
Traceless Directing Groups in Sustainable Metal-Catalyzed C–H Activation




Laboratory of Organic Chemistry, Department of Chemistry, National and Kapodistrian University of Athens, Panepistimiopolis, 15771 Athens, Greece
*
Author to whom correspondence should be addressed.
Catalysts 2021, 11(5), 554; https://0-doi-org.brum.beds.ac.uk/10.3390/catal11050554
Submission received: 30 March 2021
/
Revised: 25 April 2021
/
Accepted: 26 April 2021
/
Published: 27 April 2021
(This article belongs to the Section Catalysis in Organic and Polymer Chemistry)
Abstract
:Sustainable transformations towards the production of valuable chemicals constantly attract interest, both in terms of academic and applied research. C–H activation has long been scrutinized in this regard, given that it offers a straightforward pathway to prepare compounds of great significance. In this context, directing groups (DG) have paved the way for chemical transformations that had not been achievable using traditional reactions. Few steps, high yields, selectivity and activation of inert substrates are some of the invaluable assets of directed catalysis. Additionally, the employment of traceless directing groups (TDG) greatly improves and simplifies this strategy, enabling the realization of multi-step reactions in one-pot, cascade procedures. Cheap, abundant, readily available transition metal salts and complexes can catalyze a plethora of reactions employing TDGs, usually under low catalyst loadings—rarely under stoichiometric amounts, leading in greater atom economy and milder conditions with increased yields and step-economy. This review article summarizes all the work done on TDG-assisted catalysis with manganese, iron, cobalt, nickel, or copper catalysts, and discusses the structure-activity relationships observed, by presenting the catalytic pathways and range of transformations reported thus far.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
As the demands of the increasing world population for ample energy resources and chemical feedstock have grown over the last decades, it has become clear—perhaps more than ever—that orchestrated maneuvers must be carried out, both in industry and academia, in order to pursue a sustainable future [1,2,3]. More specifically, as the field of organic synthesis has evolved, it became clear that synthetic procedures leading to the production of all kinds of chemical compounds have to be examined, and also possibly redesigned, under the prism of sustainability. Along these lines, the 12 principles of green chemistry [4,5], also implemented by the principles of green engineering [6], provide useful insight for the amplification of sustainable, environmentally benign, and user-friendly protocols leading to the production of valuable chemicals in few synthetic steps, under mild conditions, using green solvents, sustainable starting materials, and renewable sources (e.g., solar and wind power) [7,8,9,10,11]. The aforementioned principles are well implemented in catalysis, since inherent concepts in catalyzed reactions include sustainability, mild conditions, reagent reusability, high yields, and chemo-selectivity, effected by the application of catalysts that lower kinetic barriers and promote selective reaction pathways [12].
After the initial upsurge of transition metal-catalyzed organic transformations using expensive and rare fifth- and sixth-period transition metals [13,14,15,16,17,18,19,20,21], it is now imperative to put into practice the use of low cost, earth-abundant, sustainable, transition metals found in the fourth period of the periodic table. The plethora of oxidation states and geometries that the fourth-period, d-block elements can adopt in their compounds, offers an intriguing flexibility for applications in sustainable catalysis [22]. Combined with the difficulties imposed by heavier transition metals, such as high cost, low abundance, toxicity, and incompatibility with biological systems [23,24], it is clear that there is still ground to be covered in the design of green processes via sustainable catalysis.
Construction of complex organic scaffolds more often than not requires the formation of carbon–carbon or carbon–heteroatom bonds, ring closures, and cascade reactions. Traditional synthetic procedures, upon which modern chemistry was founded, usually employ protective groups that require extra steps (protection and de-protection) which are resource consuming and often decrease overall yields [25,26]. C–H activation is an alluring alternative approach to succeed in preparing complex molecules, since one can overcome problems associated with traditional synthetic procedures. Moreover, the field is now extended towards a variety of applications, such as the synthesis of pharmaceuticals [27,28,29,30] and agrochemicals [31,32]. C–H activation has been thoroughly reviewed in the literature and the reader is referred to some recent comprehensive review articles covering the topic [33,34,35,36,37,38,39,40,41,42,43].
Motivated by the above-mentioned needs, researchers have employed the use of functional groups that significantly affect C–H activation reactions. Directing groups (DG) represent an ingenious way to orchestrate and manipulate atoms towards the synthesis of important classes of organic compounds, such as pyrroles, indoles, quinolines, indolinones, and related heterocycles [44,45,46,47,48]. Transition metals can temporarily coordinate to these specific sites on the starting materials, thus leading to the formation of key intermediates that would not form otherwise. The formation of thermodynamically favored 5- and 6-membered rings, along with the kinetic lability of 3dn ions, can then provide energetically advantageous reaction pathways towards the desired product [49,50,51,52]. The directing groups can be categorized, either by their structure (e.g., denticity, identity of the coordinating atoms and moieties), or by their utilization strategy. In the latter, one can find removable (RDG), modifiable (MDG), non-removable (NRDG), and traceless directing groups (TDG), which may be pre-installed or transiently formed. RDGs are usually removed from the target compound during a final synthetic step, while MDGs undergo an extra functionalization or cyclization step and their framework can be found in the final product. NRDGs are maintained in the final compound, thus limiting their versatility. On the other hand, TDGs make one-pot reactions viable, given that they are not incorporated in the target molecule, as they are removed during the course of the catalytic cycle. A subcategory of TDGs are transient directing groups, which are transiently formed during the reaction and removed in situ after the transformation. Each DG category has its own positive and negative characteristics and weak points and is chosen depending on the specific application [47,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67].
All aforementioned aspects of sustainable transition metal-catalyzed C–H activation reactions constitute important weapons in the arsenal of contemporary organic chemistry. In this article, enabled by our ongoing interest in the area of sustainable catalysis [40,68,69,70,71,72,73,74,75,76], we review and critically examine the use of traceless directing groups (TDG) in sustainable metal catalyzed C–H activation, highlighting the versatility provided by these categories of DGs coupled with earth-abundant, sustainable metals. In line with our previous work [40], we examine the advancements regarding the use of TDGs in transformations catalyzed by complexes of the most abundant, least toxic and biocompatible 3d metals. In the case of each metal, the variety of TDGs used, ranges from limited to very wide, therefore each case is examined separately. The literature covered is until the end of January 2021.
2. Manganese
Initial reports using Mn in C–H activation included aldehyde insertion into a C–H bond using [Mn(CO)5Br], originally as a stoichiometric [77] and later as catalytic reagent [78]. Manganese, belonging to group 7 (VIIB) of the periodic system, adopts a great variety of oxidation states and coordination numbers, rendering many of its salts and complexes successful catalysts for various chemical transformations. Manganese complexes containing the ion in higher oxidation states, namely Mn(III) and Mn(IV), have been used as oxidation catalysts (see for example [79,80] and references therein). On the other hand, the rich organometallic chemistry of lower-oxidation state Mn(I) complexes has enabled their use as a cheaper, more viable alternative to Re(I) compounds [81,82]. The mode of action of Mn(I) as a redox-neutral catalyst, operating in organometallic C–H activation via the cooperative metalation-deprotonation pathway, is now well established [40]. Even though monodentate NRDGs or RDGs are used in the majority of cases so far, there have been recent advances towards using TDGs.
Annulations via C(sp2)–H Bond Activation
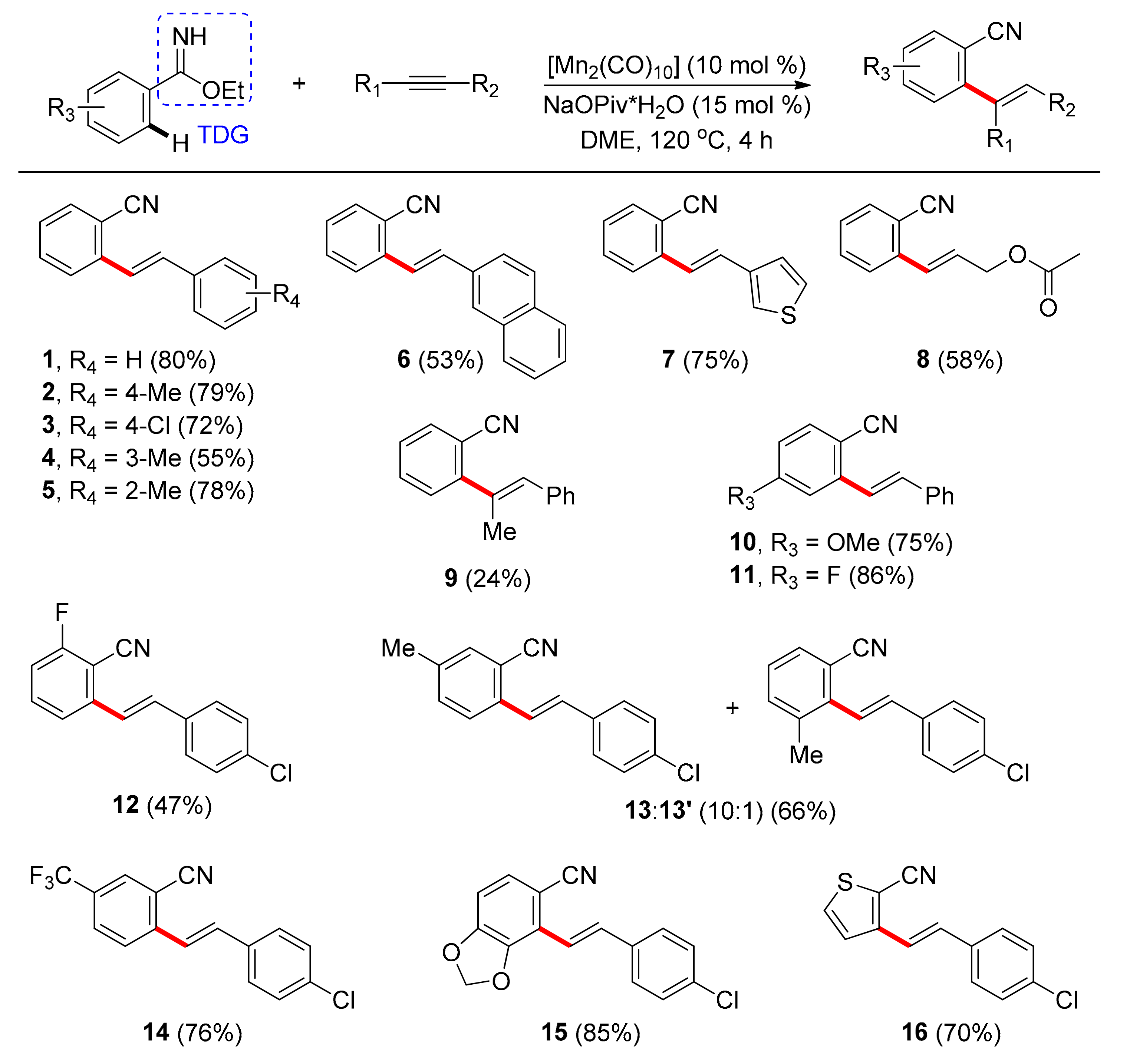
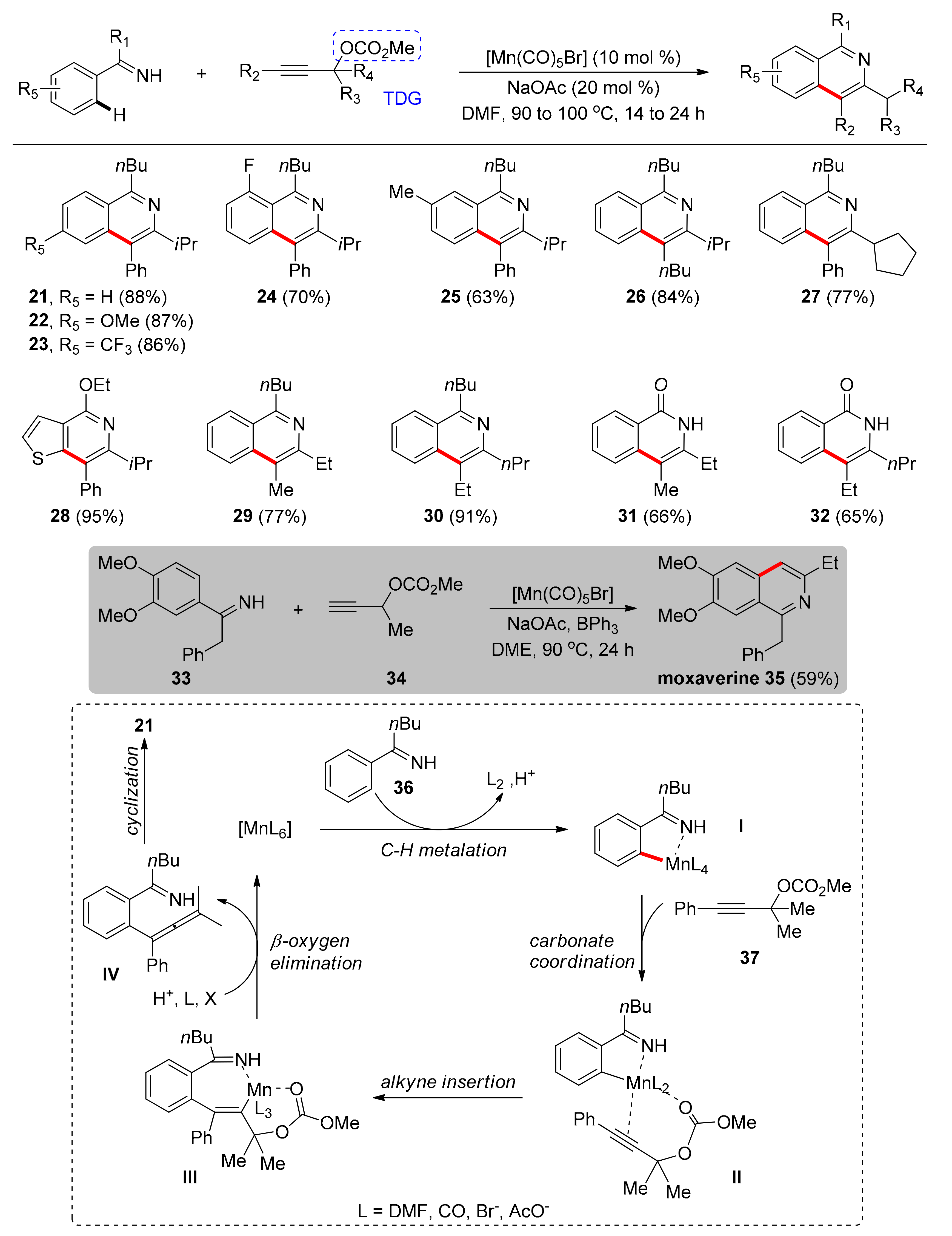
The first system found in literature that utilizes a TDG under Mn catalysis for direct C–H activation between alkynes and benzene imidates was reported by Wang and co-workers in 2016 [83]. This protocol offers access to aromatic nitriles bearing a double bond, while the cyano moiety allows for further functionalization, being a precursor towards various compounds, also found in clinical drugs, pesticides, and dyes. This system relies on the transformation of the cyano group to an imidate, since nitriles have linear configuration and, therefore, cannot aid in the directed ortho-C–H activation. After screening metal catalysts, manganese carbonyls such as pentacarbonylbromorhenium and dimanganese decacarbonyl were the catalysts that led to the expected product, with the later affording higher yields, when used only in 10 mol % catalyst loading. DME was the optimal solvent, while the catalytic addition of NaOPiv·H2O, as a weak base, was beneficial, probably by assisting cyclomanganation of benzimidate. Moreover, ethoxy benzimidate showed higher reactivity compared to other benzimidates that were tested. Notably, the doubly-alkenylated product or the Z-isomer were not detected, which reflects the high selectivity of this transformation. Under the optimal conditions, authors further examined various alkynes (Scheme 1) and found that both electron-withdrawing and -donating groups on different positions of the benzene ring performed well (1–5). In addition, aromatic alkynes, such as 2-ethynylnaphthlene and 3-ethynylthiophene, were converted, affording products 6 and 7, respectively. Aliphatic alkynes gave products in moderate yields when additional Mn(OAc)2 was used (8), while internal alkynes gave low yields with sole regioselectivity (9). Concerning the scope of imidates, para-substituted benzimidates led to high yields regardless of the electronic characteristics (10, 11) and ortho-substituted ones led to moderate yields (12). Regarding meta-substitution, the less sterically protected C–H bond was preferred with good regioselectivity (13, 13′), but the benzimidate bearing a m-CF3 group afforded only one regioisomer selectively (14). Nevertheless, product 15 was obtained in high yield after reaction at the most sterically hindered position, which could be attributed to the coordination of the neighboring oxygen atom to Mn. Finally, alkenylated product 16 was acquired in 70% yield with addition of Mn(OAc)2, when the 6-membered aromatic ring was replaced with a thiophene ring.
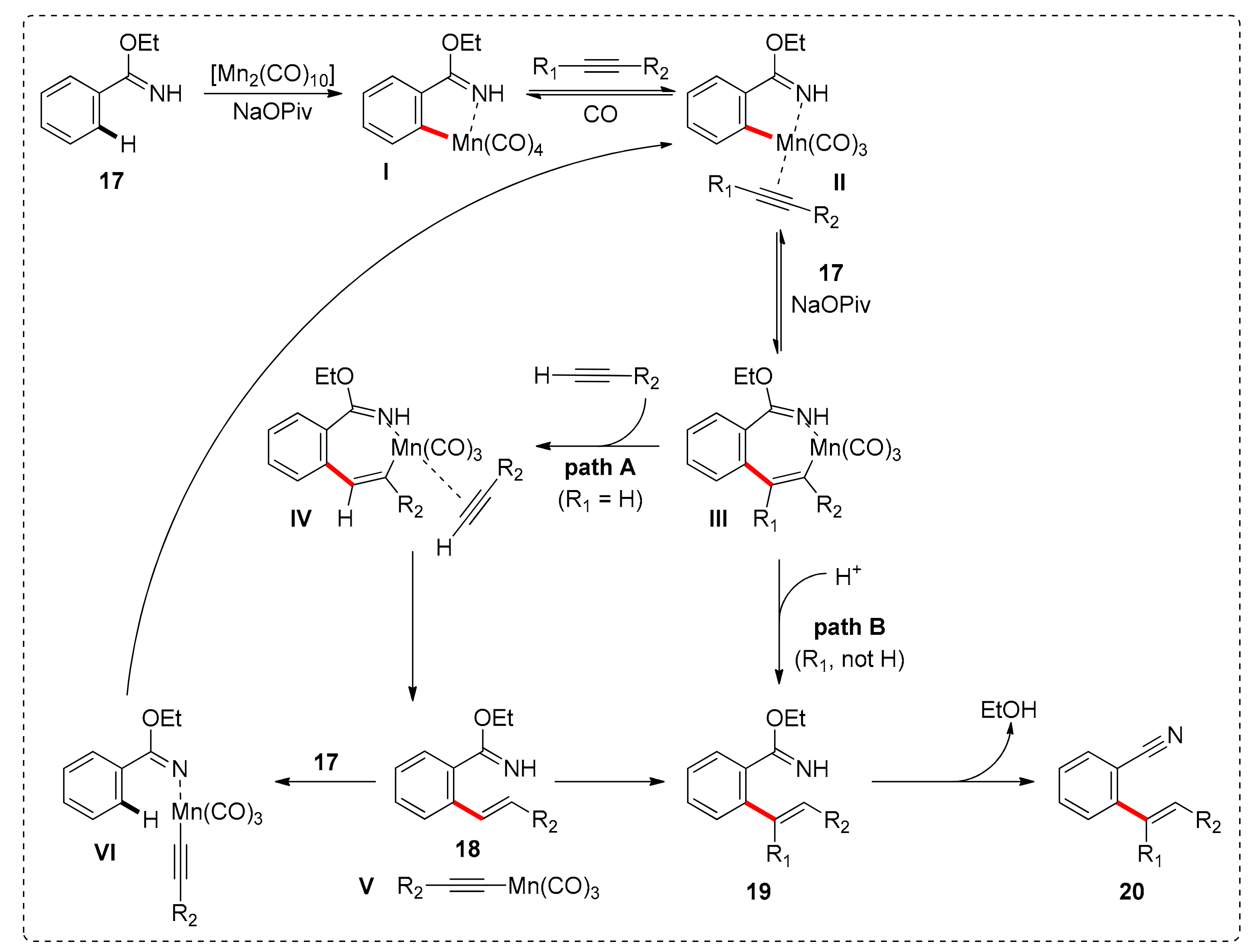
The proposed mechanism (Scheme 2), begins with the ortho-C–H activation of benzimidate 17 aided by NaOPiv, followed by alkyne-insertion in the Mn-Caryl bond. In total, two different routes can be followed towards intermediate III. For a terminal alkyne, coordination of a second alkyne is accompanied by an intramolecular H-shift. Thus, intermediate V is reached along with C–H alkenylated benzimidate 18. Then, V undergoes ligand exchange with 17 to afford VI, which undergoes C–H activation leading to the formation of intermediate II (path A). In case the alkyne is an internal one, 19 can be reached by protonation of the Mn-C bond (path B). Regardless of the followed pathway, intermediate II could regenerate from intermediate III with C–H activation of the substrate 17 with NaOPiv present. Finally, the removal of ethanol from compound 19 leads to the formation of 20.
The second system found in literature utilizing benzimines under Mn catalysis for ortho-C–H activation was reported by Glorius and co-workers in 2017 [84]. This protocol achieves the synthesis of N-heterocycles with controlled regioselectivity by using alkynes incorporating a TDG, functioning both as a chelating agent and as an internal oxidant. Therefore, unsymmetrical alkynes and unpolarized aliphatic alkynes could be used in this method, without the formation of undesired mixtures of positional isomers. It was found that the solvent had an important effect on the efficiency of the reactions, with DMF being optimal. The desired product could be isolated selectively with [MnBr(CO)5] as the catalyst, in 20 mol % catalyst loading, and sodium acetate as a base. The nature of the TDG is important as well, since acetate and hydroxy groups did not promote the transformation. Under the optimized conditions, the imine substrates were initially scrutinized (Scheme 3). It was found that alkyl aryl imines bearing either electron-withdrawing or -donating groups on p- (21–23) and o-position (24) of the benzene ring resulted in the preparation of the targeted isoquinolines in very good yields. Notably, 21 was also prepared through a gram-scale reaction. Meta-substituted alkyl aryl imine gave corresponding product 25 in 63% yield, with C–H activation taking place on the less sterically congested bond. Upon investigating the alkyne scope, the authors found that the R2 group could also be alkyl, apart from aryl (26) and sterically hindered alkyl groups (e.g., cyclopentyl) connected with the tertiary carbon of the propargyl-carbonate, with product 27 obtained in 77% yield. This method was also applicable for a thiophene-containing substrate, which led to 28 in 95% yield. Moreover, various alkyl-substituted propargyl carbonates successfully underwent this reaction, leading to high yields and regioselectivity; notably, these compounds could not be selectively prepared by previously reported C–H activation methods (29, 30). An important aspect of this report is the preparation of valuable isoquinolones, which constitute moieties of natural products and bioactive molecules, achieving regioselectivity in a two-step procedure without the need for isolation of any isoquinoline intermediate (31, 32). Using these conditions, drug moxaverine, used for treatment of functional gastrointestinal disorders, was also synthesized (35).
