
‘Oxygen-Consuming Complexes’–Catalytic Effects of Iron–Salen Complexes with Dioxygen

 ,
, 
 and
and
Abstract
:
1. Introduction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substrates | Products | Reaction | Solvent | Oxidant | Refs. |
|---|---|---|---|---|---|
| cyclohexene | cyclohexenone and cyclohexenol, epoxide was formed in small amounts | cyclohexene oxidation | MeCN | PhIO | [59] |
| cyclohexane | cyclohexanone and cyclohexanol | cyclohexane oxidation | MeCN | HOOH, TBHP | [64] |
| natural propenylbenzenes (safrol, isosafrol, anethol, eugenol, and isoeugenol) | conversion of these substrates into epoxides | propenylbenzenes epoxidations | MeCN | HOOH, TBHP | [65] |
| organic sulfur compounds | sulfoxides | oxidation | MeCN | PhIO, HOOH | [18,66,67,68] |
| various olefins | epoxides, ketones, and alcohols | oxidation | MeCN | HOOH | [69] |
| ring-substituted anilines | azobenzenes transformed to azoxybenzenes, and oligomers of anilines | oxidation | MeCN | HOOH | [19] |
| amino acids | corresponding ketimines | oxidative decarboxylation and deamination | 75% DMF–25% water solvent mixture | HOOH, t-BuOOH, PhIO, MCPB, PMS | [70] |
2. Results and Discussion
2.1. Catalytic Tests
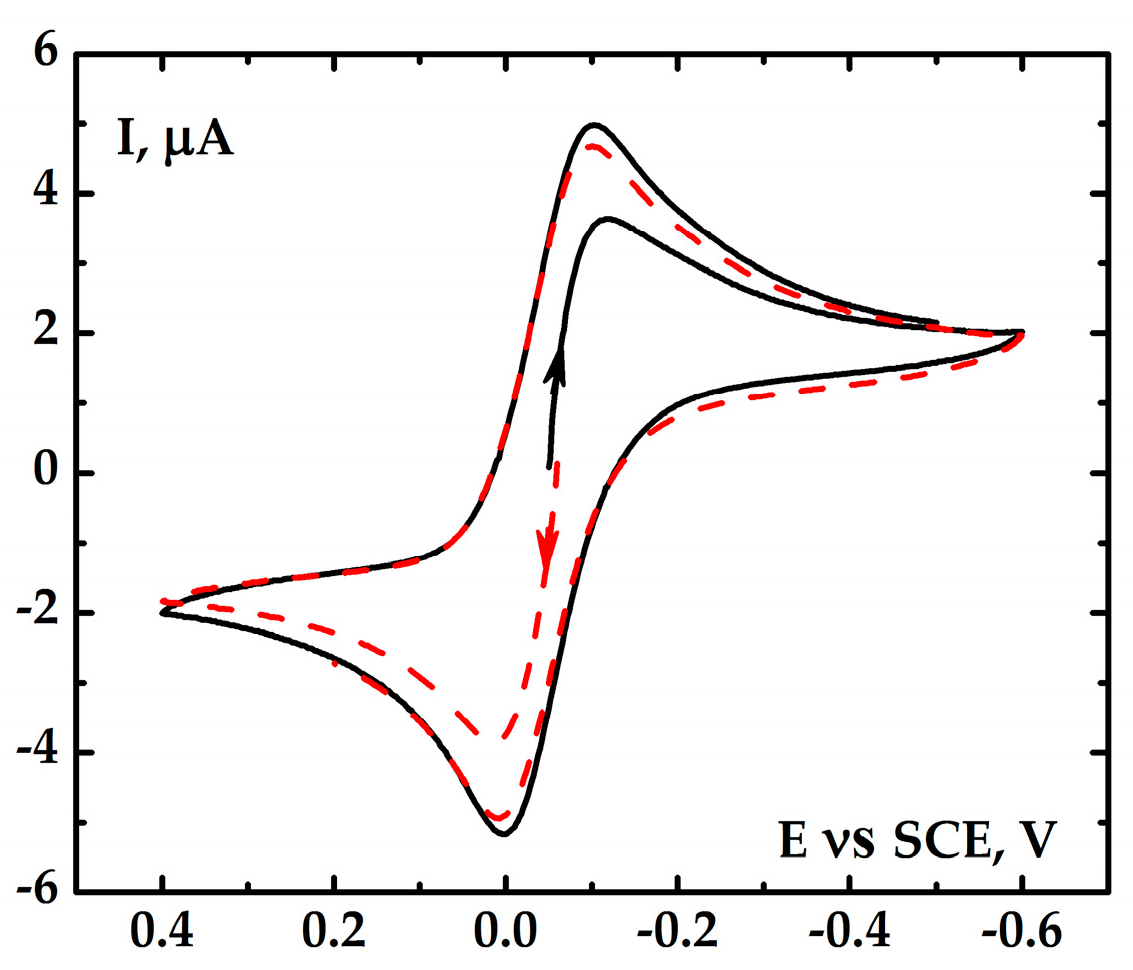
2.2. Electrochemical Research
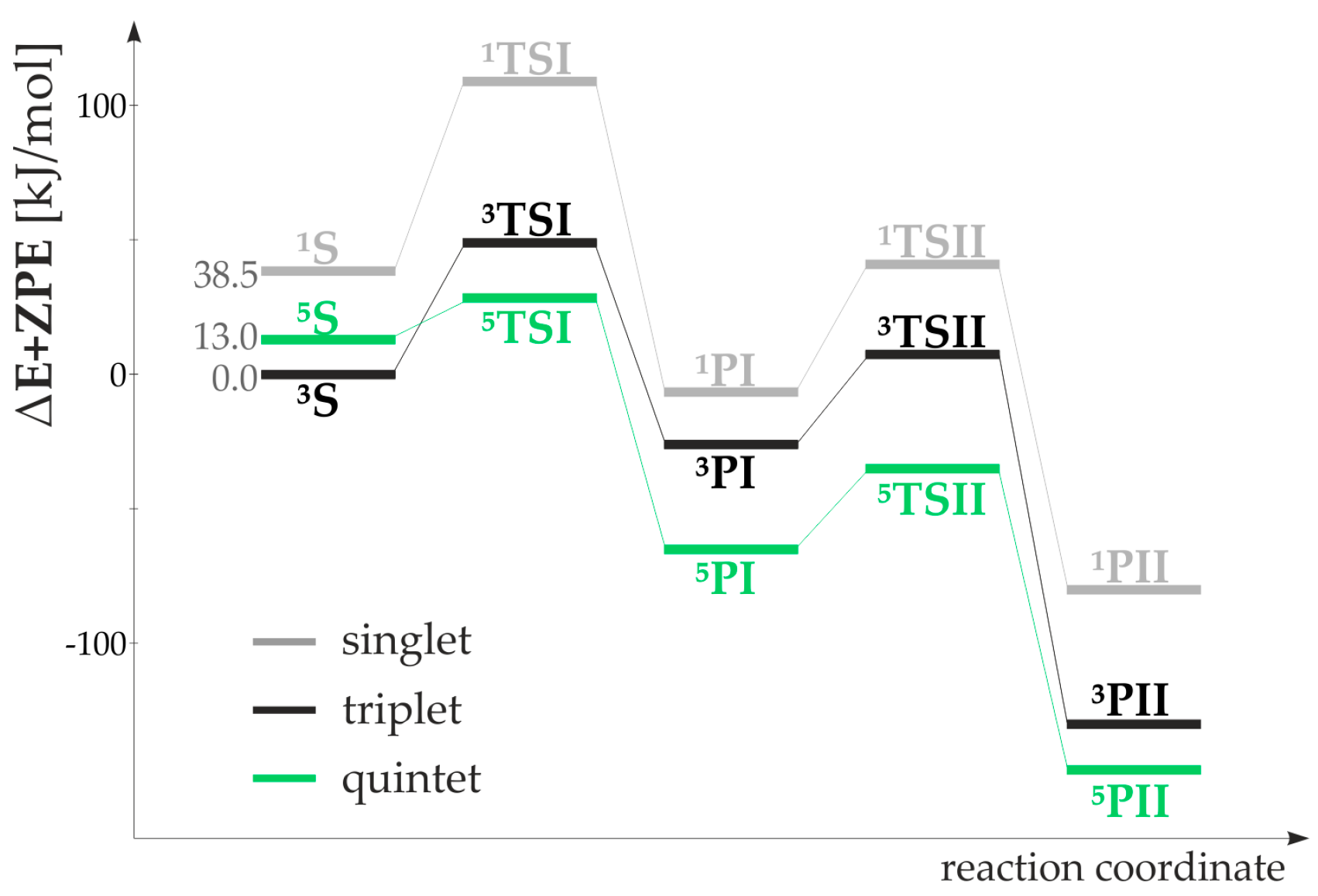
2.3. DFT Calculations
3. Materials and Methods
3.1. Chemicals
3.2. Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Waldvogel, S.R.; Selt, M. Electrochemical allylic oxidation of olefins: Sustainable and safe. Angew. Chem. Int. Ed. 2016, 55, 12578–12580. [Google Scholar] [CrossRef]
- Kim, S.; Cho, K.-B.; Lee, Y.-M.; Chen, J.; Fukuzumi, S.; Nam, W. Factors controlling the chemoselectivity in the oxidation of olefins by nonheme manganese (IV)-oxo complexes. J. Am. Chem. Soc. 2016, 138, 10654–10663. [Google Scholar] [CrossRef]
- Ammann, S.E.; Liu, W.; White, M.C. Enantioselective allylic C−H oxidation of terminal olefins to isochromans by palladium (ii)/chiral sulfoxide catalysis. Angew. Chem. Int. Ed. 2016, 55, 9571–9575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horn, E.J.; Rosen, B.R.; Chen, Y.; Tang, J.; Chen, K.; Eastgate, M.D.; Baran, P.S. Scalable and sustainable electrochemical allylic C–H oxidation. Nature 2016, 533, 77–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayeh, L.; Le, P.Q.; Tambar, U.K. Catalytic allylic oxidation of internal alkenes to a multifunctional chiral building block. Nature 2017, 547, 196–200. [Google Scholar] [CrossRef] [PubMed]
