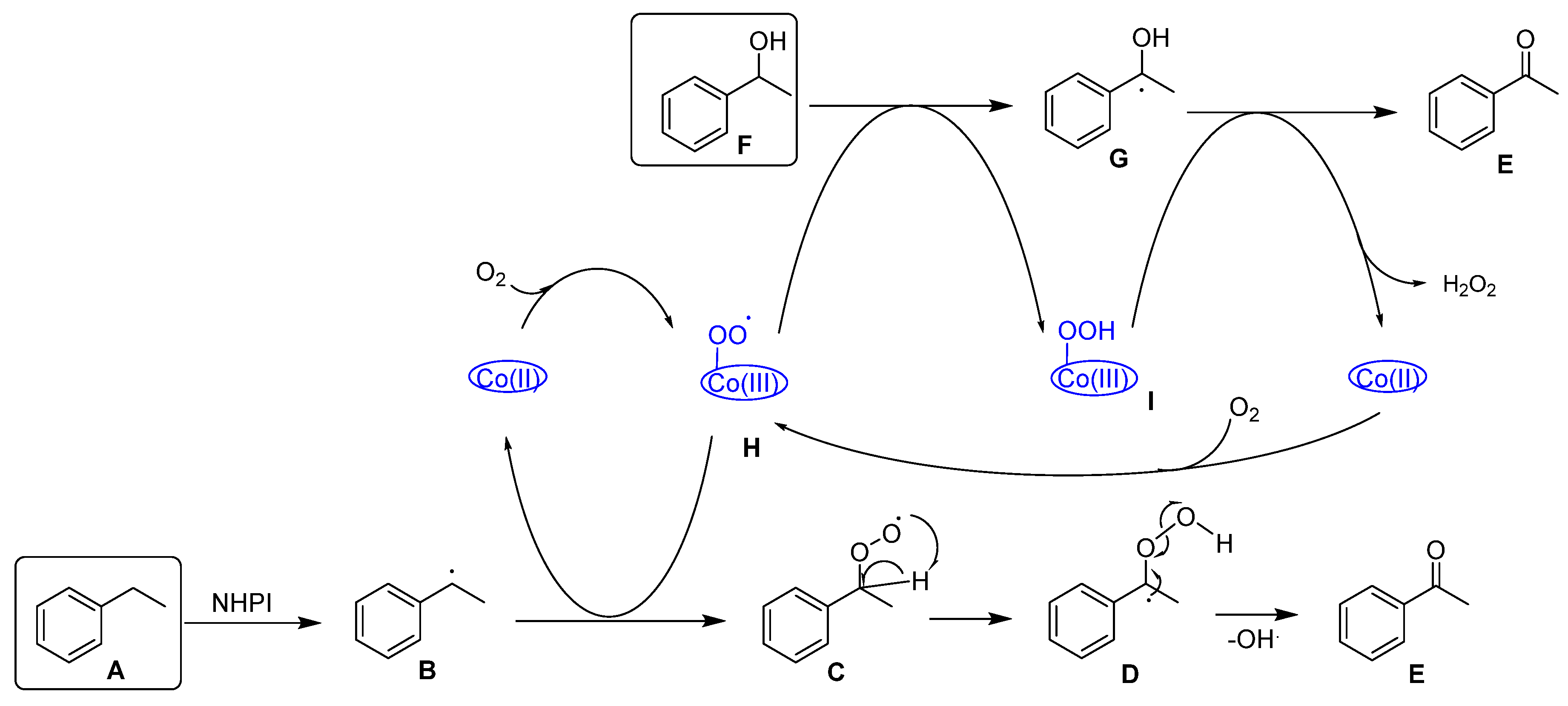
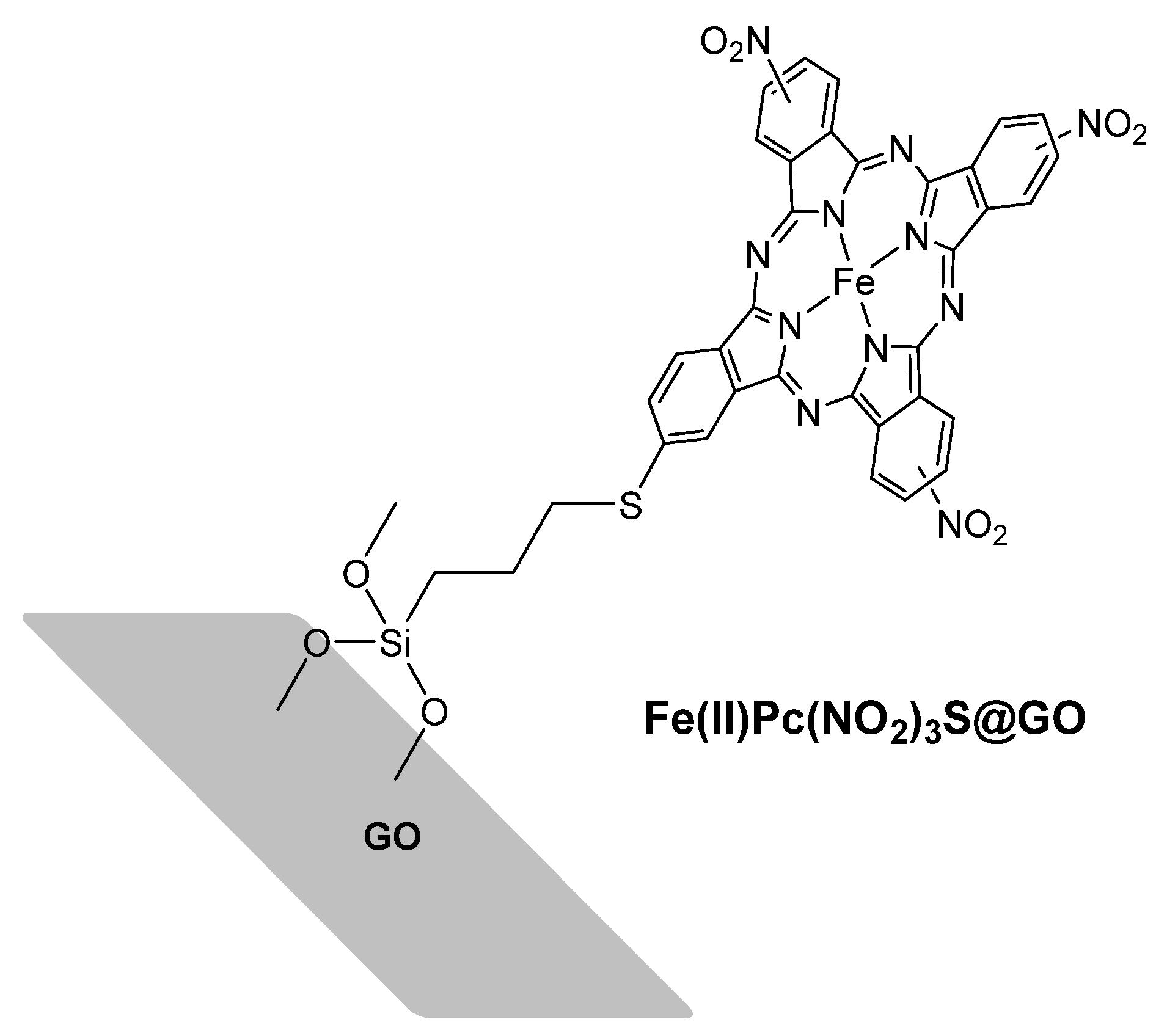
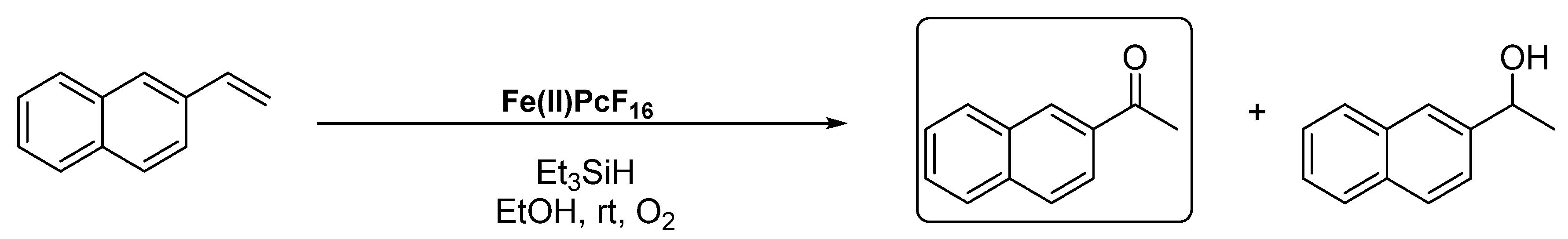
3.2. Alkyl Arenes and Alcohols
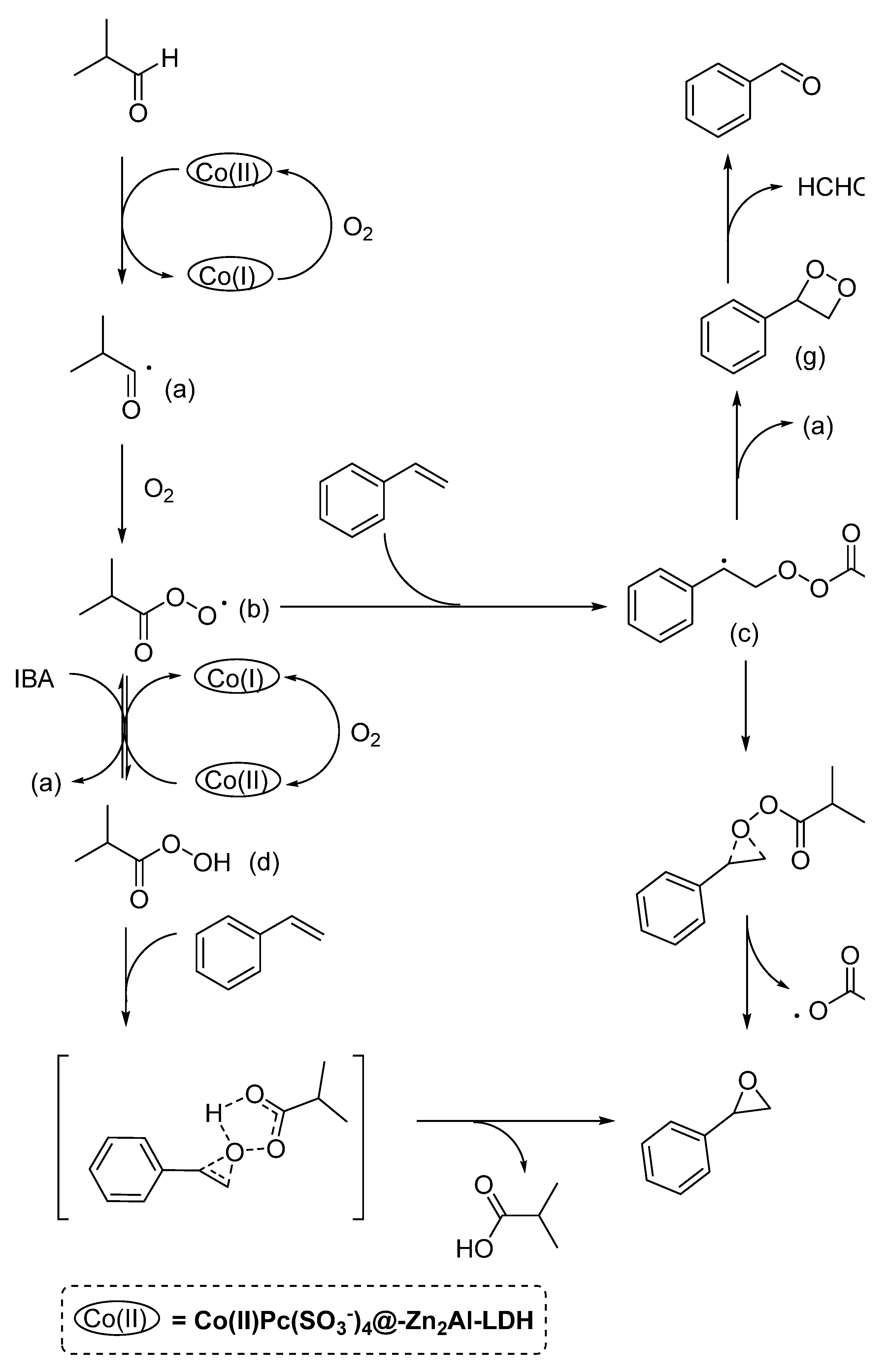
3.3. Olefins
3.4. Miscellaneous Substrates
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Braun, A.; Tcherniac, J. Über die Produkte der Einwirkung von Acetanhydrid auf Phthalamid. Ber. Dtsch. Chem. Ges. 1907, 40, 2709–2714. [Google Scholar] [CrossRef] [Green Version]
- Linstead, R.P.; Lowe, A.R. Phthalocyanines part III Preliminary experiments on the preparation of phthalocyanines from phthalonitrile. J. Chem. Soc. 1934, 1022–1027. [Google Scholar] [CrossRef]
- Robertson, J.M.; Woodward, I. 37. An X-ray study of the phthalocyanines. Part III. Quantitative structure determination of nickel phthalocyanine. J. Chem. Soc. 1937, 10, 219–230. [Google Scholar] [CrossRef]
- Moser, F.H.; Thomas, A.L. Phthalocyanine compounds. J. Chem. Educ. 1964, 41, 245. [Google Scholar] [CrossRef]
- Claessens, C.G.; Hahn, U.; Torres, T. Phthalocyanines: From outstanding electronic properties to emerging applications. Chem. Rec. 2008, 8, 75–97. [Google Scholar] [CrossRef] [PubMed]
- Gobo, N.R.S.; Brocksom, T.J.; de Oliveira, K.T. Soluble and Non-Aggregated Phthalocyanines: Synthesis, Mechanistic Aspects and Their Main Building Blocks. Curr. Org. Synth. 2017, 14, 1132–1155. [Google Scholar] [CrossRef]
- Sorokin, A.B. Phthalocyanine Metal Complexes in Catalysis. Chem. Rev. 2013, 113, 8152–8191. [Google Scholar] [CrossRef] [PubMed]
- Ferraudi, G.; Lappin, A.G. Review: Properties chemical reactivity of metallo phthalocyanine and tetramethylbenzoannulene complexes grafted into a polymer. J. Coord. Chem. 2014, 67, 3822–3839. [Google Scholar] [CrossRef]
- Stillman, M.J.; Nyokong, T. Absorption and Magnetic Circular Dichroism Spectral Properties of Phthalocyanines Part 1: Complexes of the Dianion Pc(–2). In Phthalocyanines-Properties and Applications; Leznoff, C.C., Lever, A.B.P., Eds.; VCH Publishers, Inc.: New York, NY, USA, 1989; Volume 1, pp. 133–290. [Google Scholar]
- Fitzgerald, J.P.; Haggerty, B.S.; Rheingold, A.L.; May, L.; Brewer, G.A. Iron Octaethyltetraazaporphyrins: Synthesis, Characterization, Coordination Chemistry, and Comparisons to Related Iron Porphyrins and Phthalocyanines. Inorg. Chem. 1992, 31, 2006–2013. [Google Scholar] [CrossRef]
- Koczorowski, T.; Szczolko, W.; Goslinski, T. Physicochemical Properties and Catalytic Applications of Iron Porphyrazines and Phthalocyanines. In Recent Progress in Organometallic Chemistry-IntechOpen; Rahman, M.M., Asiri, A.M., Eds.; IntechOpen Limited: London, UK, 2017. [Google Scholar] [CrossRef] [Green Version]
- Nemykin, V.N.; Lukyanets, E.A. Synthesis of substituted phthalocyanines. Arkivoc 2010, 1, 136–208. [Google Scholar] [CrossRef] [Green Version]