The proposed mechanism begins with the cyclomanganation of imidate 36 aided by the base, giving rise to manganacycle I, followed by coordination of the carbonyl oxygen atom of the carbonate 37. Insertion of the alkyne in a regioselective manner, followed by β-oxygen elimination gives allene intermediate IV, regenerating the active form of the catalyst. Eventually, the desired isoquinoline 21 is obtained via a selective intramolecular cyclization step (Scheme 3).
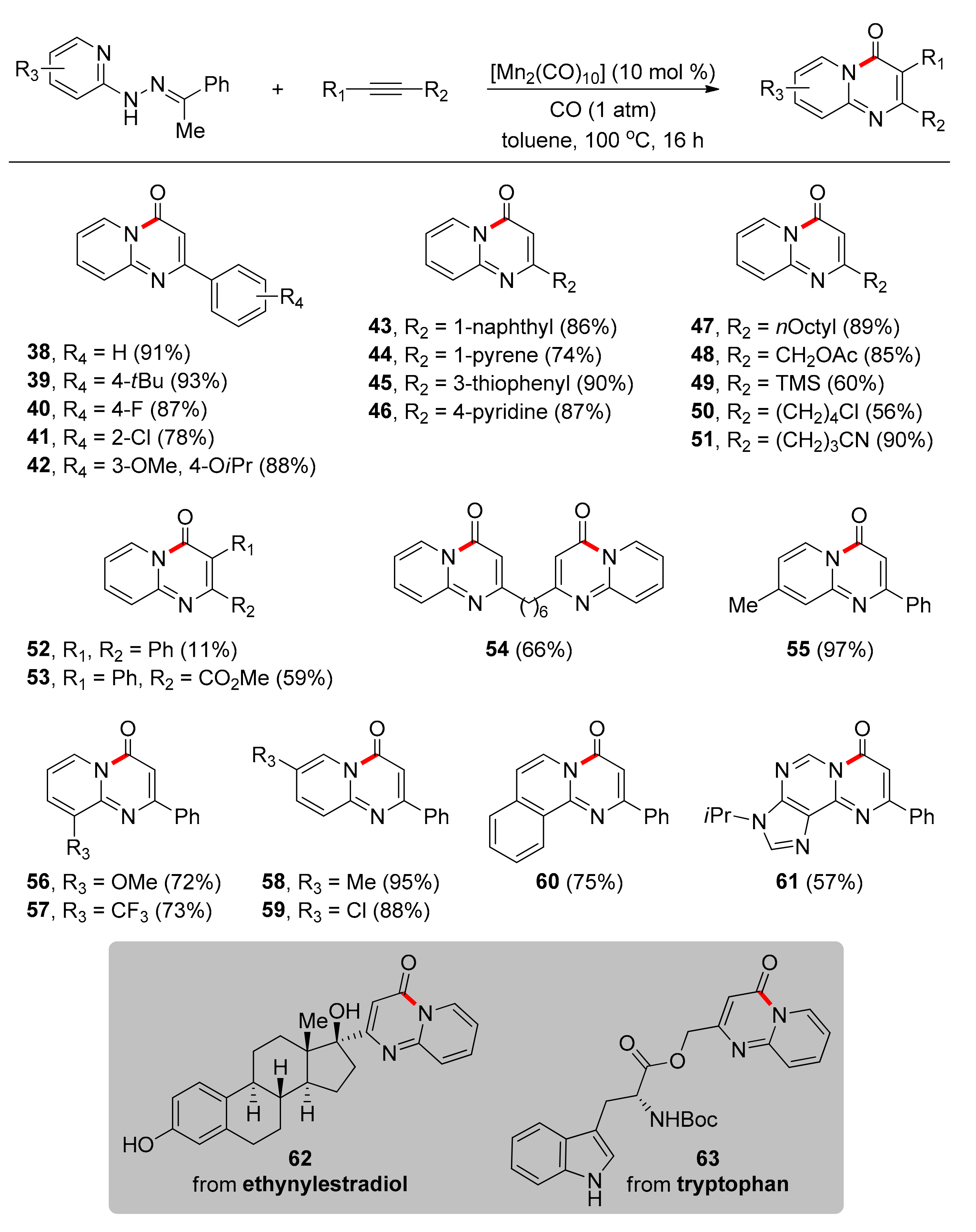
In 2018, Ackermann and colleagues reported on a Mn-catalyzed, carbonylative alkyne annulation protocol with excellent regioselectivity, which can find application in late-stage diversification of a variety of molecules with pharmaceutical interest and natural products such as amino acids, alkaloids, carbohydrates, and steroids [85]. Even though this protocol does not comprise a C–H activation, it utilizes a hydrazone as a TDG and, therefore, is presented herein as it comprises an example for future developments in TDG-assisted C–H activation. Inexpensive, nontoxic and earth-abundant transition metal Mn, specifically [Mn2(CO)10] complex, is used in 10 mol % catalytic loading, as the catalyst, which even outperforms commonly used catalysts based on Co, Ru, Re, Fe, Mo, Rh, Ni, Pd, or Ir. No toxic external metal oxidant and no additive are required, and toluene proved to be the optimal solvent. Under the optimized conditions (Scheme 4), authors investigated the substrate scope, firstly, in regard of alkynes and found that aryl substituted terminal alkynes bearing either electron-withdrawing or -donating groups on various positions of the aromatic ring afforded products in high yields (38–42). Moreover, terminal alkynes with aromatic substituents like naphthyl, pyrene, and heterocycles led to high yields (43–46), while terminal aliphatic substituted alkynes resulted in low to high yields (47–51). There were only a few examples for internal alkynes with yields being low to moderate (52, 53). Diynes proved to be viable substrates, as 54 was delivered in a single step in 66% yield. Concerning the pyridine moiety, substitution on various positions of the aromatic ring with either electron-withdrawing or electron-donating groups did not have an impact on the efficiency of the reaction (55–59). Challenging aromatic systems participated in the reaction smoothly (60, 61). Finally, this protocol was applicable to the late-stage diversification of several structurally challenging molecules related to pharmaceutical and medicinal chemistry (62, 63), a fact that is of paramount importance for the development of biologically active molecules.
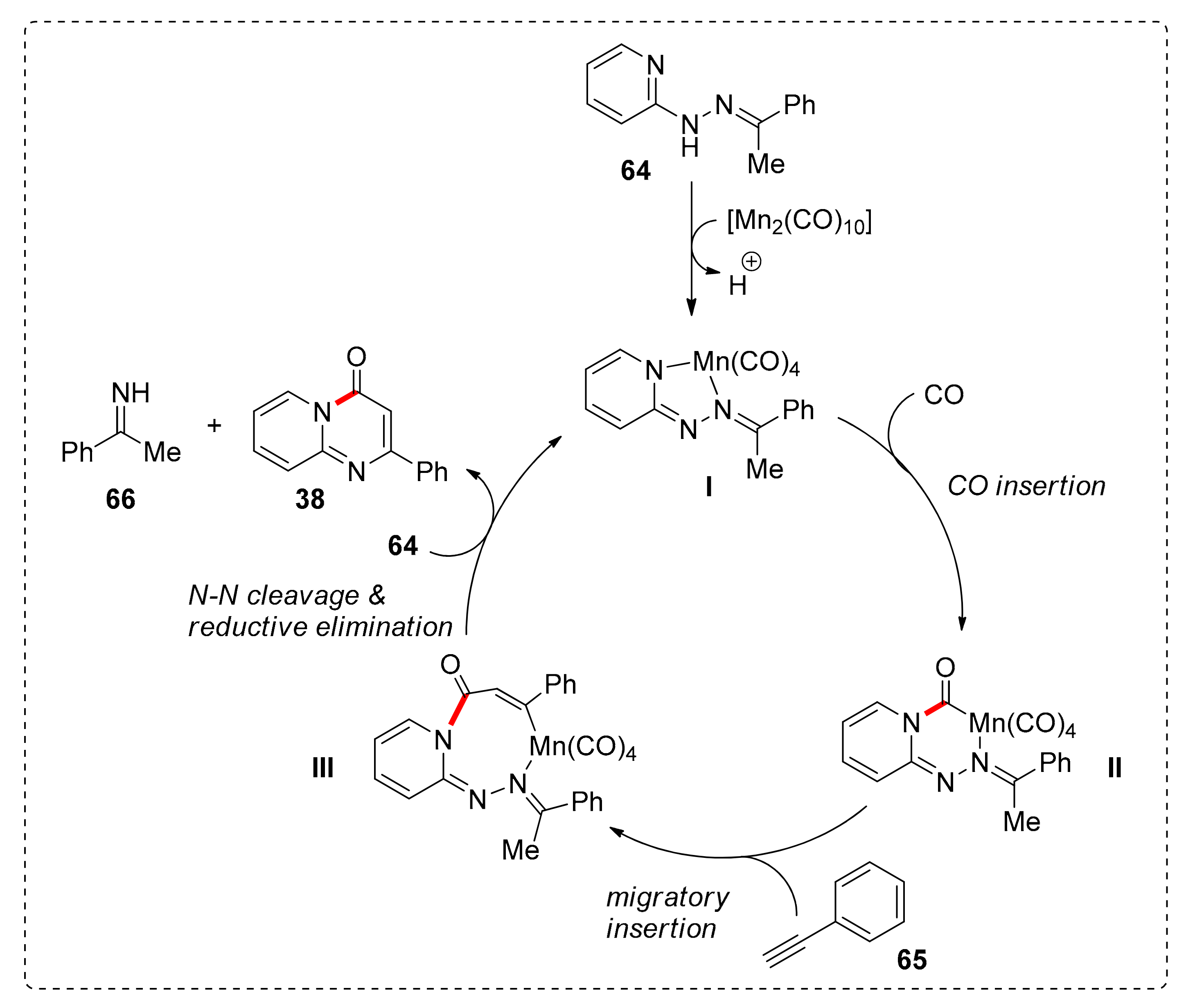
The proposed catalytic pathway (Scheme 5) commences with the coordination of the Mn catalyst to the hydrazone 64, followed by insertion of CO to the Mn–Npyrimidine bond to form intermediate II. Then, a migratory insertion of phenylacetylene leads to the eight-membered metallacycle III. N–N bond cleavage and reductive elimination completes the catalytic cycle.
3. Iron
Iron is about 56 million times more abundant than Ru and Os [86] and has the largest natural abundancy in earth’s crust, among all transition metals. It is intensively investigated for its catalytic activity in various transformations. Its low cost and biocompatibility are very attractive features, ideal properties for catalysts used in large-scale applications. Its two most stable oxidation states, Fe(II) and Fe(III), characterized as hard and borderline acids in the HSAB theory, [87] can coordinate with several donor atoms and form stable metal complexes. Iron’s abundancy and chemical versatility has been exploited by nature, which involves iron in various forms in enzymes. For example, iron in high oxidation states activate dioxygen to form oxo- and peroxo-intermediates. These intermediates can be observed in metalloenzymes, such as methane monooxygenase hydrolase (MMOH), which can activate the C–H bond in methane [88,89]. The rich chemistry of iron in C–H activation has been reviewed recently [40,90]. Herein, we discuss some representative examples of C–H activation with TDGs under iron catalysis. N-oxides are the coordinating directing group for the catalyst, despite the fact that in the literature there are cases where N-oxides undergo [3 + 2] dipolar cycloaddition reactions with activated olefins, catalyzed by iron or ruthenium complexes, leading to isoxazolidine regioisomeric mixtures [91,92].
Transformations with N-oxide as TDG
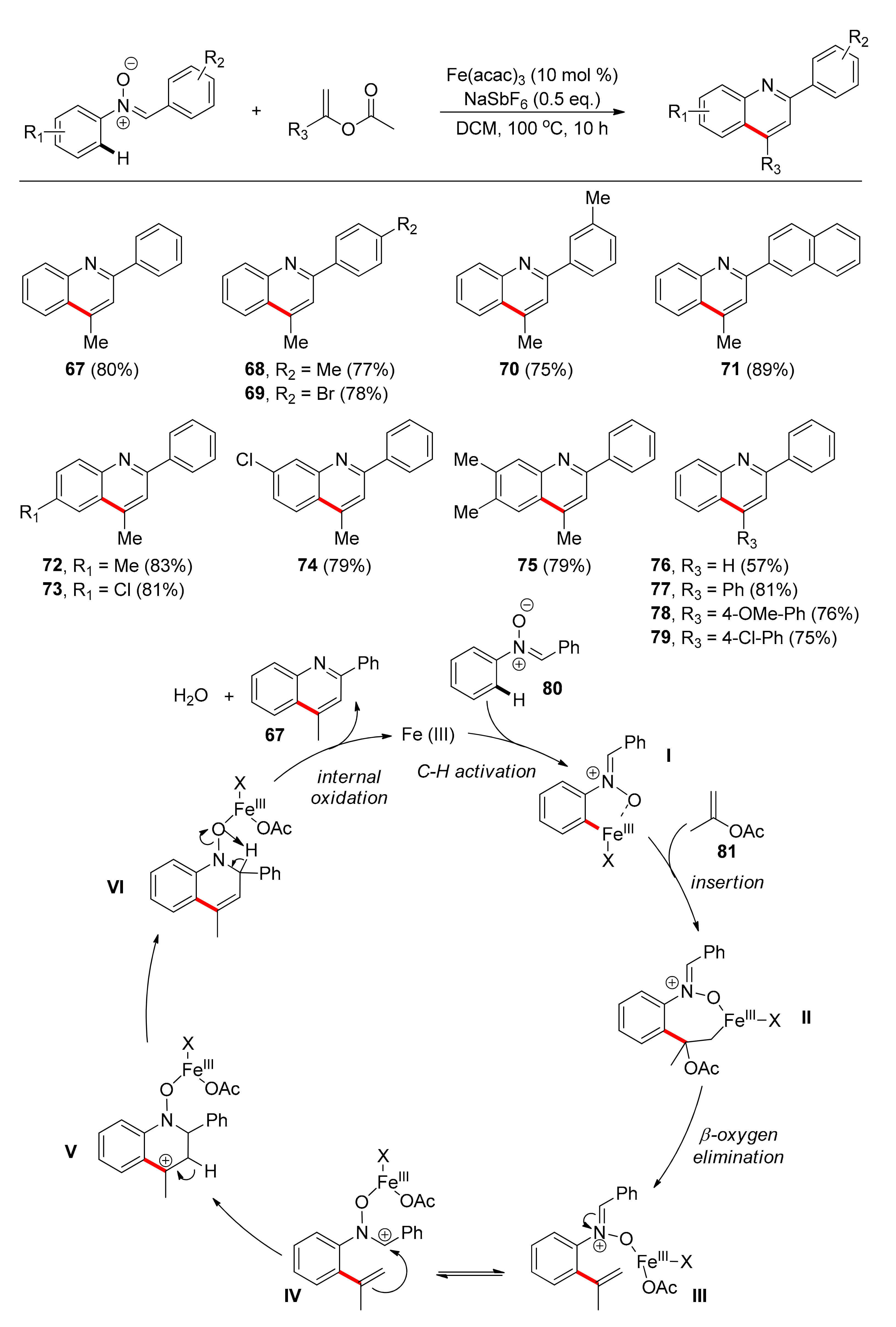
In 2016, Cheng, Shao, and colleagues developed a strategy for the intermolecular [4 + 2] arylnitrone cyclization, with geminal-substituted vinyl acetates under Fe catalysis [93]. This strategy led to 2,4-disubstituted quinolines. Mechanistic studies showed that a C–H activation process is taking place. Interestingly, in this protocol, N-oxide serves both as the directing group, which is absent in the final product, and as the internal oxidant, based on the inherent oxidizing nature of N–O bond in nitrones. The use of Fe(III) salts instead of Fe(II) was more beneficial, with the inexpensive Fe(acac)3 being the catalyst of choice in 10 mol % loading. Addition of silver salts promoted the reaction, but NaSbF6 as additive resulted in higher yields in dichloromethane. Investigation of various arylnitrones (Scheme 6), concluded that either electron-withdrawing or electron-donating groups on various positions of the phenyl-imino moieties yielded the desired quinolines in high yields (67–71). Derivatives bearing groups of either electron-withdrawing or -donating ability at either the para- or the meta- position of the phenyl ring bearing the N-O group gave the corresponding products in approximately 80% yield (72–75). Attempts with heterocyclic-substituted nitrones failed, most probably owing to the strong metal-heteroatom coordination. The same was true with substrates bearing ortho-substitution, on either phenyl ring, probably due to steric hinderance. Access to 2-alkylquinolines was also not successful. Concerning the substrate scope in regard of the vinyl acetates, which are used as acetylene equivalents, the authors found that in the absence of an aryl group the yield was moderate (76), while phenyl-substituted vinyl acetates gave yields around 80% (77–79). This discrepancy in reactivity was attributed to the formation of a stable intermediate carbocation (V) in the proposed mechanism (Scheme 6).
A plausible mechanism commences with the activation of the C–H bond by Fe(III) at the ortho- phenyl ring position of 80, with N-oxide acting as the directing group. Insertion of vinyl acetate 81 to the C-Fe bond, followed by β-oxygen elimination, affords intermediate III, which through resonance generates intermediate IV. Intramolecular nucleophilic addition between the alkene and the carbocation, followed by β-H elimination, affords the quinoline N-oxide VI. Lastly, internal oxidation leads to the cleavage of the N-O bond, affording the desired quinoline 67 and the active form of Fe(III) catalyst, completing the catalytic cycle. Water and acetic acid are the only by-products, leading to a “green” catalytic transformation (Scheme 6).
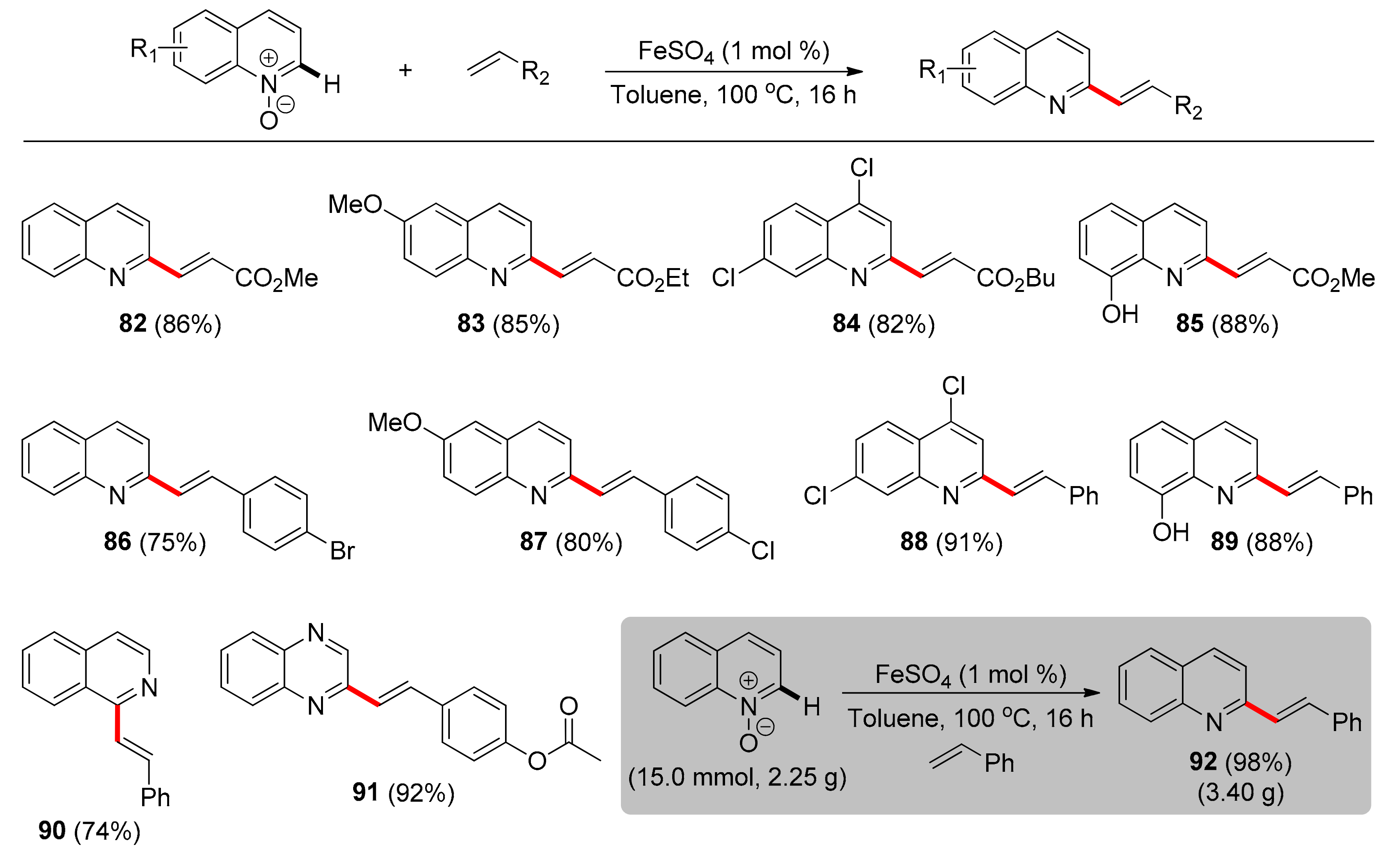
The first iron-catalyzed, C2-selective alkenylation of quinoline-N-oxides was reported by Vaccaro and co-workers in 2021 [94]. This strategy provides access to the direct alteration of an important heterocyclic substructure found both in various biologically active molecules and natural products. Easily accessible FeSO4 salt was used as a catalyst, in loading of 1 mol %, as FeSO4 outperformed different iron, manganese, cobalt, copper, and palladium catalysts, in terms of selectivity and isolated yield. Solvent-free conditions or solvents such as DMF and 1,4-dioxane resulted in decreased yields and poor selectivity in comparison to performing the reaction in a highly concentrated toluene solution (20 M), at 100 °C. An interesting fact is that external oxidants were not utilized because of the intrinsic oxidizing properties of N-oxides towards the catalyst, apart from operating as a weakly-coordinating directing group. Another fact of high importance is the high level of sustainability in this procedure, as H2O comprises the sole by-product and iron is more abundant and less hazardous compared to other metals. Under the optimal conditions (Scheme 7), the substrate scope was initially studied with regards to quinoline-N-oxides, leading to the conclusion that quinoline-N-oxides with either electron-donating or -withdrawing groups reacted regioselectively in yields ranging from 70% to 88% (82–85), while amenable groups like methoxy, chloride and hydroxy could serve as handles for further functionalization. Moreover, concerning the alkene moiety, acrylates with alkyl substituents reacted efficiently with marginal differences in product yields, while substituted or unsubstituted styrene derivatives were also converted, leading to the desired products in high yields (86–89). Notably, isoquinoline-N-oxide and quinoxaline-N-oxide were subjected to the reaction conditions and both led to corresponding products 90 and 91 with excellent selectivity. The effectiveness of this protocol is demonstrated by a multigram-scale reaction, where product 92 was obtained in excellent yield.
The proposed mechanism commences with the C–H bond activation of the ortho position of quinoline-N-oxide, with N-oxide as the directing group. Metal oxidation followed by the insertion of the alkene into the C-Fe bond and H-abstraction lead to the anticipated product, while H2O is also produced.
4. Cobalt
Cobalt can be also regarded as a relatively low-toxicity transition metal, given that it is assumes a vital role in many living organisms as a component of cobalamin (vitamin B12) and other biorelevant coordination compounds [39,95,96]. Its natural abundance, variety of stable oxidation states, rich coordination chemistry and wide availability provide ample opportunities for use in catalysis [39]. As a result, cobalt compounds have been employed as catalysts in a numerous industrially important transformations and have many uses in organic synthesis [39]. The properties of cobalt differ significantly from those of the platinum-group metals, and cobalt–carbon bonds are generally more nucleophilic [1], thus accounting for the abundance of applications of cobalt in organometallic C–H activation [39,95,96]. The first DG used in cobalt-catalyzed C–H functionalization was an imine group, facilitating the C–H carbonylation at the ortho position of the substrate, using high pressures of carbon monoxide and Co2(CO)8 [97]. This early report by Murahashi and co-workers showcased the potential of cobalt in C–H carbonylation, a transformation in which cobalt catalysis still holds a prominent position [98], notably by also utilizing TDGs. In general, cobalt has become one of the most broadly studied transition metals in C–H activation [39,95,96] and its modes of action are well understood [50], as exemplified by the rational design and use of ligands in many cases [39,95,96,99]. An important observation, especially with regards to the design of TDGs in cobalt catalysis, is that specific transformations require the use of low-valent or high-valent cobalt species [39,100]. The former may be well-defined, usually Cp*-containing Co(I) complexes in recent years [39], or they may be formed in situ by the reduction in simple Co(II) salts. The latter may also be well-defined Co(III) complexes (stabilized by Cp*-type ligands in most cases examined herein) or they may be generated in situ by oxidizing Co(II) salts, or simply by the use of a Co(III) salt [39,95,96]. Since the DG essentially acts a ligand for the active species, there are limitations with regards to the oxidation state of the metal which is most appropriate for the transformation. Co(I) requires simple N-donors, while Co(III) usually requires multidentate DGs, unless it is stabilized by Cp*-type ligands, which however is a feature that renders the catalysts far less cost-efficient as their synthesis is more demanding.
For the purposes of this review, and because of the wide variety of DGs used in Co-catalyzed C–H activation, which has been discussed in recent review articles [86,87,88], only cases where the DG is cleaved or removed during the reaction will be discussed herein. Cases in which the DG is removed after the reaction, or in a one-pot fashion, are not considered to be cases of truly traceless DGs, which we consider to be those that are either removable because of the reaction conditions and characteristics, transiently formed or incorporated in the final target after performing their task, without retaining their properties and structural characteristics. Furthermore, these groups should exhibit a directing function and not only be cleaved from the molecule during the reaction. Therefore, DGs such as N-p-methoxyphenyl (PMP) or esters, amides, imine and oxime groups, pyrimidine and any modifiable DGs that have been used in cobalt catalysis are not considered to be traceless DGs for the purposes of this review [39,95,96,101].
4.1. C–H Carbonylation with TDGs