- Litman, Z.C.; Sharma, A.; Hartwig, J.F. Oxidation of Hindered Allylic C–H Bonds with Applications to the Functionalization of Complex Molecules. ACS Catalysis 2017, 7, 1998–2001. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, R.A.; Arend, I.; Hanefeld, U. Green Chemistry and Catalysis; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2007. [Google Scholar]
- Munz, D.; Strassner, T. Alkane C–H functionalization and oxidation with molecular oxygen. Inorg. Chem. 2015, 54, 5043–5052. [Google Scholar] [CrossRef] [PubMed]
- Shul’pin, G.B.; Nesterov, D.S.; Shul’pina, L.S.; Pombeiro, A.J.L. A hydroperoxo-rebound mechanism of alkane oxidation with hydrogen peroxide catalyzed by binuclear manganese (IV) complex in the presence of an acid with involvement of atmospheric dioxygen. Inorg. Chim. Acta 2017, 455, 666–676. [Google Scholar] [CrossRef]
- Chaignon, J.; Gourgues, M.; Khrouz, L.; Moliner, N.; Bonneviot, L.; Fache, F.; Castro, I.; Albela, B. A bioinspired heterogeneous catalyst based on the model of the manganese-dependent dioxygenase for selective oxidation using dioxygen. RSC Adv. 2017, 7, 17336–17345. [Google Scholar] [CrossRef] [Green Version]
- Sawyer, D.T. Oxygen Chemistry; Oxford University Press: New York, NY, USA, 1991. [Google Scholar]
- Simándi, L.I. Catalytic Activation of Dioxygen by Metal Complexes; Springer: Cham, The Netherlands, 1992. [Google Scholar]
- Strukul, G. Catalytic Oxidations with Hydrogen Peroxide as Oxidant; Springer: Cham, The Netherlands, 1992. [Google Scholar]
- Foote, C.S.; Valentine, J.; Greenberg, A.; Liebman, J.F. Active Oxygen in Chemistry; Springer: Cham, The Netherlands, 1995. [Google Scholar]
- Sawyer, D.T.; Sobkowiak, A.; Matsushita, T. Metal [ML x; M = Fe, Cu, Co, Mn]/hydroperoxide-induced activation of dioxygen for the oxygenation of hydrocarbons: Oxygenated Fenton chemistry. Acc. Chem. Res. 1996, 29, 409–416. [Google Scholar] [CrossRef]
- Sahu, S.; Goldberg, D.P. Activation of dioxygen by iron and manganese complexes: A heme and nonheme perspective. J. Am. Chem. Soc. 2016, 138, 11410–11428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaz, A.D.N.; McGinnity, D.F.; Coon, M.J. Epoxidation of olefins by cytochrome P450: Evidence from site-specific mutagenesis for hydroperoxo-iron as an electrophilic oxidant. Proc. Natl. Acad. Sci. USA 1998, 95, 3555–3560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kryatov, S.V.; Rybak-Akimova, E.V.; Schindler, S. Kinetics and mechanisms of formation and reactivity of non-heme iron oxygen intermediates. Chem. Rev. 2005, 105, 2175–2226. [Google Scholar] [CrossRef]
