Porous Silica-Based Organic-Inorganic Hybrid Catalysts: A Review



Instituto de Tecnología Química, Universitat Politècnica de València-Consejo Superior de Investigaciones Científicas, Avenida de los Naranjos s/n, E-46022 Valencia, Spain
*
Authors to whom correspondence should be addressed.
Catalysts 2021, 11(1), 79; https://0-doi-org.brum.beds.ac.uk/10.3390/catal11010079
Submission received: 16 December 2020
/
Revised: 4 January 2021
/
Accepted: 7 January 2021
/
Published: 8 January 2021
(This article belongs to the Special Issue Nano-Porous Materials for Catalytic Systems in Green Chemistry Processes)
Abstract
:Hybrid organic-inorganic catalysts have been extensively investigated by several research groups in the last decades, as they allow combining the structural robust-ness of inorganic solids with the versatility of organic chemistry. Within the field of hybrid catalysts, synthetic strategies based on silica are among the most exploitable, due to the convenience of sol-gel chemistry, to the array of silyl-derivative precursors that can be synthesized and to the number of post-synthetic functionalization strategies available, amongst others. This review proposes to highlight these advantages, firstly describing the most common synthetic tools and the chemistry behind sol-gel syntheses of hybrid catalysts, then presenting exemplificative studies involving mono- and multi-functional silica-based hybrid catalysts featuring different types of active sites (acid, base, redox). Materials obtained through different approaches are described and their properties, as well as their catalytic performances, are compared. The general scope of this review is to gather useful information for those approaching the synthesis of organic-inorganic hybrid materials, while providing an overview on the state-of-the art in the synthesis of such materials and highlighting their capacities.