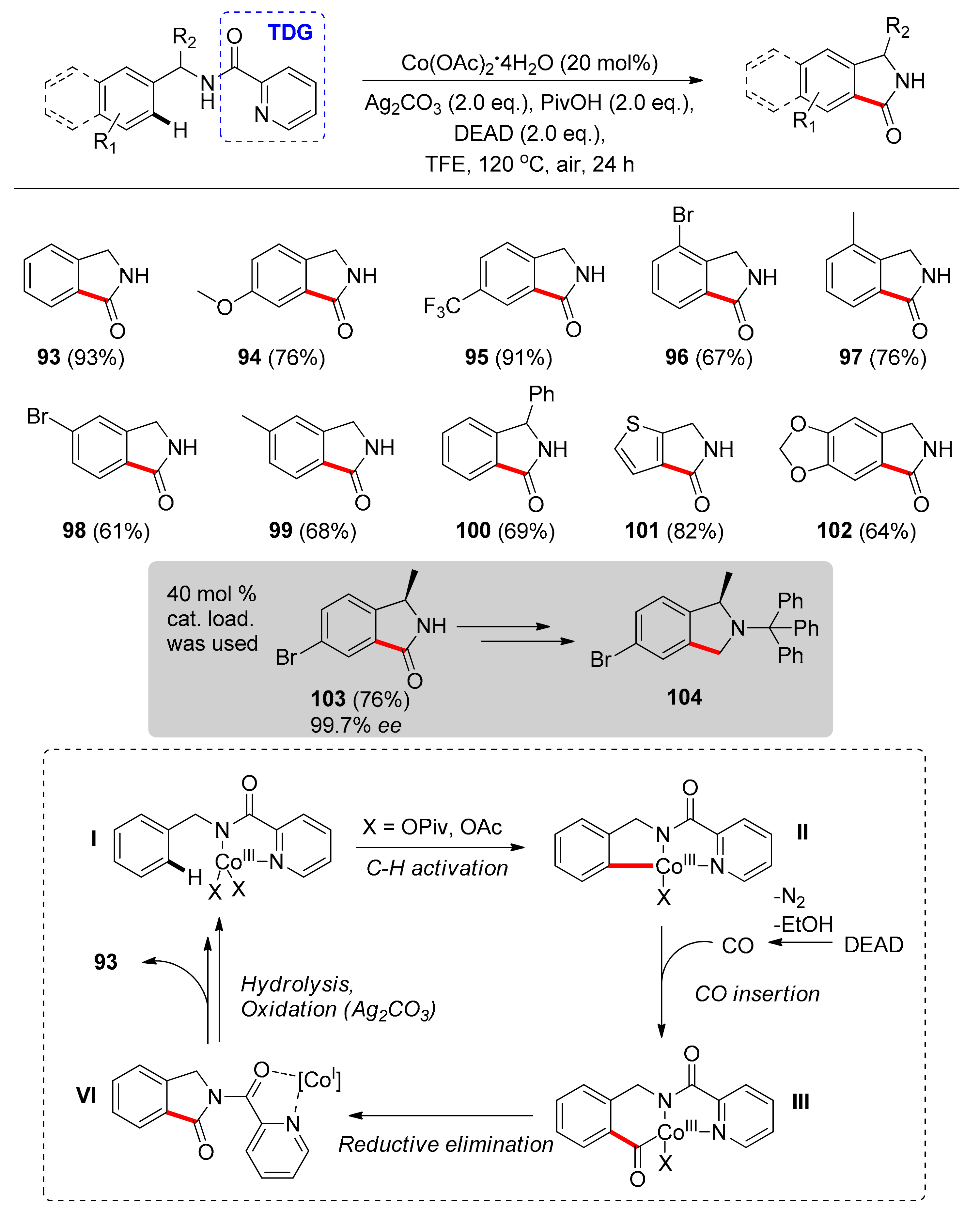
The first system to utilize a TDG for C–H carbonylation under Co catalysis, and in general under first row transition metal catalysis, was reported by Zhong and co-workers in 2017 [102]. This system relies on the use of picolinamide as a TDG, which acts as a monoanionic, bidentate ligand, stabilizing the Co(III)-based active species. Notably, the active form of the catalyst is generated in situ from the simple and cost-efficient Co(OAc)2·4H2O salt, which is used in 20 mol % catalyst loading, while the source of carbon monoxide is the DEAD (diethyl azodicarboxylate) reagent. Another advantageous feature of this system is that the reaction is performed under air; however, a silver-based oxidant is still required (Ag2CO3), which was found to be superior to other oxidants. Furthermore, pivalic acid (PivOH) is a key additive, promoting the reaction by most likely generating the active Co species and possibly aiding the hydrolysis and removal of the TDG, while trifluoroethanol (TFE) was found to be the optimal reaction solvent. Under the optimal conditions, the TDG-assisted C–H carbonylation of structurally diverse benzylamines and (hetero)aromatic amines led to various N-unsubstituted isoindolinones in moderate to high yields in a versatile way (Scheme 8). While exploring the substrate scope, the authors found that neutral or electron-withdrawing group-para-substituted benzylamines lead to higher yields than benzylamines with electron-donating aryl substituents (93–95). When the ortho or meta positions were examined, the results were the opposite (96–99); however, the difference in yield was not profound and in general the protocol is robust and tolerant towards functional groups and substitution patterns. Notably, complete regioselectivity was observed in the cases of compounds 98 and 99, a fact which was attributed to steric hindrance. Compounds bearing α-amino substituents, heteroaryl groups, or synthetically valuable functional groups were also obtained efficiently (100–102). A highlight of this report was the use of the catalytic system in order to access a valuable intermediate in the formal synthesis of (+)-garenoxacin (104), which leads to the final target molecule in two additional steps. Thus, by using a 40 mol % catalyst loading and 3.0 equivalents of DEAD, compound 103 was obtained in 76% yield and 99.7% enantiomeric excess (ee) on a gram scale, demonstrating the scalability and synthetic value of the catalytic system, as well as the practicality of C–H activation in organic synthesis.
The proposed mechanism, commencing with the in situ generation of the active Co(III) species I (Scheme 8), stabilized by the bidentate, anionic TDG. C–H metalation, followed by insertion of the in situ generated CO into the Co–C bond, and then reductive elimination and hydrolysis of the TDG lead to the targeted product and oxidation of the Co(I) species by silver, along with coordination of the substrate, regenerating the active species. Interestingly, a different pathway was postulated, involving an ester radical formation from DEAD and a Co(VI) metallacycle, leading to an intermediate with an ethyl ester group in the ortho position of the benzylamine framework. However, the postulated intermediate was synthesized and subjected to the reaction conditions, leading to only 10% conversion to the desired product, a result which suggests that the alternative pathway is unlikely to occur.
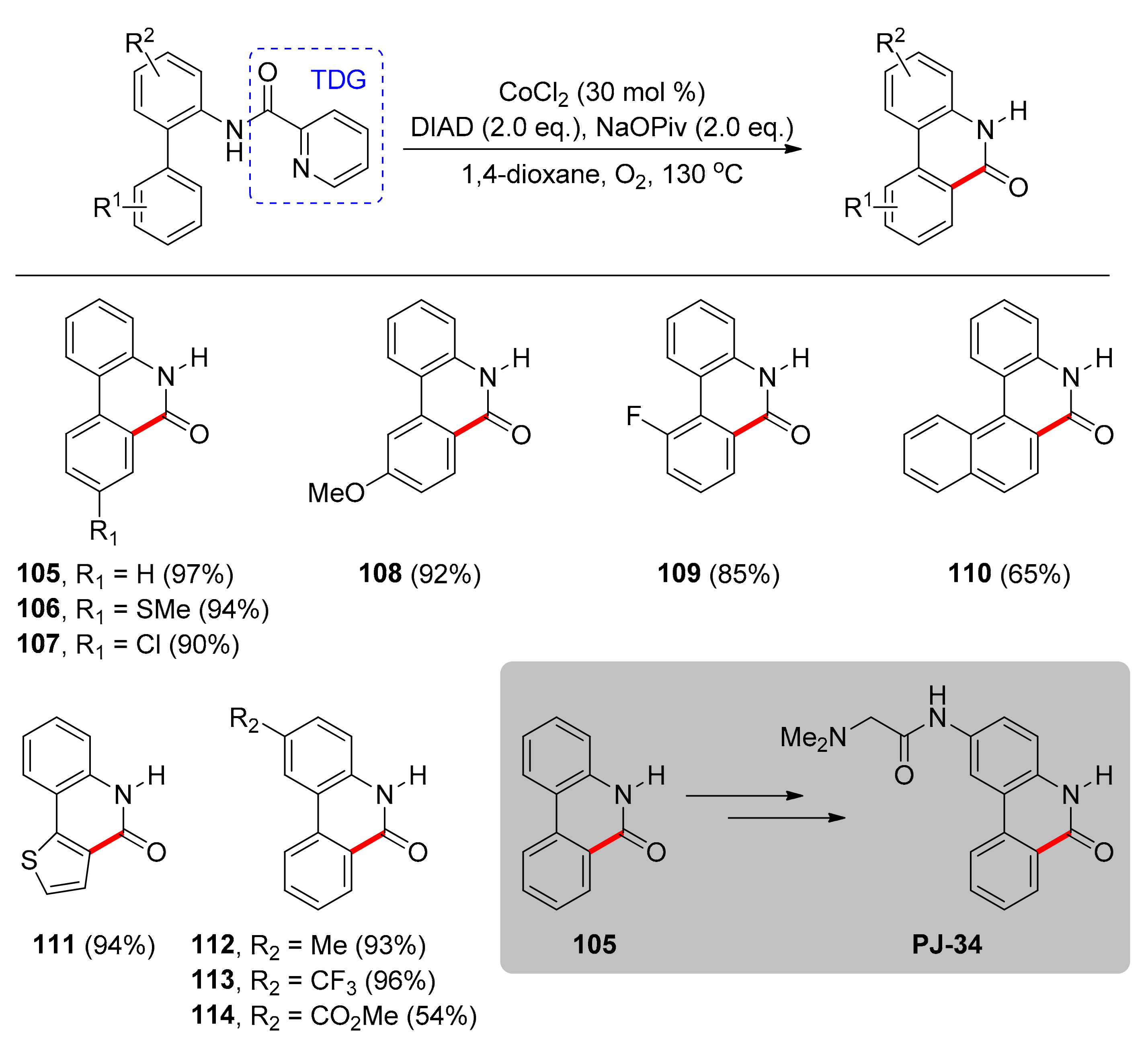
The second system found in the literature, utilizing a TDG for cobalt-catalyzed C–H carbonylation, was reported again by Zhong and collaborators in 2018 [103]. This protocol refers to synthesis of free (NH)-phenanthridinones via C–H carbonylation of o-substituted arylanilines. Phenanthridinones can be found in biologically active alkaloids and pharmaceuticals. This system relies on the use of picolinamide as a TDG, which acts as a monoanionic, bidentate ligand, stabilizing the Co(III)-based active species. Notably, the active form of the catalyst can be generated in situ from the earth abundant and cost-efficient CoCl2 salt in 30 mol % catalyst loading, after oxidation of Co(II), and Co(I), to Co(III) by oxygen. Furthermore, the source of carbon monoxide is the DIAD (diisopropyl azodicarboxylate) reagent, which is an environmentally benign carbonyl source, while sodium pivalate (NaOPiv) is a key additive, promoting the reaction by most likely generating the active Co species, acting as the base for the C–H activation step and possibly aiding subsequent steps of the catalytic circle. 1,4-Dioxane was the best solvent, although solvents had little influence on yield. Changing temperature from 130 °C did not increase the yield, therefore 130 °C was chosen as ideal, whereas blank experiments pointed out that the Co catalyst, oxygen and stoichiometric amount of the additive (2.0 equivalents) are required. Under the optimal conditions, the TDG-assisted C–H carbonylation of structurally diverse ortho-arylanilines led to various N-unsubstituted phenanthridinones in high yields (Scheme 9). Upon experimentation on the substrate scope, the authors found that o-arylanilines with neutral (105), electron-donating (106), or electron-withdrawing substituents (107) on the para position of the ortho-aryl ring provided the targeted products in 90%−97% yield. Regarding meta-substituted substrates, regioselectivity was observed and products were generated from carbonylation of the less sterically congested C–H bond (108). However, the reaction was not as competent against ortho-substituted substrates, where products were received in lower than 90% yields (109), due to steric effects. Thienyl- and naphthyl-bearing moieties could be also used, affording products 110 and 111 in 65% and 94% yields, respectively. Substitution on the aniline ring showed that electronic effects do not have a significant impact on the reaction yields (112, 113), with one exception being o-phenylaniline bearing an ester group (114). This method can be scaled-up, as demonstrated by its application to the formal synthesis of PARP inhibitor PJ-34 on a gram scale, where product 105 is an intermediate and after two additional steps (76% and 87% yield, respectively) the target molecule is obtained.
A possible mechanism was also proposed by the authors for this reaction. In the first step, Co(II) coordinates with benzylpicolinamide and then this intermediate is oxidized by oxygen to form a Co(III) complex, which is stabilized by the bidentate, anionic TDG. Then, C–H activation of the ortho-phenyl group occurs, followed by insertion of the in situ generated CO into the Co–C bond, reductive elimination, and hydrolysis of the TDG. These lead to the desired product and Co(I) species, which is oxidized by oxygen again, to the active Co(III) species, so that the catalytic cycle is completed.
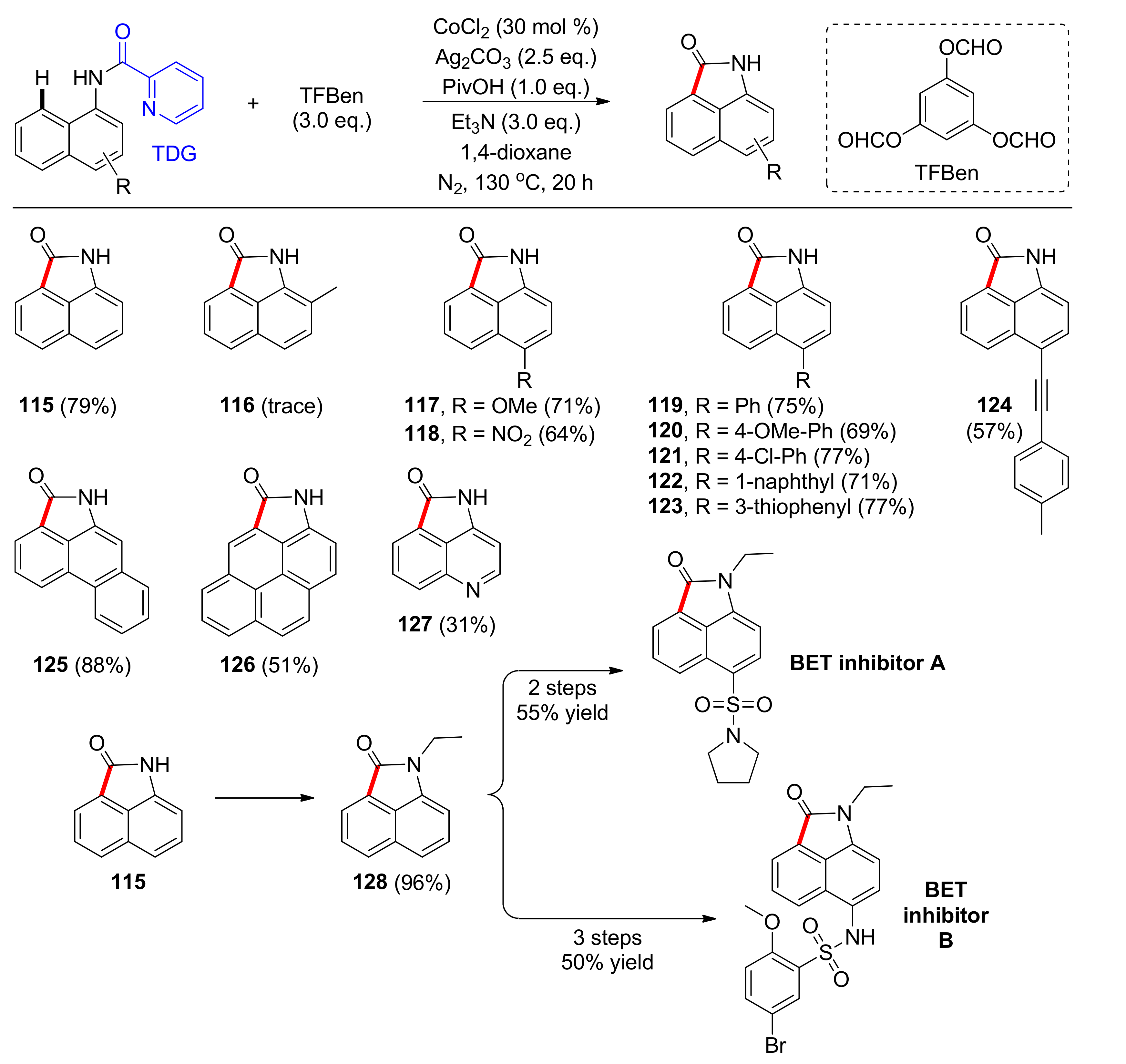
Another work concerning cobalt-catalyzed C–H carbonylation using a TDG was reported by Zhong, Wu, and colleagues in 2019 [104]. This article refers to the direct synthesis of free (NH)-benzo[cd]indol-2(1H)-ones, scaffolds found in biologically relevant compounds, from naphthylamides in moderate to high yields (Scheme 10). This method includes the picolinamide TDG, the cost-effective CoCl2 salt in 30 mol % catalyst loading and Ag2CO3 as the only oxidant under N2 atmosphere for oxidation of Co(I) and Co(II) to generate the active form of the catalyst. Moreover, the reaction takes place by utilizing benzene-1,3,5-triyl triformate (TFBen) as an easily handled, solid carbon monoxide source and the acidic additive PivOH which promotes the reaction to higher yields by most likely generating the active Co species and possibly aiding the removal of the TDG. Higher yields are also observed when using Et3N and 1,4-dioxane at 130 °C for prolonged reaction time, even though 50% conversion was achieved after 1 h. Under the optimized conditions, structurally diverse naphthylamides were tested and it was observed that a naphthylamide with a methyl group at the C2 position led to only traces of the product, implying that steric hinderance has a detrimental effect (116). On the other hand, when the C4 position of the naphthylamide is substituted, the isolated yields are moderate to high for substrates bearing substituents that are electron-donating (117) or electron-withdrawing (118), phenyl groups bearing either electron-withdrawing or -donating moieties (119–121), naphthyl groups (122), five-membered aromatic rings (123), and a synthetically valuable alkyne unit (124). For compounds having more conjugated systems (125, 126), yields are also moderate to high and, significantly, the conditions could be employed for substrates bearing either a quinoline or an isoquinoline unit (127). The versatility of this method is highlighted by a gram-scale preparation for the total synthesis of BET bromodomain inhibitors A and B, which exhibit antitumor and anti-inflammatory activities, via intermediate 115 in two and three steps, respectively. Furthermore, studies on the DG indicated that the existence of a nitrogen atom on it was crucial for the coordination of the DG with the cobalt catalyst in C–H carbonylation of naphthylamides. Finally, mechanistic investigations suggested that a radical process is not involved; therefore, it was concluded that the Co(II) catalyst coordinates with the naphthylamide. Then, it gets oxidized by Ag(I), generating the Co(III) complex. After selective C–H bond activation at the C8 position of the naphthylamide, followed by coordination of carbon monoxide generated in situ from TFBen, reductive elimination to provide the Co(I) complex, and hydrolysis that gives the product and generates the Co(I) species, that is oxidized by Ag(I) to close the cycle.
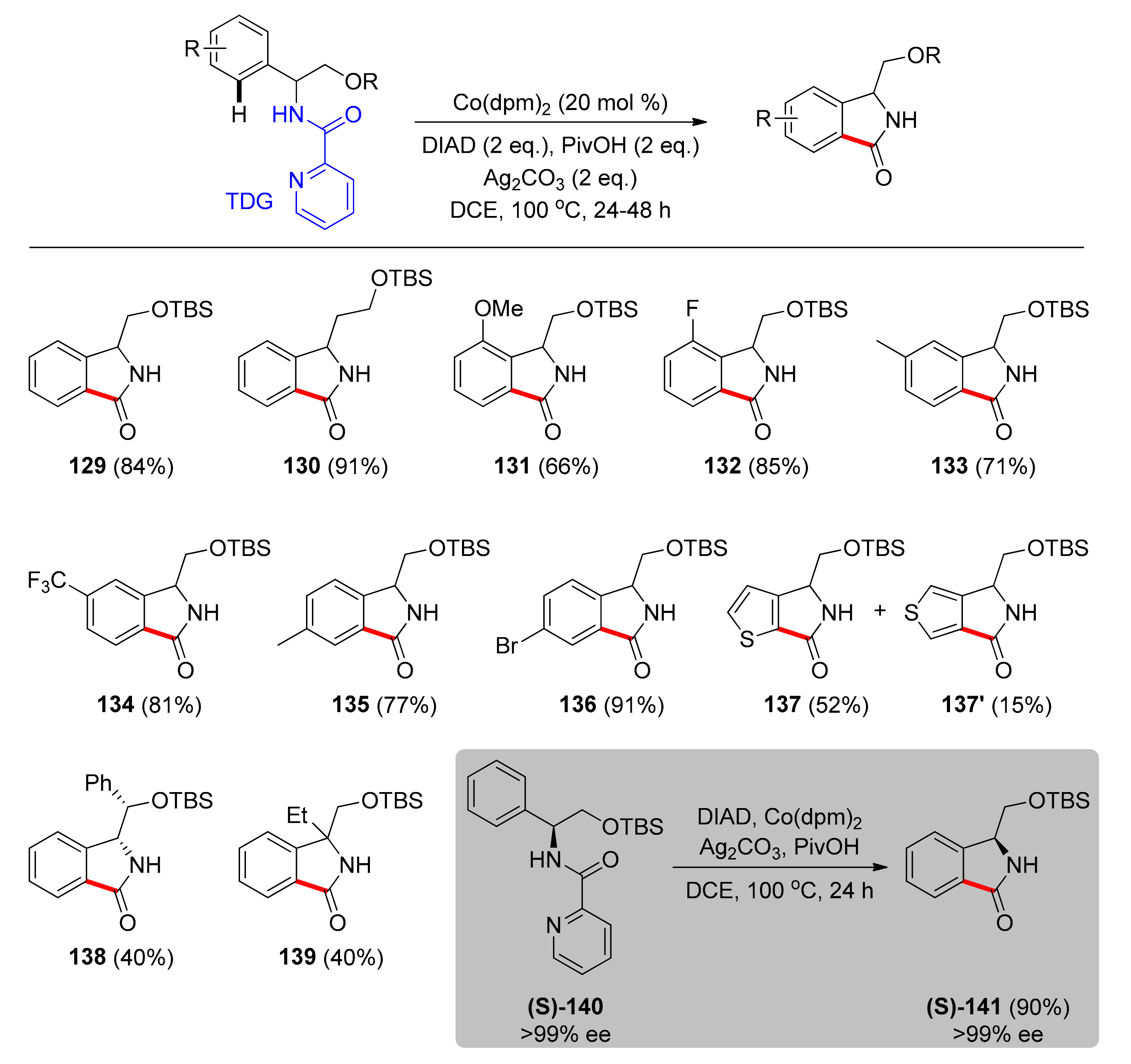
The latest report on cobalt-catalyzed C–H carbonylation concerns the synthesis of 3-hydroxymethyl isoindolinones from phenylglycinol starting materials with picolinamide as a TDG. This was a study performed by Grigorjeva and coworkers in 2020 [105]. 3-Substituted isoindolinones, especially hydroxymethyl and 3-hydroxyethyl isoindolinones, are common fragments present in natural alkaloids and biologically active compounds. Nevertheless, approaches toward the synthesis of enantiopure 3-substituted isoindolinone derivatives are limited. In this synthetic route, the DIAD (diisopropyl azodicarboxylate) reagent was found to be superior than the DEAD reagent (diethyl azodicarboxylate) as the carbon monoxide source, while commercially available [Co(dpm)2] (dpm = bis(2,2,6,6-tetramethyl-3,5-heptanedionato) was used as a catalyst in 20 mol % loading. Addition of stoichiometric amounts of Ag2CO3 as the oxidant and PivOH showed satisfying increase in reaction yield and DCE was chosen as the best performing solvent. Using the optimized conditions, the authors investigated the substrate scope (Scheme 11). Their first conclusion was that O-unprotected phenylglycinol was completely unreactive, therefore usage of O-protecting groups is necessary (129). Moreover, a β-phenylalaninol derivative was an amenable substrate that gave product 130 in excellent yield. Phenylglycinol derivatives substituted on para, meta, and ortho positions of the benzene ring, with electron-donating, electron-withdrawing, and halogenated substituents were also tested, giving moderate to high yields (131–136), a fact that reveals the wide applicability of the system. Specifically, meta-substituted substrates led to carbonylation products formed via the reaction of the less hindered C−H bond with excellent regioselectivity. In the case of a thiophene amino alcohol derivative, no selectivity was observed, and both regioisomers (137, 137΄) were isolated. Finally, a sterically hindered amide led to carbonylation product 138, as well as a quaternary amino alcohol led to 139 in synthetically useful yields. A highlight of this report is that enantiopure (S)-phenylglycinol derivative (S)-140 was subjected to the carbonylation conditions and gave product (S)-141 with completely preserved stereochemistry.
4.2. Synthesis of Isoquinolines with TDGs
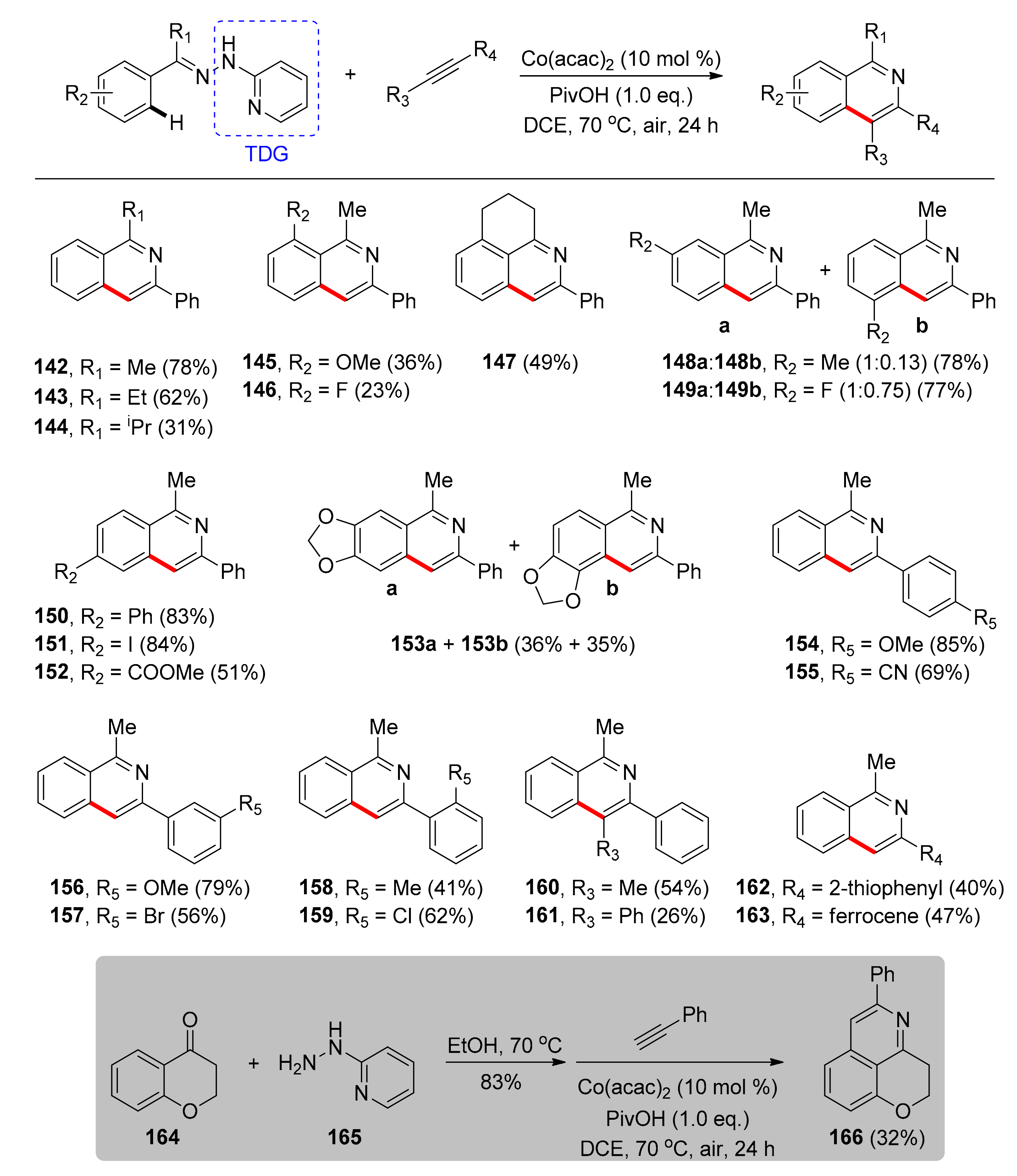
The first cobalt-catalyzed bidentate TDG-enabled synthesis of isoquinolines was reported by Zhu and co-workers in 2016 [106]. Isoquinolines were efficiently synthesized using the cost-efficient, commercially available [Co(acac)2] complex in 10 mol % catalyst loading and acidic additive PivOH, which increased the reaction yield, in DCE at 70 °C without the need for an inert atmosphere. This protocol is applicable to the synthesis of isoquinolines through 2-hydrazinylpyridine-directed coupling with terminal or internal alkynes. Under optimal conditions and based on the results of the substrate scope investigation (Scheme 12) the authors initially concluded that the steric bulk of the hydrazone group significantly affects the reaction yield (142–144). Ortho substitution led to low yields regardless of the electronic characteristics (145, 146), a characteristic attributed to the difficulty of the cobalt center to be placed in the ideal geometry. When the ortho-substituent and the hydrazone group were connected, the reactivity could partially increase (147), while in the case of meta-substitution yields lower than 80% regardless of the electronic characteristics were obtained, with regioselectivity favoring C–H activation at the less sterically congested site (148, 149). Moreover, diverse electron-donating or -withdrawing groups on the para position of the phenyl ring (150–152) showed the potential of this method, as products were mainly obtained in high yields. Disubstituted substrates afforded the products in good yields (153). Furthermore, the substrate scope for alkynes was examined and it was found that arylacetylenes bearing both electron-donating and electron-withdrawing functional groups at the para position (154, 155), as well as at the meta position (156, 157) led to products in satisfying yields. On the other hand, ortho-substituted arylacetylenes were generally less reactive (158, 159). It was found that internal alkynes (160, 161), thiophenylacetylene (162) and the bulky ferrocenylacetylene (163) were also compatible substrates for this reaction. One final example shows the high potential of this synthetic method since a natural compound, 4-chromanone 164, was used as a substrate leading to compound 166.