- Aslam, A.M.; Rajagopal, S.; Vairamani, M.; Ravikumar, M. Iron (III)–salen–H2O2 as a peroxidase model: Electron transfer reactions with anilines. Transit. Met. Chem. 2011, 36, 751–759. [Google Scholar] [CrossRef]
- Chatterjee, S.; Sengupta, K.; Samanta, S.; Das, P.K.; Dey, A. Concerted proton–electron transfer in electrocatalytic O2 reduction by iron porphyrin complexes: Axial ligands tuning H/D isotope effect. Inorg. Chem. 2015, 54, 2383–2392. [Google Scholar] [CrossRef]
- Hong, S.; Lee, Y.-M.; Ray, K.; Nam, W. Dioxygen activation chemistry by synthetic mononuclear nonheme iron, copper and chromium complexes. Coord. Chem. Rev. 2017, 334, 25–42. [Google Scholar] [CrossRef]
- Que, L. The road to non-heme oxoferryls and beyond. Acc. Chem. Res. 2007, 40, 493–500. [Google Scholar] [CrossRef]
- Friese, S.J.; Kucera, B.E.; Young, V.G.; Que, L.; Tolman, W.B. Iron (II) complexes of sterically bulky α-ketocarboxylates. Structural models for α-ketoacid-dependent nonheme iron halogenases. Inorg. Chem. 2008, 47, 1324–1331. [Google Scholar] [CrossRef]
- Weiss, J.J. Nature of the iron–oxygen bond in oxyhaemoglobin. Nature 1964, 203, 183. [Google Scholar] [CrossRef]
- Liou, Y.W.; Wang, C.M. Peroxidase mimicking: Fe (Salen) Cl modified electrodes, fundamental properties and applications for biosensing. J. Electroanal. Chem. 2000, 481, 102–109. [Google Scholar] [CrossRef]
- Emerson, J.P.; Farquhar, E.R.; Que, L., Jr. Structural “snapshots” along reaction pathways of non-heme iron enzymes. Angew. Chem. 2007, 46, 8553–8556. [Google Scholar] [CrossRef]
- Mukherjee, A.; Cranswick, M.A.; Chakrabarti, M.; Paine, T.K.; Fujisawa, K.; Münck, E.; Que, L. Oxygen activation at mononuclear nonheme iron centers: A superoxo perspective. Inorg. Chem. 2010, 49, 3618–3628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uchida, T.; Katsuki, T. Green asymmetric oxidation using air as oxidant. J. Synth. Org. Chem. Jpn. 2013, 71, 1126–1135. [Google Scholar] [CrossRef] [Green Version]
- Kurahashi, T. Reverse catalase reaction: Dioxygen activation via two-electron transfer from hydroxide to dioxygen mediated by a manganese (III) salen complex. Inorg. Chem. 2015, 54, 8356–8366. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.-W.; Kleespies, S.T.; Stout, H.D.; Meier, K.K.; Li, P.-Y.; Bominaar, E.L.; Que, L.; Münck, E.; Lee, W.-Z. Characterization of a paramagnetic mononuclear nonheme iron-superoxo complex. J. Am. Chem. Soc. 2014, 136, 10846–10849. [Google Scholar] [CrossRef] [Green Version]