1. Introduction to Hybrid Organic-Inorganic Materials
Catalysis plays a crucial role in the chemical industry since many decades. Approximately, 85–90% of the products of chemical industry are made in catalytic processes. They are indispensable in the production of transportation fuels, with about 440 oil refineries heavily relying on them (data of the year 2007) and in the production of bulk and fine chemicals in all branches of the chemical industry. They are key players in the prevention of pollution, in the abatement of automotive or industrial exhaust and they provide environmental and economic advantages by enhancing the selectivity toward the desired product, in turn avoiding or limiting the production of unwanted byproducts. Catalysts offer alternative, energetically favorable mechanisms to non-catalyzed reaction, therefore enabling processes to be carried out under industrially feasible conditions of temperature and pressure. Catalysts come in many forms, from atoms and small molecules to macromolecules such as enzymes and crystalline structures such as zeolites and metal oxides. Moreover, they can be employed in different conditions: liquid, gaseous or at the surface of solids [1].
In heterogeneous catalysis, solid materials catalyze reactions of molecules in the gas or liquid phase. They provide several advantages over homogeneous catalysts, such as the ease of separation from the reaction mixture and consequent better recyclability and reuse. In addition, solid frameworks and macromolecules bring about additional properties, compared to homogeneous catalysts, which have been exploited by scientists for several decades. Examples involve the shape selectivity of microporous zeolites that, due to the shape of the cavity in which the active site is located, allows for the catalytic activation of a specific reactant or promote the formation of a specific product by stabilizing the transition state that precedes the desired product. Nature pioneered this strategy with much greater success, specifically with enzymes, in which the way they are folded onto themselves allows for exceptional specificity toward certain reactants and selectivity toward certain products (Figure 1) [2,3,4,5]. Another instance is the synthesis of supported noble metal nanoparticles. Due to their small size, nanoparticles expose a relatively large surface area that, when they are used in homogeneous phase, cause them to aggregate and sinter, in turn lowering the surface area exposed and lowering their free energy. By depositing the nanoparticles onto solid supports (often porous), chemists have been able to limit sintering, allowing the use of these catalysts over multiple cycles and easing their separation from the reaction mixture [6,7,8]. Even further, in the last decades scientists have been able to coordinate single atoms of various metals within a solid support, with major advantages in terms of catalytic activity and atom economy [9,10].
Among the different types of heterogeneous catalysis, hybrid organic-inorganic catalysts provide some unique properties, compared to purely inorganic catalysts (e.g., zeotypes) and purely organic ones (e.g., resins). In fact, they combine the advantages of inorganic solids (high mechanical, thermal, and structural stability) and organic molecules or macromolecules (flexibility and functionality).
In general, the concept of hybrid material refers to a solid composed of two types of building blocks, an inorganic one and an organic one, mixed with each other at the nanometric scale. By cleverly combining different organic and inorganic precursors, many authors have been able to guide their self-assembly, achieving hybrids featuring unique properties for applications as advanced materials in several fields. In the last few decades, these types of materials underwent tremendous development, thanks to soft sol–gel and self-assembling micellar processes (Figure 2) [11]. Hybrid materials can be classified according to the interactions between organic and inorganic building blocks. Quite logically, the strength and the directionality of chemical bonds holding them together are crucial aspects to consider in the pursuit of designing a hybrid material with specific properties.
Sanchez and Ribot classified as Class I hybrids those in which weak bonds (hydrogen or van der Waals bonding) give cohesion to the whole structure (Figure 3). Organic dies have been imbedded in amorphous sol-gel matrixes, in order to improve their stability and avoid the formation of dimers or oligomers [12,13]. Antibacterial agents also have been deposited onto silica supports through H-bond interactions [14]. Inorganic fillers have long been imbedded into organic polymeric matrixes in order to improve their mechanical properties. However, traditional filler blending occurs under high viscosity conditions, which lead to partial agglomeration of the inorganic particles, lowering the matrix-filler interactions. By generating the inorganic particles in-situ by sol-gel chemistry, a much better dispersion can be achieved [15,16]. Wet or dry impregnation have been implemented to obtain Class I hybrids, as well [17].
In Class II materials, the two phases are linked together through strong chemical bonds (covalent, coordination or ionic bonding) [11,17,18]. This types of hybrids are of special interest for catalytic applications.
In fact, while weak bonds between organic and inorganic building blocks would be susceptible to breakage in the reaction media, with consequent loss of structural properties and catalytic activity, strong bonds would favor the preservation of the hybrid catalysts, allowing for using the catalyst over multiple cycles. There are two main families of hybrid materials belonging to class II that are commonly implemented as heterogeneous catalysts: metal-organic frameworks and mesoporous silica-based hybrids. Other types of hybrid materials have also been described, e.g., organozeolites [19], silica-graphene composites [20] and laminar metal oxides featuring organocatalysts in the interlaminar space [21].
2. Mesoporous Silica-Based Hybrid Materials
Since the development of M41S materials by the Mobil Oil Company in 1992, porous materials with large specific surface areas has been a field of extensive research, particularly with regard to potential applications in areas such as catalysis, adsorption, gas storage, chromatography, controlled drug-delivery and sensor technology. Like microporous crystalline zeolites, this class of materials feature very large specific surface areas, ordered pore systems and well-defined pore size distributions. However, unlike zeolites, M41S materials exhibit amorphous pore walls and their pore diameters is comprised between 2 and 10 nm. The most representative materials belonging to this class include silica solids MCM-41 (with a hexagonal arrangement of the mesopores), MCM-48 (with a cubic arrangement of the mesopores), and MCM-50 (with a laminar structure) (Figure 4) [22].
Not long after their discovery, researchers began attempting to incorporate organic components within an inorganic silica framework to achieve symbiosis of the properties of both components. The combination of the properties of organic and inorganic building blocks within a single material is highly attractive. In fact, this allows for the possibility of combining the enormous variety of functional groups, typical of organic chemistry, with the advantages of a thermally stable and robust inorganic substrate. This is particularly appealing for applications in heterogeneous catalysis. The symbiosis of organic and inorganic components can lead to materials with considerably different properties from those of their individual, isolated components.
Several synthetic methodologies allow to functionalize silica matrixes with specific organic functionalities such as C=C multiple bonds, alcohols, thiols, sulfonic and carboxylic acids, amines, as sol-gel chemistry provides versatile and effective ways of anchoring organic moieties to a porous solid silica support. As it will be shown below, these organic moieties (featuring acidic, basic, redox sites) can be catalytically active for a specific reaction and being selective toward the formation of one product, instead of another. Bounding catalytically active organic moieties to solid supports, opens up to the possibility of carrying out reactions under heterogeneous regime, allowing for recovery and reuse of these valuable catalyst over multiple cycles, easing product separation and purification [1,22].
Silica-based hybrids can be achieved by (i) post-synthetic modification of the pore surface of a purely inorganic silica material (grafting), (ii) co-condensation of a silica precursor and a silyl derivative bearing the functional group of interest (“one-pot” approach), (iii) condensation of bisilylated precursors featuring the functional group of interest in the linker between the two alkoxysilyl groups (alone or together with a minimal amount of tetra-alkoxysilyl precursor), leading to a material in which the organic functionalities are imbedded within the pore walls.
2.1. Post-Synthetic Modification (Grafting)
Grafting refers to the post-synthesis modification of the surfaces of mesostructured silica phases with organic groups. This process is mainly carried out by reaction of organosilanes of the type RSi(OR’)3 with the free silanol groups on the pore surfaces. Less frequently chlorosilanes ClSiR3 or silazanes HN(SiR3)2 are used (Figure 5).
In principle, by varying of the organic residue R, functionalization with different organic groups can be achieved. By implementing this approach, the mesostructure of the starting silica phase is usually retained. On the other hand, a reduction in the porosity of the hybrid, compared to the parent mesoporous silica, is usually observed, proportionally to the size of the grafted organic moieties. Furthermore, the organic silyl-derivatives might preferentially react with the surface silanol groups that they will encounter first, i.e., those located at the pore mouth or at the initial part of the mesoporous channels. This effect is modulated by several factor. Firstly, the reactivity of the alkoxy groups bounded to the silicon atom determines the ease with which hydrolysis/condensation reactions can occur.
The diffusion of the silyl-derivative compounds through the mesopores also play a crucial role and is primarily determined by its size, relative to the pores diameter, and by the chemical affinity between the organic moieties and surface silanol groups. Lastly, while in an initial stage of the grafting, silyl-derivatives are allowed to diffuse through the pore channels before the catalyst (acid or base) is added into the mixture, molecules baring acidic or base functionalities can catalyze their own hydrolysis and condensation before reaching optimal diffusion, leading to a worse distribution of organic moieties throughout the porous framework. A key advantage, unique to this method, is the fact that the templating agent used for generating the mesopores is entirely removed by calcination, prior to the grafting procedure. The grafting approach allows for introducing organic functionalities onto a pre-existing mesoporous silica framework and for that, in these hybrids, the organic moieties are expected to be pending from the pore walls, partially occupying the mesoporous channels. Grafting procedures are also employed to modify the polarity of the support’s surface. In fact, silanol-capping hydrophobic moieties, e.g., trimethyl-silyl species, can make the pore walls more hydrophobic, with beneficial effect on the properties of the hybrid materials, for certain applications.
2.2. Co-Condensation Method (“One-Pot”)
An alternative method to synthesize ordered hybrid organic-inorganic mesoporous silica phases is the co-condensation method. It is possible to prepare mesostructured silica phases by the co-condensation of tetra-alkoxysilanes, typically tetraethyl ortosilicate (TEOS) or tetramethyl ortosilicate (TMOS), together with trialkoxyorganosilanes of the type RSi(OR’)3 in the presence of structure-directing agents, obtaining materials with organic residues pending covalently from the pore walls (Figure 6).
By using structure-directing agents that are typically implemented in the synthesis of pure mesoporous silica phases (e.g., MCM or SBA silica phases), organo-functionalized silica hybrids can be prepared in such a way that the organic linkers, connecting the functional group of interest to the inorganic support, are pending directly from the pore walls, with the silicon atoms deriving from hydrolysis/condensation of the alkoxy-silyl groups being imbedded into the silica pore walls. Since the organic functionalities are direct components of the silica matrix, pore blocking is not a problem in the co-condensation method, although a slight decrease in the pore diameter can be detected. Moreover, the organic units are generally more homogeneously distributed than in hybrid mesoporous materials obtained by grafting process.
Nevertheless, the co-condensation method presents also a number of disadvantages: the degree of mesoscopic order of the resulting materials tend to decreases with increasing concentration of RSi(OR’)3 in the synthesis mixture. Consequently, the relative amount of organic moieties in the modified silica phases does not normally exceed 40 mol%. Furthermore, the yield with which the organic groups can be incorporated into the pore-wall network is generally not 100% and it decreases proportionally to the amount of organic silyl-derivative in the synthesis mixture. These observations can be explained by the fact that an increasing proportion of RSi(OR’)3 in the reaction mixture promotes homocondensation reactions, favoring the formation of oligomers. These phenomena are caused by the different hydrolysis and condensation rates of the different silyl-type precursors. It is a potential problem whenever this synthetic strategy is implemented, as the homogeneous distribution of the organic functionalities in the framework cannot be guaranteed. Moreover, exceeding in the loading of organic groups can lead to a reduction in the pore diameter, pore volume, and specific surface areas. From a purely methodological point of view, a higher degree of care needs to be taken in the removal of the surfactant, used to generate the mesopores. Calcination cannot be implemented as it would compromise the integrity of the organic groups Si-R. Consequently, template removal is commonly done by continuous extraction, carried out using a soxhlet apparatus. Although fairly effective, an incomplete removal of the template is usually observed.
An additional tool to incorporate specific organic functionalities within silica-based materials are tethering procedures. For that, firstly, mono-silanes RSi(OR’)3 are incorporated into a silica support (either by co-condensation or grafting). Afterwards, in a post-synthetic step, an organocatalyst featuring the active site of interest and a specific functional group R* is reacted with the hybrid material. R and R* need to be chosen properly, so that they can react and form a covalent bond that can guarantee the anchoring of the organocatalyst. Example of such R/R* pairs include primary amine and isocyanate, reacting to form urea group; thiol and terminal alkene, reacting in presence of a radical initiator to form a thioether; a halide (Br or I) functioning as good leaving group in a SN2 reaction and a strong nucleophile.
2.3. Periodic Mesoporous Organosilicas (PMOs)
PMOs are a class of silica-based organic–inorganic hybrid materials obtained by hydrolysis and condensation reactions of bridged silsesquioxane precursors of general formula (R’O)3Si-R-Si(OR’)3, used as the only silicon source [23,24]. In contrast with the two methods previously reported (grafting and direct synthesis), in this case, the organic building blocks bear two alkoxy-silyl groups that can largely improve their incorporation in the three-dimensional network as well as their homogeneous distribution throughout the solid (Figure 7A). Moreover, a major difference between PMOs and silica-based hybrids obtained by co-condensation and grafting is that in the former, active sites are imbedded into the pore walls, rather than pending from the surface of the support. As in the case of the synthesis of pure silica mesophases, the use of a surfactant in the presence of bi-silylated organosilica precursors allows obtaining materials featuring ordered mesopores, narrow pore size distribution and in which the organic bridges are integral components of the silica network [22].
Although presenting several advantages over grafting and co-condensation, the synthesis of PMOs also bring about challenging aspects, the most notable being the synthesis of the suitable bi-silylated precursors. In fact, alkoxy-silyl groups are highly reactive species and synthesizing complex molecules baring two of such groups and a specific organic bridged group R, is often a challenging task.
Nevertheless, bridged polysilsesquioxanes featuring a wide range of bridging groups have been prepared. In 1999, three research groups independently reported on mesoporous organosilica hybrids from bisilylated precursors baring methyl or ethyl groups as organic bridges [23,24,26]. After that, several PMOs were prepared featuring ethylene, ethenylene, phenylene and thiophene as R groups. Rigid unsaturated precursors able to form weak interaction, such as π−π can self-assemble in the synthesis mixture. When hydrolysis/condensation take place, this order can be preserved and layered “crystal-like” structural sub-domains can be included in the final PMO-type materials. The first materials featuring 1,4-phenylene bridges was prepared in 2002. Upon varying the size of the R bridged unit, materials featuring layers of different width were obtained (5.6–11.9 Å) [27]. Building upon these early studies, a variety of organic groups, ranging from hydrocarbons and heteroaromatics to metal complexes, have been successfully incorporated into PMO hybrids (Figure 7B) [22,25,28].
3. Sol-Gel Chemistry and Hybrid Materials
In this section, a description of the sol-gel chemistry of silica is reported, with a specific focus on aspects that need to be considered when designing and preparing silica-based hybrid materials. In a general definition, a sol is defined as a colloidal dispersion of particles in a liquid, while a gel consists of a continuous solid skeleton enclosing a continuous liquid phase. The formal definition of sol-gel processing is therefore the growth of colloidal particles and their linking together to form a gel.
3.1. Hydrolysis Reactions
Hydrolyzed silica in aqueous solution is constituted by orthosilicic acid, Si(OH)4. Protonation of silanols to form cationic species ≡Si(OH2)+ can occur below pH 2, while further hydrolysis involves the deprotonation of a silanol group to form an anionic species Si(OH)3O− above pH 7, and Si(OH)2O22− above pH 12 (Figure 8). Silicate gels are most often synthesized by acid/base-catalyzed hydrolysis of alkoxides dissolved in water and/or their parent alcohols [29,30]. Reaction 1 in Figure 8 describes the hydrolysis of an alkoxide precursor molecule.
The hydrolysis reaction of alkoxy-silyl precursors can be acid or base catalyzed. Acid catalysts protonate the alkoxide group, making a better leaving group (ROH) that does not require a proton transfer in the transition state. Base catalysts contributes to the reaction rate by increasing the concentration of strongly nucleophilic OH− species, which attack the electrophilic silicon atom, with release of a −OR species.
3.2. Condensation Reactions
Concerning the condensation of silicic units, Iler [31] divides the polymerization process into three approximate pH domains: pH < 2, 2–7, and >7. At pH 2, a boundary is present, due the point of zero charge, i.e., surface charge is zero, and the isoelectric point, i.e., electrical mobility of the silica particles, is also zero. A pH of 7, represents another boundary, because both the solubility and dissolution rates are maximized at or above pH 7, and because above pH 7 the silica particles are appreciably ionized. Consequently, particle growth occurs without aggregation or gelation.
Below pH 2, condensation involves the protonation of a silanol (or an alkoxide), followed by nucleophilic attack on the silicon atom by a ≡SiOH species (reactions (2) and (3) in Figure 8). In this case, the reaction rate is favored by the presence of a better leaving group (H2O or ROH). Moreover, like for acid-catalyzed hydrolysis, the reaction does not require a proton transfer in the transition state.
Above pH 2, condensation involves a bi-molecular nucleophilic condensation mechanism (SN2-Si), featuring the attack of hydrolyzed, anionic species (≡SiO−) on neutral species. Due to inductive effects, the most acidic silanols, which are the most likely to be deprotonated, are the most highly condensed species. Consequently, condensation occurs preferentially between more condensed species and less condensed, neutral, species. In other words, monomers and oligomers feed the growth of bigger particles. More specifically, once tetramers are formed, they tend to cyclize due to the proximity of chain ends and further growth occurs by addition of monomer and other low-molecular-weight species to cyclic species to form particles, and by aggregation of particles to form bigger networks.
3.3. Particle Growth and Si-O-Si Hydrolysis
The hydrolysis rate of siloxane bond (Si-O-Si) increases above pH 4 and at very low pH. In general, siloxane networks are formed under those conditions in which depolymerization is less likely to occur, i.e., when the condensation is irreversible and siloxane bonds cannot be hydrolyzed once they are formed.
Particle growth above pH 7 differs from that below pH 7 by at least two factors. Firstly, while near the isoelectric point no electrostatic repulsion is present, so that growth and aggregation processes occur altogether, at pH above 7 particles are appreciably charged, which makes particle aggregation unlikely. Secondly, above pH 7, the size dependence of the solubility of silica increases. Consequentially, particle growth is mediated by Ostwald ripening, a process in which smaller, more soluble particles dissolve and feed the growth on larger, less soluble particles. The growth ceases when the difference in solubility between the largest and smallest particles becomes negligible [32].
In Figure 9, a schematic representation of hydrolysis and condensation reaction of silica, under acid or base catalysis, is reported.
Overall, it has been observed that acid catalyzed sol-gel processes lead to the formation of bigger particles with less defects, while base catalyzed syntheses lead to the formation of smaller particles featuring more defects. The solubility of silica can be influenced also by changing the reaction medium. For example, by replacing water with a 90% methanol 10% water mixture, the solubility of silica is reduced by a factor of 28 and hydrolysis of siloxane bond is less likely.
3.4. Fluoride-Catalyzed Hydrolysis and Condensation
Besides acid and base catalysis, there is a less common way to catalyze sol-gel reactions. F− ions have been reported to efficiently catalyze hydrolysis/condensation of alkoxy silanes decades ago [33]. Hydrolysis/condensation reactions follow the pathways reported in Figure 10. In the hydrolysis reaction, a nucleophilic fluorine ion approaches a molecule of alkoxy silane in solution. A SN2 reaction occurs, forming a partially fluorinated silicon alkoxide that in presence of H3O+, yields water and alcohol as by-products. Due to strong inductive effect, fluorine withdraws electron density from the silicon atom, making it more susceptible to nucleophilic attack from H2O. Consequentially, a pentacovalent complex is formed in the presence of water. The complex reacts to form a partially hydrated silicon alkoxide and hydrofluoric acid. The condensation process occurs by formation of a hexacovalent intermediate.
Iler postulated that the effectiveness of fluorine in the polymerization reaction is due to the smaller ionic radius of the fluorine anion, compared to that of the hydroxyl. Other anions, such as Cl−, Br−, I−, NO3−, SO4− are all bulkier than hydroxyl and for that, are much less effective than hydroxyl in catalyzing these reactions by nucleophilic attack [34].
Under optimal conditions, fluoride catalyzed sol-gel synthesis can lead to mesoporous materials featuring high surface area and relatively narrow pore size distribution. Reale et al. studied the influence of Si/F− ratio on the textural properties of pure silica materials. For that, they used NH4F as fluoride source, as NH4+/NH3 equilibrium in water ensured the presence of protons in the reaction medium, needed for the mechanism described in Figure 10. Varying the Si/F− ratio between 15 and 320, they were able to obtain materials featuring specific surface area values (BET) ranging from 500 to 1000 m2/g and mean pore size (BJH) ranging between 75 and 30 Å. The authors explained these variations by considering that the larger the mesoporous diameter, the larger the average silica particle radius. This could denote that the nucleation of silica particles is highly disfavored at high fluoride concentration, which would result in a low concentration of viable nuclei that can grow at the expenses of the silica precursor, in solution. Accordingly, high fluoride concentration would promote the formation silica particles featuring large diameter and large inter-particle void. The authors also studied the effect of the alcohol used as co-solvent by preparing materials using a 1:1 mixture of water and one among methanol, ethanol and 2-propanol. Upon increase of the alcohol size, surface area decreased and mean pore size increased. Nucleophilic attack of water to the fluorinated silicon intermediate, which is the rate determining step in the TMOS hydrolysis, is less favored by more polar solvents. Consequentially, a lower amount of nuclei is formed as consequence of the slower hydrolysis kinetics. Coherently, the silica material prepared in 2-butanol resembles that prepared using the highest fluoride concentration [35].
It is important to highlight that these syntheses are carried out at neutral pH, 36 °C, using low amount of solvent and in the absence of templating agents. By tuning the composition of the synthesis mixture, it is possible to achieve highly porous materials, with textural properties that are tuned for a specific application. Moreover, F− sol-gel syntheses allow to incorporate Al and Ti within the network as well as organic building blocks, by substituting a fraction of TMOS with an equimolar amount of either an Al (or Ti) precursor or a silyl-derivative of the type RSi(OR`)3, baring a functional group R of interest.
3.5. Surfactant-Mediated Sol-Gel Synthesis
As mentioned before, surfactants are widely implemented in sol-gel synthesis to achieve mesoporous silica materials, for applications in several fields. Supramolecular aggregates of ionic surfactants (long-chain alkyltrimethylammonium halides) and non-ionic triblock copolymers have been used as structure-directing agents (SDAs) in many synthetic procedures. In solution, under certain conditions, they form of a lyotropic liquid-crystalline phase, which can guide the assembly of ordered mesostructured materials through hydrolysis and condensation of the silica precursors under basic conditions. The mesoporous materials are obtained by removal of the surfactant by extraction or calcination.
Typically, a lyotropic liquid-crystalline phase is formed above a certain concentration of surfactant. However, a cooperative self-assembly between the SDA and the silica precursors can lead to a liquid-crystal phase with hexagonal, cubic, or laminar arrangement, even at lower concentration of surfactant. An essential condition to ensure this cooperative self-assembly and avoid phase separation is the presence of attractive interactions between the template and the silica precursor. Such interactions vary, depending on the type of surfactant and on the pH of the solution. If the reaction takes place under basic conditions, silica species will be present in the form of anions. If a cationic quaternary ammonium surfactant is used as the SDA, electrostatic interactions such as those depicted in Figure 11A will be produced. However, if the preparation takes place below pH 2, silica species will be positively charged and to produce an interaction with the cationic surfactant, it is necessary to add a counter ion X− (usually a halide, Figure 11B). Although less commonly, negatively charged surfactants (e.g., long-chain alkyl phosphates) can also be used as the SDA. Compared to positively charged surfactant, a reverse situation is expected, with protonated silicon species interacting with the negatively charged functional groups of the surfactant molecules, at pH below 2 (Figure 11C), and a mediating positive ion M+ needed under basic conditions (Figure 11D). Lastly, when non-ionic surfactants are used (e.g., long chain amines), attractive interaction between SDA molecules and silica precursors are provided by hydrogen bonds. Such interactions can be promoted by adding to the mixture either polar molecules (e.g., polyethylene oxide, depicted as triangles in Figure 11E) or ion pairs (Figure 11F) [22].
3.6. Silyl-Derivatives Precursors
The most implemented precursors for sol-gel synthesis of silica are tetraethylortholisicate (TEOS) and tetremethylorthosilicate (TMOS). Bernards et al. studied the hydrolizability of siloxane featuring alkoxy residues of different length. They found that the hydrolyzability of the silanes in an acidic environment was inversely proportional to the size of the alkoxy terminal groups (TMOS > TEOS > TPOS, Figure 12). They explained that the effect of the alcohols on the hydrolysis rate of each of the silanes was due to differences in degree of dissociation of HCl in each one of the alcohols (methanol > ethanol, 1-propanol > 2-propanol) [36].
Alkoxy groups are highly reactive species. They can easily undergo partial hydrolysis and condensation leading to oligomers. This is particularly problematic considering that, in many cases, they are used as precursors of materials in which the functional groups R need to be homogeneously dispersed throughout the material (heterogeneous catalysts, adsorbents, sensors etc.). Silyl derivatives are very moisture-sensitive. For that, the design of synthetic routes usually aims to incorporate the alkoxysilyl group in the last step of the preparation of the building block. Whenever this is not possible, silyl derivatives need to be protected from humidity, by storing them under inert atmosphere and by always carrying out reactions that involve them under anhydrous conditions, especially if acid or bases are also present in the reaction mixture. Additional care needs to be taken in the work-up procedure, as distillation at high temperature can cause spontaneous condensation of alkoxy groups. The size of the alkoxy group also affects the kinetic of hydrolysis with TMOS containing synthesis mixture, reaching the sol-gel transition after shorter time, compared to TEOS-based mixtures. This is in line with what can be deduced by observing the mechanism depicted in Figure 9 and the ball and stick structures depicted in Figure 12. Ethoxy substituents are bulkier than methoxy ones and therefore they partially hinder the approach of the nucleophile to the silicon [29]. Consequentially, methoxysilyl groups are expected to be less stable and more reactive than ethoxysilyl groups.
4. Silica-Based Hybrids in Heterogeneous Catalysis
With the aim of highlighting the broad scope of application of silica-based hybrid catalysts, a general overview of different type of active sites bounded onto silica supports is described in this section (summarized in Table 1), together with few representative examples of their synthesis and applications.
4.1. Acid Catalysts
Many reactions in the chemical industry are carried out under acid catalysis conditions. Purely inorganic microporous zeolites are perhaps the most notable example of heterogeneous catalysts, being them implemented in several chemical processes [37]. Nevertheless, the small pore size limits their application to certain reactions, while others require catalysts in which the active sites are accessible to bulkier molecules. To overcome the pore size limitations, inorganic acids such as polyphosphoric acid [38], perchloric acid [39], fluoroboric acid [40] and sulfuric acid [41] have been supported onto silica. Although effective in overcoming some of the limitations of microporous acid catalysts, these purely inorganic materials present little margin for modifications and fine tuning of their properties for a specific catalyzed reaction. Another solution to this problem is functionalizing mesoporous silica supports with organic molecules baring Brönsted acid groups [42]. Considering the different possibilities, the two main classes of materials developed through this approach are silica-supported heteropolyacids and silica-supported sulfonic acids.
Heteropolyacids (HPAs) are polyoxometalates constituted by MOx polyhedra (usually distorted octahedra) which surround one or more heteroatoms (Figure 13) [43]. Several authors reported on silica-supported HPAs for applications in heterogeneous catalysis [44,45,46,47,48]. Sato et al. supported three types of HPA over differently porous silica by wet impregnation: phosphotungstic acid (H3PW12O40·nH2O), silicotungstic acid (H4SiW12O40·24H2O), and phosphomolybdic acid (H3PMo12O40·nH2O). The catalysts were used in the vapor-phase dehydration of glycerol to acrolein. It was found that silico-tungstic acid, supported on silica materials featuring mesopores of 10 nm, showed the best catalytic activity, with 98 mol% conversion and an acrolein selectivity higher than 85 mol%. Moreover, they observed an influence of the meropores size on the catalytic activity, with a very steep deactivation of the catalysts when the HPA were supported onto a silica material having mesopores of 3 nm [49]. In another study, Blanco et al. used silica supported H3PMo12O40·nH2O and H3PW12O40·nH2O for dehydration reactions of different alcohols. Specifically, the dehydration of 1,2-diphenylethanol, 1-3,4-dimethoxyphenyl-2-phenylethanol and cholesterol was investigated. The authors found both catalysts to be highly active and selective with no appreciable loss of the catalytic activity over multiple cycles [50]. Laurenti et al. prepared silica supported H4SiW12O40·nH2O and H3PW12O40·nH2O by wet impregnation and tested them for the alkylation of 3-methylthiophene with 2-methyl-2-butene, used as model reaction for gasoline desulfurization. Their performances were shown to be superior to those of zeolites USY, Beta and MCM-22. Supported H3PW12O40·nH2O was also tested in a real feed of FCC gasoline with promising results [51]. Although highly active for some specific activation, polyoxometales such as HPAs provide little room for fine tuning of the chemical structure and consequential optimization of the catalytic activity of the heterogeneous catalysts.
Perhaps, the most versatile way to incorporate strong Brönsted acid sites into mesoporous silica matrixes is the synthesis of supported sulfonic acid groups. The acidity of the sulfonic acid proton is strongly influenced by its chemical make-up, which allows for a fine tuning of the acidity of the sulfonic proton by exploiting the versatility of organic synthesis [52].
Since 1998, sulfonic acid groups have been supported onto mesoporous silicas [53,54]. In the first studies on these hybrid acid catalysts, two approaches were typically implemented: (i) post synthetic grafting of 3-mercaptopropyltrimethoxysilane (MPTMS) followed by oxidation of the thiol groups to sulfonic acid moieties and (ii) co-condensation of MPTMS with TEOS or TMOS, also followed by oxidation. Since their introduction, many advances have been made, such as increasing the MPTMS loading through a coating method [52], replacing calcination with extraction methods [55], and co-functionalization of MPTMS and octyl substituents to enhance the catalytic activity by increasing the hydrophobicity of the hybrid catalyst [56]. Supported sulfonic acid catalysts are primarily employed in biomass conversion [52]. However, their versatility allowed their successful implementation in other catalytic processes such as synthesis of 2,2-bis(5-methylfuryl)propane by condensation of acetone with 2-methylfuran [53], esterification of D-sorbitol with lauric acid [57], synthesis of monolaurin by direct esterification of glycerol with lauric acid [58], Fries and Beckmann rearrangements [59,60], syntheses of polyhydroquinoline derivatives [61], synthesis of β-aminocarbonyls through Mannich reactions [62], and others [52].
Several substrates featuring sulfonic acid groups have been implemented as precursors for the sulfonation of mesoporous materials. From the point of view of the inclusion of sulfonic acid groups into the final hybrid materials, different approaches can be followed: (i) implementation of commercial sulfur-containing organosiloxanes and eventual conversion of the sulfur-based functional groups to sulfonic acid moieties, usually carried out after incorporation into the solid material (e.g., MPTMS); (ii) synthesis of new silyl-derivatives containing sulfonic acid groups; (iii) functionalization of supported organic moieties with sulfuric acid [52,63,64]. In Figure 14, some organic moieties that have been successfully used to synthesize silica-supported sulfonic acid catalysts are reported, including alkyl-sulfonic acids (precursors 1, 2), aryl-sulfonic acids (precursors 3, 4, 8, 10, 11), S-containing heterocycles (precursors 5–7 and 12) and bridged species (precursor 13). Unsurprisingly, using different siloxane precursors influences the properties of the final catalyst, across the board. A few examples are reported below.
An increase in hydrophobicity, and consequently hindered catalyst deactivation by water, and an increase in the mass transfer of hydrophobic compounds have been achieved by using phenylsulfonic acid derivatives instead of propylsulfonic acid moieties [52]. Phenylsulfonic acid siloxane-based precursors have been used by several authors to synthesize supported sulfonic acid materials that have shown superior catalytic properties when compared both with others non-commerial/non-aromatic catalysts and with commercial resins such as Amberlyst-15 (Figure 14, precursors 8, 10, 11) [65,66,67,68,69]. Lindlar et al. reported the synthesis of MCM-41-Ph-SO3H (Figure 14, precursor 11) [52] with pore diameters of up to 6 nm by implementing swelling agents. Firstly, the phenyl groups were grafted onto the silica surface; subsequently, chlorosulfonic acid was used for sulfonation. However, chlorosulfonic acid could also react with free silanols. To avoid this phenomenon, capping agents, such as trimethoxymethylsilane, were grafted after functionalization with the phenyl precursor. For the sulfonation of mesoporous silica materials, grafting is the most widely used method, due to the advantages it has over co-condensation method, the main one being SDA removal by calcination rather than by extraction, which is usually not completely effective and would imply the use of a solvent. Nevertheless, Gedeona et al. found that after extraction, further calcination with slow heating ramp up to 200 °C could completely remove P123 from the extracted sample [70].
The functionalization of fluorinated sulfonic acid precursors inside the mesopores was first reported by Harmer et al. in 1997 [71]. In these hybrid mesoporous materials, the acidity of the sulfonic acid groups is strongly enhanced by the presence of electronegative fluorine atoms in its proximity. However, the preparation and stability of these hybrids are major drawbacks as they would often undergo leaching and consequent deactivation [65]. 1,2,2-Trifluoro-2-hydroxy-1-trifluoromethylethane sulfonic acid sultones (Figure 14, precursor 6) have been directly anchored onto silica surfaces through a direct synthetic strategy [72]. The same authors reported another strategy for the preparation of fluorinated supported sulfonic acids through a platinum-catalyzed hydrosilylation procedure. The resulting silyl-derivative could then be anchored onto the silica surface through a co-condensation reaction or grafting [73]. In our group, we recently prepared two silica-supported aryl-sulfonic acid catalysts, one of them featuring a fluorinated aromatic ring (starting from precursors 3 and 4, Figure 14). After an in-depth characterization, the hybrids were tested in the acetal formation reaction between benzaldehyde and ethylene glycol. Although, both catalysts showed excellent performances, the fluorinated hybrid was able to retain its catalytic activity over three catalytic cycles, while the non-fluorinated one showed a minimal but progressive loss of catalytic activity. A hydrophobic version of the catalysts, in which surface-silanols were capped with hexamethyl disilazane, was prepared and tested in the same catalytic process. The hydrophobic catalysts showed a drop in their catalytic activity, compared to the hydrophilic hybrids, indicating a beneficial role of the polar surface of the silica support on the overall catalytic activity [74].
Recently, supported N-propylsulfamic acid groups have drawn significant interest. Sulfamic acid-based catalysts can be described as strong acids that are zwitterionic in the absence of water (Figure 14; precursor 9) [75,76]. These catalysts are easily separable from reaction mixtures and can be recycled different times if supported on the surface of mesopores. Under alkaline or neutral pH, sulfamic acid derivatives can be boiled without appreciable hydrolysis. However, they slowly hydrolyze under acid conditions, giving a supported primary amine and releasing a molecule of sulfuric acid [77].