- Ghani, F.; Kristen, J.; Riegler, H. Solubility Properties of Unsubstituted Metal Phthalocyanines in Different Types of Solvents. J. Chem. Eng. Data 2012, 57, 439–449. [Google Scholar] [CrossRef]
- Boucher, L.J. Metal Complexes of Phthalocyanines. In Coordination Chemistry of Macrocyclic Compounds; Melson, G.A., Ed.; Springer: Boston, MA, USA, 1979; pp. 461–516. [Google Scholar] [CrossRef]
- Kobak, R.Z.U.; Ari, M.U.; Tekin, A.; Gul, A. Aggregation behavior in unsymmetrically substituted metal-free phthalocyanines. Chem. Phys. 2015, 448, 91–97. [Google Scholar] [CrossRef]
- Nyokong, T.; Gasyna, Z.; Stillman, M.J. Phthalocyanine. pi.-cation-radical species: Photochemical and electrochemical preparation of [ZnPc(-1).+ in solution. Inorg. Chem. 1987, 26, 548–553. [Google Scholar] [CrossRef]
- Milaeva, E.R.; Speier, G.; Lever, A.B.P. The Redox Chemistry of Metallophthalocyanines in Solution; Wiley-VCH: New York, NY, USA, 1993; Volume 3, pp. 3–69. [Google Scholar]
- Silva, A.M.S.; Neves, M.G.P.M.S.; Martins, R.R.L.; Cavaleiro, J.A.S.; Boschi, T.; Tagliatesta, P. Photo-oxygenation of meso-tetraphenylporphyrin derivatives: The influence of the substitution pattern and characterization of the reaction products. J. Porphyr. Phthalocyanines 1998, 2, 45–51. [Google Scholar] [CrossRef]
- D’Alessandro, N.; Tonucci, L.; Dragani, L.K.; Morvillo, A.; Bressan, M. Fate of nickel and cobalt sulfophthalocyanines under oxidizing conditions: A spectroscopic investigation. J. Porphyr. Phthalocyanines 2003, 7, 484–492. [Google Scholar] [CrossRef]
- D’Alessandro, N.; Tonucci, L.; Morvillo, A.; Dragani, L.K.; Di Deo, M.; Bressan, A. Thermal stability and photostability of water solutions of sulfophthalocyanines of Ru(II), Cu(II), Ni(II), Fe(III) and Co(II). J. Organomet. Chem. 2005, 690, 2133–2141. [Google Scholar] [CrossRef]
- Jones, C.W. On the Stability and Recyclability of Supported Metal-Ligand Complex Catalysts: Myths, Misconceptions and Critical Research Needs. Top. Catal. 2010, 53, 942–952. [Google Scholar] [CrossRef]
- Bressan, M.; Celli, N.; d’Alessandro, N.; Liberatore, L.; Morvillo, A.; Tonucci, L. Ruthenium sulfophthalocyanine catalyst for the oxidation of chlorinated olefins with hydrogen peroxide. J. Organomet. Chem. 2000, 593, 416–420. [Google Scholar] [CrossRef]
- Wang, G.; Ramesh, N.; Hsu, A.; Chu, D.; Chen, R. Density functional theory study of the adsorption of oxygen molecule on iron phthalocyanine and cobalt phthalocyanine. Mol. Simul. 2008, 34, 1051–1056. [Google Scholar] [CrossRef]
- Shi, Z.; Zhang, J.J. Density functional theory study of transitional metal macrocyclic complexes’ dioxygen-binding abilities and their catalytic activities toward oxygen reduction reaction. J. Phys. Chem. C 2007, 111, 7084–7090. [Google Scholar] [CrossRef] [Green Version]
- Dolotova, O.V.; Bundina, N.I.; Kaliya, O.L.; Lukyanets, E.A. Manganese Phthalocyanine Coordination Chemistry: Recent Results and Present Status. J. Porphyr. Phthalocyanines 1997, 1, 355–366. [Google Scholar] [CrossRef]