- Farquhar, E.R.; Koehntop, K.D.; Emerson, J.P.; Que, L., Jr. Post-translational self-hydroxylation: A probe for oxygen activation mechanisms in non-heme iron enzymes. Biochem. Biophys. Res. Commun. 2005, 338, 230–239. [Google Scholar] [CrossRef]
- Shan, X.; Que, L., Jr. High-valent nonheme iron-oxo species in biomimetic oxidations. J. Inorg. Biochem. 2006, 100, 421–433. [Google Scholar] [CrossRef]
- Thibon, A.; England, J.; Martinho, M.; Young, V.G., Jr.; Frisch, J.R.; Guillot, R.; Girerd, J.-J.; Münck, E.; Que, L., Jr.; Banse, F. Proton-and Reductant-Assisted Dioxygen Activation by a Nonheme Iron (II) Complex to Form an Oxoiron (IV) Intermediate. Angew. Chem. Int. Ed. 2008, 47, 7064–7067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiedler, A.T.; Que, L. Reactivities of Fe (IV) complexes with oxo, hydroxo, and alkylperoxo ligands: An experimental and computational study. Inorg. Chem. 2009, 48, 11038–11047. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Zhang, M.; Bühlmann, P.; Que, L. Redox Potential and C−H Bond Cleaving Properties of a Nonheme FeIV=O Complex in Aqueous Solution. J. Am. Chem. Soc. 2010, 132, 7638–7644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakazawa, H.; Itazaki, M. Fe–H Complexes in Catalysis. In Iron Catalysis; Topics in Organometallic Chemistry; Plietker, B., Ed.; Springer: Berlin/Heidelberg, Germany, 2011; Volume 33. [Google Scholar]
- Junge, K.; Schröder, K.; Beller, M. Homogeneous catalysis using iron complexes: Recent developments in selective reductions. Chem. Commun. 2011, 47, 4849–4859. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Lutz, M.; Milan, M.; Costas, M.; Otte, M.; Klein Gebbink, R.J.M. Non-Heme Iron Catalysts with a Rigid Bis-Isoindoline Backbone and Their Use in Selective Aliphatic C−H Oxidation. Adv. Synth. Catal. 2017, 359, 2590–2595. [Google Scholar] [CrossRef]
- Hage, R.; de Boer, J.W.; Gaulard, F.; Maaijen, K. Advances in Inorganic Chemistry; van Eldik, R., Hubbard, C.D., Eds.; Academic Press: Cambridge, MA, USA, 2013; Volume 65, pp. 85–116. [Google Scholar]
- Duarte, F.M.; Maldonado-Hódar, F.J.; Madeira, L.M. Influence of the iron precursor in the preparation of heterogeneous Fe/activated carbon Fenton-like catalysts. Appl. Catal. A Gen. 2013, 458, 39–47. [Google Scholar] [CrossRef]
- Su, B.; Cao, Z.-C.; Shi, Z.-J. Exploration of earth-abundant transition metals (Fe, Co, and Ni) as catalysts in unreactive chemical bond activations. Acc. Chem. Res. 2015, 48, 886–896. [Google Scholar] [CrossRef] [PubMed]
- Balaraman, E.; Nandakumar, A.; Jaiswal, G.; Sahoo, M.K. Iron-catalyzed dehydrogenation reactions and their applications in sustainable energy and catalysis. Catal. Sci. Technol. 2017, 7, 3177–3195. [Google Scholar] [CrossRef]