Supporting complex sulfonic-functionalized moieties is not a trivial task, especially when green chemistry standards need to be met. Phenyl-sulfonic acid catalysts often require using either corrosive agents for sulfonation of aromatic precursors or expensive pre-sulfonated reactants, to begin with. Even less trivial is the synthesis of fluorinated compounds, with the few silyl-derivatives reported being synthesized through low-yielding steps, starting from expensive reactants. There is room for improvement also in the synthetic strategies implemented to obtain the silica-supported hybrid catalysts. The use of expensive surfactants and of acid/base reactants in the sol-gel processes, as well as the high calcination temperatures, are also aspects that need to be kept into account when designing a hybrid catalyst with potential scaling-up perspectives.
Sulfonated Periodic Mesoporous Organosilicas (PMOs)
As mentioned before, many studies have been carried out with the scope of tuning the hydrophilic/hydrophobic nature of supported sulfonic acid materials, due to the influence that this parameter has on their catalytic activity. This is particularly important, because one of the main catalytic applications of supported sulfonic acid moieties involves the use of hydrophobic fatty acids as reactants [78,79]. Silanol-capping with hydrophobic groups and implementation of hydrophobic sulfonated silyl-derivatives are viable alternatives, which nevertheless present disadvantages such as decreasing the pore size and consequent mass transfer limitations in the case of bulky compounds [80]. Periodic mesoporous organosilicas (PMOs) are often regarded as the next generation of these catalysts, with sufficient hydrophobicity and crystal-like pore walls. The first sulfonated benzene-bridged PMO (Ph-PMO-SO3H) was synthesized through the condensation of 1,4-bis(triethoxysilyl)benzene (BTEB) in presence of specific SDAs. The solid obtained was then sulfonated with a 25% solution of SO3/H2SO4. The same authors also carried out a co-condensation of BTEB and MPTMS under basic conditions, in the presence of a SDA, achieving a Ph-PMO with pendant thiol groups, subsequently converted to sulfonic acid by treatment with HNO3 at room temperature for 24 h [81].
The use of acidic SBA-15-like PMOs, in which the location of the acid sites could be controlled has been proposed and tested in acid-catalyzed reactions in hot water [82]. The hybrids were prepared either by the condensation of BTEB, followed by sulfonation using chlorosulfonic acid (catalyst 1) or by MPTMS condensation and further oxidation with H2O2 (catalyst 2).
The acidity of the materials was studied by calorimetric measurements of the adsorption of ammonia. The catalytic activity of the materials was tested in the gas-phase dehydration of isopropanol, used as a model reaction, and compared with the reference acid sulfonated resin, i.e., Amberlyst-15. While the oxidation step of MPTMS thiol groups to sulfonic acid groups in H2O2 did not affect the mesoscopic structure, the treatment with chlorosulfonic acid, to incorporate sulfonic acid moieties in the structural aromatic units, did affect it. In spite of the partially compromised structure, catalyst 1 (obtained by treatment with chlorosulfonic acid) showed catalytic activity in the gas-phase dehydration of isopropanol equivalent to that of Amberlyst-15, although with progressive partial loss of the activity due to the leaching of sulfur species. On the other hand, SBA-15-like PMOs obtained by H2O2 oxidation of the thiol groups (catalyst 2) showed lower catalytic activity in the gas-phase dehydration of isopropanol. However, catalyst 2 did not show any significant leaching of sulfur species was observed.
The catalytic performances of these materials were also evaluated in biomass valorization, such as cellobiose hydrolysis in hot water. At 150 °C the catalysts showed significant leaching of sulfur species with the catalytic activity observed mainly due to species performing in homogeneous phase. Extensive washing of the catalysts prior to the treatment somehow improved their stability and recyclability. The washing step probably acted by removing the labile species that leached in the catalytic tests. Consequently and unsurprisingly, this led to lower catalytic activity in the first cycle [82].
In another example, Jerome et al. also studied different types of silica supported sulfonic acid materials and monitored the catalytic activities for the aqueous synthesis of bis(indolyl)methanes [83]. They extensively studied the effect of hydrophobicity on the catalytic activities and found that an increase in hydrophobicity led to better catalytic performances for this reaction. By comparing Ph-PMO-Pr-SO3H and SBA-15-PrSO3 catalysts, the former, in which phenyl groups are present within the pore walls, showed higher catalytic activity [84].
The versatility of silica-based synthesis, allows for the incorporation of multiple functionalities, either to improve the catalytic efficiency of a specific active site or to carry out multi-step cascade reactions. As an example of the former, in some catalytic processes, the activity of sulfonic acid catalysts benefits from the presence of amine-type functionalities.
Thiel et al. introduced amine groups via post-synthesis functionalization of a BTEB-based PMO-SO3H obtaining a bi-functional PMO (Figure 15) [85]. Firstly, BTEB was reacted with tert-butyloxycarbonyl(Boc)-protected APTS in the presence of SDA molecules. The resulting PMO structure was then sulfonated with chlorosulfonic acid. Finally, the protecting group was removed from the catalyst. The catalyst was tested in nitroaldol condensation reactions, with excellent yields. The authors also tested the catalytic activity of N-protected PMO-SO3H/-NHBoc in nitroaldol condensation reaction, recording no conversion. Interestingly, the protected system exhibited remarkable activity in the conversion of dimethoxyphenyl-methane to benzaldehyde. This catalyst was also compared with monofunctionalized mesoporous materials (SBA-NH2 and PMO-SO3H) with both catalysts performing worse than the bi-functional catalyst PMO-SO3H/-NH2.
In another relevant study Alauzun et al. reported the synthesis a bi-functional mesoporous material containing two antagonist functions, i.e., an acidic group in the framework and a basic one in the channel pores. The material was synthesized by co-condensation of a mixture of a bridged disilane featuring a disulfur group–(MeO)3Si-(CH2)3-S-S-(CH2)3-Si(OMe)3 (S2); tetraethyl orthosilicate (TEOS); a silyl-derivative featuring a protected amine group–(MeO)3Si-(CH2)3-NHBoc. The synthesis was carried out in the presence of non-ionic surfactant P123. The final bi-functional hybrid catalyst was obtained by post-synthetic reduction of disulfide units followed by their oxidation to SO3H groups and deprotection of amino groups [86]. Overall, relatively few reports on sulfonic acid-functionalized PMOs are reported in the scientific literature; synthesis and purification of molecules featuring a sulfonic acid group and two alkoxy-silyl functionalities, to work as bridged precursor, are demanding tasks that pose severe limitations, related to the strong tendency of bisilylated moieties to undergo spontaneous hydrolysis/condensation (higher than that of monosilylated ones), together with the harsh conditions that are usually required for the preparation of platform molecules featuring sulfonic acids.
4.2. Base Catalysts
Another important class of catalysts are those carrying base functionalities, able to activate certain functional groups by electron-donation. Within the realm of silica-supported hybrids, the majority of these catalysts consist of N-based compounds (amines and N-cycles of various nature) and consequentially this class of hybrids have been vastly studied. Bigi et al. indicated a strong correlation between the activity scale of supported amines and that of the corresponding homogeneous counterparts, the order of activity being primary > secondary > tertiary in nitroaldol condensation reactions at 90 °C [87]. However, this general trend has not always been confirmed. For example, Kitayama et al. reported on mesoporous silica FSM-16 functionalized with secondary amines being more active for the aldol reaction than FSM-16 functionalized with primary amines [88]. In another study, Katz et al. found that acid–base synergy between surface silanols and primary amines could also affect the catalytic activity and that such cooperativity enhanced the selectivity to unsaturated products in base-catalyzed reactions, such as the Michael, Henry and Knoevenagel reactions [89].
Shantz et al. prepared a series of surface-anchored amines by either grafting or tethering reactions and compared their activity in the Henry reaction between nitrobenzaldehyde and nitromethane (Figure 16A). The authors found the primary amine (catalyst 1) to be the less active than secondary and tertiary amine. They also observed differences in selectivity. The primary amine appeared to follow the imine formation mechanism, favoring the formation of the nitrostyrene product. On the other hand, the samples containing secondary amines resulted to be highly selective to the nitroalcohol product. Interestingly, the selectivity to the nitroalcohol product increased proportional to the amine loading. Among the secondary amines, the less sterically hindered one (catalyst 2) appeared to be the most active. Clearly, the activity of supported amines cannot be rationalized only upon their basic strength, in terms of pKa, but other aspects such as nucleophilicity and the reaction mechanisms need to be considered [90,91,92].
Silica-supported guanidine moieties have also been synthesized by several authors and implemented in a number of catalytic reactions. Dupont et al. synthetized a guanidine-based silyl-derivative by reacting 3-aminopropyltrimethoxysilane and dicyclohexylcarbodiimide (Figure 16B). The silyl-derivative was grafted onto silica and the hybrid was tested for the production of biodiesel. Conversions up to 98% were obtained for soybean oil methanolysis at 353 K for 3 h. The catalytic system was transposed to a semi-pilot scale in a continuous flow reactor [93]. In another study, Sartori et al. used a guanidine-based hybrid catalyst in the cycloaddition of epoxides, to produce cyclic carbonates at 140 °C. Different epoxides were used, with yield at 70 h ranging between 55 and 95 %. The catalyst was less active than the homogeneous counterpart, although it could be reused over three catalytic cycles [94].
Another relevant class of base hybrid catalysts are supported imidazolium based moieties. In an exemplificative study, Hesemann et al. developed a mesoporous SBA-15 type silica, bearing ionic imidazolium substructures, via post-synthesis grafting reactions using bisilylated imidazolium precursors (Figure 16C). Passivated versions of the hybrids were prepared and the four catalysts were tested in Henry reactions and cycloadditions of carbon dioxide to epichlorohydrin. In both types of reaction, the passivated versions showed higher catalytic activity, although to a lesser extent in the case of cycloaddition reactions.
The authors speculated a beneficial synergistic effect involving the imidazolium cation, its counter-ion and the hydrophobic groups introduced during the passivation of the material [95]. Gianotti et al. prepared a hybrid catalyst using proton sponges as organic building blocks. The proton sponges, i.e., diamines that exhibit very high basicity, were reacted to obtain bi-silylated building blocks (Figure 16D). The resulting precursors were co-condensed with TMOS under fluoride catalyzed conditions. The hybrid was tested in the Knoevenagel condensation between benzaldehyde and ethyl cyanoacetate, showing high activity and recyclability over four catalytic cycles [96].
The capacity for electron rich atoms, such as N, to coordinate electrophilic atoms, together with the versatility of organic syntheses, allows for the possibility of functionalizing silica supports with chiral amines to carry out asymmetric catalytic reactions. Wang et al. developed a silica supported pyrrolidine-based chiral ionic liquid capable of catalyzing Michael addition reaction of ketones with nitrostyrenes in good yields, excellent enantioselectivities and high diastereoselectivities. The catalyst could be reused up to six times (Figure 16E) [97]. In another study, Llopis et al. reported on a new chiral mesoporous hybrid material based on pyrrolidine units integrated into an organic–inorganic structure, obtained from a bi-silylated precursor. The mesoporous hybrid was prepared by fluoride sol-gel methodology under soft synthesis conditions and in the absence of structural directing agents. The chiral solid was tested as asymmetric catalyst in the Michael addition reaction between a wide range of linear aldehydes to nitroalkenes with excellent yields as well as high enantioselectivities. The hybrid catalyst was stable, easily separable and could be reused several times (Figure 16F) [98]. From this very broad overview, the potential of hybrid organic-inorganic catalysts in heterogeneous catalysis is very clear. Organic species featuring base active sites of different nature, strength (pKa), reactivity (nucleophilicity) and stereo-selectivity can be supported onto silica supports for applications as heterogeneous catalysts in industrially relevant reactions. To highlight the multi-disciplinary character of this field, it should be pointed out that the synthesis and reactivity of many amines have been extensively studied by organic chemists throughout the decades and, as a welcomed consequence, the body of literature that resulted provide many key tools that have been implemented in the synthesis of silyl-derivatives, featuring the optimal active site for a given catalytic process.
Base–H-Bond Donor Cooperative Catalysis
Silica-based materials functionalized with amine groups are known to efficiently catalyze C-C coupling reactions, especially when weak acid sites are present in close proximity to amine function. Cooperative effect of multiple active sites is well known. Undoubtedly, the most notable example are enzymes, in which multiple active sites (e.g., acid, base, H-bond donor) co-catalyze a given reaction. The complex enzyme architecture allows the catalytic sites and substrates to adopt a favorable conformation to promote the cooperative interaction, as functional groups placement and overall flexibility of the structure are optimal for such synergy to occur. Slight modifications of these parameters can have a major impact on mechanistic pathways and catalytic rates [99].
Accounting for all variables contributing to this synergy and properly understanding the cooperative action occurring between the two sites are crucial aspects in the rational design of a catalyst [100]. Moreover, such insights can improve the rational design of hybrid catalysts featuring additional functional groups, such as metal nanoparticles or clusters. Many studies have been conducted by several authors, focused on understanding the effect of several variables on the activity of such catalysts in C-C bond formation reactions, such as aldol and nitro-aldol additions. Key feature that influence this cooperativity have been highlighted by the studies below described.
Firstly, silanol groups appeared to be the ideal partner for cooperative catalysis carried out by amines. Brunelli et al. prepared a series of materials containing low loadings (0.36 mmol/g) of primary amine, to ensure the presence of enough silanol groups to act cooperatively with the base sites. The materials were prepared by grafting the silyl-derivatives onto the surface of the silica in a sequential fashion, firstly reacting the 3-aminopropyl triethoxysilane and then the protected carboxylic acid organosilane derivative. A fraction of the amino-functionalized silica was not functionalized with carboxylic acid groups and was used for comparison. By following the procedure, the authors ensured to have the same amount of amine grafted onto the catalysts. The catalysts were tested in the reaction between 4-nitrobenzaldehyde and acetone. The amine–silanol cooperative catalyst (Figure 17, cat. 1) showed an initial TOF of 3.0 h−1, which the authors reported as high catalytic activity for primary amines, confirming the efficient acid–base synergy. By introducing the protected carboxylic acid, the catalytic activity dropped, compared to the amine-silanol material, with an initial TOF of 1.5 h−1 (Figure 17, cat. 2). Finally, unprotecting the carboxylic acid provided an amine–carboxylic acid organosiliceous material which showed the lowest catalytic activity of the three catalysts with an initial TOF of 1.2 h−1 (Figure 17, cat. 3). Surface silanols proved to be better cooperative partners than carboxylic acids. Such trend was ascribed to differences in pKa values for silanols and carboxylic acids, with the former being optimal for cooperative catalysis. HMDS-capping of surface silanols induced a drop of the initial TOF to 0.8 h−1 (Figure 17, cat. 4) [101].
The length of the alkyl linker bounding the amine to the silica support is also an important parameter. Brunelli et al. studied the effect of the length of such linker on the performances of the hybrid porous organosiliceous materials. They prepared an array of silica-supported amines, bounded to the surface through linkers of different size (Figure 18A). Silanol-capped versions of the catalysts were also prepared. Firstly, the materials were tested for CO2 capture (Figure 18B), showing a much lower adsorption capacity for linkers shorter than C3. Additionally, no further increase in the adsorption was observed for materials featuring butyl or pentyl linkers. For HMDS-capped mate-rials, the CO2 adsorption capacity was significantly lower for solids featuring C3 or longer linkers, as the capping molecule blocked the cooperative interaction needed for efficient CO2 adsorption. Non-passivated and passivated hybrids were also tested for their catalytic activity in the aldol condensation of 4-nitrobenzaldehyde with acetone (Figure 18C–E). Upon increasing the length of the carbon chain, up to C3, a surge in the reaction rate was observed. Further increase in the linker length beyond propyl resulted in identical catalytic rates. Lastly, as for CO2 adsorption, silanol-capping proved to be detrimental for the performances of the hybrids, once again confirming the beneficial cooperative effect of base sites and surface silanols. The importance of the linker length lies in the fact that it affects the distance between surface silanols and amine groups, ultimately influencing the simultaneous interaction of the two sites can have with the substrate molecules.
The distance between amine functional groups and surface silanol groups can also be modulated by varying the pore size. In fact, larger pores will feature a lower surface curvature, which will locate surface silanols farther from the amines than they would be, were they anchored on a support featuring smaller pores. Accordingly, supporting amines on support with larger pores will require anchoring the base sites through longer alkyl linkers [102].
At a mechanistic level, the beneficial effect of silanol groups lies in their ability to activate the carbonyl bond of the substrate through hydrogen-bridge interactions which promote the nucleophilic attack by the base. Moreover, they favor diffusion of polar molecules throughout the pore system and promote their adsorption, which in turn increase their local concentration, with beneficial effects on the reaction rate [103,104,105].
4.3. Organometallic Catalysts
The possibility of covalently incorporate organic moieties within inorganic insoluble structures, allowed several research groups to heterogenize organometallic complexes. This is perhaps one of the most promising area of research within the field of hybrid catalysts preparation, as transition metal complexes can catalyze a number of reactions including C-C, C-N, C-O bonding formations, oxidations, reductions, as well as different kind of asymmetric reactions. Considering the vast amount of literature on this topic [106,107,108,109], rather than thoroughly describing the state-of-the-art on silica-supported metal complexes, in this section, relevant examples involving silica-supported complexes of different metals are shown, with the scope of describing common synthetic approaches implemented to obtain them.
Suzuki-Miyaura, Heck and Sonogashira coupling reactions are among the most efficient routes for the formation of C-C bonds. Palladium catalysts have been long known to be highly active in these coupling reactions and several types of ligands have been used to support palladium onto silica matrixes [110]. Luo et al. synthesized a mesoporous silica-supported palladium imino-pyridine complex by implementing a multi-step strategy. Firstly, they carried out a nucleophilic substitution reaction between imino-pyridine ligand and 3-chloropropyl triethoxysilane, achieving a silyl-derivative with coordination capacity. Secondly, they grafted the silyl-derivative onto MCM-41. Lastly, after quantifying the amount of ligand in the solid, they obtained the supported complex by suspending the solid in a palladium acetate solution in acetone, at room temperature (Figure 19). The catalyst was found to be an efficient for Suzuki-Miyaura coupling reactions in aqueous medium. The reactions of various aryl bromides with arylboronic acids were carried out, using low Pd loading in the reaction mixture (0.1 mol %), with good to excellent yields and very low Pd leaching (<0.2 ppm) [111].
Varma et al. prepared a Pd-NHC-based organosilica, synthesizing firstly the silyl-derivative of the carbene, then promoting the coordination of the metal and then carrying out a sol-gel process catalyzed by NH4F (Figure 20). The integrity of the organic building block was confirmed by 13C CP/MAS NMR. However, the catalyst exhibited a BET surface area of only 5 m2/g. As mentioned above, bridged silsesquioxane are prone to assemble in an ordered fashion whenever they can act as rigid building blocks, i.e., whenever they are constituted by unsaturated bonds that block the rotation of the atoms around the bond axes. It appears that in the case of the silyl-derivatives reported in this study, the flexible propyl linkers allowed conformational rearrangements of such building blocks, which favored the formation of non-porous solids.
The hybrid catalyst was tested in Heck and Suzuki coupling using different substrates, showing good catalytic activity. However, it should be pointed out that considering the non-porous nature of the solid, the majority of the active sites was most likely inaccessible by the substrate molecules [112].
Sanchez et al. prepared mesoporous silica-supported chiral ruthenium NHC-based catalysts for asymmetric reactions. Neutral unsymmetrical pyridine pincer-type ligands with lateral (S)-prolinamide donor function and a N-heterocyclic carbene (NHC) moiety were prepared. The prolinamide substituents of the CNN-pincer backbone were functionalized with a triethoxysilyl group. The incorporation of ruthenium was achieved by Lin’s method of transmetalation from intermediate silver (I) complexes. Lastly, the silyl-derivative Ru complexes were grafted by heating at reflux in toluene for 24 h (Figure 21). The heterogenized catalysts were observed to be highly active, selective, and recyclable in the asymmetric hydrogenation of alkenes and showed promising results in the cyclopropanation of styrenes [113].
Corma et al. reported on the synthesis of periodic mesoporous organosilicas prepared using a bisilylated vanadyl Schiff base complexes as building block. The synthesis of the catalysts was carried out following the scheme reported in Figure 22. Firstly, a Suzuki coupling between p-vinylphenylboronic acid and 4-bromosalicylaldehyde derivatives was carried out (step b), followed by the diimine formation using ethylenediamine or (1R,2R)-(−)-l,2-diaminocyclohexane (step c). Subsequently, the formation of the vanadyl complex was performed (step d). The authors reported that an alternative strategy in which vanadyl complexation was carried out after the introduction of the trimethoxysilane groups on the ligand led to extensive hydrolysis and the formation of untreatable mixtures. Then, radical coupling between 3-mercaptopropyltrimethoxysilane and the p-styryl residues was performed, to achieve the bisilylated vanadyl complex. Lastly, the organic building blocks were co-condensed with TEOS, in different proportions, to obtain the hybrid PMOs. The templating agent was removed by extraction.
The catalysts were tested in the room-temperature cyanosilylation of aldehydes in chloroform, using trimethylsilyl cyanide as reagent, showing high conversions and complete selectivities to the desired silylated cyanohydrins. Although ee of 30% was observed for the cyanosilylation of benzaldehydes, the result was much lower than that observed for the same reaction performed using the vanadyl complex in homogeneous phase [114,115].
The incorporation of delicate organic molecules within inorganic structures often requires custom-made synthetic strategies that allow preserving their integrity. This is particularly true for supported organometallic complexes. For example, while in some cases the best strategy is the incorporation of the ligand within the support and subsequent coordination with the metal, in other cases this approach will lead to the formation of supported nanoparticles, as some metals tend to aggregate more than others. In other cases, the alkoxy-silyl groups could be susceptible to reactants that are needed to synthesize some organometallic complexes (e.g., strong hydrated bases) and a strategy that would consider this aspect would be in need. Overall, knowledge about sol-gel synthesis and organometallic chemistry are both crucial to successfully prepare silica-supported organometallic complexes.
Organic-inorganic solids have also been cleverly implemented to incorporate metal nanoparticles within silica structures [116,117]. In a representative example, Budroni et al. prepared Au nanoparticles embedded in silica. Firstly, ionic gold was reduced with hydrazine, in the presence of a mixture of 1-dodecanethiol and 3-mercaptopropyltrimethoxysilane (MPMS). Alkanethiols lowers the surface energy of the Au particles and consequently their tendency to aggregate. Alkanethiol-stabilized gold nanoparticles were then dispersed into an ethanol solution, together with TEOS. Hydrolysis and condensation of the alkoxysilyl groups was carried out using NH4F as catalysts. 1-dodecanethiol molecules act as physical spacers between the particles and the silica shell. The method proposes to include the nanoparticles into a shell of silica that will prevent them from aggregation at high temperature, at the same time without limiting the accessibility of the reactant to the catalytic sites [118].
In another study, the same authors prepared a material composed of palladium nanoparticles embedded into amorphous silica. Firstly, they prepared Pd nanoparticles in an inverse microemulsion system, reducing PdCl2 with hydrazine and stabilizing the metal particles with a mixture of two thiols, 1-dodecanethiol (DT) and 3-mercaptopropyltrimethoxysilane (MPMS). Subsequent co-condensation of TEOS in presence of the stabilized nanoparticles allowed to incorporate the Pd NPs into a silica matrix. The alkoxy silyl groups of the MPMS molecules promoted the intimate mixture of palladium nanoparticles and the silica precursor. Lastly, calcination in air and reduction in H2/N2 mixture led to the final material (Figure 23A) [117].
Barau et al. reported on the synthesis of silica-supported palladium nanoparticles, achieved through different approaches. Firstly, they prepared a catalyst by adding the palladium precursor in the synthetic mixture during the preparation of the mesoporous support (co-gelation); then, they prepared two more catalysts by wet impregnation using a Pd-containing solution and either a pure silica support or a mesoporous silica previously functionalized with aminopropyl groups. The catalyst prepared by wet impregnation using an aminopropyl-functionalized support showed superior catalytic activity in the Heck reaction between iodobenzene and methylacrylate. The authors reported that by preparing the catalyst in such manner, a homogeneously distributed nanoparticle system, stabilized by the pending organic moieties, should be achieved [119].
Johnston et al. immobilized Pd nanoparticles onto silica-based mesocellular foams (MCFs). MCF feature a three-dimensional structure with high surface area (500–800 m2/g) and interconnected pores of size tunable between 25 and 42 nm, interconnected by 9–22 nm windows [120]. The authors used a support featuring 29 nm pores and 15 nm windows; moreover, the silica support was functionalized with aminopropyl groups, in order to promote the interactions between palladium nanoparticles and the support. TEM microscopy on the resulting material showed a narrow particle size distribution (1–2 nm, Figure 23B). The hybrid was found to be a highly efficient heterogeneous catalyst for the oxidation of alcohols. Primary and secondary benzylic alcohols were oxidized to the corresponding carbonyl compounds in high yields, using air as the source of molecular oxygen. The catalyst was found to be highly stable and was recycled several times without any leaching of the metal [121].
A rather interesting approach to the incorporation of Pd nanoparticles was reported by Ma and co-workers. They used an amino-functionalized SBA-16 as support and reduced Pd(OAc)2 precursor by chemical reduction with NaBH4 (Figure 23C). SBA-16 is an ordered mesoporous material with cage-like structures [122]. This mesoporous silica has a tunable cage size (4–10 nm) and pore entrance size generally smaller than 4 nm. Each nanocage is interconnected by eight neighboring pore entrances in three special dimensions. Besides hosting metal complexes, isolated nanocages of SBA-16 can limit the growth of metal nanoparticles by the spatial restrictions [123,124]. Based on the literature on the topic, it appears that -NH2 groups play a crucial role in stabilizing small Pd nanoparticles. Their ability to coordinate palladium through the N lone electron pair and the steric blockage, provided by the aminopropyl pendant fragments as a whole, limits particle migration on the surface of the support, ultimately hindering particle aggregation and leaching. Moreover, the base character of the amine groups opens up to the use of these hybrids as multi-functional catalysts for cascade reactions.
5. Silica-Based Multi-Functional Hybrid Catalysts
The possibility of incorporating active sites within solid structures, fixing them in specific regions of the material opens up to the possibility of designing heterogeneous catalysts featuring multiple active sites, to carry out cascade or tandem reactions, using only one recoverable catalytic system. This is no trivial task, considering that catalytically active sites are, by definition, reactive functional groups and that different active sites can interact with each other (or annihilate) in the synthesis mixture preventing their homogeneous distribution or causing their mutual inactivation. To avoid this, clever synthetic strategies are in need. Approaches based on silica are particularly advantageous for this purpose, for various reasons. The versatility of sol-gel chemistry and the reactivity of the surface of silica materials make it possible to introduce different functionalities in subsequent steps (e.g., co-condensation and grafting). Silica supports are also generally cheap and easy to synthetize. Moreover, their thermal and mechanical stability, their high surface area, tunable pore size diameter and the ease with which their surface can be modified in a post-synthesis step (introducing additional functional groups or varying their hydrophilic/hydrophobic properties) are welcomed features for inorganic supports. In this section, representative examples describing some synthetic strategies used for preparing multi-functional silica-based heterogeneous catalysts are reported.
Gianotti et al. prepared bi-functional mesoporous hybrid materials, containing both acid groups and proton sponges. Co-condensation of TMOS, 3-mercaptopropyltriethoxysilane (MPTES) and bisilylated 1,8-bis(dimethylamino)naphthalene (DMAN-2Sil), was carried out under NH4F catalysis, at low temperature and neutral pH (Figure 24A). After the synthesis, in order to oxidize thiol groups to sulfonic acids, the hybrid was stirred in 30% H2O2 solution. The authors also prepared ordered mesoporous organosilica featuring the same functional groups. They achieved that by implementing a surfactant-mediated one-pot co-condensation of MPTES, disilylated-DMAN and TEOS, in presence of hexadecylamine (HDA). Template removal was performed by ethanol extraction using a Soxhlet apparatus and the thiol groups were oxidized to sulfonic acid groups with a H2O2 solution. Lastly, the bifunctional hybrids were also prepared by multi-step synthesis (Figure 24B). In this case, firstly, both ordered and non-ordered containing MPTES silicas were synthesized following a template route and a NH4F co-condensation route at room temperature. Secondly, after extraction of the template and oxidation of the thiol groups to sulfonic acids, monosilylated DMAN (DMAN-1Sil) was grafted both on ordered and non-ordered materials. The hybrid catalysts were tested in the one-pot cascade reaction involving an acid-catalyzed acetal hydrolysis followed by a base-catalyzed Knoevenagel or nitroaldol (Henry) reaction. Interestingly, non-ordered hybrids, prepared by NH4F catalyzed route, showed superior catalytic activity, compared to the other hybrids.
This was particularly true when a more demanding methylene compound (such as diethylmalonate or nitromethane) was used as substrate. The authors tentatively ascribed the enhanced activity to the higher structural flexibility of the non-ordered silica network and to the higher amount of silanol groups that can perform a cooperative electrophilic activation [63].
In another study, Asefa et al. prepared bi-functional mesoporous organosilicas baring Pd-diamino and propylamino residues. The synthesis was carried out following the scheme reported in Figure 25, using MCM-41 and SBA-15 as supports. For both hybrids, firstly, 3-aminopropyltrimethoxysilane was grafted onto the silica support. Then, a silyl-functionalized Pd complex was prepared by mixing at room temperature PdII acetate and N-(2-aminoethyl)-3-aminopropyltrimethoxysilane (1:1 mol ratio). The silyl-derivative was then grafted onto the amino-functionalized mesoporous silica, in toluene, to achieve the bi-functional hybrid catalyst. The authors attributed great importance to using the appropriate solvent in each of the grafting procedures carried out. Specifically, by using isopropanol, they were able to achieve a more homogenous dispersion of the active sites throughout the surface of the support, at the same time leaving enough free silanol groups to perform a second grafting. For that, they used toluene, which is reportedly a good solvent for grafting processes. The hybrid catalysts were tested in the one-pot tandem Sonogashira-Henry reactions (Figure 25, inset). The catalysts showed to be active in the tandem reaction with a 60% yield of the Sonogashira-Henry product, after 5 h [125].
Another interesting possibility is the coexistence of acid/base functionalities together with metal nanoparticles, to perform tandem or cascade reactions. In an interesting study, Bäckvall et al. prepared an artificial metalloenzyme with two different modes of reactivity: one exhibited by a nanometallic component and one by an enzyme. The presence of the two functionalities in the same cavity of a mesoporous heterogeneous support created an environment in which these active sites could work in close proximity to one another in a cooperative fashion, to perform tandem catalysis. In this work, siliceous mesocellular foams (MCFs) were used as inorganic supports. MCFs are a class of porous siliceous materials featuring high pore volume and cavities, which make them the ideal platform for supporting macromolecules such as enzymes. The approach used for the multi-step synthesis was based on leaving the incorporation of the enzyme as the last one, as by doing otherwise the macromolecule would have risked to be affected by the strongly basic condition in which the metal nanoparticles could be obtained. An amino-functionalized MCF was used as support for Pd nanoparticles deposition (1–2 nm in diameter) [121]. The material was then suspended in aqueous glutaraldehyde in phosphate buffer, to react the remaining amine groups, not interacting with the Pd. The enzyme Candida antarctica lipase B (CALB) was then reacted with the pendant aldehyde groups, to achieve the bi-functional catalyst (Figure 26). The catalyst was tested in the dynamic kinetic resolution (DKR) of a primary amine. In this reaction a lipase enzyme selectively transforms only one enantiomer of the starting amine into the corresponding amide. The racemization reaction allows interconversion of the amine enantiomers and continuously feeds the enzyme catalyzed reaction with more reactive enantiomer (Figure 26B,C). The hybrid catalyst proved to be exceptionally active for the DKR of 1-phenylethylamine and ethyl methoxy acetate with quantitative yield and ee of 99%.
Layered structures also provide interesting synthetic alternatives for preparing multifunctional catalysts. Corma et al. prepared a bifunctional acid-base hybrid catalyst starting from laminar MWW zeolite (Figure 27). For that, firstly, they proceeded with swelling of the layered zeolitic precursors, in presence of cetyltrimethylammonium hydroxide (CTMA). The positively charged alkylammonium groups in the surfactant molecules facilitated the interaction with the negative charges of the zeolitic framework. Secondly, the swollen material was suspended in a dioxane solution of 1,4-bis(triethoxysilyl) benzene (BTEB). This aryl-bridged silsesquioxane molecules were able to diffuse in the interlayer space between the zeolites layers and to covalently bind them through the alkoxide groups. The swelling agents were removed by acid extraction. X-ray diffraction allowed to observe the presence of dimeric pillaring moieties constituted by two condensed BTEB molecules located in the interlayer space. Lastly, the aromatic bridging moieties were functionalized with amine groups, to obtain the bi-functional catalyst. Specifically, the pillared hybrid MWW-BTEB material was reacted in a HNO3/H2SO4 mixture to incorporate -NO2 groups, which were then reduced to -NH2 through reaction with SnCl2. The final material featured a hierarchical system of pores and the presence of the two active sites in different pore frameworks. Specifically, the acid sites were located within the micropores of the zeolitic layers and the basic amino groups were located in the mesopores formed by intercalation of the organic pillaring agents.
The catalyst was tested in a cascade reaction, involving an acetal hydrolysis followed by a Knoevenagel condensation. Specifically, benzaldehyde dimethylacetal was hydrolyzed to produce benzaldehyde, which was reacted with malononitrile to give benzylidene malononitrile. The results showed high activity of the catalyst in the tandem reaction, with a product yield of 96% after 7.2 h of reaction and a selectivity of 99% [126].
In another study, Gaona et al. prepared functional acid-base hybrid materials based on (organo)aluminosilicate layers prepared through direct hydrothermal synthesis processes in the presence of aluminum-rich alkaline media and two combined types of organosiloxane precursors. They were able to achieve isolated aluminosilicate sheets with amino and/or sulfonic groups homogeneously distributed along their external surface, located between the layers. The NH2-functionalized hybrid material was prepared by hydrothermal synthesis in which triethoxymethylsilane (TEMS) and 3-aminopropyl trimethoxysilane (APTMS) were implemented as silicon precursors together with sodium aluminate (NaAlO2) and NaOH. The slurry was stirred at room temperature until gelation occurred and it was then transferred into a stainless steel autoclave and heated at 135 °C for 9 days under autogenous pressure and static conditions. Finally, the solid was washed with distilled water and dried. A catalyst featuring HSO3 moieties was prepared, as well, by using as silicon precursors a mixture of TEMS and 3-mercaptopropyltrimethoxysilane (MPTMS). The thiol groups resulting from the use of the MPTMS were oxidized to sulfonic acids by stirring the material in an acid ethanolic solution (0.02 M H2SO4) at room temperature for 24 h. Finally, by using TEMS, APTMS and MPTMS as silicon sources, the authors were able to prepare laminar materials featuring both NH2 and HSO3 groups. Individual nanolayers were able to interact with each other through multiple types of interactions, such as Van der Waals, hydrogen or ionic interactions, allowing to obtain layered crystalline materials without using a structure directing agent. Different catalytic reactions were implemented to test the activity of the different hybrids. The condensation of carbonyl compounds with active methylenic substrates, in reactions such as Knoevenagel and Henry was used to probe the activity of NH2-functionalized catalyst, whereas acetalization of benzaldehyde with methanol to 1,1-dimethoxytoluene was used to test the activity of the HSO3-functionalized material. The performances of the bi-functional acid-base catalysts were evaluated in one-pot two-step process involving an acid-catalyzed acetal hydrolysis followed by a base-catalyzed Knoevenagel reaction, with a product yield of 92%. This study provided a valid alternative to the typical mesoporous silica for applications as inorganic supports for the synthesis of multi-functional hybrid organic-inorganic catalysts [127].