- Wariishi, H.; Gold, M.H. Lignin peroxidase compound III. Mechanism of formation and decomposition. J. Biol. Chem. 1990, 265, 2070–2077. [Google Scholar] [CrossRef]
- Lever, A.B.P.; Wilshire, J.P.; Quan, S.K. Oxidation of manganese(II) phthalocyanine by molecular-oxygen. Inorg. Chem. 1981, 20, 761–768. [Google Scholar] [CrossRef]
- Uchida, K.; Soma, M.; Naito, S.; Onishi, T.; Tamaru, K. Manganese phthalocyanine as a model of tryptophan-2,3-dioxygenase. Chem. Lett. 1978, 7, 471–474. [Google Scholar] [CrossRef]
- Moxon, N.T.; Fielding, P.E.; Gregson, A.K. Manganese 4,4’,4″,4′″-tetrasulfonated phthalocyanine, its dioxygen adduct, and electron-spin-resonance evidence for intramolecular electron-transfer in solution. J. Chem. Soc. Chem. Commun. 1981, 98–99. [Google Scholar] [CrossRef]
- Ercolani, C.; Gardini, M.; Pennesi, G.; Rossi, G. Dioxygen Activation and Catalytic Oxidation of triphenylphosphine by iron phthalocyanine compounds. J. Mol. Catal. 1985, 30, 135–144. [Google Scholar] [CrossRef]
- Kontarinis, D.; Paraskevas, S.M.; Paraskevas, M.S. Oxygen adducts of Co-phthalocyanine. Synth. React. Inorg. Met. Org. Chem. 2003, 33, 1381–1389. [Google Scholar] [CrossRef]
- Hassanein, M.; Abdo, M.; Gerges, S.; El-Khalafy, S. Study of the oxidation of 2-aminophenol by molecular oxygen catalyzed by cobalt(II) phthalocyaninetetrasodiumsulfonate in water. J. Mol. Catal. A Chem. 2008, 287, 53–56. [Google Scholar] [CrossRef]
- Schutten, J.H.; Piet, P.; German, A.L. Some observations on complexes of a cobalt phthalocyanine with poly(vinylamine) and their catalytic activity in the autoxidation of thiols. Die Makromol. Chem. 1979, 180, 2341–2350. [Google Scholar] [CrossRef]
- Pan, Y.; Chen, W.X.; Lu, S.F.; Zhang, Y.F. Novel aqueous soluble cobalt phthalocyanine: Synthesis and catalytic activity on oxidation of 2-mercaptoethanol. Dye. Pigment. 2005, 66, 115–121. [Google Scholar] [CrossRef]
- Geraskin, I.M.; Luedtke, M.W.; Neu, H.M.; Nemykin, V.N.; Zhdankin, V.V. Organic iodine(V) compounds as terminal oxidants in iron(III) phthalocyanine catalyzed oxidation of alcohols. Tetrahedron Lett. 2008, 49, 7410–7412. [Google Scholar] [CrossRef]
- Kivits, P.; Debont, R.; Vanderveen, J. Vanadyl phthalocyanine: An organic material for optical data recording. Appl. Phys. A 1981, 26, 101–105. [Google Scholar] [CrossRef]
- Ramadan, A.J.; Rochford, L.A.; Moffat, J.; Mulcahy, C.; Ryan, M.P.; Jones, T.S.; Heutz, S. The morphology and structure of vanadyl phthalocyanine thin films on lithium niobate single crystals. J. Mater. Chem. C 2016, 4, 348–351. [Google Scholar] [CrossRef] [Green Version]
- Wong, E.W.Y.; Walsby, C.J.; Storr, T.; Leznoff, D.B. Phthalocyanine as a Chemically Inert, Redox-Active Ligand: Structural and Electronic Properties of a Nb(IV)-Oxo Complex Incorporating a Highly Reduced Phthalocyanine(4-) Anion. Inorg. Chem. 2010, 49, 3343–3350. [Google Scholar] [CrossRef]
