
Synergistic Effect of Neighboring Fe and Cu Cation Sites Boosts FenCum-BEA Activity for the Continuous Direct Oxidation of Methane to Methanol

 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Result and Discussion
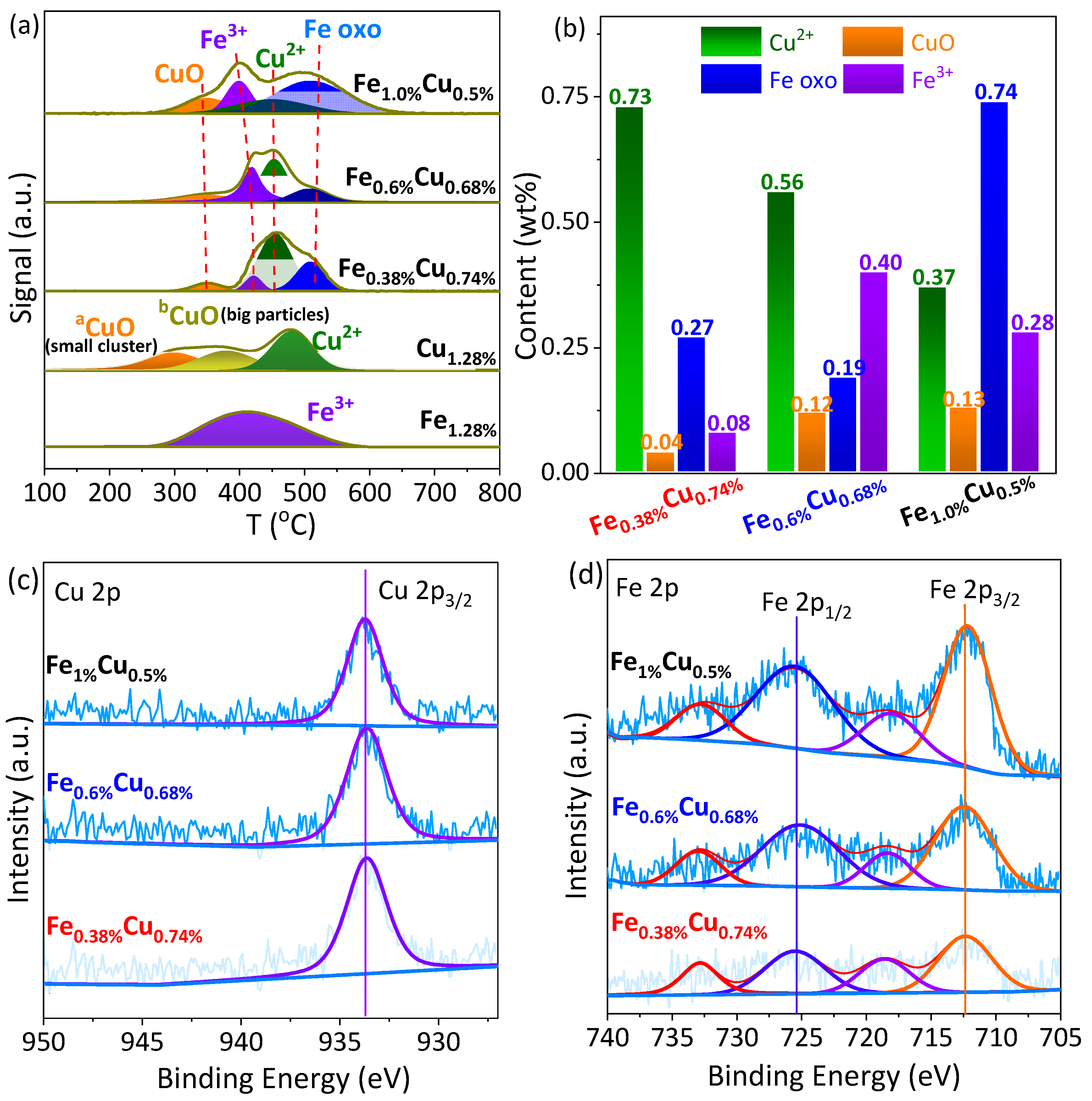
2.1. Physicochemical Property Characterizations
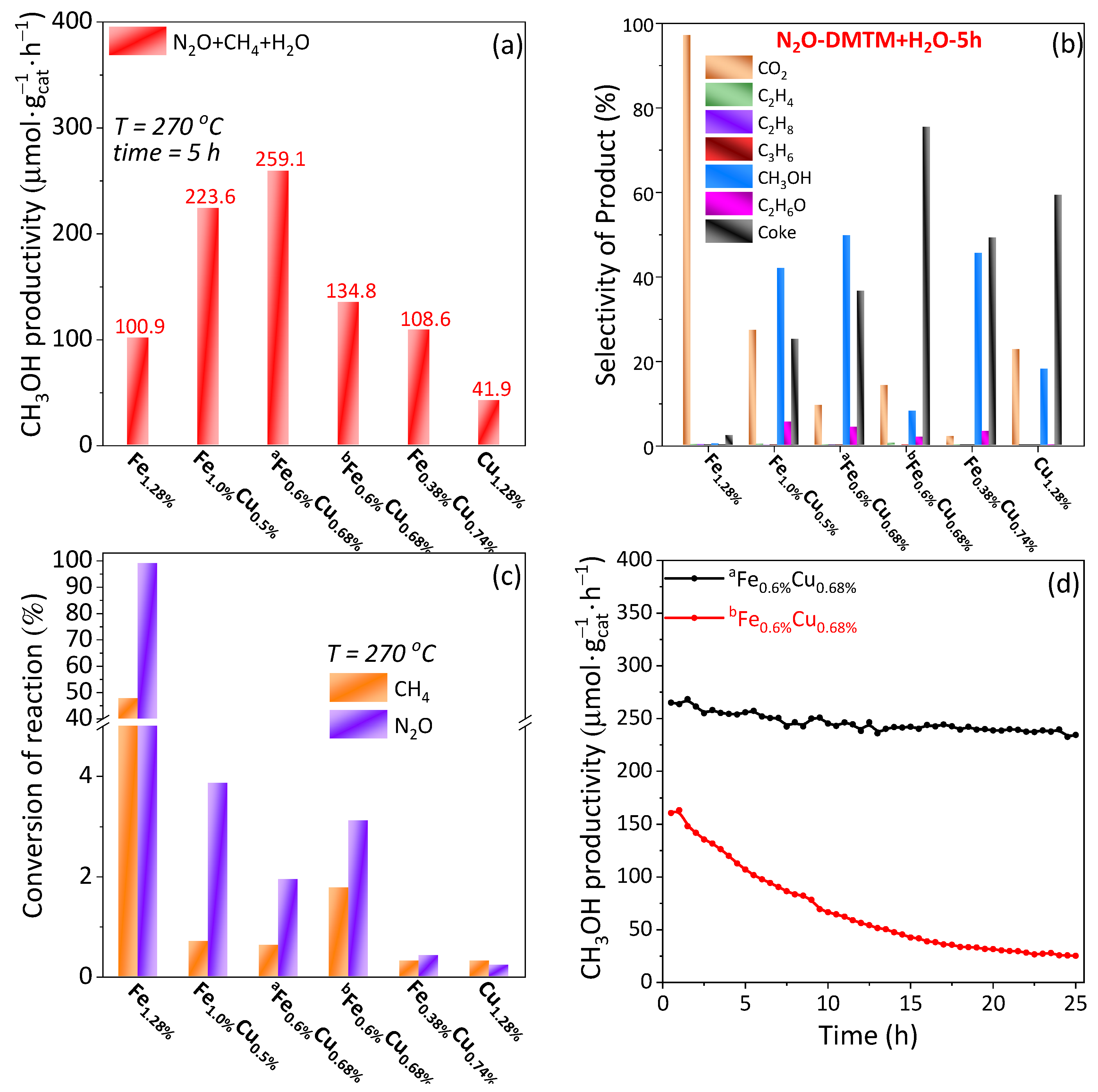
2.2. Activity Measurement
2.2.1. Activity Comparison with Monomeric Fe- and Cu-BEA
2.2.2. Activity Comparison among FenCum-BEA
2.3. Evolution of the Bimetal Active Center of [Fe-O-Cu] and Thermodynamics Stability Analysis
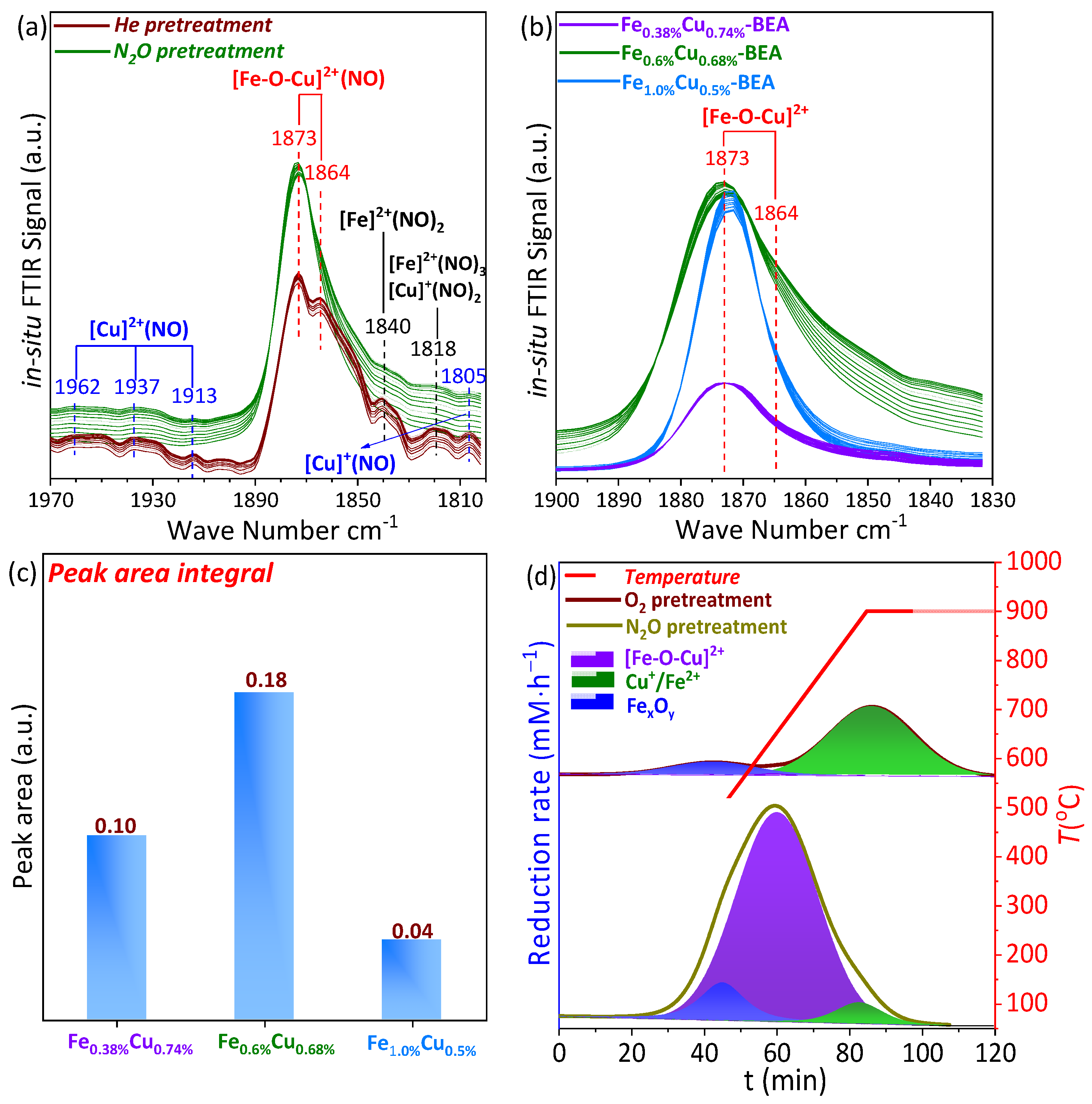
2.3.1. NO In Situ FTIR
2.3.2. H2-TPR after N2O and O2 Pretreatment
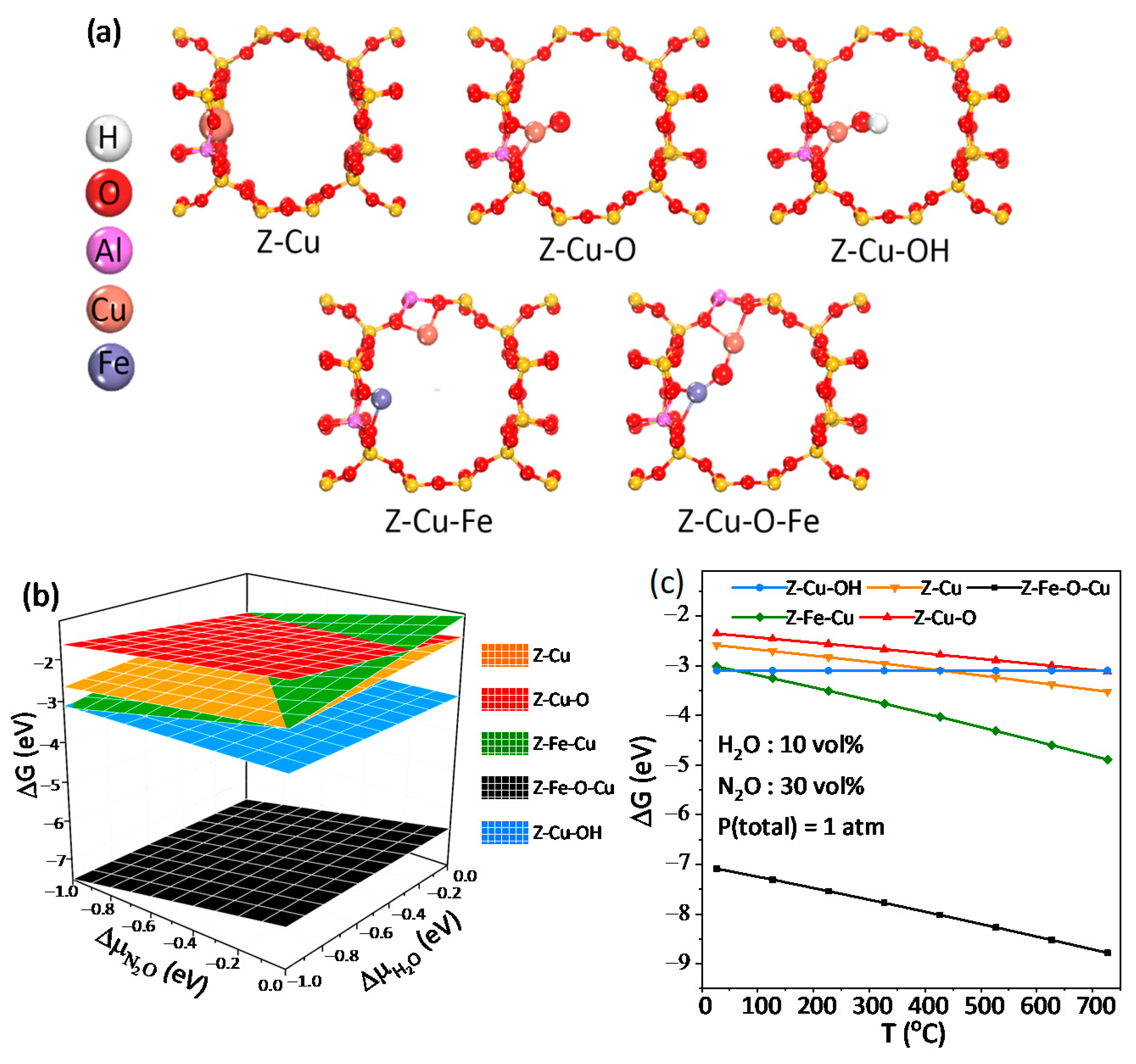
2.3.3. Ab Initio Thermodynamics (AIT) Analysis
2.4. Mechanistic Insight into Synergetic Effect of Neighboring Fe and Cu Cations during H2O-Mediated N2O DMTM
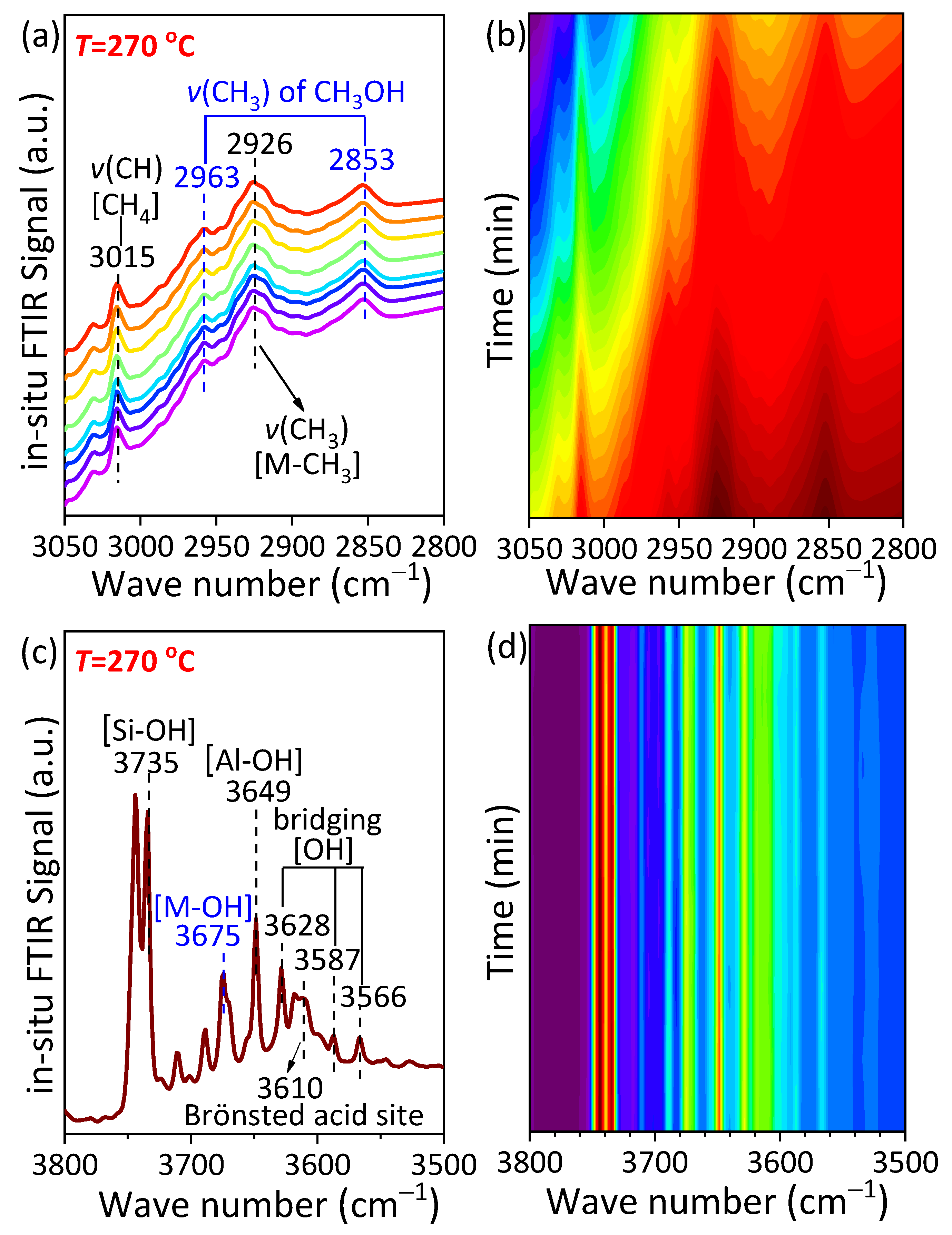
2.4.1. Experimental Mechanism Study
- (a)
- In situ FTIR
- (b)
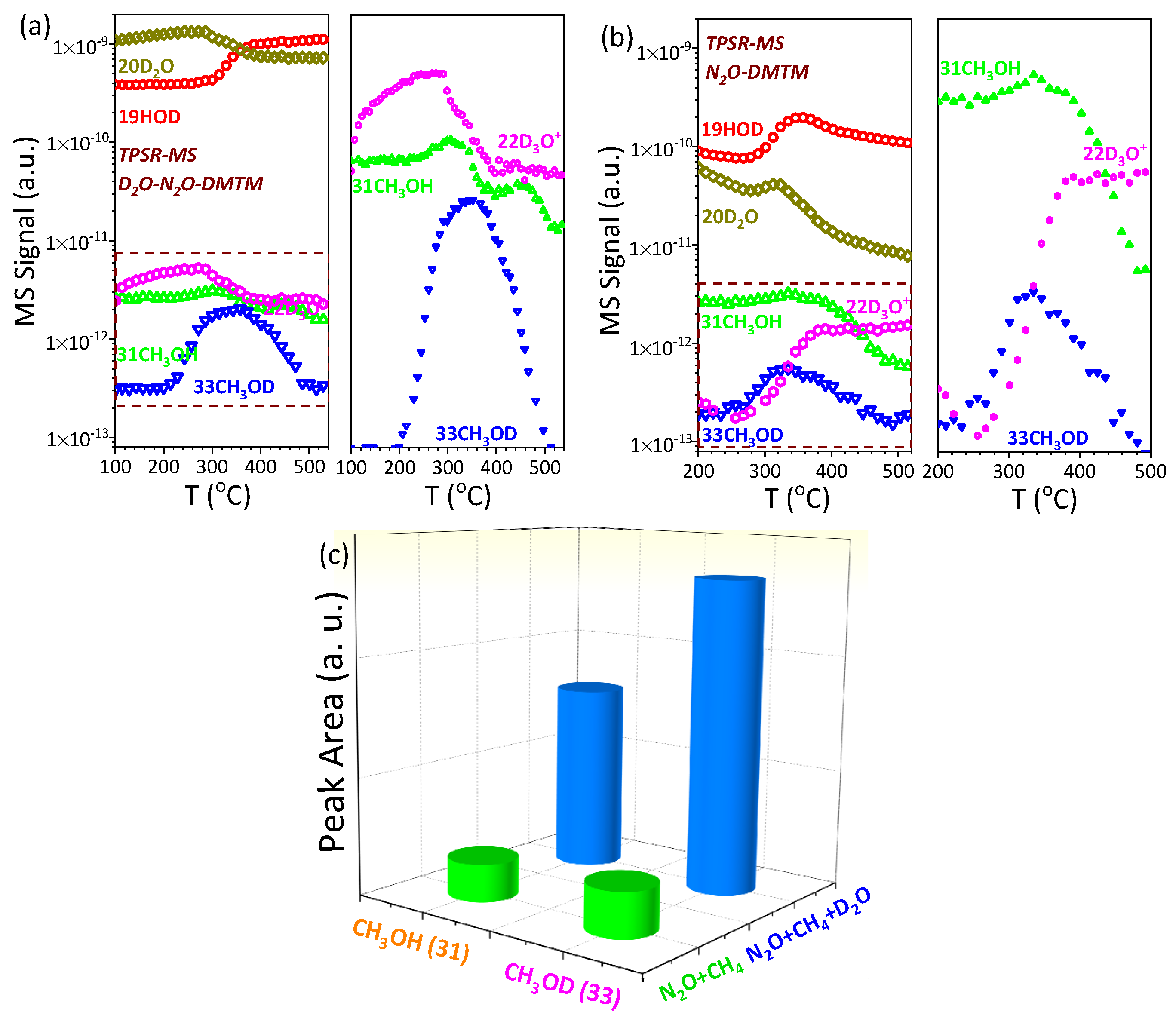
- D2O isotopic tracer experiment
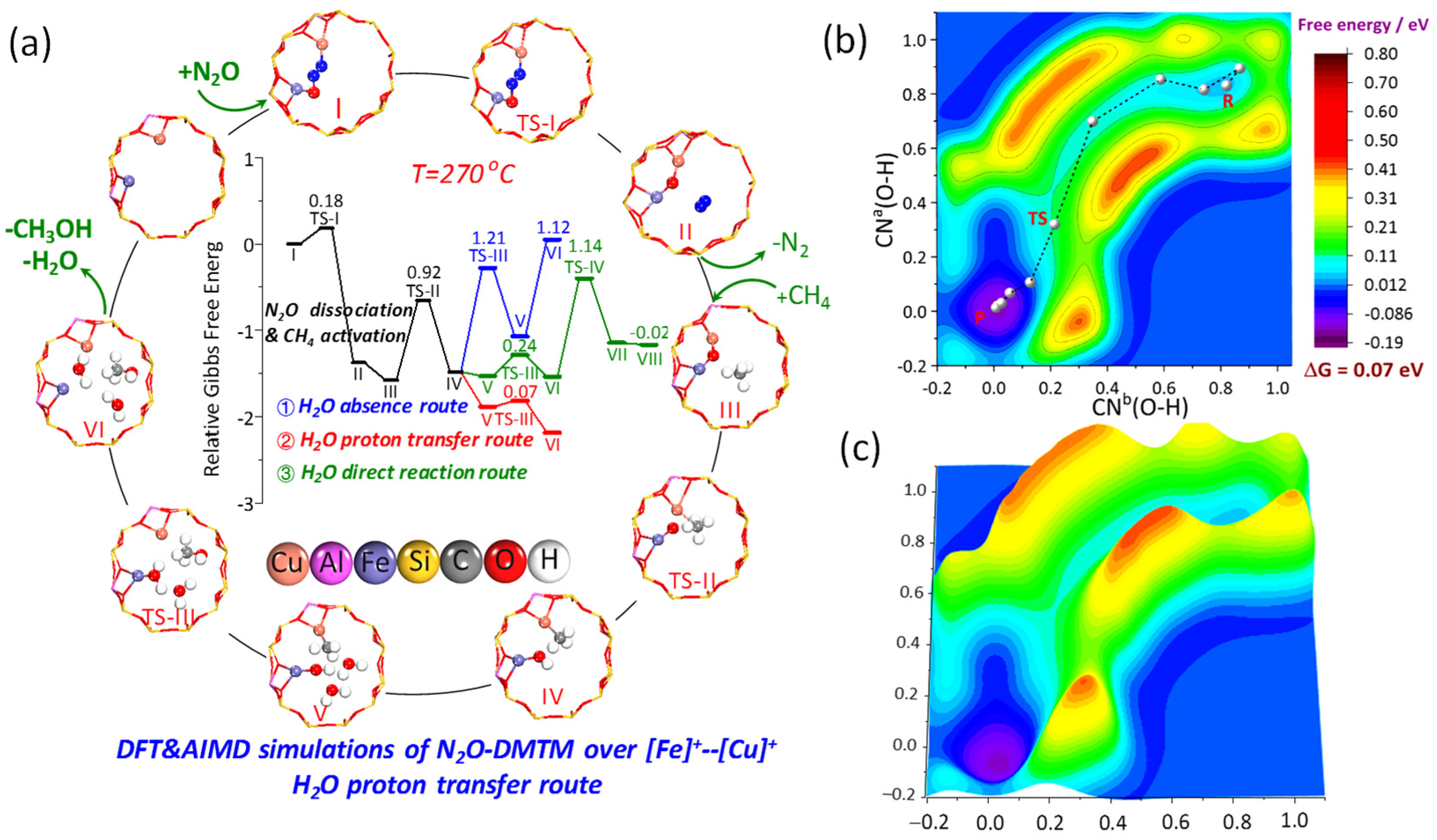
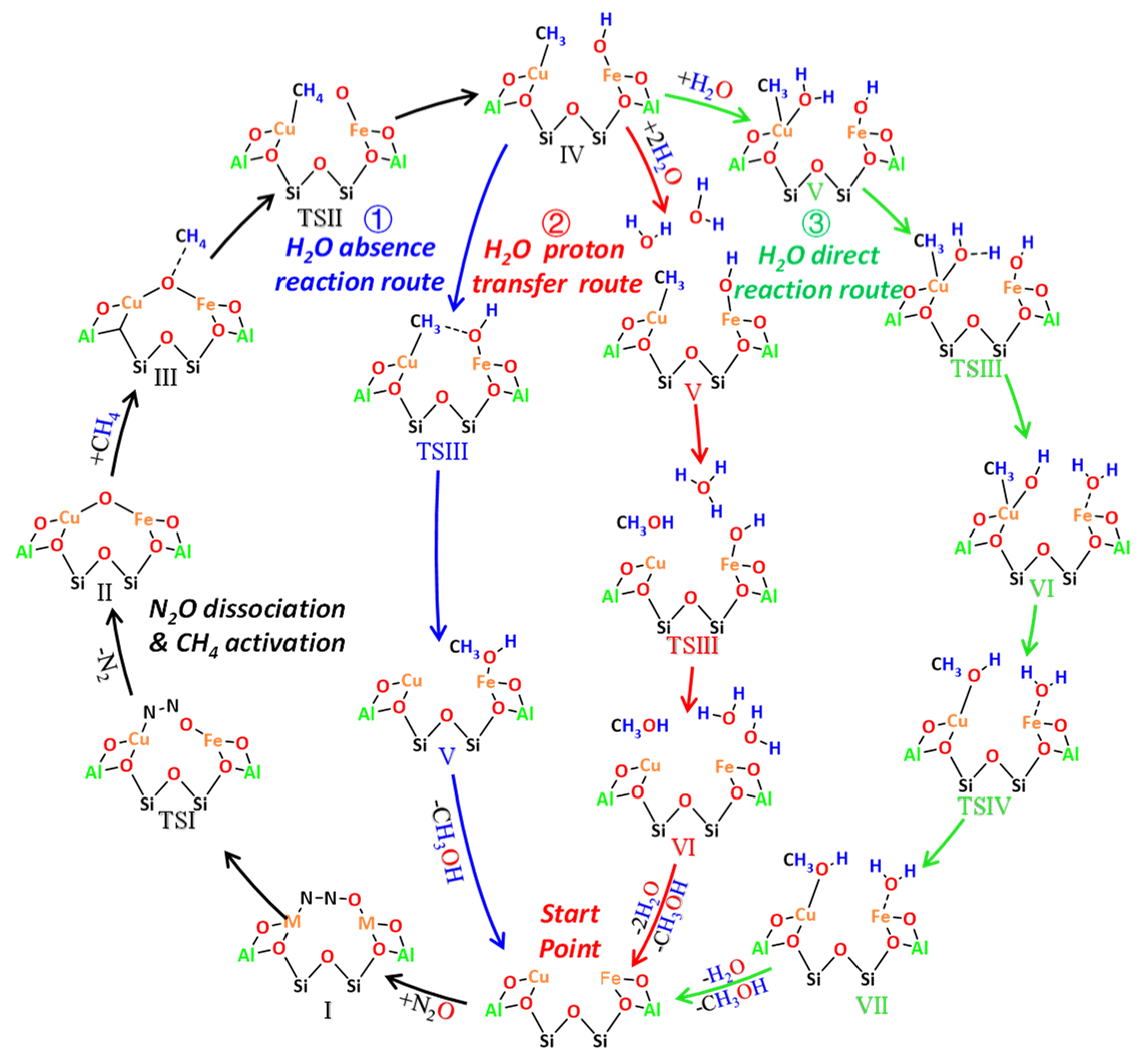
2.4.2. Theoretical Mechanism Simulations by DFT and AIMD
- (a)
- N2O dissociation and CH4 activation steps
- (b)
- Further reaction of [Fe-OH]--[Cu-CH3] in the absence and presence of H2O
- (c)
- Reaction mechanism comparisons
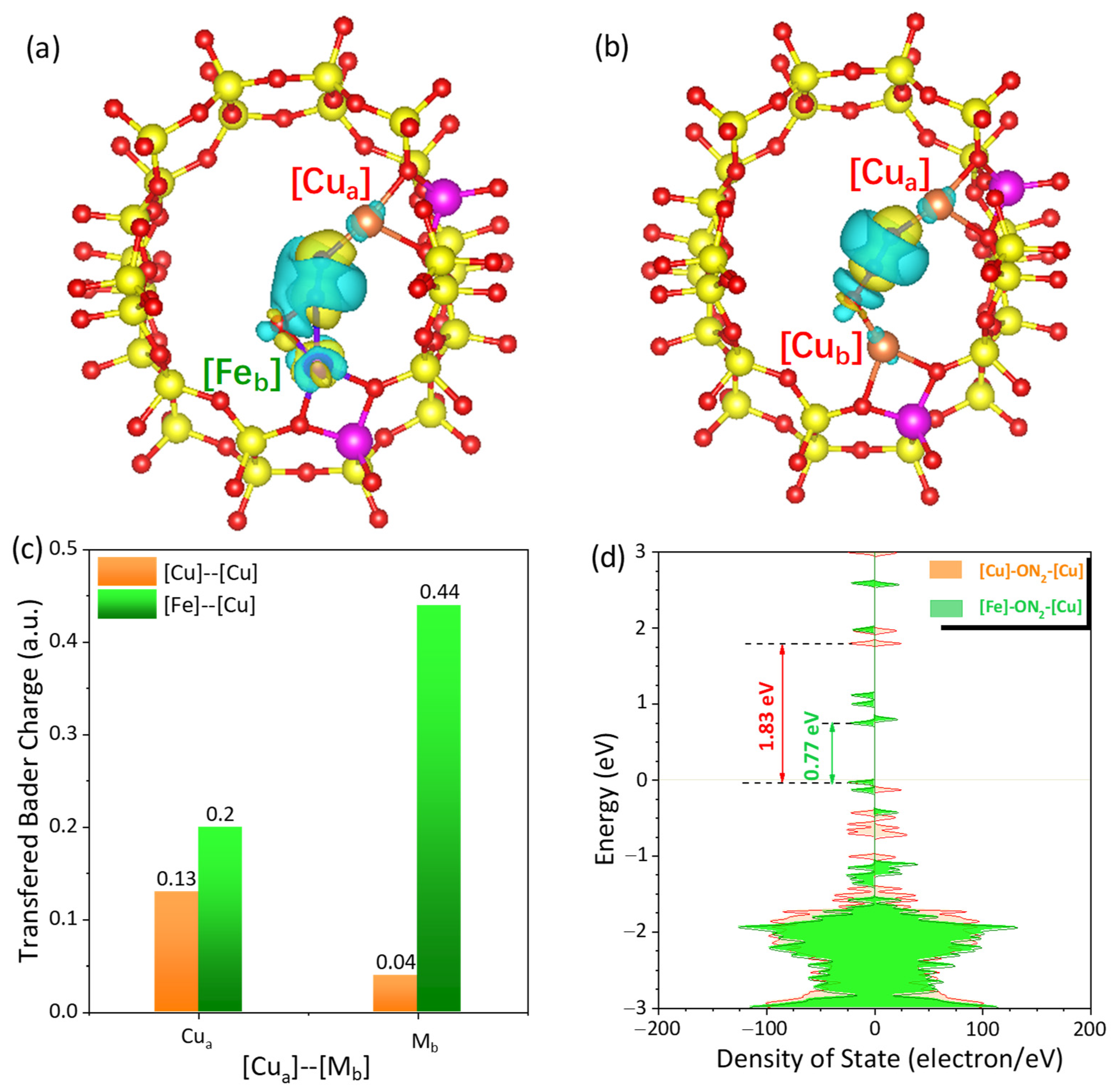
2.4.3. Illustration of the Synergistic Effect of Neighboring Fe and Cu Cations