Mechanistic experiments showed that [Co(acac)2] initially reacts with oxygen to generate a catalytically active species which coordinates with the substrate. Then, ortho-C–H activation takes place, followed by insertion of the alkyne to give rise to an alkenyl-cobalt intermediate, and concurrent nucleophilic attack of the Co–C bond on the nitrogen atom linked to the hydrazone carbon atom via a double bond. N–N bond cleavage and regeneration of the cobalt catalyst close the catalytic cycle [106].
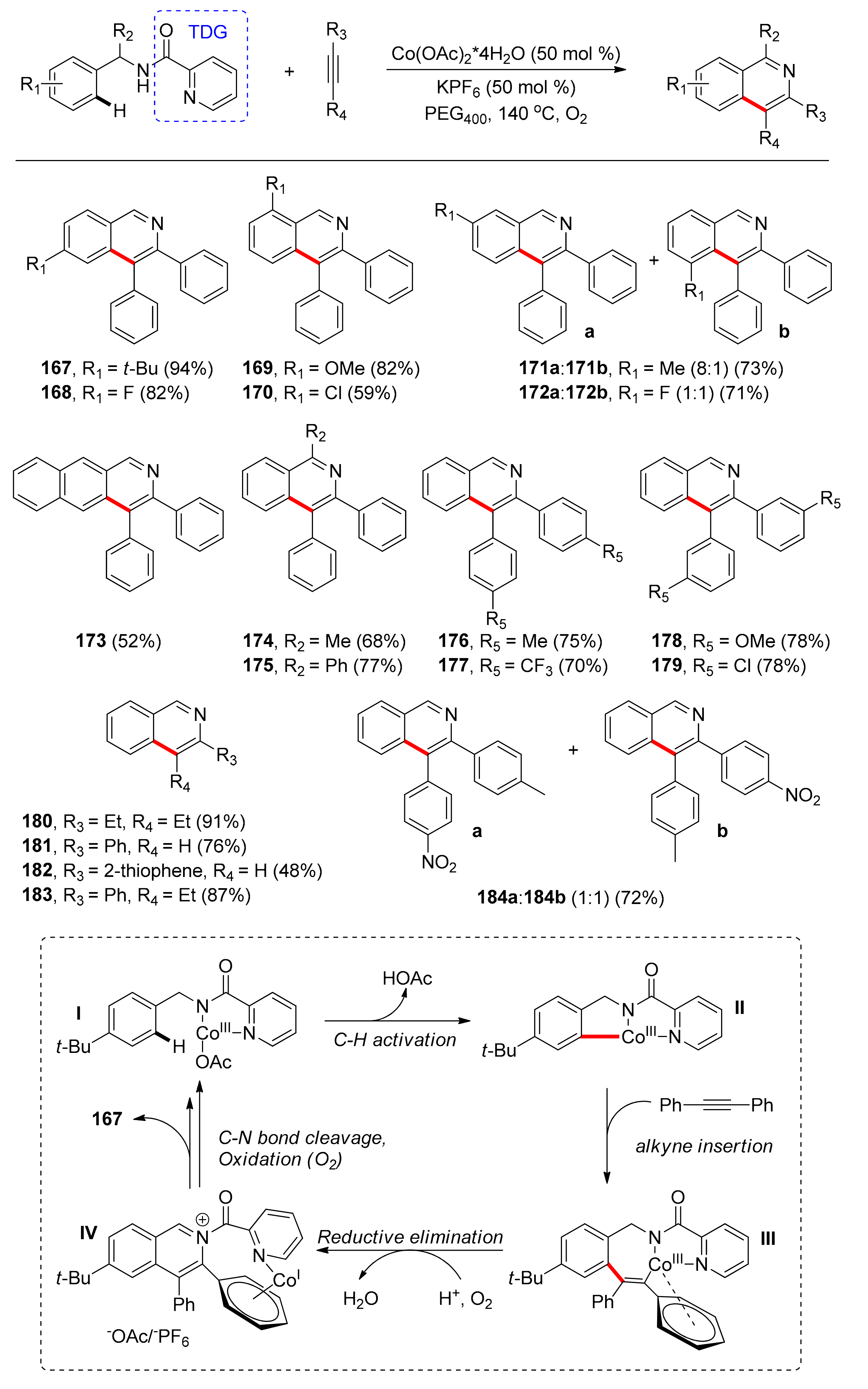
The second report in the literature referring to cobalt-catalyzed, TDG-assisted oxidative annulation of benzylamides with alkynes for the synthesis of isoquinolines through C–H/N–H bonds activation was reported by Cui and co-workers in 2017 [107]. This method employs picolinamide as the TDG and oxygen as the oxidant. After screening a number of Co sources, Co(OAc)2·4H2O afforded the optimal results in 50 mol % catalyst loading, while PEG-400 (polyethylene glycol-400) as the solvent and KPF6 as an additive increased the reaction yield at 140 °C. Under the optimal conditions, the authors investigated the scope of benzylamides (Scheme 13) and found that electron-donating groups on the ortho or para position of the benzylamides led to increased yields (167, 169), while electron-withdrawing groups had the opposite effect (168, 170). Yields were moderate to high, whereas the efficiency of the reaction was not sterically hindered by substituents on the ortho position. Meta-substituted substrates led to regioisomeric mixtures in good yields (71–79%) with the higher regioselectivity observed when the substituent was a methyl group (8:1) rather than a fluoro group (1:1), possibly due to steric effects (171, 172). Moreover, 2-naphthamide (173), as well as α-substituted substrates (174, 175) reacted smoothly under the reaction conditions allowing facile access to more complicated isoquinolines. While exploring the scope of alkynes, the authors found that para and meta substituted symmetric diarylacetylenes bearing both electron-donating and electron-withdrawing groups afforded the products in higher than 70% yields (176–179). In the case of ortho substitution, products were not obtained, probably due to steric effects. Dialkyl-substituted symmetrical alkynes (180) showed excellent reactivity, while terminal arynes (181, 182) and unsymmetrical internal alkynes with one phenyl group (183) afforded the desired products in good yields and high regioselectivity. Furthermore, unsymmetrical internal aryl alkynes with two aryl groups (184) led to a regioisomeric mixture (1:1) in 72% total yield.
Further studies led to the conclusion that the reaction does not involve a mechanism progressing via free radicals. The reaction was proposed (Scheme 13) to initiate with the in situ oxidation of the Co(II) species to Co(III) by oxygen and coordination of the active form of the catalyst with benzylamide (I). Picolinamide stabilizes the Co(III) species as a bidentate, anionic TDG. C–H metalation (II) and alkyne insertion into the Co–C bond lead to a seven-membered intermediate (III), which undergoes reductive elimination and dehydrogenation aided by oxygen, affording isoquinolinium salt (IV). Subsequent in situ C–N bond scission affords isoquinoline 167 and the Co(I) species is then oxidized by oxygen to give the active Co(III) species (Scheme 13).
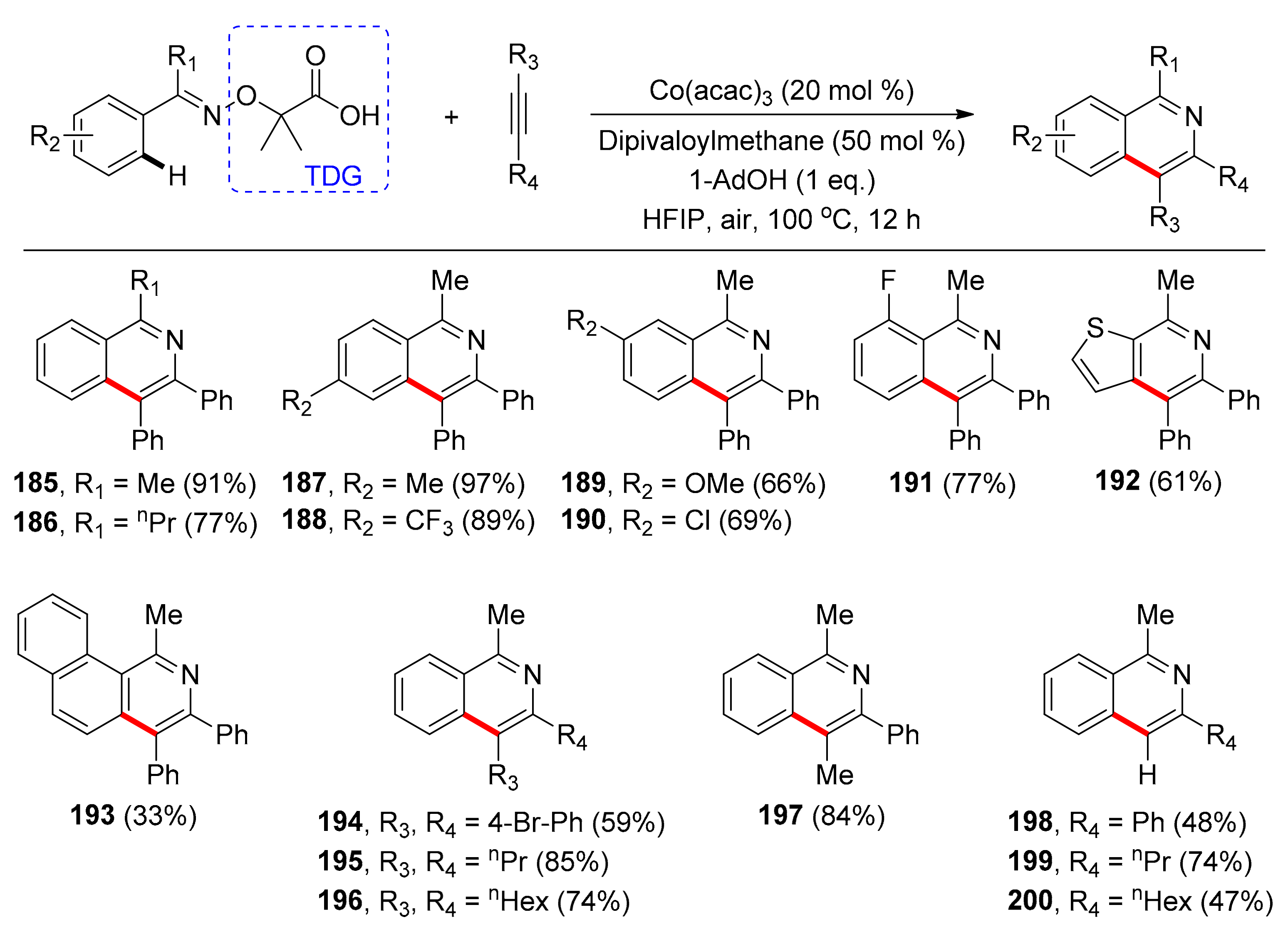
Another protocol towards the Cp*-free, cobalt-catalyzed, TDG-assisted, synthesis of isoquinolines was reported by Song and co-workers in 2019 [108]. Substituted isoquinolines are important heterocyclic compounds found in chiral ligands and pharmaceuticals, amongst others. For their synthesis, the cheap and readily available Co(acac)3 salt was used as the catalyst in 20 mol % loading. Alternative cobalt sources including Cp*Co(III) were unreactive. Addition of 1-adamantane carboxylic acid (1 equivalent) and dipivaloylmethane (0.5 equivalents) are advantageous probably because the first serves as both a ligand and proton donor and the latter as a ligand. Among the examined solvents, hexafluoroisopropanol (HFIP) afforded the best results at 100 °C in air, while under an argon atmosphere yields decreased significantly. Studies on the directing group showed that the reaction was not feasible with monodentate moieties. When the carboxylic acid functionality was displaced by a carboxylic ester group the product was not detected, a fact that highlights the necessity of the weakly coordinating carboxylate-directing motif. Under the optimized conditions, a plethora of derivatized α-imino-oxy acids was tested (Scheme 14) with methyl group being the optimal substituent for the α-imino position (185, 186). Moreover, para-substituted α-imino-oxy acids afforded products in high yields regardless of their electronic characteristics (187, 188), while meta-substituted α-imino-oxy acids led to products in moderate to high yields with C–H activation taking place exclusively on the less hindered position (189, 190). The sole example of ortho substitution is o-fluoro-substituted α-imino-oxy acid which afforded the desired product in 77% yield (191). The heteroarene 192 and the bicyclic α-imino-oxy acid 193 were also compatible with this reaction protocol. Concerning the reaction scope with respect to alkynes, it was found that symmetrical, both diaryl and dialkyl alkynes could participate in the reaction, leading to moderate to high yields (194–196). The examined unsymmetrical internal alkyne led to only one of the regioisomeric products in 84% yield (197). Finally, upon using terminal alkynes, the corresponding cyclization derivatives were obtained in low to good yields and high regioselectivities (198–200).
A possible mechanism was also proposed: it involved ligand exchange of Co(acac)3 to generate a catalytically active species, and subsequent coordination of Co- by the substrate. Then, C–H activation takes place, while the Co(III) species is stabilized by the N,O-bidentate, anionic directing group. Coordination and alkyne insertion into the C–Co bond provides the seven-membered cobaltacyclic intermediate, which can undergo substitution intramolecularly, giving product 185 and side product 2-hydroxyisobutyric acid (or to the release of CO2 and acetone) together with the regeneration of the active form of the catalyst. External metal oxidants were not required, as the N–O bond can act as an internal oxidant.
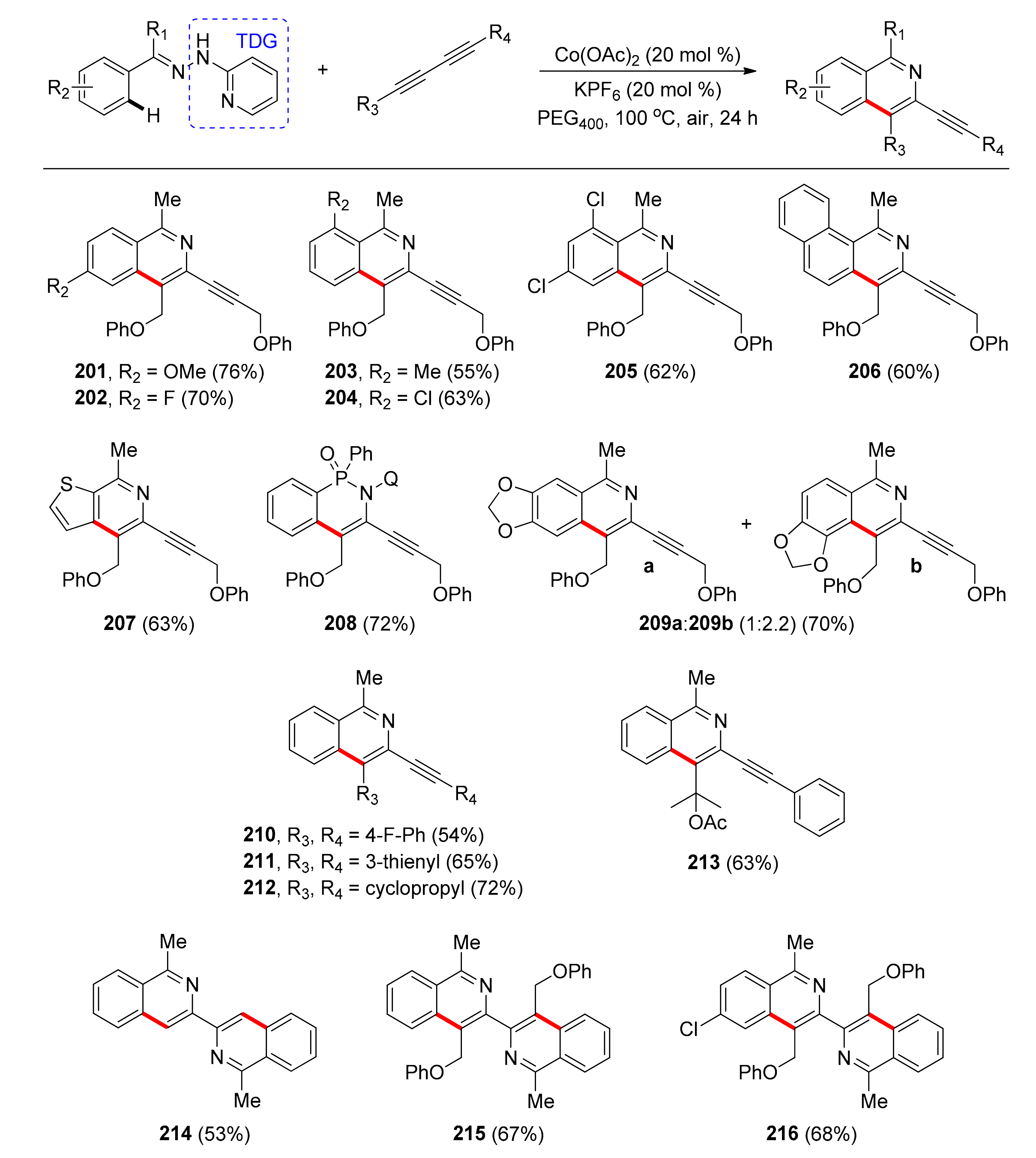
The most recent protocol that utilizes a TDG for C–H activation and [4 + 2] annulation under Co catalysis was reported by Volla and co-workers in 2020 [109]. The protocol refers to the synthesis of 3-alkynylated isoquinolines and biisoquinolines, which are scaffolds found in natural products, numerous bioactive molecules, as well as functional materials. This system engages 2-aminopyridine as TDG, which stabilizes the Co(III)-based active species, given that it is a bidentate group simultaneously acting as an internal oxidant that lifts the necessity for any external oxidants. The active form of the catalyst is generated in situ from the inexpensive and earth-abundant Co(OAc)2 salt, which is used in 20 mol % catalyst loading. Optimization studies showed that addition of KPF6 in a benign and cheap solvent like polyethylene glycol-400 (PEG400) at 100 °C enhanced the yield further. With the optimal conditions in hand, the authors investigated the substrate scope (Scheme 15) and concluded that both electron-donating and electron-withdrawing functional groups at different positions on the aryl ring afforded products in moderate yields (201–205). Moreover, 1-naphthyl substituted (206) and 2-thienyl substituted hydrazones (207), as well as 8-aminoquinoline derived phosphinamide 208 reacted smoothly under the reaction conditions, while an 1,3-dioxole substituted hydrazone provided mixtures of products in terms of regioisomers (209a+209b) in 70% yield. Concerning the 1,3-diyne substrate, symmetrical aryl- and heteroaryl-substituted 1,3-diynes afforded products in moderate yields (210, 211), whereas alkyl-substituted 1,3-diynes were more reactive (212). Unsymmetrical aryl alkyl diynes were also employed under the reaction conditions and resulted in regioselective formation of the products (213), as the coordinative insertion of organocobalt intermediate takes place selectively with the aliphatic alkyne. Finally, the authors attempted the synthesis of biisoquinolines, hence symmetrical (214, 215) and unsymmetrical bisheterocycles (216) were obtained in moderate yields in a one-pot, sequential approach.
A plausible mechanism was proposed, which commences with the in situ oxidation of the Co(II) species to the Co(III) active species by oxygen. This coordinates with the substrate and is stabilized by the bidentate TDG. C–H metalation, 1,3-diyne coordination, and insertion into the Co–C bond affords a seven-membered cobaltacycle, which undergoes reductive elimination and N–N bond cleavage, leading to the targeted product and regenerating the active Co(III) species.
4.3. Other TDG-Assisted Transformations
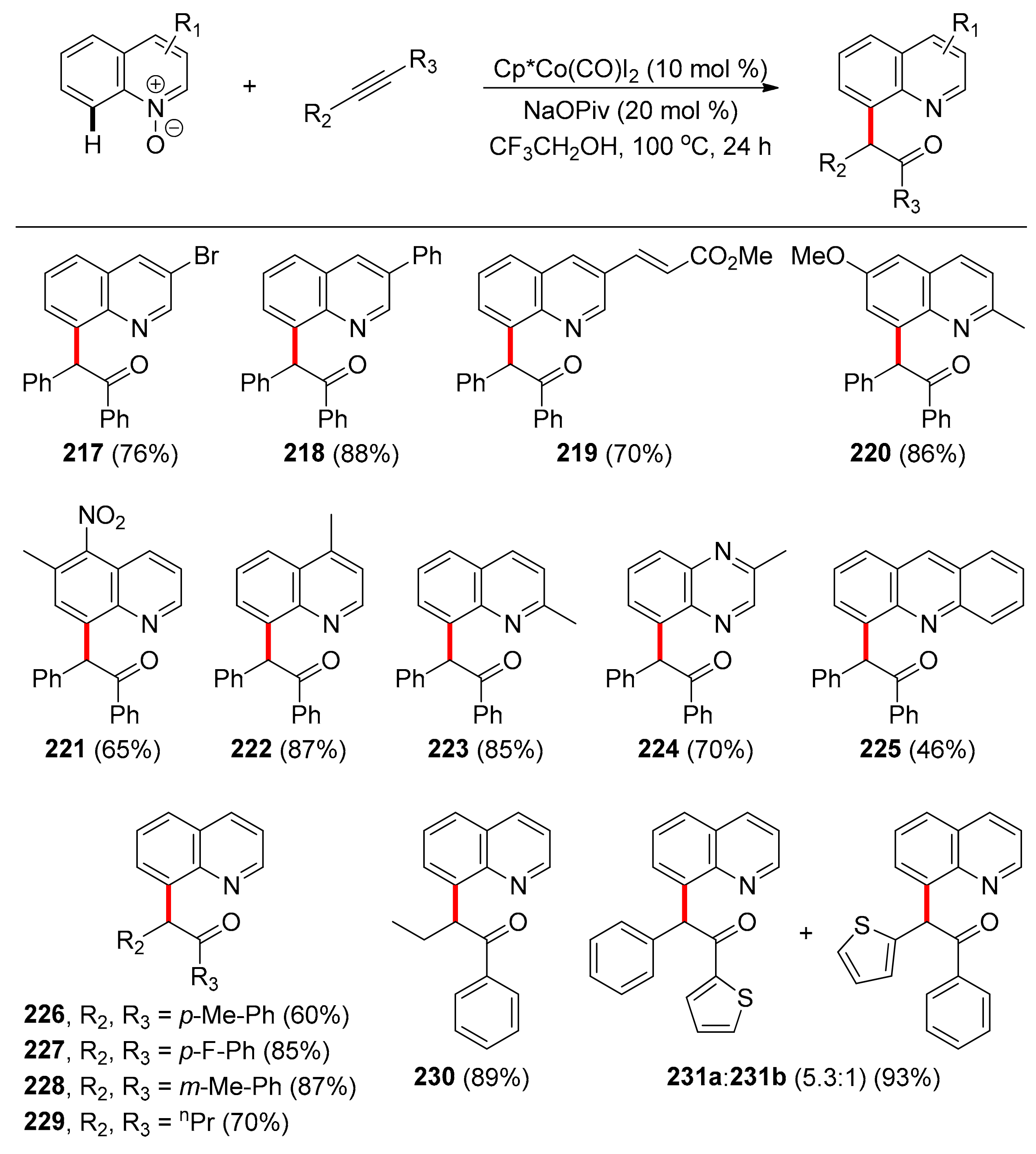
The first protocol that relies on cobalt catalysis using N-oxide as TDG concerns a C–H and C–O coupling, via C–H activation/oxygen atom transfer reaction of quinoline N-oxide and internal alkynes (Sundararaju and co-workers, 2016) [110]. Quinolines are heterocycles affording versatile molecules such as natural products, as well as products related to medicine and agrochemicals. However, most of the literature describes functionalization on the C2 position of quinolines, which underlines the importance of this report, dealing with functionalization on the C8 position. In this transformation, cobalt catalyst Cp*Co(CO)I2 is used in 10 mol % catalyst loading, without the need of external oxidants, while NaOPiv is necessary for the reaction. Trifluoroethanol was the optimal solvent, at 100 °C, with the alkyne being used as the limiting reactant. This protocol could not proceed utilizing other cobalt sources of Co(III) or Co(II), or even when using Rh(III), Ir(III), or Ru(II) sources. Various substituted quinoline-N-oxides were tested (Scheme 16) and it was found that mono- and di-substituted quinoline-N-oxides, at various positions of the heteroarene, were compatible with this protocol and products were obtained in moderate to high yields (217–223). Interestingly, sensitive functionalities, such as multiple bonds, ketones, or nitro groups, remained intact after reaction. Other heterocycles were also amenable to this protocol (224, 225), while a gram-scale reaction using only 5 mol % of [Cp*Co(CO)I2] highlighted the scalability of this transformation, with the product obtained in 94% yield. Concerning the alkyne substrate, studies showed that symmetrical alkynes could easily react (226–229). Especially, product 229 was obtained in 70% yield with Co(III), while the Ir(III) catalyst tested did not promote this reaction and Rh(III) gave only 15% yield, emphasizing the unique reactivity of Co(III) over the other group 9 metals. Unsymmetrical internal alkynes bearing one alkyl and one aromatic substituent provided a single regioisomer (230) and unsymmetrical biaryl-substituted alkynes gave mixtures of regioisomers in high yields (231).
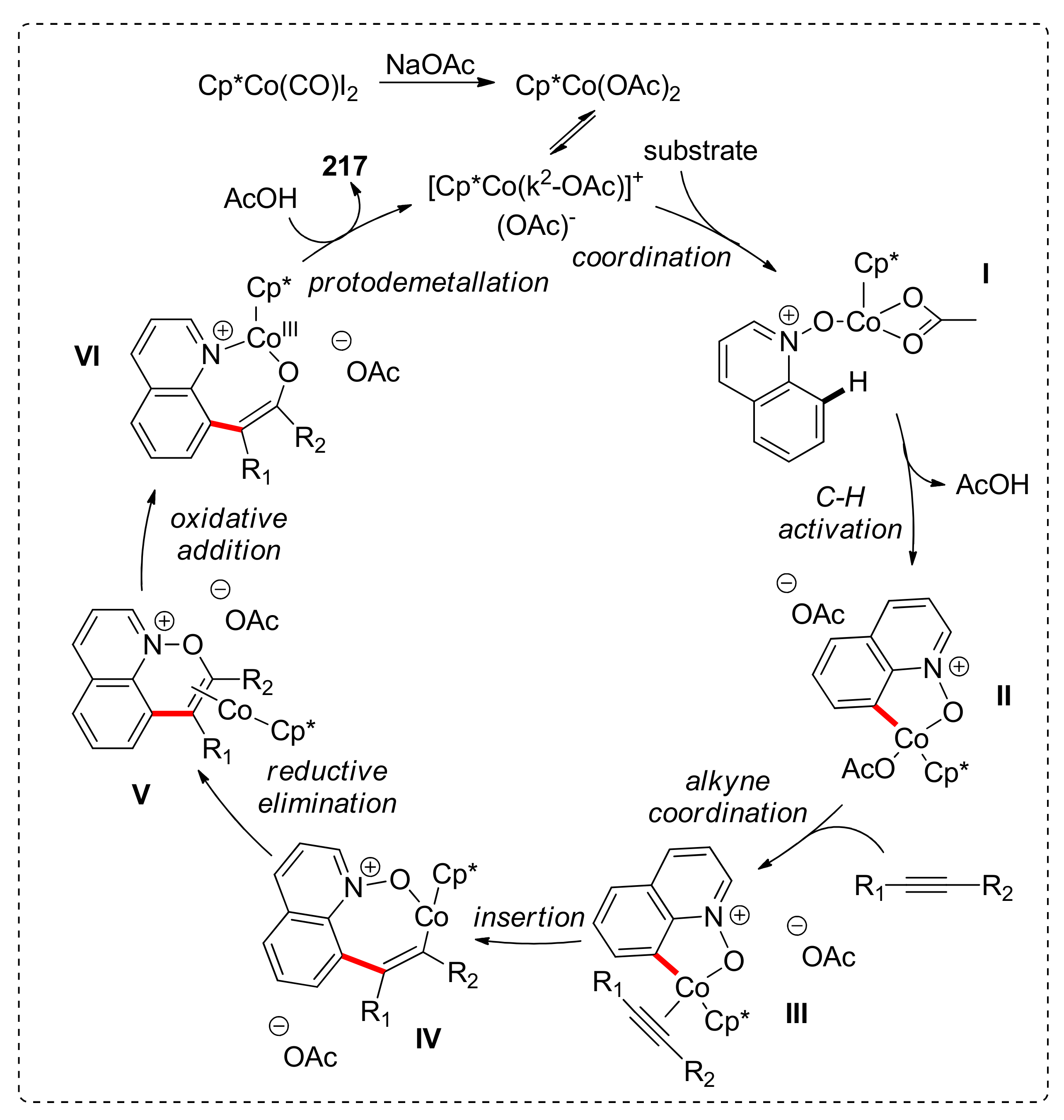
The proposed mechanism for this reaction commences with the cobalt catalyst been coordinated with the quinoline-N-oxide substrate, followed by C–H activation at the C8 position. After alkyne coordination and migratory insertion into the Co–C bond, alkenyl cobaltacyclic intermediate IV is formed. Reductive elimination leads to the Co(I) intermediate V, followed by oxidative addition of the metal to the N–O bond and protodemetallation to C8 functionalized quinoline 217, with the concomitant regeneration of the active catalyst (Scheme 17).
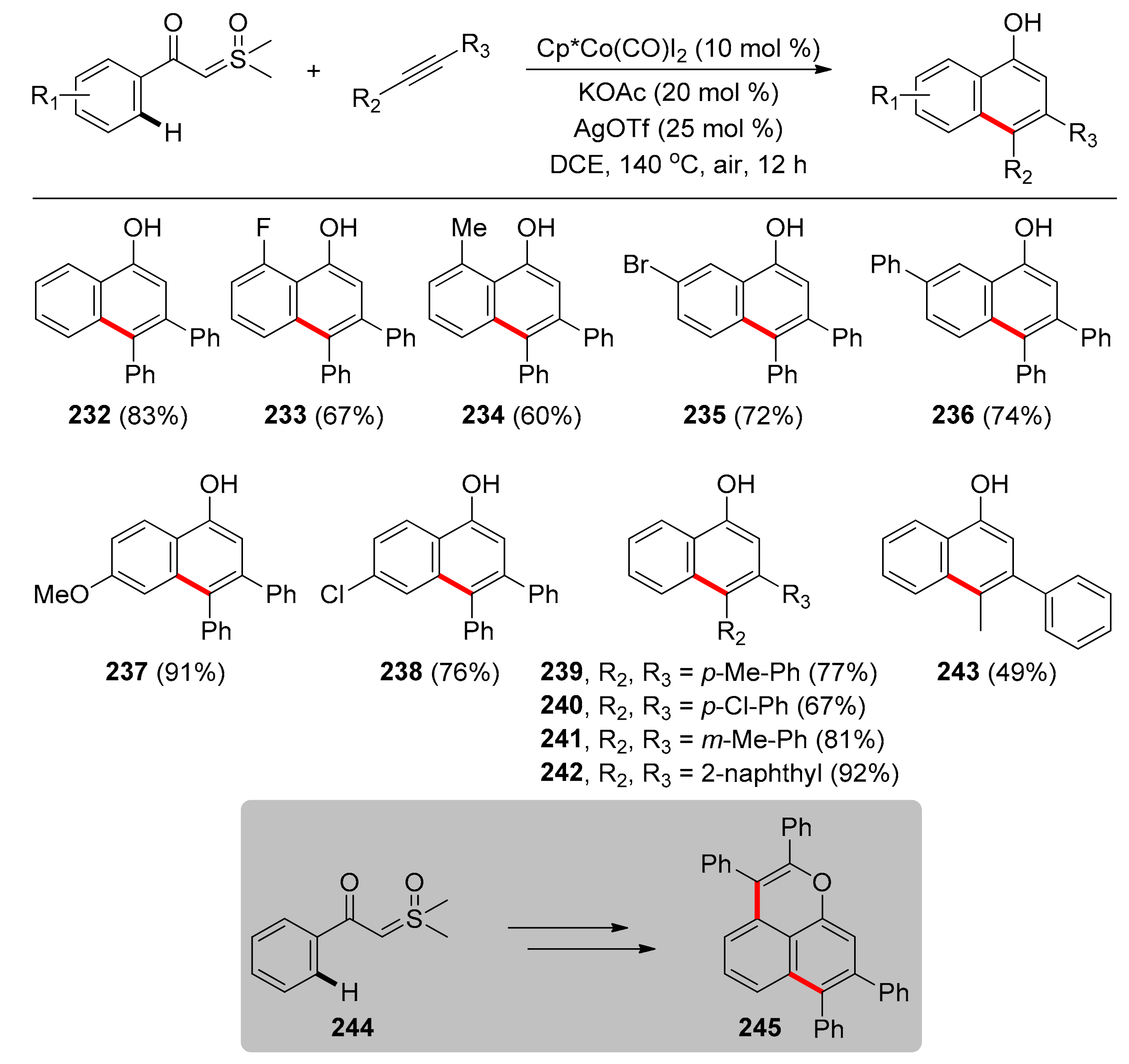
Another protocol catalyzed by Cp*Co(III) species was reported by Tan and co-workers in 2019, enabling C–H activation and [4 + 2] cyclization of readily available sulfoxonium ylides with alkynes for the synthesis of 1-naphthols [111]. 1-Naphthols are scaffolds widely found in pharmaceuticals and bioactive natural products; therefore, the direct synthesis of these compounds via earth-abundant, cheap, and biocompatible first-row transition metal catalysis is of great interest. In this transformation, sulfoxonium ylide functions as a TDG, while [Cp*Co(CO)I2] is the optimal Co pre-catalyst, used in 10 mol % loading. The active catalyst is generated in situ after treatment with AgOTf and KOAc, while the reaction proved to be more high-yielding in DCE, at 140 °C under air. Under the optimized conditions, examination of sulfoxonium ylides (Scheme 18) revealed that when they bear neutral, electron-withdrawing, or electron-donating groups at various positions of the phenyl ring gave the corresponding products in moderate to high yields (232–238), with ortho-substitution leading to comparatively lower yields, possibly due to steric hindrance (233, 234). Meta-substituted sulfoxonium ylides reacted regioselectively, with the C–H activation taking place exclusively at the less-hindered position (235, 236) and para-substituted substrates afforded products in the highest yields (237, 238). As far as the alkyne substrate is concerned, symmetrical diphenylacetylenes substituted on para and meta position were tested, providing the products in low to high yields (239–242), with electron-donating substituents on the diphenylacetylenes leading to higher yields. Alkyne 1-phenyl-1-propyne afforded 243 with great regioselectivity. Notably, this protocol was applied in the one-pot tandem reaction for synthesis of 2,3,7,8-tetraphenylbenzo[de]chromene 245 in 22% yield through two consecutive steps of C–H activation, employing the hydroxyl group of the product of the first step as the directing group for the second step.