- Harris, D.L. High-valent intermediates of heme proteins and model compounds. Curr. Opin. Chem. Biol. 2001, 5, 724–735. [Google Scholar] [CrossRef]
- Dolphin, D. The Electronic Configurations of Catalases and Peroxidases in their High Oxidation States: A Definitive Assessment. Isr. J. Chem. 1981, 21, 67–71. [Google Scholar] [CrossRef]
- Jiang, G.; Chen, J.; Thu, H.Y.; Huang, J.S.; Zhu, N.; Che, C.M. Ruthenium porphyrin-catalyzed aerobic oxidation of terminal aryl alkenes to aldehydes by a tandem epoxidation-isomerization pathway. Angew. Chem. Int. Ed. 2008, 47, 6638–6642. [Google Scholar] [CrossRef]
- Collman, J.P.; Barnes, C.E.; Brothers, P.J.; Collins, T.J.; Ozawa, T.; Gallucci, J.C.; Ibers, J.A. Oxidation of ruthenium(II) and ruthenium(III) porphyrins. Crystal structures of .mu.-oxo-bis[(p-methylphenoxo)(meso-tetraphenylporphyrinato)ruthenium(IV)] and ethoxo(meso-tetraphenylporphyrinato)(ethanol)ruthenium(III)-bisethanol. J. Am. Chem. Soc. 1984, 106, 5151–5163. [Google Scholar] [CrossRef]
- Baek, H.K.; Vanwart, H.E. Elementary Steps in the Formation of Horseradish Peroxidase Compound I: Direct Observation of Compound 0, a New Intermediate with a Hyperporphyrin Spectrum. Biochemistry 1989, 28, 5714–5719. [Google Scholar] [CrossRef]
- Zucca, P.; Neves, C.M.B.; Simoes, M.M.Q.; Neves, M.G.P.M.S.; Cocco, G.; Sanjust, E. Immobilized Lignin Peroxidase-Like Metalloporphyrins as Reusable Catalysts in Oxidative Bleaching of Industrial Dyes. Molecules 2016, 21, 964. [Google Scholar] [CrossRef] [Green Version]
- Meunier, B.; Sorokin, A. Oxidation of pollutants catalyzed by metallophthalocyanines. Acc. Chem. Res. 1997, 30, 470–476. [Google Scholar] [CrossRef]
- Sorokin, A.B.; Kudrik, E.V. Phthalocyanine metal complexes: Versatile catalysts for selective oxidation and bleaching. Catal. Today 2011, 159, 37–46. [Google Scholar] [CrossRef]
- Sorokin, A.; Meunier, B. Oxidative Degradation of Polychlorinated Phenols Catalyzed by Metallosulfophthalocyanines. Chem. Eur. J. 1996, 2, 1308–1317. [Google Scholar] [CrossRef]
- Floris, B.; Donzello, M.P.; Ercolani, C. Single-atom bridged dinuclear metal complexes with emphasis on phthalocyanine systems. In The Porphyrin Handbook; Kadish, K.M., Smith, K.M., Guilard, R., Eds.; Academic Press: San Diego, CA, USA, 2003; pp. 1–62. [Google Scholar]
- Sorokin, A.B. Recent progress on exploring µ-oxo bridged binuclear porphyrinoid complexes in catalysis and material science. Coord. Chem. Rev. 2019, 389, 141–160. [Google Scholar] [CrossRef]
- Sorokin, A.B.; Tuel, A. Metallophthalocyanine functionalized silicas: Catalysts for the selective oxidation of aromatic compounds. Catal. Today 2000, 57, 45–59. [Google Scholar] [CrossRef]
- Sorokin, A.B.; Kudrik, E.V.; Bouchu, D. Bio-inspired oxidation of methane in water catalyzed by N-bridged diiron phthalocyanine complex. Chem. Comm. 2008. [Google Scholar] [CrossRef]