3. Materials and Methods
3.1. Experimental Methods
3.1.1. Catalyst Preparation
3.1.2. Catalyst Characterizations
3.1.3. Activity Measurement
3.2. Computational Methods
3.2.1. Constructed Model
3.2.2. Computational Method
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Agarwal, N.; Freakley, S.J.; McVicker, R.U.; Althahban, S.M.; Dimitratos, N.; He, Q.; Morgan, D.J.; Jenkins, R.L.; Willock, D.J.; Taylor, S.H.; et al. Aqueous Au-Pd colloids catalyze selective CH4 oxidation to CH3OH with O2 under mild conditions. Science 2017, 358, 223–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, L.; Luo, J.; Li, H.; Ren, F.; Zhang, Y.; Liu, A.; Li, W.-X.; Zeng, J. Water enables mild oxidation of methane to methanol on gold single-atom catalysts. Nat. Commun. 2021, 12, 1218. [Google Scholar] [CrossRef] [PubMed]
- Ravi, M.; Ranocchiari, M.; Van Bokhoven, J.A. The Direct Catalytic Oxidation of Methane to Methanol-A Critical Assessment. Angew. Chem. Int. Ed. 2017, 56, 16464–16483. [Google Scholar] [CrossRef]
- Li, M.; Shan, J.; Giannakakis, G.; Ouyang, M.; Cao, S.; Lee, S.; Allard, L.F.; Flytzani-Stephanopoulos, M. Single-step selective oxidation of methane to methanol in the aqueous phase on iridium-based catalysts. Appl. Catal. B Environ. 2021, 292, 120124. [Google Scholar] [CrossRef]
- Kulkarni, A.R.; Zhao, Z.-J.; Siahrostami, S.; Nørskov, J.K.; Studt, F. Monocopper Active Site for Partial Methane Oxidation in Cu-Exchanged 8MR Zeolites. ACS Catal. 2016, 6, 6531–6536. [Google Scholar] [CrossRef]