- Bedford, R.B.; Bruce, D.W.; Frost, R.M.; Goodby, J.W.; Hird, M. Iron (III) salen-type catalysts for the cross-coupling of aryl Grignards with alkyl halides bearing β-hydrogens. Chem. Commun. 2004, 2822–2823. [Google Scholar] [CrossRef]
- Routier, S.; Vezin, H.; Lamour, E.; Bernier, J.-L.; Catteau, J.-P.; Bailly, C. DNA cleavage by hydroxy-salicylidene-ethylendiamine-iron complexes. Nucleic Acids Res. 1999, 27, 4160–4166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woldemariam, G.A.; Mandal, S.S. Iron (III)-salen damages DNA and induces apoptosis in human cell via mitochondrial pathway. J. Inorg. Biochem. 2008, 102, 740–747. [Google Scholar] [CrossRef] [PubMed]
- Nejat Dehkordi, M.; Lincoln, P. Comprehensive study on the binding of iron Schiff base complex with DNA and determining the binding mode. J. Fluoresc. 2013, 23, 813–821. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, N.; Pratheek, B.M.; Garai, A.; Kumar, A.; Meena, V.S.; Ghosh, S.; Singh, S.; Kumari, S.; Chandrashekar, T.K.; Goswami, C.; et al. Induction of apoptosis by Fe(salen)Cl through caspase-dependent pathway specifically in tumor cells. Cell Biol. Int. 2014, 38, 1118–1131. [Google Scholar] [CrossRef] [PubMed]
- Nworie, F.S. Bis (salicylidene) ethylenediamine (salen) and bis (salicylidene) ethylenediamine-metal complexes: From structure to biological activity. J. Anal. Pharm. Res. 2016, 3, 00076. [Google Scholar]
- Zhang, R.; Ma, J.; Wang, W.; Wang, B.; Li, R. Zeolite-encapsulated M (Co, Fe, Mn)(SALEN) complexes modified glassy carbon electrodes and their application in oxygen reduction. J. Electroanal. Chem. 2010, 643, 31–38. [Google Scholar] [CrossRef]
- Bonometti, V.; Labbé, E.; Buriez, O.; Mussini, P.; Amatore, C. Exploring the first steps of an electrochemically-triggered controlled polymerization sequence: Activation of alkyl-and benzyl halide initiators by an electrogenerated FeIISalen complex. J. Electroanal. Chem. 2009, 633, 99–105. [Google Scholar] [CrossRef]
- Gazi, S.; Rajakumar, A.; Singh, N.D. Photodegradation of organic dyes in the presence of [Fe (III)-salen] Cl complex and H2O2 under visible light irradiation. J. Hazard. Mater. 2010, 183, 894–901. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Cheng, F.; Wang, S.; Zhang, T.; Chen, J. M (Salen)-derived nitrogen-doped M/C (M = Fe, Co, Ni) porous nanocomposites for electrocatalytic oxygen reduction. Sci. Rep. 2014, 4, 4386. [Google Scholar] [CrossRef] [Green Version]