- Colomban, C.; Kudrik, E.V.; Afanasiev, P.; Sorokin, A.B. Degradation of chlorinated phenols in water in the presence of H2O2 and water-soluble mu-nitrido diiron phthalocyanine. Catal. Today 2014, 235, 14–19. [Google Scholar] [CrossRef]
- Colomban, C.; Tobing, A.H.; Mukherjee, G.; Sastri, C.V.; Sorokin, A.B.; de Visser, S.P. Mechanism of Oxidative Activation of Fluorinated Aromatic Compounds by N-Bridged Diiron-Phthalocyanine: What Determines the Reactivity? Chem. Eur. J. 2019, 25, 14320–14331. [Google Scholar] [CrossRef]
- Basu, B.; Satapathy, S.; Bhatnagar, A.K. Merox and Related Metal Phthalocyanine Catalyzed Oxidation Processes. Catal. Rev. Sci. Eng. 1993, 35, 571–609. [Google Scholar] [CrossRef]
- Zhou, X.R.; Li, J.; Wang, X.N.; Jin, K.; Ma, W. Oxidative desulfurization of dibenzothiophene based on molecular oxygen and iron phthalocyanine. Fuel Process. Technol. 2009, 90, 317–323. [Google Scholar] [CrossRef]
- Chen, S.C.; Lu, W.Y.; Yao, Y.Y.; Chen, H.X.; Chen, W.X. Oxidative desulfurization of dibenzothiophene with molecular oxygen catalyzed by carbon fiber-supported iron phthalocyanine. React. Kinet. Mech. Catal. 2014, 111, 535–547. [Google Scholar] [CrossRef]
- Price, B.K.; Tour, J.M. Functionalization of Single-Walled Carbon Nanotubes “On Water”. J. Am. Chem. Soc. 2006, 128, 12899–12904. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, J.; Ren, T.; Hu, Y.; Ge, J.; Zhao, D. Oxidative desulfurization of dibenzothiophene based on air and cobalt phthalocyanine in an ionic liquid. RSC Adv. 2014, 4, 3206–3210. [Google Scholar] [CrossRef]
- Chauhan, D.K.; Patnam, P.L.; Ganguly, S.K.; Jain, S.L. A two in one approach: Renewable support and enhanced catalysis for sweetening using chicken feather bound cobalt(II) phthalocyanine under alkali free environment. RSC Adv. 2016, 6, 51983–51988. [Google Scholar] [CrossRef]
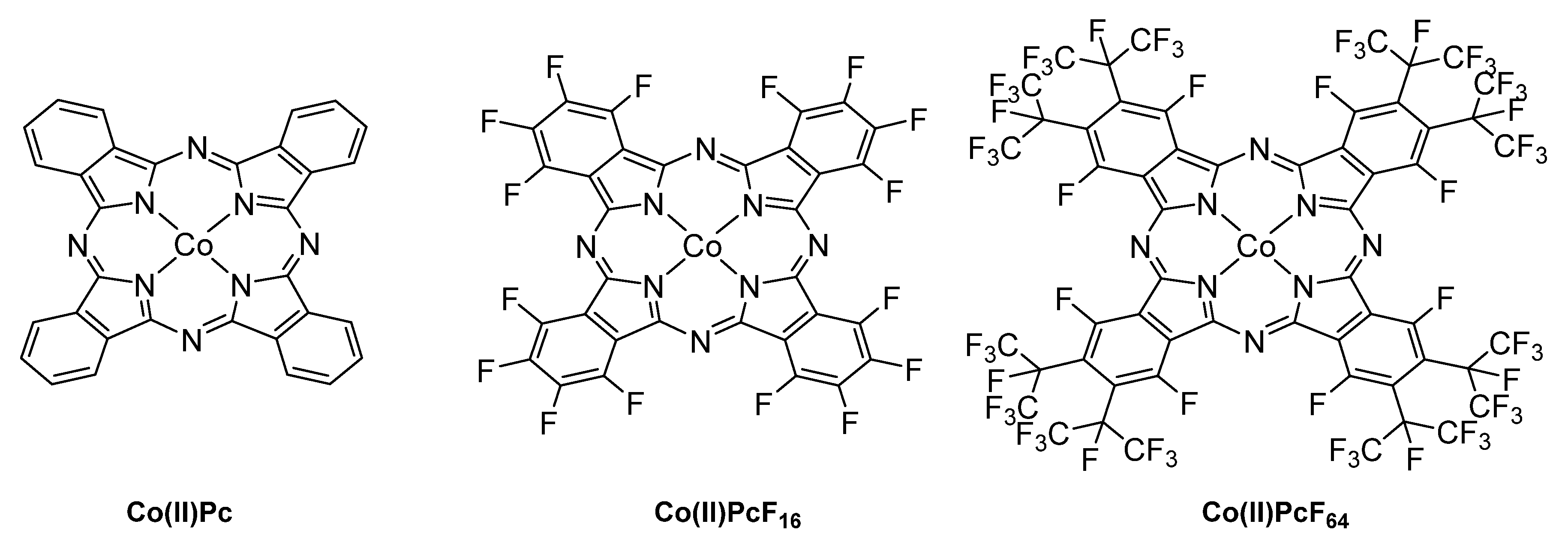
- Reid, N.; Barat, R. Impact of Fluorinated Cobalt(II) Phthalocyanine Catalysts on Aerobic Thiol Oxidation Kinetics. Chem. Eng. Commun. 2016, 203, 714–723. [Google Scholar] [CrossRef]
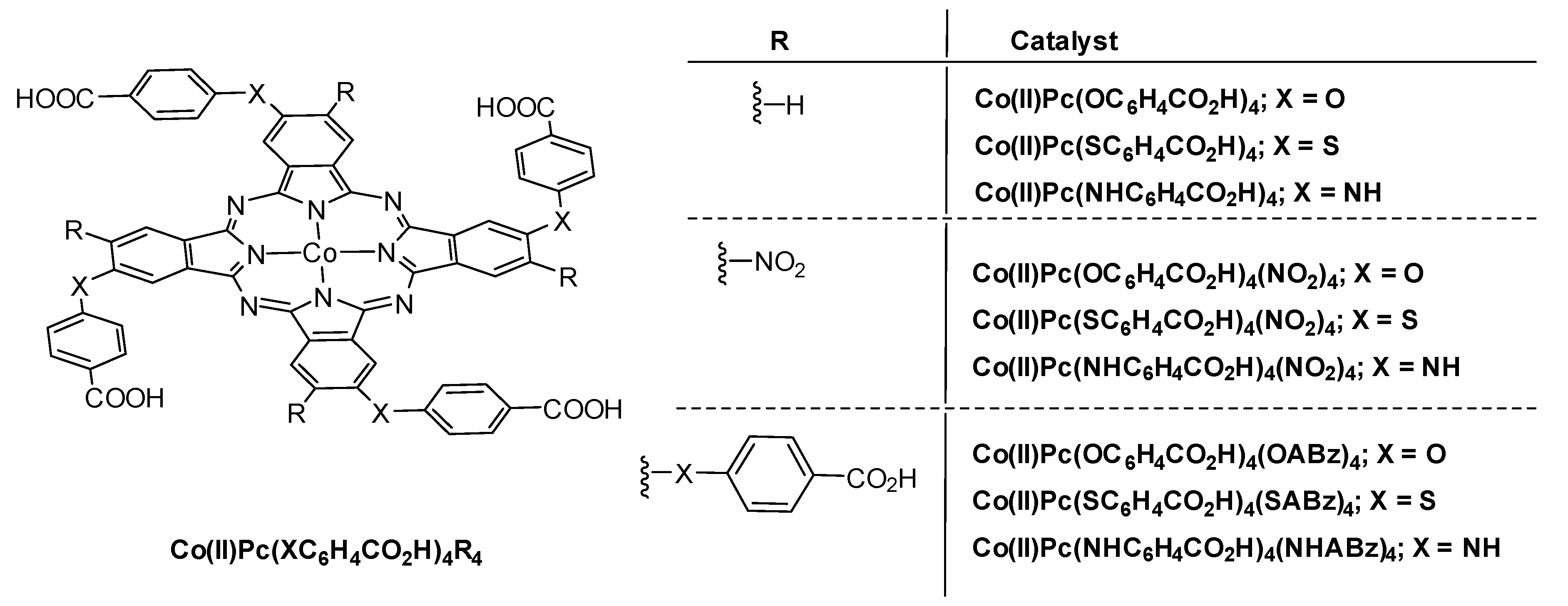
- Vashurin, A.; Maizlish, V.; Kuzmin, I.; Znoyko, S.; Morozova, A.; Razumov, M.; Koifman, O. Symmetrical and difunctional substituted cobalt phthalocyanines with benzoic acids fragments: Synthesis and catalytic activity. J. Porphyr. Phthalocyanines 2017, 21, 37–47. [Google Scholar] [CrossRef]
- Filippova, A.; Vashurin, A.; Znoyko, S.; Kuzmin, I.; Razumov, M.; Chernova, A.; Shaposhnikov, G.; Koifman, O. Novel Co(II) phthalocyanines of extended periphery and their water-soluble derivatives. Synthesis, spectral properties and catalytic activity. J. Mol. Struct. 2017, 1149, 17–26. [Google Scholar] [CrossRef]
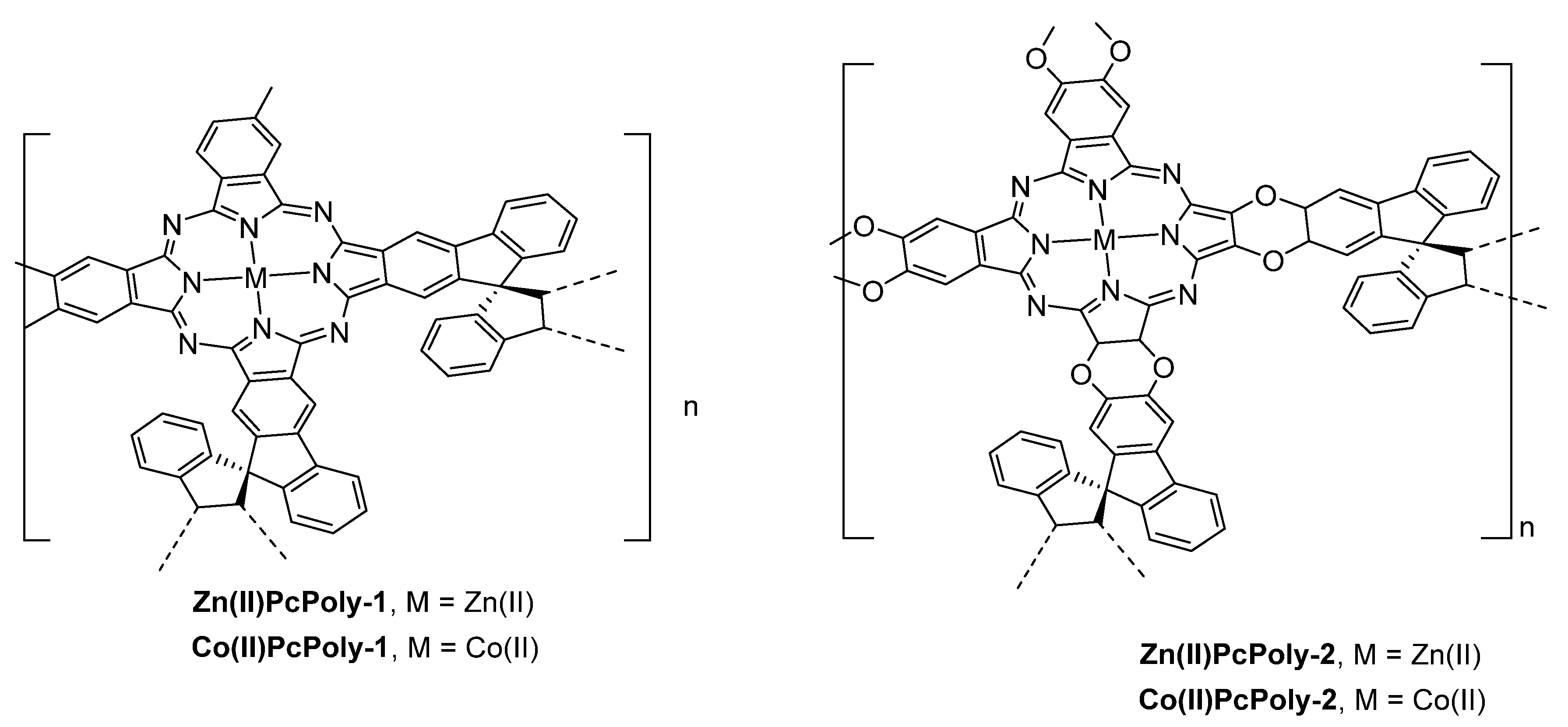
- Tamura, R.; Kawata, T.; Hattori, Y.; Kobayashi, N.; Kimura, M. Catalytic Oxidation of Thiols within Cavities of Phthalocyanine Network Polymers. Macromolecules 2017, 50, 7978–7983. [Google Scholar] [CrossRef]