- Dinh, K.T.; Sullivan, M.; Serna, P.; Meyer, R.J.; Dincă, M.; Román-Leshkov, Y. Viewpoint on the Partial Oxidation of Methane to Methanol Using Cu- and Fe-Exchanged Zeolites. ACS Catal. 2018, 8, 8306–8313. [Google Scholar] [CrossRef] [Green Version]
- Jeong, Y.R.; Jung, H.; Kang, J.; Han, J.W.; Park, E.D. Continuous Synthesis of Methanol from Methane and Steam over Copper-Mordenite. ACS Catal. 2021, 11, 1065–1070. [Google Scholar] [CrossRef]
- Hammond, C.; Forde, M.; Ab Rahim, M.H.; Thetford, A.; He, Q.; Jenkins, R.L.; Dimitratos, N.; Lopez-Sanchez, J.A.; Dummer, N.; Murphy, D.; et al. Direct Catalytic Conversion of Methane to Methanol in an Aqueous Medium by using Copper-Promoted Fe-ZSM-5. Angew. Chem. Int. Ed. 2012, 51, 5129–5133. [Google Scholar] [CrossRef]
- Hammond, C.; Dimitratos, N.; Jenkins, R.L.; Lopez-Sanchez, J.A.; Kondrat, S.A.; ab Rahim, M.H.; Forde, M.M.; Thetford, A.; Taylor, S.H.; Hagen, H.; et al. Elucidation and Evolution of the Active Component within Cu/Fe/ZSM-5 for Catalytic Methane Oxidation: From Synthesis to Catalysis. ACS Catal. 2013, 3, 689–699. [Google Scholar] [CrossRef]
- Xu, J.; Armstrong, R.D.; Shaw, G.; Dummer, N.; Freakley, S.; Taylor, S.H.; Hutchings, G.J. Continuous selective oxidation of methane to methanol over Cu- and Fe-modified ZSM-5 catalysts in a flow reactor. Catal. Today 2016, 270, 93–100. [Google Scholar] [CrossRef] [Green Version]