- Khare, S.; Chokhare, R. Synthesis, characterization and catalytic activity of Fe (Salen) intercalated α-zirconium phosphate for the oxidation of cyclohexene. J. Mol. Catal. A Chem. 2011, 344, 83–92. [Google Scholar] [CrossRef]
- Rydel-Ciszek, K.; Charczuk, M.; Pacześniak, T.; Chmielarz, P. Manganese (II) complexes with Bn-tpen as powerful catalysts of cyclohexene oxidation. Chem. Pap. 2017, 71, 2085–2093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sobkowiak, A.; Naróg, D.; Sawyer, D.T. Iron (III, II)-induced activation of dioxygen for the oxygenation of cyclohexene and related unsaturated hydrocarbons. J. Mol. Catal. A Chem. 2000, 159, 247–256. [Google Scholar] [CrossRef]
- Naróg, D.; Szczepanik, A.; Sobkowiak, A. Iron (II, III)-catalyzed oxidation of limonene by dioxygen. Catal. Lett. 2008, 120, 320–325. [Google Scholar] [CrossRef]
- Naróg, D.; Lechowicz, U.; Pietryga, T.; Sobkowiak, A. Iron (II, III)-catalyzed oxidative N-dealkylation of amines with dioxygen. J. Mol. Catal. A Chem. 2004, 212, 25–33. [Google Scholar] [CrossRef]
- Matsushita, T.; Sawyer, D.T.; Sobkowiak, A. MnIIILx/t-BuOOH-induced activation of dioxygen for the oxygenation of cyclohexene. J. Mol. Catal. A Chem. 1999, 137, 127–133. [Google Scholar] [CrossRef]
- Biswas, A.N.; Das, P.; Kandar, S.K.; Agarwala, A.; Bandyopadhyay, D.; Bandyopadhyay, P. Chiral iron (III)-salen-catalyzed oxidation of hydrocarbons. Catal. Commun. 2009, 10, 708–711. [Google Scholar] [CrossRef]
- Sankaralingam, M.; Lee, Y.-M.; Nam, W.; Fukuzumi, S. Selective oxygenation of cyclohexene by dioxygen via an iron (V)-oxo complex-autocatalyzed reaction. Aerobic oxidation of hydrocarbons catalyzed by electronegative iron salen complexes. Inorg. Chem. 2017, 56, 5096–5104. [Google Scholar] [CrossRef] [PubMed]
- Böttcher, A.; Grinstaff, M.W.; Labinger, J.A.; Gray, H.B. Aerobic oxidation of hydrocarbons catalyzed by electronegative iron salen complexes. J. Mol. Catal. A Chem. 1996, 113, 191–200. [Google Scholar] [CrossRef]
- Olivo, G.; Lanzalunga, O.; Di Stefano, S. Non-Heme Imine-Based Iron Complexes as Catalysts for Oxidative Processes. Adv. Synth. Catal. 2016, 358, 843–863. [Google Scholar] [CrossRef]
- Hakkoum, A.; Ameur, N.; Bachir, R.; Bedrane, S. Activity of Bimetallic Gold-Iron Catalysts in Adipic Acid Production by Direct Oxidation of Cyclohexene with Molecular Oxygen. Ann. Chim.-Sci. Matériaux 2019, 43, 299–304. [Google Scholar] [CrossRef]
- Salomão, G.C.; Olsen, M.H.N.; Drago, V.; Fernandes, C.; Cardozo Filho, L.; Antunes, O.A.C. Oxidation of cyclohexane promoted by [Fe (III)(Salen) Cl] and [Mn (III)(Salen) Cl]. Catal. Commun. 2007, 8, 69–72. [Google Scholar] [CrossRef]