- Huang, H.; Ash, J.; Kang, J.Y. Base-controlled Fe(Pc)-catalyzed aerobic oxidation of thiols for the synthesis of S-S and S-P(O) bonds. Org. Biomol. Chem. 2018, 16, 4236–4242. [Google Scholar] [CrossRef]
- Motahari, K.; Salabat, A.; Ahmadi, H. Co (II) phthalocyanine-amine functionalized graphene oxide as a solid base catalyst for the oxidation of thiols. Fuller. Nanotub. Carbon Nanostruct. 2018, 26, 342–350. [Google Scholar] [CrossRef]
- Scott, D.W.; Myers, D.L.; Hill, H.; Omadoko, O. Sodium cobalt(II) tetrasulfophthalocyanine and catalytic oxidation of ethanethiol. Fuel 2019, 242, 573–579. [Google Scholar] [CrossRef]
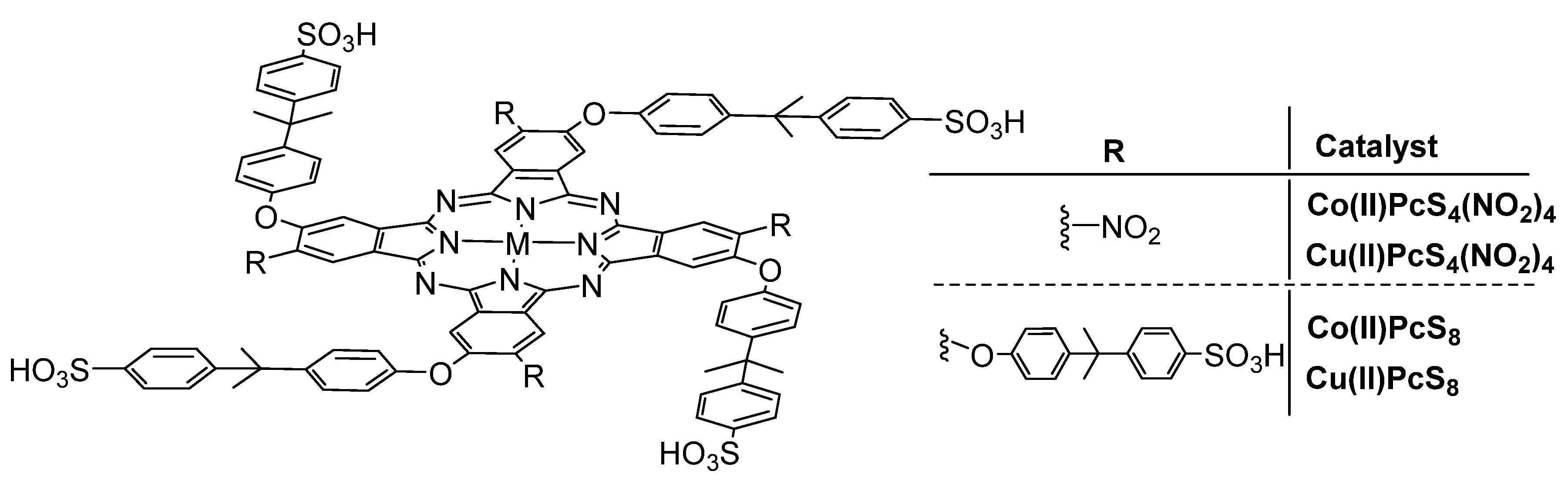
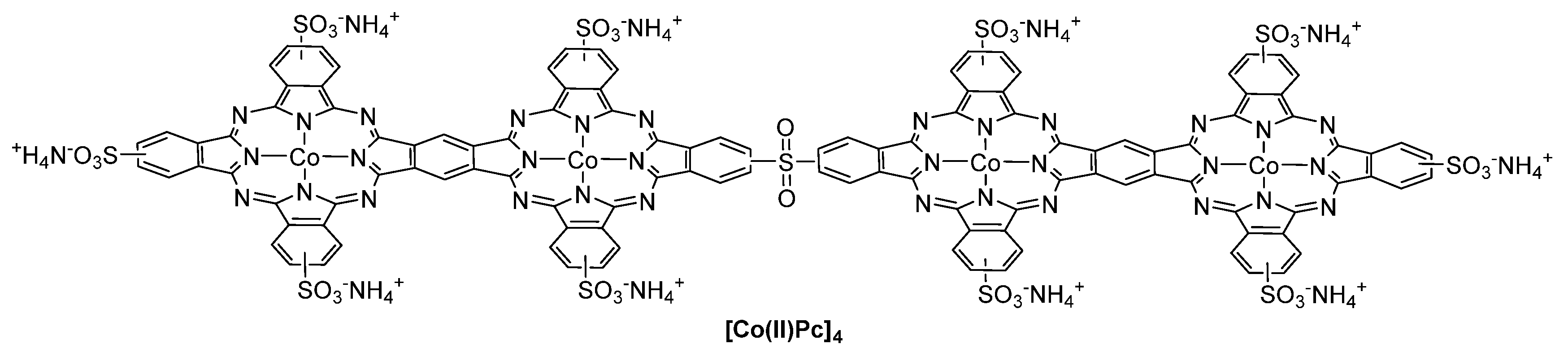
- Li, Y.; Zhang, H.Z.; Jiang, Y.; Shi, M.; Bawa, M.; Wang, X.H.; Zhu, S.Y. Assembly of metallophthalocyanine-polyoxometalate hybrid for highly efficient desulfurization of organic and inorganic sulfur under aerobic conditions. Fuel 2019, 241, 861–869. [Google Scholar] [CrossRef]
- Makarov, S.G.; Ketkov, S.Y.; Wohrle, D. A planar binuclear cobalt(ii) phthalocyanine as a highly efficient catalyst for the oxidation of a mercaptan. Chem. Comm. 2020, 56, 5653–5656. [Google Scholar] [CrossRef] [PubMed]
- Adolfsson, H. Transition Metal-Catalyzed Epoxidation of Alkenes. In Modern Oxidation Methods; Bäckvall, J.-E., Ed.; Wiley-VCH Verlag GmbH & Co.: Weinheim, Germany, 2004; pp. 21–49. [Google Scholar] [CrossRef]
- Pernicone, N.; Cerboni, M.; Prelazzi, G.; Pinna, F.; Fagherazzi, G. An investigation on Pd/C industrial catalysts for the purification of terephthalic acid. Catal. Today 1998, 44, 129–135. [Google Scholar] [CrossRef]
- Sorokin, A.B. Catalytic Transformations in the Presence of Metal Phthalocyanine Complexes and Their Analogs. In Handbook of Porphyrin Science; World Scientific: Singapore, 2016; pp. 193–322. [Google Scholar] [CrossRef]
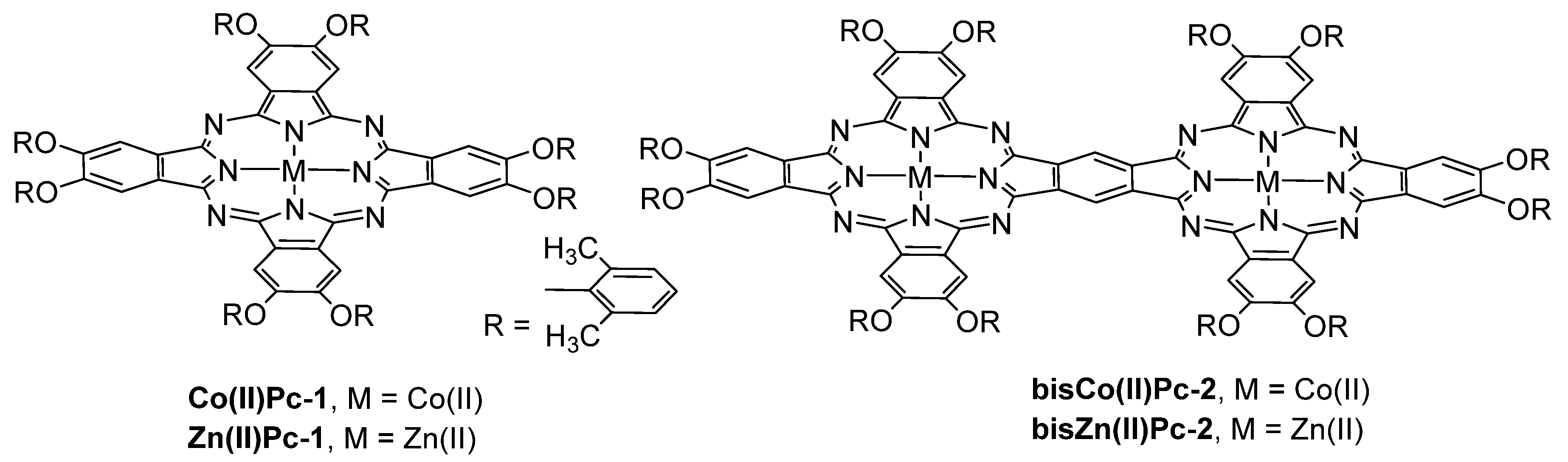
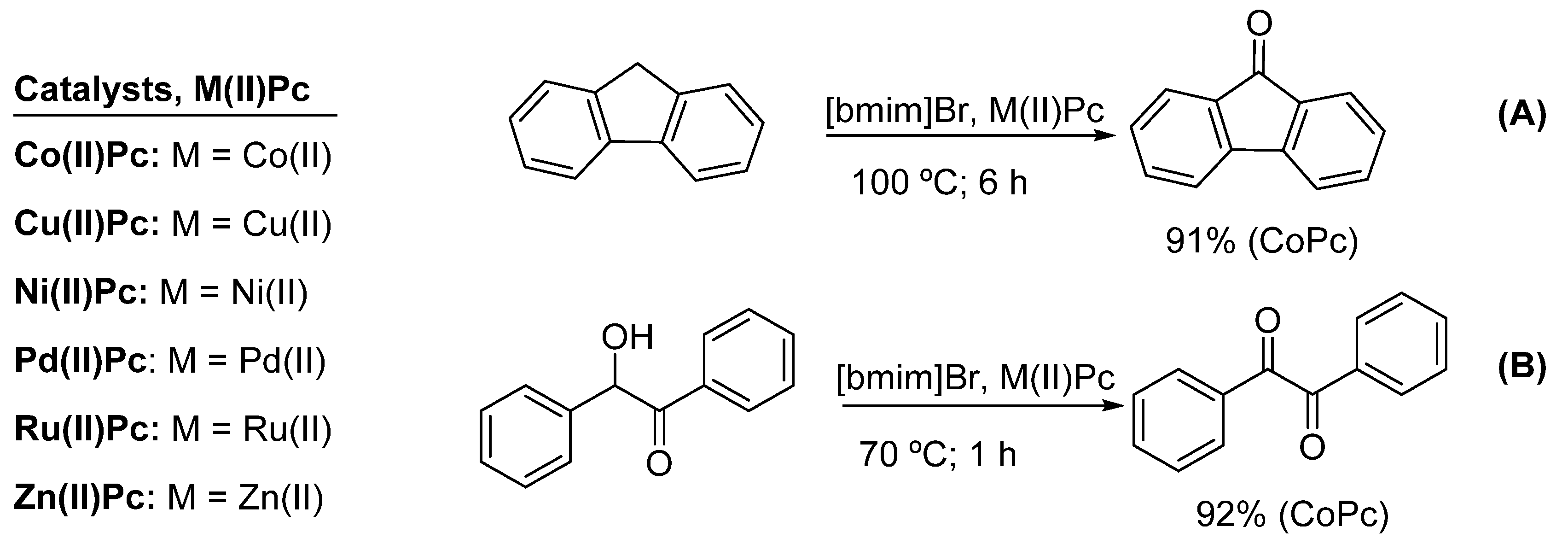
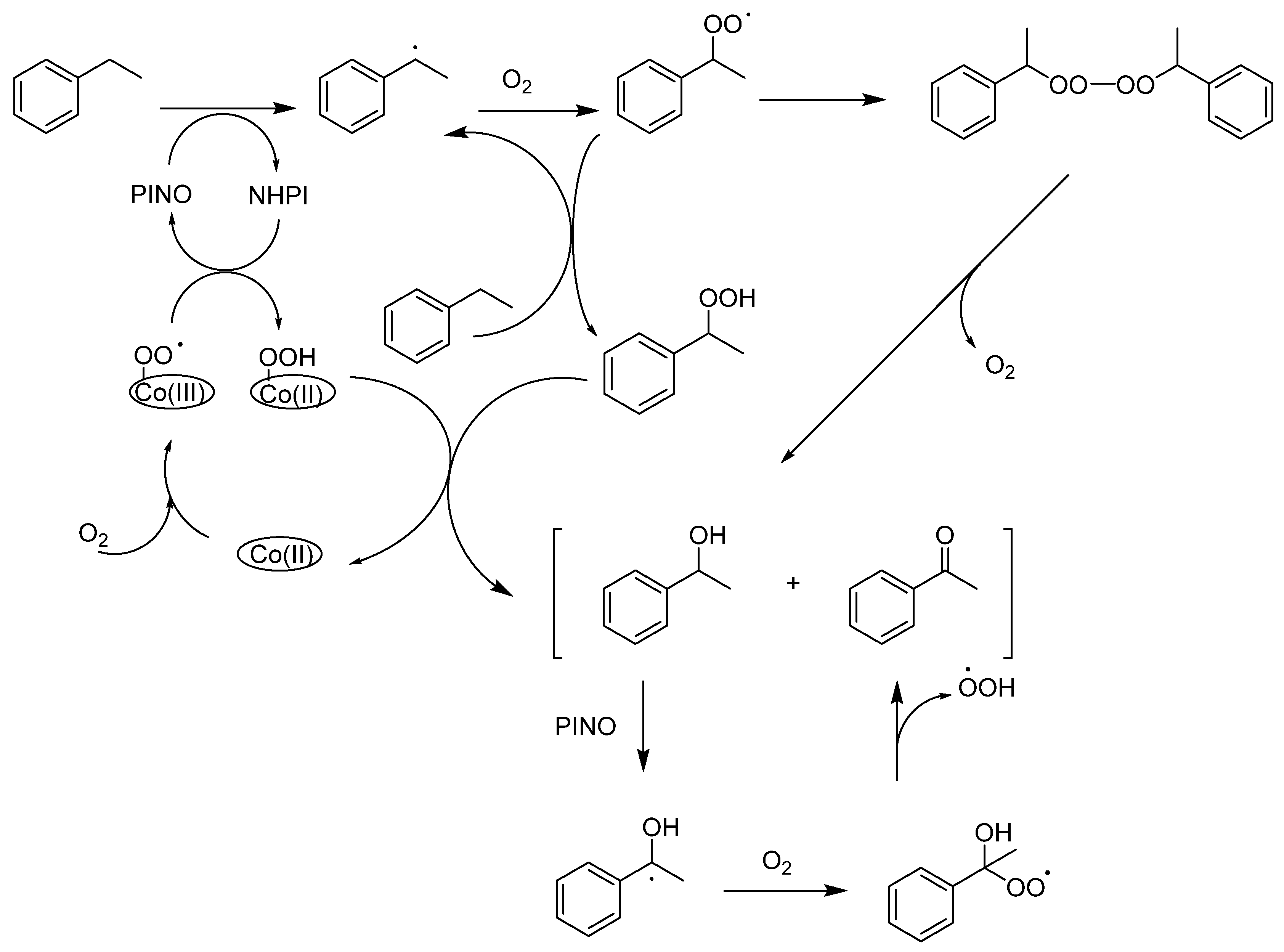
- Shaabani, A.; Farhangi, E.; Rahmati, A. Aerobic oxidation of alkyl arenes and alcohols using cobalt(II) phthalocyanine as a catalyst in 1-butyl-3-methyl-imidazolium bromide. Appl. Catal. A Gen. 2008, 338, 14–19. [Google Scholar] [CrossRef]
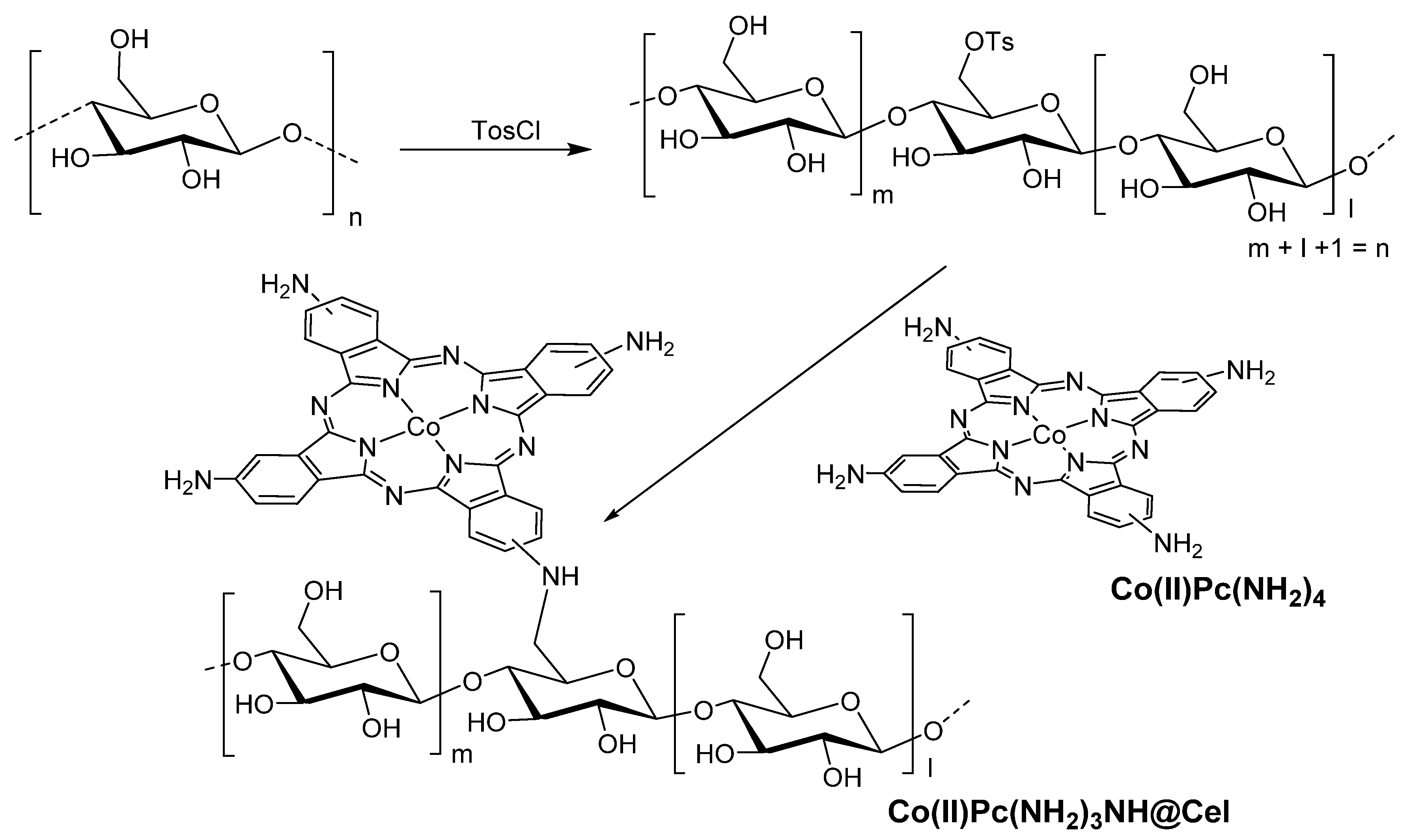
- Shaabani, A.; Keshipour, S.; Hamidzad, M.; Shaabani, S. Cobalt(II) phthalocyanine covalently anchored to cellulose as a recoverable and efficient catalyst for the aerobic oxidation of alkyl arenes and alcohols. J. Mol. Catal. A Chem. 2014, 395, 494–499. [Google Scholar] [CrossRef]
- Mahyari, M.; Shaabani, A. Graphene oxide-iron phthalocyanine catalyzed aerobic oxidation of alcohols. Appl. Catal. A Gen. 2014, 469, 524–531. [Google Scholar] [CrossRef]