- Yu, T.; Li, Z.; Lin, L.; Chu, S.; Su, Y.; Song, W.; Wang, A.; Weckhuysen, B.M.; Luo, W. Highly Selective Oxidation of Methane into Methanol over Cu-Promoted Monomeric Fe/ZSM-5. ACS Catal. 2021, 11, 6684–6691. [Google Scholar] [CrossRef]
- Wang, L.; Li, Z.; Wang, Z.; Chen, X.; Song, W.; Zhao, Z.; Wei, Y.; Zhang, X. Hetero-Metallic Active Sites in Omega (MAZ) Zeolite-Catalyzed Methane Partial Oxidation: A DFT Study. Ind. Eng. Chem. Res. 2021, 60, 2400–2409. [Google Scholar] [CrossRef]
- Dandu, N.K.; Adeyiga, O.; Panthi, D.; Bird, S.A.; Odoh, S.O. Performance of density functional theory for describing hetero-metallic active-site motifs for methane-to-methanol conversion in metal-exchanged zeolites. J. Comput. Chem. 2018, 39, 2667–2678. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Huang, L.; Chen, W.; Zhou, J.; Zheng, A. Rationally designing mixed Cu–(μ-O)–M (M = Cu, Ag, Zn, Au) centers over zeolite materials with high catalytic activity towards methane activation. Phys. Chem. Chem. Phys. 2018, 20, 26522–26531. [Google Scholar] [CrossRef] [PubMed]
- Sazama, P.; Moravkova, J.; Sklenak, S.; Vondrova, A.; Tabor, E.; Sadovska, G.; Pilar, R. Effect of the Nuclearity and Coordination of Cu and Fe Sites in β Zeolites on the Oxidation of Hydrocarbons. ACS Catal. 2020, 10, 3984–4002. [Google Scholar] [CrossRef]
- Tang, X.; Wang, L.; Yang, B.; Fei, C.; Yao, T.; Liu, W.; Lou, Y.; Dai, Q.; Cai, Y.; Cao, X.-M.; et al. Direct oxidation of methane to oxygenates on supported single Cu atom catalyst. Appl. Catal. B Environ. 2021, 285, 119827. [Google Scholar] [CrossRef]