The proposed mechanism for this transformation begins with the reaction of [Cp*Co(CO)I2] with AgOTf and KOAc to generate in situ the active cationic Co(III) complex, coordinated to the sulfoxonium ylide via the carbonyl oxygen. Then, C–H bond activation at the ortho position affords a five-membered cobaltacyclic species. Then, alkyne coordination followed by migratory insertion leads to a seven-membered intermediate, which undergoes O- to C-tautomerization and α-elimination of DMSO. Migratory insertion of the cobalt-carbenic intermediate into the C–Co bond and protonolysis by acetic acid leads to 1-naphthol, concomitantly regenerating the catalyst.
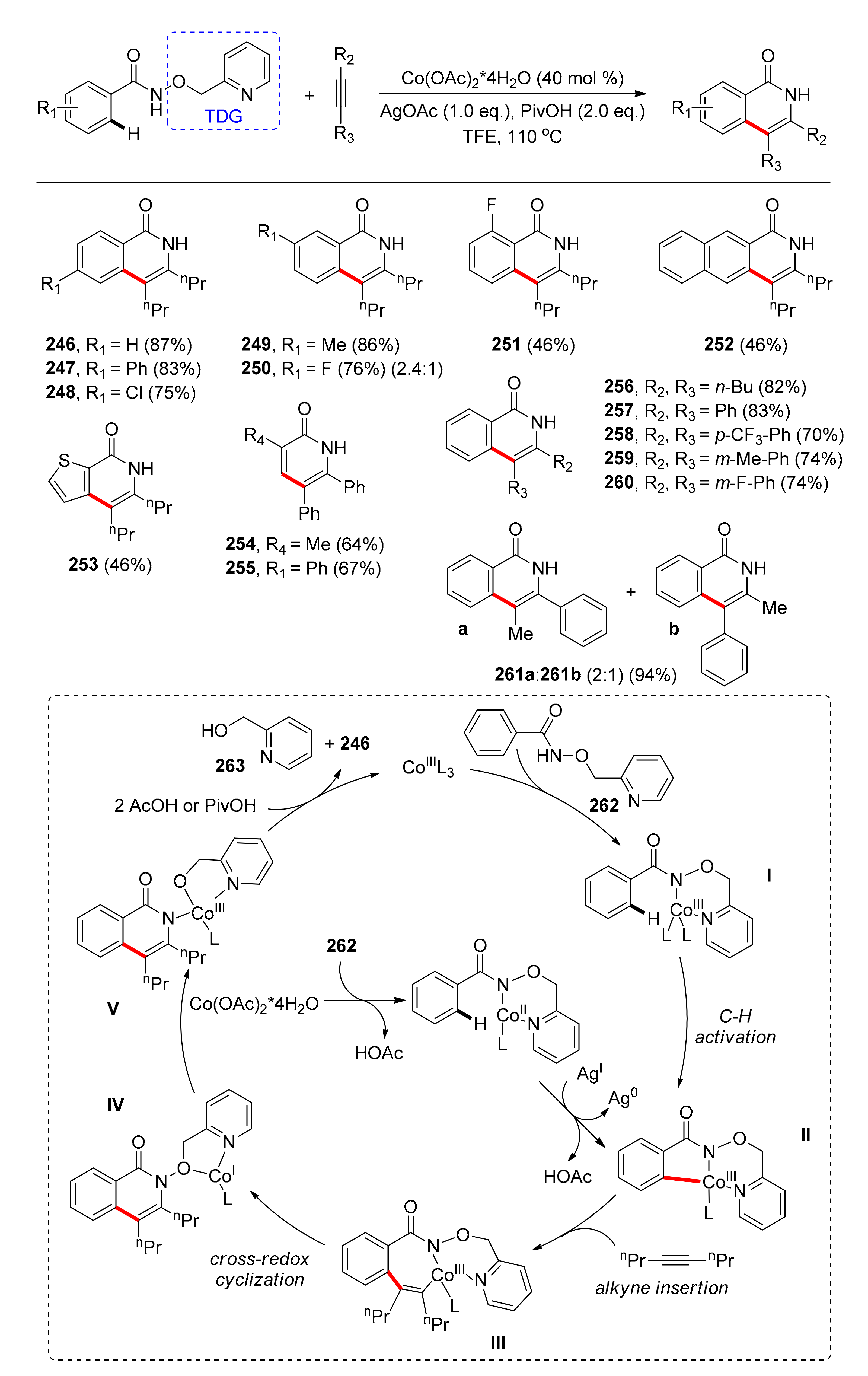
The latest protocol concerning cobalt-catalyzed C–H activation using a TDG was reported by Yang, Song, and co-workers in 2020 and refers to the synthesis of isoquinolinones [46]. Isoquinolinone and its derivatives are scaffolds found in pharmaceuticals with anticancer and antihypertensive properties. This synthesis utilizes 2-(hydroxymethyl)pyridine as a TDG and the Co(OAc)2·4H2O salt as the low-cost and earth-abundant, Cp*-free, pre-catalyst. The pre-catalyst is used in 40 mol % loading, since coordination of cobalt with the 2-(hydroxymethyl)pyridine formed in the reaction inhibits its catalytic activity. The AgOAc reagent is used for oxidizing Co(II) to Co(III), while addition of PivOH increases the yield. Among the tested solvents, 2,2,2-trifluoroethanol gave the optimum results. While exploring the substrate scope (Scheme 19), under the optimized conditions, that para-substituted benzamides with electron-donating groups afforded products in excellent yields, while benzamides with electron-withdrawing groups gave lower yields (246–248). Meta-substituted benzamides also reacted in good yields; moreover, the higher the steric bulk of the meta-substituent was, the higher the selectivity towards the product formed on the less obstructed position, providing good regioselectivity (249, 250). The authors reported only one example of ortho-substitution which suggests that steric hindrance affected the efficiency of the reaction (251). Analogous results were also observed when employing a bicyclic system (252) and heteroarenes (253), while terminal olefin substrates resulted in yields lower than 70% (254, 255). Concerning the alkyne substrate, many symmetrical alkynes were tested, such as alkynes with longer alkyl chains and phenylacetylenes, para-substituted with electron-withdrawing groups, or meta-substituted either with electron-donating or -withdrawing substituents (256–260), all of which led to good yields. Additionally, 1-phenylpropyne afforded a regioisomeric mixture in 94% total yield (261).
A possible mechanism was also proposed, which commences with the coordination of the Co(OAc)2·4H2O salt to the substrate (262), which is then oxidized to cobalt(III) by AgOAc. The Co(III) species activates the C–H bond on the ortho position, an event which is followed by the coordination and the migratory insertion of the alkyne into the C–Co bond, giving the seven-membered cobaltacyclic intermediate III. Afterwards, cross-redox cyclization affords intermediate IV, where the Co(I) species inserts into the N–O bond, providing V. Protodemetallation of V releases the product (246) and Co(III) to start a new catalytic cycle (Scheme 19).
5. Nickel
Nickel is ranked third in terms of abundancy in earth’s crust among those examined in the present review (84 ppm, after iron and manganese [86]). It forms salts bearing a plethora of possible counter-ions, thus offering a wide range of solubilities and coordinating abilities to choose from. The intriguing catalytic properties of nickel compounds is demonstrated by its important role in biological redox processes, as exemplified in enzymes, such as hydrogenases, ureases, and superoxide dismutases [112]. The activity of such metalloenzymes and its prospects in catalytic applications is demonstrated by biomimetic nickel complexes that can reach high turnover frequencies of 250 s-1 in the electrocatalytic hydrogen evolution reaction [113]. Regarding nickel-catalyzed reactions involving organic substrates, nickel has attracted interest as evidenced by recent review articles addressing the subject [114,115,116,117].
Thiolation via C(sp2)–H Bond Activation
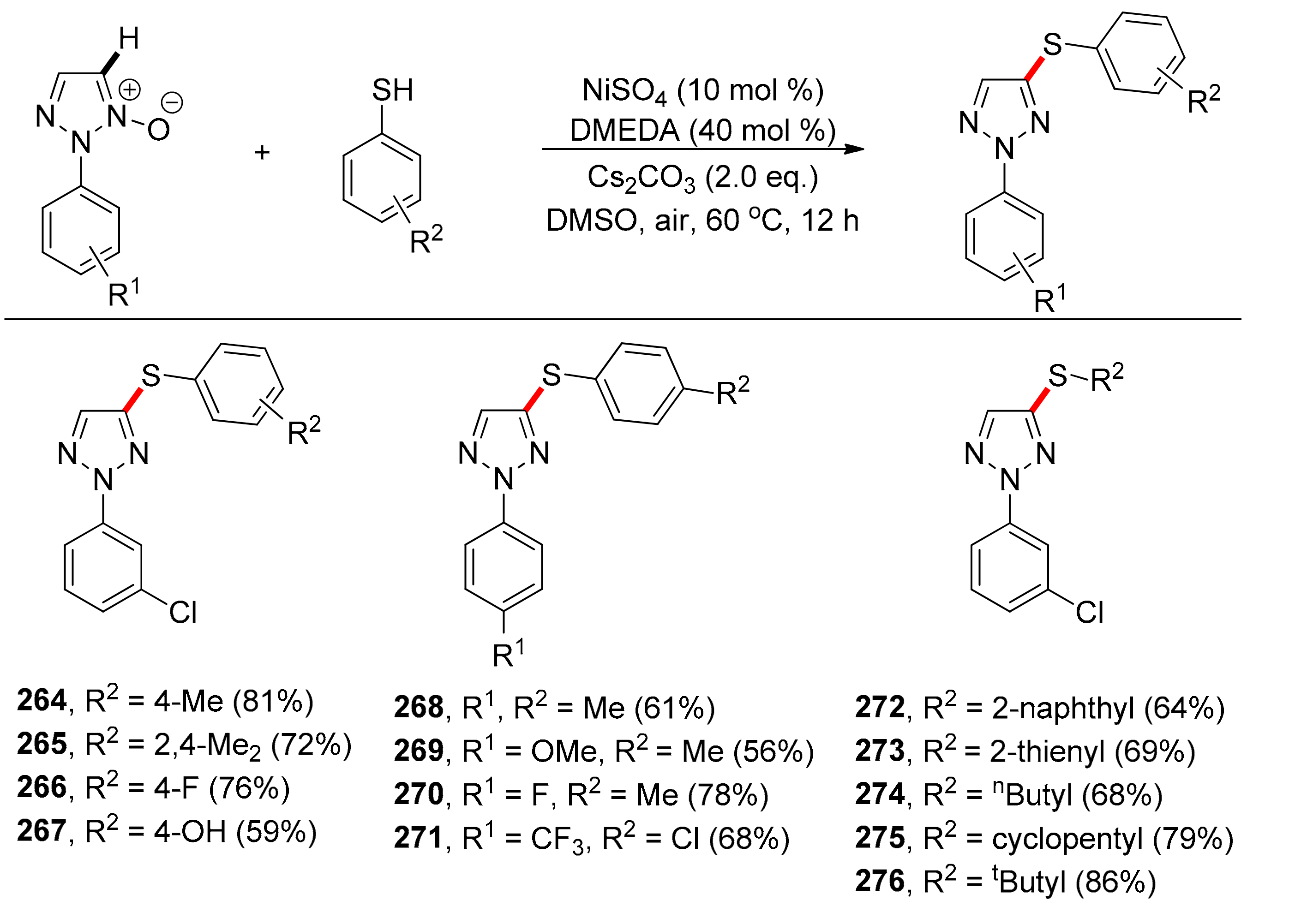
In 2015, Chen, Liu and co-workers [118] reported on the nickel sulfate-catalyzed C–H activation and thiolation of 1,2,3-triazole N-oxide C–H bonds, with thiolating agents such as aryl or alkyl thiols, or diphenyl disulfide. In this case, the N-oxide acts as a TDG (Scheme 20). Initial optimization involved testing various metal salts, ligands, bases, and solvents. 2-(3-Chlorophenyl)-2H-1,2,3-triazole 1-oxide and 4-methyl-benzenethiol were used as model substrates. Among all transition–metal salts employed, NiSO4 (0.1 equivalents) exhibited the best activity. In blank experiments carried out without NiSO4, product formation was not detected. DMSO was the best solvent and among the various ligands tested (PPh3, pyridine, TMEDA, DMEDA) the optimal results were obtained with DMEDA. The activity of DMEDA, reaching 81% yield in contrast to 53% for TMEDA, can be attributed to its chelating, but less bulky nature. Optimized conditions involved 10 mol % NiSO4 (metal source), 40 mol % DMEDA (ligand), DMSO as solvent, Cs2CO3 (base), at 60 °C for 12 h.
Subsequent substrate scope studies, including unsubstituted, alkyl-substituted, and halogenated thiols gave products 264–267 in very good yields ranging between 70% and 81% (Scheme 20). Other thiols, such as naphthalene-2-thiol and thiophene-2-thiol also performed well, yielding the desired products 272 and 273 in good yields. Primary, secondary, or tertiary thiols also provided thiolation products 274–276 in good to excellent yields. 4,5-Dimethylthiazole-3-oxide also yielded the 4-substituted methyl- or bromo-benzene thioether in high yields (78% and 63%, respectively). When 1,3-propane-dithiol was used, the bis-thiolated product was obtained in 48% yield.
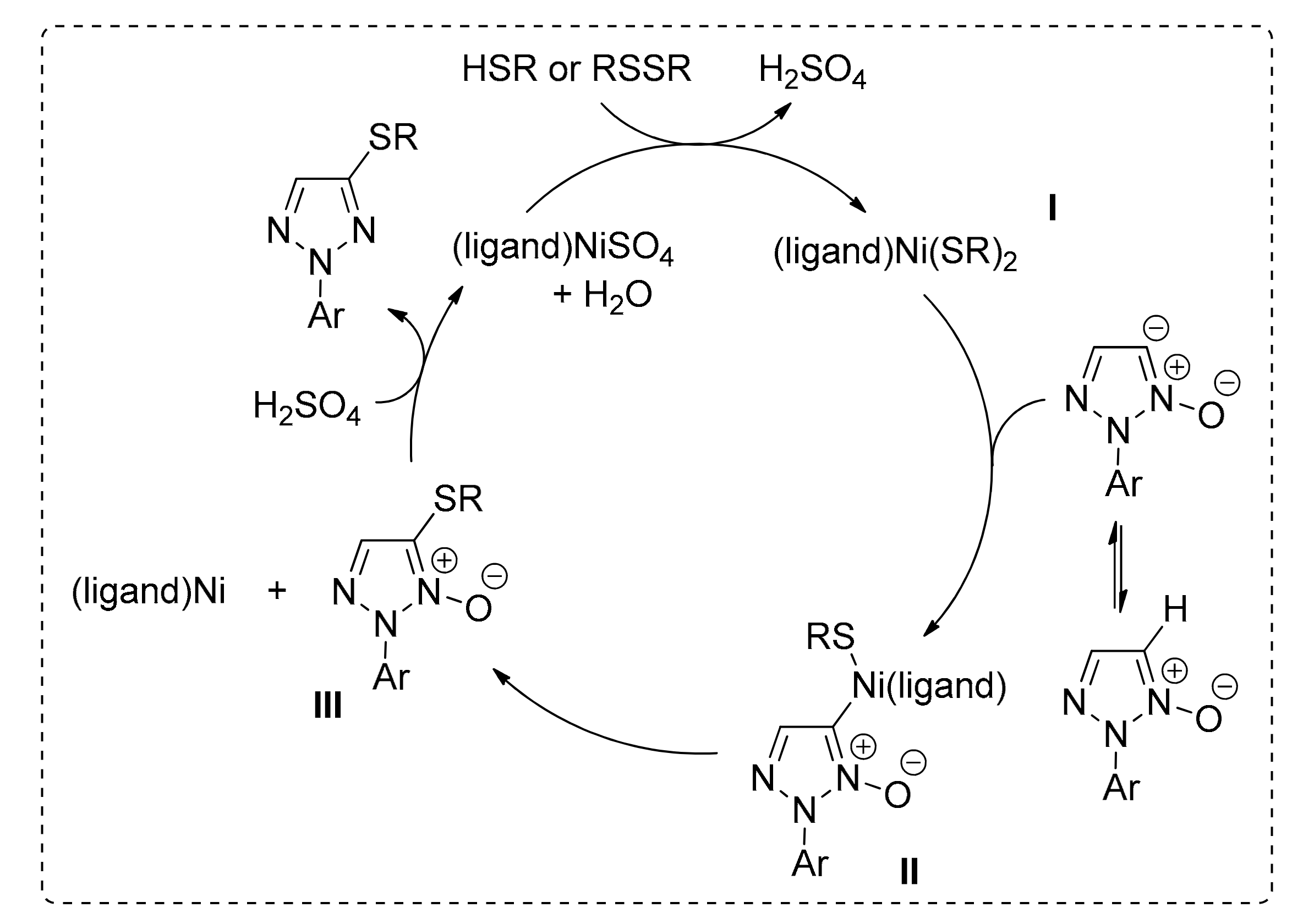
Mechanistic experiments revealed that molecular O2 was not necessary for the completion of the reaction. The de-oxygenation of 1,2,3-triazole occurs after the C–S bond formation step, while kinetic isotope effect experiments revealed that the desired C–H bond cleavage is involved in the rate-determining step. The proposed mechanism is shown in Scheme 21. After S-coordination of the thiol on Ni(II) to yield I, a deprotonated triazole-oxide undergoes metal insertion to give II. Reductive elimination of nickel in II can then yield intermediate III, which upon oxidation provides the starting nickel complex and the coupled thioether product.
6. Copper
Copper-catalyzed transformations involving C–H activation [119,120] are lately increasing, underlining the growing interest in expanding the gamut of earth-abundant metals used in contemporary synthetic challenges. Recent publications [121,122] dealing with the most common formal oxidation states reached by copper, namely Cu(I), Cu(II), and Cu(III), indicate the ongoing investigation of the role of copper in fundamental properties of various compounds that appear as well-defined catalysts or catalytic intermediates during Cu-catalysis.
6.1. Etheration via C(sp2)–H Bond Activation
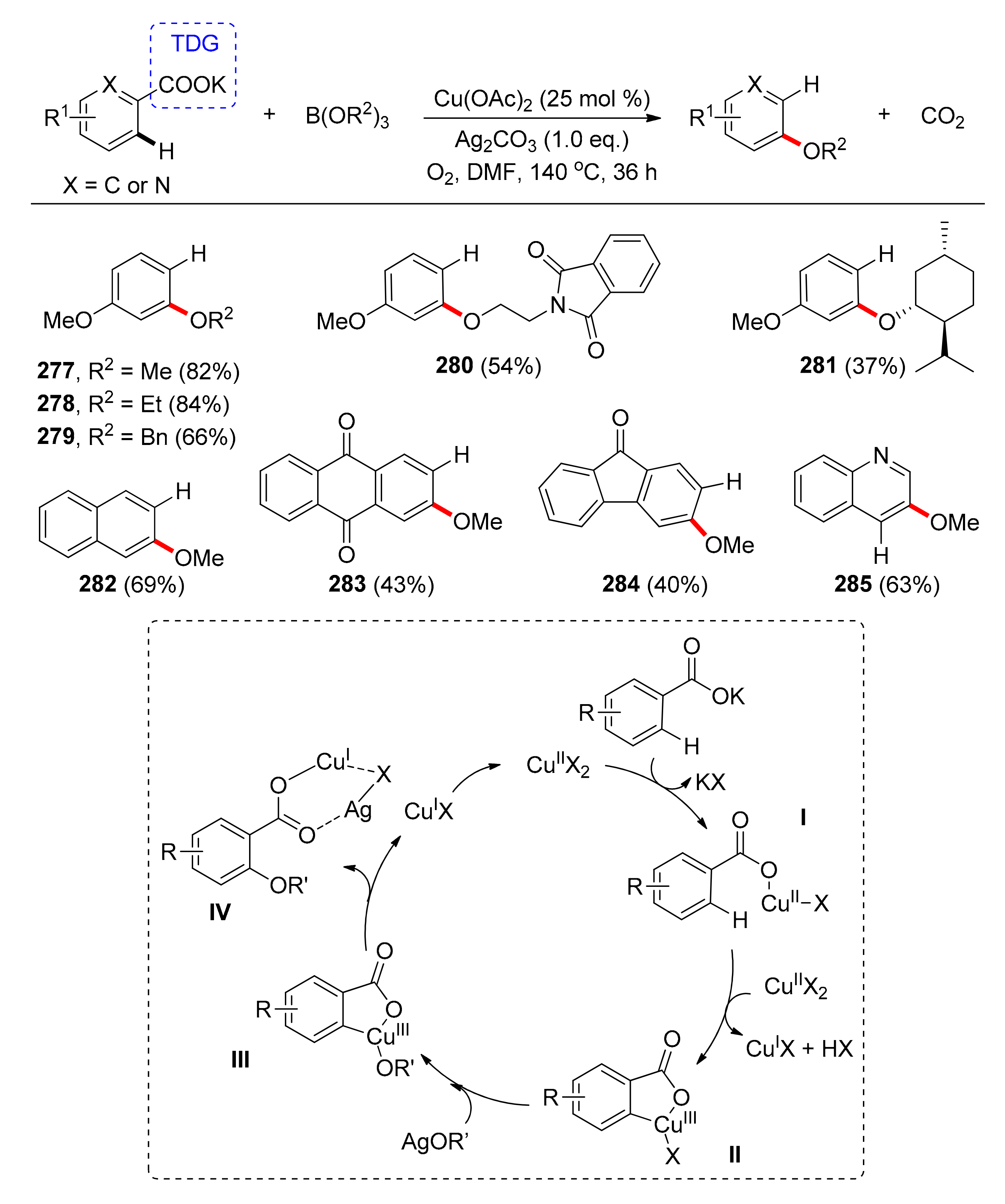
In 2013, Gooβen and co-workers [123] reported on the copper-catalyzed synthesis of aryl ethers from benzoates with a carboxylate TDG. Initial catalyst and condition optimization involved the reaction of potassium p-methoxy-benzoate with trimethoxy borate with copper(II) acetate and silver(II) carbonate, under oxygen, in DMF at 140 °C. The desired m-dimethoxy-benzene was obtained in 84% yield, after 36 h. In the absence of either silver or copper salts, the reaction did not proceed. Investigation with various substituents on different positions of the potassium benzoate or with fused aromatics, proceeded smoothly with yields up to 84% (Scheme 22). Chiral alkoxide 281 retained its configuration, while both electron-withdrawing and -donating groups on the substrates underwent conversion. Nitro, cyano, and sulfonyl substituents were successfully tolerated. Notably, anthraquinone and fluorenone derivatives 283 and 284 that are not accessible via traditional routes were also prepared in moderate yields (~40%).
The proposed catalytic cycle consists of two pathways; an ortho-alkoxylation cycle realized by Cu(II) including several redox events with alternating copper oxidation states, and a silver-mediated, decarboxylation cycle (Scheme 22). Initially, the benzoate is coordinate to Cu(I) and after oxidation and C–H activation, intermediate II is formed. After an alkoxide salt is transferred to Cu(III) via intermediate II, IV is formed in the presence of Ag co-catalyst, while re-oxidation of Cu(I) affords the active Cu(II) catalyst.
Mechanistic investigations revealed that the C–H scission step is rate-limiting. Use of phenolic substrates excluded the presence of phenols as intermediates, thus suggesting that the metalated arene and alkoxide lead to C–O bond formation.
6.2. Benzofuran Synthesis via C(sp2)–H Bond Activation
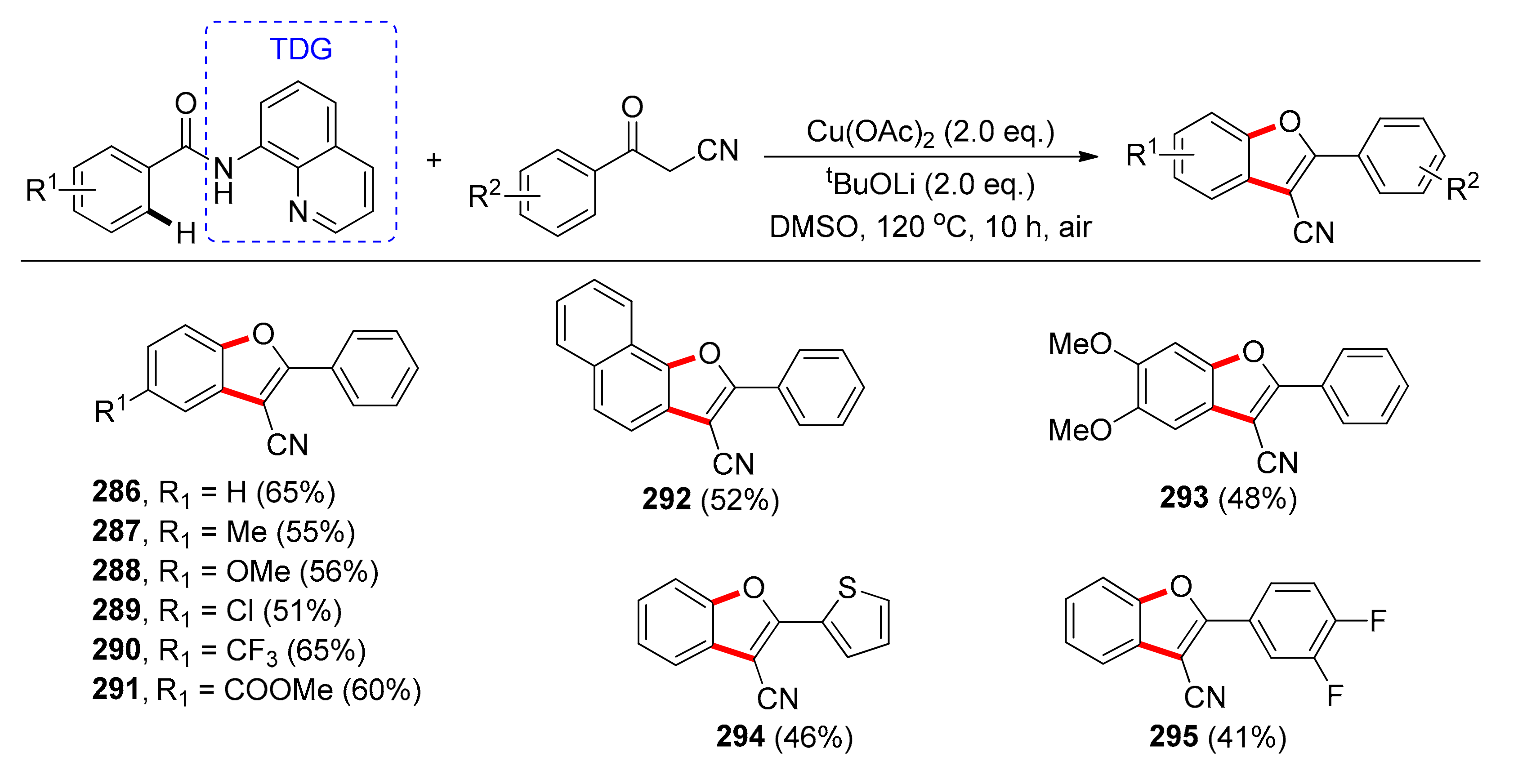
In 2019, Zhang and coworkers reported a copper-catalyzed cascade coupling reaction between benzamides and benzoylacetonitriles for the preparation of benzofurans [124], using an amide traceless directing group, in a one-pot procedure (Scheme 23). Lithium t-butoxide was employed as the optimum base among alkali metal carbonate salts. Different copper sources or the use of Pd or Rh and Ni salts did not afford the benzofuran at satisfactory yields, or no reaction occurred whatsoever. Varying substituents on the benzene ring of the benzamide, including methyl, methoxy, alkyl, aryl, halogen, nitro, or cyano groups also afforded the target compounds 286–291 in moderate to very good yields (36%–67%). Electron-donating groups on the p-position of the arene yielded benzofurans 287 and 288 in reduced yields, but electron-withdrawing groups on the arene ring afforded 290 and 291 in good yields. Halogen-groups (289), enabling further possible transformations, were also tolerated. Various groups of either electron-donating or -withdrawing nature on the benzoylacetonitriles’ (292–295) benzene ring did not affect the reaction.
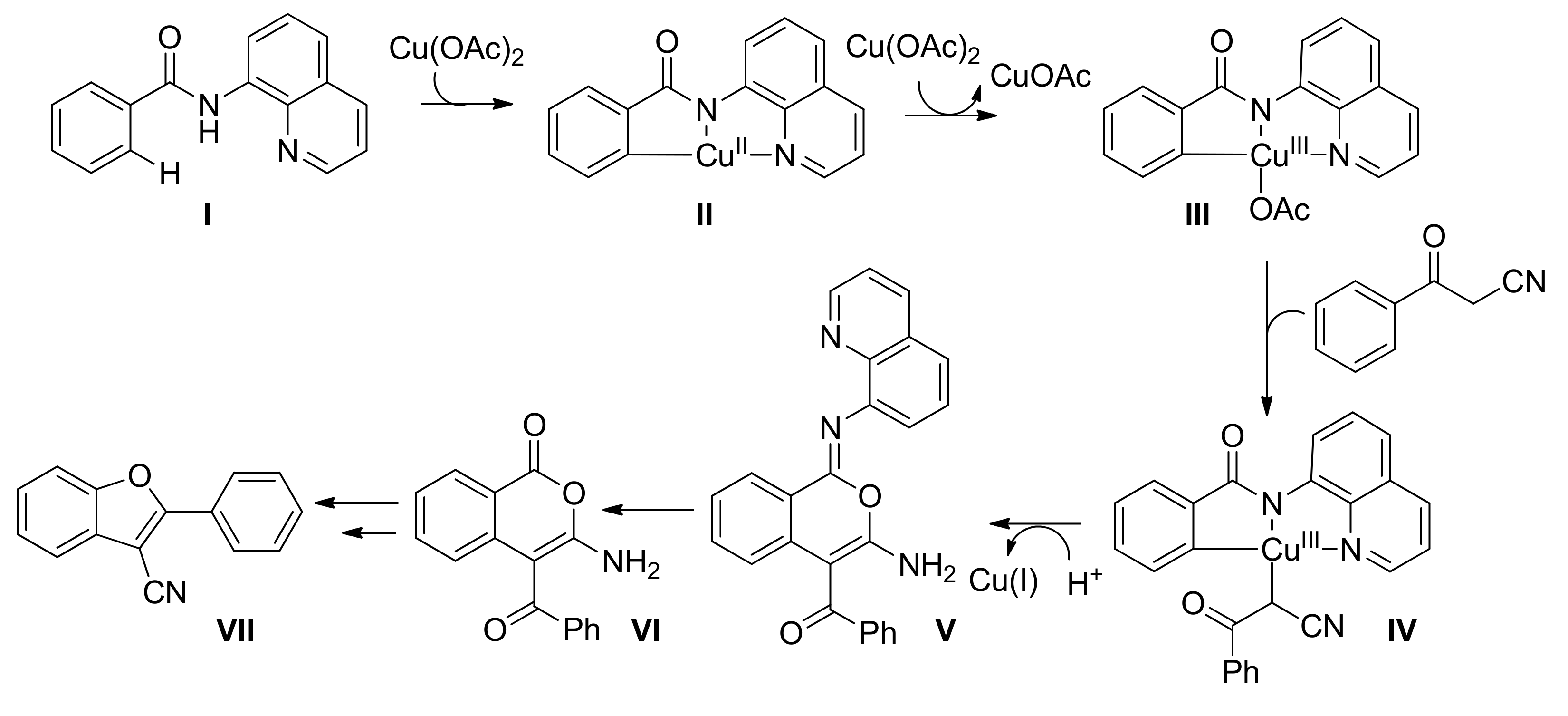
Mechanistic experiments led to the proposition that during the first reaction of quinolone-benzamide (I, Scheme 24) with copper(II) acetate, a tridentate Cu(II) complex is formed after C–H activation (II, Scheme 24). Disproportionation of coordinated or acetate Cu(II) in the reaction generates the corresponding Cu(III) complex. Given that TEMPO and BHT had a detrimental effect on the reaction yield, a single electron transfer was proposed to occur during the transformation of the intermediate amino-benzoyl-isochromenone. Interestingly, when compound VI was employed under the optimal reaction conditions, benzofuran VII was obtained, suggesting that VI is the key intermediate to afford the final product.
6.3. Amination via C(sp2)–H Bond Activation
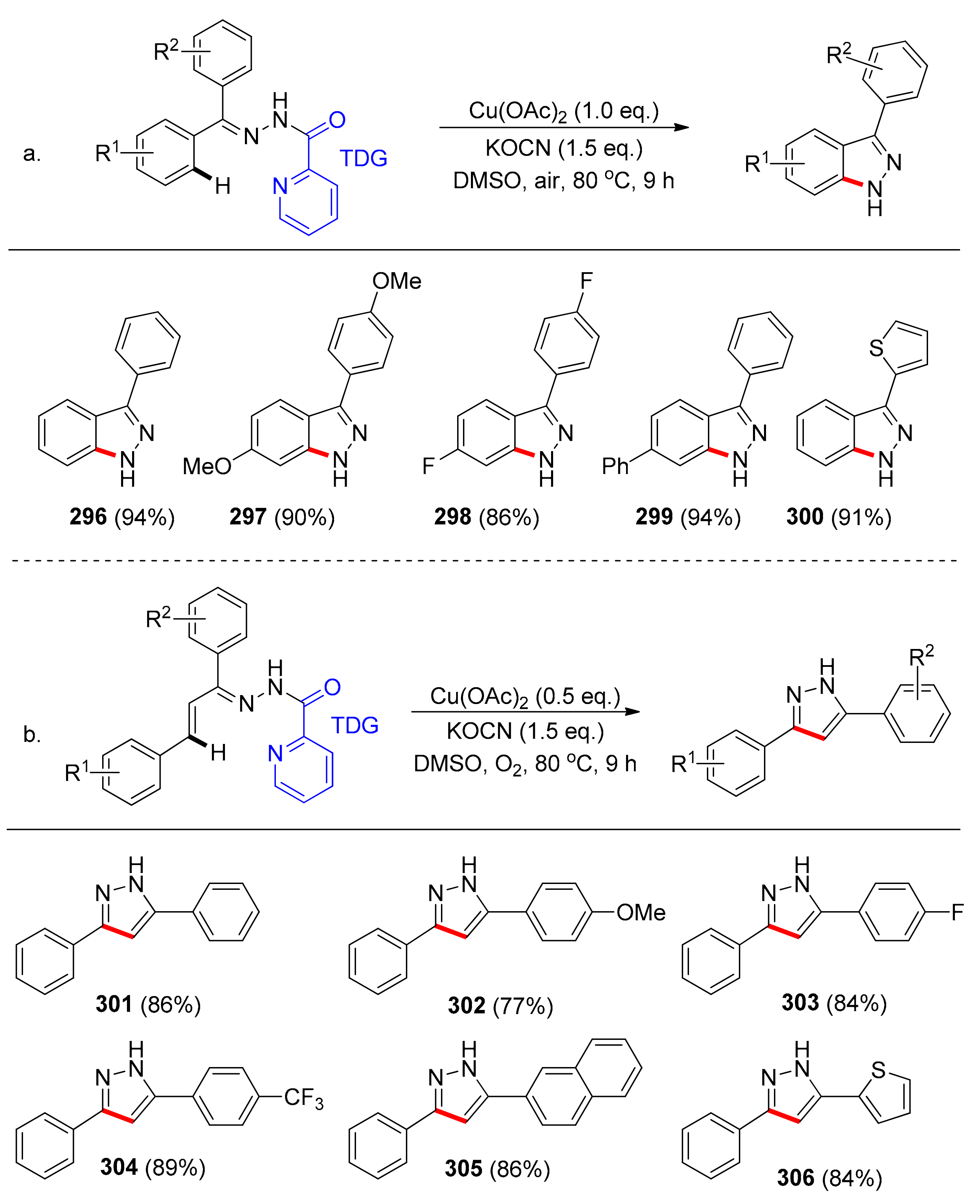
Ding and coworkers [125] reported on a facile preparation of 1H-indazoles and 1H-pyrazoles via a Cu-catalyzed intramolecular C–H amination of hydrazones with a 2-amidopyridyne TDG (Scheme 25). These heterocycles are of great interest as they exhibit biological activity, among others encompassing anti-cancer and anti-viral properties. Of all copper and other transition metal sources employed during initial reaction optimization, copper(II) acetate in DMSO with KOCN as a base under aerobic conditions (80 °C, 9 h), were found to be optimal. Different groups on the amide DG such as benzoyl, hydrogen, p-toluenesulfonyl, acetyl, and diethyl phosphate, led to low yields or no reaction at all. Substituents of either electron-withdrawing or -donating nature did not have an influence on the yield of the amination to afford the desired indazoles 296–300 (Scheme 25a).
Different substituents on either of the rings of the starting hydrazone (e.g., methyl, methoxy, halogen, aryl) in both cases (Scheme 25a,b) produced the desired compounds in very high yields (up to 96%). Substrates bearing naphthyl or thienyl functionalities also reacted satisfyingly yielding the desired products 305 and 306.
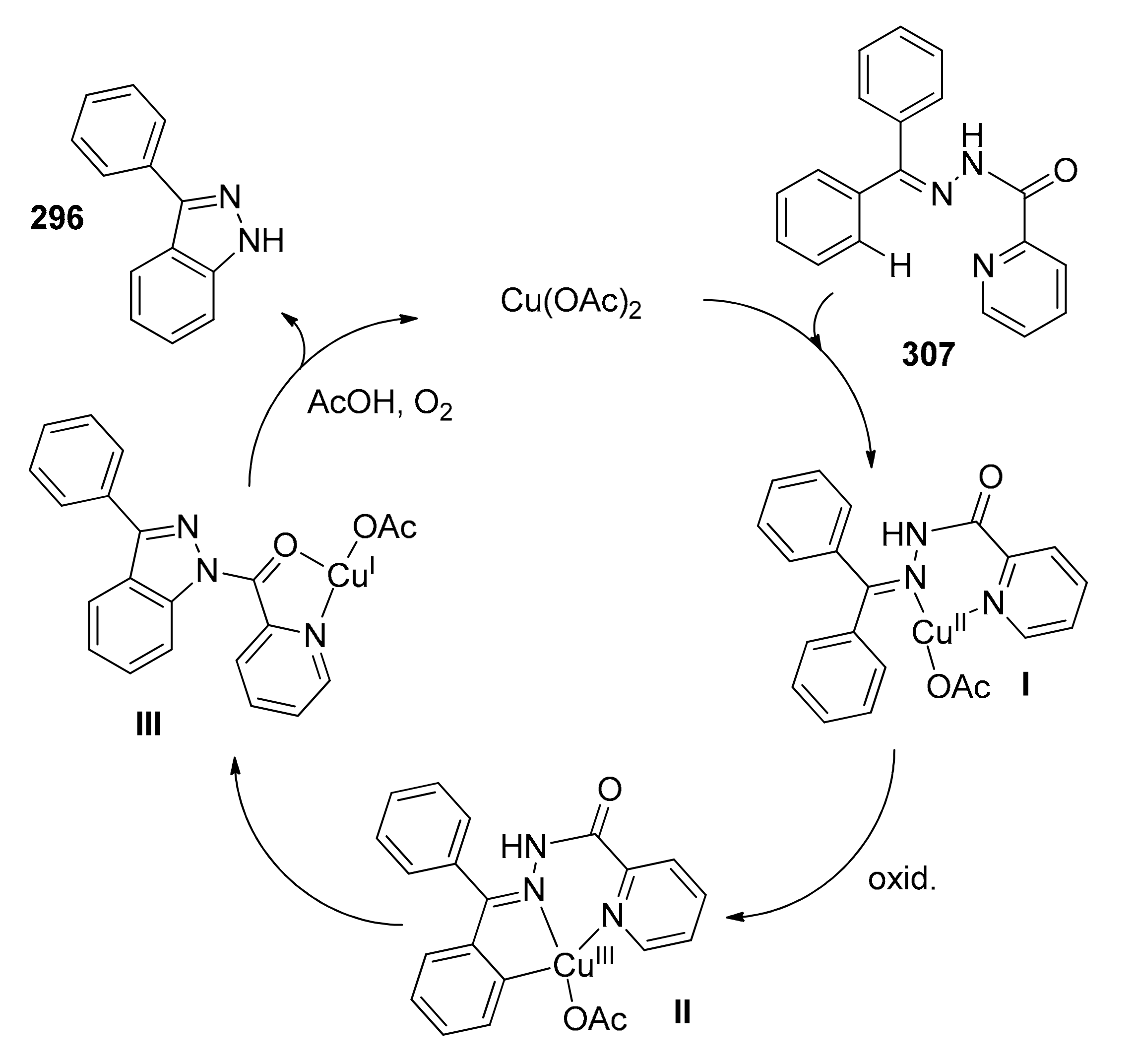
Control experiments without copper(II) acetate led to no product formation, while in the absence of KOCN only a 20% yield was reached. Use of radical scavengers (TEMPO) slightly decreased the yield, indicating that a radical pathway may contribute to the process. The catalytic cycle the authors propose commences with coordination of copper in a bidentate manner on the hydrazone to afford I (Scheme 26). After successive C–H activation (II) and reductive elimination, Cu(I) intermediate (III) hydrolyzes to produce the desired benzofuran and copper(II) acetate after air oxidation.
7. Conclusions and Outlook