- Chauhan, P.; Yan, N. Nanocrystalline cellulose grafted phthalocyanine: A heterogeneous catalyst for selective aerobic oxidation of alcohols and alkyl arenes at room temperature in a green solvent. RSC Adv. 2015, 5, 37517–37520. [Google Scholar] [CrossRef]
- Panwar, V.; Kumar, P.; Ray, S.S.; Jain, S.L. Organic inorganic hybrid cobalt phthalocyanine/polyaniline as efficient catalyst for aerobic oxidation of alcohols in liquid phase. Tetrahedron Lett. 2015, 56, 3948–3953. [Google Scholar] [CrossRef]
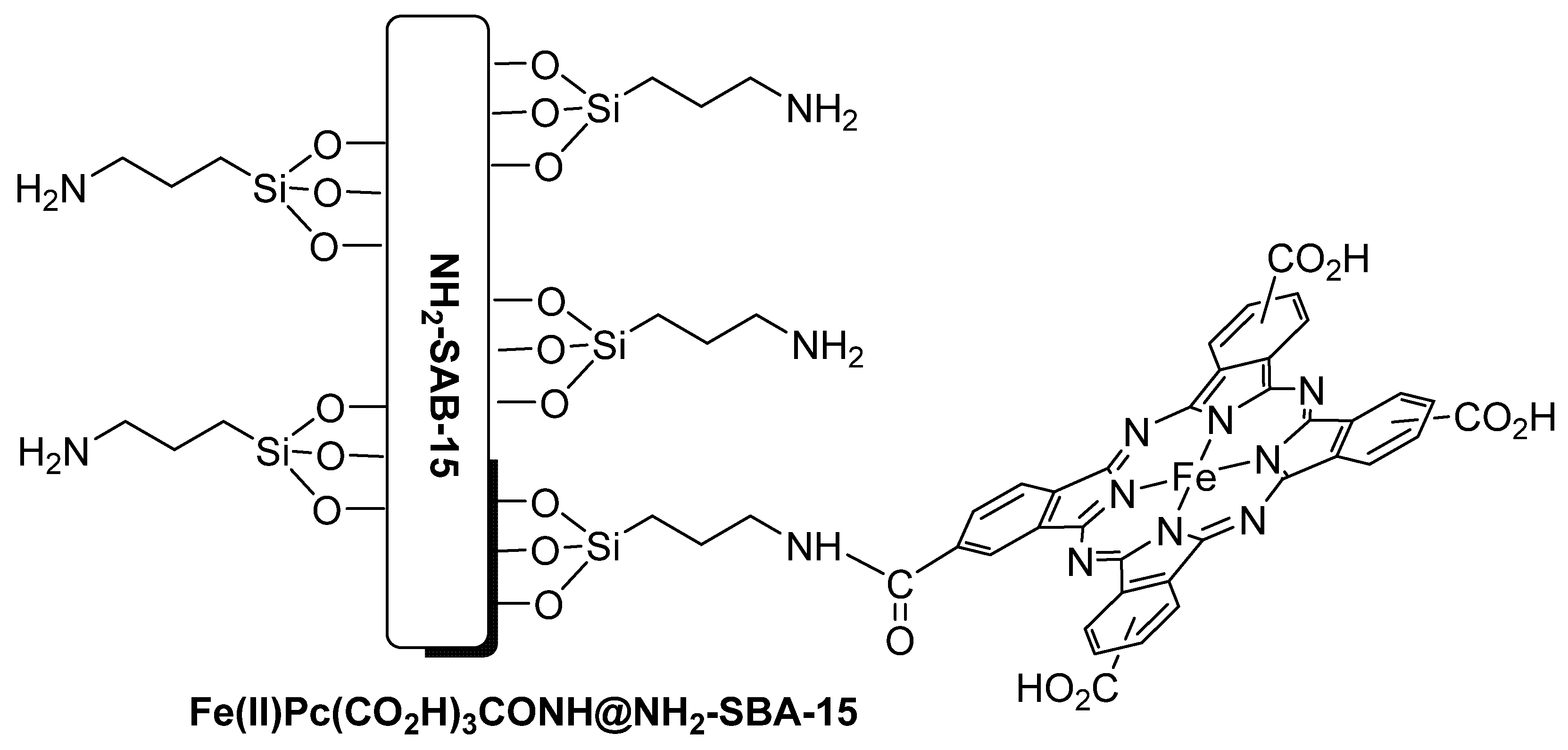
- Huang, C.; Liu, R.; Yang, W.Y.; Zhang, C.T.; Zhu, H.J. Iron(II) phthalocyanine immobilized SBA-15 catalysts: Preparation, characterization and application for toluene selective aerobic oxidation. Inorg. Chim. Acta 2017, 467, 307–315. [Google Scholar] [CrossRef]
- Huang, C.; Liu, R.; Zhang, C.T.; Cheng, Q.P.; Zhu, H.J. Iron(II) and copper(II) phthalocyanine-catalyzed synthesis of 2-nitro-4-methylsulfonylbenzoic acid under mild conditions. J. Chem. Sci. 2017, 129, 1587–1594. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Dai, X.; Chen, Y.; Sun, F.A.; He, M.; Chen, Q. Synergistic Catalytic Oxidation of Ethylbenzene to Acetophenone by Metallophthalocyanine Intercalated Layered Double Hydroxide with Oxygen. ChemistrySelect 2018, 3, 566–572. [Google Scholar] [CrossRef]
- Zhou, W.Y.; Zhou, J.C.; Chen, Y.; Cui, A.J.; Sun, F.A.; He, M.Y.; Xu, Z.X.; Chen, Q. Metallophthalocyanine intercalated layered double hydroxides as an efficient catalyst for the selective epoxidation of olefin with oxygen. Appl. Catal. A Gen. 2017, 542, 191–200. [Google Scholar] [CrossRef]
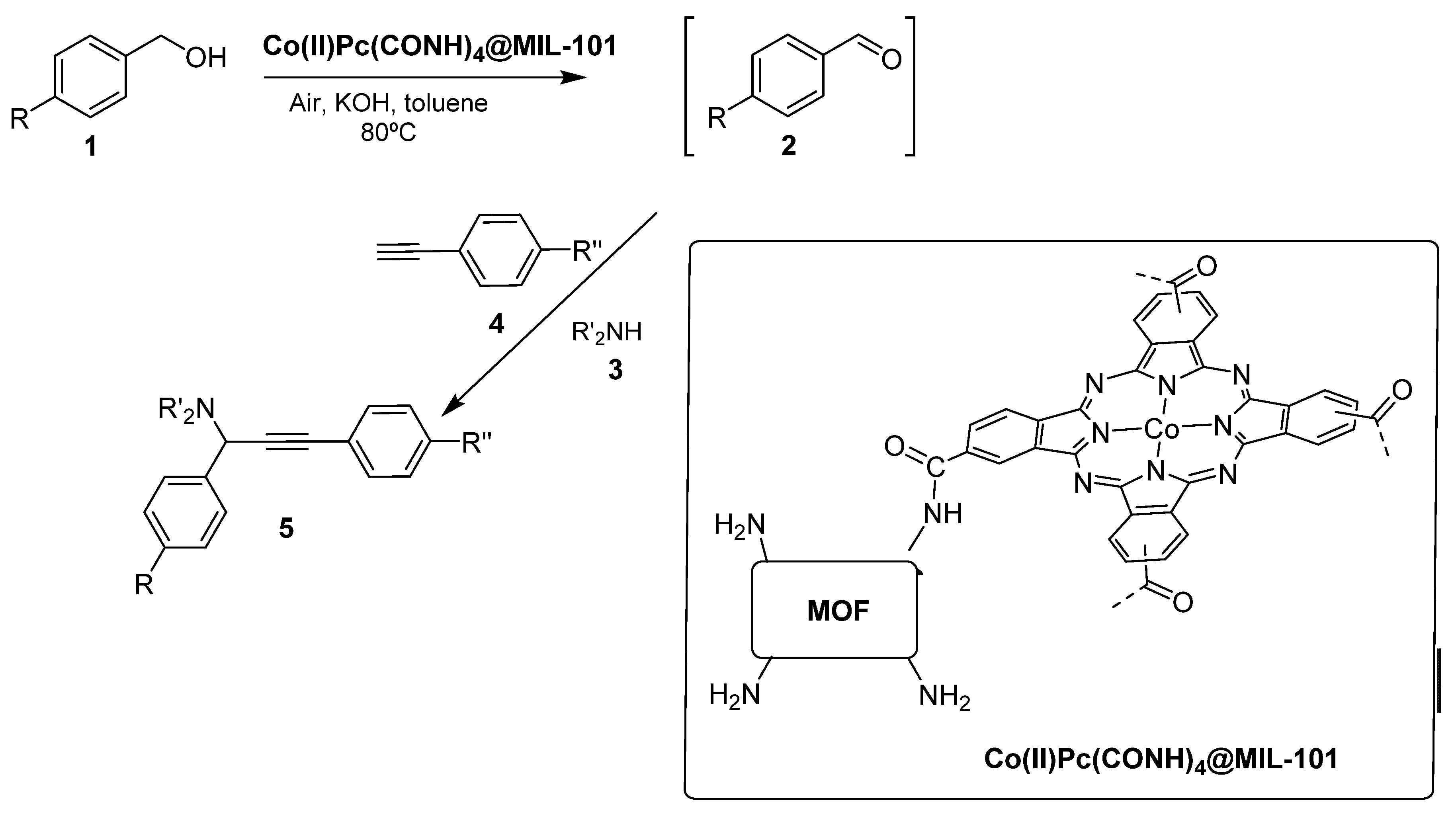
- Shaabani, A.; Mohammadian, R.; Hashemzadeh, A.; Afshari, R.; Amini, M.M. Amine-functionalized MIL-101(Cr) embedded with Co(ii) phthalocyanine as a durable catalyst for one-pot tandem oxidative A(3) coupling reactions of alcohols. New J. Chem. 2018, 42, 4167–4174. [Google Scholar] [CrossRef]
- Li, F.; Tang, S.; Tang, Z.L.; Ye, L.J.; Li, H.H.; Niu, F.F.; Sun, X.L. Synergistic Catalytic Effect of N-Hydroxyphthalimide/Cobalt Tetraamide Phthalocyanine and Its Application for Aerobic Oxidation of Hydrocarbons and Alcohols. Catal. Lett. 2020. [Google Scholar] [CrossRef]
- Puls, F.; Knolker, H.J. Conversion of Olefins into Ketones by an Iron-Catalyzed Wacker-type Oxidation Using Oxygen as the Sole Oxidant. Angew. Chem. Int. Ed. 2018, 57, 1222–1226. [Google Scholar] [CrossRef]
- Castro, K.; Figueira, F.; Paz, F.A.A.; Tome, J.P.C.; da Silva, R.S.; Nakagaki, S.; Neves, M.G.P.M.S.; Cavaleiro, J.A.S.; Simoes, M.M.Q. Copper-phthalocyanine coordination polymer as a reusable catechol oxidase biomimetic catalyst. Dalton Trans. 2019, 48, 8144–8152. [Google Scholar] [CrossRef]
- Brandolese, A.; Ragno, D.; Di Carmine, G.; Bernardi, T.; Bortolini, O.; Giovannini, P.P.; Pandoli, O.G.; Altomare, A.; Massi, A. Aerobic oxidation of 5-hydroxymethylfurfural to 5-hydroxymethyl-2-furancarboxylic acid and its derivatives by heterogeneous NHC-catalysis. Org. Biomol. Chem. 2018, 16, 8955–8964. [Google Scholar] [CrossRef] [PubMed]




