- Zhang, R.; Liu, N.; Lei, Z.; Chen, B. Selective Transformation of Various Nitrogen-Containing Exhaust Gases toward N2 over Zeolite Catalysts. Chem. Rev. 2016, 116, 3658–3721. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Zhang, R.; Chen, B.; Li, Y.; Li, Y. Comparative study on the direct decomposition of nitrous oxide over M (Fe, Co, Cu)–BEA zeolites. J. Catal. 2012, 294, 99–112. [Google Scholar] [CrossRef]
- Zhao, G.; Benhelal, E.; Adesina, A.A.; Kennedy, E.M.; Stockenhuber, M. Comparison of Direct, Selective Oxidation of Methane by N2O over Fe-ZSM-5, Fe-Beta, and Fe-FER Catalysts. J. Phys. Chem. C 2019, 123, 27436–27447. [Google Scholar] [CrossRef]
- Zhao, G.; Adesina, A.A.; Kennedy, E.M.; Stockenhuber, M. Formation of Surface Oxygen Species and the Conversion of Methane to Value-Added Products with N2O as Oxidant over Fe-Ferrierite Catalysts. ACS Catal. 2020, 10, 1406–1416. [Google Scholar] [CrossRef]
- Parfenov, M.V.; Starokon, E.V.; Pirutko, L.V.; Panov, G.I. Quasicatalytic and catalytic oxidation of methane to methanol by nitrous oxide over FeZSM-5 zeolite. J. Catal. 2014, 318, 14–21. [Google Scholar] [CrossRef]
- Fan, L.; Cheng, D.-G.; Song, L.; Chen, F.; Zhan, X. Direct conversion of CH4 to oxy-organics by N2O using freeze-drying FeZSM-5. Chem. Eng. J. 2019, 369, 522–528. [Google Scholar] [CrossRef]
- Ipek, B.; Lobo, R.F. Catalytic conversion of methane to methanol on Cu-SSZ-13 using N2O as oxidant. Chem. Commun. 2016, 52, 13401–13404. [Google Scholar] [CrossRef]