- Barbosa Souza, D.P.; Fricks, A.T.; Alvarez, H.M.; Salomão, G.C.; Neves Olsen, M.H.; Filho, L.C.; Fernandes, C.; Antunes, O.A.C. Epoxidation of natural propenylbenzenes catalyzed by [FeIII(Salen)Cl] and [FeIII(TPP)Cl]. Catal. Commun. 2007, 8, 1041–1046. [Google Scholar] [CrossRef]
- Sivasubramanian, V.K.; Ganesan, M.; Rajagopal, S.; Ramaraj, R. Iron (III)−salen complexes as enzyme models: Mechanistic study of oxo (salen) iron complexes oxygenation of organic sulfides. J. Org. Chem. 2002, 67, 1506–1514. [Google Scholar] [CrossRef] [PubMed]
- Venkataramanan, N.S.; Kuppuraj, G.; Rajagopal, S. Metal–salen complexes as efficient catalysts for the oxygenation of heteroatom containing organic compounds—synthetic and mechanistic aspects. Coord. Chem. Rev. 2005, 249, 1249–1268. [Google Scholar] [CrossRef]
- Jayaseeli, A.M.I.; Rajagopal, S. [Iron (III)–salen] ion catalyzed H2O2 oxidation of organic sulfides and sulfoxides. J. Mol. Catal. A Chem. 2009, 309, 103–110. [Google Scholar] [CrossRef]
- Mirkhani, V.; Moghadam, M.; Tangestaninejad, S.; Mohammadpoor-Baltork, I.; Rasouli, N. A comparative study of oxidation of alkanes and alkenes by hydrogen peroxide catalyzed by Cu(salen) complex covalently bound to a Keggin type polyoxometalate and its neat counterpart. Inorg. Chem. Commun. 2007, 10, 1537–1540. [Google Scholar] [CrossRef]
- Góger, S.; Bogáth, D.; Baráth, G.; Simaan, A.J.; Speier, G.; Kaizer, J. Bio-inspired amino acid oxidation by a non-heme iron catalyst. J. Inorg. Biochem. 2013, 123, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Lloret, F.; Moratal, J.; Faus, J. Solution chemistry of NN′-ethylenebis (salicylideneiminato) iron (III). Part 1. Deprotonation equilibria and reversible decomposition in acid medium of NN′-ethylenebis (salicylideneimine). Stability constant of NN′-ethylenebis (salicylideneiminato) iron (III). J. Chem. Soc. Dalton Trans. 1983, 8, 1743–1748. [Google Scholar]
- Liang, Y.; Duan, R.-L.; Hu, C.-Y.; Li, L.-L.; Pang, X.; Zhang, W.-X.; Chen, X.-S. Salen-iron complexes: Synthesis, characterization and their reactivity with lactide. Chin. J. Polym. Sci. 2018, 36, 185–189. [Google Scholar] [CrossRef]
- Gallagher, K.J.; Webster, R.L. Room temperature hydrophosphination using a simple iron salen pre-catalyst. Chem. Commun. 2014, 50, 12109–12111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsushita, T.; Kono, H.; Nishino, M.; Shono, T. Reactions of Chloroiron (III) Schiff Base Complexes with Superoxide Ion in Dimethyl Sulfoxide. Bull. Chem. Soc. Jpn. 1982, 55, 2581–2587. [Google Scholar] [CrossRef] [Green Version]