The thorough investigation of catalytic systems employing low-cost, earth-abundant, first-row transition metals has allowed for the achievement of unprecedented advances in sustainable catalysis. After several initial scattered reports, the field of directing group-assisted catalysis has now matured, encompassing contemporary prerequisites to advance modern chemistry, accomplishing sustainability and a low environmental footprint for the production of high-value fine chemicals, in demand by chemical and pharmaceutical industries. traceless directing groups (TDG) are a step towards ideal C–H functionalization reactions, especially under sustainable metal catalysis; however, there are still many challenges ahead for most sustainable metals, as the use of truly traceless DGs is still underdeveloped or inhibited by the characteristics and properties of the complexes used. For manganese, carboxylate-based or hydrazone and imine-based DGs can be targeted, based on the examples discussed herein. For iron, the use of N-oxides, in recent works, is also a promising direction, albeit one that bears inherent limitations. The case of cobalt is a flourishing field in terms of established and newly emerging TDGs. However, the transformations developed so far are limited to almost exclusively annulations of N-containing substrates. Exploration of more and diverse electrophilic reaction partners, even with the currently used TDGs and substrates, could lead to new transformations. The nature of these transformations is frequently compatible with ambient conditions, a paramount feature for further design; therefore, more TDGs could be targeted, as formation and hydrolysis/cleavage of bonds are feasible under the reaction conditions. Copper is by far the most widely used sustainable metal in C–H activation, yet still, the use of TDGs is lacking. The hydrazone group seems optimal in the case of copper, as bidentate, N-based DGs perform excellently in this field, and, therefore, future advancements are expected to follow this direction. Undoubtedly, carboxylate-assisted protocols are another promising avenue. The advancements highlighted in this review are complementary to those discussed in other recent review articles dealing with sustainable C–H activation. It is evident that the field is being approached from multiple fronts, given that many points need to be covered in order to achieve the principles of Green Chemistry [126].
Author Contributions
Conceptualization, A.Z., N.V.T., and G.C.V.; resources, G.C.V.; writing—original draft preparation, A.Z., I.Z., and N.V.T.; writing—review and editing, G.C.V.; supervision, G.C.V.; project administration, G.C.V.; funding acquisition, N.V.T. and G.C.V. All authors have read and agreed to the published version of the manuscript.
Funding
This research was co-financed by Greece and the European Union (European Social Fund—ESF) through the Operational Programme «Human Resources Development, Education and Lifelong Learning 2014–2020» in the context of the project “Sustainable catalytic systems in Organic Synthesis” (MIS: 5047938).
Data Availability Statement
Data sharing not applicable. No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Jenck, J.F.; Agterberg, F.; Droescher, M.J. Products and Processes for a Sustainable Chemical Industry: A Review of Achievements and Prospects. Green Chem. 2004, 6, 544–556. [Google Scholar] [CrossRef]
- Poliakoff, M.; Fitzpatrick, J.M.; Farren, T.R.; Anastas, P.T. Green Chemistry: Science and Politics of Change. Science 2002, 297, 807–810. [Google Scholar] [CrossRef] [Green Version]
- Armor, J.N. A History of Industrial Catalysis. Catal. Today 2011, 163, 3–9. [Google Scholar] [CrossRef]
- Anastas, P.T.; Kirchhoff, M.M. Origins, Current Status, and Future Challenges of Green Chemistry. Acc. Chem. Res. 2002, 35, 686–694. [Google Scholar] [CrossRef]
- Anastas, P.; Eghbali, N. Green Chemistry: Principles and Practice. Chem. Soc. Rev. 2010, 39, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Anastas, P.T.; Zimmerman, J.B. Design through the 12 Principles of Green Engineering. Environ. Sci. Technol. 2003, 37, 94–101. [Google Scholar] [CrossRef] [Green Version]
- Turner, J.A. A Realizable Renewable Energy Future. Science 1999, 285, 687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Styring, S. Artificial Photosynthesis for Solar Fuels. Faraday Discuss. 2012, 155, 357–376. [Google Scholar] [CrossRef] [PubMed]
- Saeedmanesh, A.; Mac Kinnon, M.A.; Brouwer, J. Hydrogen Is Essential for Sustainability. Curr. Opin. Electrochem. 2018, 12, 166–181. [Google Scholar] [CrossRef]
- Frontana-Uribe, B.A.; Little, R.D.; Ibanez, J.G.; Palma, A.; Vasquez-Medrano, R. Organic Electrosynthesis: A Promising Green Methodology in Organic Chemistry. Green Chem. 2010, 12, 2099–2119. [Google Scholar] [CrossRef]
- Lewis, N.S.; Nocera, D.G. Powering the Planet: Chemical Challenges in Solar Energy Utilization. Proc. Natl. Acad. Sci. USA 2006, 103, 15729. [Google Scholar] [CrossRef] [Green Version]
- Behr, A.; Neubert, P. Applied Homogeneous Catalysis; John Wiley & Sons: Weinheim, Germany, 2012; ISBN 3-527-32641-3. [Google Scholar]
- Lersch, M.; Tilset, M. Mechanistic Aspects of C−H Activation by Pt Complexes. Chem. Rev. 2005, 105, 2471–2526. [Google Scholar] [CrossRef] [PubMed]
- Mkhalid, I.A.I.; Barnard, J.H.; Marder, T.B.; Murphy, J.M.; Hartwig, J.F. C-H Activation for the Construction of C-B Bonds. Chem. Rev. 2010, 110, 890–931. [Google Scholar] [CrossRef]
- Satoh, T.; Miura, M. Oxidative Coupling of Aromatic Substrates with Alkynes and Alkenes under Rhodium Catalysis. Chem. Eur. J. 2010, 16, 11212–11222. [Google Scholar] [CrossRef] [Green Version]
- Lyons, T.W.; Sanford, M.S. Palladium-Catalyzed Ligand-Directed C-H Functionalization Reactions. Chem. Rev. 2010, 110, 1147–1169. [Google Scholar] [CrossRef] [Green Version]
- Kuninobu, Y.; Takai, K. Organic Reactions Catalyzed by Rhenium Carbonyl Complexes. Chem. Rev. 2011, 111, 1938–1953. [Google Scholar] [CrossRef]
- Arockiam, P.B.; Bruneau, C.; Dixneuf, P.H. Ruthenium(II)-Catalyzed C-H Bond Activation and Functionalization. Chem. Rev. 2012, 112, 5879–5918. [Google Scholar] [CrossRef]
- Rao, Y.; Shan, G.; Yang, X. Some Recent Advances in Transition-Metal-Catalyzed Ortho SP2 C-H Functionalization Using Ru, Rh, and Pd. Sci. China Chem. 2014, 57, 930–944. [Google Scholar] [CrossRef]
- Motevalli, S.; Sokeirik, Y.; Ghanem, A. Rhodium-Catalysed Enantioselective C-H Functionalization in Asymmetric Synthesis. Eur. J. Org. Chem. 2016, 2016, 1459–1475. [Google Scholar] [CrossRef]
- Vougioukalakis, G.C.; Grubbs, R.H. Ruthenium-Based Heterocyclic Carbene-Coordinated Olefin Metathesis Catalysts. Chem. Rev. 2010, 110, 1746–1787. [Google Scholar] [CrossRef]
- Bullock, R.M. Catalysis without Precious Metals; Wiley: Weinheim, Germany, 2011; ISBN 978-3-527-63240-4. [Google Scholar]
- Clavier, H.; Grela, K.; Kirschning, A.; Mauduit, M.; Nolan, S.P. Sustainable Concepts in Olefin Metathesis. Angew. Chem. Int. Ed. 2007, 46, 6786–6801. [Google Scholar] [CrossRef] [PubMed]
- Vougioukalakis, G.C. Removing Ruthenium Residues from Olefin Metathesis Reaction Products. Chem. Eur. J. 2012, 18, 8868–8880. [Google Scholar] [CrossRef] [PubMed]
- Newhouse, T.; Baran, P.S.; Hoffmann, R.W. The Economies of Synthesis. Chem. Soc. Rev. 2009, 38, 3010–3021. [Google Scholar] [CrossRef]
- Gaich, T.; Baran, P.S. Aiming for the Ideal Synthesis. J. Org. Chem. 2010, 75, 4657–4673. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Cue, B.W. Green Techniques for Organic Synthesis and Medicinal Chemistry, 2nd ed.; Wiley: Hoboken, NJ, USA, 2018; ISBN 978-1-119-28817-6. [Google Scholar]
- Caro-Diaz, E.J.E.; Urbano, M.; Buzard, D.J.; Jones, R.M. C–H Activation Reactions as Useful Tools for Medicinal Chemists. Bioorg. Med. Chem. Lett. 2016, 26, 5378–5383. [Google Scholar] [CrossRef]
- Wu, G.; Zhao, T.; Kang, D.; Zhang, J.; Song, Y.; Namasivayam, V.; Kongsted, J.; Pannecouque, C.; De Clercq, E.; Poongavanam, V.; et al. Overview of Recent Strategic Advances in Medicinal Chemistry. J. Med. Chem. 2019, 62, 9375–9414. [Google Scholar] [CrossRef]
- Abrams, D.J.; Provencher, P.A.; Sorensen, E.J. Recent Applications of C–H Functionalization in Complex Natural Product Synthesis. Chem. Soc. Rev. 2018, 47, 8925–8967. [Google Scholar] [CrossRef]
- Choi, J.; Wang, D.Y.; Kundu, S.; Choliy, Y.; Emge, T.J.; Krogh-Jespersen, K.; Goldman, A.S. Net Oxidative Addition of C(sp3)-F Bonds to Iridium via Initial C-H Bond Activation. Science 2011, 332, 1545–1548. [Google Scholar] [CrossRef]
- Wu, X.F. Transition Metal.-Catalyzed Heterocycle Synthesis via C-H Activation; Wiley: Weinheim, Germany, 2016; ISBN 978-3-527-33888-7. [Google Scholar]
- Prendergast, A.M.; McGlacken, G.P. Transition Metal Mediated C–H Activation of 2-Pyrones, 2-Pyridones, 2-Coumarins and 2-Quinolones. Eur. J. Org. Chem. 2018, 2018, 6068–6082. [Google Scholar] [CrossRef]
- Cano, R.; Mackey, K.; McGlacken, G.P. Recent Advances in Manganese-Catalysed C-H Activation: Scope and Mechanism. Catal. Sci. Technol. 2018, 8, 1251–1266. [Google Scholar] [CrossRef]
- Vásquez-Céspedes, S.; Wang, X.; Glorius, F. Plausible Rh(V) Intermediates in Catalytic C-H Activation Reactions. ACS Catal. 2018, 8, 242–257. [Google Scholar] [CrossRef]
- Gensch, T.; James, M.J.; Dalton, T.; Glorius, F. Increasing Catalyst Efficiency in C−H Activation Catalysis. Angew. Chem. Int. Ed. 2018, 57, 2296–2306. [Google Scholar] [CrossRef] [Green Version]
- Nesterov, D.S.; Nesterova, O.V.; Pombeiro, A.J.L. Homo- and Heterometallic Polynuclear Transition Metal Catalysts for Alkane C–H Bonds Oxidative Functionalization: Recent Advances. Coord. Chem. Rev. 2018, 355, 199–222. [Google Scholar] [CrossRef]
- Sauermann, N.; Meyer, T.H.; Qiu, Y.; Ackermann, L. Electrocatalytic C–H Activation. ACS Catal. 2018, 8, 7086–7103. [Google Scholar] [CrossRef]
- Gandeepan, P.; Müller, T.; Zell, D.; Cera, G.; Warratz, S.; Ackermann, L. 3d Transition Metals for C-H Activation. Chem. Rev. 2019, 119, 2192–2452. [Google Scholar] [CrossRef] [PubMed]
- Tzouras, N.V.; Stamatopoulos, I.K.; Papastavrou, A.T.; Liori, A.A.; Vougioukalakis, G.C. Sustainable Metal Catalysis in CH Activation. Coord. Chem. Rev. 2017, 343, 25–138. [Google Scholar] [CrossRef]
- Trowbridge, A.; Walton, S.M.; Gaunt, M.J. New Strategies for the Transition-Metal Catalyzed Synthesis of Aliphatic Amines. Chem. Rev. 2020, 120, 2613–2692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Q.; Poisson, T.; Pannecoucke, X.; Besset, T. The Transient Directing Group Strategy: A New Trend in Transition-Metal-Catalyzed C–H Bond Functionalization. Synthesis 2017, 49, 4808–4826. [Google Scholar]
- Chen, Z.; Wang, B.; Zhang, J.; Yu, W.; Liu, Z.; Zhang, Y. Transition Metal-Catalyzed C–H Bond Functionalizations by the Use of Diverse Directing Groups. Org. Chem. Front. 2015, 2, 1107–1295. [Google Scholar] [CrossRef]
- Ujwaldev, S.M.; Harry, N.A.; Divakar, M.A.; Anilkumar, G. Cobalt-Catalyzed C–H Activation: Recent Progress in Heterocyclic Chemistry. Catal. Sci. Technol. 2018, 8, 5983–6018. [Google Scholar] [CrossRef]
- Prakash, S.; Kuppusamy, R.; Cheng, C.-H. Cobalt-Catalyzed Annulation Reactions via C−H Bond Activation. ChemCatChem 2018, 10, 683–705. [Google Scholar] [CrossRef]
- Liu, M.; Niu, J.-L.; Yang, D.; Song, M.-P. Development of a Traceless Directing Group: Cp*-Free Cobalt-Catalyzed C–H Activation/Annulations to Access Isoquinolinones. J. Org. Chem. 2020, 85, 4067–4078. [Google Scholar] [CrossRef]
- Kommagalla, Y.; Chatani, N. Cobalt(II)-Catalyzed CH Functionalization Using an N,N′-Bidentate Directing Group. Coord. Chem. Rev. 2017, 350, 117–135. [Google Scholar] [CrossRef]
- Dutta, C.; Choudhury, J. C–H Activation-Annulation on the N-Heterocyclic Carbene Platform. RSC Adv. 2018, 8, 27881–27891. [Google Scholar] [CrossRef] [Green Version]
- Martínez de Salinas, S.; Sanjosé-Orduna, J.; Odena, C.; Barranco, S.; Benet-Buchholz, J.; Pérez-Temprano, M.H. Weakly Coordinated Cobaltacycles: Trapping Catalytically Competent Intermediates in Cp*CoIII Catalysis. Angew. Chem. Int. Ed. 2020, 59, 6239–6243. [Google Scholar] [CrossRef] [PubMed]
- Sanjosé-Orduna, J.; Benet-Buchholz, J.; Pérez-Temprano, M.H. Unravelling Molecular Aspects of the Migratory Insertion Step in Cp*CoIII Metallacyclic Systems. Inorg. Chem. 2019, 58, 10569–10577. [Google Scholar] [CrossRef]
- Sanjosé-Orduna, J.; Mudarra, Á.L.; Martínez de Salinas, S.; Pérez-Temprano, M.H. Sustainable Knowledge-Driven Approaches in Transition-Metal-Catalyzed Transformations. ChemSusChem 2019, 12, 2882–2897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanjosé-Orduna, J.; Gallego, D.; Garcia-Roca, A.; Martin, E.; Benet-Buchholz, J.; Pérez-Temprano, M.H. Capturing Elusive Cobaltacycle Intermediates: A Real-Time Snapshot of the Cp*CoIII-Catalyzed Oxidative Alkyne Annulation. Angew. Chem. Int. Ed. 2017, 56, 12137–12141. [Google Scholar] [CrossRef] [PubMed]
- Sambiagio, C.; Schönbauer, D.; Blieck, R.; Dao-Huy, T.; Pototschnig, G.; Schaaf, P.; Wiesinger, T.; Zia, M.F.; Wencel-Delord, J.; Besset, T.; et al. A Comprehensive Overview of Directing Groups Applied in Metal-Catalysed C–H Functionalisation Chemistry. Chem. Soc. Rev. 2018, 47, 6603–6743. [Google Scholar] [CrossRef] [Green Version]
- Rasheed, O.K.; Sun, B. Advances in Development of C–H Activation/Functionalization Using a Catalytic Directing Group. ChemistrySelect 2018, 3, 5689–5708. [Google Scholar] [CrossRef]
- Zhang, F.; Spring, D.R. Arene C–H Functionalisation Using a Removable/Modifiable or a Traceless Directing Group Strategy. Chem. Soc. Rev. 2014, 43, 6906–6919. [Google Scholar] [CrossRef]
- Wei, D.; Zhu, X.; Niu, J.-L.; Song, M.-P. High-Valent-Cobalt-Catalyzed C−H Functionalization Based on Concerted Metalation–Deprotonation and Single-Electron-Transfer Mechanisms. ChemCatChem 2016, 8, 1242–1263. [Google Scholar] [CrossRef]
- Mahmudov, K.T.; Gurbanov, A.V.; Guseinov, F.I.; Guedes da Silva, M.F.C. Noncovalent Interactions in Metal Complex Catalysis. Coord. Chem. Rev. 2019, 387, 32–46. [Google Scholar] [CrossRef]
- Rej, S.; Chatani, N. Rhodium-Catalyzed C(sp2)- or C(sp3)−H Bond Functionalization Assisted by Removable Directing Groups. Angew. Chem. Int. Ed. 2019, 58, 8304–8329. [Google Scholar] [CrossRef]
- Gandeepan, P.; Ackermann, L. Transient Directing Groups for Transformative C–H Activation by Synergistic Metal Catalysis. Chem 2018, 4, 199–222. [Google Scholar] [CrossRef] [Green Version]
- De Sarkar, S.; Liu, W.; Kozhushkov, S.I.; Ackermann, L. Weakly Coordinating Directing Groups for Ruthenium(II)-Catalyzed C-H Activation. Adv. Synth. Catal. 2014, 356, 1461–1479. [Google Scholar] [CrossRef]