Li et al. prepared a series of catalysts by immobilizing Pd nanoparticles onto amino-functionalized SBA-15. Firstly, the hybrid support was prepared by one-pot co-condensation of TEOS and aminopropyltrimethoxysilane (APTMS) in presence of block copolymer P123; then, palladium nanoparticles were supported by adsorption/reduction procedure. The amino groups present on the silica support were designed for playing two roles: stabilizing the Pd NPs against aggregation and leaching, both by weak interaction between the N lone pair and the empty orbitals of the Pd atoms and by partial occlusion of the pore channels, and acting as Brønsted base sites in a cascade reaction (Figure 28). The bi-functional catalyst provided full yield of the hydrogenated product. To access the key role played by each of the two active sites, mono-functional catalysts featuring only one of the two sites were prepared. As expected, NH2-functionalized SBA-15 led to a 100% yield of product B1 (Figure 28B) and no conversion was observed using Pd NPs supported onto pure silica SBA-15.
Interestingly, a physical mixture of the two mono-functional catalysts also gave a 100% yield of the desired product C1. However, while the bi-functional catalyst was able to retain its catalytic activity, with 88% yield of product C1 in the 5th catalytic cycle, the physical mixture of the two mono-functional catalysts suffered from fast deactivation, with only a 26% yield of product C1 obtained in the third catalytic cycles. Presumably, the interactions between the Pd NPs and the surface of the silica support, without any organic groups, are relatively weak and cannot prevent aggregation and leaching of the Pd species during the catalysis. On the other hand, the presence of pendant aminopropyl moieties stabilized Pd nanoparticles within the mesoporous channels, allowing the hybrid to retain its performances over more catalytic cycles. TEM images recorded on the bifunctional hybrid after five catalytic cycles enforce this hypothesis (Figure 28C): the nanoparticles appear to be well dispersed throughout the porous channels of the support and no macro-aggregates are visible [128].
Besides these representative examples, many other articles and reviews have been published dealing with silica-based multi-functional catalysts featuring various combinations of active sites [18,64,129,130,131,132,133].
The field of multi-functional organic-inorganic hybrid catalysts is still in a relatively early stage of development. However, the reported literature suggests that, likely, the full potential of this field is still largely unexplored. Coexistence of acid and base functionalities in the reaction mixture was not possible before the development of organic-inorganic hybrids. Nowadays, catalytic processes involving subsequent acid + base catalyzed steps carried out in homogeneous phase require neutralization and purification of the intermediates, which contemplate the use corrosive reactants and great energy consumption. Moreover, acid/base catalytic steps followed by a redox step are present in the majority of production processes in the fine chemicals and pharmaceuticals industries. Proof-of-concept studies such as those reported in this review opens up the development or more and more multi-functional catalysts to be implemented in the chemical industry. Lastly, as the field of artificial enzymes develops [134], the versatility of silica-based approaches to supporting organic molecules, opens up to the possibility of supporting a higher number of these macro-biomolecules to achieve higher stability and reusability [135].
6. Conclusions and Perspectives
In the last decades, silica-based hybrid organic-inorganic materials have been proposed by ever more researchers as viable alternative to traditional heterogeneous catalysts. In the first part of this review article, after a briefly introducing the concept of hybrid catalyst, the focus was put on silica-based organocatalysts. Different synthetic strategies commonly implemented to obtain them were described, such as co-condensation, grafting and PMOs synthesis. Then, an in-depth description of the chemistry behind sol-gel processes was presented. Specific focus was put on the effect of the pH and on the catalyst used (acid, base, fluoride ions). The interactions between silica precursors and templating agents of different nature were also described, as well as the most commonly implemented alkoxysilyl precursors.
In the second part of this review, the main classes of organic-inorganic hybrid catalysts were introduced. Relevant exemplificative studies, drawn from the scientific literature, were presented, with specific focus on the synthetic strategies implemented to obtain them and on the effect they turned out to have on the properties of the hybrid catalyst. Acid catalysts were firstly described, specifically heteropolyacids (HPAs) and supported-sulfonic acids. These hybrids offer to overcome size and diffusion limitation that microporous zeolites (by far the most widely implemented class of heterogeneous acid catalysts) encounter when the catalytic process involves bulky substrates. Although HPA-based catalysts showed promising results for a number of catalytic applications, supported-sulfonic acid groups seem to offer better perspectives in term of their versatility. In fact, the acidity of the sulfonic acid proton can be tuned by changing the nature of the substituent onto which the sulfonic acid group is attached. However promising, silica-supported sulfonic acid catalysts offer room for improvements, as they are typically obtained from costly substrates and, in many cases, from multi-step, low-yielding syntheses. Then, silica-supported base catalysts were described. Specifically, representative studies involving the use of silica-supported amines of different type were described, e.g., alkyl-amines, guanidines, proton sponges. After this overview, the cooperative effect occurring between supported amines and the polar silanol groups present on the surface of the porous supports was accessed in details. Supported amines are usually employed as heterogeneous catalysts in processes involving C-C bond formation reactions starting from substrates carrying a carbonyl group (e.g., Knoevenagel condensation, Henry reaction, Michael addition). For these reactions, surface silanol groups play a key role by favoring the adsorption of substrate molecules and activating the carbonyl group by H-bond interaction with the oxygen atom. Different strategies to prepare, silica-supported organometallic catalysts were then described. The incorporation of such delicate functional groups have been achieved by methodologies involving, amongst others, (i) the anchoring of the ligand and subsequent metal coordination; (ii) the synthesis of the organometallic complex starting from a silylated pincer-type ligand and subsequent grafting onto the silica support; (iii) the synthesis of the organometallic complex through coordination by multiple silylated ligand molecules and subsequent grafting. The possibility of using hybrid organic-inorganic catalysts as scaffold for stabilizing metal nanoparticles was also described. In the last section, reports on the synthesis of silica-based multi-functional hybrid catalysts are reported. By means of custom-designed synthetic strategies, several authors were able to incorporate multiple functional groups, sometimes incompatible with each other, within a single solid porous catalyst.
As this review hopefully demonstrated, the versatility of sol-gel chemistry of silica, thermal and mechanical and textural properties of silica-based materials and the reactivity of their surface are all features that can be taken advantage of, in the rational design of a functional material. The chemistry of silyl-derivatives allows preparing precursors carrying the functional group of interest, e.g., catalytic sites, ligands, hydrophobic moieties. Finally, post-synthetic modification of the hybrid materials opens up to the possibility of anchoring precursors that can be then converted to the functional groups of interest (e.g., nitro to amine; thiol to sulfonic acid), which can make the incorporation of reactive functional groups that would be otherwise impossible.
However, for this field to develop further within industries, some drawbacks should be overcome. Firstly, to achieve sustainability on an industrial scale, cheaper and greener way to synthesize silyl-derivatives precursors are in need (e.g., sulfonic acid catalysts precursors); Secondly, a better understanding of deactivation mechanisms would allow designing heterogeneous catalysts and catalytic processes that could work over more cycles. Lastly, as it was shown in Section 4.2, understanding the interaction between the silica support and the pendant organic molecules can provide key information to maximize the catalytic activity of a given catalyst. Although this has been extensively studied for Base–H-bond donor catalysts, the interaction between other type of organic functionalities and silica supports is still relatively unexplored.
Author Contributions
Conceptualization, A.E. and U.D.; original draft preparation, A.E.; editing and review, A.E. and U.D.; supervision, U.D. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Spanish Government (MAT2017-82288-C2-1-P and Severo Ochoa Excellence Program SEV-2016-0683) and MULTY2HYCAT (EU-Horizon 2020 funded project under grant agreement no. 720783).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
A.E. acknowledge “la Caixa” foundation for the PhD scholarship.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Chorkendorff, I.; Niemantsverdriet, J.W. Concepts of Modern Catalysis and Kinetics, 2nd ed.; Wiley-VCH: Hoboken, NJ, USA, 2007. [Google Scholar]
- Gallego, E.M.; Paris, C.; Cantín, Á.; Moliner, M.; Corma, A. Conceptual Similarities between Zeolites and Artificial Enzymes. Chem. Sci. 2019, 10, 8009–8015. [Google Scholar]
- Gallego, E.M.; Portilla, M.T.; Paris, C.; León-Escamilla, A.; Boronat, M.; Moliner, M.; Corma, A. “Ab Initio” Synthesis of Zeolites for Preestablished Catalytic Reactions. Science 2017, 355, 1051–1054. [Google Scholar]
- Wohlfahrt, G.; Witt, S.; Hendle, J.; Schomburg, D.; Kalisz, H.M.; Hecht, H.J. 1.8 and 1.9 Å Resolution Structures of the Penicillium Amagasakiense and Aspergillus Niger Glucose Oxidases as a Basis for Modelling Substrate Complexes. Acta Crystallogr. Sect. D Biol. Crystallogr. 1999, 55, 969–977. [Google Scholar]
- Donoeva, B.; De Jong, K.P. Torn between Two Sites. Nat. Mater. 2020, 19, 5–6. [Google Scholar]
- Kamat, P.V. Photophysical, Photochemical and Photocatalytic Aspects of Metal Nanoparticles. J. Phys. Chem. B 2002, 106, 7729–7744. [Google Scholar]
- Jain, P.K.; Huang, X.; El-Sayed, I.H.; El-Sayed, M.A. Review of Some Interesting Surface Plasmon Resonance-Enhanced Properties of Noble Metal Nanoparticles and Their Applications to Biosystems. Plasmonics 2007, 2, 107–118. [Google Scholar]
- Liu, L.; Corma, A. Metal Catalysts for Heterogeneous Catalysis: From Single Atoms to Nanoclusters and Nanoparticles. Chem. Rev. 2018, 118, 4981–5079. [Google Scholar]
- Qiao, B.; Wang, A.; Yang, X.; Allard, L.F.; Jiang, Z.; Cui, Y.; Liu, J.; Li, J.; Zhang, T. Single-Atom Catalysis of CO Oxidation Using Pt/FeOx. Nat. Chem. 2011, 3, 634–641. [Google Scholar] [PubMed]
- Wang, A.; Li, J.; Zhang, T. Heterogeneous Single-Atom Catalysis. Nat. Rev. Chem. 2018, 2, 65–81. [Google Scholar]
- Sanchez, C.; Julián, B.; Belleville, P.; Popall, M. Applications of Hybrid Organic-Inorganic Nanocomposites. J. Mater. Chem. 2005, 15, 3559–3592. [Google Scholar]
- Martins Estevão, B.; Miletto, I.; Marchese, L.; Gianotti, E. Optimized Rhodamine B Labeled Mesoporous Silica Nanoparticles as Fluorescent Scaffolds for the Immobilization of Photosensitizers: A Theranostic Platform for Optical Imaging and Photodynamic Therapy. Phys. Chem. Chem. Phys. 2016, 18, 9042–9052. [Google Scholar] [PubMed]
- Parejo, P.G.; Alvarez-Herrero, A.; Zayat, M.; Levy, D. Thermo-Optic Properties of Hybrid Sol–Gel Thin Films Doped with Rhodamine 6G at High Vacuum Conditions. J. Mater. Sci. 2015, 50, 6677–6687. [Google Scholar]
- Catauro, M.; Barrino, F.; Poggetto, G.D.; Crescente, G.; Piccolella, S.; Pacifico, S. New SiO2/Caffeic Acid Hybrid Materials: Synthesis and Characterization. Materials 2020, 13, 394. [Google Scholar]
- Toki, M.; Chow, T.Y.; Ohnaka, T.; Samura, H.; Saegusa, T. Structure of Poly(Vinylpyrrolidone)-Silica Hybrid. Polym. Bull. 1992, 29, 653–660. [Google Scholar]
- Saegusa, T.; Chujo, Y. Organic polymer hybrids with silica gel formed by means of the sol-gel method. Polym. Prep. 1989, 1, 39. [Google Scholar]
- Sanchez, C.; Ribot, F. Design of Hybrid Organic-Inorganic Materials Synthesized via Sol-Gel Chemistry. New J. Chem. 1994, 18, 1007–1047. [Google Scholar]
- Díaz, U.; Brunel, D.; Corma, A. Catalysis Using Multifunctional Organosiliceous Hybrid Materials. Chem. Soc. Rev. 2013, 42, 4083–4097. [Google Scholar]
- Ghiaci, M.; Abbaspur, A.; Kia, R.; Seyedeyn-Azad, F. Equilibrium Isotherm Studies for the Sorption of Benzene, Toluene, and Phenol onto Organo-Zeolites and as-Synthesized MCM-41. Sep. Purif. Technol. 2004, 40, 217–229. [Google Scholar]
- Yang, K.; Chen, B.; Zhu, L. Graphene-Coated Materials Using Silica Particles as a Framework for Highly Efficient Removal of Aromatic Pollutants in Water. Sci. Rep. 2015, 5, 11641. [Google Scholar]
- Gaona, A.; Moreno, J.M.; Velty, A.; Díaz, U.; Corma, A. One-Pot Synthesis of Hierarchical Porous Layered Hybrid Materials Based on Aluminosilicate Sheets and Organic Functional Pillars. J. Mater. Chem. A 2014, 2, 19360–19375. [Google Scholar]
- Hoffmann, F.; Cornelius, M.; Morell, J.; Fröba, M. Silica-Based Mesoporous Organic-Inorganic Hybrid Materials. Angew. Chemie Int. Ed. 2006, 45, 3216–3251. [Google Scholar] [CrossRef] [PubMed]
- Asefa, T.; MacLachlan, M.J.; Coombs, N.; Ozin, G.A. Periodic Mesoporous Organosilicas with Organic Groups inside the Channel Walls. Nature 1999, 402, 867–871. [Google Scholar] [CrossRef]
- Inagaki, S.; Guan, S.; Fukushima, Y.; Ohsuna, T.; Terasaki, O. Novel Mesoporous Materials with a Uniform Distribution of Organic Groups and Inorganic Oxide in Their Frameworks. J. Am. Chem. Soc. 1999, 121, 9611–9614. [Google Scholar] [CrossRef]
- Fujita, S.; Inagaki, S. Self-Organization of Organosilica Solids with Molecular-Scale and Mesoscale Periodicities. Chem. Mater. 2008, 20, 891–908. [Google Scholar] [CrossRef]
- Melde, B.J.; Holland, B.T.; Blanford, C.F.; Stein, A. Mesoporous Sieves with Unified Hybrid Inorganic/Organic Frameworks. Chem. Mater. 1999, 11, 3302–3308. [Google Scholar] [CrossRef]
- Inagaki, S.; Guan, S.; Ohsuna, T.; Terasaki, O. An Ordered Mesoporous Organosilica Hybrid Material with a Crystal-like Wall Structure. Nature 2002, 416, 304–307. [Google Scholar] [CrossRef] [PubMed]
- Mizoshita, N.; Tani, T.; Inagaki, S. Syntheses, Properties and Applications of Periodic Mesoporous Organosilicas Prepared from Bridged Organosilane Precursors. Chem. Soc. Rev. 2011, 40, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Osterholtz, F.D.; Pohl, E.R. Kinetics of the Hydrolysis and Condensation of Organofunctional Alkoxysilanes: A Review. J. Adhes. Sci. Technol. 1992, 6, 127–149. [Google Scholar] [CrossRef]
- Schmidt, H.; Scholze, H.; Kaiser, A. Principles of Hydrolysis and Condensation Reaction of Alkoxysilanes. J. Non. Cryst. Solids 1984, 63, 1–11. [Google Scholar] [CrossRef]
- Iler, R.K. Polymerization of Silica. In The Chemistry of Silica: Solubility, Polymerization, Colloid and Surface Properties and Biochemistry of Silica; Wiley: Hoboken, NJ, USA, 1979; pp. 172–311. [Google Scholar]
- Jeffrey Brinker, C. Sol-Gel Processing of Silica. In Colloidal Silica: Fundamentals and Applications; CRC Press: Boca Raton, FL, USA, 2005; pp. 615–635. [Google Scholar]
- Winter, R.; Chan, J.B.; Frattini, R.; Jonas, J. The Effect of Fluoride on the Sol-Gel Process. J. Non. Cryst. Solids 1988, 105, 214–222. [Google Scholar] [CrossRef]
- Pope, E.J.A.; Mackenzie, J.D. Sol-Gel Processing of Silica. II. The Role of the Catalyst. J. Non. Cryst. Solids 1986, 87, 185–198. [Google Scholar] [CrossRef]
- Reale, E.; Leyva, A.; Corma, A.; Martínez, C.; García, H.; Rey, F. A Fluoride-Catalyzed Sol-Gel Route to Catalytically Active Non-Ordered Mesoporous Silica Materials in the Absence of Surfactants. J. Mater. Chem. 2005, 15, 1742–1754. [Google Scholar] [CrossRef]
- Bernards, T.N.M.; Van Bommel, M.J.; Boonstra, A.H. Hydrolysis-Condensation Processes of the Tetra-Alkoxysilanes TPOS, TEOS and TMOS in Some Alcoholic Solvents. J. Non. Cryst. Solids 1991, 134, 1–13. [Google Scholar] [CrossRef]
- Yilmaz, B.; Müller, U. Catalytic Applications of Zeolites in Chemical Industry. Top. Catal. 2009, 52, 888–895. [Google Scholar] [CrossRef] [Green Version]
- Aoyama, T.; Takido, T.; Kodomari, M. Silica Gel-Supported Polyphosphoric Acid (PPA/SiO2) as an Efficient and Reusable Catalyst for Conversion of Carbonyl Compounds into Oxathioacetals and Dithioacetals. Synlett 2004, 2, 2307–2310. [Google Scholar] [CrossRef]
- Khatik, G.L.; Sharma, G.; Kumar, R.; Chakraborti, A.K. Scope and Limitations of HClO4-SiO2 as an Extremely Efficient, Inexpensive, and Reusable Catalyst for Chemoselective Carbon-Sulfur Bond Formation. Tetrahedron 2007, 63, 1200–1210. [Google Scholar] [CrossRef]
- Bandgar, B.P.; Patil, A.V.; Chavan, O.S. Silica Supported Fluoroboric Acid as a Novel, Efficient and Reusable Catalyst for the Synthesis of 1,5-Benzodiazepines under Solvent-Free Conditions. J. Mol. Catal. A Chem. 2006, 256, 99–105. [Google Scholar] [CrossRef]
- Rajput, V.K.; Roy, B.; Mukhopadhyay, B. Sulfuric Acid Immobilized on Silica: An Efficient Reusable Catalyst for Selective Hydrolysis of the Terminal O-Isopropylidene Group of Sugar Derivatives. Tetrahedron Lett. 2006, 47, 6987–6991. [Google Scholar] [CrossRef]
- Kaur, M.; Sharma, S.; Bedi, P.M.S. Silica Supported Brönsted Acids as Catalyst in Organic Transformations: A Comprehensive Review. Cuihua Xuebao Chin. J. Catal. 2015, 36, 520–549. [Google Scholar] [CrossRef]
- Kourasi, M.; Wills, R.G.A.; Shah, A.A.; Walsh, F.C. Heteropolyacids for Fuel Cell Applications. Electrochim. Acta 2014, 127, 454–466. [Google Scholar] [CrossRef]
- Rafiee, E.; Rashidzadeh, S.; Azad, A. Silica-Supported Heteropoly Acids: Highly Efficient Catalysts for Synthesis of α-Aminonitriles, Using Trimethylsilyl Cyanide or Potassium Cyanide. J. Mol. Catal. A Chem. 2007, 261, 49–52. [Google Scholar] [CrossRef]
- Xiong, J.; Zhu, W.; Ding, W.; Yang, L.; Zhang, M.; Jiang, W.; Zhao, Z.; Li, H. Hydrophobic Mesoporous Silica-Supported Heteropolyacid Induced by Ionic Liquid as a High Efficiency Catalyst for the Oxidative Desulfurization of Fuel. RSC Adv. 2015, 5, 16847–16855. [Google Scholar] [CrossRef]
- Kozhevnikov, I.V. Friedel-Crafts Acylation and Related Reactions Catalysed by Heteropoly Acids. Appl. Catal. A Gen. 2003, 256, 3–18. [Google Scholar] [CrossRef]
- Li, J.; Yang, Z.; Li, S.; Jin, Q.; Zhao, J. Review on Oxidative Desulfurization of Fuel by Supported Heteropolyacid Catalysts. J. Ind. Eng. Chem. 2020, 82, 1–16. [Google Scholar] [CrossRef]
- Ren, Y.; Yue, B.; Gu, M.; He, H. Progress of the Application of Mesoporous Silica-Supported Heteropolyacids in Heterogeneous Catalysis and Preparation of Nanostructured Metal Oxides. Materials 2010, 3, 764–785. [Google Scholar] [CrossRef] [Green Version]
- Tsukuda, E.; Sato, S.; Takahashi, R.; Sodesawa, T. Production of Acrolein from Glycerol over Silica-Supported Heteropoly Acids. Catal. Commun. 2007, 8, 1349–1353. [Google Scholar] [CrossRef]
- Vázquez, P.; Pizzio, L.; Cáceres, C.; Blanco, M.; Thomas, H.; Alesso, E.; Finkielsztein, L.; Lantaño, B.; Moltrasio, G.; Aguirre, J. Silica-Supported Heteropolyacids as Catalysts in Alcohol Dehydration Reactions. J. Mol. Catal. A Chem. 2000, 161, 223–232. [Google Scholar] [CrossRef]
- Arias, M.; Laurenti, D.; Geantet, C.; Vrinat, M.; Hideyuki, I.; Yoshimura, Y. Gasoline Desulfurization by Catalytic Alkylation over Silica-Supported Heteropolyacids: From Model Reaction to Real Feed Conversion. Catal. Today 2008, 130, 190–194. [Google Scholar] [CrossRef]
- Doustkhah, E.; Luque, R.; Kim, J.; Lin, J.; Ide, Y.; Yamauchi, Y.; Rostamnia, S.; Wu, K.C.-W.; Len, C.; Luo, X.; et al. Development of Sulfonic-Acid-Functionalized Mesoporous Materials: Synthesis and Catalytic Applications. Chem. A Eur. J. 2018, 25, 1614–1635. [Google Scholar] [CrossRef]
- Van Rhijn, W.M.; De Vos, D.E.; Sels, B.F.; Bossaert, W.D.; Jacobs, P.A. Sulfonic Acid Functionalised Ordered Mesoporous Materials as Catalysts for Condensation and Esterification Reactions. Chem. Commun. 1998, 3, 317–318. [Google Scholar] [CrossRef]
- Lim, M.H.; Blanford, C.F.; Stein, A. Synthesis of Ordered Microporous Silicates with Organosulfur Surface Groups and Their Applications as Solid Acid Catalysts. Chem. Mater. 1998, 10, 467–470. [Google Scholar] [CrossRef]
- Boveri, M.; Aguilar-Pliego, J.; Pérez-Pariente, J.; Sastre, E. Optimization of the Preparation Method of HSO3-Functionalized MCM-41 Solid Catalysts. Catal. Today 2005, 107, 868–873. [Google Scholar] [CrossRef]
- Dacquin, J.P.; Cross, H.E.; Brown, D.R.; Düren, T.; Williams, J.J.; Lee, A.F.; Wilson, K. Interdependent Lateral Interactions, Hydrophobicity and Acid Strength and Their Influence on the Catalytic Activity of Nanoporous Sulfonic Acid Silicas. Green Chem. 2010, 12, 1383–1391. [Google Scholar] [CrossRef]
- Pérez-Pariente, J.; Díaz, I.; Mohino, F.; Sastre, E. Selective Synthesis of Fatty Monoglycerides by Using Functionalised Mesoporous Catalysts. Appl. Catal. A Gen. 2003, 254, 173–188. [Google Scholar] [CrossRef]
- Bossaert, W.D.; De Vos, D.E.; Van Rhijn, W.M.; Bullen, J.; Grobet, P.J.; Jacobs, P.A. Mesoporous Sulfonic Acids as Selective Heterogeneous Catalysts for the Synthesis of Monoglycerides. J. Catal. 1999, 182, 156–164. [Google Scholar] [CrossRef]
- Van Grieken, R.; Melero, J.A.; Morales, G. Fries Rearrangement of Phenyl Acetate over Sulfonic Modified Mesostructured SBA-15 Materials. Appl. Catal. A Gen. 2005, 289, 143–152. [Google Scholar] [CrossRef]
- Zhao, W.; Salame, P.; Launay, F.; Gédéon, A.; Hao, Z. Sulfonic Acid Functionalised SBA-15 as Catalysts for Beckmann Rearrangement and Esterification Reaction. J. Porous. Mater. 2008, 15, 139–143. [Google Scholar] [CrossRef] [Green Version]
- Rostamnia, S.; Pourhassan, F. The SBA-15/SO3H Nanoreactor as a Highly Efficient and Reusable Catalyst for Diketene-Based, Four-Component Synthesis of Polyhydroquinolines and Dihydropyridines under Neat Conditions. Chin. Chem. Lett. 2013, 24, 401–403. [Google Scholar] [CrossRef]
- Srinivas, D.; Saikia, L. Functionalized SBA-15 and Its Catalytic Applications in Selective Organic Transformations. Catal. Surv. Asia. 2008, 12, 114–130. [Google Scholar] [CrossRef]
- Gianotti, E.; Diaz, U.; Velty, A.; Corma, A. Designing Bifunctional Acid-Base Mesoporous Hybrid Catalysts for Cascade Reactions. Catal. Sci. Technol. 2013, 3, 2677–2688. [Google Scholar] [CrossRef]
- Zeidan, R.K.; Hwang, S.J.; Davis, M.E. Multifunctional Heterogeneous Catalysts: SBA-15-Containing Primary Amines and Sulfonic Acids. Angew. Chemie Int. Ed. 2006, 45, 6332–6335. [Google Scholar] [CrossRef] [PubMed]
- Zuo, D.; Lane, J.; Culy, D.; Schultz, M.; Pullar, A.; Waxman, M. Applied Catalysis B: Environmental Sulfonic Acid Functionalized Mesoporous SBA-15 Catalysts for Biodiesel Production. Appl. Catal. B Environ. 2013, 129, 342–350. [Google Scholar] [CrossRef]
- Parambadath, S.; Chidambaram, M.; Singh, A.P. Synthesis, Characterization and Catalytic Properties of Benzyl Sulphonic Acid Functionalized Zr-TMS Catalysts. Catal. Today 2004, 97, 233–240. [Google Scholar] [CrossRef]
- Requies, J.; Güemez, M.B.; Arias, P.L. Applied Catalysis B: Environmental Dehydration of d-Xylose to Furfural Using Selective and Hydrothermally Stable Arenesulfonic SBA-15 Catalysts. Appl. Catal. B Environ. 2014, 145, 34–42. [Google Scholar]
- Wang, X.; Cheng, S.; Chan, J.C.C.; Chao, J.C.H. Template-Free Synthesis of Mesoporous Phenylsulfonic Acid Functionalized Silica. Micropor. Mesopor. Mat. 2006, 96, 321–330. [Google Scholar] [CrossRef]
- Krawiec, P.; Kockrick, E.; Borchardt, L.; Geiger, D.; Corma, A.; Kaskel, S. Ordered Mesoporous Carbide Derived Carbons: Novel Materials for Catalysis and Adsorption. J. Phys. Chem. C 2009, 113, 7755–7761. [Google Scholar] [CrossRef]
- Habib, S.; Salam, P.; Launay, F.; Semmer-herledan, V.; Marie, O.; Zhao, W.; Antoine, G. Investigation of the Catalytic Activity of Extracted and Smoothly Calcined Arenesulfonic Modified SBA-15 Materials. J. Mol. Catal. A 2007, 271, 117–125. [Google Scholar] [CrossRef]
- Harmer, M.A.; Sun, Q.; Michalczyk, M.J.; Yang, Z. Unique Silane Modified Perfluorosulfonic Acids as Versatile Reagents for New Solid Acid Catalysts. Chem. Comm. 1997, 18, 1803–1804. [Google Scholar] [CrossRef]
- Alvaro, M.; Corma, A.; Das, D.; Fornés, V.; García, H. “ Nafion ” -Functionalized Mesoporous MCM-41 Silica Shows High Activity and Selectivity for Carboxylic Acid Esterification and Friedel–Crafts Acylation Reactions. J. Catal. 2005, 231, 48–55. [Google Scholar] [CrossRef]
- Macquarrie, D.J.; Tavener, S.J.; Harmer, M.A. Novel Mesoporous Silica-Perfluorosulfonic Acid Hybrids as Strong Heterogeneous Brønsted Catalysts. Chem. Commun. 2005, 18, 2363–2365. [Google Scholar] [CrossRef]
- Erigoni, A.; Paul, G.; Meazza, M.; Hernández-Soto, M.C.; Miletto, I.; Rios, R.; Segarra, C.; Marchese, L.; Raja, R.; Rey, F.; et al. Acid Properties of Organosiliceous Hybrid Materials Based on Pendant (Fluoro)Aryl-Sulfonic Groups through a Spectroscopic Study with Probe Molecules. Catal. Sci. Technol. 2019, 9, 6308–6317. [Google Scholar] [CrossRef]
- Rostami, A.; Tahmasbi, B.; Gholami, H.; Taymorian, H. Supported N-Propylsulfamic Acid on Magnetic Nanoparticles Used as Recoverable and Recyclable Catalyst for the Synthesis of 2,3-Dihydroquinazolin- 4(1H)-Ones in Water. Chin. Chem. Lett. 2013, 24, 211–214. [Google Scholar] [CrossRef]
- Hajjami, M.; Ghorbani, F.; Bakhti, F. MCM-41-N-Propylsulfamic Acid: An Efficient Catalyst for One-Pot Synthesis of 1-Amidoalkyl-2-Naphtols. Appl. Catal. A Gen. 2014, 470, 303–310. [Google Scholar] [CrossRef]
- Clapp, L.B. Sulfamic Acid and Its Uses. J. Chem. Educ. 1943, 20, 189–191. [Google Scholar] [CrossRef]
- Pirez, C.; Lee, A.F.; Jones, C.; Wilson, K. Can Surface Energy Measurements Predict the Impact of Catalyst Hydrophobicity upon Fatty Acid Esterification over Sulfonic Acid Functionalised Periodic Mesoporous Organosilicas? Catal. Today 2014, 234, 167–173. [Google Scholar] [CrossRef]
- Mbaraka, I.K.; Shanks, B.H. Design of Multifunctionalized Mesoporous Silicas for Esterification of Fatty Acid. J. Catal. 2005, 229, 365–373. [Google Scholar] [CrossRef]
- Karimi, B.; Zareyee, D. Design of a Highly Efficient and Water-Tolerant Sulfonic Acid Nanoreactor Based on Tunable Ordered Porous Silica for the von Pechmann Reaction. Org. Lett. 2008, 10, 3989–3992. [Google Scholar] [CrossRef]
- Yang, Q.; Kapoor, M.P.; Inagaki, S. Sulfuric Acid-Functionalized Mesoporous Benzene-Silica with a Molecular-Scale Periodicity in the Walls. J. Am. Chem. Soc. 2002, 124, 9694–9695. [Google Scholar] [CrossRef]
- Karaki, M.; Karout, A.; Toufaily, J.; Rataboul, F.; Essayem, N.; Lebeau, B. Synthesis and Characterization of Acidic Ordered Mesoporous Organosilica SBA-15: Application to the Hydrolysis of Cellobiose and Insight into the Stability of the Acidic Functions. J. Catal. 2013, 305, 204–216. [Google Scholar] [CrossRef]
- Karam, A.; Alonso, J.C.; Gerganova, T.I.; Ferreira, P.; Bion, N.; Barrault, J.; Jérôme, F. Sulfonic Acid Functionalized Crystal-like Mesoporous Benzene-Silica as a Remarkable Water-Tolerant Catalyst. Chem. Commun. 2009, 45, 7000–7002. [Google Scholar] [CrossRef] [Green Version]
- Kapoor, M.P.; Yanagi, M.; Kasama, Y.; Yokoyama, T.; Inagaki, S.; Shimada, T.; Nanbu, H.; Juneja, L.R. Self-Assembly of Cubic Phenylene Bridged Mesoporous Hybrids from Allylorganosilane Precursors. J. Mater. Chem. 2006, 16, 3305–3311. [Google Scholar] [CrossRef]
- Shylesh, S.; Wagener, A.; Seifert, A.; Ernst, S.; Thiel, W.R. Mesoporous Organosilicas with Acidic Frameworks and Basic Sites in the Pores: An Approach to Cooperative Catalytic Reactions. Angew. Chem. Int. Ed. 2010, 49, 184–187. [Google Scholar] [CrossRef] [PubMed]
- Alauzun, J.; Mehdi, A.; Reyé, C.; Corriu, R.J.P. Mesoporous Materials with an Acidic Framework and Basic Pores. A Successful Cohabitation. J. Am. Chem. Soc. 2006, 128, 8718–8719. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Shantz, D.F. Nitroaldol Reactions Catalyzed by Amine-MCM-41 Hybrids. J. Catal. 2010, 271, 170–177. [Google Scholar] [CrossRef]
- Shimizu, K.; Suzuki, H.; Hayashi, E.; Kodama, T.; Tsuchiya, Y. Catalytic Direct 1, 4-Conjugate Addition of Aldehydes to Vinylketones on Secondary-Amines Immobilised in FSM-16 Silica Lated FSM-16 Mesoporous Silica, Which Can Be Regarded as a Novel Heterogeneous Catalysis for a Practical C–C Bond. Chem. Commun. 2002, 2, 16–17. [Google Scholar]
- Bass, J.D.; Solovyov, A.; Pascall, A.J.; Katz, A. Acid-Base Bifunctional and Dielectric Outer-Sphere Effects in Heterogeneous Catalysis: A Comparative Investigation of Model Primary Amine Catalysts. J. Am. Chem. Soc. 2006, 128, 3737–3747. [Google Scholar] [CrossRef]
- Konuray, A.O.; Fernández-Francos, X.; Ramis, X. Analysis of the Reaction Mechanism of the Thiol-Epoxy Addition Initiated by Nucleophilic Tertiary Amines. Polym. Chem. 2017, 8, 5934–5947. [Google Scholar] [CrossRef] [Green Version]
- Borch Jacobsen, C.; Meldal, M.; Diness, F. Mechanism and Scope of Base-Controlled Catalyst-Free N-Arylation of Amines with Unactivated Fluorobenzenes. Chem. A Eur. J. 2017, 23, 846–851. [Google Scholar] [CrossRef]
- Khoi, V.N.; Morrill, L.C.; Smith, A.D.; Romo, D. Catalytic Generation of Ammonium Enolates and Related Tertiary Amine-Derived Intermediates: Applications, Mechanism, and Stereochemical Models (n → p*). Lewis Base Catal. Org. Synth. 2016, 2, 527–653. [Google Scholar]
- Balbino, J.M.; De Menezes, E.W.; Benvenutti, E.V.; Cataluña, R.; Ebeling, G.; Dupont, J. Silica-Supported Guanidine Catalyst for Continuous Flow Biodiesel Production. Green Chem. 2011, 13, 3111–3116. [Google Scholar] [CrossRef]
- Barbarini, A.; Maggi, R.; Mazzacani, A.; Mori, G.; Sartori, G.; Sartorio, R. Cycloaddition of CO2 to Epoxides over Both Homogeneous and Silica-Supported Guanidine Catalysts. Tetrahedron Lett. 2003, 44, 2931–2934. [Google Scholar] [CrossRef]
- Motos-Pérez, B.; Roeser, J.; Thomas, A.; Hesemann, P. Imidazolium-Functionalized SBA-15 Type Silica: Efficient Organocatalysts for Henry and Cycloaddition Reactions. Appl. Organomet. Chem. 2013, 27, 290–299. [Google Scholar] [CrossRef]
- Gianotti, E.; Diaz, U.; Colucci, S.; Corma, A. Hybrid Organicinorganic Catalytic Mesoporous Materials with Proton Sponges as Building Blocks. Phys. Chem. Chem. Phys. 2011, 13, 11702–11709. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Wang, L.; Zhang, Y.; Wang, G. Silica Gel Supported Pyrrolidine-Based Chiral Ionic Liquid as Recyclable Organocatalyst for Asymmetric Michael Addition to Nitrostyrenes. Tetrahedron 2008, 64, 7633–7638. [Google Scholar] [CrossRef]
- Llopis, S.; García, T.; Cantín, Á.; Velty, A.; Díaz, U.; Corma, A. Chiral Hybrid Materials Based on Pyrrolidine Building Units to Perform Asymmetric Michael Additions with High Stereocontrol. Catal. Sci. Technol. 2018, 8, 5835–5847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunelli, N.A.; Didas, S.A.; Venkatasubbaiah, K.; Jones, C.W. Tuning Cooperativity by Controlling the Linker Length of Silica-Supported Amines in Catalysis and CO2 Capture. J. Am. Chem. Soc. 2012, 134, 13950–13953. [Google Scholar] [CrossRef]
- Lauwaert, J.; Moschetta, E.G.; Van Der Voort, P.; Thybaut, J.W.; Jones, C.W.; Marin, G.B. Spatial Arrangement and Acid Strength Effects on Acid-Base Cooperatively Catalyzed Aldol Condensation on Aminosilica Materials. J. Catal. 2015, 325, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Brunelli, N.A.; Venkatasubbaiah, K.; Jones, C.W. Cooperative Catalysis with Acid-Base Bifunctional Mesoporous Silica: Impact of Grafting and Co-Condensation Synthesis Methods on Material Structure and Catalytic Properties. Chem. Mater. 2012, 24, 2433–2442. [Google Scholar] [CrossRef]
- Brunelli, N.A.; Jones, C.W. Tuning Acid-Base Cooperativity to Create next Generation Silica-Supported Organocatalysts. J. Catal. 2013, 308, 60–72. [Google Scholar] [CrossRef]
- Lauwaert, J.; De Canck, E.; Esquivel, D.; Van Der Voort, P.; Thybaut, J.W.; Marin, G.B. Effects of Amine Structure and Base Strength on Acid-Base Cooperative Aldol Condensation. Catal. Today 2015, 246, 35–45. [Google Scholar] [CrossRef]
- Erigoni, A.; Hernández-Soto, M.C.; Rey, F.; Segarra, C.; Díaz, U. Highly Active Hybrid Mesoporous Silica-Supported Base Organocatalysts for C–C Bond Formation. Catal. Today 2020, 345, 227–236. [Google Scholar] [CrossRef]
- Kandel, K.; Althaus, S.M.; Peeraphatdit, C.; Kobayashi, T.; Trewyn, B.G.; Pruski, M.; Slowing, I.I. Substrate Inhibition in the Heterogeneous Catalyzed Aldol Condensation: A Mechanistic Study of Supported Organocatalysts. J. Catal. 2012, 291, 63–68. [Google Scholar] [CrossRef]
- Jezequel, M.; Jose, M.; Carrillo-hermosilla, F.; Neugebauer, U.; Niccolai, G.P.; Corker, J.; Fiddy, S.; Evans, J.; Broyer, J.; Malinge, J.; et al. Supported Metallocene Catalysts by Surface Organometallic Chemistry. Synthesis, Characterization, and Reactivity in Ethylene Polymerization of Oxide-Supported Mono- and Biscyclopentadienyl Zirconium Alkyl Complexes: Establishment of Structure / Reactivity Relationship. J. Am. Chem. Soc. 2001, 123, 3520–3540. [Google Scholar]
- Sommer, W.J.; Weck, M. Supported N-Heterocyclic Carbene Complexes in Catalysis. Coord. Chem. Rev. 2007, 251, 860–873. [Google Scholar] [CrossRef]
- Habeche, F.; Hachemaoui, M.; Mokhtar, A.; Chikh, K.; Benali, F.; Mekki, A.; Zaoui, F.; Cherifi, Z.; Boukoussa, B. Recent Advances on the Preparation and Catalytic Applications of Metal Complexes Supported-Mesoporous Silica MCM-41 (Review). J. Inorg. Organomet. Polym. Mater. 2020, 30, 4245–4268. [Google Scholar] [CrossRef]
- Conley, M.P.; Copéret, C.; Thieuleux, C. Mesostructured Hybrid Organic-Silica Materials: Ideal Supports for Well-Defined Heterogeneous Organometallic Catalysts. ACS Catal. 2014, 4, 1458–1469. [Google Scholar] [CrossRef]
- Zamboulis, A.; Moitra, N.; Moreau, J.J.E.; Cattoën, X.; Wong Chi Man, M. Hybrid Materials: Versatile Matrices for Supporting Homogeneous Catalysts. J. Mater. Chem. 2010, 20, 9322–9338. [Google Scholar] [CrossRef]
- Zhang, Q.; Su, H.; Luo, J.; Wei, Y. Recyclable Palladium(II) Imino-Pyridine Complex Immobilized on Mesoporous Silica as a Highly Active and Recoverable Catalyst for Suzuki-Miyaura Coupling Reactions in Aqueous Medium. Tetrahedron 2013, 69, 447–454. [Google Scholar] [CrossRef]
- Polshettiwar, V.; Varma, R.S. Pd-N-Heterocyclic Carbene (NHC) Organic Silica: Synthesis and Application in Carbon-Carbon Coupling Reactions. Tetrahedron 2008, 64, 4637–4643. [Google Scholar] [CrossRef]
- Del Pozo, C.; Corma, A.; Iglesias, M.; Sánchez, F. Recyclable Mesoporous Silica-Supported Chiral Ruthenium-(NHC)NN-Pincer Catalysts for Asymmetric Reactions. Green Chem. 2011, 13, 2471–2481. [Google Scholar] [CrossRef]
- Baleizão, C.; Gigante, B.; Das, D.; Alvaro, M.; Garcia, H.; Corma, A. Synthesis and Catalytic Activity of a Chiral Periodic Mesoporous Organosilica (ChiMO). Chem. Commun. 2003, 3, 1860–1861. [Google Scholar] [CrossRef] [PubMed]
- Baleizão, C.; Gigante, B.; Das, D.; Álvaro, M.; Garcia, H.; Corma, A. Periodic Mesoporous Organosilica Incorporating a Catalytically Active Vanadyl Schiff Base Complex in the Framework. J. Catal. 2004, 223, 106–113. [Google Scholar] [CrossRef]
- Jiang, Y.; Gao, Q. Heterogeneous Hydrogenation Catalyses over Recyclable Pd(0) Nanoparticle Catalysts Stabilized by PAMAM-SBA-15 Organic-Inorganic Hybrid Composites. J. Am. Chem. Soc. 2006, 128, 716–717. [Google Scholar] [CrossRef] [PubMed]
- Budroni, G.; Corma, A.; García, H.; Primo, A. Pd Nanoparticles Embedded in Sponge-like Porous Silica as a Suzuki-Miyaura Catalyst: Similarities and Differences with Homogeneous Catalysts. J. Catal. 2007, 251, 345–353. [Google Scholar] [CrossRef]
- Budroni, G.; Corma, A. Gold-Organic-Inorganic High-Surface-Area Materials as Precursors of Highly Active Catalysts. Angew. Chem. Int. Ed. 2006, 45, 3328–3331. [Google Scholar] [CrossRef]
- Barau, A.; Budarin, V.; Caragheorgheopol, A.; Luque, R.; MacQuarrie, D.J.; Prelle, A.; Teodorescu, V.S.; Zaharescu, M. A Simple and Efficient Route to Active and Dispersed Silica Supported Palladium Nanoparticles. Catal. Lett. 2008, 124, 204–214. [Google Scholar] [CrossRef]
- Schmidt-Winkel, P.; Lukens, W.W.; Yang, P.; Margolese, D.I.; Lettow, J.S.; Ying, J.Y.; Stucky, G.D. Microemulsion Templating of Siliceous Mesostructured Cellular Foams with Well-Defined Ultralarge Mesopores. Chem. Mater. 2000, 12, 686–696. [Google Scholar] [CrossRef]
- Johnston, E.V.; Verho, O.; Kärkäs, M.D.; Shakeri, M.; Tai, C.W.; Palmgren, P.; Eriksson, K.; Oscarsson, S.; Bäckvall, J.E. Highly Dispersed Palladium Nanoparticles on Mesocellular Foam: An Efficient and Recyclable Heterogeneous Catalyst for Alcohol Oxidation. Chem. A Eur. J. 2012, 18, 12202–12206. [Google Scholar] [CrossRef]
- Kim, T.W.; Ryoo, R.; Kruk, M.; Gierszal, K.P.; Jaroniec, M.; Kamiya, S.; Terasaki, O. Tailoring the Pore Structure of SBA-16 Silica Molecular Sieve through the Use of Copolymer Blends and Control of Synthesis Temperature and Time. J. Phys. Chem. B 2004, 108, 11480–11489. [Google Scholar] [CrossRef]
- Yang, H.; Han, X.; Li, G.; Wang, Y. N-Heterocyclic Carbene Palladium Complex Supported on Ionic Liquid-Modified SBA-16: An Efficient and Highly Recyclable Catalyst for the Suzuki and Heck Reactions. Green Chem. 2009, 11, 1184–1193. [Google Scholar] [CrossRef]
- Ma, Z.; Yang, H.; Qin, Y.; Hao, Y.; Li, G. Palladium Nanoparticles Confined in the Nanocages of SBA-16: Enhanced Recyclability for the Aerobic Oxidation of Alcohols in Water. J. Mol. Catal. A Chem. 2010, 331, 78–85. [Google Scholar] [CrossRef]
- Sharma, K.K.; Biradar, A.V.; Das, S.; Asefa, T. Bifunctional Mesoporous Silica Catalyst for C-C Bond Forming Tandem Reactions. Eur. J. Inorg. Chem. 2011, 21, 3174–3182. [Google Scholar] [CrossRef]
- Corma, A.; Díaz, U.; García, T.; Sastre, G.; Velty, A. Multifunctional Hybrid Organic-Inorganic Catalytic Materials with a Hierarchical System of Well-Defined Micro- and Mesopores. J. Am. Chem. Soc. 2010, 132, 15011–15021. [Google Scholar] [CrossRef] [PubMed]
- Gaona, A.; Díaz, U.; Corma, A. Functional Acid and Base Hybrid Catalysts Organized by Associated (Organo)Aluminosilicate Layers for C-C Bond Forming Reactions and Tandem Processes. Chem. Mater. 2017, 29, 1599–1612. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Liu, H.; Yu, Y.; Cao, C.Y.; Song, W.G. One-Pot Multistep Cascade Reactions over Multifunctional Nanocomposites with Pd Nanoparticles Supported on Amine-Modified Mesoporous Silica. Chem. An Asian J. 2013, 8, 2459–2465. [Google Scholar] [CrossRef]
- Huh, S.; Chen, H.T.; Wiench, J.W.; Pruski, M.; Lin, V.S.Y. Cooperative Catalysis by General Acid and Base Bifunctionalized Mesoporous Silica Nanospheres. Angew. Chem. Int. Ed. 2005, 44, 1826–1830. [Google Scholar] [CrossRef]
- Patankar, S.C.; Dodiya, S.K.; Yadav, G.D. Cascade Engineered Synthesis of Ethyl Benzyl Acetoacetate and Methyl Isobutyl Ketone (MIBK) on Novel Multifunctional Catalyst. J. Mol. Catal. A Chem. 2015, 409, 171–182. [Google Scholar] [CrossRef]
- Huang, Y.; Trewyn, B.G.; Chen, H.T.; Lin, V.S.Y. One-Pot Reaction Cascades Catalyzed by Base- and Acid-Functionalized Mesoporous Silica Nanoparticles. New J. Chem. 2008, 32, 1311–1313. [Google Scholar] [CrossRef]
- Fernandes, A.E.; Jonas, A.M. Design and Engineering of Multifunctional Silica-Supported Cooperative Catalysts. Catal. Today 2019, 334, 173–186. [Google Scholar] [CrossRef]
- Li, P.; Cao, C.Y.; Liu, H.; Yu, Y.; Song, W.G. Synthesis of a Core-Shell-Shell Structured Acid-Base Bifunctional Mesoporous Silica Nanoreactor (MS-SO3H@MS@MS-NH2) and Its Application in Tandem Catalysis. J. Mater. Chem. A 2013, 1, 12804–12810. [Google Scholar] [CrossRef]
- Schwizer, F.; Okamoto, Y.; Heinisch, T.; Gu, Y.; Pellizzoni, M.M.; Lebrun, V.; Reuter, R.; Köhler, V.; Lewis, J.C.; Ward, T.R. Artificial Metalloenzymes: Reaction Scope and Optimization Strategies. Chem. Rev. 2018, 118, 142–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carceller, J.M.; Mifsud, M.; Climent, M.J.; Iborra, S.; Corma, A. Production of Chiral Alcohols from Racemic Mixtures by Integrated Heterogeneous Chemoenzymatic Catalysis in Fixed Bed Continuous Operation. Green Chem. 2020, 22, 2767–2777. [Google Scholar] [CrossRef]
Figure 1.
(A): Stereoview of the active site of glucose oxidase with the modelled substrate β-d-glucose [4]. Reproduced with permission of the International Union of Crystallography. (B): The structure of γ-valerolactone within zeolite pores as obtained using in situ X-ray diffraction data [5]. Reprinted with permission from [5]. Copyright 2020, Nature Springer. (C): Single Pt atoms bounded to N- rich or S- rich regions in doped carbonaceous supports [10]. Reprinted with permission from [10]. Copyright 2018, Nature Springer.
Figure 1.
(A): Stereoview of the active site of glucose oxidase with the modelled substrate β-d-glucose [4]. Reproduced with permission of the International Union of Crystallography. (B): The structure of γ-valerolactone within zeolite pores as obtained using in situ X-ray diffraction data [5]. Reprinted with permission from [5]. Copyright 2020, Nature Springer. (C): Single Pt atoms bounded to N- rich or S- rich regions in doped carbonaceous supports [10]. Reprinted with permission from [10]. Copyright 2018, Nature Springer.