{kind=link}
{kind=link}
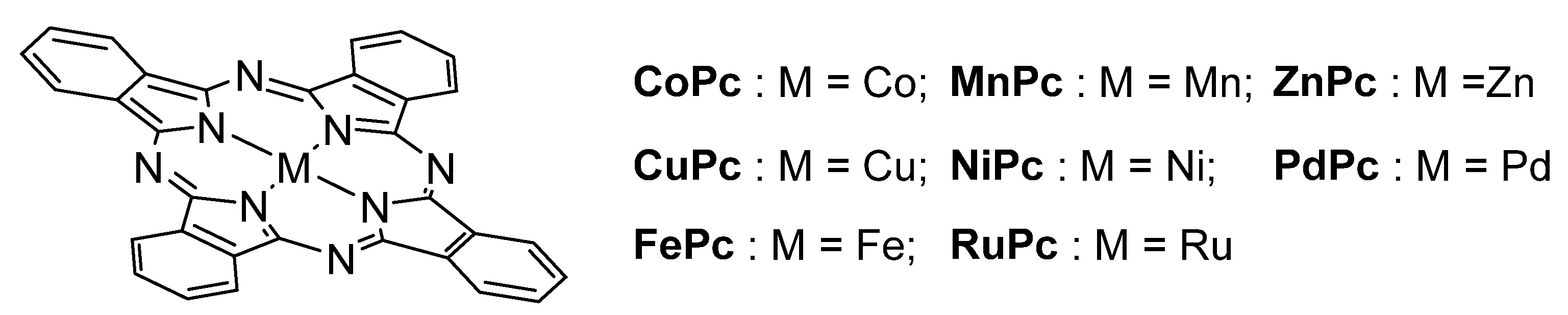{kind=link}
{kind=link}
{kind=link}
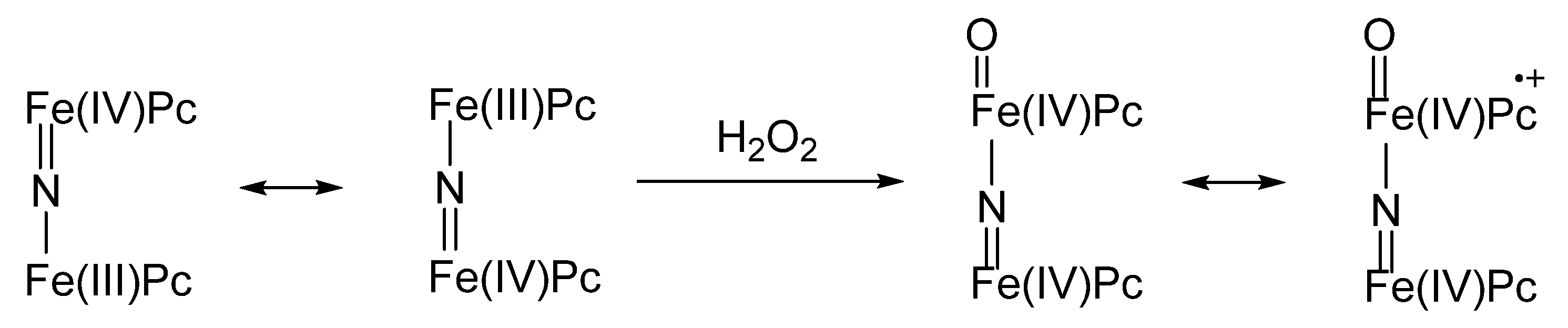{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
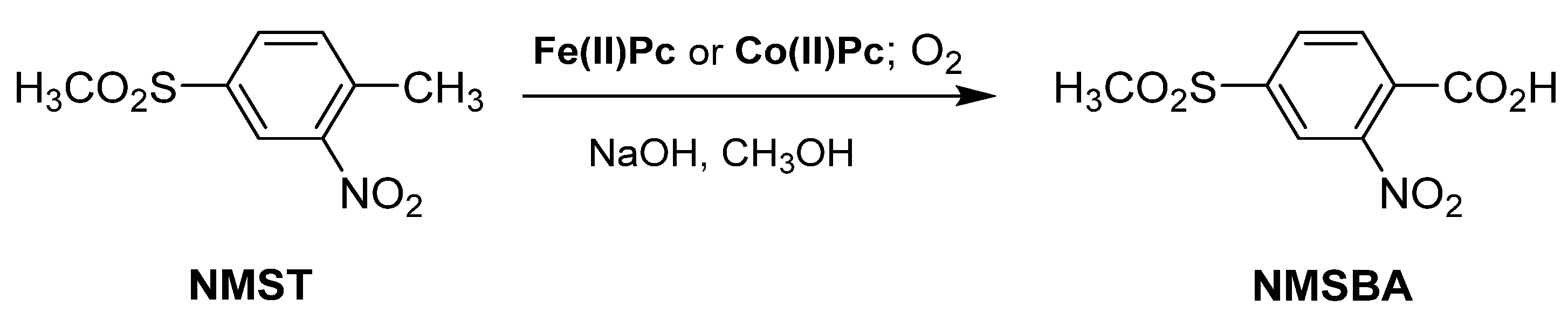{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Mercaptan | Dissulfide | Time (min) | Conv. (%) | Yield (%) |
|---|---|---|---|---|---|
| 1 |  |  | 30 | 98.5 | 96 |
| 2 |  |  | 35 | 97 | 92 |
| 3 |  |  | 45 | 96 | 93 |
| 4 |  |  | 55 | 96 | 93 |
| 5 |  |  | 65 | 91 | 89 |
| 6 |  |  | 120 | 72 | 68 |
| Entry | Mercaptan | Dissulfide | Conv. (%) | Yield (%) |
|---|---|---|---|---|
| 1 |  |  | - | 99 |
| 2 |  |  | 98 | - |
| 3 |  |  | 97 | - |
| 4 |  |  | 99 | 99 |
| 5 |  |  | 91 | - |
| 6 |  |  | 98 | - |
| 7 |  |  | 99 | - |
| 8 |  |  | 99 | - |
| Entry | Mercaptan | H-Phosphonate/H-phosphine | Product | Isolated Yield (%) |
|---|---|---|---|---|
| 1 |  |  |  | 90 |
| 2 |  |  |  | 73 |
| 3 |  |  |  | 63 |
| 4 |  |  |  | 88 |
| 5 |  |  |  | 52 |
| 6 |  |  |  | 57 |
| 7 |  |  |  | 79 |
| 8 |  |  |  | 77 |
| 9 |  |  |  | 45 |
| Entry | Substrate | Product | Conversion (%) | Selectivity (%) |
|---|---|---|---|---|
| 1 |  |  | 92 (R = OMe) 76 (R = Br) 55 (R = NO2) 0 (R = NH2) | 99 (R = OMe) 99 (R = Br) 99 (R = NO2) - (R = NH2) |
| 2 |  |  | 82 | 99 |
| 3 |  |  | 80 | 99 |
| 4 |  |  | 79 | 99 |
| 5 |  |  | 90 | 99 |
| 6 |  |  | 86 | 99 |
| 7 |  |  | 0 | - |
| 8 |  |  | 48 | 15 85 |
| Entry | Substrate | Product | Rection Time (h) | Conv. (%) | Yield (%) |
|---|---|---|---|---|---|
| 1 |  |  | 1.5 1.5 2 | 100 (R = H) 100 (R = CH3) 98 (R = F) | 90 (R = H) 82 (R = CH3) 85 (R = F) |
| 2 |  |  | 2 | 100 | 81 |
| 3 |  |  | 2 | 98 | 89 |
| 4 |  |  | 2 | 98 | 99 |
| 5 |  |  | 2 | 99 | 99 |
| 6 |  |  | 2 4 | 98 (n = 2) 86 (n = 4) | 99 (n = 2) 99 (n = 4) |
| 7 |  |  | 2 | 100 | 82 |
| 8 |  |  | 2 | 99 | 90 |
| 9 |  |  | 4 | 58 | 98 |
| 10 |  |  | 1,5 | 100 | 99 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pereira Monteiro, C.J.; Ferreira Faustino, M.A.; Pinho Morgado Silva Neves, M.d.G.; Quialheiro Simões, M.M.; Sanjust, E. Metallophthalocyanines as Catalysts in Aerobic Oxidation. Catalysts 2021, 11, 122. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11010122
Pereira Monteiro CJ, Ferreira Faustino MA, Pinho Morgado Silva Neves MdG, Quialheiro Simões MM, Sanjust E. Metallophthalocyanines as Catalysts in Aerobic Oxidation. Catalysts. 2021; 11(1):122. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11010122
Chicago/Turabian StylePereira Monteiro, Carlos J., Maria Amparo Ferreira Faustino, Maria da Graça Pinho Morgado Silva Neves, Mário M. Quialheiro Simões, and Enrico Sanjust. 2021. "Metallophthalocyanines as Catalysts in Aerobic Oxidation" Catalysts 11, no. 1: 122. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11010122