- Xu, R.; Liu, N.; Dai, C.; Li, Y.; Zhang, J.; Wu, B.; Yu, G.; Chen, B. H2O-Built Proton Transfer Bridge Enhances Continuous Methane Oxidation to Methanol over Cu-BEA Zeolite. Angew. Chem. Int. Ed. 2021, 60, 16634–16640. [Google Scholar] [CrossRef] [PubMed]
- Memioglu, O.; Ipek, B. A potential catalyst for continuous methane partial oxidation to methanol using N2O: Cu-SSZ-39. Chem. Commun. 2020, 57, 1364–1367. [Google Scholar] [CrossRef]
- Narsimhan, K.; Iyoki, K.; Dinh, K.; Román-Leshkov, Y. Catalytic Oxidation of Methane into Methanol over Copper-Exchanged Zeolites with Oxygen at Low Temperature. ACS Cent. Sci. 2016, 2, 424–429. [Google Scholar] [CrossRef] [Green Version]
- Sushkevich, V.L.; Palagin, D.; Ranocchiari, M.; Van Bokhoven, J.A. Selective anaerobic oxidation of methane enables direct synthesis of methanol. Science 2017, 356, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Palagin, D.; Sushkevich, V.L.; van Bokhoven, J.A. Water Molecules Facilitate Hydrogen Release in Anaerobic Oxidation of Methane to Methanol over Cu/Mordenite. ACS Catal. 2019, 9, 10365–10374. [Google Scholar] [CrossRef]
- Liu, Z.; Huang, E.; Orozco, I.; Liao, W.; Palomino, R.M.; Rui, N.; Duchoň, T.; Nemšák, S.; Grinter, D.C.; Mahapatra, M.; et al. Water-promoted interfacial pathways in methane oxidation to methanol on a CeO 2 -Cu 2 O catalyst. Science 2020, 368, 513–517. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhang, H.; Yan, Y. High efficiency of phenol oxidation in a structured fixed bed over Cu-ZSM-5/PSSF prepared by ion-exchanged method. Chem. Eng. J. 2020, 380, 122466. [Google Scholar] [CrossRef]