Figure 2.
Scheme of the main chemical routes for the synthesis of organic-inorganic hybrids [11]. Reprinted with permission from [11]. Copyright 2005, Royal Society of Chemistry.

Figure 3.
(A): Schematic representation of the porous structure of a phenyl modified silica matrix doped with Rhodamine 6G molecules [13]. Reprinted with permission from [13]. Copyright 2015, Nature Springer. (B): Structure of PVP-silica hybrid material [15]. Reprinted with permission from [15]. Copyright 1992, Nature Springer.
Figure 3.
(A): Schematic representation of the porous structure of a phenyl modified silica matrix doped with Rhodamine 6G molecules [13]. Reprinted with permission from [13]. Copyright 2015, Nature Springer. (B): Structure of PVP-silica hybrid material [15]. Reprinted with permission from [15]. Copyright 1992, Nature Springer.

Figure 4.
Structures of mesoporous M41S materials: MCM-41 (A), MCM-48 (B) and MCM-50 (C) [22]. Reprinted with permission from [22]. Copyright 2006, John Wiley and Sons.

Figure 5.
Grafting of a generic silyl-derivative RSi(OR’)3 onto a mesoporous silica pore walls [22]. Reprinted with permission from [22]. Copyright 2006, John Wiley and Sons.

Figure 6.
Co-condensation between tetraethyl ortosilicate (TEOS) and a generic silyl-derivative compound, RSi(OR’)3, followed by calcination/extraction to achieve hybrid organic-inorganic mesoporous silica [22]. Reprinted with permission from [22]. Copyright 2006, John Wiley and Sons.

Figure 7.
(A): General synthetic pathway to PMOs [22]. Reprinted with permission from [22]. Copyright 2006, John Wiley and Sons. (B): Representative bridged silsesquioxanes [25]. Reprinted with permission from [25]. Copyright 2008, American Chemical Society.

Figure 8.
Protonation/deprotonation of orthosilicic acid. Hydrolysis and condensation reactions of silica precursors.
Figure 8.
Protonation/deprotonation of orthosilicic acid. Hydrolysis and condensation reactions of silica precursors.

Figure 9.
Schematic representation of hydrolysis/condensation reactions of tetramethyl ortosilicate (TMOS) under acid and base catalysis.
Figure 9.
Schematic representation of hydrolysis/condensation reactions of tetramethyl ortosilicate (TMOS) under acid and base catalysis.

Figure 10.
Fluoride-catalyzed hydrolysis (A) and condensation (B) of tetramethyl ortosilicate [34]. Reprinted with permission from [34]. Copyright 1986, Elsevier.

Figure 11.
Schematic representation of the possible interactions between silica precursors and surfactant molecules [22]. Reprinted with permission from [22]. Copyright 2006, John Wiley and Sons.

Figure 12.
Ball and stick representation of tetramethyl ortosilicate (TMOS), tetraethyl orthosilicate (TEOS) and tetrapropyl orthosilicate (TPOS).
Figure 12.
Ball and stick representation of tetramethyl ortosilicate (TMOS), tetraethyl orthosilicate (TEOS) and tetrapropyl orthosilicate (TPOS).

Figure 13.
Ball and stick representations of two of the most common HPA variants. P (black), Mo (dark grey), O (light grey) [43]. Reprinted with permission from [43]. Copyright 2014, Elsevier.

Figure 14.
List of some of the precursors implemented in the synthesis of silica supported sulfonic acids [52].
Figure 14.
List of some of the precursors implemented in the synthesis of silica supported sulfonic acids [52].

Figure 15.
Synthesis of a bi-functionalized PMO for nitroaldol catalytic reactions [52,84]. Reprinted with permission from [52]. Copyright 2018, John Wiley and Sons.

Figure 16.
(A): Schematic representation of the grafted amines [87]. Reprinted with permission from [87]. Copyright 2010, Elsevier. (B): Synthesis of guanidine-based silyl-derivative [93]. Reprinted with permission from [93]. Copyright 2011, Royal Society of Chemistry. (C): Imidazolium-based silyl-derivatives [95]. Reprinted with permission from [95]. Copyright 2013, John Wiley and Sons. (D): 1,8-bis(dimethylamino)naphthalene (DMAN)-based silyl-derivative. Reprinted with permission from [96]. Copyright 2011, Royal Society of Chemistry. (E): Silica-supported pyrrolidine-based chiral ionic liquid [97]. Reprinted with permission from [97]. Copyright 2008, Elsevier. (F): Silica-functionalized pyrrolidine-based chiral hybrid catalyst. Reprinted with permission from [98]. Copyright 2018, Royal Society of Chemistry.
Figure 16.
(A): Schematic representation of the grafted amines [87]. Reprinted with permission from [87]. Copyright 2010, Elsevier. (B): Synthesis of guanidine-based silyl-derivative [93]. Reprinted with permission from [93]. Copyright 2011, Royal Society of Chemistry. (C): Imidazolium-based silyl-derivatives [95]. Reprinted with permission from [95]. Copyright 2013, John Wiley and Sons. (D): 1,8-bis(dimethylamino)naphthalene (DMAN)-based silyl-derivative. Reprinted with permission from [96]. Copyright 2011, Royal Society of Chemistry. (E): Silica-supported pyrrolidine-based chiral ionic liquid [97]. Reprinted with permission from [97]. Copyright 2008, Elsevier. (F): Silica-functionalized pyrrolidine-based chiral hybrid catalyst. Reprinted with permission from [98]. Copyright 2018, Royal Society of Chemistry.

Figure 17.
Hybrid bi-functional catalysts tested in the reaction of 4-nitrobenzaldehyde with acetone [101]. Reprinted with permission from [101]. Copyright 2012, American Chemical Society.

Figure 18.
(A): Silica-supported alkylamines used as adsorbent and as heterogeneous catalysts. (B): Amine efficiencies for CO2 adsorption (400 ppm in He). (C): Schematic representation of the aldol condensation between 4-nitrobenzaldehyde and acetone. Performances of the non-passivated (D) and passivated (E) hybrids in the reaction between of 4-nitrobenzaldehyde and acetone [99]. Reprinted with permission from [99]. Copyright 2012, American Chemical Society.
Figure 18.
(A): Silica-supported alkylamines used as adsorbent and as heterogeneous catalysts. (B): Amine efficiencies for CO2 adsorption (400 ppm in He). (C): Schematic representation of the aldol condensation between 4-nitrobenzaldehyde and acetone. Performances of the non-passivated (D) and passivated (E) hybrids in the reaction between of 4-nitrobenzaldehyde and acetone [99]. Reprinted with permission from [99]. Copyright 2012, American Chemical Society.

Figure 19.
Schematic representation of the preparation of MCM-41 supported Pd complex [111]. Reprinted with permission from [111]. Copyright 2013, Elsevier.

Figure 20.
Preparation of Pd-NHC organosilica [112]. Reprinted with permission from [112]. Copyright 2008, Elsevier.

Figure 21.
Schematic representation of the synthesis of silica-supported chiral Ru complexes [113]. Reprinted with permission from [113]. Copyright 2011, Royal Society of Chemistry.

Figure 22.
Schematic representation of the synthesis of PMOs containing vanadyl Schiff base complexes [115]. Reprinted with permission from [115]. Copyright 2004, Elsevier.

Figure 23.
(A): Schematic representation of the synthesis of silica-supported Pd NPs. Reprinted with permission from [117]. Copyright 2007, Elsevier. (B): TEM images of MCF-supported Pd NPs [121]. Reprinted with permission from [121]. Copyright 2012, John Wiley and Sons. (C): Schematic representation of the synthesis of Pd/SBA-16. Reprinted with permission from [124]. Copyright 2010, Elsevier.
Figure 23.
(A): Schematic representation of the synthesis of silica-supported Pd NPs. Reprinted with permission from [117]. Copyright 2007, Elsevier. (B): TEM images of MCF-supported Pd NPs [121]. Reprinted with permission from [121]. Copyright 2012, John Wiley and Sons. (C): Schematic representation of the synthesis of Pd/SBA-16. Reprinted with permission from [124]. Copyright 2010, Elsevier.

Figure 24.
(A): One-pot synthesis of bi-functional mesoporous hybrids. (B): DMAN grafting on mesoporous silicas containing sulfonic acids. Reprinted with permission from [63]. Copyright 2013, Royal Society of Chemistry.
Figure 24.
(A): One-pot synthesis of bi-functional mesoporous hybrids. (B): DMAN grafting on mesoporous silicas containing sulfonic acids. Reprinted with permission from [63]. Copyright 2013, Royal Society of Chemistry.

Figure 25.
Schematic representation of the synthesis of bi-functional hybrid catalyst baring amino functionalities and Pd-diamino complexes. Inset: Sonogashira-Henry tandem reaction used for testing the activity of the hybrid catalyst. Reprinted with permission from [125]. Copyright 2011, John Wiley and Sons.
Figure 25.
Schematic representation of the synthesis of bi-functional hybrid catalyst baring amino functionalities and Pd-diamino complexes. Inset: Sonogashira-Henry tandem reaction used for testing the activity of the hybrid catalyst. Reprinted with permission from [125]. Copyright 2011, John Wiley and Sons.

Figure 26.
(A): Synthesis of the bi-functional hybrid catalyst. (B): Schematic representation of a general dynamic kinetic resolution (DKR) of a primary amine. (C): DKR of an amine with a bi-functional biomimetic catalyst in which Pd nanoparticles and a lipase (CALB) are co-immobilized in the same pore of MCF. Reprinted with permission from [121]. Copyright 2012, John Wiley and Sons.
Figure 26.
(A): Synthesis of the bi-functional hybrid catalyst. (B): Schematic representation of a general dynamic kinetic resolution (DKR) of a primary amine. (C): DKR of an amine with a bi-functional biomimetic catalyst in which Pd nanoparticles and a lipase (CALB) are co-immobilized in the same pore of MCF. Reprinted with permission from [121]. Copyright 2012, John Wiley and Sons.

Figure 27.
(A): Synthetic procedure implemented for the synthesis of the pillared hybrid zeolitic materials from MWW precursors. (B): Layered hybrid material obtained by pillaring with BTEB silsesquioxane molecules (MWW-BTEB). Reprinted with permission from [126]. Copyright 2010, American Chemical Society.
Figure 27.
(A): Synthetic procedure implemented for the synthesis of the pillared hybrid zeolitic materials from MWW precursors. (B): Layered hybrid material obtained by pillaring with BTEB silsesquioxane molecules (MWW-BTEB). Reprinted with permission from [126]. Copyright 2010, American Chemical Society.

Figure 28.
(A): Synthesis procedure for the SBA-15-NH2/Pd-p catalyst. (B): One-pot Knoevenagel condensation/hydrogenation multi-step cascade reaction sequence between benzaldehyde and ethyl cyanoacetate. (C): TEM image of the SBA-15-NH2/Pd-p catalyst after five catalytic cycles. Reprinted with permission from [128]. Copyright 2010, John Wiley and Sons.
Figure 28.
(A): Synthesis procedure for the SBA-15-NH2/Pd-p catalyst. (B): One-pot Knoevenagel condensation/hydrogenation multi-step cascade reaction sequence between benzaldehyde and ethyl cyanoacetate. (C): TEM image of the SBA-15-NH2/Pd-p catalyst after five catalytic cycles. Reprinted with permission from [128]. Copyright 2010, John Wiley and Sons.