- Ellman, J.A.; Ackermann, L.; Shi, B.-F. The Breadth and Depth of C–H Functionalization. J. Org. Chem. 2019, 84, 12701–12704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ackermann, L. Robust Ruthenium(II)-Catalyzed C-H Arylations: Carboxylate Assistance for the Efficient Synthesis of Angiotensin-II-Receptor Blockers. Org. Process. Res. Dev. 2015, 19, 260–269. [Google Scholar] [CrossRef]
- Ma, W.; Gandeepan, P.; Li, J.; Ackermann, L. Recent Advances in Positional-Selective Alkenylations: Removable Guidance for Twofold C-H Activation. Org. Chem. Front. 2017, 4, 1435–1467. [Google Scholar] [CrossRef]
- Ma, W.; Kaplaneris, N.; Fang, X.; Gu, L.; Mei, R.; Ackermann, L. Chelation-Assisted Transition Metal-Catalysed C–H Chalcogenylations. Org. Chem. Front. 2020, 7, 1022–1060. [Google Scholar] [CrossRef]
- Rej, S.; Ano, Y.; Chatani, N. Bidentate Directing Groups: An Efficient Tool in C–H Bond Functionalization Chemistry for the Expedient Construction of C–C Bonds. Chem. Rev. 2020, 120, 1788–1887. [Google Scholar] [CrossRef]
- Pichette Drapeau, M.; Gooßen, L.J. Carboxylic Acids as Directing Groups for C−H Bond Functionalization. Chem. A Eur. J. 2016, 22, 18654–18677. [Google Scholar] [CrossRef] [PubMed]
- Font, M.; Quibell, J.M.; Perry, G.J.P.; Larrosa, I. The Use of Carboxylic Acids as Traceless Directing Groups for Regioselective C–H Bond Functionalisation. Chem. Commun. 2017, 53, 5584–5597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinaka, A.; Vougioukalakis, G.C. Using Sustainable Metals to Carry out “Green” Transformations: Fe- and Cu-Catalyzed CO2 Monetization. Coord. Chem. Rev. 2015, 288, 69–97. [Google Scholar] [CrossRef]
- Liori, A.A.; Stamatopoulos, I.K.; Papastavrou, A.T.; Pinaka, A.; Vougioukalakis, G.C. A Sustainable, User-Friendly Protocol for the Pd-Free Sonogashira Coupling Reaction. Eur. J. Org. Chem. 2018, 2018, 6134–6139. [Google Scholar] [CrossRef]
- Papastavrou, A.T.; Pauze, M.; Gómez-Bengoa, E.; Vougioukalakis, G.C. Unprecedented Multicomponent Organocatalytic Synthesis of Propargylic Esters via CO2 Activation. ChemCatChem 2019, 11, 5379–5386. [Google Scholar] [CrossRef]
- Voutyritsa, E.; Triandafillidi, I.; Tzouras, N.V.; Nikitas, N.F.; Pefkianakis, E.K.; Vougioukalakis, G.C.; Kokotos, C.G. Photocatalytic Atom Transfer Radical Addition to Olefins Utilizing Novel Photocatalysts. Molecules 2019, 24, 1644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milenković, M.R.; Papastavrou, A.T.; Radanović, D.; Pevec, A.; Jagličić, Z.; Zlatar, M.; Gruden, M.; Vougioukalakis, G.C.; Turel, I.; Anđelković, K.; et al. Highly-Efficient N-Arylation of Imidazole Catalyzed by Cu(II) Complexes with Quaternary Ammonium-Functionalized 2-Acetylpyridine Acylhydrazone. Polyhedron 2019, 165, 22–30. [Google Scholar] [CrossRef]
- Tzouras, N.V.; Neofotistos, S.P.; Vougioukalakis, G.C. Zn-Catalyzed Multicomponent KA2 Coupling: One-Pot Assembly of Propargylamines Bearing Tetrasubstituted Carbon Centers. ACS Omega 2019, 4, 10279–10292. [Google Scholar] [CrossRef] [Green Version]
- Zorba, L.P.; Vougioukalakis, G.C. The Ketone-Amine-Alkyne (KA2) Coupling Reaction: Transition Metal-Catalyzed Synthesis of Quaternary Propargylamines. Coord. Chem. Rev. 2021, 429, 213603. [Google Scholar] [CrossRef]
- Adejumo, T.T.; Tzouras, N.V.; Zorba, L.P.; Radanović, D.; Pevec, A.; Grubišić, S.; Mitić, D.; Anđelković, K.K.; Vougioukalakis, G.C.; Čobeljić, B.; et al. Synthesis, Characterization, Catalytic Activity, and DFT Calculations of Zn(II) Hydrazone Complexes. Molecules 2020, 25, 4043. [Google Scholar] [CrossRef]
- Neofotistos, S.P.; Tzouras, N.V.; Pauze, M.; Gómez-Bengoa, E.; Vougioukalakis, G.C. Manganese-Catalyzed Multicomponent Synthesis of Tetrasubstituted Propargylamines: System Development and Theoretical Study. Adv. Synth. Catal. 2020, 362, 3872–3885. [Google Scholar] [CrossRef]
- Kuninobu, Y.; Nishina, Y.; Takeuchi, T.; Takai, K. Manganese-Catalyzed Insertion of Aldehydes into a C-H Bond. Angew. Chem. Int. Ed. 2007, 46, 6518–6520. [Google Scholar] [CrossRef]
- Kuninobu, Y.; Fujii, Y.; Matsuki, T.; Nishina, Y.; Takai, K. Rhenium-Catalyzed Insertion of Nonpolar and Polar Unsaturated Molecules into an Olefinic C−H Bond. Org. Lett. 2009, 11, 2711–2714. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Jiang, Z.; Fukuzumi, S.; Nam, W.; Wang, B. Artificial Nonheme Iron and Manganese Oxygenases for Enantioselective Olefin Epoxidation and Alkane Hydroxylation Reactions. Coord. Chem. Rev. 2020, 421, 213443. [Google Scholar] [CrossRef]
- Chandra, P.; Ghosh, T.; Choudhary, N.; Mohammad, A.; Mobin, S.M. Recent Advancement in Oxidation or Acceptorless Dehydrogenation of Alcohols to Valorised Products Using Manganese Based Catalysts. Coord. Chem. Rev. 2020, 411, 213241. [Google Scholar] [CrossRef]
- Liu, W.; Ackermann, L. Manganese-Catalyzed C–H Activation. ACS Catal. 2016, 6, 3743–3752. [Google Scholar] [CrossRef]
- Wang, C. Manganese-Mediated C–C Bond Formation via C–H Activation: From Stoichiometry to Catalysis. Synlett 2013, 24, 1606–1613. [Google Scholar] [CrossRef]
- Yang, X.; Jin, X.; Wang, C. Manganese-Catalyzed Ortho-C−H Alkenylation of Aromatic N−H Imidates with Alkynes: Versatile Access to Mono-Alkenylated Aromatic Nitriles. Adv. Synth. Catal. 2016, 358, 2436–2442. [Google Scholar] [CrossRef]
- Lu, Q.; Greßies, S.; Cembellín, S.; Klauck, F.J.R.; Daniliuc, C.G.; Glorius, F. Redox-Neutral Manganese(I)-Catalyzed C−H Activation: Traceless Directing Group Enabled Regioselective Annulation. Angew. Chem. Int. Ed. 2017, 56, 12778–12782. [Google Scholar] [CrossRef]
- Liang, Y.-F.; Steinbock, R.; Münch, A.; Stalke, D.; Ackermann, L. Manganese-Catalyzed Carbonylative Annulations for Redox-Neutral Late-Stage Diversification. Angew. Chem. Int. Ed. 2018, 57, 5384–5388. [Google Scholar] [CrossRef]
- Haynes, W.M. CRC Handbook of Chemistry and Physics, 97th ed.; CRC Press: Boca Raton, FL, USA, 2016; ISBN 978-1-4987-5429-3. [Google Scholar]
- Pearson, R.G. Hard and Soft Acids and Bases. J. Am. Chem. Soc. 1963, 85, 3533–3539. [Google Scholar] [CrossRef]
- Kroneck, P.M.H.; Torres, M.E.S. Metal Ions in Life Sciences. In Sustaining Life on Planet. Earth: Metalloenzymes Mastering Dioxygen and Other Chewy Gases; Springer International Publishing: Cham, Switzerland, 2015; ISBN 978-3-319-12415-5. [Google Scholar]
- Battistella, B.; Ray, K. O2 and H2O2 Activations at Dinuclear Mn and Fe Active Sites. Coord. Chem. Rev. 2020, 408, 213176. [Google Scholar] [CrossRef]
- Shang, R.; Ilies, L.; Nakamura, E. Iron-Catalyzed C–H Bond Activation. Chem. Rev. 2017, 117, 9086–9139. [Google Scholar] [CrossRef]
- Viton, F.; Bernardinelli, G.; Kündig, E.P. Iron and Ruthenium Lewis Acid Catalyzed Asymmetric 1,3-Dipolar Cycloaddition Reactions between Nitrones and Enals. J. Am. Chem. Soc. 2002, 124, 4968–4969. [Google Scholar] [CrossRef] [PubMed]
- Bădoiu, A.; Bernardinelli, G.; Mareda, J.; Kündig, E.P.; Viton, F. Iron- and Ruthenium-Lewis Acid Catalyzed Asymmetric 1,3-Dipolar Cycloaddition Reactions between Enals and Diaryl Nitrones. Chem. Asian J. 2008, 3, 1298–1311. [Google Scholar] [CrossRef] [PubMed]
- Zhong, M.; Sun, S.; Cheng, J.; Shao, Y. Iron-Catalyzed Cyclization of Nitrones with Geminal-Substituted Vinyl Acetates: A Direct [4 + 2] Assembly Strategy Leading to 2,4-Disubstituted Quinolines. J. Org. Chem. 2016, 81, 10825–10831. [Google Scholar] [CrossRef]
- Ferlin, F.; Zangarelli, A.; Lilli, S.; Santoro, S.; Vaccaro, L. Waste-Minimized Synthesis of C2 Functionalized Quinolines Exploiting Iron-Catalysed C–H Activation. Green Chem. 2021, 23, 490–495. [Google Scholar] [CrossRef]
- Moselage, M.; Li, J.; Ackermann, L. Cobalt-Catalyzed C–H Activation. ACS Catal. 2016, 6, 498–525. [Google Scholar] [CrossRef]
- Mei, R.; Dhawa, U.; Samanta, R.C.; Ma, W.; Wencel-Delord, J.; Ackermann, L. Cobalt-Catalyzed Oxidative C−H Activation: Strategies and Concepts. ChemSusChem 2020, 13, 3306–3356. [Google Scholar] [CrossRef]
- Murahashi, S. Synthesis of Phthalimidines from Schiff Bases and Carbon Monoxide. J. Am. Chem. Soc. 1955, 77, 6403–6404. [Google Scholar] [CrossRef]
- Lukasevics, L.; Grigorjeva, L. Cobalt-Catalyzed Carbonylation of the C–H Bond. Org. Biomol. Chem. 2020, 18, 7460–7466. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Meng, G.; Nolan, S.P.; Szostak, M. N-Heterocyclic Carbene Complexes in C–H Activation Reactions. Chem. Rev. 2020, 120, 1981–2048. [Google Scholar] [CrossRef]
- Gao, K.; Yoshikai, N. Low-Valent Cobalt Catalysis: New Opportunities for C–H Functionalization. Acc. Chem. Res. 2014, 47, 1208–1219. [Google Scholar] [CrossRef]
- Rani, G.; Luxami, V.; Paul, K. Traceless Directing Groups: A Novel Strategy in Regiodivergent C–H Functionalization. Chem. Commun. 2020, 56, 12479–12521. [Google Scholar] [CrossRef] [PubMed]
- Ling, F.; Ai, C.; Lv, Y.; Zhong, W. Traceless Directing Group Assisted Cobalt-Catalyzed C−H Carbonylation of Benzylamines. Adv. Synth. Catal. 2017, 359, 3707–3712. [Google Scholar] [CrossRef]
- Ling, F.; Zhang, C.; Ai, C.; Lv, Y.; Zhong, W. Metal-Oxidant-Free Cobalt-Catalyzed C(Sp2)–H Carbonylation of Ortho-Arylanilines: An Approach toward Free (NH)-Phenanthridinones. J. Org. Chem. 2018, 83, 5698–5706. [Google Scholar] [CrossRef] [PubMed]
- Ying, J.; Fu, L.-Y.; Zhong, G.; Wu, X.-F. Cobalt-Catalyzed Direct Carbonylative Synthesis of Free (NH)-Benzo[Cd]Indol-2(1H)-Ones from Naphthylamides. Org. Lett. 2019, 21, 5694–5698. [Google Scholar] [CrossRef] [PubMed]
- Lukasevics, L.; Cizikovs, A.; Grigorjeva, L. Synthesis of 3-Hydroxymethyl Isoindolinones via Cobalt-Catalyzed C(Sp2)–H Carbonylation of Phenylglycinol Derivatives. Org. Lett. 2020, 22, 2720–2723. [Google Scholar] [CrossRef]
- Zhou, S.; Wang, M.; Wang, L.; Chen, K.; Wang, J.; Song, C.; Zhu, J. Bidentate Directing-Enabled, Traceless Heterocycle Synthesis: Cobalt-Catalyzed Access to Isoquinolines. Org. Lett. 2016, 18, 5632–5635. [Google Scholar] [CrossRef]
- Kuai, C.; Wang, L.; Li, B.; Yang, Z.; Cui, X. Cobalt-Catalyzed Selective Synthesis of Isoquinolines Using Picolinamide as a Traceless Directing Group. Org. Lett. 2017, 19, 2102–2105. [Google Scholar] [CrossRef]
- Li, X.-C.; Du, C.; Zhang, H.; Niu, J.-L.; Song, M.-P. Cp*-Free Cobalt-Catalyzed C–H Activation/Annulations by Traceless N,O-Bidentate Directing Group: Access to Isoquinolines. Org. Lett. 2019, 21, 2863–2866. [Google Scholar] [CrossRef]
- Dey, A.; Volla, C.M.R. Traceless Bidentate Directing Group Assisted Cobalt-Catalyzed sp2-C–H Activation and [4 + 2]-Annulation Reaction with 1,3-Diynes. Org. Lett. 2020, 22, 7480–7485. [Google Scholar] [CrossRef] [PubMed]
- Barsu, N.; Sen, M.; Premkumar, J.R.; Sundararaju, B. Cobalt(III) Catalyzed C-8 Selective C–H and C–O Coupling of Quinoline N-Oxide with Internal Alkynes via C–H Activation and Oxygen Atom Transfer. Chem. Commun. 2016, 52, 1338–1341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.; Wu, Q.; Liu, D.; Yu, L.; Tan, Z.; Zhu, G. Synthesis of 1-Naphthols via Cp*Co(III)-Catalyzed C–H Activation and Cyclization of Sulfoxonium Ylides with Alkynes. Org. Chem. Front. 2019, 6, 3868–3873. [Google Scholar] [CrossRef]
- Sigel, A.; Sigel, H.; Sigel, R.K.O. Metal Ions in Life Sciences. In Nickel and Its Surprising Impact in Nature; Wiley: Chichester, UK, 2007; ISBN 978-0-470-02812-4. [Google Scholar]
- Brazzolotto, D.; Gennari, M.; Queyriaux, N.; Simmons, T.R.; Pécaut, J.; Demeshko, S.; Meyer, F.; Orio, M.; Artero, V.; Duboc, C. Nickel-Centred Proton Reduction Catalysis in a Model of [NiFe] Hydrogenase. Nat. Chem. 2016, 8, 1054–1060. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, J.; Muto, K.; Itami, K. Nickel-Catalyzed Aromatic C–H Functionalization. Top. Curr. Chem. 2016, 374, 55. [Google Scholar] [CrossRef] [PubMed]
- Harry, N.A.; Saranya, S.; Ujwaldev, S.M.; Anilkumar, G. Recent Advances and Prospects in Nickel-Catalyzed C–H Activation. Catal. Sci. Technol. 2019, 9, 1726–1743. [Google Scholar] [CrossRef]
- Khake, S.M.; Chatani, N. Nickel-Catalyzed C−H Functionalization Using A Non-Directed Strategy. Chem 2020, 6, 1056–1081. [Google Scholar] [CrossRef]
- Khake, S.M.; Chatani, N. Chelation-Assisted Nickel-Catalyzed C−H Functionalizations. Trends Chem. 2019, 1, 524–539. [Google Scholar] [CrossRef]
- Zhu, J.; Chen, Y.; Lin, F.; Wang, B.; Chen, Z.; Liu, L. NiSO4-Catalyzed C–H Activation/C–S Cross-Coupling of 1,2,3-Triazole N-Oxides with Thiols. Org. Biomol. Chem. 2015, 13, 3711–3720. [Google Scholar] [CrossRef]
- Li, Z.-K.; Jia, X.-S.; Yin, L. Recent Advances in Copper(II)-Mediated or -Catalyzed C–H Functionalization. Synthesis 2018, 50, 4165–4188. [Google Scholar] [CrossRef]
- Tonis, E.; Stein, F.; Stamatopoulos, I.K.; Stubbe, J.; Zarkadoulas, A.; Sarkar, B.; Vougioukalakis, G.C. A Palladium-Free Sonogashira Coupling Protocol Employing an In Situ Prepared Copper/Chelating 1,2,3-Triazolylidene System. Synlett 2020. [Google Scholar] [CrossRef]
- DiMucci, I.M.; Lukens, J.T.; Chatterjee, S.; Carsch, K.M.; Titus, C.J.; Lee, S.J.; Nordlund, D.; Betley, T.A.; MacMillan, S.N.; Lancaster, K.M. The Myth of d8 Copper(III). J. Am. Chem. Soc. 2019, 141, 18508–18520. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Liu, H.; Liu, S.; Lu, Z.; Lu, C.; Leng, X.; Lan, Y.; Shen, Q. C(sp3)-CF3 Reductive Elimination from a Five-Coordinate Neutral Copper(III) Complex. J. Am. Chem. Soc. 2020, 142, 9785–9791. [Google Scholar] [CrossRef] [PubMed]
- Bhadra, S.; Dzik, W.I.; Gooßen, L.J. Synthesis of Aryl Ethers from Benzoates through Carboxylate-Directed C-H-Activating Alkoxylation with Concomitant Protodecarboxylation. Angew. Chem. Int. Ed. 2013, 52, 2959–2962. [Google Scholar] [CrossRef]
- Yu, S.; Lv, N.; Liu, Z.; Zhang, Y. Cu(II)-Mediated C−C/C−O Bond Formation via C−H/C−C Bond Cleavage: Access to Benzofurans Using Amide as a Traceless Directing Group. Adv. Synth. Catal. 2020, 362, 118–125. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; Fan, Q.; Zhao, Y.; Ding, C. Copper-Promoted Oxidative Intramolecular C–H Amination of Hydrazones to Synthesize 1H-Indazoles and 1H-Pyrazoles Using a Cleavable Directing Group. Eur. J. Org. Chem. 2019, 2019, 5801–5806. [Google Scholar] [CrossRef]
- Dalton, T.; Faber, T.; Glorius, F. C–H Activation: Toward Sustainability and Applications. ACS Cent. Sci. 2021, 7, 245–261. [Google Scholar] [CrossRef]
Scheme 1.
Mn-catalyzed, C–H alkenylation of aromatic N–H imidates with alkynes.