- Xia, Y.; Zhan, W.; Guo, Y.; Guo, Y.; Lu, G. Fe-Beta zeolite for selective catalytic reduction of NOx with NH3: Influence of Fe content. Chin. J. Catal. 2016, 37, 2069–2078. [Google Scholar] [CrossRef]
- Xiao, P.; Osuga, R.; Wang, Y.; Kondo, J.N.; Yokoi, T. Bimetallic Fe–Cu/beta zeolite catalysts for direct hydroxylation of benzene to phenol: Effect of the sequence of ion exchange for Fe and Cu cations. Catal. Sci. Technol. 2020, 10, 6977–6986. [Google Scholar] [CrossRef]
- Gao, F.; Kollar, M.; Kukkadapu, R.K.; Washton, N.M.; Wang, Y.; Szanyi, J.; Peden, C.H. Fe/SSZ-13 as an NH3-SCR catalyst: A reaction kinetics and FTIR/Mössbauer spectroscopic study. Appl. Catal. B Environ. 2015, 164, 407–419. [Google Scholar] [CrossRef] [Green Version]
- Berlier, G.; Lamberti, C.; Rivallan, M.; Mul, G. Characterization of Fe sites in Fe-zeolites by FTIR spectroscopy of adsorbed NO: Are the spectra obtained in static vacuum and dynamic flow set-ups comparable? Phys. Chem. Chem. Phys. 2010, 12, 358–364. [Google Scholar] [CrossRef]
- He, M.; Zhang, J.; Sun, X.-L.; Chen, B.-H.; Wang, Y.-G. Theoretical Study on Methane Oxidation Catalyzed by Fe/ZSM-5: The Significant Role of Water on Binuclear Iron Active Sites. J. Phys. Chem. C 2016, 120, 27422–27429. [Google Scholar] [CrossRef]