{kind=link}
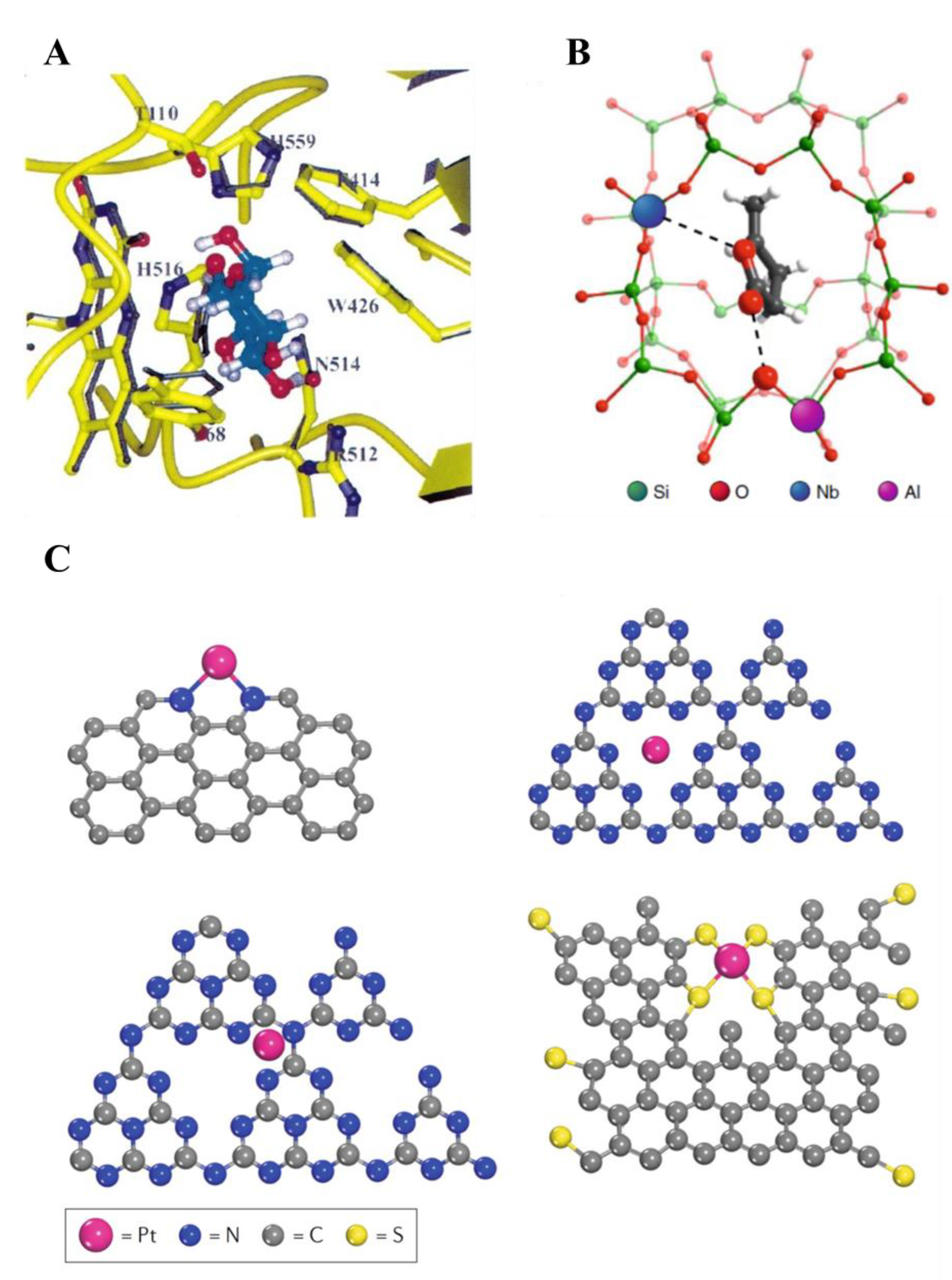{kind=link}
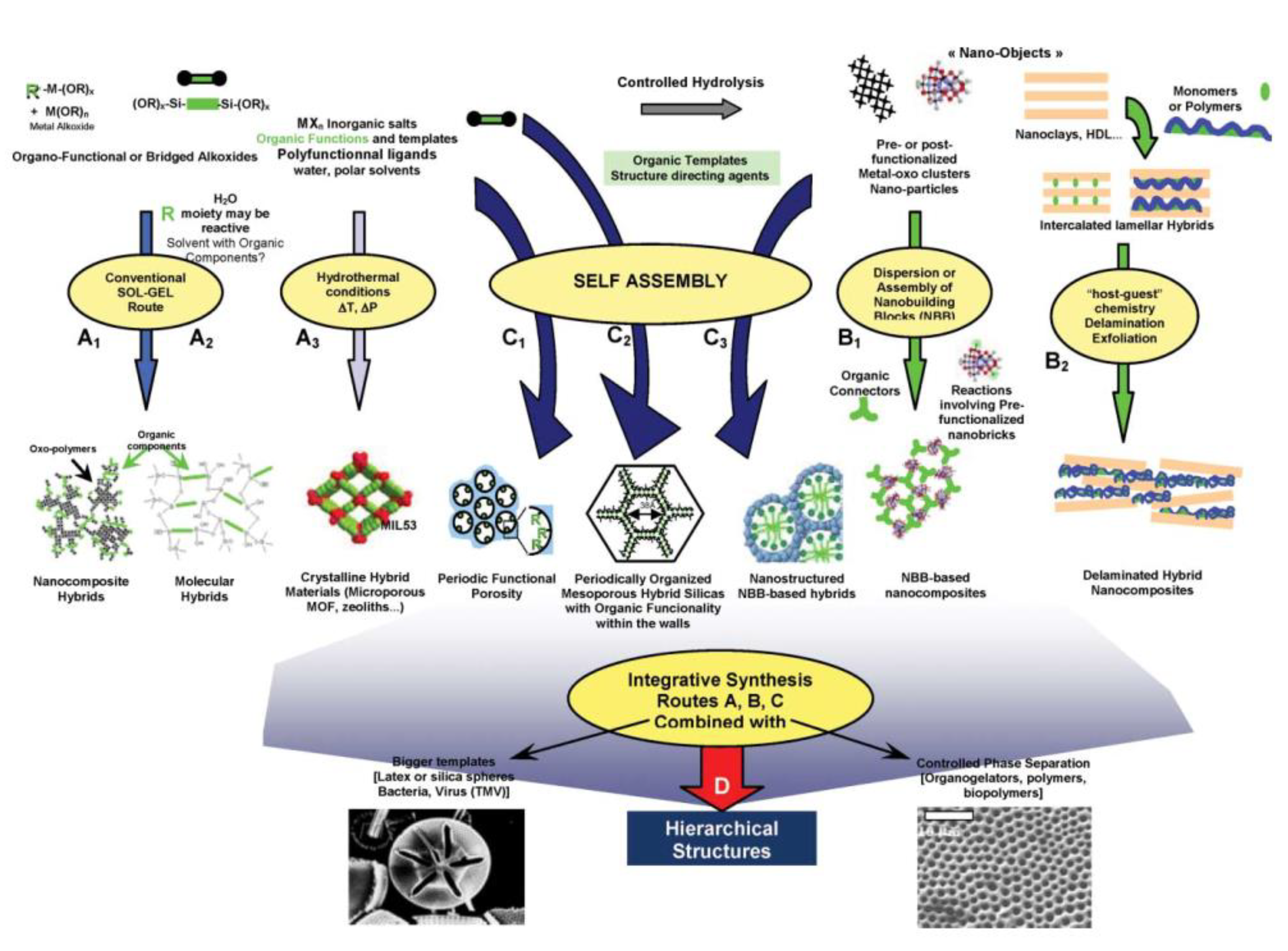{kind=link}
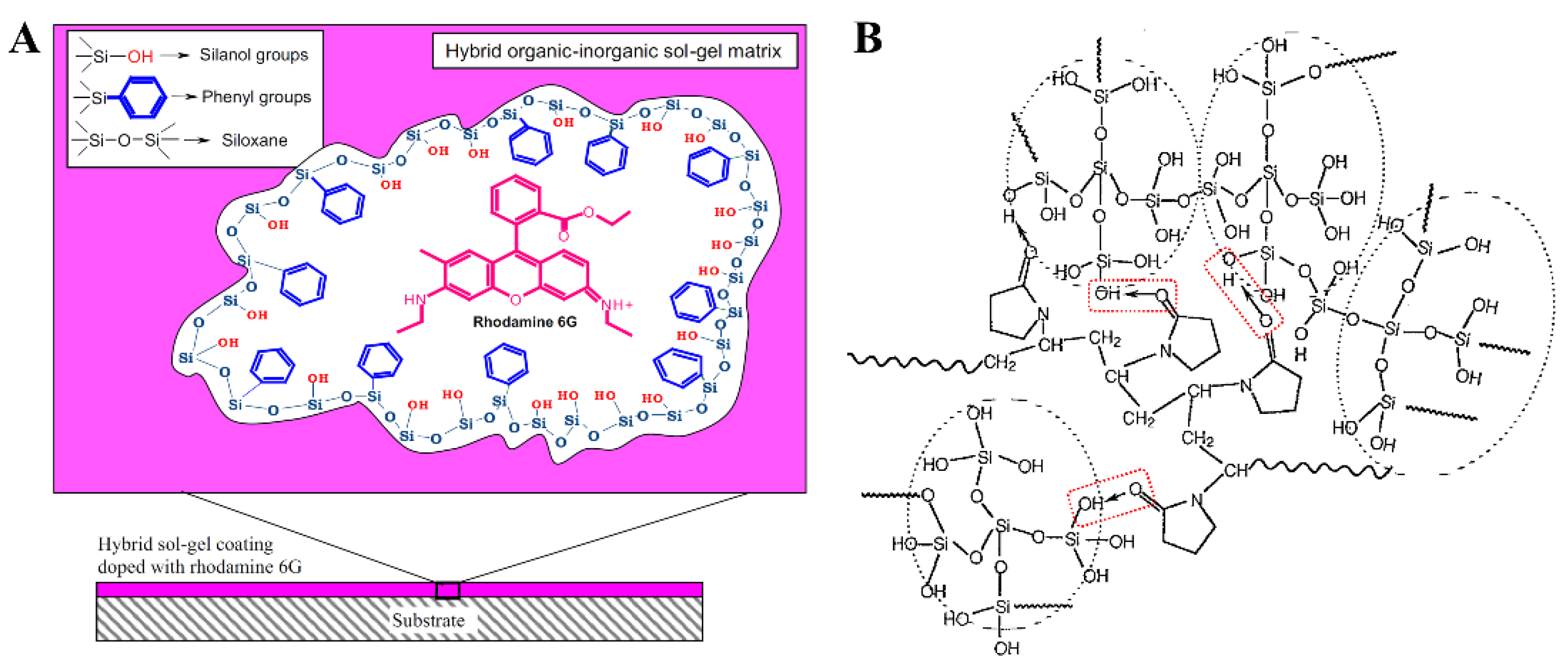{kind=link}
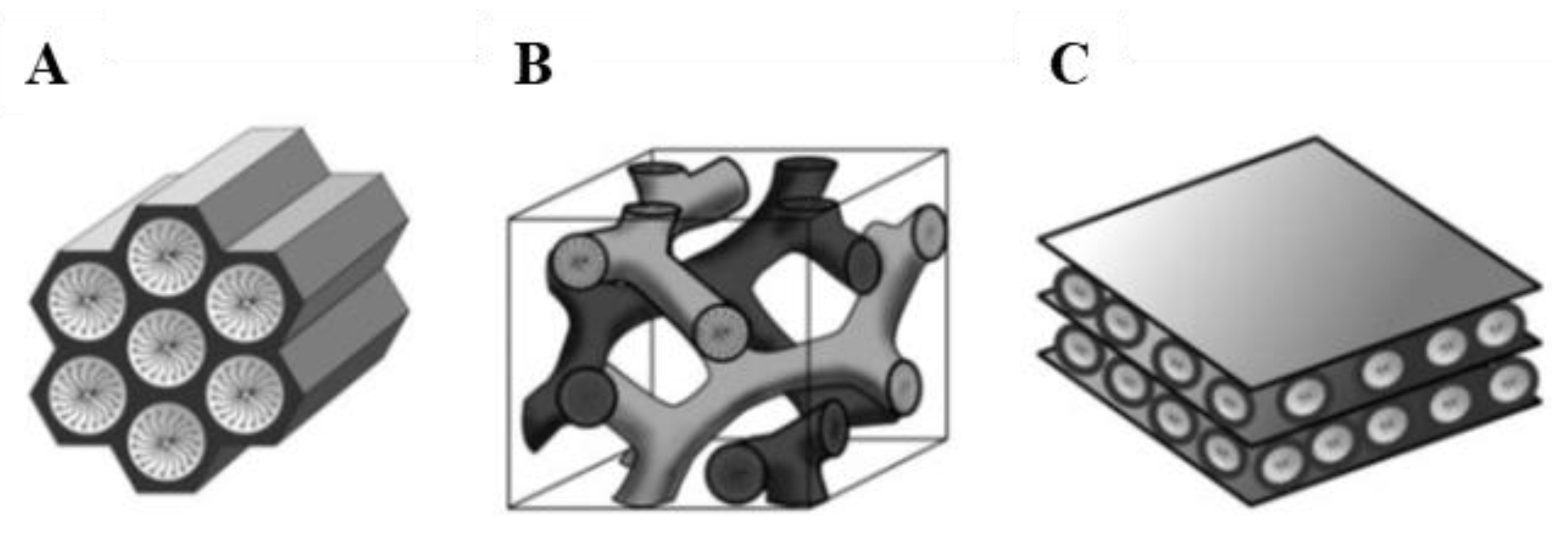{kind=link}
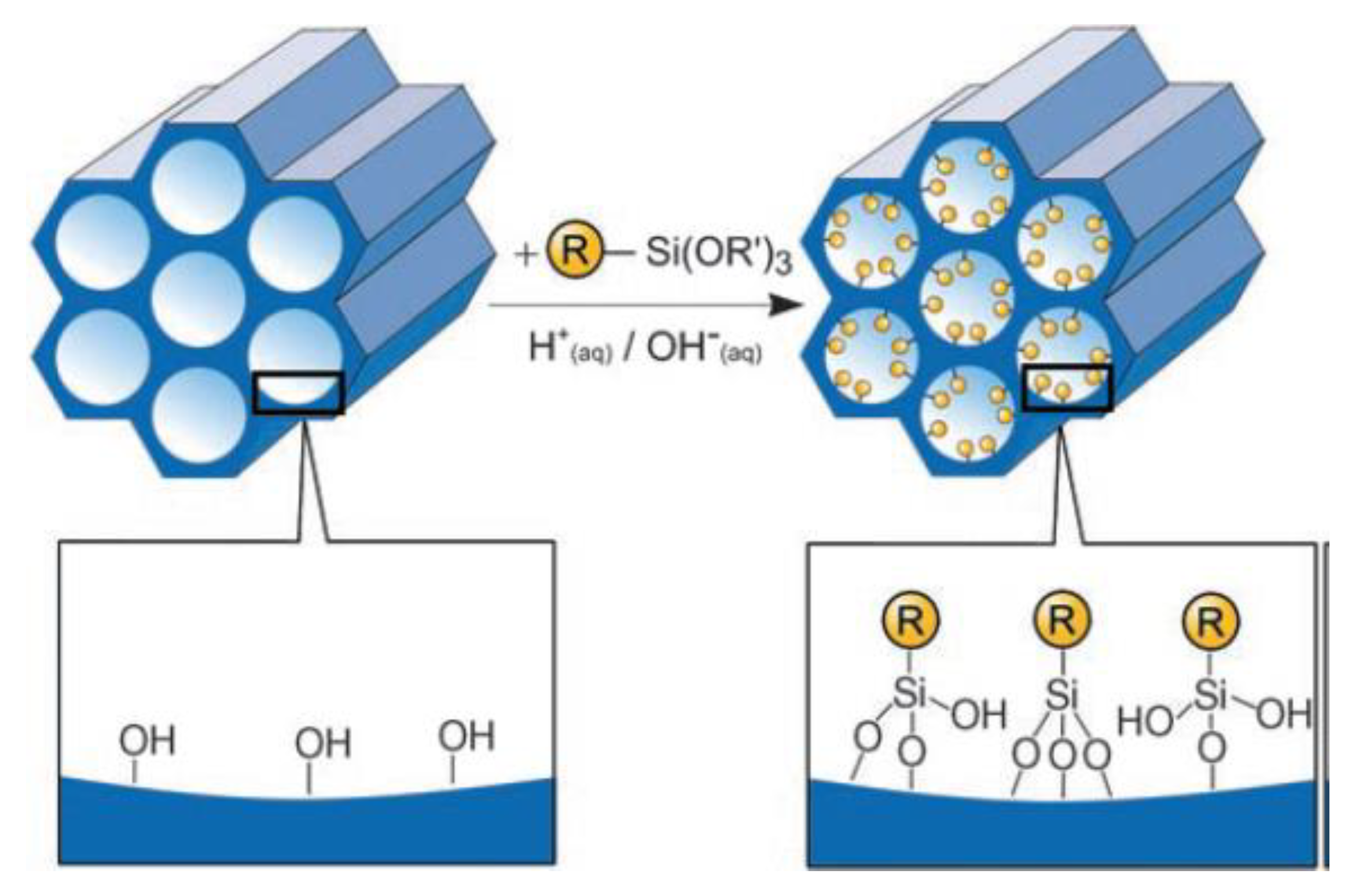{kind=link}
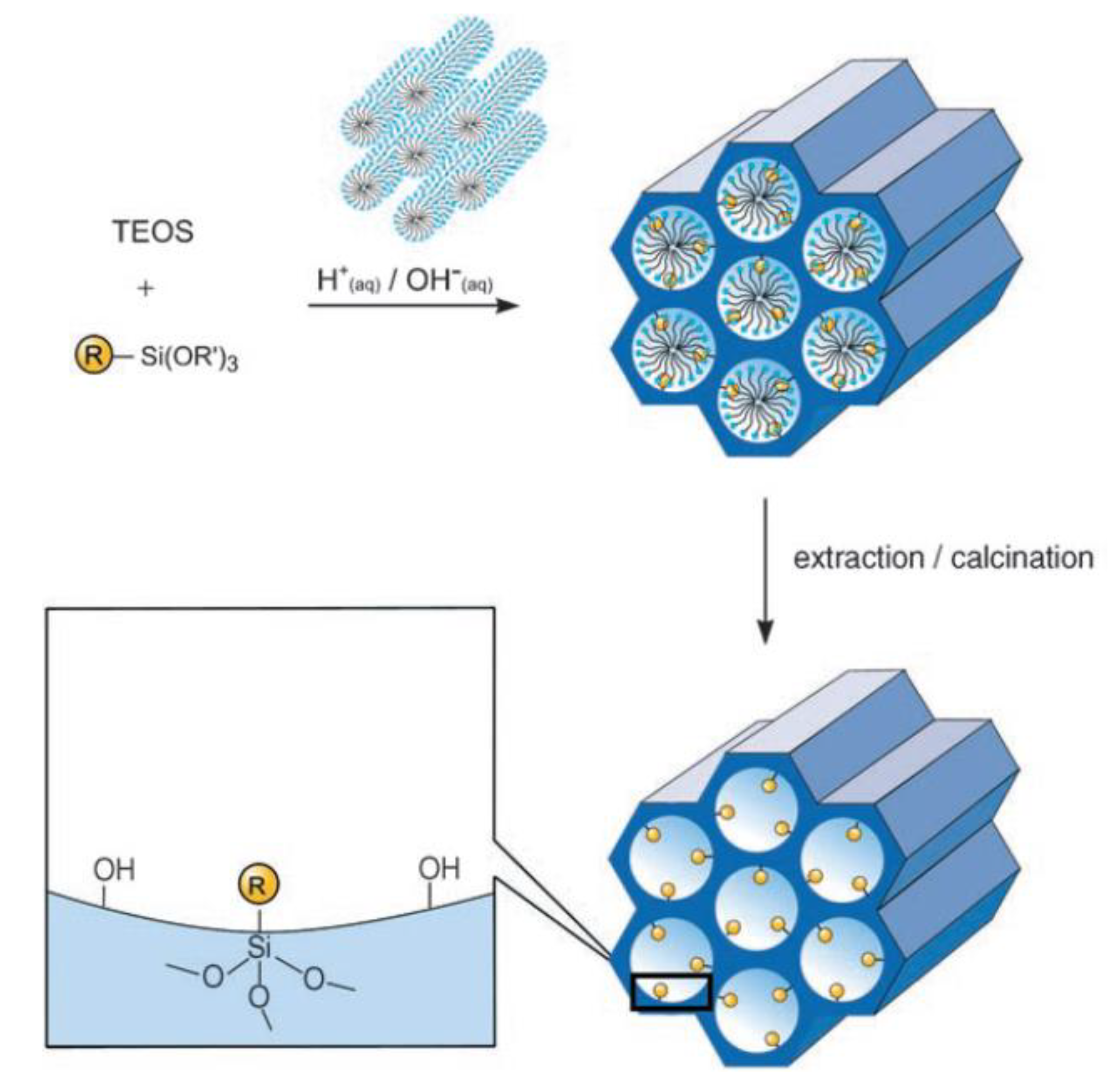{kind=link}
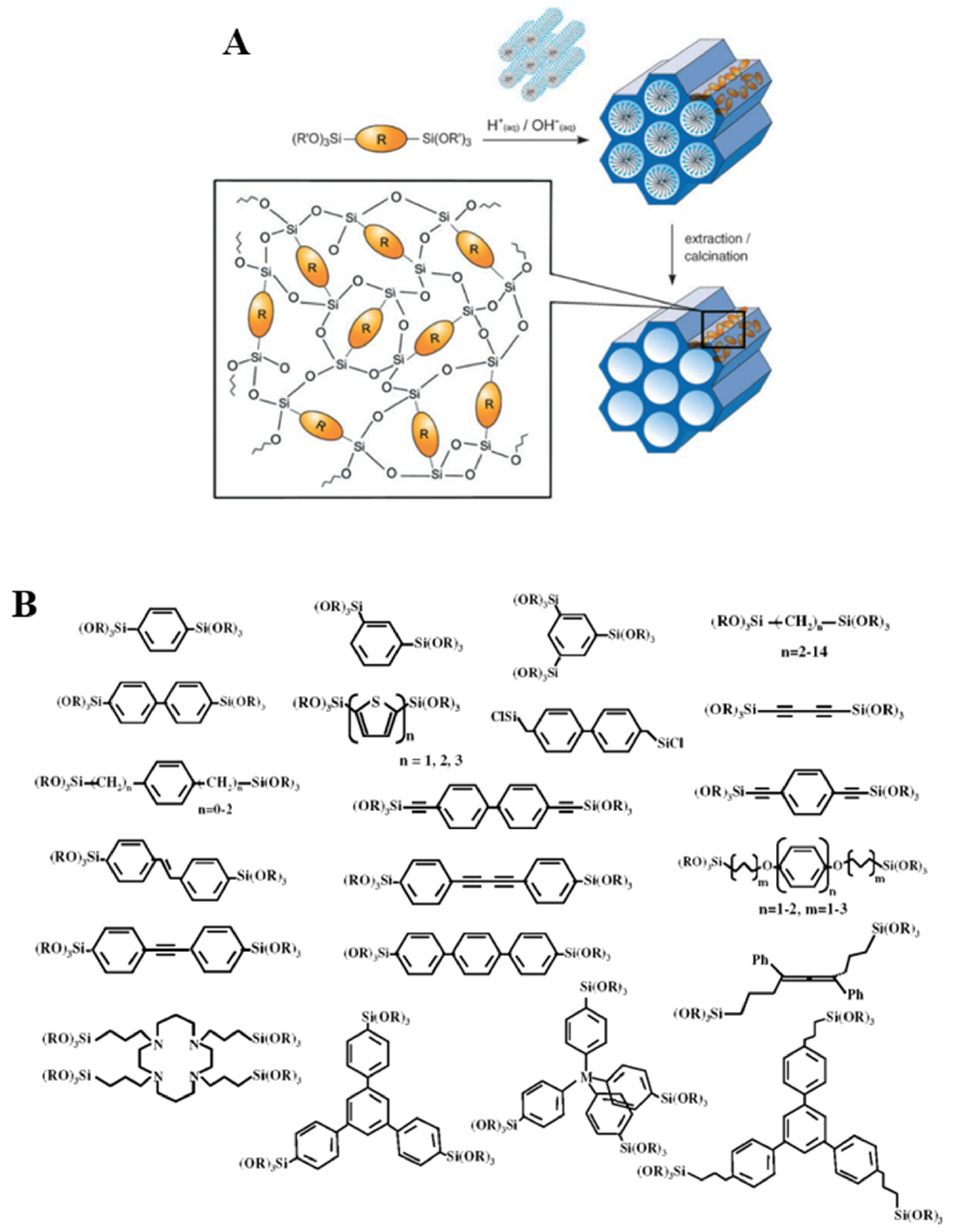{kind=link}
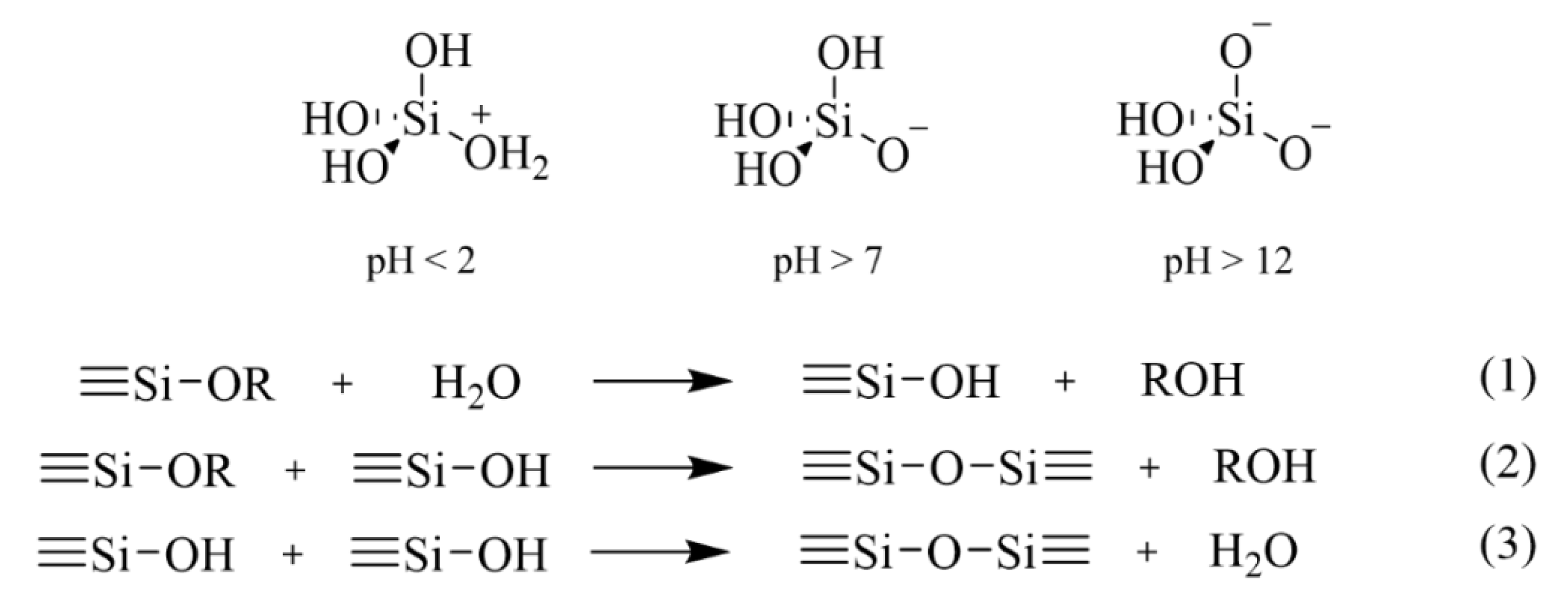{kind=link}
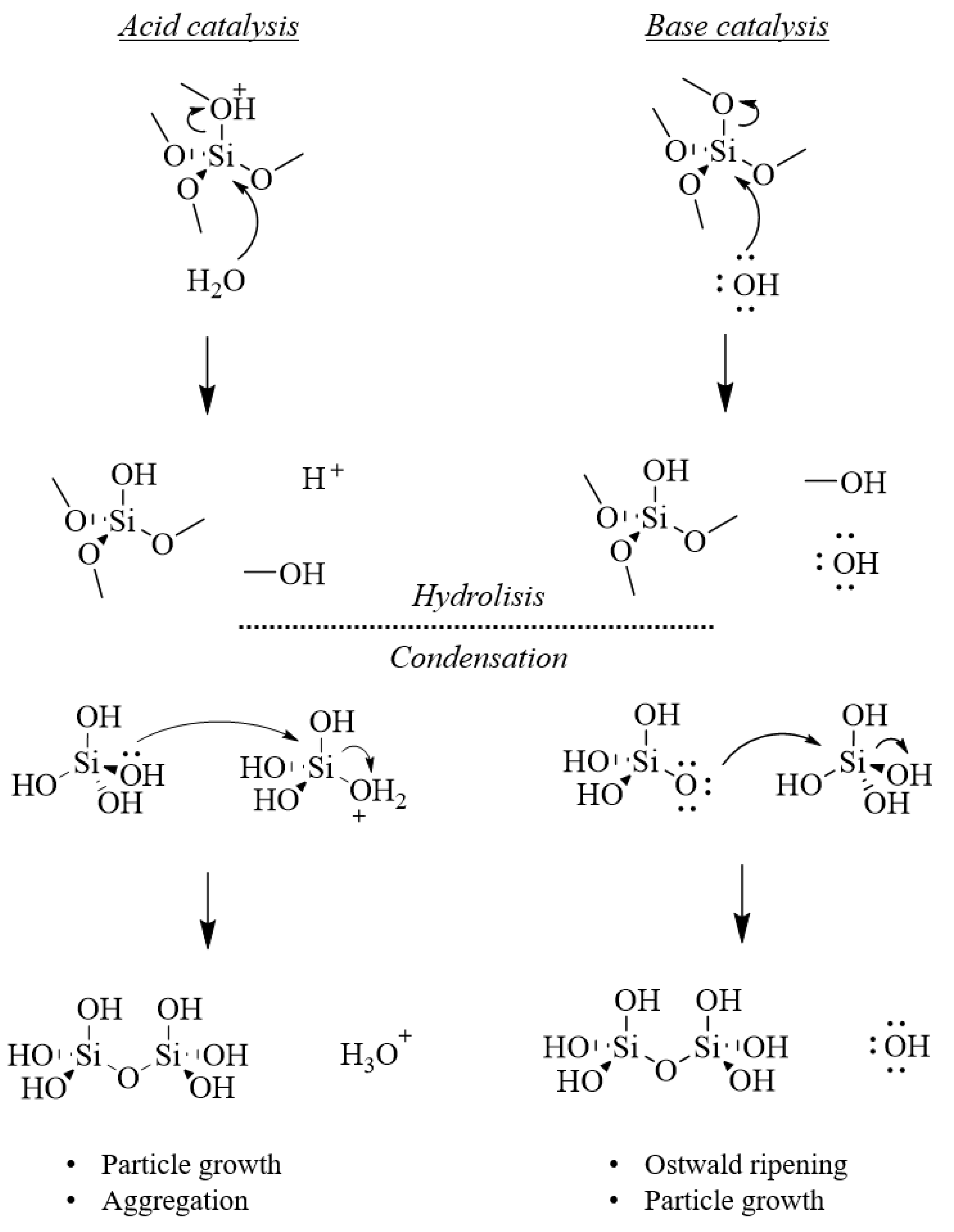{kind=link}
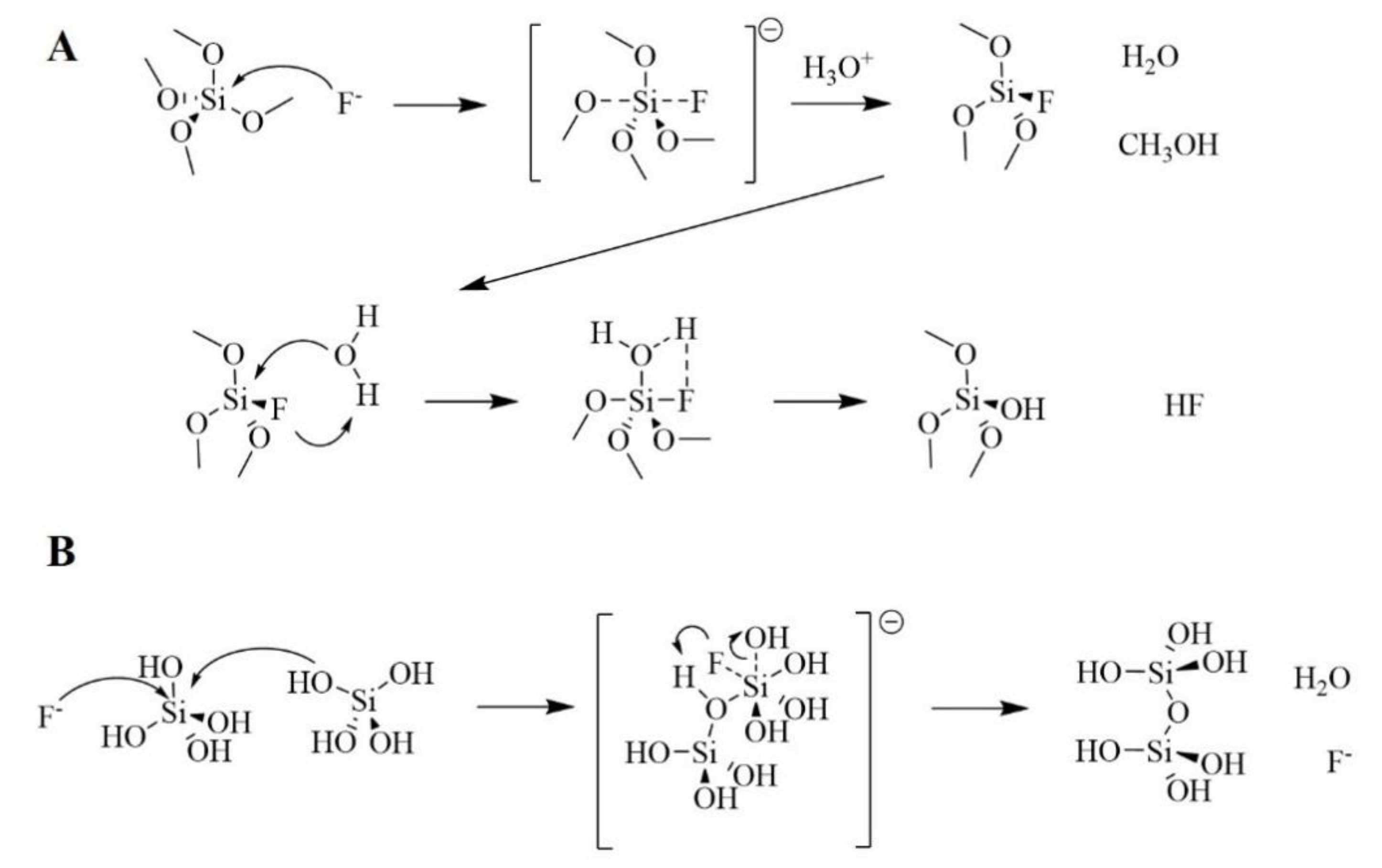{kind=link}
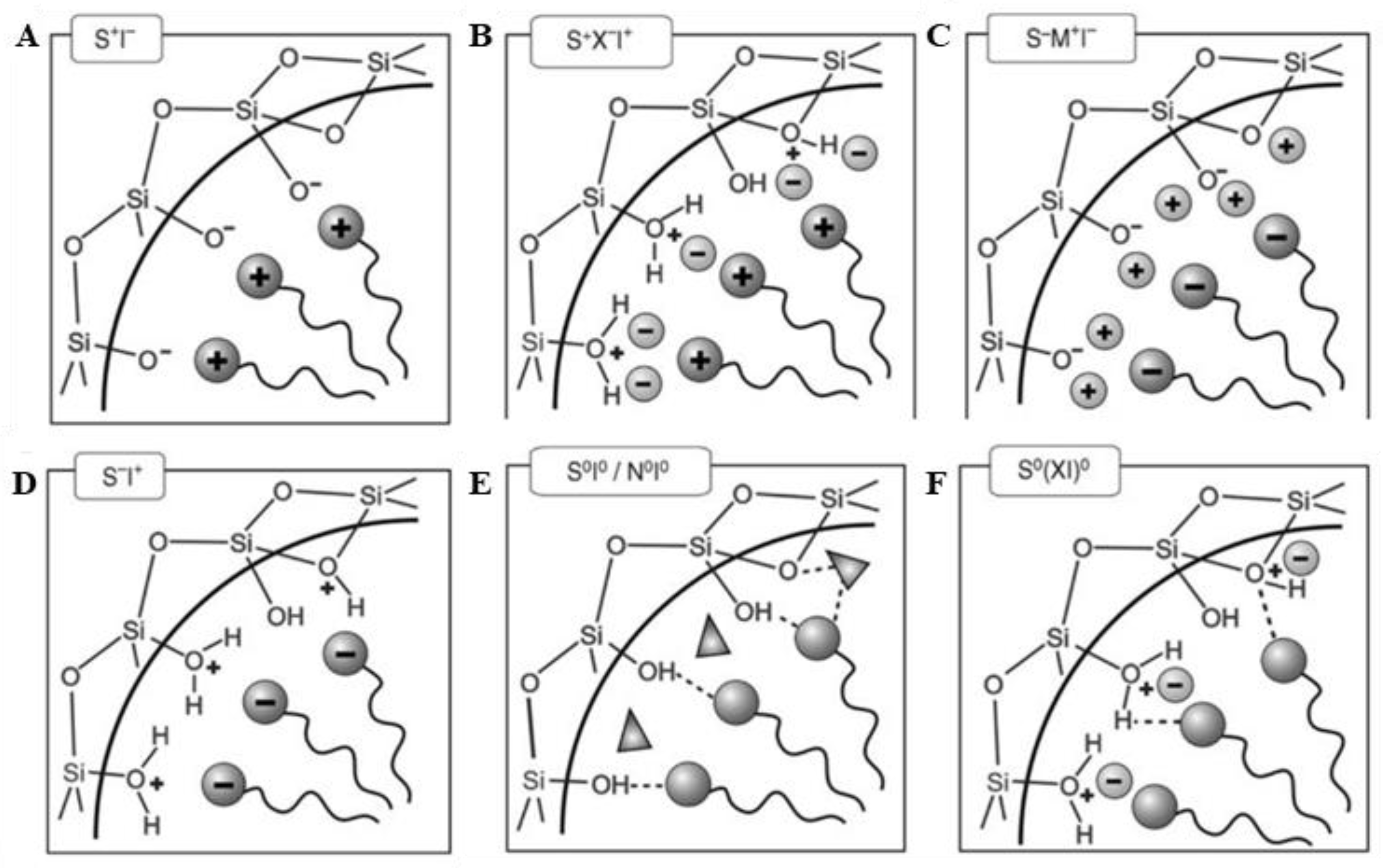{kind=link}
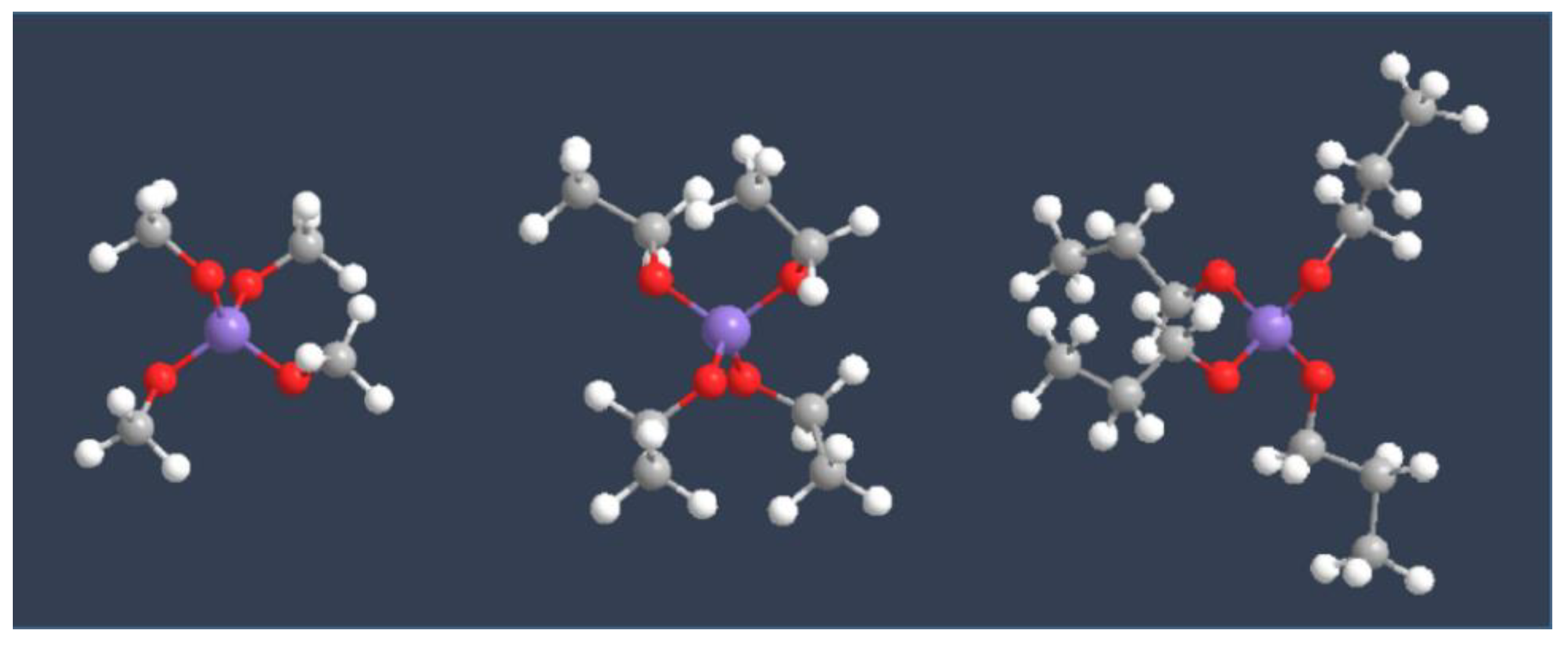{kind=link}
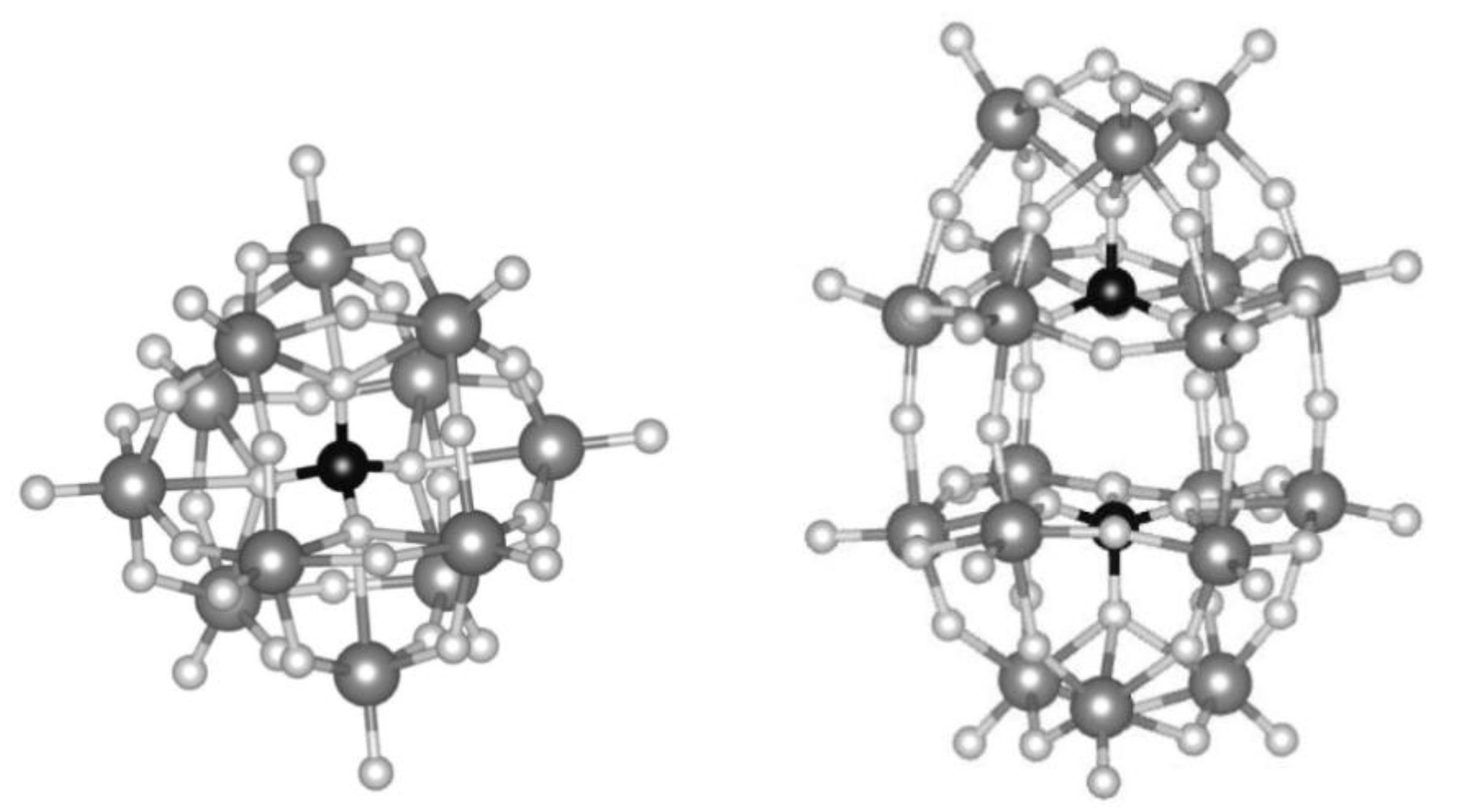{kind=link}
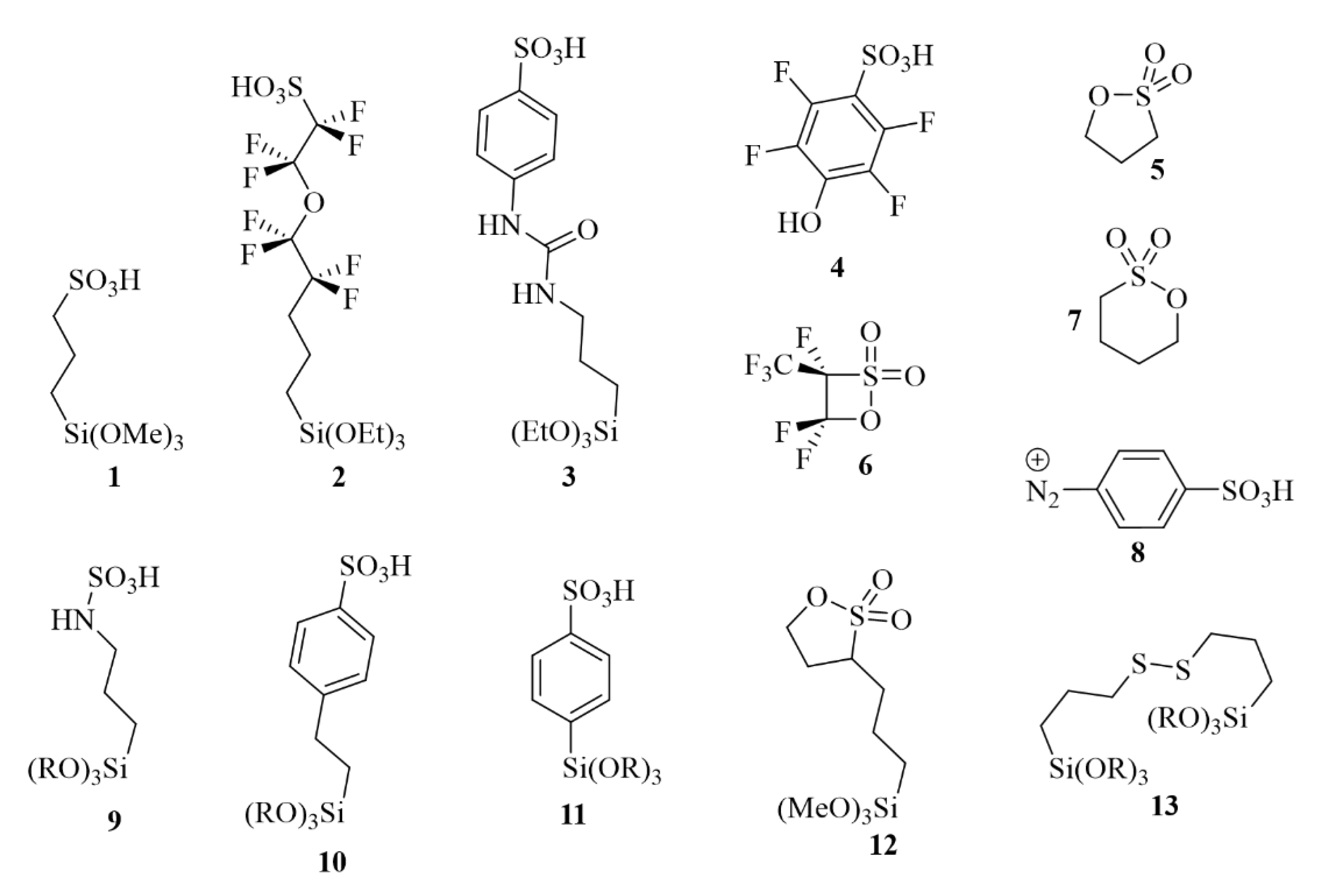{kind=link}
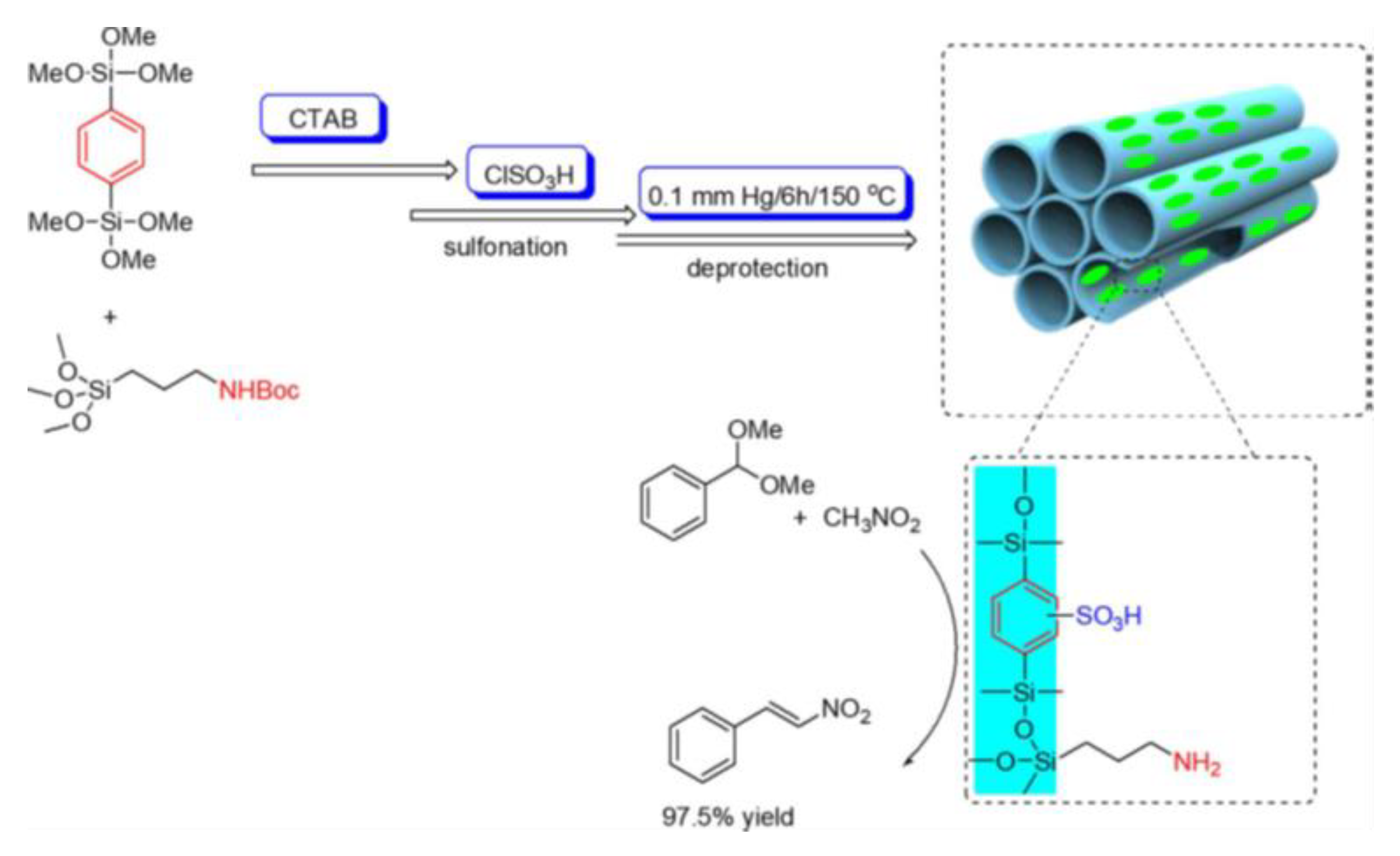{kind=link}
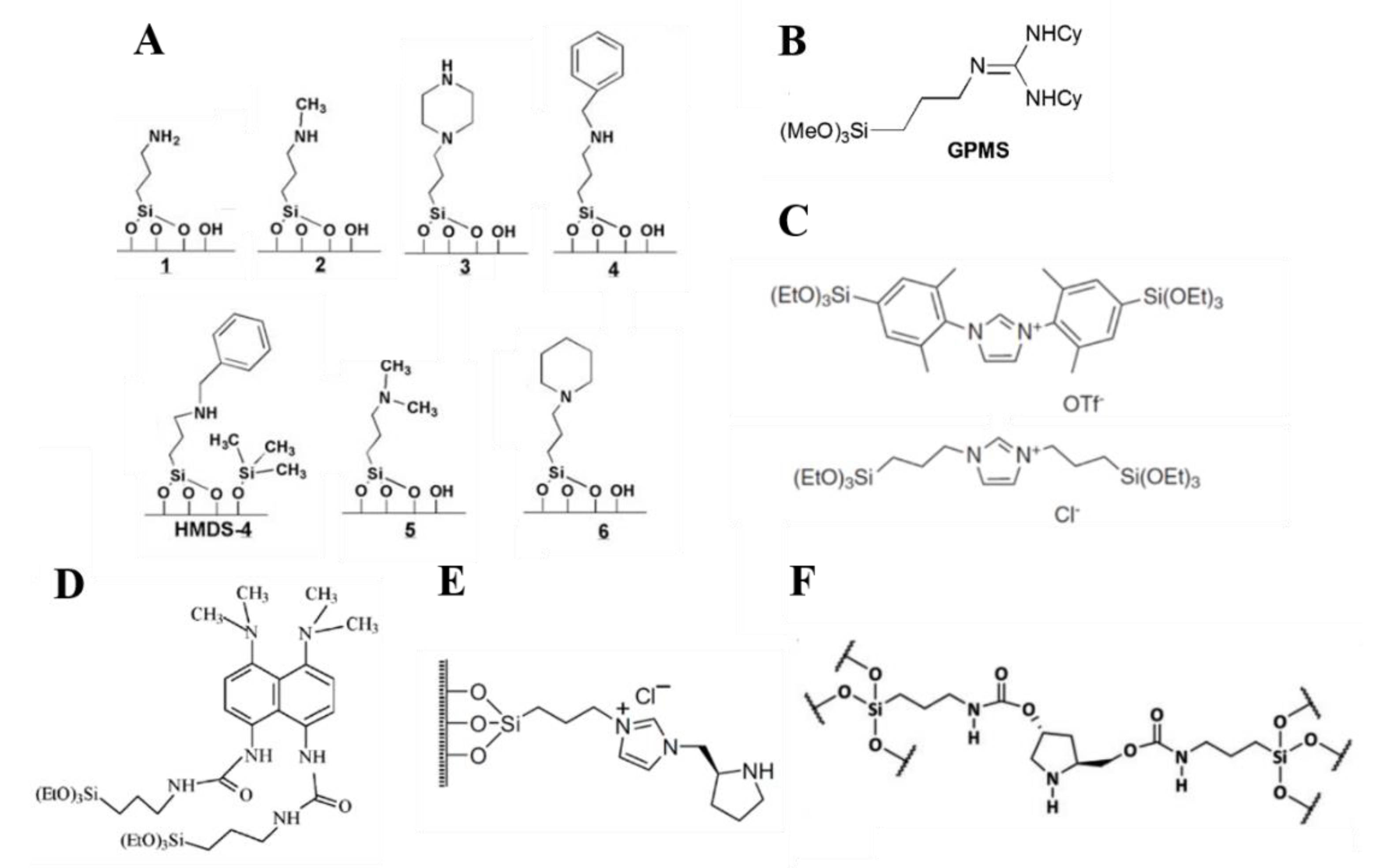{kind=link}