- Cozzi, P.G. Metal–Salen Schiff base complexes in catalysis: Practical aspects. Chem. Soc. Rev. 2004, 33, 410–421. [Google Scholar] [CrossRef]
- Rohde, J.-U.; Stubna, A.; Bominaar, E.L.; Münck, E.; Nam, W.; Que, L. Nonheme Oxoiron(IV) Complexes of Tris(2-pyridylmethyl)amine with cis-Monoanionic Ligands. Inorg. Chem. 2006, 45, 6435–6445. [Google Scholar] [CrossRef] [PubMed]
- Lane, B.S.; Burgess, K. Metal-catalyzed epoxidations of alkenes with hydrogen peroxide. Chem. Rev. 2003, 103, 2457–2474. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, D.T.; Sobkowiak, A.; Roberts, J.L. Electrochemistry for Chemists, 2nd ed.; Wiley: New York, NY, USA, 1995. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision B.01; Gaussian. Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Becke, A.D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]








| Substrate Concentration, M | Ketone, mM | Alcohol, mM | Epoxide, mM | TON |
|---|---|---|---|---|
| (a) | ||||
| 0.5 | 9.4 | 4.2 | 0.1 | 13.7 |
| 1 | 53.6 | 26.1 | 0.9 | 78.8 |
| 2 | 90.5 | 38.0 | 0.6 | 129.2 |
| 3 | 128.6 | 49.9 | 0.6 | 179.1 |
| 4 | 190.4 | 53.1 | 1.0 | 244.4 |
| (b) | ||||
| 0.5 | 2.8 | 0.7 | 0.0 | 3.2 |
| 1 | 27.8 | 12.6 | 0.4 | 40.8 |
| 2 | 56.5 | 24.5 | 0.6 | 81.6 |
| 3 | 111.9 | 47.4 | 1.0 | 160.2 |
| 4 | 215.0 | 82.5 | 1.3 | 298.8 |
| Line | Reactions: | ∆rG [kJ/mol] |
|---|---|---|
| 1 | [(salen)Fe]OO [(salen)Fe]OOH | −307.9 |
| 2 | (Cl)[(salen)Fe]OO (Cl)[(salen)Fe]OOH | −280.3 |
| 3 | (ClO4)[(salen)Fe]OO (ClO4)[(salen)Fe]OOH | −300.0 |
| 4 | [(salen)Fe]=O [(salen)Fe]OH | −385.3 |
| 5 | (Cl)[(salen)Fe]=O (Cl)[(salen)Fe]OH | −371.1 |
| 6 | (ClO4)[(salen)Fe]=O (ClO4)[(salen)Fe]OH | −384.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rydel-Ciszek, K.; Pacześniak, T.; Miłaczewska, A.; Chmielarz, P.; Sobkowiak, A. ‘Oxygen-Consuming Complexes’–Catalytic Effects of Iron–Salen Complexes with Dioxygen. Catalysts 2021, 11, 1462. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11121462
Rydel-Ciszek K, Pacześniak T, Miłaczewska A, Chmielarz P, Sobkowiak A. ‘Oxygen-Consuming Complexes’–Catalytic Effects of Iron–Salen Complexes with Dioxygen. Catalysts. 2021; 11(12):1462. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11121462
Chicago/Turabian StyleRydel-Ciszek, Katarzyna, Tomasz Pacześniak, Anna Miłaczewska, Paweł Chmielarz, and Andrzej Sobkowiak. 2021. "‘Oxygen-Consuming Complexes’–Catalytic Effects of Iron–Salen Complexes with Dioxygen" Catalysts 11, no. 12: 1462. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11121462