- Mahyuddin, M.H.; Staykov, A.; Shiota, Y.; Yoshizawa, K. Direct Conversion of Methane to Methanol by Metal-Exchanged ZSM-5 Zeolite (Metal = Fe, Co, Ni, Cu). ACS Catal. 2016, 6, 8321–8331. [Google Scholar] [CrossRef]
- Available online: http://www.iza-structure.org/databases/ (accessed on 5 November 2021).
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Hutter, J.; Iannuzzi, M.; Schiffmann, F.; VandeVondele, J. cp2k: Atomistic simulations of condensed matter systems. WIREs Comput. Mol. Sci. 2014, 4, 15–25. [Google Scholar] [CrossRef] [Green Version]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Liu, N.; Dai, C.; Xu, R.; Yu, G.; Wang, N.; Zhang, J.; Chen, B. Synergistic Effect of Neighboring Fe and Cu Cation Sites Boosts FenCum-BEA Activity for the Continuous Direct Oxidation of Methane to Methanol. Catalysts 2021, 11, 1444. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11121444
Li Y, Liu N, Dai C, Xu R, Yu G, Wang N, Zhang J, Chen B. Synergistic Effect of Neighboring Fe and Cu Cation Sites Boosts FenCum-BEA Activity for the Continuous Direct Oxidation of Methane to Methanol. Catalysts. 2021; 11(12):1444. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11121444
Chicago/Turabian StyleLi, Yan, Ning Liu, Chengna Dai, Ruinian Xu, Gangqiang Yu, Ning Wang, Jie Zhang, and Biaohua Chen. 2021. "Synergistic Effect of Neighboring Fe and Cu Cation Sites Boosts FenCum-BEA Activity for the Continuous Direct Oxidation of Methane to Methanol" Catalysts 11, no. 12: 1444. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11121444