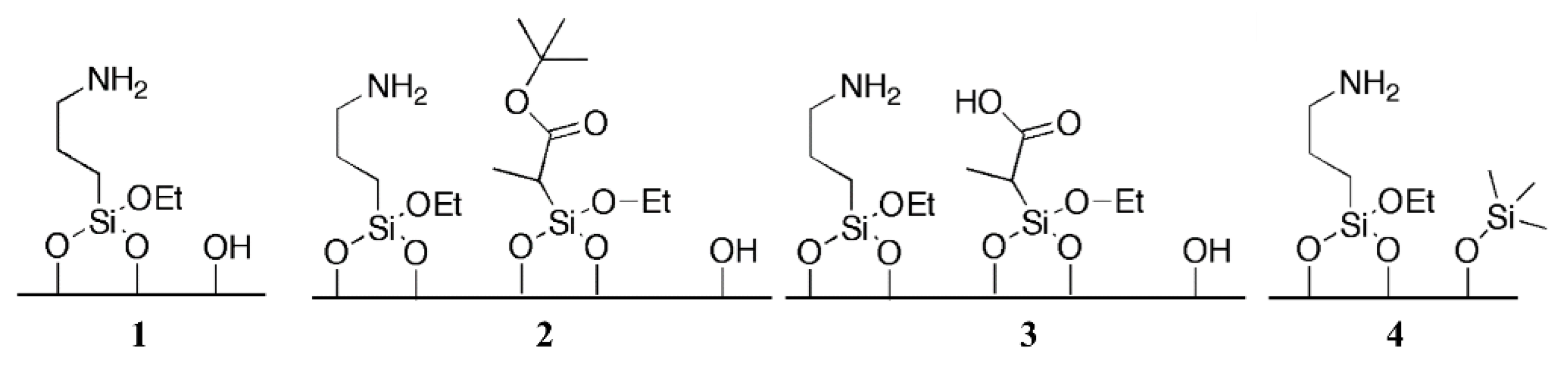{kind=link}
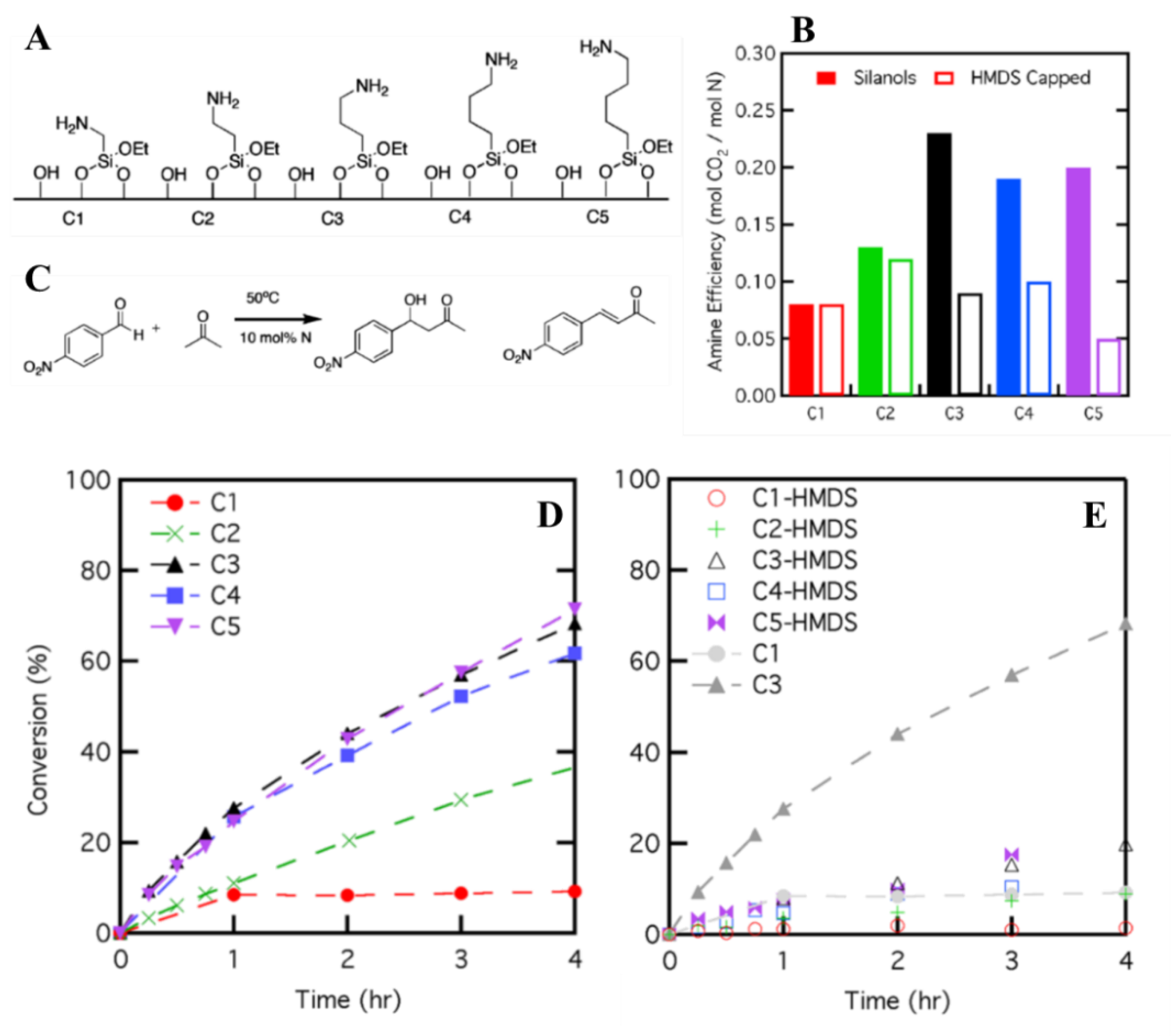{kind=link}
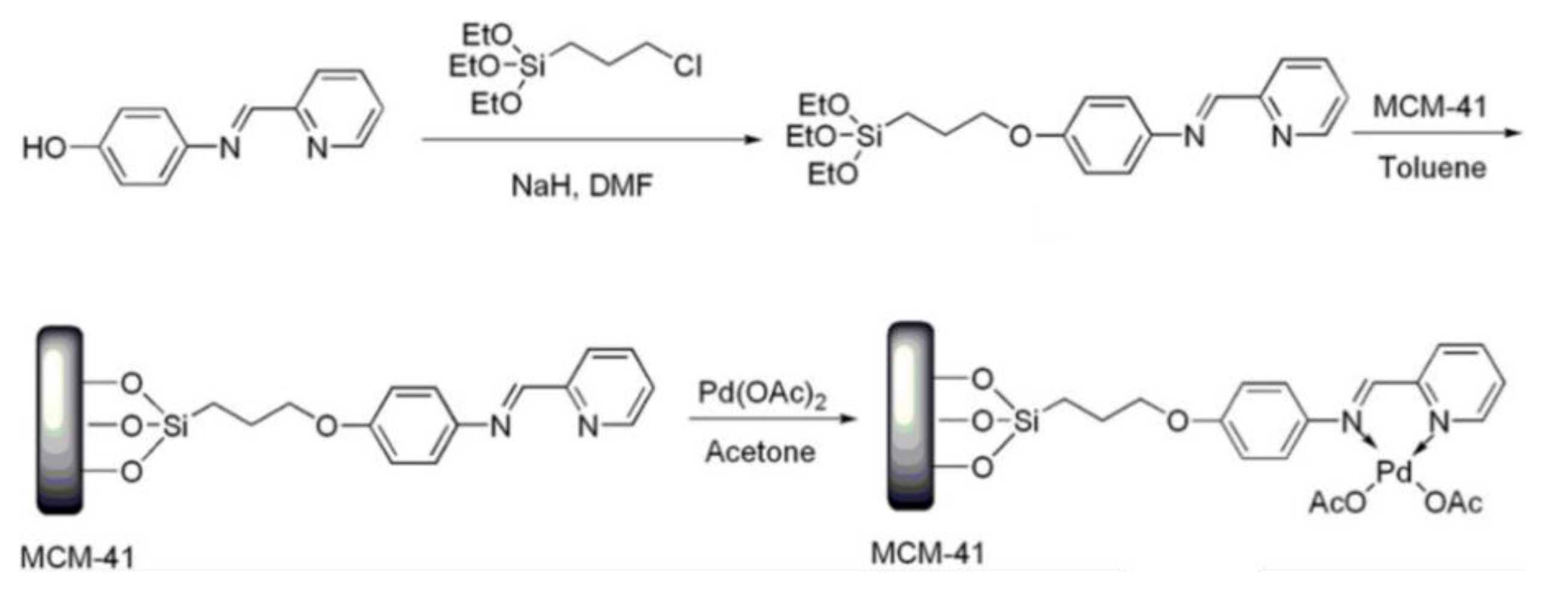{kind=link}
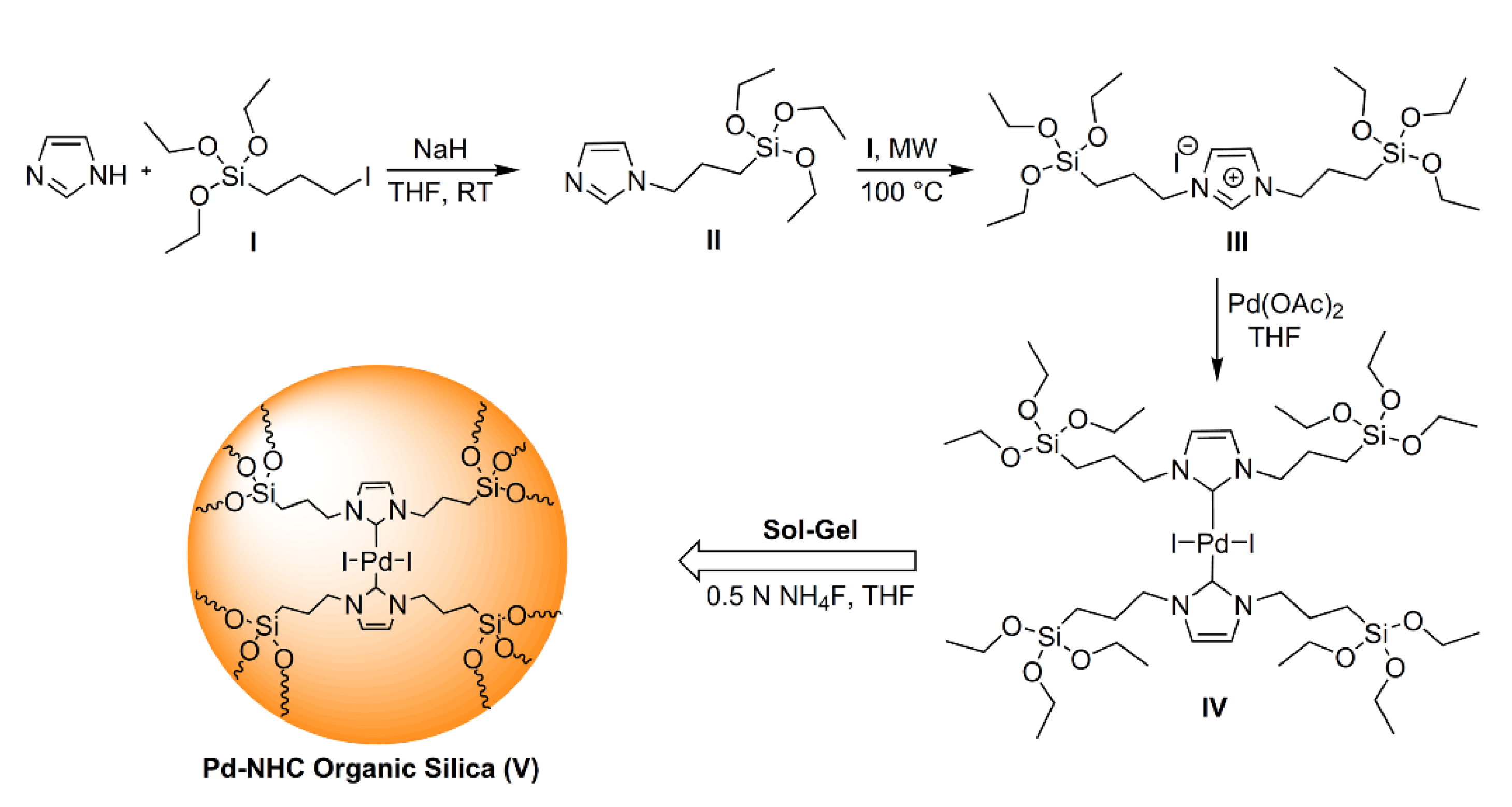{kind=link}
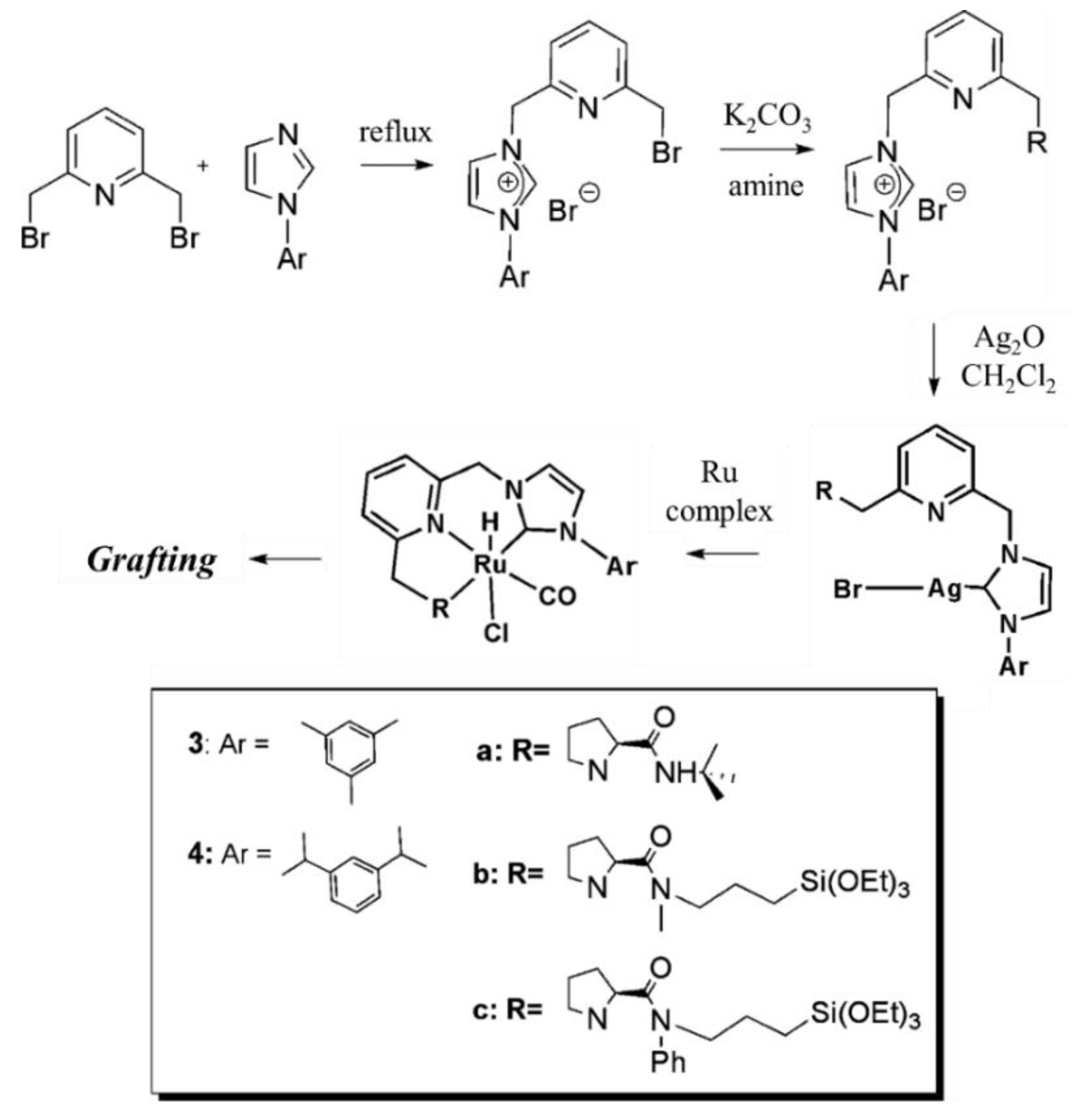{kind=link}
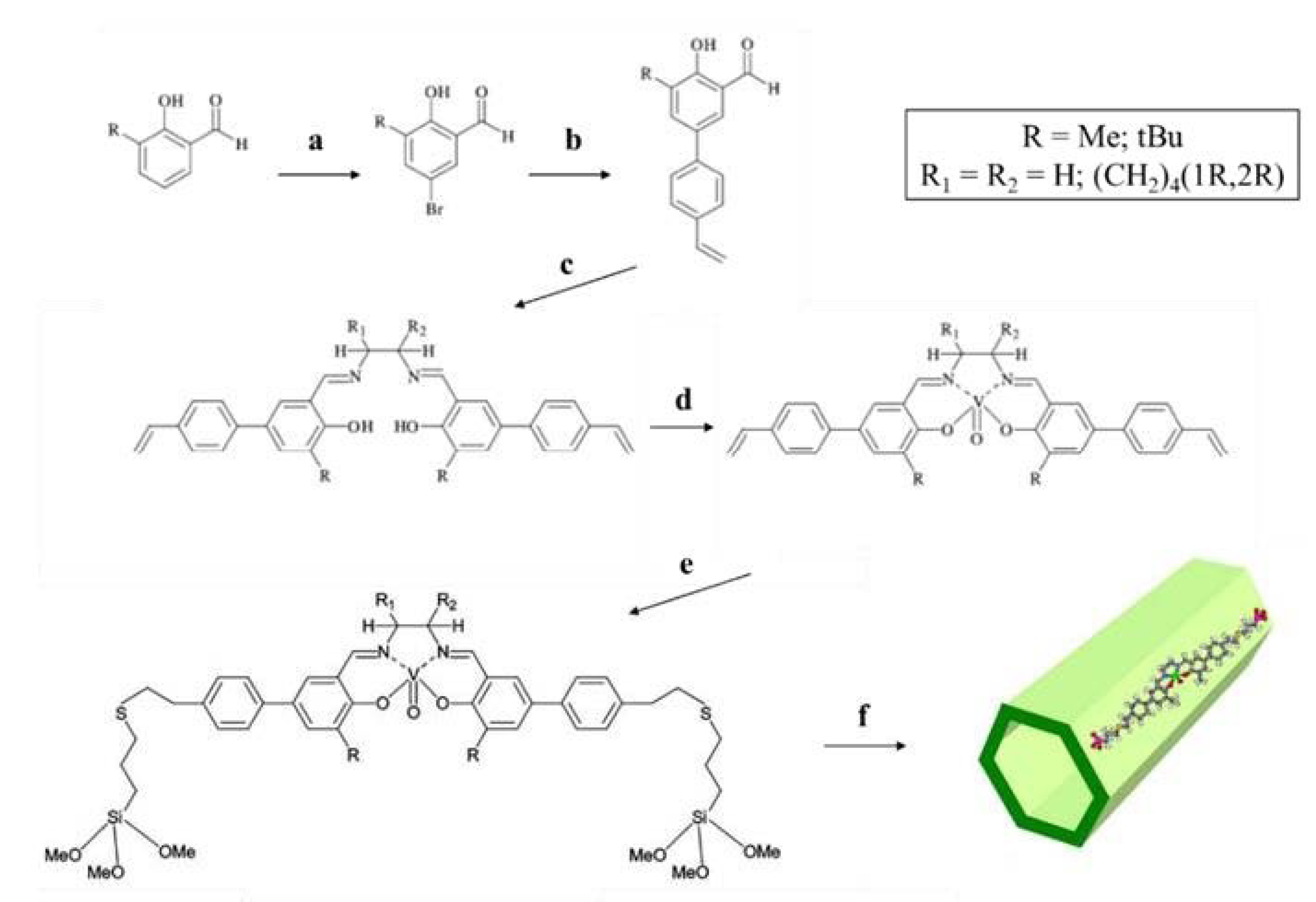{kind=link}
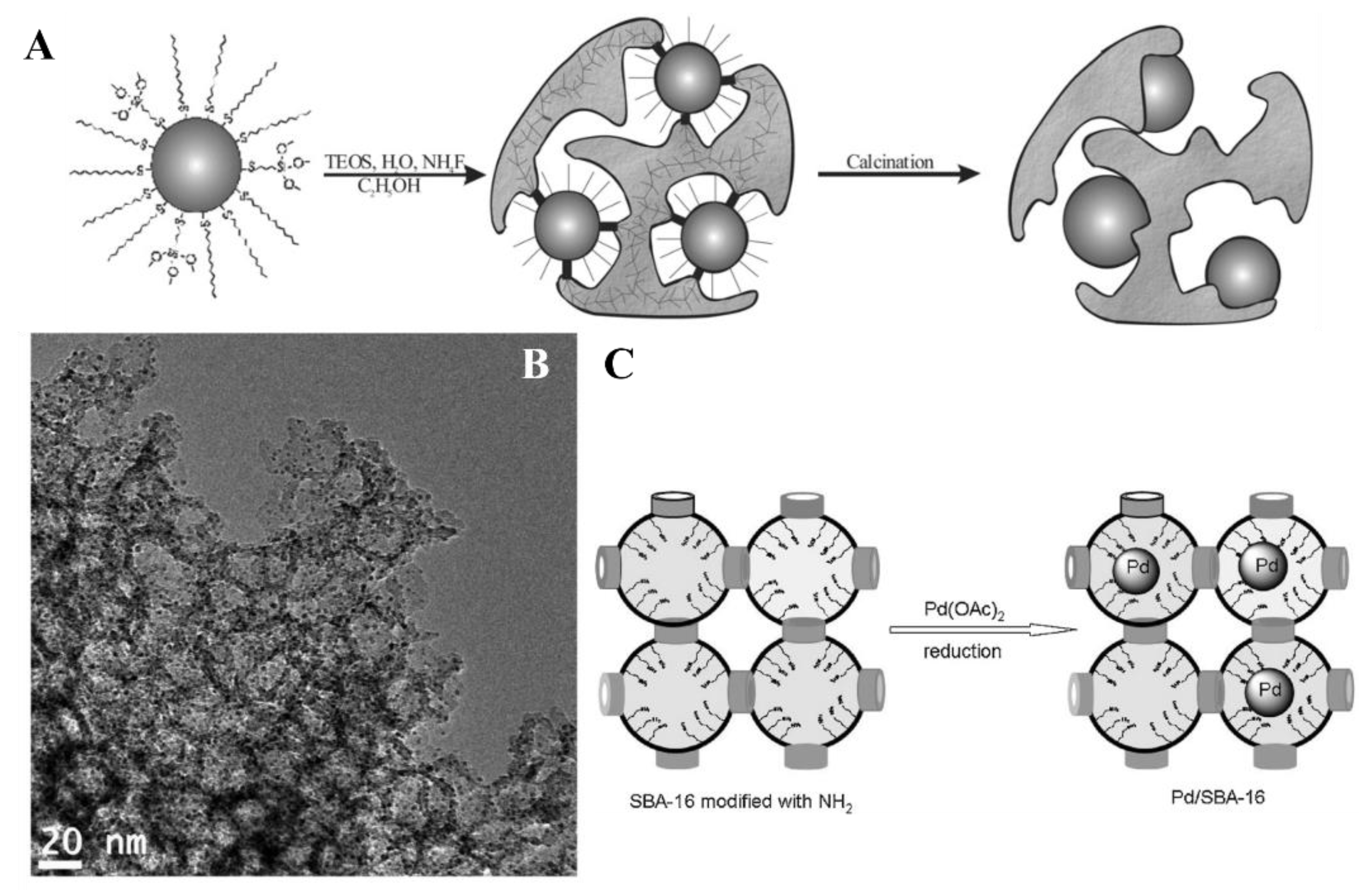{kind=link}
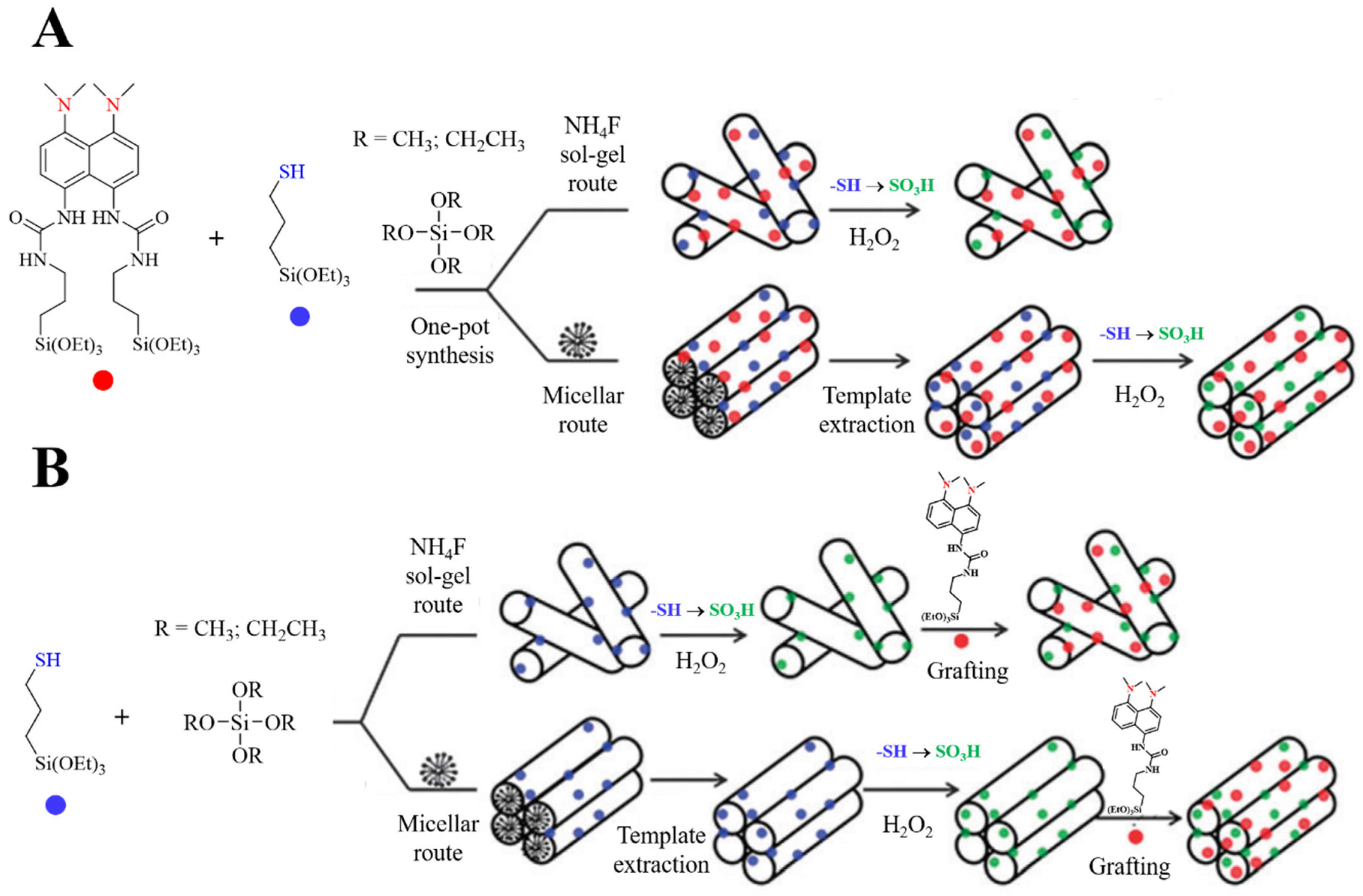{kind=link}
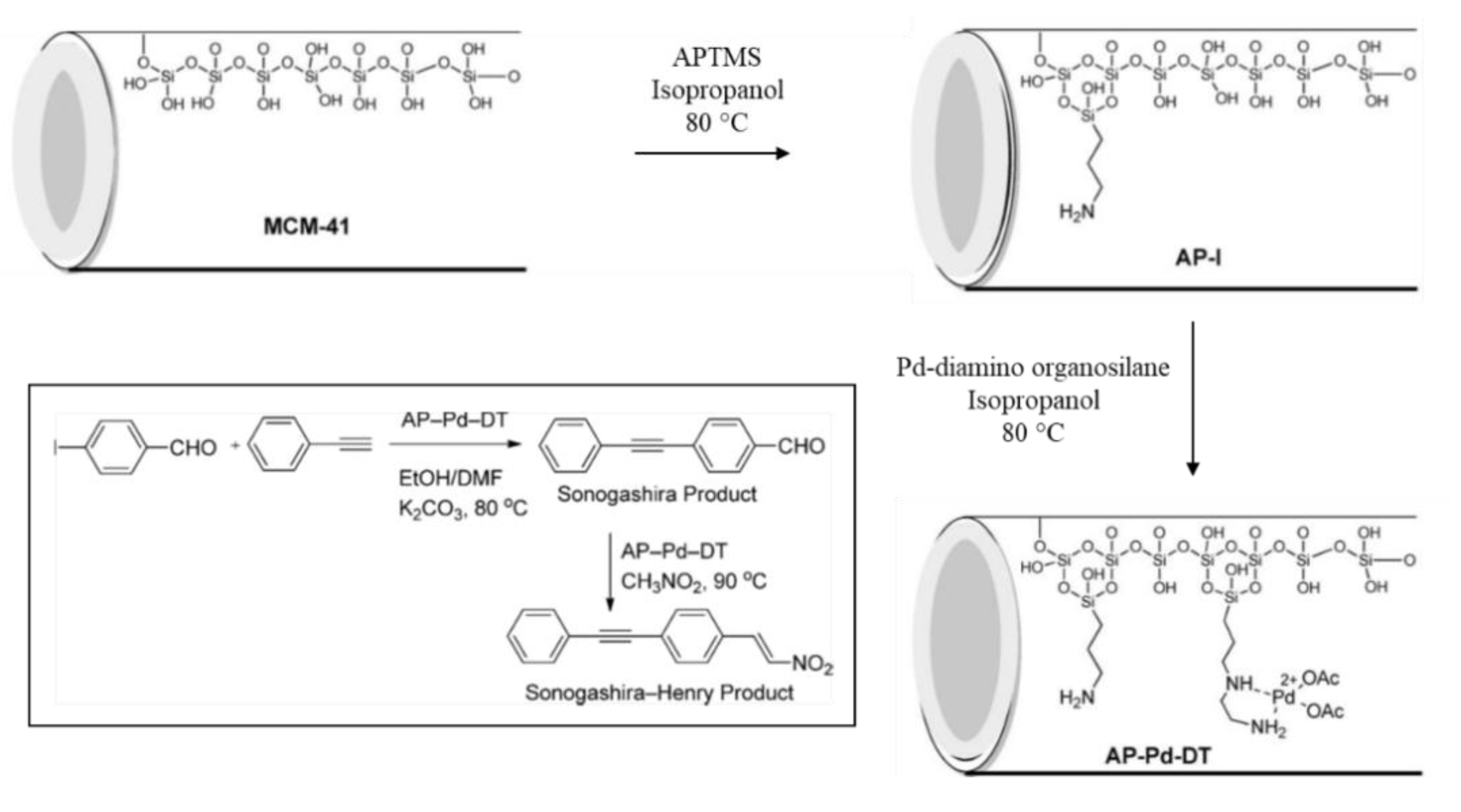{kind=link}
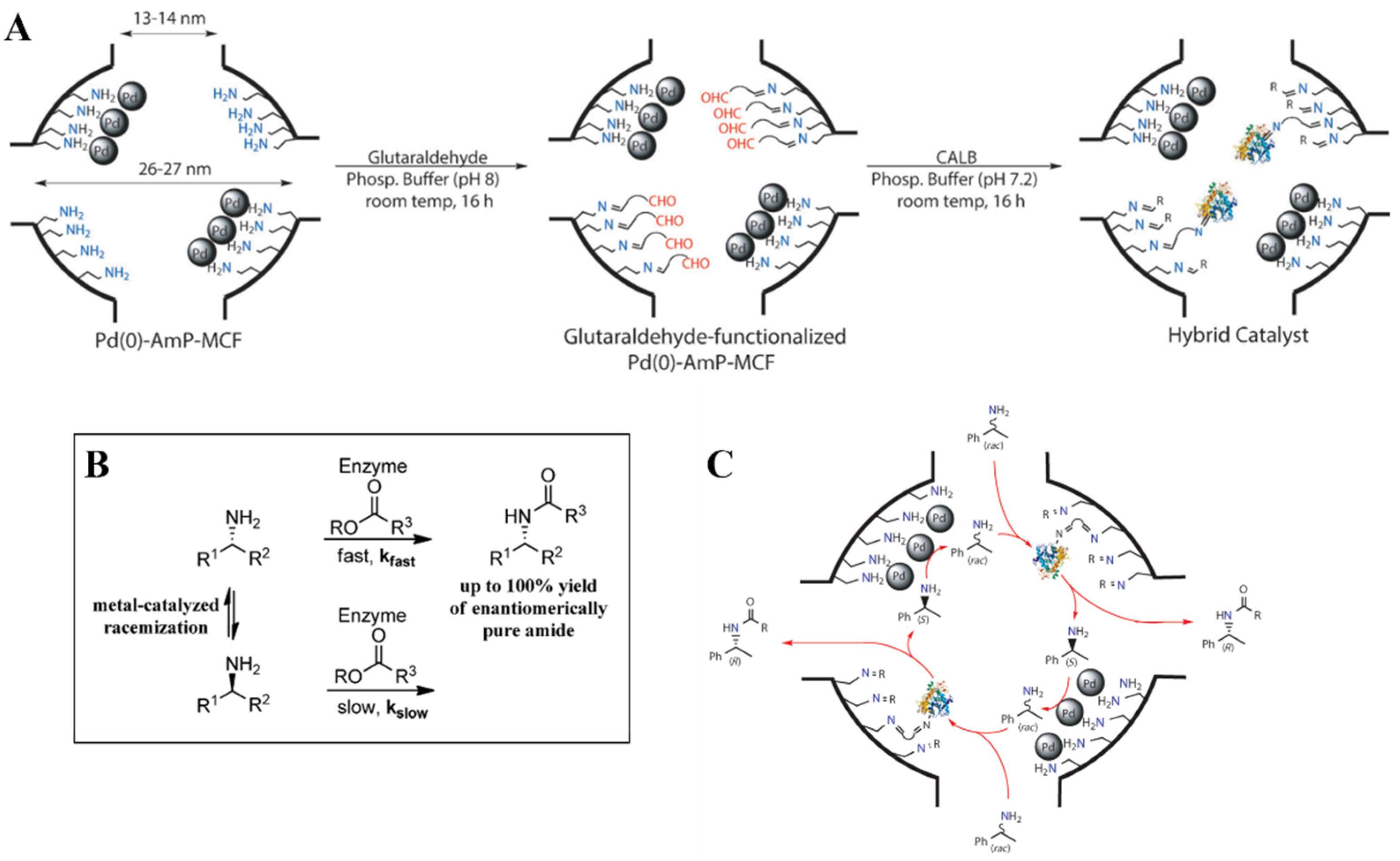{kind=link}
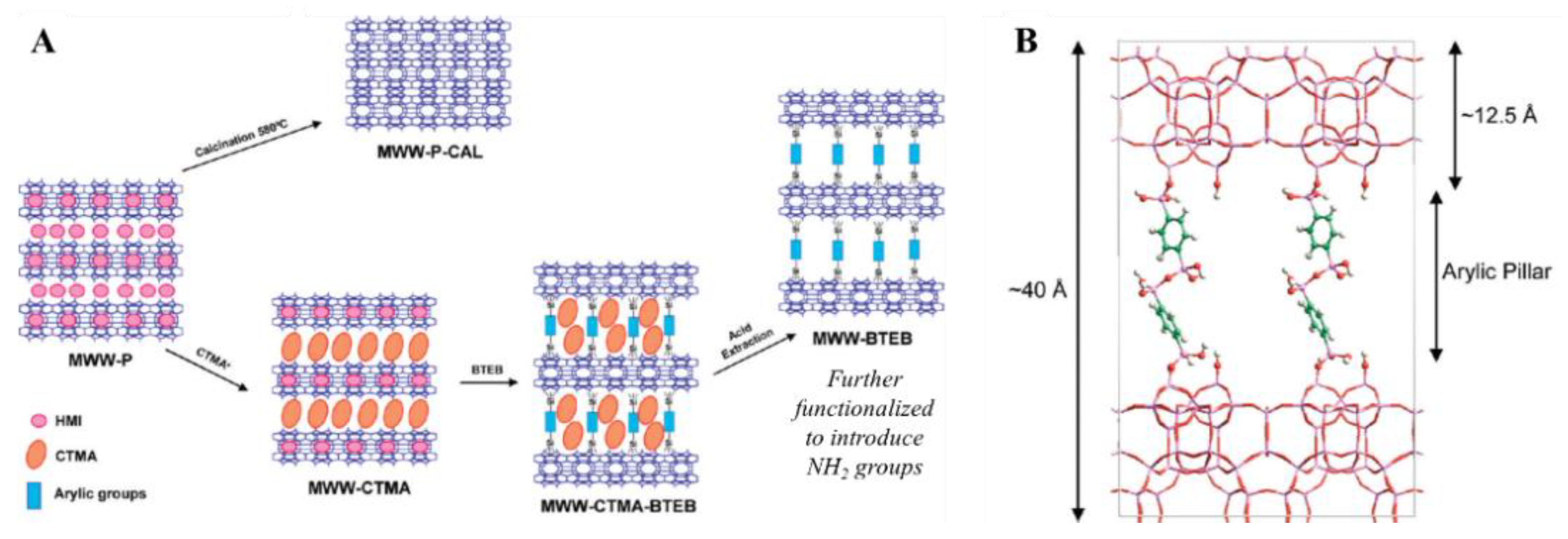{kind=link}
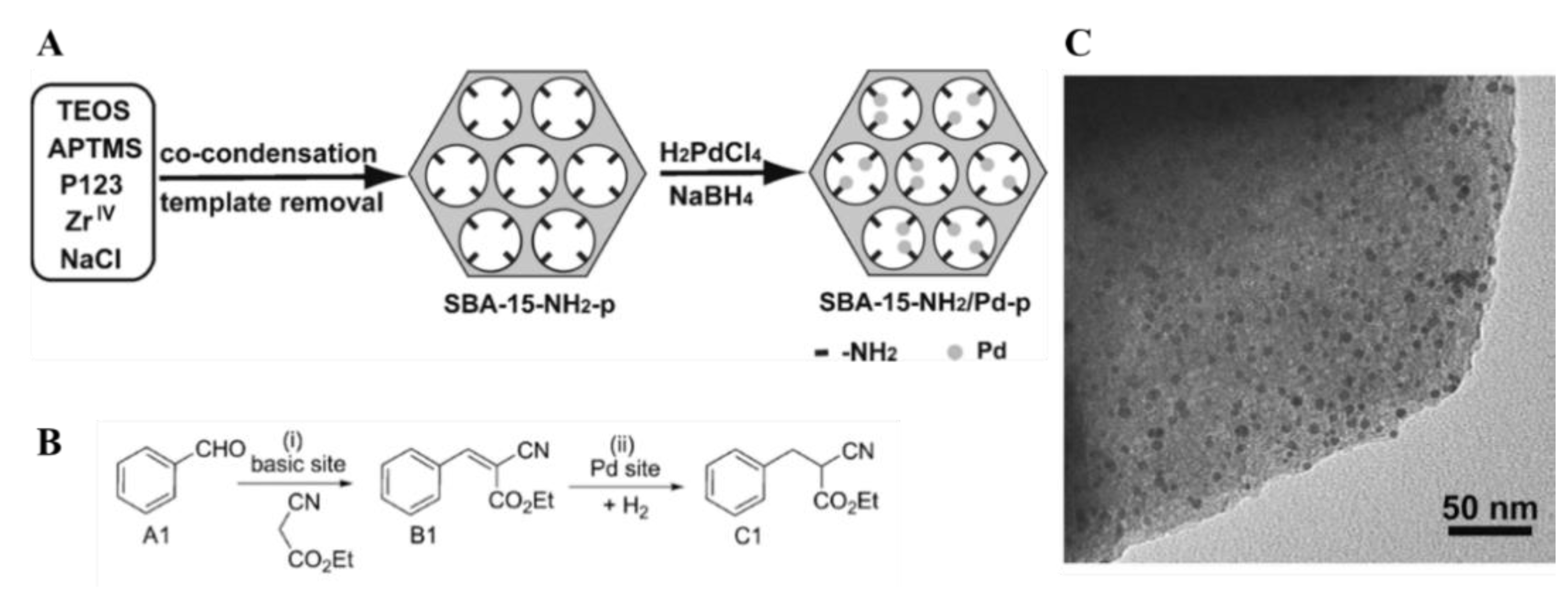{kind=link}
Table 1.
Families of hybrid catalysts described in this review article.
| Catalyst | Active Site |
|---|---|
| Heteropolyacids (HPAs) | Acid |
| Sulfonic acids | Acid |
| Amines | Base |
| N-cycles | Base |
| Organometallic complexes | Redox |
| Metal nanoparticles | Redox |
| Multi-functional catalysts | Multiple |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Erigoni, A.; Diaz, U. Porous Silica-Based Organic-Inorganic Hybrid Catalysts: A Review. Catalysts 2021, 11, 79. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11010079
AMA Style
Erigoni A, Diaz U. Porous Silica-Based Organic-Inorganic Hybrid Catalysts: A Review. Catalysts. 2021; 11(1):79. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11010079
Chicago/Turabian StyleErigoni, Andrea, and Urbano Diaz. 2021. "Porous Silica-Based Organic-Inorganic Hybrid Catalysts: A Review" Catalysts 11, no. 1: 79. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11010079
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.