Scheme 2.
Proposed catalytic cycle.

Scheme 3.
Mn(I)-catalyzed, C–H activation of benzimines for regioselective coupling with alkynes bearing a TDG. Synthesis of moxaverine and proposed catalytic cycle.
Scheme 3.
Mn(I)-catalyzed, C–H activation of benzimines for regioselective coupling with alkynes bearing a TDG. Synthesis of moxaverine and proposed catalytic cycle.

Scheme 4.
Mn-catalyzed carbonylative alkyne annulation strategy towards synthesis of pyrido[1,2-a]pyrimidin-4-ones. Late-stage diversification of structurally challenging molecules.
Scheme 4.
Mn-catalyzed carbonylative alkyne annulation strategy towards synthesis of pyrido[1,2-a]pyrimidin-4-ones. Late-stage diversification of structurally challenging molecules.

Scheme 5.
Proposed catalytic cycle.

Scheme 6.
Fe-catalyzed, intermolecular [4 + 2] cyclization of arylnitrones with geminal-substituted vinyl acetates.
Scheme 6.
Fe-catalyzed, intermolecular [4 + 2] cyclization of arylnitrones with geminal-substituted vinyl acetates.

Scheme 7.
Fe-catalyzed synthesis of C2 functionalized quinolines and application in a multigram scale reaction.
Scheme 7.
Fe-catalyzed synthesis of C2 functionalized quinolines and application in a multigram scale reaction.

Scheme 8.
Co-catalyzed, picolinamide-assisted C–H carbonylation of (het)arylamines. Synthesis of valuable intermediates in the formal synthesis of (+)-garenoxacin on gram scale and proposed catalytic cycle.
Scheme 8.
Co-catalyzed, picolinamide-assisted C–H carbonylation of (het)arylamines. Synthesis of valuable intermediates in the formal synthesis of (+)-garenoxacin on gram scale and proposed catalytic cycle.

Scheme 9.
Co-catalyzed, picolinamide-assisted C–H carbonylation of ortho-arylanilines and formal synthesis of PARP inhibitor PJ-34 on a gram scale.
Scheme 9.
Co-catalyzed, picolinamide-assisted C–H carbonylation of ortho-arylanilines and formal synthesis of PARP inhibitor PJ-34 on a gram scale.

Scheme 10.
Co-catalyzed, picolinamide-assisted C–H carbonylation of naphthylamides and its application in the total synthesis of BET bromodomain inhibitors A and B.
Scheme 10.
Co-catalyzed, picolinamide-assisted C–H carbonylation of naphthylamides and its application in the total synthesis of BET bromodomain inhibitors A and B.

Scheme 11.
Co-catalyzed, picolinamide-assisted C–H carbonylation of phenylglycinol derivatives and its application on an enantiopure derivative. For the synthesis of 138, 30 mol % of [Co(dpm)2] was used, while for 139 were used 60 mol % of [Co(dpm)2] and 4 equivalents of Ag2CO3.
Scheme 11.
Co-catalyzed, picolinamide-assisted C–H carbonylation of phenylglycinol derivatives and its application on an enantiopure derivative. For the synthesis of 138, 30 mol % of [Co(dpm)2] was used, while for 139 were used 60 mol % of [Co(dpm)2] and 4 equivalents of Ag2CO3.

Scheme 12.
Co-catalyzed, 2-hydrazinylpyridine-directed C–H alkenylation with alkynes and its application on a naturally-occurring substrate.
Scheme 12.
Co-catalyzed, 2-hydrazinylpyridine-directed C–H alkenylation with alkynes and its application on a naturally-occurring substrate.

Scheme 13.
Co-catalyzed, picolinamide-assisted C–H activated synthesis of isoquinolines.

Scheme 14.
Co-catalyzed, N,O-bidentate TDG-assisted, C–H activated synthesis of isoquinolines.

Scheme 15.
Co-catalyzed, 2-aminopyridine-assisted C–H functionalization and [4 + 2] annulation of hydrazones and 1,3-diynes.
Scheme 15.
Co-catalyzed, 2-aminopyridine-assisted C–H functionalization and [4 + 2] annulation of hydrazones and 1,3-diynes.

Scheme 16.
Co-catalyzed, C(8)-H bond functionalization of quinoline with internal alkynes, using quinoline-N-oxide as a TDG.
Scheme 16.
Co-catalyzed, C(8)-H bond functionalization of quinoline with internal alkynes, using quinoline-N-oxide as a TDG.

Scheme 17.
Proposed catalytic cycle.

Scheme 18.
Co-catalyzed, C–H activation and [4 + 2] cyclization between sulfoxonium ylides and alkynes for synthesis of 1-naphthols and its application on a one-pot tandem reaction through two consecutive steps of C–H activation.
Scheme 18.
Co-catalyzed, C–H activation and [4 + 2] cyclization between sulfoxonium ylides and alkynes for synthesis of 1-naphthols and its application on a one-pot tandem reaction through two consecutive steps of C–H activation.

Scheme 19.
Co-catalyzed, C–H activation and annulation towards isoquinolinones.

Scheme 20.
Ni-catalyzed C–H thiolation of 1,2,3-triazole-oxides.

Scheme 21.
Proposed mechanism for the Ni-catalyzed C–H thiolation of 1,2,3-triazole-oxides.

Scheme 22.
Cu-catalyzed synthesis of aryl ethers from substituted potassium benzoates.

Scheme 23.
Cu-catalyzed copper-mediated cascade coupling of benzamides.

Scheme 24.
Proposed mechanism of benzamide coupling.

Scheme 25.
(a) Indazole and (b) pyrazole synthesis using hydrazones.

Scheme 26.
Proposed mechanism of indazole synthesis from hydrazones.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Zarkadoulas, A.; Zgouleta, I.; Tzouras, N.V.; Vougioukalakis, G.C. Traceless Directing Groups in Sustainable Metal-Catalyzed C–H Activation. Catalysts 2021, 11, 554. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11050554
AMA Style
Zarkadoulas A, Zgouleta I, Tzouras NV, Vougioukalakis GC. Traceless Directing Groups in Sustainable Metal-Catalyzed C–H Activation. Catalysts. 2021; 11(5):554. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11050554
Chicago/Turabian StyleZarkadoulas, Athanasios, Ioanna Zgouleta, Nikolaos V. Tzouras, and Georgios C. Vougioukalakis. 2021. "Traceless Directing Groups in Sustainable Metal-Catalyzed C–H Activation" Catalysts 11, no. 5: 554. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11050554
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.